

Abstract
Natural killer (NK) cells are innate lymphoid cells that have adopted exquisite activating and inhibitory signaling mechanisms to be tolerant to normal cells, but distinguish and eliminate tumor cells and virus-infected cells. Inhibitory signaling through killer cell immunoglobulin (Ig)-like receptors (KIRs) is critical for maintaining tolerance, so the mechanisms by which they suppress NK cell activation are of great interest. In this issue of Science Signaling, Matalon et al. advance our understanding of how KIRs disrupt activation signaling by stimulating dephosphorylation of the adaptor protein linker for activation of T cells (LAT) and PLC-γ by Src homology 2 domain–containing protein tyrosine phosphatase 1 (SHP-1), and the ubiquitylation of LAT by Cbl family E3 ubiquitin ligases.
Natural killer (NK) cells are a subset of innate lymphoid cells that provide an ideal cellular model system with which to study signal transduction crosstalk (1). They are regulated by a dynamic balance of positive and negative signals transduced from a wide range of germline-encoded activating, inhibitory, and adhesion receptors. As NK cells probe the surfaces of adjacent cells in the body, many activating receptors can be triggered by their ligands, but an array of inhibitory receptors simultaneously engage with major histocompatibility class I (MHC-I) molecules on the healthy cells to dominantly disrupt adhesion and suppress activation signaling. Whereas most healthy cells express MHC-I, tumor cells or virus-infected cells commonly reduce the cell surface abundance of MHC-I and thereby avoid detection by cytolytic T cells. In so doing, however, these abnormal MHC-I–deficient cells become prime targets for NK cell–mediated attack, because they lack inhibitory receptor ligands. Activation signaling predominates when NK cells encounter an MHC-I–deficient target cell or a target cell with an overwhelming abundance of activating receptor ligands. Activation signaling initiates strong adhesion, cytoskeletal polarization, and the targeted mobilization of lytic granules to release perforins and granzymes toward the NK immunological synapse (NKIS), which efficiently and selectively initiates apoptosis of the abnormal cell (2).
Killer cell immunoglobulin (Ig)-like receptors (KIRs) are a family of highly polymorphic inhibitory and activating receptors that bind to MHC-I and are expressed on most NK cells and some T cells in humans (1). Different inhibitory KIR family members recognize distinct subsets of the diverse allotypes of MHC-I in humans (HLA-A, HLA-B, and HLA-C), and distinct KIR family members are differentially expressed on individual NK cells in an individual to create a diverse cell repertoire with disparate KIR expression profiles and MHC-I detection capacities. Expression of a KIR that recognizes an endogenous MHC-I molecule not only tolerizes a mature NK cell to prevent indiscriminant cytolysis of normal cells, but also “educates” or “licenses” a developing NK cell to become functionally mature. Therefore, inhibitory KIRs are key regulators of human NK cell function, and both functional properties depend upon downstream inhibitory signaling pathways.
Like those on T and B cells, most activating receptors on NK cells transduce signals based upon protein tyrosine kinase (PTK)–mediated signaling, much of which is initiated through immunoreceptor tyrosine-based activation motifs (ITAMs) (3). The predominant PTKs that initiate activation signaling are believed to be Src family kinases (SFKs), especially Lck and Fyn, which phosphorylate ITAMs on activating receptor-associated transmembrane adaptor proteins, such as DNAX-activation protein of 12 kDa (DAP12), TCR-ζ, or FcR-γ, which can then recruit and activate zeta-associated protein of 70 kDa (ZAP-70) or Syk family PTKs. Activation of these PTKs initiates a cascade of tyrosine phosphorylation-centric downstream signaling events to drive degranulation and the subsequent production of an array of cytokines and chemokines, notably interferon-γ (IFN-γ) and tumor necrosis factor α (TNF-α), to promote inflammatory responses.
The central element of inhibitory signaling by KIRs is their recruitment of the protein tyrosine phosphatases Src homology 2 (SH2) domain–containing protein tyrosine phosphatase 1 (SHP-1) and SHP-2, to two phosphorylated cytoplasmic immunoreceptor tyrosine-based inhibitory motifs (ITIMs; V/IxYxxL) upon engagement with MHC-I on target cells (3). Whereas SHP-1 recruitment appears to require SFK-mediated phosphorylation of both tyrosines in the ITIM, evidence indicates that SHP-2 can bind weakly to KIR even in the unphosphorylated state, but more avidly upon phosphorylation of the N-terminal ITIM (4). Although Lck and SHP-1 are rapidly recruited to the center of the inhibitory NKIS (iNKIS) (5), the tyrosine phosphorylation of KIRs appears to be remarkably transient and occurs with Lck in microclusters at the iNKIS, as opposed to uniformly across the entire interface (6). Furthermore, Abeyweera et al. showed that the initiation of inhibitory KIR signaling induces rapid disruption of activating receptor microclusters, remodeling of the actin cytoskeleton, and retraction of the NK cell from the target cell (7).
A key question has dogged the field for a long time: what are the critical substrates that are dephosphorylated by SHP-1 and SHP-2 to efficiently terminate activation signaling? Early work to answer this question used substrate-trapping mutants of SHP-1 to capture putative substrates. In vitro studies by Binstadt et al. identified SH2 domain-containing Leukocyte Protein of 76 kDa (SLP-76) as the predominant tyrosine-phosphorylated protein to bind to a recombinant substrate-trapping SHP-1 protein in NK cell lysates (8). Subsequently, Stebbins et al. used a KIR-trapping SHP-1 chimeric receptor as an in vivo approach to identify the SHP-1 substrate, Vav-1, upon KIR engagement with MHC-I on target cells (9). Vav-1 is considered to be a critical target for the early termination of the NK cell activation pathway by inhibitory KIRs because it is an essential guanine nucleotide exchange factor (GEF) that activates the Rho family guanosine triphosphatase (GTPase) Rac-1 to initiate polymerization of the actin cytoskeleton, promote adhesion with the target cell, and instigate the recruitment of lytic granules to the iNKIS. However, the dephosphorylation of a single effector protein by SHP-1 has seemed too simplistic, and a larger repertoire of substrates has long been suspected and anticipated.
The article by Matalon et al. in this issue of Science Signaling (10) makes a substantial advance in expanding our understanding of the molecular mechanism of inhibitory KIR signaling (Fig. 1). The authors present an elegant set of experiments using standard protein biochemistry, short inhibitory RNA (siRNA)–mediated silencing and reconstitution methods, and fluorescence resonance energy transfer (FRET) technology in NK cell lines and primary NK cells. This work provides evidence that SHP-1 directly associates with and dephosphorylates LAT, PLC-γ1, and PLC-γ2 to disrupt their aggregation when the inhibitory receptor is engaged with ligand (HLA-Cw4) on target cells. LAT is a transmembrane adaptor protein that nucleates PLC-γ1, PLC-γ2, SLP-76, and the adaptor Grb2 through phosphotyrosine-mediated interactions, and activation of PLC-γ initiates downstream Ca2+ signaling, which is required for degranulation responses by cytolytic lymphocytes. These findings substantially expand upon earlier observations (11) to support a mechanism by which inhibitory KIRs terminate early tyrosine phosphorylation-dependent activation signaling events to disrupt the assembly of an essential protein complex.
Fig. 1. Proposed model for the negative effects of inhibitory KIRs on LAT–PLC-γ signaling.
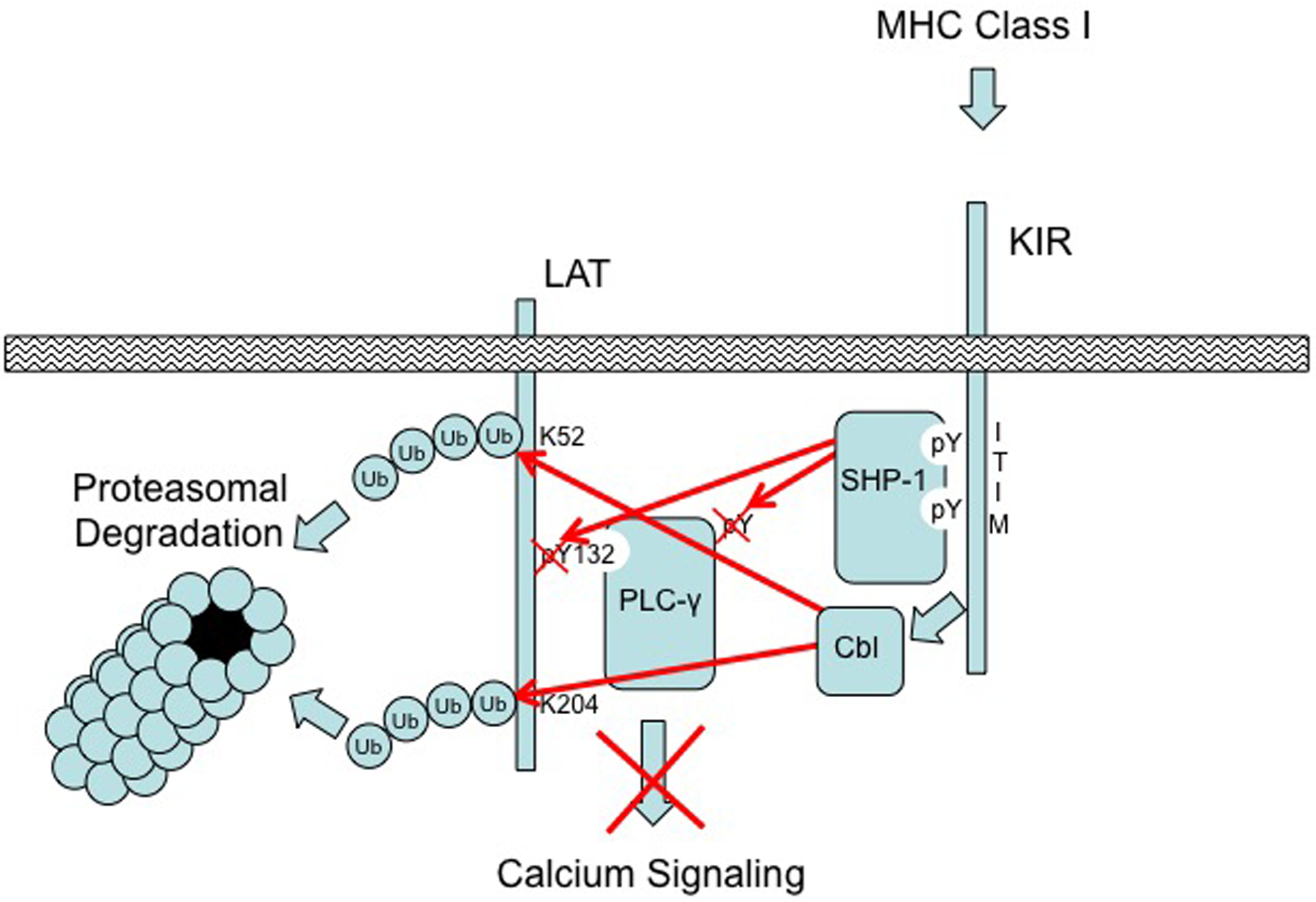
Activating receptors engaged by ligands on a target cell stimulate the phosphorylation of Tyr132 of LAT to recruit and activate PLC-γ through tyrosine phosphorylation (pY), which results in Ca2+ signaling. Co-engagement of KIR with MHC-I on the target cell stimulates the SFK–mediated phosphorylation of cytoplasmic ITIMs, which results in the recruitment and activation of SHP-1. Activated SHP-1 dephosphorylates PLC-γ and Tyr132 of LAT to disrupt its association with PLC-γ and abrogate enzymatic function. In addition, Cbl-b and c-Cbl are activated by the engagement of inhibitory KIRs with MHC-I to polyubiquitylate (Ub) LAT at Lys52, Lys204, or both, which stimulates the subsequent proteasomal degradation of the adaptor protein to further diminish activation signaling capacity. Figure is adapted from Fig. S8 of Matalon et al. (10).
In addition, the work of Matalon et al. further uncovered another critical piece of the puzzle by which engagement of inhibitory KIRs disrupts the assembly of the LAT–PLC-γ signaling complex through ubiquitylation of LAT by the Cbl family E3 ubiquitin ligases. Previous work by Peterson and Long established that activation signaling in human NK cells results in the physical association between the tyrosine-phosphorylated Vav-1 and c-Cbl, and that this association is disrupted in a SHP-1–dependent manner when inhibitory KIRs are engaged by ligand (12). Demonstrating the strong suppressive effect of Cbl family members, disruption of Cbl-b activity in NK cells markedly potentiates their anti-tumor response (13). Matalon et al. have extended these observations by showing that LAT is ubiquitylated on lysine residues when KIRs are engaged with MHC-I, and siRNA-mediated knockdown of both Cbl-b and the related c-Cbl abrogated LAT ubiquitylation. Furthermore, the authors showed that simultaneous siRNA-mediated silencing of both E3 ligases effectively abolished the capacity of KIR to inhibit both Ca2+ signaling and cytotoxicity toward MHC-I ligand–bearing target cells.
In summary (Fig. 1), this study by Matalon et al. has revealed two key elements of the mechanism by which inhibitory KIRs suppress activating signaling pathways in human NK cells: (i) the tyrosine-dephosphorylation of LAT and PLC-γ by SHP-1 to disrupt their association and the activation of PLC-γ; and (ii) the ubiquitylation of LAT by Cbl family E3 ligases. Although additional work is necessary to further dissect the interplay between these newly defined pathways and the means by which Cbl-b and c-Cbl are activated, our understanding of the molecular mechanisms suppressing the killer instinct of NK cells has come of age.
Funding:
Supported by NCI grants R01-CA083859 (K.S.C.) and CA06947 (FCCC) and a Health Research Formula Fund (CURE) grant from the PA Department of Health.
References and Notes
- 1.Campbell KS, Hasegawa J, Natural killer cell biology: an update and future directions. The Journal of allergy and clinical immunology 132, 536–544 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orange JS, Formation and function of the lytic NK-cell immunological synapse. Nat Rev Immunol 8, 713–725 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.MacFarlane AW 4th, Campbell KS, Signal transduction in natural killer cells. Current topics in microbiology and immunology 298, 23–57 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Yusa S, Campbell KS, Src Homology 2-containing protein tyrosine phosphatase-2 (SHP-2) can play a direct role in the inhibitory function of killer cell Ig-like receptors in human NK cells. J. Immunol 170, 4539–4547 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Vyas YM, Maniar H, Dupont B, Cutting edge: Differential segregation of the Src homology 2-containing protein tyrosine phosphatase-1 within the early NK cell immune synapse distinguishes noncytolytic from cytolytic interactions. J. Immunol 168, 3150–3154 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Treanor B, Lanigan PM, Kumar S, Dunsby C, Munro I, Auksorius E, Culley FJ, Purbhoo MA, Phillips D, Neil MA, Burshtyn DN, French PM, Davis DM, Microclusters of inhibitory killer immunoglobulin-like receptor signaling at natural killer cell immunological synapses. The Journal of cell biology 174, 153–161 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abeyweera TP, Merino E, Huse M, Inhibitory signaling blocks activating receptor clustering and induces cytoskeletal retraction in natural killer cells. The Journal of cell biology 192, 675–690 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Binstadt BA, Billadeau DD, Jevremovic D, Williams BL, Fang N, Yi T, Koretzky GA, Abraham RT, Leibson PJ, SLP-76 is a direct substrate of SHP-1 recruited to killer cell inhibitory receptors. J. Biol. Chem 273, 27518–27523 (1998). [DOI] [PubMed] [Google Scholar]
- 9.Stebbins CC, Watzl C, Billadeau DD, Leibson PJ, Burshtyn DN, Long EO, Vav1 dephosphorylation by the tyrosine phosphatase SHP-1 as a mechanism for inhibition of cellular cytotoxicity. Mol Cell Biol 23, 6291–6299 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matalon O, Fried S, Ben-Shmuel A, Pauker MH, Joseph N, Keizer D, Piterburg M, Barda-Saad M, Natural killer cell cytotoxicity depends on the adaptor LAT and phospholipase Cγ, which are targets of the phosphatase SHP-1. Sci Signal 9, raxx (2016). [DOI] [PubMed] [Google Scholar]
- 11.Valiante NM, Phillips JH, Lanier LL, Parham P, Killer cell inhibitory receptor recognition of human leukocyte antigen (HLA) class I blocks formation of a pp36/PLC-g signaling complex in human natural killer (NK) cells. J. Exp. Med 184, 2243–2250 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peterson ME, Long EO, Inhibitory receptor signaling via tyrosine phosphorylation of the adaptor Crk. Immunity 29, 578–588 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paolino M, Choidas A, Wallner S, Pranjic B, Uribesalgo I, Loeser S, Jamieson AM, Langdon WY, Ikeda F, Fededa JP, Cronin SJ, Nitsch R, Schultz-Fademrecht C, Eickhoff J, Menninger S, Unger A, Torka R, Gruber T, Hinterleitner R, Baier G, Wolf D, Ullrich A, Klebl BM, Penninger JM, The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 507, 508–512 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]