

Abstract
Formation and retrieval of conditioned fear memories critically depend on the amygdala. Here, we identify an inhibitory projection from somatostatin-positive neurons in the central amygdala to parvalbumin-positive neurons in the zona incerta that is required for both recent and remote fear memories. Thus, amygdala inhibitory input to parvalbumin-positive neurons in the zona incerta, a nucleus not previously implicated in fear memory, is an essential component of the fear-memory circuitry.
Pavlovian fear conditioning is widely used to examine the neural basis of associative learning1,2. Studies of the circuit mechanisms of fear conditioning have identified the amygdala as a key hub in a network of other important brain regions2. However, our understanding of fear memory remains incomplete. Indeed, recent experiments revealed that previously unrecognized brain regions are essential for fear memory3–6. Moreover, most studies to date examined recent fear memory; even less is known about remote fear memory7.
The Zona incerta (ZI) has been implicated in diverse behaviors8–11. We first tested different behaviors of mice after we bilaterally injected the medial ZI with AAVs expressing enhanced green-fluorescent protein (EGFP) and tetanus toxin light chain (TetTox), which completely blocks evoked synaptic transmission12 (Figure 1a, 1e, Supplementary Figure 1a). Littermates injected with AAVs expressing only EGFP were used as controls. Mice expressing TetTox in the medial ZI were grossly normal, but showed strongly impaired fear conditioning, increased anxiety and altered rotarod motor learning (Figure 1e–1h, Supplementary Figure 2). The fear conditioning impairment was particularly interesting because the ZI was not previously implicated in fear memories, although such a role has been suggested11,13. Moreover, deep brain stimulation of the subthalamic nucleus (STN), a region adjoining the ZI, impairs fear recognition14 and induces fear and panic15 in humans.
Figure 1: Synaptic signaling by ZI PV+-neurons is required for fear memory acquisition and recall of remote fear memory.
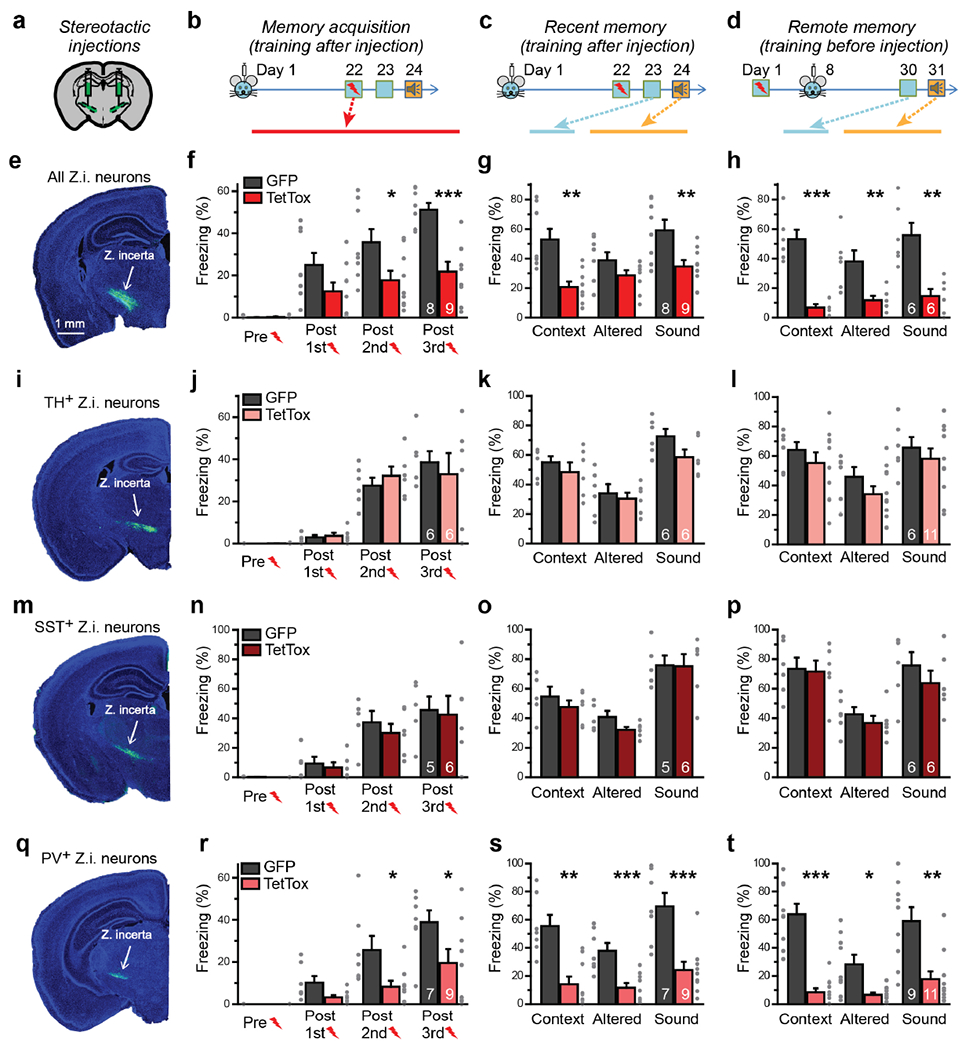
(a-d) Experimental design for fear memory experiments (a, stereotactic injection strategy of AAVs encoding TetTox into the medial ZI; b-d, experimental sequence of injections, fear conditioning training, and freezing behavior observations).
(e-h) Inactivation of all synaptic outputs from the medial ZI (e) impairs fear conditioning training (f; P = 0.8, Mann-Whitney test; P = 0.09, 0.03, 0.00014, two-sided unpaired t-tests), recent fear memory (g; P = 0.00101, 0.13, 0.009, two-sided unpaired t-tests), and remote memory when outputs are inactivated after training (h; P = 4 × 10−4, 0.009, 0.0017, two-sided unpaired t-tests).
(i-l) Inactivation of dopaminergic neurons in the ZI by injection of double-floxed TetTox into TH-Cre mice (i) has no effect on fear conditioning training (j; P = 1, Mann-Whitney test; P = 0.7, 0.4, 0.6, two-sided unpaired t-tests), recent memory (k; P = 0.4, 0.6, 0.08, two-sided unpaired t-tests), or remote memory (l; P = 0.4, 0.2, 0.5, two-sided unpaired t-tests).
(m-p) Inactivation of SST+-neurons in the ZI by injection of double-floxed TetTox into SST-Cre mice (m) has no effect on fear conditioning training (n; P = 0.9, Mann-Whitney test; P = 0.6, 0.5, 0.8, two-sided unpaired t-tests), recent memory (o; P = 0.4, 0.08, 0.9, two-sided unpaired t-tests), or remote memory (p; P = 0.9, 0.4, 0.4, two-sided unpaired t-tests).
(q-t) Inactivation of PV+-neurons in the ZI by injection of double-floxed TetTox into PV-Cre mice (q) impairs fear conditioning training (r; P = 1, Mann-Whitney test; P = 0.07, 0.02, 0.049, two-sided unpaired t-tests), recent fear memory (s; P = 0.002, Mann-Whitney test; P = 8 × 10−4, 9 × 10−4, two-sided unpaired t-tests), and remote memory when outputs are inactivated after training (t; P = 3 × 10−5, 0.014, two-sided unpaired t-tests; P = 0.003, Mann-Whitney test) similar to the inactivation of all ZI outputs.
Quantitative data are means ± SEM. P < 0.05 was considered significant (*P < 0.05, **P < 0.01, ***P < 0.001). All numbers of mice used are presented in right bars. For TetTox or EGFP group in all memory acquisition results (f, j, n, r), P < 0.05, F > 7, one-way repeated measures ANOVA.
In these fear conditioning experiments, we quantified three aspects of fear memory. First, we measured freezing during training (memory acquisition) 21 days after the injections; second, we measured recall in the same context and in an altered training context at one and two days after training, respectively, with the tone delivered in the altered context; third, we performed the injections 7 days after fear-memory training, and measured recall 22 days after the injections (or 29 days after training) to test the effect of ZI manipulations on memory that had already been formed12 (Figure 1b–1d). Already during training, inactivation of synaptic outputs from the ZI impaired freezing, suggesting that the subsequent impairment in recent fear memory is due to impaired fear-memory acquisition (Figure 1f, 1g). Strikingly, TetTox expression in the medial ZI also impaired remote fear-memory retrieval when the ZI outputs were inactivated 7 days after training, demonstrating that even with intact fear-memory acquisition, inactivation of synaptic outputs from ZI neurons still impaired retrieval of remote fear memory (Figure 1h). Both cued and contextual fear memories were affected. The TetTox-induced reduction in freezing was not caused by changes in locomotion or foot shock responses (Supplementary Figure 2c, Supplementary Figure 3a). Together, these results suggest that the ZI is essential both for the initial acquisition of fear memory and for the remote retrieval of previously acquired fear memory.
To assess which ZI neuron type(s) contribute to fear-memory conditioning, we injected double-floxed, Cre-dependent TetTox into the ZI of Cre-driver mouse lines expressing Cre-recombinase selectively in tyrosine hydroxylase-positive (TH+), somatostatin-positive (SST+) or parvalbumin-positive (PV+) neurons. TH+-neurons of the ZI were proposed to modulate responses to aversive stimuli16; however, silencing ZI TH+-neurons caused no significant impairment of fear conditioning (Figure 1i–1l). SST+-neurons in the ZI are thought to function in cortical neuronal development17. Inactivating ZI SST+-neurons also did not affect fear conditioning (Figure 1m–1p). PV+-neurons, located in the ventral part of medial ZI (Figure 1q, Supplementary Figure 1b), have no known functions. Interestingly, bilateral silencing of ZI PV+-neurons largely reproduced the behavioral impairments produced by silencing all ZI neurons (Figure 1q–1t, Supplementary Figure 3b, 4). Importantly, these mice froze normally during innate fear responses (Supplementary Figure 3c, 3d). Together, these results suggest that ZI PV+-neurons are uniquely essential for fear-conditioning memory acquisition and remote retrieval.
To map the presynaptic inputs to ZI PV+-neurons, we performed retrograde monosynaptic tracing experiments using rabies virus (Figure 2a–c). We identified multiple input brain regions, including the central amygdala (CeA), a critical region in fear conditioning behavior2. Other robustly labeled brain regions included the primary and supplementary motor and somatosensory cortices, substantia innominata, ventral posteromedial nucleus of the thalamus, substantia nigra, superior colliculus (SC), midbrain reticular nucleus, dorsal raphe nucleus, and dentate nucleus (Figure 2c, Supplementary Figure 5). We found that most of the CeA neurons projecting to ZI PV+-neurons express SST (90 ± 3% of GFP+ neurons, n = 3 mice.) but not protein kinase C delta (PKCdelta, 9 ± 3% of GFP+ neurons; Figure 2d), which mark two of the major CeA cell types that play distinct roles in fear conditioning18,19. We also conducted retrograde tracing using cholera toxin subunit B (CTB), which confirmed all of the presynaptic regions identified by rabies tracing (Supplementary Figure 6a–6j) and that most of the ZI-projecting neurons in CeA are SST+ (Supplementary Figure 6k–6r, 98 ± 1% of green neurons are SST+, 10 ± 1% of green neurons are PKCdelta+, n = 3 mice).
Figure 2. The central nucleus of the amygdala forms mono-synaptic connections with ZI PV+-neurons that are essential for memory acquisition and remote memory recall.
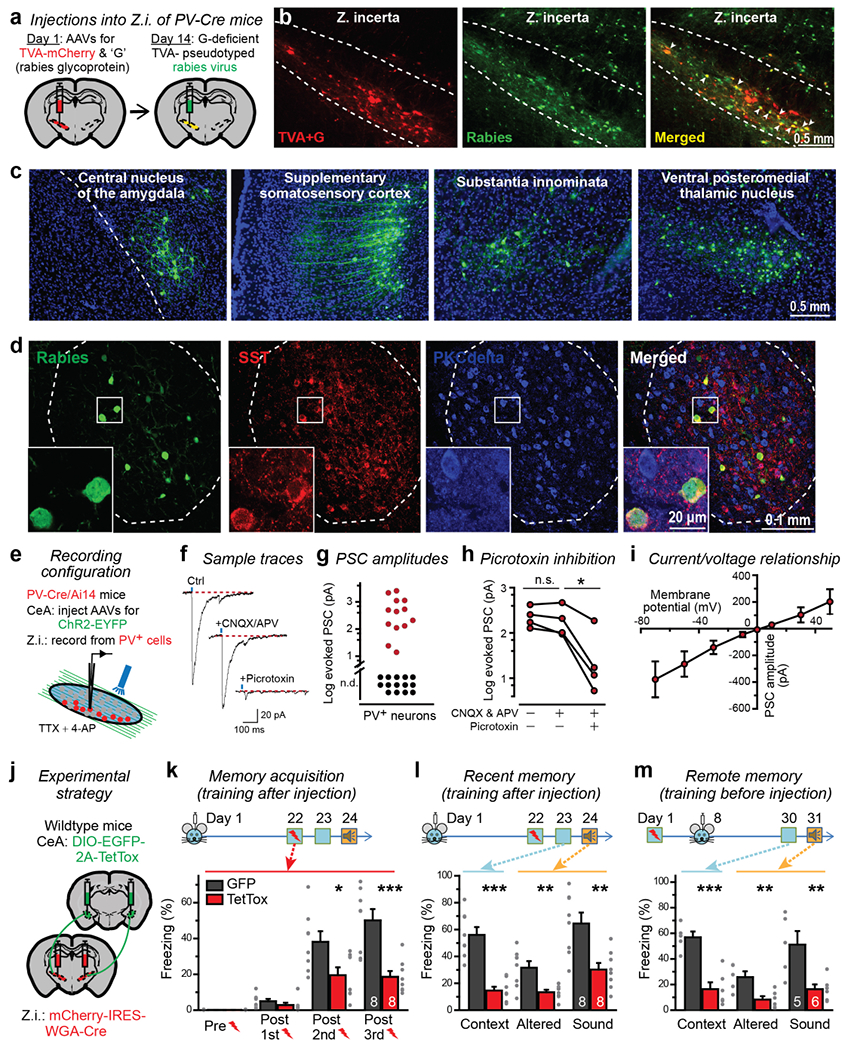
(a-d) Rabies tracing experiments identify synaptic projections to ZI PV+-neurons from CeA SST+-neurons and other brain regions (a, rabies virus tracing strategy; b, illustration of ZI infections with AAVs encoding TVA-mCherry (red) and rabies-virus glycoprotein and with GFP expressing rabies virus (green). Arrowheads indicate starter cells. Dashed lines indicate approximate nuclei boundaries; c, representative images illustrating presynaptic neurons identified by rabies virus tracing; d, representative low and high magnification (boxed area) images of presynaptic green neurons in CeA co-immunostained with SST and PKCdelta). All images are on the ipsilateral side of injection and have the same orientation as shown in a. Experiments in b-c and d were independently repeated with similar results five and three times, respectively.
(e-i) CeA neurons form mono-synaptic inhibitory connections with ZI PV+-neurons (e, experimental strategy; f, sample traces of postsynaptic currents evoked by optogenetic stimulation in the presence of tetrodotoxin (TTX) and 4-Aminopyridine (4-AP) before and after addition of CNQX and APV, followed by picrotoxin; g, synaptic response amplitude plot (n.d.: not detected); h, amplitudes of synaptic responses in four neurons that were sequentially exposed to CNQX and APV followed by picrotoxin (two-sided paired t-test, P = 0.2, 0.038); i, current/voltage plot of synaptic responses, n = 5 cells).
(j-m) Synaptic projections from the CeA onto ZI PV+-neurons are essential for memory acquisition and remote memory recall (j, experimental strategy; k-m, tests of memory acquisition (k; P = 1, 0.2, Mann-Whitney test; P = 0.02, 6 × 10−4, two-sided unpaired t-tests), recent memory (l; P = 2 × 10−5, 0.007, 0.003, two-sided unpaired t-tests), and remote memory (m; P = 3 × 10−4, 0.007, 0.009, two-sided unpaired t-tests)). Quantitative data are means ± SEM. P < 0.05 was considered significant (*P < 0.05, **P < 0.01, ***P < 0.001). Numbers of mice used are presented in right bars. One-way repeated measures ANOVA test was used to analyze fear acquisition results in k. TetTox group, P < 0.01, F = 13.7. EGFP group, P < 0.001, F = 37.1.
Given the central role of the CeA in fear memory18,20, we functionally characterized the connection between CeA and ZI PV+-neurons. We injected AAVs expressing channelrhodopsin (ChR2-EYFP) into the CeA of the PV-reporter mice in which PV+-neurons are labeled with tdTomato (Figure 2e). Three weeks later, we used whole-cell voltage-clamp recordings from PV+-neurons in the ZI to record optogenetically-induced postsynaptic currents (Figure 2f). These recordings were performed in the presence of tetrodotoxin and 4-Aminopyridine to exclude polysynaptic connections. 13 of 27 recorded cells exhibited reliable light-evoked postsynaptic currents (Figure 2g), confirming a monosynaptic connection between the CeA and the ZI PV+-neurons. In a separate cohort of mice in which we co-expressed TetTox with channelrhodopsin in CeA, 0 of 21 recorded cells exhibited light-evoked postsynaptic currents, confirming that TetTox blocks synaptic transmission (Supplementary Figure 7). Using sequentially applied inhibitors of glutamate receptors (CNQX + APV) and of GABA- and glycine-receptors (picrotoxin), we found that only picrotoxin had a significant effect on synaptic responses (Figure 2f, 2h), suggesting that the CeA–ZI projection is inhibitory. Consistent with this conclusion and previous findings that CeA SST+ neurons are inhibitory18, the current/voltage relationship of the synaptic responses suggested a reversal potential of 0 mV (with high-CI internal solution for patch-clamp recordings) (Figure 2i).
To assess whether the CeA–ZI projection is required for recent and remote fear memory, we expressed wheat-germ agglutinin-Cre (WGA-Cre) bilaterally in the medial ZI, and Cre-dependent TetTox in the CeA (Figure 2j, Supplementary Figure 8). The WGA-Cre expressed in ZI neurons will be trans-synaptically transferred to presynaptic inputs3, including inputs from the CeA. As a result, the synaptic outputs from CeA neurons projecting to the ZI will be inactivated, enabling us to test the role of this projection in fear memory. Mice with this manipulation exhibited a dramatic impairment in both memory acquisition and remote fear memory (Figure 2k–2m), similar to that observed upon silencing ZI PV+-neurons. Together, these results suggest that the inhibitory synaptic projection of CeA SST+-neurons to ZI PV+-neurons is essential for both recent and remote fear memory.
Preliminary mapping of the synaptic outputs from ZI PV+-neurons using a Cre-dependent version of SynaptoTag3 that expresses an EGFP-synaptobrevin-2 (Syb2) fusion protein to label presynaptic terminals revealed strong labeling of presynaptic terminals in the ventral thalamus, the lateral and deeper layer of the SC, and the lateral PAG, as well as locally in the dorsal and ventral ZI, suggesting that these brain regions receive inputs from ZI PV+-neurons (Supplementary Figure 9). Using the same retrograde and intersectional viral infection methods, we silenced the synaptic projection from medial ZI to lateral SC or PAG, and found that neither of these two manipulations replicate the major fear-memory deficits we observed after silencing ZI PV+-neurons (Supplementary Figure 10, 11).
Our study reveals a previously unrecognized function of the ZI in fear-memory acquisition and remote retrieval. Moreover, we identified a direct connection between CeA SST+-neurons and ZI PV+-neurons that is essential for recent and remote fear memory. CeA SST+-neurons are critical for fear-memory acquisition and expression/retrieval18. Our results suggest a new mechanism by which the CeA SST+-neurons contribute to fear-memory processing. Our anterograde SynaptoTag tracing results show that ZI PV+-neurons provide both local and long-range outputs (Supplementary Figure 9). Given that silencing the medial ZI projections to SC and PAG, two important nodes in fear expression, have limited effects on fear memory (Supplementary Figure 10, 11), we propose that ZI PV+-neurons may engage in fear-memory processing through its local outputs within the ZI. Indeed, data from Allen Brain Institute mouse connectivity data portal showed that ZI neurons project to fear effectors such as lateral hypothalamus, PAG, and pontine reticular nucleus, and involvement of rostral ZI GABAergic neurons in conditioned freezing response was recently reported11. Moreover, our rabies retrograde tracing results show that besides the CeA, ZI PV+-neurons also receive inputs from cholinergic, dopaminergic and serotoninergic neuromodulatory systems (Figure 2c, Supplementary Figure 5d, 5f). Therefore, we propose that ZI PV+-neurons serve as an important node between the CeA and downstream effectors and could be dynamically regulated by an animal’s behavioral state.
An interesting observation of our study is that blocking synaptic inhibition from the CeA to the ZI (presumably enhancing ZI neuron activity) and blocking ZI synaptic outputs onto downstream targets both produced behavioral deficits (Figure 1f–1h, 2k–2m). We think these seemingly contradictory results are informative because they suggest that not only the strength of synaptic output from ZI PV+ neurons, but also its precise pattern, which is subject to modulation by CeA inputs, are essential components of fear memory. Blocking the information flow in either node will adversely affect the fear memory. Consistent with this hypothesis, we found that optogenetic activation of ZI PV+-neurons could not directly induce freezing (Supplementary Figure 12). Future studies probing the physiological activities of ZI PV+-neurons during different phases of conditioned fear memory will provide further insight into how information flow within the ZI circuitry supports fear memory.
METHODS
Animals.
C57BL/6J, PV-Cre, SST-Cre, TH-Cre and Ai14 reporter mice were purchased from the Jackson Laboratory. They were maintained on a C57BL/6J background. Mice were group housed (maximum 5 mice in a cage) on a 12 hours light/dark cycle (7 am to 7 pm, light) with food and water freely available. Male mice 45-60 days of age were used for all the experiments. Animal experiments were conducted following protocols approved by Administrative Panel on Laboratory Animal Care at Stanford University.
Vector construction and AAV preparation:
In the DIO-SynaptoTag vector, the following components were arranged sequentially downstream of left-ITR of AAV2: synapsin promoter, loxp2272, loxp, inverted mCherry-IRES-Syb2-EGFP, loxp2272, loxp, WPRE, hGH poly A sequence and right ITR. AAV DIO-SynaptoTag was packaged with AAV-DJ capsids for high efficiency in vivo neuronal infection. Virus was prepared with a procedure as previously described21. Briefly, the DIO-SynaptoTag construct was cotransfected with pHelper and pRC-DJ into HEK293 cells. 72 hr later, cells were collected, lysed and loaded onto iodixanol gradient for centrifugation at 80,000g for 2 hrs. The fraction with 40% iodixanol of the gradient was collected, washed and concentrated with 100,000 MWCO tube filter. The genomic titer of virus was measured by qPCR.
Stereotaxic injection:
All the AAV viruses, except DIO-SynaptoTag, were produced by the Stanford Virus Core. They were all in AAV-DJ serotype. The concentrations of virus used for stereotaxic injection were adjusted to 1.0 × 109 infectious units/ml. The rabies viruses were purchased from the Janelia Virus Service Facility. Alexa-Fluor 488-conjugated CTB was purchased from Invitrogen and used at 0.5% in PBS. Mice were anesthetized with tribromoethanol (300 mg/kg) and head-fixed with a stereotaxic device (KOPF model 1900). Viruses or CTB-488 were injected using a glass micropipette attached to a 10 μl Hamilton syringe. The pipette tips were beveled to be sharp and smooth. 0.2 μl of AAV or CTB were injected at a flow rate of 0.2 μl/min bilaterally for behavior experiments and on the left site for tracing experiments. We waited for four minutes both before and after each injection. AAVs were allowed to express for 3 weeks (7 weeks for AAV DIO-SynaptoTag) and CTB were allowed to express for 1 week. The coordinates used for the medial ZI were 1.8 mm posterior to Bregma, 1.3 mm from the midline, 4.6 mm below dura. The coordinates used for the central amygdala were 1.15 mm posterior to Bregma, 2.9 mm from the midline, 4.5 mm below dura. The coordinates used for the lateral SC were 4.08 mm posterior to Bregma, 1.5 mm from the midline, 2.1 mm below dura. The coordinates used for the lateral PAG were 4.38 mm posterior to Bregma, 0.75 mm from the midline, 2.4 mm below dura. For rabies tracing experiments, 0.2 μl of a 1:1 volume mixture of AAV5-CAG-DIO-TVA-mCherry and AAV8-CAG-DIO-G was injected into the left ZI of 6-8 weeks old PV-Cre mice22. Two weeks later, 0.3 μl RVdG was injected into the same brain region. One week later, mice were perfused and processed for imaging.
Behavior:
Behavior experiments started three weeks after AAV injections. Mice were handled daily for three days before their first behavior test. Mice were tested in either fear conditioning related assays alone or all the other various tests (minimum 4 days interval between each two tests) in the sequence of open field, Y maze, elevated plus maze, novel object recognition, three-chamber social interaction, acoustic startle, prepulse inhibition, rotarod and water T maze. The TetTox and EGFP mice were coded and blinded to the experimenters. After behavior tests were finished, mice were perfused, sectioned and imaged to verify injection sites.
Fear conditioning:
On training day, mice were individually placed in fear conditioning chamber (Coulbourn Instruments) located in the center of a sound attenuating cubicle. The conditioning chamber was cleaned with 10% ethanol to provide a background odor. A ventilation fan provided a background noise at ~55 dB. After a 2 min exploration period, 3 tone-foot shock pairings separated by 1 min intervals were delivered. The 85 dB 2 kHz tone lasted for 30 s and the foot shock was 0.75 mA and lasted for 2 s. The foot shocks co-terminated with the tone. The mice remained in training chamber for another 60 s before being returned to home cages. In context test, mice were placed back into the original conditioning chamber for 5 min. During altered context test, the conditioning chamber were modified by changing its metal grid floor to a plastic sheet, white metal side walls to plastic walls decorated with red stripes, background odor of 10% ethanol to 1% vanilla. Mice were placed in the altered chamber for 5 min to measure the freeze level in the altered context and after this 5-min period a tone (85 dB, 2 kHz) was delivered for 1 min to measure the freeze to tone. The behavior of the mice was recorded with the Freezeframe software and analyzed with Freezeview software (Coulbourn Instruments). Motionless bouts lasting more than 1 s were considered as freeze. On the training day, freezing percentage in the 2 min exploration period and 1 min period after each foot shock were summarized as an indication of fear memory acquisition. The average running speed of mice during each foot shock and in the 5-s period before and after each foot shock were analyzed with a tracking software Viewer III (BIOBSERVE).
Visually induced freezing:
This behavior paradigm was modified from a previous study23. An LCD monitor (14 inch × 11 inch, 60 Hz refresh rate, Samsung) was placed facing downwards 43 cm away from the ground. Mice were individually placed in a new housing cage (31 cm × 20 cm) on the ground beneath the monitor. The behavior of the mice was recorded with the Freezeframe software and analyzed with Freezeview software. Before the visual stimuli, mice were given 5 minutes to become habituated to the new environment. After 5 mins, the visual stimuli were triggered when the animal’s location was approximately at the center of the cage. Animals were videotaped for another 2 mins after delivery of visual stimuli. The visual stimuli were consisted of two sweeps (separated by 3s) of a 6 cm black disk that appeared at a corner of the monitor and then moved smoothly to the diagonally opposite corner. The two sweeps move across the monitor over 0.5 s (fast) and 10 s (slow) respectively. In our hands, this two-sweeps stimuli reliably induce animals’ freezing behavior for about 30–60 s. The 100-s period before and after the visual stimuli were used to quantify the freezing percentage.
Predator odor induced freezing:
The same mice used for visually induced freezing experiments were used in this behavior paradigm, which was modified from a previous study24 In short, mice were individually placed in a clean housing cage, inside which a small piece of kimwipe (2 × 2 cm) was taped to the wall. The cage was placed inside a fume hood to prevent odors from spreading. Each mouse was habituated to the cage for 5 min, after which 20 μl of 2,5-dihydro-2,4,5-trimethylthiazoline (TMT, SRQ Bio), a component of fox feces, were applied to the kimwipe piece. The lid was closed immediately. The animal’s behavior was monitored with a webcam (c920, Logitech) placed above the cage for another 10 min. Animals’ freezing behavior was manually measured. The 5 min habituation period was used to calculate baseline freezing percentage. The period between 5 min and 10 min after TMT application was used to calculate TMT-induced freezing percentage.
Open field:
White colored plastic boxes were used as the open field chambers (dimension: 34 × 34 × 40 cm). Mice were individually placed into the center of the chambers and allowed to freely explore for 10 min. The locomotion and exploratory behaviors of mice were recorded with a tracking system Viewer III (BIOBSERVE). The traveling distance and the time spent in center area were analyzed. The center area was defined as the 17 × 17 cm central section of the chambers. The total traveling distance was used to evaluate locomotor activity and the time spent in center area was commonly used to estimate the anxiety level in an open environment.
Spontaneous alternation Y maze:
A light grey plastic Y maze was used to evaluate spatial working memory. The maze consisted of three arms separated by 120 degrees (dimensions of each arm: 40 × 10 × 17 cm). Mice were individually placed in the distal end of one arm and allowed to freely explore the whole maze for 10 min. Completed arm entry was defined as the entering of a whole mouse including its tail into an arm. The sequences and total numbers of arm entries were recorded and analyzed with Viewer III tracking system. Visiting all three different arms consecutively was termed a ‘correct’ trial, and visiting one arm twice or more in three consecutive entries was termed wrong trial. We calculated the correct alternation percentage as (number of correct trials/total number of correct and wrong trials) × 100.
Elevated plus maze:
The grey-painted maze had four 30 × 8 cm arms. Two of them were open arms without walls and other two were enclosed by 10 cm high walls. The maze was elevated 40 cm over the floor. At the beginning of the tests, mice were individually placed at the junction of an open and a closed arms, facing the closed arm. Then the mice were allowed to freely move in the whole maze for 10 min. The time durations mice spent in the open vs. the closed arms were recorded and analyzed with Viewer III tracking system.
Novel object recognition:
The same boxes used for open field test were used. On day 1, mice were given 10 min of habituation time individually in an empty box. On day 2, mice were individually placed in the box for 10 min with two identical objects, either T75 cell culture flask filled with bedding material or blocks of Legos. On day 3, one object was replaced with a novel object. Object location in chamber was randomized. Exploration behavior was recorded and analyzed with Viewer III tracking system. Recognition index was defined as the time spent on the novel object (or the left object for training phase) divided by the time spent on both objects.
Three-chamber social interaction:
Social interaction was evaluated in a transparent three-chamber box (The dimension of each chamber was 60 × 30 × 30 cm). Mice were individually placed in the central chamber for a 5-min habituation. After the habituation, another mouse (intruder) was placed inside an upside-down wire mesh pencil cup located in one of the side chambers (named as social chamber). The intruders were of the same gender, same species and at similar age to the test mouse. An identical pencil cup was placed in the other side chamber (named as non-social chamber). After that, the doors to the side chambers were opened and the test mouse was allowed to freely travel among the three chambers for 10 min. The time durations that the mice spent in each of the chambers were recorded and analyzed with Viewer III tracking system.
Acoustic startle response and pre-pulse inhibition:
Kinder Scientific startle reflex system was used and data were analyzed with Startle Monitor II software. Mice were individually placed in a small cage within a sound attenuation cabinet. Mice had restricted space in the cage but could turn around. For acoustic startle response experiments, 50 ms noise at 75 dB, 85 dB, 95 dB, 105 dB and 115 dB were given. For pre-pulse inhibition experiments, 50 ms 120 dB noise were given with preceding noise at 0, 68 dB, 71 dB or 77 dB. For both experiments, mice were given 3 min of habituation time before delivery of sound. The stimulus sequence and inter-stimulus interval were both pseudo-randomized. Each stimulus was repeated for 10 times. The peak values of the absolute force mice placed on the bottom of the cage were measured as the startle response. Background noise was 65 dB.
Rotarod:
An accelerating rotarod designed for mice (IITC Life Science) was used. The test consisted of three trials per day over the course of 3 days. The rotarod was activated after placing mice on the motionless rod. The rod accelerated from 4 to 40 revolutions per min in 5 min. Each trial ended when a mouse fell off, made one complete revolution while hanging on, or reached 300 s.
Water T maze:
The customized T maze consisted of a long arm and two shorter arms (18.5 cm × 5 cm). On day 1, mice were individually placed into the starting arm and allowed to swim freely without any platform for 1 min. The arms that each mouse entered first were recorded. On day 2 and day 3, a platform was placed in the arm that the mouse did not visit first on day 1. Mice were individually placed into the starting arm and allowed 1 min to find the platform. Mice were gently guided to the platform and allowed to rest for 10 s if they could not find the platform within 1 min. Mice were given 10 trials each day. For each trial (excluding the first trial on day 2), the time it took for the mice to find the platform and the error number (number of entries into the wrong arm plus number of entries into the correct arm without finding the platform) were manually recorded. Water at room temperature was filled to a depth of 13 cm, 1 cm above the surface of the platform. Nonfat milk was added to make the water milky and hide the platform. Mice were excluded from experiments if they have difficulty swimming.
Brain slice electrophysiology:
P21 PV-Ai14 mice were injected with 0.2 μl either AAV ChR2 (H134R)-EYFP alone, or a 1:1 mixture of AAV ChR2 (H134)-EYFP and AAV TetTox-EGFP in left CeA. Three weeks later, coronal brain slices (300 μm) were cut with a vibratome (VT1200S, Leica) in ice cold solution (in mM): 228 Sucrose, 2.5 KCl, 1 NaH2PO4, 7 MgSO4, 0.5 CaCl2, 26 NaHCO3, 11 D-glucose saturated with 95% O2/5% CO2 and recovered in oxygenated artificial cerebrospinal fluid (ACSF) containing (in mM): 119 NaCl, 2.5 KCl, 1 NaH2PO4, 1.3 MgSO4, 2.5 CaCl2, 26 NaHCO3, 10 D-glucose. After slices were transferred to a recording chamber, oxygenated ACSF was continually perfused (1 ml/min, maintained at 32°C) in the presence of 1 μM TTX and 100 μM 4-AP. Whole-cell voltage-clamp recordings were made with 2-3 MΩ pipettes filled with internal solution containing (in mM): 135 CsCI, 10 FIEPES, 1 EGTA, 1 Na-GTP, 4 Mg-ATP. The whole-cell current signals were recorded with MultiClamp 700B and Clampex 10.4 data acquisition software (Molecular Device, LLC). A customized DMD based photostimulation optogenetical system was used for light evoked post synaptic current (PSC) recordings. Only area of ZI was given blue light. Recordings were selectively made from red fluorescent cells in left ZI under visualization with an upright microscope (BX51WI, Olympus). After establishment of the whole-cell configuration and equilibration of the intracellular pipette solution with the cytoplasm, laser intensity was adjusted to get a maximal response without over stimulation. 1 ms blue light was applied five repetitions at 10-s intervals with the cells clamped respectively at −70 mV, −50 mV, −30 mV, −10 mV, 0 mV, +10 mV, +30 mV and +50 mV for I-V relationship. Junction potential under our internal solution and ACSF composition was 4.5 mV and was corrected for the I-V recordings. Membrane potential was held at −70 mV for the other recordings. 20 μM CNQX and 50 μM APV were perfused followed by additional 50 μM picrotoxin to check whether the light-evoked PSCs were excitatory or inhibitory.
In vivo optogenetic stimulation:
PV-Cre mice were bilaterally implanted with optic fiber cannulae (200 μm core, 0.22 NA, Thorlabs) during the same surgery procedure for viral injection of AAV DI0-ChR2 (H134R)-EYFP. The tips of optical fibers were placed 0.2 mm dorsal to the viral injection site. The optical fibers were secured to the skull with light-cured dental adhesive cement (Geristore paste, DenMat). Mice were habituated to body restraint and their cannulae connected to optical fiber sleeves (Thorlabs) once a day for three days before behavioral testing. On experiment day, the cannulae of mice were connected via patch cables and a 1 × 2 rotary joint (Doric Lenses) to a 473 nm laser (OEM Laser Systems). The behavior of mice were tested in the fear conditioning chamber. No foot shock or sound were delivered. After 2 mins of habituation, 30-s of laser stimulation (5 ms pulse, 15 Hz) was triggered (Master 8, A.M.P.I) and repeated 5 times with an inter-stimuli-interval of 2 mins. Different frequency from 1 Hz to 50 Hz were tested. The power of the laser was calibrated to be ~10 mW at the tip of the optic fiber. Freezing behavior was measured during a 30-s period immediately before the delivery of light pulses (light off), and the 30-s period of light pulses (light on).
Histology:
Mice were deep anesthetized with tribromoethanol and perfused with 10 ml of PBS followed by 10 ml of fixative (4% paraformaldehyde diluted in PBS). The brains were removed and post-fixed in 4°C overnight and then immersed in 30% sucrose solution for two days before being sectioned at 50 pm thicknesses on a cryostat (Leica CM3050 S). The free-floating brain sections were collected in PBS. For injection site verification, the sections were directly mounted onto glass slides with Vectashield mounting medium with DAPI. For tracing experiments, one out of every five sections were collected for the whole brain. For immunohistochemistry, standard procedures3 were followed. We used primary antibodies to SST (rabbit, BMA Biomedicals, cat. no. T-4103, 1:1000)25 and PKCdelta (mouse, BD Biosciences, cat. no. 610398, 1:1000)19. A scanning microscope (BX61VS, Olympus) was used to scan fluorescent images for whole brain slices and a confocal microscope (Nikon A1) was used for higher resolution imaging. To image green signals in CTB-488 and SST-Ai14 co-localization experiments, a 500–530 nm virtual filter (spectral detector mode) was used instead of the default 500–550 nm filter (standard PMTs) in order to prevent leakage of SST-AM4 red signals into the green channel.
Statistics:
All results are presented as mean ± s.e.m. and were analyzed by the OriginPro 8 software (OriginLab Corp.). No statistical methods were used to predetermine sample sizes but our sample sizes are similar to those reported in previous publications3,18. Normality tests and F tests for equality of variance were performed before choosing the statistical test. Unless otherwise indicated, statistics were based on two-sided unpaired t-tests or Mann-Whitney tests (for datasets that were not normally distributed) for two-group comparisons. P < 0.05 was considered significant (*P < 0.05, **P < 0.01, ***P < 0.001). Freezing expressions during the acquisition phase were analyzed with one-way repeated measures ANOVA. Animals in the same litter were randomly assigned to different treatment groups and blinded to experimenters in various experiments. Injection sites and viral expression were confirmed for all animals. Mice showing incorrect injection sites or optic fiber placement were excluded from data analysis.
Supplementary Material
Acknowledgements.
We thank K. Beier for invaluable help with rabies tracing experiments; J. Liang, S. Lu, R. Land for assistance; and L. Chen for feedback on manuscript. This study was supported by NIH/NIMH (MH052804 to T.C.S.) and NIH/NINDS (1K01NS105155 to M.Z.).
Footnotes
Competing Interests. The authors declare no competing interests.
Reporting Summary. Further information on experimental design is available in the Life Sciences Reporting Summary linked to this article.
Accession code: DNA sequence of the DIO-SynaptoTag plasmid have been deposited in GenBank under accession number MH883617.
Data availability: The data that support the findings of this study are available from corresponding authors upon reasonable request.
REFERENCES
- 1.Maren S Annual review of neuroscience 24, 897–931 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Fanselow MS & Poulos AM Annual review of psychology 56, 207–234 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Xu W & Sudhof TC Science 339, 1290–1295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Do-Monte FH, Quinones-Laracuente K & Quirk GJ Nature 519, 460–463 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Penzo MA, et al. Nature 519, 455–459 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vetere G, et al. Neuron 94, 363–374 e364 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Frankland PW & Bontempi B Nature reviews neuroscience 6, 119–130 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Mitrofanis J Neuroscience 130, 1–15 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Liu K, et al. Nature 548, 582–587 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X & van den Pol AN Science 356, 853–859 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chou XL, et al. Nature communications 9, 1151 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu W, et al. Neuron 73, 990–1001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wheeler AL, et al. PLoS computational biology 9, e1002853 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biseul I, et al. Neuropsychologia 43, 1054–1059 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Okun MS, et al. Journal of neurology, neurosurgery, and psychiatry 75, 921–923 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Essig J & Felsen G Trends in neurosciences 39, 2–4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J & Kriegstein AR Science 350, 554–558 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, et al. Nature neuroscience 16, 332–339 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haubensak W, et al. Nature 468, 270–276 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilensky AE, et al. J Neurosci 26, 12387–12396 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zolotukhin S, et al. Gene therapy 6, 973–985 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Beier KT, et al. Cell 162, 622–634 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Franceschi G, et al. Current biology 26, 2150–2154 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Otsuka S, et al. J Neurosci 36, 11801–11816 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stephenson-Jones M, et al. Nature 539, 289–293 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.