

Significance
We previously demonstrated that intrahepatic portal hypertension is caused in part by an endothelialopathy in which a number of G protein-coupled receptor (GPCR) regulatory proteins are dysregulated in injured sinusoidal endothelial cells (SECs). Here, we identified a fundamental molecular mechanism controlling eNOS function. We show that β-arrestin2 (β-Arr2) is an integral component of the eNOS signaling complex, which includes eNOS/GIT1, and that this module becomes dysregulated after injury-reduced levels of β-Arr2 lead to a reduction in eNOS phosphorylation/activation and NO production in injured SECs. The physiological and functional importance of β-Arr2 is demonstrated in vivo and ex vivo, and molecular mechanisms underlying its behavior are uncovered. These findings have important therapeutic implications for a number of vascular disorders.
Keywords: cirrhosis, liver, portal hypertension
Abstract
Endothelial cell nitric oxide (NO) synthase (eNOS), the enzyme responsible for synthesis of NO in endothelial cells, is regulated by complex posttranslational mechanisms. Sinusoidal portal hypertension, a disorder characterized by liver sinusoidal endothelial cell (SEC) injury with resultant reduced eNOS activity and NO production within the liver, has been associated with defects in eNOS protein–protein interactions and posttranslational modifications. We and others have previously identified novel eNOS interactors, including G protein-coupled receptor (GPCR) kinase interactor 1 (GIT1), which we found to play an unexpected stimulatory role in GPCR-mediated eNOS signaling. Here we report that β-arrestin 2 (β-Arr2), a canonical GPCR signaling partner, localizes in SECs with eNOS in a GIT1/eNOS/NO signaling module. Most importantly, we show that β-Arr2 stimulates eNOS activity, and that β-Arr2 expression is reduced and formation of the GIT1/eNOS/NO signaling module is interrupted during liver injury. In β-Arr2–deficient mice, bile duct ligation injury (BDL) led to significantly reduced eNOS activity and to a dramatic increase in portal hypertension compared to BDL in wild-type mice. Overexpression of β-Arr2 in injured or β-Arr2–deficient SECs rescued eNOS function by increasing eNOS complex formation and NO production. We also found that β-Arr2–mediated GIT1/eNOS complex formation is dependent on Erk1/2 and Src, two kinases known to interact with and be activated by β-Arr2 in response to GCPR activation. Our data emphasize that β-Arr2 is an integral component of the GIT1/eNOS/NO signaling pathway and have implications for the pathogenesis of sinusoidal portal hypertension.
Liver fibrosis and portal hypertension cause significant morbidity and mortality. The pathogenesis of portal hypertension includes both extrahepatic and intrahepatic factors (1) and intrahepatic portal hypertension appears to be related to endothelialopathy in injured sinusoidal endothelial cells (SECs) (2). SECs are highly specialized endothelial cells that play a prominent role in maintaining overall liver homeostasis (3, 4). Endothelial nitric oxide synthase (eNOS) expressed in liver SECs plays an essential role in the regulation of endothelial function and acts as a master regulator of vascular tone and liver blood flow through the generation of nitric oxide (NO) in liver (2). Appropriate NO synthesis by eNOS is regulated by complex posttranslational modifications and protein–protein interactions, where the balance between eNOS stimulatory and inhibitory pathways controls NO production in SECs (5). Precise control of eNOS activity is critical in regulation of portal blood flow and portal pressure in human liver disease and in animal models of liver injury (6–8).
We previously reported on a G protein-coupled receptor (GPCR) signaling pathway in SECs in which endothelin B (ETB) receptor-mediated eNOS activation and NO production are dependent on G protein βγ subunit-mediated activation of PI3K/Akt to regulate eNOS (6). After liver injury, G protein-coupled receptor kinase 2 (GRK2), an important GPCR signaling partner, is up-regulated and interacts with Akt to blunt eNOS activity (7). Additionally, we have shown that the G protein-coupled receptor kinase-interacting protein 1 (GIT1) serves as a scaffold protein to stimulate eNOS enzymatic activity, but is down-regulated after injury (8). Together, these data suggest that multiple G protein signaling pathway components are tightly integrated to regulate eNOS in health and disease.
β-Arrestins are ubiquitously expressed signaling molecules that have diverse functions in the regulation of GPCR signaling cascades. Together with GRKs, arrestins are key mediators of GPCR desensitization, endocytosis, intracellular trafficking, and resensitization (9, 10). The two β-arrestin proteins β-arrestin1 (β-Arr1) and β-arrestin2 (β-Arr2) also function as scaffolds regulating important downstream intracellular signaling cascades (11), including MAPK/Erk1/2 (12–14), Src (11, 15), and Akt (16–18). Although β-Arr2 has been previously suggested to serve as a scaffold for functional coupling of eNOS (19–21), the role of β-Arr2 in regulating eNOS interactions is not understood. In this study, we show that β-Arr2 works together with other GPCR partners to regulate eNOS function, but also that β-Arr2 down-regulation in injured SECs following liver injury contributes to reduced NO production and portal hypertension in that state. The data have distinct implications for the pathogenesis and treatment of sinusoidal portal hypertension.
Materials and Methods
Animal Model of Liver Injured and Portal Hypertension.
Sprague-Dawley rats (450 to 500 g male) were purchased from Charles River Laboratory, and β-Arr1 and β-Arr2 knockout (KO) (22) and wild-type (WT) male littermate mice on a C57BL/6J background aged 16 to 18 wk, were used in all experiments. Mice were bred using heterozygous breeding pairs to obtain KO and WT littermates. Liver injury and portal hypertension were induced by performing bile duct ligation in rats and mice as described previously (7, 23, 24). Briefly, common bile duct ligation (BDL) ligation was performed during laparotomy under anesthesia by isolating and ligating the common bile duct. Control animals were subjected to sham laparotomy without bile duct ligation. At 10 to 14 d after surgery, this animal model leads to liver injury/fibrosis and portal hypertension. All animals were cared for in the Medical University of South Carolina (MUSC) animal facility and received humane care according to the National Institutes of Health Guidelines; all experimental procedures were approved by the MUSC Institutional Animal Care and Use Committee.
Cell Isolation.
SECs and hepatic stellate cells (HSCs) were isolated from male rats or male β-Arr2 WT and KO mice as described (23, 25). In brief, the liver was perfused and digested with 1 mg/mL pronase (Roche Molecular Biochemicals) followed by 0.13 mg/mL collagenase (Worthington Biochemical Corporation) and the digested liver was agitated in a solution containing 0.2 mg/mL pronase (Roche) and 0.0375 mg/mL DNase (Roche); HSCs and SECs were separated by density gradient centrifugation after layering cells on a gradient of 8.2% and 15.6% Accudenz (Accurate Chemical and Scientific). HSCs were removed and cultured as described (25). SECs were further purified by centrifugal elutriation at an 18 mL/min flow rate and were plated on rat-tail collagen-coated dishes and grown in standard 199OR medium containing 20% serum (10% horse/calf). The purity of SECs and HSCs was assessed as described previously (24, 25). Cultures of only more than 95% purity were used for study.
Adenovirus.
Recombinant adenoviruses encoding mouse WT full-length β-Arr2, and GIT1 were constructed by Vector Biolabs. Briefly, mouse β-Arr2 or GIT1 were subcloned into a pAAV-cis plasmid, followed by preparation of the pAAV cis-plasmid and complimentary plasmids using standard cloning protocols. Adenoviruses encoding mouse WT full-length β-Arr2 (Ad β-Arr2) or GIT1 (Ad-GIT1) were grown in HEK293 cells and purified using CsCl centrifugation as described (8, 26). Adenovirus without insert DNA (Ad-empty vector [EV]) was used as a control adenovirus. For cell culture experiments, adenoviral constructs were used at a multiplicity of infection (MOI) of 250. In in vivo BDL animal experiments, adenovirus at a concentration of 1 × 1010 pfu/kg in 200 μL of phosphate-buffered saline was administered via the femoral vein. The efficiency of adenovirus infection of SECs in vivo was as previously described (26).
Isolation of Cell Fractions.
Primary SECs isolated from normal and injured (BDL) rat or from β-Arr2 WT and KO mice were harvested after cultured for 24 h. Cell compartment proteins were then separated into cytosolic (C) and membrane (M) fractions using the Fraction-PREP Cell Fractionation System (BioVision) according to the manufacturer’s protocol. Immunoblotting was performed on indicated fractions.
Immunoprecipitation Assay.
Immunoprecipitation assays were used to investigate the interaction of eNOS with GIT1 and with tyrosine phosphorylated GIT1 in SECs freshly isolated from β-Arr2 WT and KO mice. Briefly, cell lysates from SECs (200 μg of total protein) were incubated with rabbit anti-GIT1 antibody (AB) (catalog H-170, Santa Cruz Biotechnology) overnight. After GIT1 immunoprecipitation (using Protein A/G Plus-Agarose; Santa Cruz Biotechnology), immunoprecipitates were subjected to immunoblotting with mouse monoclonal anti-eNOS antibody (BD Transduction Laboratories) or to detect tyrosine phosphorylation, mouse monoclonal anti-phosphotyrosine antibody (clone 4G10, Millipore Sigma). An immunoprecipitation with IgG was utilized in each experiment as an additional control. Immunoprecipitated proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS/PAGE).
Immunoblot Analysis.
A total of 25 to 50 μg protein lysate from primary SECs (or HSCs, used as a control) were separated by SDS/PAGE and transferred to nitrocellulose and probed by the indicated primary antibodies followed by respective horseradish peroxidase (HRP)-conjugated secondary antibodies. Primary antibodies used in immunoblotting included anti-eNOS antibody (BD Transduction Laboratories), anti–phospho-eNOS-Ser1177 (BD Transduction Laboratories or Cell Signaling Technology), anti–β-arrestin1/2 (Cell Signaling), anti–β-Arr1 (Abcam), anti–β-Arr2 (Santa Cruz Biotechnology), anti-GIT1 antibody H-170 (Santa Cruz), anti-desmin antibody (Sigma), anti–phospho-Erk1/2 antibody (Cell Signaling); total Erk1/2 antibody (Cell Signaling) and HRP-conjugated secondary antibodies. Immunoreactive bands were visualized using Super Signal West Pico chemiluminescent substrate (Thermo Scientific) as per the manufacturer’s instruction and scanned and quantitated with standard software. Immunoblot images shown are representative of at least three experiments.
Immunohistochemistry and Immunofluorescence Microscopy.
Briefly, frozen rat or mouse liver sections were fixed in methanol for 5 min, permeabilized in 0.1% Triton X-100, and incubated in blocking reagent. Primary antibodies (anti–β-Arr2 [B-4] mouse monoclonal AB from Santa Cruz Biotechnology, anti–β-Arr2 goat polyclonal antibody from Abcam, anti–β-Arr1 rabbit polyclonal antibody from Cell Signaling Technology, anti-eNOS rabbit monoclonal antibody from Cell Signaling Technology, anti-eNOS mouse monoclonal antibody from BD Pharmingen, anti-ALB [P-20] goat polyclonal antibody from Santa Cruz Biotechnology, or anti-desmin rabbit polyclonal antibody from Thermo Scientific) were incubated with slices at 4 °C overnight. After washing, sections were incubated with secondary fluorescent antibodies conjugated to Alexa Fluor 555 (red, Invitrogen), Alexa Fluor 488 (green, Invitrogen), or Alexa Fluor 647 (blue, Abcam). Fluorescent images were acquired on a Zeiss Axio Observer Z1 fluorescence microscope (Carl Zeiss) with 20×/0.8 Zeiss Plan-Apochromat objective, and channel images were overlaid with Zeiss LSM Image Browser software. Immunostaining was also used to observe the colocalization of eNOS with GIT1, and eNOS with GIT1 and β-Arr2 in SECs. Briefly, primary SECs isolated from β-Arr2 WT and KO mice after transduction with the indicated adenovirus, or from rat liver, were fixed and permeabilized and then labeled with primary antibodies followed by Alexa Fluor 488 (green, Invitrogen), Alexa Fluor 555 (red, Invitrogen), or Alexa Fluor 647 (blue, Abcam). eNOS and GIT1 expression and localization were visualized using an LSM-510 Zeiss confocal microscope. eNOS with GIT1 and β-Arr2 were visualized using a Zeiss LSM 880 laser scanning confocal microscope with a 63 numerical aperture (NA), 1.4 oil-immersion plan-apochromat objective, and channel images were overlaid using Zeiss LSM Image Browser software.
Three-Dimensional Superresolution Imaging.
β-Arr2 and eNOS were immunolabeled with anti–β-Arr2 and anti-eNOS antibody as described above. β-Arr2 (Alexa Fluor 568, magenta) or eNOS (Alexa Fluor 647, green) were visualized in horizontal cross-section (X-Y) and cross-section (X-Z) with an SR-200 inverted microscope (Vutara, Inc.) with 60×/1.2 NA Olympus water immersion objective. Samples were imaged based on the single molecule localization (SML) biplane fluorescence photoactivation localization microscopy (FPALM) technology as described (27, 28) and data were analyzed using Vutara SRX software (version 3.16).
Colocalization Analysis.
Colocalization of β-Arr1 and eNOS, β-Arr1 and albumin, β-Arr1 and β-Arr2, β-Arr2 and eNOS, β-Arr2 and VE-cadherin, and β-Arr2 with GIT1 and eNOS in fluorescence images from liver sections or SECs was analyzed from the confocal images of double- or triple-labeled liver sections using the Coloc2 tool and/or scatterplots in FIJI (a version of ImageJ, http://fiji.sc), to determine the correlation between the intensities of the two-color channels at each pixel, and quantified by Pearson correlation coefficient (PCC) (29, 30). Values of PCC close to unity indicate strong colocalization, while values near zero indicate minimal colocalization.
Portal Pressure Measurement.
Portal pressure measurement was as described (7, 31). In brief, after anesthesia (ketamine 50 mg/kg i.p.), laparotomy was performed, and an i.v. catheter (18 gauge for rat, 23 gauge for mouse, Becton Dickinson Vascular Access) was inserted into the portal vein. A calibrated SPR-1000 pressure catheter (SPR-1000, ADInstruments) was directly attached to a blood-filled portal vein, and portal pressure was recorded continuously via a PowerLab 4/35 Channel Recorder (PL 3504, ADInstruments).
Nitric Oxide Measurement.
NO production was measured in conditioned medium by measuring total nitrite concentration, measured with a colorimetric Nitric Oxide Assay Kit (Abcam) as per the manufacturer’s instructions. Briefly, nitrate in the conditional medium was converted to nitrite by adding nitrate reductase and enzyme cofactor to the medium. After addition of an enhancer and Griess Reagent R1 and R2, changes in absorbance were measured at 550 nm with a microplate reader. The amount of nitrite was expressed as nanomole of nitrite per milligram protein.
NOS Activity Assay.
3H-labeled l-arginine conversion to 3H-labeled l-citrulline was assessed as described per the manufacturer’s instructions (Cayman Chemical Company). To determine NOS activity, triplicate samples of cell lysates were incubated with a reaction buffer at room temperature for 30 min, then 37 °C for 30 min in the presence or absence of 1 mM l-NAME, an inhibitor of eNOS. The reaction was terminated by the addition of stop buffer and passed over a Dowex AG 50WX-8 resin column. Radiolabeled counts per minute of generated l-citrulline were measured and used to determine specific l-NAME–inhibitable NOS activity. Each experiment includes total counts and background counts.
Statistical Analyses.
All experiments were performed in replicates using cells isolated from different rats or mice. All results were expressed as mean ± SD from at least three independent experiments. Differences between two groups were evaluated using the two-tailed Student’s t test, and comparisons among multiple groups were performed with one-way ANOVA. Statistical analysis was performed with Prism 8.0 software (GraphPad Software). No data were excluded from the analyses; P < 0.05 was considered statistically significant.
Data Sharing.
To allow others to replicate data and/or build on the published work, the authors will provide readers access to the data, associated protocols, code, and materials in the paper upon request. Unique Materials (e.g., cloned DNAs) will be made promptly available on request by qualified researchers for their own use.
Results
β-Arr2 Localization and Expression in Normal and Injured Liver.
To clarify the cellular expression and localization of β-Arr1 and β-Arr2 in whole liver, we performed immunohistochemistry in rat liver tissue utilizing cell-specific markers (albumin for hepatocytes; eNOS and VE-cadherin for SECS). Dual immunofluorescence imaging revealed that β-Arr1 and eNOS colocalized minimally (SI Appendix, Fig. S1A), with β-Arr1 being found primarily in what appear to be hepatocyte nuclei, consistent with previous data (32), while there was substantial colabeling of β-Arr1 and albumin (SI Appendix, Fig. S1B). eNOS or VE-cadherin (found only in endothelial cells in the liver) colocalized extensively with β-Arr2 (SI Appendix, Fig. S1C). These data suggest that in the liver, β-Arr1 is found primarily in hepatocytes, while β-Arr2 is found primarily in SECs.
In order to validate the specificity of anti–β-Arr1 and anti–β-Arr2 antibodies, we performed double immunofluorescence studies on livers from WT, β-Arr1 KO, and β-Arr2 KO mice. As with colabeling of β-Arr1 and eNOS (SI Appendix, Fig. S1A), there was little colocalization of β-Arr1 and β-Arr2 (SI Appendix, Fig. S2, Left). In β-Arr1 knockout mice, only background labeling of β-Arr1 was identified (SI Appendix, Fig. S2, Middle), and β-Arr2 was found in a sinusoidal pattern, typical of SEC labeling (similar to SI Appendix, Fig. S1 A and C). In β-Arr2 knockout mice, β-Arr1 was found in hepatocyte nuclei (similar to SI Appendix, Fig. S1 A and B), while β-Arr2 was not identified (SI Appendix, Fig. S2, Right). These data confirm the cellular specificity of β-Arr1 and β-Arr2 antibodies used in our studies.
To determine whether specific arrestin isoforms signaled to eNOS, we overexpressed β-Arr1 and β-Arr2 in SECs from normal rat livers and then measured eNOS activation. We found that β-Arr2 overexpression increased eNOS Ser1177 activation-associated phosphorylation, while β-Arr1 did not (SI Appendix, Fig. S3A). Conversely, eNOS activation was significantly reduced in β-Arr2 KO SECs compared to WT, in whole cell lysates and in cytosolic and membrane fractions (SI Appendix, Fig. S3 B and C).
We next measured β-Arr2 expression in SECs isolated from normal and BDL-injured livers, and found that β-Arr2 expression was dramatically reduced in SECs after liver injury (Fig. 1A, total cell lysates, and SI Appendix, Fig. S3D, primarily in cytosolic fractions). β-Arr2 phosphorylation at Ser412 was also correspondingly reduced by injury (SI Appendix, Fig. S3D). Additionally, eNOS enzymatic activity was reduced in injured cells compared to sham SECs (Fig. 1B). These data suggest that reduced β-Arr2 directly impacts eNOS function in injured SECs, and that β-Arr2 normally regulates eNOS.
Fig. 1.

β-Arr2 expression and localization in SECs in normal and injured livers. (A) SECs were isolated from normal (sham operated) and injured (by BDL) rat livers as in Materials and Methods. Total SEC protein (50 μg) was subjected to immunoblotting to detect β-Arr2 and β-actin. Specific bands corresponding to β-Arr2 were quantitated and are presented graphically as the ratio of β-Arr2 to β-actin, shown on the Bottom (n = 4/group). (B) SECs were isolated as in A, eNOS enzymatic activity was measured in cell lysates and normalized to that of normal SECs and presented graphically (the activity in normal rat SECs was set at 100; n = 4/group). (C) Immunofluorescence labeling was performed in normal and injured (BDL) rat liver sections with mouse monoclonal anti–β-Arr2/Alex Fluor 488-label (green) and rabbit polyclonal anti-eNOS/Alex Fluor 555-label (red), as described in Materials and Methods. The Left images depict representative normal liver sections, while the Right images depict representative injured liver sections. (Scale bars, 100 μm.) Images are representative of at least 10 fields in each liver. (D and E) The assessment of protein expression and colocalization of β-Arr2 and eNOS as in C were quantitated using Fiji (ImageJ) software. The integrated density (D) and PCC (E) as described in Materials and Methods are shown; changes in normal and injured livers are presented graphically (n = 6/group). Statistical significance for A, B, D, and E was evaluated using an unpaired two-tailed Student’s t test. Data are mean ± SD, *P < 0.05, **P < 0.01 for differences between indicated groups; ns, no significant difference.
We next mapped the localization and expression of β-Arr2 in situ in normal and injured whole rat liver (Fig. 1C). We found that β-Arr2 appeared reduced in the injured liver, while eNOS expression was unchanged compared to normal liver. β-Arr2 normally colocalized with eNOS in a pattern typical of SECs (in hepatic sinusoids), but there was less colabeling of β-Arr2 and eNOS after liver injury (Fig. 1 C and D). To further investigate the cellular expression of β-Arr2 in the liver, we colabeled β-Arr2 (green) and desmin (red), a marker for HSCs (33). While β-Arr2 expression was reduced after liver injury (SI Appendix, Fig. S4 A, Upper), desmin labeling was increased, consistent with stellate cell proliferation. There was minimal colabeling of β-Arr2 with desmin in either normal or injured liver (SI Appendix, Fig. S4B). We also examined β-Arr2 protein expression in HSCs and found little to no β-Arr2 protein expression or phosphorylation in either normal or injured HSCs (SI Appendix, Fig. S2C). In aggregate, the data suggest that β-Arr2 is abundantly expressed in SECs in the normal liver, but its expression is remarkably reduced in SECs in the injured liver.
Three-Dimensional Superresolution Imaging of β-Arr2/eNOS Colocalization in Normal and Injured SECs.
To examine β-Arr2 and eNOS colocalization more closely, we performed superresolution imaging of individual SECs. β-Arr2 (green) and eNOS (magenta) colocalized (white, X-Z section) in the cytoplasm of normal SECs (Fig. 2, Upper). However, β-Arr2 expression was reduced after liver injury, and eNOS and β-Arr2 interaction (white, X-Z section) was markedly reduced after liver injury (Fig. 2, Lower). Thus, these data indicate that β-Arr2 normally closely associates with eNOS.
Fig. 2.

Superresolution imaging of β-Arr2/eNOS in normal and injured SECs. SECs were isolated form normal (A) and injured (BDL, B) rat livers. β-Arr2 and eNOS were immmunolabeled with mouse monoclonal anti–β-Arr2 and rabbit polyclonal anti-eNOS antibody. β-Arr2 (Alexa Fluor 647, green) and eNOS (Alexa Fluor 568, magenta) were visualized in horizontal cross-section (X-Y), and in cross-section (X-Z) with an SR-200 inverted microscope (Vutara, Inc.) (60×/1.2 NA Olympus water immersion objective). Samples were imaged based on the SML biplane FPALM technology as described. Representative white field (Left) with the portion of the cell that is imaged at high resolution; X-Y section with eNOS labeled in magenta and β-Arr2 in green; and X-Z section images with β-Arr2 and eNOS colocalization appearing white from normal SECs (Upper) and injured SECs (Lower) are shown. (Scale bars, 1 μm.)
β-Arr2 Overexpression in Injured SECs Rescues NO Production.
Given this finding of reduced β-Arr2 expression in injured SECs, and the known reduced eNOS phosphorylation and enzymatic activity that occurs in SECs after liver injury, we attempted to rescue eNOS phosphorylation by overexpressing β-Arr2 in SECs using β-Arr2 adenovirus. We found that overexpression of β-Arr2 enhanced NO production in both normal SECs (Fig. 3A) and injured SECs (though not surprisingly, the effect was less prominent in injured SECs, Fig. 3B).
Fig. 3.

Overexpression of β-Arr2 in SECs after liver injury. SECs isolated from normal (A) and injured (BDL, B) rat livers were infected with adenovirus (MOI 250) encoding β-Arr2 (Ad-β-Arr2), an empty vector (Ad-EV), or without any treatment for 36 h. Conditioned media was collected and nitrite levels were measured as in Materials and Methods, and quantitative data are shown graphically (n = 3/group). Statistical significance for A and B was evaluated using an unpaired two-tailed Student’s t test. Data are mean ± SD, *P < 0.05, **P < 0.01 for differences between indicated groups.
β-Arr2 Deficiency Has Prominent Effects on Sinusoidal Dynamics and Physiologic Function.
To specifically address the in vivo relevance of β-Arr2 in SECs, we performed in vivo microscopy experiments (34) in β-Arr2 gene KO mice (35). The diameter of sinusoids in β-Arr2 KO mice was reduced before and (to a greater extent) after injury compared to controls (Fig. 4 A, Lower). The disruption in sinusoidal structure and function was most prominent in KO mice after liver injury (Fig. 4 A, Right Lower); quantitative analysis of the sinusoidal area revealed a significant reduction in KO mice (Fig. 4B). Additionally, portal pressure was elevated in both normal and injured β-Arr2–deficient mice (Fig. 4C). These data point to dramatic abnormalities in sinusoidal dynamics in β-Arr2–deficient mice.
Fig. 4.
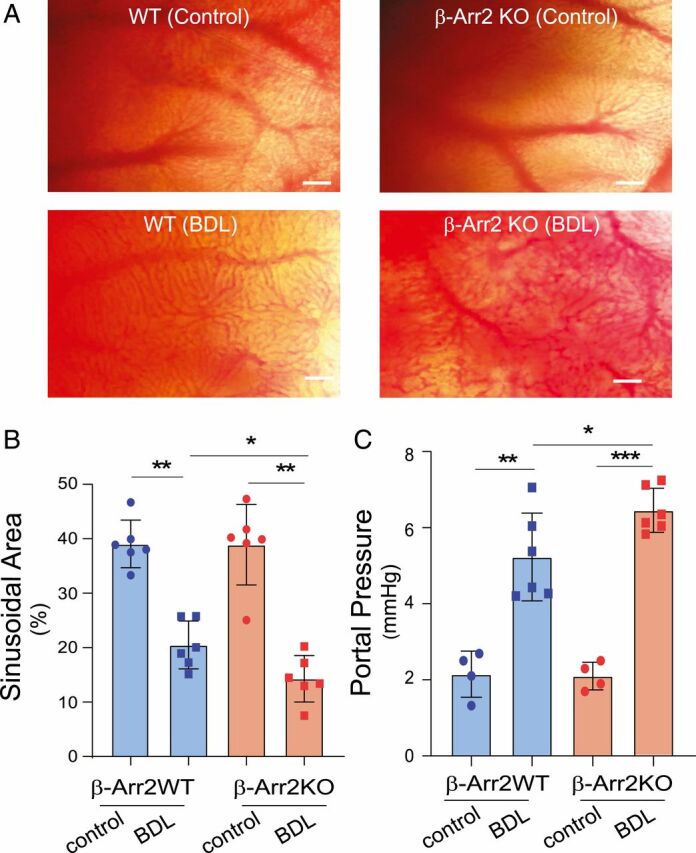
Physiologic effects of β-Arr2 deficiency. (A) Normal or injured (imaging was performed 14 d after BDL) β-Arr2 WT or β-Arr2 KO mice were anesthetized and liver lobes were examined using in vivo microscopy; images of hepatic sinusoids were obtained as described in Materials and Methods. (Scale bars, 50 μm [images are representative of more than 10 fields in each liver, from three different mice]). (B) Sinusoidal volume as in A was quantitated as described in Materials and Methods (n = 6/group). (C) Portal pressure in normal or injured β-Arr2 WT or β-Arr2 KO mice as in A was measured as described in Materials and Methods. Sham surgery was performed as a control (n = 6/group). Statistical significance for B and C was evaluated by one-way ANOVA. Data are mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.005 for differences between indicated groups.
ET-1 Stimulates β-Arr2 Translocation.
Because β-Arr2 translocates to and is activated by agonist-stimulated G protein-coupled receptors (36, 37), we examined the effect of ET-1 stimulation on β-Arr2 translocation. SECs isolated from normal livers were exposed to ET-1, leading to rapid β-Arr2 translocation from the cytoplasm to the cell membrane (Fig. 5 A, Left). However, β-Arr2 translocation was blunted in injured SECs after exposure to ET-1 (Fig. 5 A, Right). We used superresolution imaging to examine β-Arr2 (green) and eNOS (magenta) colocalization after exposure to ET-1 in normal SECs (Fig. 5 B, Left) and injured SECs (Fig. 5 B, Right). Quantitative analysis demonstrated that ET-1 stimulation increased β-Arr2 and eNOS colocalization in normal SECs, while this was significantly reduced in cells isolated after liver injury (Fig. 5C). We next examined ET-1 stimulation of eNOS phosphorylation in β-Arr2 KO mice. Lack of β-Arr2 led to blunting of ET-1 stimulation of eNOS phosphorylation (Fig. 5D). Finally, the ability of ET-1 to stimulate eNOS enzymatic activity was reduced in β-Arr2–deficient mice (Fig. 5E).
Fig. 5.

ET-1 activation of β-Arr2 and eNOS. (A) SECs from normal (Left two panels) and BDL injured (Right two panels) rat livers were cultured for 24 h and exposed to ET-1 (20 nM) for 30 min. Cells were fixed and labeled with antibody to β-Arr2 (Upper red) and nuclei were labeled with DAPI (Lower, blue). Representative images (of more than 10 others) of single and merged channels are shown. (Scale bars, 10 μm.) (B) SECs isolated from normal rat livers (Left two panels) and BDL injured (Right two panels) rat livers were cultured for 24 h and exposed to ET-1 as in A. β-Arr2 and eNOS were immmunolabeled with anti–β-Arr2 and anti-eNOS antibodies. β-Arr2 (Alexa Fluor 647, green) and eNOS (Alexa Fluor 568, magenta) were visualized in horizontal cross-section (X-Y), in cross-section (X-Z) with an SR-200 inverted microscope (Vutara, Inc.) (60×/1.2 NA Olympus water immersion objective). Samples were imaged based on the SML biplane FPALM technology as described. Representative X-Y section (Upper) and X-Z (Bottom) section images from control (Left) and exposed to ET-1 (Right) are shown. (Scale bar, 1 μm.) (C) β-Arr2 and eNOS colocalization in normal and injured SECs as in B was quantified as in Materials and Methods; the changes in normal and injured SECs are presented graphically (1 for complete and 0 for no colocalization [n = 3/group]). (D) SECs isolated from β-Arr2 WT and KO mice were exposed to ET-1 (20 nM) for 30 min. Cell lysates were subjected to immunoblotting with the indicated antibodies. Specific bands corresponding to P-eNOS were quantified and are presented as the ratio of P-eNOS to eNOS, shown on the Right (n = 3/group). (E) eNOS enzymatic activity was measured in cell lysates treated as in D and NOS activity was normalized to that of control cells from β-Arr2 WT mice without ET-1 exposure and is presented graphically (the activity in SECs from β-Arr2 WT mice without ET-1 exposure was set at 100; n = 3/group). Statistical significance for C–E was evaluated by ordinary one-way ANOVA. Data are mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.005 for differences between indicated groups; n.s., no significant difference.
GIT1 Activation of eNOS Is β-Arr2 Dependent.
We previously identified GIT1 as a potent stimulator of eNOS activity (8). In the context of these previous findings, we tested the hypothesis that β-Arr2 might facilitate GIT1 interaction with eNOS and stimulation of eNOS activity.
We initially performed localization experiments to evaluate the possibility that these three proteins co-colocalized. In triple immunofluorescence studies, eNOS, GIT1, and β-Arr2 colocalized in both whole liver sections (in a sinusoidal pattern, SI Appendix, Fig. S5A) and in isolated SECs (SI Appendix, Fig. S5B), suggesting that these three proteins are physically associated. Next, SECs isolated from β-Arr2 WT and KO mice were transduced with full-length GIT1 or β-Arr2 adenoviruses (Ad-GIT1 or Ad-β-Arr2). In these experiments, we first confirmed that overexpression of β-Arr2 in SECs led to increased eNOS activity (Fig. 6A); this was the case for SECs isolated from either WT or β-Arr2 KO mice. Remarkably, while overexpression of GIT1 in SECs from WT mice dramatically increased eNOS activation as expected (24), transduced GIT1 in β-Arr2–deficient SECs did not restore eNOS activity (Fig. 6 A, Right), suggesting that β-Arr2 is necessary for GIT1 augmentation of eNOS activity.
Fig. 6.

GIT1 regulation of eNOS/NO activity is β-Arr2 dependent. (A) SECs isolated from β-Arr2 WT and KO mice were infected with adenovirus (MOI 250) encoding β-Arr2 (Ad-β-Arr2, Left), GIT1 (Ad-GIT1, Right) or empty virus (Ad-EV) for 36 h and NOS activity was measured in cell lysates (NOS activity was normalized to that of control cells from β-Arr2 WT mice and is presented graphically) (the activity of cells from β-Arr2 WT mice was arbitrarily set to 100; n = 3/group). (B) SECs were isolated from β-Arr2 WT and KO mice, seeded in 96-well plates, and then infected with Ad-β-Arr2 or empty virus (MOI 250) as indicated. Thirty-six hours later, cells were fixed, labeled with anti-eNOS (Alexa Fluor 488, green) and anti-GIT1 (Alexa Fluor 552, red) antibodies as indicated. Representative images (of >10 others) of single and merged channels are shown. (Scale bars, 10 μm.) (C) Colocalization of GIT1 and eNOS as in B was quantified using Fiji (ImageJ) software and PCC as described in Materials and Methods and changes are presented graphically (n = 5/group). (D) SECs were isolated from β-Arr2 WT and KO mice and infected with Ad-β-Arr2 and Ad-GIT1 or Ad-EV as in A. Protein levels of phospho-eNOS (P-eNOS), phosphor-Erk1/2(P-Erk1/2), and total Erk1/2 were assessed by immunoblotting with antibodies to P-eNOS, P-Erk1/2, and total Erk1/2 (Left). Specific bands of P-eNOS (Right, Upper) or P-Erk1/2 (Right, Lower) were quantified and normalized to the level of total eNOS or Erk1/2 and presented in the graph (n = 3/group). (E) SECs isolated from β-Arr2 WT and KO mice were infected with Ad-β-Arr2 or Ad-EV as in C for 36 h. Cells were harvested, and cell lysates were subjected to immunoprecipitation (IP) with antibody to GIT1 followed by immunoblotting with antibodies to eNOS (Upper), 4G-10 (Middle), and GIT1 (Lower). On the Right, bands corresponding to eNOS-GIT1 complex (Upper) and GIT1 tyrosine phosphorylation with 4G-10 expression (Lower) were quantified and normalized to the level of immunoprecipitated GIT1 (n = 3/group). Statistical significance for A and C–E was evaluated by ordinary one-way ANOVA. Data are mean ± SD, *P < 0.05, **P < 0.01 for differences between indicated groups; n.s, no significant difference.
We examined the effect of β-Arr2 on the GIT1–eNOS interaction using dual immunolabeling; we found that eNOS and GIT1 colocalization was prominent in normal WT SECs (Fig. 6 B, Left), but reduced in β-Arr2 KO cells (Fig. 6 B, Middle). This eNOS–GIT1 colocalization was rescued by reintroduction of β-Arr2 in KO SECs (Fig. 6 B, Right and Fig. 6C). β-Arr2 transduces a number of GPCR signals, in part by acting as a scaffold for activation of the Erk1/2 (12, 38–40) and Src (9, 11) pathways. GIT1 is also a known scaffold for Erk1/2 activation (39, 41, 42) and a prominent Src substrate (43, 44), and serves to facilitate c-Src-dependent activation of MEK1-Erk1/2 in response to both GPCRs and tyrosine kinase-coupled receptors (TKRs) (41, 45). Therefore, we examined Erk1/2, and found that Erk1/2 and eNOS phosphorylation were reduced in β-Arr2–deficient cells (SI Appendix, Fig. S6A). Overexpression of GIT1 or β-Arr2 in normal SECs increased Erk1/2 and eNOS phosphorylation (Fig. 6 D, Left). However, in β-Arr2–deficient SECs, β-Arr2 partially restored Erk1/2 and eNOS phosphorylation, whereas GIT1 was unable to restore these (Fig. 6 D, Right).
To further explore the molecular mechanism underlying regulation of GIT1 and β-Arr2 interaction, we examined GIT1 tyrosine phosphorylation and eNOS–GIT1 interaction. We hypothesized that β-Arr2 might work through Erk1/2 to regulate GIT1 tyrosine phosphorylation, thus regulating eNOS–GIT1 interaction and eNOS activation. Both GIT1 tyrosine phosphorylation and eNOS–GIT1 interaction were reduced when SECs were exposed to the MEK/Erk1/2 inhibitor PD98059 (SI Appendix, Fig. S6B). Further, GIT1 tyrosine phosphorylation was reduced in β-Arr2–deficient cells, and this reduction in GIT1 tyrosine phosphorylation was rescued by overexpression of β-Arr2 (Fig. 6E). We have previously demonstrated that Src regulates GIT1 tyrosine phosphorylation and GIT1/eNOS/NO signaling (24). These data suggest that β-Arr2 works with Erk1/2 to mediate GIT1 tyrosine phosphorylation and subsequent GIT1-tyrosine–mediated GIT1–eNOS interaction that leads to eNOS activation.
Discussion
G protein-coupled receptor regulation of eNOS activity and NO production in liver SECs is traditionally conceived as being mediated primarily by the Akt/PI3K and calcium/calmodulin pathways, both of which may act at some distance from the receptor(s) that activate them (21, 46–49). Our previous studies have identified additional signaling connections between GPCRs and eNOS that are mediated by receptor regulatory proteins including GRK2 and GIT1 (6, 8, 24). In this work, we demonstrate that an additional GPCR regulatory protein, β-Arr2, is also a key player in eNOS regulation by GPCRs. That these numerous GPCR regulatory proteins appear to all act together to regulate eNOS suggests the formation of an extended signalosome around the activated receptor that directs multiple signals to eNOS within this complex to regulate its activity.
GPCR signaling to eNOS is thought to occur primarily through convergence of two distinct pathways: the G protein-dependent release of calcium (usually Gαq-PLCβ-IP3/DAG-mediated) to activate calmodulin, and the G protein-dependent activation of Akt (usually Gβγ-PI3Kγ-PIP3-PDK–mediated) to phosphorylate eNOS at Ser1177 (50) However, eNOS also is known to undergo posttranslational modification by additional protein kinases, such as Src and PKC, and to bind to other regulatory partner proteins such as caveolin-1 (51). In particular, we have shown that in SECs, eNOS activity is stimulated directly by association with GIT1 (8) and this association is promoted by tyrosine phosphorylation of GIT1 by Src kinases (24). β-Arr2 is a GPCR-regulated scaffolding protein that is intimately involved in numerous aspects of GPCR signaling, and many of these likely contribute to the ability of GPCR activators such as endothelin-1 to activate eNOS. First, β-Arr2 is a negative regulator of GPCR signaling and is known to desensitize and internalize activated endothelin receptors (52). This effect should limit eNOS activation by ET-1, and must therefore be counterbalanced by additional signaling events. Second, β-Arr2 also functions to transduce GPCR signaling pathways that are independent of G proteins, by scaffolding signaling molecules such as Src (11, 53) and MEK/Erk (54, 55). β-Arr2-facilitated endothelin receptor activation of Src family members (56) and MEK/Erk (57) are known. Interestingly, an additional pathway from β-Arr1 through ILK to Akt has been reported (58); the ability of β-Arr2 to utilize such a mechanism remains unknown, although a recent study indicated that β-Arr2 is critical for shear stress-induced Akt/eNOS activation in human vascular endothelial cells (59). Third, β-Arr2 has been reported to directly bind to eNOS in an in vitro binding assay (19), although the significance of this for eNOS function has not yet been assessed. Thus, β-Arr2 may potentially affect eNOS activity through multiple mechanisms.
Previously, we have demonstrated that ET-1 and the ETB receptor couple to the heterotrimeric G protein αq/11 (Gαq/11), leading to PI3K activation and stimulation of eNOS activity (6). We previously also found that ET-1 signaling promotes Src kinase tyrosine phosphorylation of GIT1 to activate eNOS/NO; the Src kinase inhibitor PP2 blocked ET-1-induced Src kinase activity, GIT1 tyrosine phosphorylation, and GIT1–eNOS binding (24). But we did not know how Src was regulated. In this study, we examined β-Arr2, and found that ET-1 stimulates β-Arr2 colocalization with eNOS, and that overexpression of β-Arr2 promoted GIT1–eNOS interaction in both normal and injured SECs (Fig. 5). We find that ET-1-stimulated tyrosine phosphorylation of GIT1 is β-Arr2-dependent, suggesting that the β-Arr2-Src kinase complex mediates ET-1 signaling to GIT1–eNOS to induce eNOS stimulation. β-Arr2 also scaffolds the MEK/Erk1/2 mitogen-activated kinase cascade (60). In this study, we found that ERK1/2 phosphorylation of eNOS is dependent on β-Arr2 (Fig. 6). Further, GIT1 is downstream of β-Arr2, since overexpression of GIT1 in WT SECs increases ERK1/2 and eNOS phosphorylation and eNOS activity, but GIT1 expression in β-Arr2-KO SECs is unable to augment phosphorylation or activity (Fig. 6). Additionally, overexpression of β-Arr2 together with GIT1 enhanced eNOS activity (Fig. 6). These findings suggest that the efficient GIT1–eNOS phosphorylation itself is also dependent upon on Erk1/2 activity and that Erk1/2 is critical in β-Arr2–mediated eNOS activation. Interestingly, GIT1 has also been reported to scaffold MEK/Erk downstream of GPCRs (particularly for the angiotensin II AT1A receptor) (41, 61), but does not appear to function as such a scaffold in this instance.
Our current working model is that in normal SECs, β-Arr2 facilitates GIT1 activation through Src phosphorylation, which in turn stimulates GIT1–eNOS association, forming a ternary complex of β-Arr2, GIT1, and eNOS that leads to normal NO production. This is supported by a number of pieces of data. First, we have demonstrated that β-Arr2, GIT1, and eNOS colocalize (SI Appendix, Fig. S5). Additionally, GIT1 and eNOS colocalization is reduced in β-Arr2–deficient SECs (SI Appendix, Fig. S5). In contrast, in injured SECs, down-regulation of β-Arr2 leads to reduced GIT1–eNOS interaction and therefore reduced NO production (Fig. 7). Because injury also reduces the level of GIT1, supplementation of injured SECs with β-Arr2 alone leads to only a partial rescue of activity (Figs. 3 and 5). In this model, the multiplex interactions among several receptor-proximal signaling elements (β-Arr2, GIT1, GRK2) strongly suggests that these proteins form a multiprotein signalosome to activate eNOS, and leads to further questions about how such a multiprotein complex forms and is regulated.
Fig. 7.
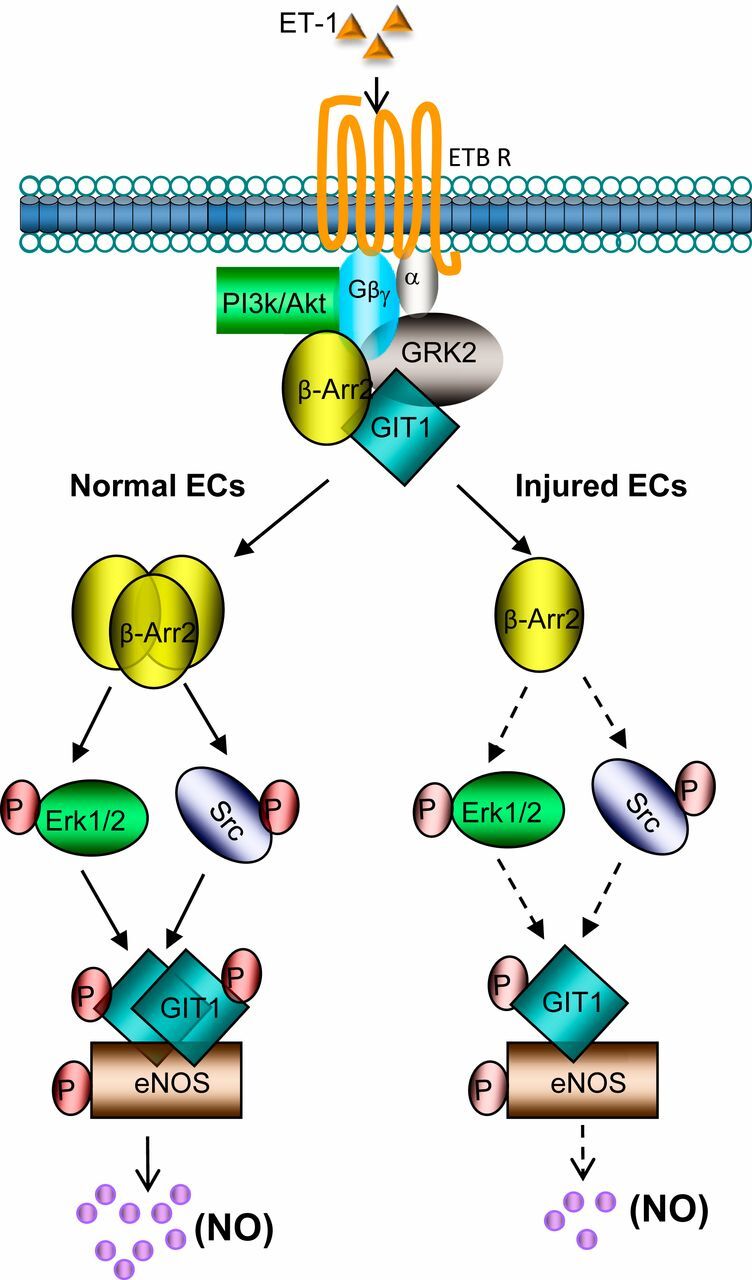
A model of β-Arr2–mediated eNOS activation through GIT1–eNOS signaling in SECs. In SECs, ET-1 activates its cognate G protein-coupled receptor (ETB receptor) and causes Gα and Gβγ activation and dissociation. In normal SECs, ETB receptor-mediated β-Arr2 recruitment of Src and activation of Erk1/2 after β-Arr2 replaces GRK2. Activated Src phosphorylates GIT1 (24). Phosphorylated GIT1 then associates with eNOS to facilitate its activating phosphorylation at Ser1177 by Akt. Receptor-activated Gβγ simultaneously stimulates Akt (at Ser473) activation through PI3-kinase and this activation may be amplified by other signaling pathways, likely through reduced GRK2/Akt interaction. In injured SECs, the reduction of β-Arr2 likely limits Src and Erk1/2-mediated GIT1 phosphorylation, with subsequent downstream effects on eNOS activity and NO production.
Here, we have shown not only that β-Arr2 is a critical mediator of eNOS activation, but also that β-Arr2 levels are regulated after liver injury. β-Arr2 expression is reduced following cholestatic injury to contribute to dysfunctional reduced eNOS activity in injured liver sinusoids. Repletion of β-Arr2 expression in injured liver partially restored eNOS activity. The mechanism underlying the down-regulation of β-Arr2 in injured SECs remains unknown, but it has been previously shown that β-Arr2 expression is transcriptionally regulated by such transcription cofactors as p300, cAMP-response element-binding protein (CREB) (62, 63), and the glucocorticoid receptor (64). Alternatively, β-Arr2 may be posttranscriptionally regulated (65), as emphasized by data showing that β-Arr2 can be up-regulated by the proteasome pathway, a process that seems to be mediated by competing with GRK2 (66). It is noteworthy that our data are consistent with previous data demonstrating that β-Arr2 levels are altered in other disease states (20, 67–70).
In summary, we have shown that β-Arr2 is a critical component of the multiprotein GPCR–eNOS signaling complex that promotes eNOS activation (Fig. 7). Additionally, the down-regulation of β-Arr2 (along with other receptor signaling complex components) after liver injury plays an important role in the reduced NO production and dysregulation of sinusoidal blood flow characteristic of the injured liver. The data indicate that dysregulated β-Arr2 is important in the pathogenesis of portal hypertension and further that it could be targeted to improve sinusoidal blood flow in disease.
Supplementary Material
Acknowledgments
We thank Katherine Robinson and Courtney Haycraft for valuable assistance with mouse colony breeding and genotyping mice; Yingyu Ren for primary cell isolation; Manasa Gudheti for assistance with Vutara three-dimensions superresolution images; Mi-Hye Lee and Li Li for assistance with confocal microscopy; Martin Romeo for providing technical support with imaging studies; Loretta Jophlin, Serhan Karvar, and Zengdun Shi for technical support; Loretta Jophlin and Shewta Singh for help with animal models; and Rebecca Lee and Shewta Singh for technical help with immunohistochemistry. This work was supported by the NIH (Grant R01 DK 113159 to D.C.R.).
Footnotes
Competing interest statement: Editor R.J.L. has coauthored papers with L.M.L. in 2019 and 2020.
This article is a PNAS Direct Submission.
This article contains supporting information online at https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.1922608117/-/DCSupplemental.
Change History
August 26, 2022: The competing interests footnote has been updated; please see accompanying Correction for details.
References
- 1.Gracia-Sancho J., Marrone G., Fernández-Iglesias A., Hepatic microcirculation and mechanisms of portal hypertension. Nat. Rev. Gastroenterol. Hepatol. 16, 221–234 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Iwakiri Y., Shah V., Rockey D. C., Vascular pathobiology in chronic liver disease and cirrhosis - current status and future directions. J. Hepatol. 61, 912–924 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Poisson J., et al., Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 66, 212–227 (2017). [DOI] [PubMed] [Google Scholar]
- 4.McCuskey R. S., Morphological mechanisms for regulating blood flow through hepatic sinusoids. Liver 20, 3–7 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Heiss E. H., Dirsch V. M., Regulation of eNOS enzyme activity by posttranslational modification. Curr. Pharm. Des. 20, 3503–3513 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu S., Premont R. T., Kontos C. D., Huang J., Rockey D. C., Endothelin-1 activates endothelial cell nitric-oxide synthase via heterotrimeric G-protein betagamma subunit signaling to protein jinase B/Akt. J. Biol. Chem. 278, 49929–49935 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Liu S., Premont R. T., Kontos C. D., Zhu S., Rockey D. C., A crucial role for GRK2 in regulation of endothelial cell nitric oxide synthase function in portal hypertension. Nat. Med. 11, 952–958 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Liu S., Premont R. T., Rockey D. C., G-protein-coupled receptor kinase interactor-1 (GIT1) is a new endothelial nitric-oxide synthase (eNOS) interactor with functional effects on vascular homeostasis. J. Biol. Chem. 287, 12309–12320 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luttrell L. M., Lefkowitz R. J., The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 115, 455–465 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Benovic J. L., et al., Functional desensitization of the isolated beta-adrenergic receptor by the beta-adrenergic receptor kinase: Potential role of an analog of the retinal protein arrestin (48-kDa protein). Proc. Natl. Acad. Sci. U.S.A. 84, 8879–8882 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luttrell L. M., et al., Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 283, 655–661 (1999). [DOI] [PubMed] [Google Scholar]
- 12.Cassier E., et al., Phosphorylation of β-arrestin2 at Τhr383 by MEK underlies β-arrestin-dependent activation of Erk1/2 by GPCRs. eLife 6, e23777 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Azuma N., et al., Endothelial cell response to different mechanical forces. J. Vasc. Surg. 32, 789–794 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Bao X., Lu C., Frangos J. A., Mechanism of temporal gradients in shear-induced ERK1/2 activation and proliferation in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 281, H22–H29 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Wang S., et al., Src is required for mechanical stretch-induced cardiomyocyte hypertrophy through angiotensin II type 1 receptor-dependent β-arrestin2 pathways. PLoS One 9, e92926 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rakesh K., et al., beta-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci. Signal. 3, ra46 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Brien W. T., et al., Glycogen synthase kinase-3 is essential for β-arrestin-2 complex formation and lithium-sensitive behaviors in mice. J. Clin. Invest. 121, 3756–3762 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beaulieu J. M., et al., An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261–273 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Ozawa K., et al., S-nitrosylation of beta-arrestin regulates beta-adrenergic receptor trafficking. Mol. Cell 31, 395–405 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taguchi K., et al., Tonic inhibition by G protein-coupled receptor kinase 2 of Akt/endothelial nitric-oxide synthase signaling in human vascular endothelial cells under conditions of hyperglycemia with high insulin levels. J. Pharmacol. Exp. Ther. 349, 199–208 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Taguchi K., Matsumoto T., Kamata K., Kobayashi T., G protein-coupled receptor kinase 2, with β-arrestin 2, impairs insulin-induced Akt/endothelial nitric oxide synthase signaling in ob/ob mouse aorta. Diabetes 61, 1978–1985 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gesty-Palmer D., El Shewy H., Kohout T. A., Luttrell L. M., beta-Arrestin 2 expression determines the transcriptional response to lysophosphatidic acid stimulation in murine embryo fibroblasts. J. Biol. Chem. 280, 32157–32167 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Liu S., Premont R. T., Singh S., Rockey D. C., Caveolin 1 and G-protein-coupled receptor kinase-2 coregulate endothelial nitric oxide synthase activity in sinusoidal endothelial cells. Am. J. Pathol. 187, 896–907 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu S., Premont R. T., Rockey D. C., Endothelial nitric-oxide synthase (eNOS) is activated through G-protein-coupled receptor kinase-interacting protein 1 (GIT1) tyrosine phosphorylation and Src protein. J. Biol. Chem. 289, 18163–18174 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh S., Liu S., Rockey D. C., Caveolin-1 is upregulated in hepatic stellate cells but not sinusoidal endothelial cells after liver injury. Tissue Cell 48, 126–132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu Q., Que L. G., Rockey D. C., Adenovirus-mediated gene transfer to nonparenchymal cells in normal and injured liver. Am. J. Physiol. Gastrointest. Liver Physiol. 282, G565–G572 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Juette M. F., et al., Three-dimensional sub-100 nm resolution fluorescence microscopy of thick samples. Nat. Methods 5, 527–529 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Mlodzianoski M. J., Juette M. F., Beane G. L., Bewersdorf J., Experimental characterization of 3D localization techniques for particle-tracking and super-resolution microscopy. Opt. Express 17, 8264–8277 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Sezgin E., et al., A comparative study on fluorescent cholesterol analogs as versatile cellular reporters. J. Lipid Res. 57, 299–309 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zinchuk V., Grossenbacher-Zinchuk O., Quantitative colocalization analysis of fluorescence microscopy images. Curr. Protoc. Cell Biol. 62, Unit 4 19 11-14 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Xie C., Wei W., Zhang T., Dirsch O., Dahmen U., Monitoring of systemic and hepatic hemodynamic parameters in mice. J. Vis. Exp. 4, e51955 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scott M. G., et al., Differential nucleocytoplasmic shuttling of beta-arrestins. Characterization of a leucine-rich nuclear export signal in beta-arrestin2. J. Biol. Chem. 277, 37693–37701 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Rockey D. C., Boyles J. K., Gabbiani G., Friedman S. L., Rat hepatic lipocytes express smooth muscle actin upon activation in vivo and in culture. J. Submicrosc. Cytol. Pathol. 24, 193–203 (1992). [PubMed] [Google Scholar]
- 34.Edwards C., Feng H. Q., Reynolds C., Mao L., Rockey D. C., Effect of the nitric oxide donor V-PYRRO/NO on portal pressure and sinusoidal dynamics in normal and cirrhotic mice. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G1311–G1317 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Bohn L. M., et al., Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 286, 2495–2498 (1999). [DOI] [PubMed] [Google Scholar]
- 36.Zhang J., et al., Cellular trafficking of G protein-coupled receptor/beta-arrestin endocytic complexes. J. Biol. Chem. 274, 10999–11006 (1999). [DOI] [PubMed] [Google Scholar]
- 37.Wang P., et al., Beta-arrestin 2 functions as a G-protein-coupled receptor-activated regulator of oncoprotein Mdm2. J. Biol. Chem. 278, 6363–6370 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Shenoy S. K., et al., beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J. Biol. Chem. 281, 1261–1273 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Yin G., Zheng Q., Yan C., Berk B. C., GIT1 is a scaffold for ERK1/2 activation in focal adhesions. J. Biol. Chem. 280, 27705–27712 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Luttrell L. M., et al., Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc. Natl. Acad. Sci. U.S.A. 98, 2449–2454 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yin G., Haendeler J., Yan C., Berk B. C., GIT1 functions as a scaffold for MEK1-extracellular signal-regulated kinase 1 and 2 activation by angiotensin II and epidermal growth factor. Mol. Cell. Biol. 24, 875–885 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Nieuw Amerongen G. P., et al., GIT1 mediates thrombin signaling in endothelial cells: Role in turnover of RhoA-type focal adhesions. Circ. Res. 94, 1041–1049 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Haendeler J., et al., GIT1 mediates Src-dependent activation of phospholipase Cgamma by angiotensin II and epidermal growth factor. J. Biol. Chem. 278, 49936–49944 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Kawachi H., Fujikawa A., Maeda N., Noda M., Identification of GIT1/Cat-1 as a substrate molecule of protein tyrosine phosphatase zeta/beta by the yeast substrate-trapping system. Proc. Natl. Acad. Sci. U.S.A. 98, 6593–6598 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rui Z., et al., GIT1Y321 phosphorylation is required for ERK1/2- and PDGF-dependent VEGF secretion from osteoblasts to promote angiogenesis and bone healing. Int. J. Mol. Med. 30, 819–825 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Lymperopoulos A., Rengo G., Funakoshi H., Eckhart A. D., Koch W. J., Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat. Med. 13, 315–323 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Cannavo A., Koch W. J., GRK2 as negative modulator of NO bioavailability: Implications for cardiovascular disease. Cell. Signal. 41, 33–40 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cannavo A., Liccardo D., Koch W. J., Targeting cardiac β-adrenergic signaling via GRK2 inhibition for heart failure therapy. Front. Physiol. 4, 264 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pronin A. N., Satpaev D. K., Slepak V. Z., Benovic J. L., Regulation of G protein-coupled receptor kinases by calmodulin and localization of the calmodulin binding domain. J. Biol. Chem. 272, 18273–18280 (1997). [DOI] [PubMed] [Google Scholar]
- 50.Sessa W. C., eNOS at a glance. J. Cell Sci. 117, 2427–2429 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Chen F., et al., PKC-dependent phosphorylation of eNOS at T495 regulates eNOS coupling and endothelial barrier function in response to G+ -toxins. PLoS One 9, e99823 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Freedman N. J., et al., Phosphorylation and desensitization of human endothelin A and B receptors. Evidence for G protein-coupled receptor kinase specificity. J. Biol. Chem. 272, 17734–17743 (1997). [DOI] [PubMed] [Google Scholar]
- 53.Yang F., et al., Allosteric mechanisms underlie GPCR signaling to SH3-domain proteins through arrestin. Nat. Chem. Biol. 14, 876–886 (2018). [DOI] [PubMed] [Google Scholar]
- 54.DeFea K. A., et al., beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol. 148, 1267–1281 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ge L., Ly Y., Hollenberg M., DeFea K., A beta-arrestin-dependent scaffold is associated with prolonged MAPK activation in pseudopodia during protease-activated receptor-2-induced chemotaxis. J. Biol. Chem. 278, 34418–34426 (2003). [DOI] [PubMed] [Google Scholar]
- 56.Imamura T., et al., beta -Arrestin-mediated recruitment of the Src family kinase Yes mediates endothelin-1-stimulated glucose transport. J. Biol. Chem. 276, 43663–43667 (2001). [DOI] [PubMed] [Google Scholar]
- 57.Cramer H., et al., Coupling of endothelin receptors to the ERK/MAP kinase pathway. Roles of palmitoylation and G(alpha)q. Eur. J. Biochem. 268, 5449–5459 (2001). [DOI] [PubMed] [Google Scholar]
- 58.Cianfrocca R., et al., Beta-arrestin-1 mediates the endothelin-1-induced activation of Akt and integrin-linked kinase. Can. J. Physiol. Pharmacol. 88, 796–801 (2010). [DOI] [PubMed] [Google Scholar]
- 59.Carneiro A. P., Fonseca-Alaniz M. H., Dallan L. A. O., Miyakawa A. A., Krieger J. E., β-arrestin is critical for early shear stress-induced Akt/eNOS activation in human vascular endothelial cells. Biochem. Biophys. Res. Commun. 483, 75–81 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Tohgo A., Pierce K. L., Choy E. W., Lefkowitz R. J., Luttrell L. M., beta-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J. Biol. Chem. 277, 9429–9436 (2002). [DOI] [PubMed] [Google Scholar]
- 61.Zhang N., Cai W., Yin G., Nagel D. J., Berk B. C., GIT1 is a novel MEK1-ERK1/2 scaffold that localizes to focal adhesions. Cell Biol. Int. 34, 41–47 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang X., He J. Q., Ding L., Paré P. D., Sandford A. J., Promoter polymorphism and expression of beta-arrestin 2 in neutrophils. Clin. Chim. Acta 385, 79–80 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Ma L., Pei G., Beta-arrestin signaling and regulation of transcription. J. Cell Sci. 120, 213–218 (2007). [DOI] [PubMed] [Google Scholar]
- 64.Oakley R. H., Revollo J., Cidlowski J. A., Glucocorticoids regulate arrestin gene expression and redirect the signaling profile of G protein-coupled receptors. Proc. Natl. Acad. Sci. U.S.A. 109, 17591–17596 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Becuwe M., Herrador A., Haguenauer-Tsapis R., Vincent O., Léon S., Ubiquitin-mediated regulation of endocytosis by proteins of the arrestin family. Biochem. Res. Int. 2012, 242764 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Penela P., Elorza A., Sarnago S., F. Mayor, Jr, Beta-arrestin- and c-Src-dependent degradation of G-protein-coupled receptor kinase 2. EMBO J. 20, 5129–5138 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thathiah A., et al., β-arrestin 2 regulates Aβ generation and γ-secretase activity in Alzheimer’s disease. Nat. Med. 19, 43–49 (2013). [DOI] [PubMed] [Google Scholar]
- 68.Sun W. Y., et al., Down-regulation of β-arrestin2 promotes tumour invasion and indicates poor prognosis of hepatocellular carcinoma. Sci. Rep. 6, 35609 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grange-Midroit M., et al., G protein-coupled receptor kinases, beta-arrestin-2 and associated regulatory proteins in the human brain: Postmortem changes, effect of age and subcellular distribution. Brain Res. Mol. Brain Res. 101, 39–51 (2002). [DOI] [PubMed] [Google Scholar]
- 70.McCrink K. A., et al., β-Arrestin2 improves post-myocardial infarction heart failure via sarco(endo)plasmic reticulum Ca2+-ATPase-dependent positive inotropy in cardiomyocytes. Hypertension 70, 972–981 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.