

Abstract
BACKGROUND
Arrhythmogenic cardiomyopathy (ACM) is a variably penetrant disease increasingly identified in young patients.
OBJECTIVES
This study sought to describe the diverse phenotype, genotype, and outcomes in pediatric and adolescent patients.
METHODS
Records from 1999 to 2016 were reviewed for individuals age <21 years with a consistent personal or family history. Patients were categorized by right ventricular (RV), left dominant (LD), or biventricular subtypes using 2010 Task Force Criteria or proposed features of LD disease, encompassing electrocardiographic, structural, histological, and arrhythmic characteristics. Genetic variants classified as pathogenic and/or likely pathogenic by 2015 American College of Medical Genetics and Genomics criteria in recognized disease-associated genes were included.
RESULTS
Manifest disease was evident in 32 patients (age 15.1 ± 3.8 years), of whom 22 were probands, including 16 RV, 7 LD, and 9 biventricular ACM. Nondiagnostic features were seen in 5 of 15 family members. RV disease was associated with cardiac arrest and ventricular tachycardia (p = 0.02) and prevalence of PKP2 variants (p < 0.01), whereas biventricular disease was associated with a younger age of onset (p = 0.02). LD ACM was associated with variants in DSP and LMNA, and biventricular ACM with more a diverse etiology in desmosomal genes. Cardiac arrest was observed in 5 probands (age 15.3 ± 1.9 years) and ventricular tachycardia in 10 (age 16.6 ± 2.7 years), 6 probands, and 4 family members. Features suggestive of myocardial inflammation were seen in 6 patients, with ventricular tachycardia and/or cardiac arrest in 3 patients. Cardiac transplantation was performed in 10 patients. There were no deaths. In RV and biventricular disease, electrocardiographic preceded imaging features, whereas the reverse was seen in LD disease.
CONCLUSIONS
ACM in the young has highly varied phenotypic expression incorporating life-threatening arrhythmia, heart failure, and myocardial inflammation. Increased awareness of early onset, aggressive disease has important implications for patient management and familial screening.
Keywords: arrhythmogenic right ventricular cardiomyopathy, desmosomes, diagnostic criteria, genetics, pediatrics, phenotype
Arrhythmogenic cardiomyopathy (ACM) is a hereditable disorder characterized by an early propensity to ventricular arrhythmia initially disproportionate to the degree of ventricular dysfunction, with subsequent deterioration of ventricular function (1). Initially identified as a disease of the right ventricle (RV) and named accordingly as arrhythmogenic right ventricular cardiomyopathy (ARVC) or ARV dysplasia, left dominant (LD) and biventricular subtypes have since been recognized (2,3). This variability suggests an extensive morphological disease spectrum primarily linked by genetic association to the 5 desmosomal proteins fundamental to intercar-diomyocyte adhesion and communication. A similar phenotype has also been associated with nondesmosomal proteins and genes including the intermediate filament desmin (DES), the nuclear envelope constituents lamin A/C (LMNA), and transmembrane protein-43 (TMEM-43) (4–6). The electrocardiographic (ECG), structural, and arrhythmic abnormalities associated with both desmosomal and nondesmosomal genes show variable penetrance and expressivity, with a clinical phenotype determined by genetic, environmental, and stochastic components (7).
ACM displays age-related penetrance with the disease typically only becoming manifest in the fourth decade of life. Rare cases are reported in probands presenting during childhood and adolescence, with only small case series reported in the published reports. Although rare, the disease is recognized in children, and one-sixth of the overall disease population may show features during adolescence with a significant burden of life-threatening arrhythmias (8,9). Prior studies to date have focused on RV disease (8) or disease evolution in family members (10). Given the increased understanding of the phenotypic variability of ACM, we sought to assess the clinical manifestations and associated genetic findings in all patients with a consistent personal or familial phenotype who presented under the age of 21 years.
METHODS
Databases at Boston Children’s Hospital were reviewed for probands or patients screened due to family members with a history consistent with a clinical syndrome of ACM and who were <21 years of age at the time of initial clinical contact. Investigations included pedigree analysis; resting, exercise, and ambulatory ECG; echocardiography, cardiac magnetic resonance (CMR) imaging; and RV fluoroscopic angiography. Individuals are referenced by family letter and patient number.
PHENOTYPIC CHARACTERIZATION.
Individuals were characterized phenotypically using standard ECG, imaging, and histological investigations (Online Table 1) and categorized as affected or unaffected. Affected individuals were divided into 3 subtypes as follows (3):
Classic, right-sided ACM (referred to subsequently as ARVC), defined as isolated RV disease or left ventricular (LV) involvement in the presence of notable RV enlargement and/or dysfunction. Diagnosis was established using the 2010 International Task Force criteria (TFC) (11).
Left dominant ACM, with prominent LV manifestations in the setting of relatively mild right-sided disease. Patients with at least 2 of the proposed diagnostic criteria were included (12).
Biventricular ACM, characterized by equal, bilateral involvement with no apparent predilection for either ventricle, with features of either the 2010 TFC or proposed LD diagnostic criteria.
LV involvement was defined by identification of at least 1 of the following: ejection fraction (EF) <55%; epicardial late gadolinium enhancement (LGE); end-diastolic volume ≥120 ml/m2; wall motion abnormalities; and T-wave inversion in leads 1, aVL, and V5 and V6 (3). Patient age at LV involvement was also recorded. For patients categorized with LD or biventricular disease, RV involvement was considered as at least 1 of the following: RVEF <40%; epicardial LGE; end-diastolic volume ≥120 ml/m2 in male and ≥110 ml/m2 in female subjects; wall motion abnormalities; and T-wave inversion in leads V2 and V3.
The arrhythmia phenotype was defined for each patient as the most significant arrhythmia documented, defined in escalating order as ≥500 premature ventricular complexes per 24 h, nonsustained VT of at least 3 beats at ≥120 beats/min, sustained monomorphic VT (≥30 s), and ventricular fibrillation and/or cardiac arrest (3). The patient age at appearance of repolarization or depolarization abnormalities on 12-lead resting ECG, structural features on echocardiography or CMR, and arrhythmic features as defined by either the TFC or LD criteria was recorded. Features recorded within 1 month of each other were considered simultaneous. A clinical syndrome of myocardial inflammation was inferred from chest pain with elevated serum troponin with or without ST-segment changes on ECG, in the absence of fever and an infectious or other identified etiology.
Family members identified with the same assumed disease-causing sequence variant as the clinically affected proband were assessed for nondiagnostic disease features (i.e., not meeting full 2010 TFC or <2 LD criteria).
The follow-up duration was from the date of first evaluation to most recent clinical encounter for those under continued clinical care or was censored at the date of last contact or cardiac transplantation.
GENETIC SEQUENCING.
Genetic sequencing was performed in Clinical Laboratory Improvement Amendments-certified laboratories between 2008 and 2017 using contemporaneous sequencing technology. Genetic variants considered potentially causative in the following genes were included: the desmosomal genes plakophilin-2 (PKP2), desmoplakin (DSP), desmocollin-2 (DSC2), desmoglein-2 (DSG2), and plakoglobin (JUP); and the nondesmosomal genes transmembrane protein 43 (TMEM-43), desmin (DES), and lamin A/C (LMNA) in cases where the individual or familial phenotype was consistent with ACM. Variant classification was performed using the 2015 American College of Medical Genetics and Genomics guidelines to ensure uniform methodology (13), and only pathogenic and likely pathogenic variants were included as part of the 2010 TFC.
STATISTICAL ANALYSIS.
Continuous variables are expressed as mean ± SD and categorical variables as number (percentage). Differences between disease subtypes were assessed using analysis of variance for continuous variables and Fisher exact test for categorical variables. The Student’s t-test was used to assess statistical differences between 2 groups. A p value of <0.05 was considered statistically significant.
RESULTS
In total, 47 patients (24 female) from 36 families were identified; 22 were probands and 25 family members. A clinical diagnosis of ARVC, LD ACM, or biventricular ACM was made in 32 patients at age 15.1 ± 3.8 years. An additional 15 family members were evaluated and did not fulfill diagnostic criteria. Twenty-seven individuals were symptomatic (all probands and 5 family members), with the mean age at first symptom of 14.4 ± 4.0 years. A family history of ARVC or ACM was present in 31 of 36 families, a history of sudden cardiac death in 7 families, and death from heart failure in 2 families.
The phenotype identified was ARVC in 16 patients, LD ACM in 7 patients, and biventricular in 9 patients, the specific details of which are discussed herein. The age at diagnosis was significantly younger in biventricular ACM than in LD or ARVC (p = 0.02) (Central Illustration). Electrocardiographic disease features were evident prior to or concordant with imaging features in all cases of ARVC or biventricular disease. In patients with LD disease, imaging features preceded ECG findings in 5 of 7 individuals.
CENTRAL ILLUSTRATION. Phenotype and Genotype Data From 32 Patients With Manifest Arrhythmogenic Cardiomyopathy.

The 3 cardiac phenotypes are shown, including arrhythmogenic right ventricular cardiomyopathy (ARVC) with thinning of the ventricular myocardium, right ventricular dilatation, and aneurysms; left dominant arrhythmogenic cardiomyopathy (ACM) with thinning of the left ventricular myocardium; and biventricular ACM with involvement of both ventricles including marked left ventricular dilatation. The specific demographic and phenotypic features along with the pertinent genetic findings are shown for each subtype. *Variant of unknown significance. +1 patient represented in both cardiac arrest and ventricular tachycardia groups.
Cardiac arrest occurred in 5 patients (all probands) at 15.3 ± 1.9 years of age and sustained VT in 14 patients (10 probands) at 16.7 ± 2.7 years of age. Both arrhythmias were more prevalent in ARVC (12 of 16; 75%) than in LD (3 of 7; 43%) or biventricular (2 of 9; 22%) disease (p = 0.03). A pathogenic or likely pathogenic variant in PKP2 was more prevalent in ARVC (12 of 12; 100%) than in LD (0 of 6; 0%) or biventricular (3 of 9; 33%) ACM (p < 0.01). The genetic and clinical findings are summarized in the Central Illustration and Tables 1 and 2.
TABLE 1.
Genetic Findings in Patients Categorized by Clinical Phenotype
| Demographics |
Genetic Variants |
||||||
|---|---|---|---|---|---|---|---|
| Family | Patient # | Status | Family History | Gene | Nucleotide | Amino Acid | Classification ACMG 2015 |
| ARVC | |||||||
| A | 1 | P | Yes | PKP2 | c.1755_1756insTTGACTCA | L586fsX658 | Pathogenic |
| B | 1 | P | Yes | PKP2 | c.2203C>T | R735X | Pathogenic |
| C | 1 | P | Yes | PKP2 | c.1821dupT | V608CfsX6 | Pathogenic |
| C | 2 | FM | Yes | PKP2 | c.1821dupT | V608CfsX6 | Pathogenic |
| D | 1 | FM | Yes | PKP2 | c.1613G>A | W538X | Pathogenic |
| E | 1 | P | No | ||||
| F | 1 | FM | Yes | PKP2 | c.1511–1 G>A | Likely pathogenic | |
| PKP2 | c.976 G>A | A326T | VUS | ||||
| G | 1 | P | Yes | PKP2 | c.2489+1 G>A | Pathogenic | |
| DSC2 | c.1028_1030deLTTA | ILe343deL | VUS | ||||
| H | 1 | P | Yes | PKP2 | c.2062T>C | S688P | VUS |
| H | 2 | FM | Yes | Not tested | |||
| I | 1 | P | Yes | PKP2 | c.1237C>T | R413X | Pathogenic |
| J | 1 | P | Yes | PKP2 | c.1237C>T | R413X | Pathogenic |
| DSC2 | c.996T>G | Y332X | Likely pathogenic | ||||
| K | 1 | P | Yes | PKP2 | c.951deLG | H318TfsX2 | Likely pathogenic |
| L | 1 | P | Yes | PKP2 | c.1237C>T | R413X | Pathogenic |
| M | 1 | P | No | ||||
| N | 1 | FM | Yes | PKP2 | c.2489+1 G>A | Pathogenic | |
| LD ACM | |||||||
| O | 1 | P | Yes | DSP | c.3526deLG | V1176fsX20 | Likely pathogenic |
| DSG2 | c.1003A>G | T355A | VUS | ||||
| P | 1 | FM | Yes | Not tested | |||
| Q | 1 | P | No | DSP | c.2920deLA | T974fsX3 | Likely pathogenic |
| R | 1 | FM | Yes | LMNA | c.1621 C>T | R541C | Likely pathogenic |
| R | 2 | FM | Yes | LMNA | c.1621 C>T | R541C | Likely pathogenic |
| S | 1 | FM | Yes | LMNA | c.637C>T | R225X | Pathogenic |
| T | 1 | P | Yes | DSP | c.3434deLC | A1145fsX14 | Likely pathogenic |
| BiV ACM | |||||||
| U | 1 | P | Yes | DSP | c.1873C>T | Q625X | Likely pathogenic |
| DSP | c.6442G>A | A2148T | VUS | ||||
| V | 1 | P | Yes | DSG | c.152G>T | W51L* | VUS |
| V | 2 | FM | Yes | DSG2 | c.152G>T | W51L* | VUS |
| W | 1 | P | Yes | PKP2 | c.2509deLA | S837VfsX94* | Pathogenic |
| X | 1 | P | No | PKP2 | c.1034+1 G>T | Pathogenic | |
| Y | 1 | P | No | DSP | c.478C>T | R160X | Pathogenic |
| DSP | c.8020G>A | A2647T | |||||
| Z | 1 | P | Yes | DSC2 | c.631–2 A>G | Pathogenic | |
| AA | 1 | P | Yes | PKP2 | c.1162C>T | R388W | VUS |
| PKP2 | c.2301deLC | G769LfsX31 | Likely pathogenic | ||||
| BB | 1 | P | No | DES | c.347A>G | N116S | Likely pathogenic |
| Unaffected family members | |||||||
| CC | 1 | FM | Yes | DSP | c.3474_3475insA | G1159RfsX3 | Pathogenic |
| DD | 1 | FM | Yes | DSP | c.478C>T | R160X | Pathogenic |
| DD | 2 | FM | Yes | DSP | c.478C>T | R160X | Pathogenic |
| EE | 1 | FM | Yes | PKP2 | c.1211_1212insT | V406SfsX4 | Pathogenic |
| EE | 2 | FM | Yes | PKP2 | c.1211_1212insT | V406SfsX4 | Pathogenic |
| FF | 1 | FM | Yes | PKP2 | c.2146–1 G>C | Pathogenic | |
| GG | 1 | FM | Yes | DES | c.735 +1G>A | Likely pathogenic | |
| HH | 1 | FM | Yes | TMEM43 | c.1073C>T | S358L | Pathogenic |
| W | 2 | FM | Yes | PKP2 | c.2509deLA | S837VfsX94 | Pathogenic |
| W | 3 | FM | Yes | PKP2 | c.2509deLA | S837VfsX94 | Pathogenic |
| U | 2 | FM | Yes | DSP | c.1873C>T | Q625X | Likely pathogenic |
| II | 2 | FM | Yes | LMNA | c.961C>T | R321X | Pathogenic |
| LMNA | c.992G>A | R331G | VUS | ||||
| JJ | 1 | FM | Yes | LMNA | c.1621 C>T | R541C | Likely pathogenic |
| J | 2 | FM | Yes | DSC2 | c.996T>G | Y332X | Likely pathogenic |
| C | 3 | FM | Yes | PKP2 | c.1821dupT | V608CfsX6 | Pathogenic |
Homozygous.
ACM = arrhythmogenic cardiomyopathy; ACMG = American College of Medical Genetics and Genomics; ARVC = arrhythmogenic right ventricular cardiomyopathy; BiV = biventricular; DES =desmin; DSC2 =desmocollin-2; DSP =desmoplakin; FM = family member; LD = left dominant; LMNA =lamin A/C; P = proband; PKP2 =plakophilin-2; TMEM43 =transmembrane protein 43; VUS = variant of unknown significance.
TABLE 2.
Clinical Findings in Patients Categorized by Clinical Phenotype
|
ARVC |
||||||||
|
Demographics |
2010 Task Force Criteria (11) |
|||||||
| Family | Patient # | Age at Diagnosis, yrs | Structural | Tissue | Repolarization | Depolarization | Arrhytdmia | Family History and Genetics |
| A | 1 | 13.9 | ++ | ++ | + | ++ | ||
| B | 1 | 15.2 | ++ | + | + | ++ | ||
| C | 1 | 16.8 | ++ | ++ | + | + | ++ | |
| C | 2 | 18.3 | + | + | ++ | |||
| D | 1 | 15.0 | ++ | + | + | ++ | ||
| E | 1 | 14.5 | ++ | ++ | + | + | ||
| F | 1 | 22.1 | + | ++ | + | + | ++ | |
| G | 1 | 14.2 | ++ | + | + | ++ | ||
| H | 1 | 20.2 | + | + | ++ | |||
| H | 2 | 21.8 | + | ++ | + | + | ||
| I | 1 | 18.4 | ++ | + | + | ++ | ||
| J | 1 | 12.0 | ++ | + | + | + | ++ | |
| K | 1 | 16.5 | ++ | ++ | + | ++ | ||
| L | 1 | 14.3 | ++ | + | + | ++ | ||
| M | 1 | 17.6 | ++ | + | + | |||
| N | 1 | 16.1 | + | + | ++ | |||
|
LD ACM |
||||||||
|
Clinical Features of LD ACM (3) |
||||||||
| ECG | Arrhythmia | Imaging | CMR | Biopsy | Genetics | |||
| O | 1 | 13.1 | † | † | † | † | ||
| P | 1 | 12.9 | † | † | ||||
| Q | 1 | 16.8 | † | † | † | † | ||
| R | 1 | 14.6 | † | † | † | † | † | † |
| R | 2 | 13.0 | † | † | † | † | ||
| S | 1 | 17.6 | † | † | † | † | ||
| T | 1 | 16.9 | † | † | † | |||
|
BiV ACM |
||||||||
|
2010 Task Force Criteria (11) |
||||||||
| Structural | Tissue | Repolarization | Depolarization | Arrhythmia | Family History and Genetics | |||
| U | 1 | 16.0 | ++ | ++ | ++ | + | + | ++ |
| V | 1 | 15.0 | ++ | ++ | + | + | ||
| V | 2 | 18.8 | ++ | ++ | + | + | ||
| W | 1 | 4.9 | ++ | + | + | + | ++ | |
| X | 1 | 16.3 | ++ | ++ | + | ++ | + | ++ |
| Y | 1 | 8.9 | ++ | ++ | + | ++ | ||
| Z | 1 | 14.7 | ++ | ++ | ++ | + | + | ++ |
| AA | 1 | 5.6 | + | + | ++ | |||
| BB | 1 | 11.6 | ++ | ++ | ++ | + | ||
Repolarization indicates abnormalities of repolarization on ECG, and depolarization indicates abnormalities of depolarization on ECG.
= Major criteria;
= minor criteria;
= LD ACM criteria present
CMR = cardiac magnetic resonance; ECG = electrocardiography; other abbreviations as in Table 1.
PHENOTYPIC CHARACTERIZATION ARVC.
Sixteen patients (6 female) presented at 14.9 ± 2.0 years of age. There were 11 probands, whose age at first symptom was 14.7 ± 2.2 years, including 3 with cardiac arrest, 5 with VT, 2 with arrhythmic syncope, and 1 with palpitations. All 16 patients fulfilled TFC (excluding genetic findings) at age 16.7 ± 2.9 years and probands at 15.1 ± 2.4 years compared with family members at 18.7 ± 3.2 years. Arrhythmia was documented prior to diagnosis in only 1 proband (H1) (Figure 1). An additional 2 patients had nonsustained VT documented during follow-up. Four patients, all family members, were asymptomatic at diagnosis. Features of LV disease were seen in 5 probands and 2 family members, present at diagnosis in 6, with no difference in age at diagnosis in those with (16.3 ± 3.4 years) compared with those without (17.0 ± 2.7 years) LV involvement. Features suggestive of myocardial inflammation (troponin T 2.16 ng/ml; normal <0.01) were seen in a 14-year old female proband (G1) who presented with VT with manifest disease meeting diagnostic criteria, with chronic inflammation and fibrosis evident on myocardial biopsy, and biventricular LGE on CMRI imaging.
FIGURE 1. Arrhythmic Phenotype Preceding ECG Features in Classical Right-Sided ACM.

(A) A 12-lead electrocardiograph (ECG) from a 17-year-old Haitian American male patient (H1) who presented following recurrent syncopal episodes, with normal depolarization and repolarization parameters. Cardiac magnetic resonance showed normal ventricular volumes and systolic function with nonspecific changes in the right ventricular free wall. An electrophysiology study including programmed ventricular stimulation in the presence of isoproterenol was negative, and a loop recorder was implanted. (B) He experienced a further pre-syncopal episode with documented monomorphic ventricular tachycardia at 300 beats/min. On subsequent investigation, his sister fulfilled the International Task Force criteria for classical right-sided disease. ACM = arrhythmogenic cardiomyopathy.
LD ACM.
LD ACM was seen in 7 patients (3 female) who were 15.0 ± 2.1 years of age at diagnosis. Three probands first presented with symptoms at the age of 14.9 ± 3.5 years, 2 of whom (O1, Q1) presented with symptoms consistent with recurrent myocardial inflammation, associated with repeated cardiac arrest in 1 case (Q1) (Figures 2A to 2D). Both were diagnosed with myocarditis at initial presentation. Other presentations included VT in 1 case, exertional dyspnea in 1, and cascade screening in 3. At presentation, LV function was mildly depressed in 6 patients (EF: 46.0 i 4.4%) and normal in 1 proband, who initially presented at 11 years of age subsequently developed LV dysfunction (EF: 41%) 3 years later. In 6 patients who underwent CMR LGE imaging, there was extensive enhancement exclusively affecting the LV, with a distribution pattern ranging from epicardial to transmural. No patient had features of structural RV disease.
FIGURE 2. ECG and Imaging Manifestations of Myocardial Inflammation Associated With a Desmoplakin Truncating Variant.
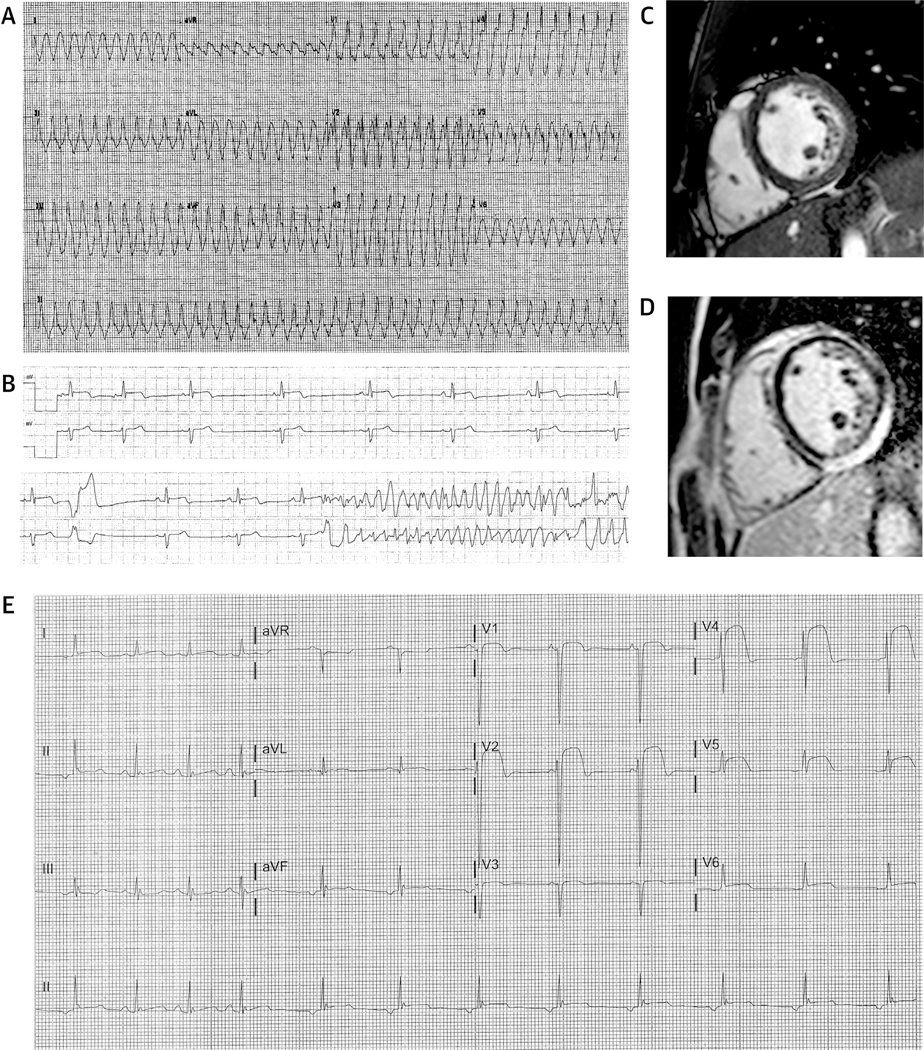
(A) A 12-lead electrocardiogram (ECG) recorded from a 16-year-old-male patient (Q1) who presented with a cardiac arrest after a week-long history of chest pain, diagnosed as myocarditis. The ECG shows a broad complex tachycardia with right bundle morphology consistent with left ventricular tachycardia. (B) He represented 5years later with further chest pain and elevated troponin levels (troponin T: 2.95 ng/ml) and experienced an in-hospital cardiac arrest. Note ST-segment changes in the top rhythm strip. (C, D) Cardiac magnetic resonance performed shortly after showed mild leftventricular dilatation and with appropriate systolic wall thickening (mid-ventricular short-axis cine image in end-diastole [C]) with a large region of epicardial and/or mid-myocardial late gadolinium enhancement in the inferior and inferolateral segments of the left ventricle. (E) A 12-lead ECG from a 10-year-old male proband (BB1) who presented with chest pain and troponin release (troponin T: 2.75 ng/ml; normal: <0.01 ng/ml) demonstrating severe ST-segment elevation across numerous leads.
Arrhythmia findings were evident in all cases: ventricular fibrillation in 1; VT in 2; and nonsustained VT in 4. Two patients (R1, R2) developed cardiac failure necessitating cardiac transplantation 2.6 and 3.9 years after initial presentation, with severe LV dilatation, extensive transmural fibrosis, and wall thinning evident on explant pathology.
Biventricular ACM.
Biventricular ACM was present in 8 probands (4 females), who met 2010 International TFC at 11.6 ± 4.6 years of age. A single family member presented with diagnostic features at 18 years of age. LV dysfunction (EF: 29 ± 15%) was evident in all at initial evaluation. CMR was performed in 5 patients with extensive biventricular LGE and associated regional wall motion abnormalities present in 4 (Figures 3A to 3D). Arrhythmic findings included sustained in 2 cases and nonsustained VT in 7 cases.
FIGURE 3. Biventricular ACM.

Cardiac magnetic resonance cine images in end-diastole from a 14-year-old male patient (Z1) who presented with severe cardiac failure: (A) 4-chamber view; (B) 2-chamber left ventricular (LV) view; and (C) 3-chamber right ventricular view. There was severe biventricular enlargement (LV end-diastolic volume: 247 ml/m2, right ventricular end-diastolic volume: 340 ml/m2) and severe biventricular systolic dysfunction (LV and right ventricular ejection fractions: <10%). (D) There were multiple segments with late gadolinium enhancement and wall thinning including the LV apex. After support with an LV device, he underwent successful cardiac transplantation. (E, F) Gross pathological assessment showed extensive chamber dilatation (note LV assistive device site) with marked myocardial thinning and fibrofatty replacement. (G) Additional histological analysis (hematoxylin and eosin stain) again showed extensive fibrofatty replacement. (H) Genetic testing identified a previously described splice acceptor variant in desmocollin-2, which exposed a cryptic splice acceptor site with subsequent premature truncation (33). His asymptomatic mother with a normal clinical evaluation carried the same variant (Table 3). ACM = arrhythmogenic cardiomyopathy; EA = extracellular anchor domain; EC1 = extracellular domain 1; IA = intracellular anchor domain; ICS = intracellular cadherin-typical sequence; SSPro = signal peptide/propetide sequence; TM = transmembrane domain.
Cardiac transplant was performed in all 8 probands at 12.5 ± 4.2 years of age, 4 of whom required prior mechanical support. Explant pathology in 7 cases showed extensive biventricular dilatation and wall thinning with extensive and frequently transmural fibrofatty infiltration and fibrosis (Figures 3E to 3G).
Features consistent with myocardial inflammation were seen in 3 patients and was the first manifestation in a 10-year-old Hispanic male (BB1) (Figure 2E), who presented with recurrent symptomatic episodes with subsequent development of marked biventricular dysfunction, regional dyskinesia, and LGE. Endocardial and epicardial myocardial biopsies demonstrated extensive myocyte degeneration with chronic inflammatory changes, and on electron microscopy, irregular pale desmosomes in clusters and loss of close cell-to-cell apposition at the intercellular junction, features previously associated with the disease (14). In 2 others (U1, AA1), troponin elevation suggestive of myocardial inflammation was evident in the setting of advanced disease.
Two patients presented in the neonatal period. The first (W1), homozygous for a pathogenic PKP2 truncating variant, had severe biventricular dysfunction following uneventful congenital heart surgery to correct a ventricular septal defect and aortic coarctation. Her older brother died from intractable heart failure reportedly associated with the same congenital lesions at age 5 months, although his genetic status was unknown. A maternal uncle died suddenly in his 30s. At 3 years of age, she represented with atrial fibrillation and developed rapidly progressive heart failure necessitating biventricular assist devices and transplantation at age 4 years. A second patient (AA1) required extracorporeal support at age 3 days for cardiogenic shock associated with LV noncompaction (non-compacted-to-compacted ratio: 5:1). After initial recovery, there was gradual deterioration in biventricular function from the age of 5 years, with recurrent chest pain and elevated troponin levels (troponin I: 2.57 ng/ml; normal <0.04 ng/ml) consistent with myocardial inflammation from the age of 6, and resultant cardiac transplantation at 10 years of age. Genetic testing identified both truncating and missense variants in PKP2.
NONDIAGNOSTIC FINDINGS.
There were 15 (10 female) unaffected family members age 15.7 ± 5.2 years who did not reach diagnostic criteria. All were asymptomatic and identified by definition through cascade screening, first performed at age 10.1 ± 4.3 years with follow-up over 4.9 ± 3.0 years. Two individuals had terminal activation delay >55 ms on ECG, and 3 had coarse, curled hair in the setting of a cardiocutaneous phenotype in multiple family members.
GENETIC SEQUENCING.
Genetic sequencing was performed in 45 patients (96%), using single gene in 3 patients, desmosomalin 9 patients, or broadercardiomyopathy panels in 11 patients when testing probands. Of 54 variants identified, 42 were pathogenic or likely pathogenic in 40 patients (18 probands [82%]), of which 37 (88%) inferred loss of function, in the heterozygous (31 probands [76%]), compound heterozygous (5 probands [12%]), digenic (2 probands [5%]), and homozygous (3 probands [7%]) state.
Individual variants not classified as pathogenic or likely pathogenic included a homozygous DSG2 missense variant in 2 siblings both with a biventricular phenotype, inferred to be autosomal recessive inheritance as both parents were asymptomatic but declined further investigations. A PKP2 missense variant (S688P) was considered of unknown significance although has been associated with the disease previously (15,16).
DISCUSSION
This study demonstrates a number of important features pertinent to ACM in children and young adults (Central Illustration). First, while clinical expression typically occurs in the fourth decade of life, ACM may be fully manifest in young individuals with highly varied phenotype including ventricular arrhythmia, features consistent with recurrent myocardial inflammation, and cardiac failure. Second, there is marked variability in the underlying phenotype characterized by right, left, and biventricular disease variants; this necessitates a high degree of clinical suspicion, appropriate diagnosis, and management in patients presenting with consistent clinical features, specifically given the risk of life-threatening arrhythmias, which may be heightened in those with LV involvement and associated DSP variants (17). Finally, consistent with the experience in adult patients (12,15), the phenotypic variability appears to be concordant with genetic findings, where ARVC is associated with a high proportion of PKP2 variants, whereas other subtypes are associated with a more diffuse genetic etiology, specifically DSP and LV disease. Collectively these data suggest phenotypic variability results from genetic heterogeneity and the respective roles of proteins within the desmosomal complex and interaction with other molecular components of the cardiac intercalated disk.
NATURAL HISTORY.
The natural history of the disease has been considered to consist of 4 sequential clinicopathological phases (18), suggesting the earlier phases would be more evident in younger patients. The first, a “concealed” phase that occurs prior to the onset of classical ECG, histological, and structural changes, is characterized by acute exacerbations of myocardial inflammation and life-threatening ventricular arrhythmias (12,19,20).
In this cohort, features consistent with myocardial inflammation were evident in 6 patients with associated sustained VT and with cardiac arrest in 3 patients, although this was not confirmed pathologically. Importantly, features of myocardial inflammation were seen not only as an initial disease manifestation, but also in the presence of advanced cardiomyopathy, suggesting ongoing inflammation may be an important contributor to disease progression and arrhythmogenesis. The role of inflammation is increasingly recognized as a fundamental component of disease pathogenesis (12,16). Elevated levels of myocardial and circulating inflammatory mediators have been identified in clinically affected individuals with ACM, and recapitulation of plakoglobin cytoplasmic translocation has been demonstrated in functional models by the addition of interleukins-6 and −17 and tumor necrosis factor-α (21,22).
Potential arrhythmia mechanisms in concealed disease have been studied in detail. In murine models, PKP2 haploinsufficiency leads to ultrastructural changes, altered sodium channel kinetics and potentially impaired wave front conduction and arrhythmia (23). A reduced immunoreactive signal for sodium channels, connexin-43, and the desmosomal protein plakoglobin have been identified in human myocardium (24) and may represent an early and integral step in the disease process prior to ECG and structural changes (22). More recently, PKP2 has been identified as critical to the transcription of several cardiomyocyte proteins key to cellular calcium homeostasis, suppression of which promotes an arrhythmogenic state that can be suppressed with flecainide (25). The clinical correlate of this phenomenon could be confirmed by the identification of pathogenic genetic variants in sudden death cases with a normal autopsy (26), although ultrastructural and cellular changes or wider familial evaluation supporting variant pathogenicity have been described to date. In this study, and that of Te Riele et al. (8), a diagnosis of ACM was evident in all cases except 1 at the time of either cardiac arrest or VT—even in the youngest case who experienced a cardiac arrest age 11—suggesting that in the vast majority of cases identified to date, life-threatening arrhythmias occur in the presence rather than absence of structural and electrocardiographic changes.
Subsequent stages include a second, arrhythmic phase characterized by diffuse myocardial fibrosis with functional and structural RV remodeling, and a third phase of RV dysfunction and failure (17). A small minority progress to a fourth stage of biventricular dilatation and congestive heart failure. Our experience suggests that in the young, ACM represents a broad clinical spectrum with different clinicopathological features presenting at varying times in the clinical course, rather than a progressive process.
Two patients in our cohort presented with neonatal cardiac failure, a period of recovery, and then later disease manifestation, raising the question of whether their initial presentation was a first, early manifestation of ACM. Homozygous ablation of PKP2 in embryonic mice is associated with gestational lethality secondary to grossly abnormal cardiac morphogenesis and altered trabeculation within the primitive ventricular myocardium, implicating PKP2 as key regulator in embryonic cardiac development (27). Congenital cardiac anomalies have been reported in another child homozygous for a PKP2 loss of function variant (28). Whether a significant reduction or complete loss of PKP2 function could lead to abnormal cardiac morphogenesis or inadequate compaction of ventricular myocardium is intriguing, although to date neither sequence variation in PKP2 nor copy number variants encompassing 12p11.21 have been identified in patients with congenital heart disease or convincingly implicated in associated developmental pathways (29).
DIAGNOSIS IN CHILDREN.
In agreement with prior studies, we found existing criteria accurately reflect the underlying ECG and structural changes in the young and therefore appear appropriate for diagnostic purposes (8,9). We included patients with variants in LMNA and DES reflecting the wider spectrum of the disease beyond desmosomal genes, and although both may be associated with a highly variable cardiac phenotype, all variants included in this study have been previously associated with a clinical picture consistent with ACM (4,5).
In both ARVC and biventricular disease, ECG features were present either with, or before, structural changes evident on cardiac imaging, suggesting that in the absence of ECG features the utility of cardiac imaging is limited in identifying early disease expression. Conversely in LD ACM, mild LV dysfunction, epicardial LGE, and wall motion abnormalities often preceded repolarization abnormalities on the surface ECG, consistent with the findings of a prior study that identified LV epicardial LGE prior to any other morphological or kinetic abnormalities (9). These findings would be better validated by serial evaluation of individuals at risk by virtue of familial or genetic predisposition.
GENETIC FINDINGS.
We identified a high prevalence of associated genetic variants across the different subtypes of ACM in a young population, a finding consistent with a prior study in young patients (8) that is considerably higher than adult populations (15), potentially explained by the lack of phenocopies such as sarcoid. In a number of families (Table 3), an asymptomatic parent was identified who carried the same genetic variant, with no identifiable phenotype or only very subtle features consistent with the notoriously low disease penetrance (Figure 3H). Why expression should be evident in younger rather than older individuals remains unclear, although this is predicated on the assumption that the identified variant was truly causal.
TABLE 3.
Families With Affected Children and Unaffected, Asymptomatic Parents With Concordant Genetic Findings
| Family | Proband Age, yrs (Sex) | ACM Subtype | Clinical Features | Parental Age, yrs (Sex) | Genetic Variants |
|---|---|---|---|---|---|
| G | 14(F) | ARVC | Sustained ventricular tachycardia; myocardial inflammation on biopsy | 52 (F) |
PKP2 c.2189+1G>A DSC2 Ile343del |
| J | 11 (F) | ARVC | Cardiac arrest | 51 (F) |
PKP2 R413X DSC2 Y332X |
| L | 14 (M) | ARVC | Syncope | 41 (M) | PKP2 R413X |
| O | 12 (F) | Left dominant | Features of myocardial inflammation, NSVT and decreased LV function | 40 (F) |
DSP V1176fsX20 DSG2 T355A |
| Z | 14 (M) | Biventricular | Cardiac failure and transplantation | 45 (F) | DSG2 c.631–2A>G |
| X | 16 (M) | Biventricular | Cardiac failure and transplantation | 52 (F) | PKP2 c.1034+1 G>T |
DSG2 = desmoglein-2; F = female; LV = left ventricular; M = male; NSVT = nonsustained ventricular tachycardia; other abbreviations as in Table 1.
Various studies have addressed the differing contributions of desmosomal and other genetic variants together with environmental influences in disease penetrance and arrhythmic risk (7). The role of endurance athletics as an environmental modifier has recently been implicated in both adults and children (30), although the duration and intensity of exercise needed to increase arrhythmic risk varies significantly between studies (31). The retrospective nature of our study made formal analysis of exercise intensity and duration complex, with the potential for significant recall bias. Although a proportion of patients reported here were high-school level athletes, many were not, and so the degree to which endurance athletics can be implicated in such early disease expression is difficult to separate from other influences. The negative influence of ascertainment bias in determining disease severity and survival in inherited heart disease related to founder mutations has recently been described (32), so to truly assess the relative effects of differing genetic and environmental factors, comprehensive genetic and clinical analysis needs to be performed across a wide spectrum of individuals.
STUDY LIMITATIONS.
Referrals to our center may be biased such that more severely affected cases are seen, and therefore overemphasize the clinical and genetic spectrum of disease in families where a younger patient is affected compared with others with disease onset evident in adult life. Ascertainment of cases was performed in a nonrandom, retrospective manner and is therefore subject to significant bias and inclusion of only select cases. Less severely affected probands may have been undetected or misdiagnosed, thereby underestimating the disease severity and variability. Additionally, cascade screening across families was incomplete, such that the genetic and clinical status of many family members is unknown. A family history and presence of disease-associated genetic variants are a component of the international TFC, allowing family members to reach a definitive diagnosis with a less evident or severe phenotype. We attempted to limit this by using clinical criteria alone, and ultimately only 1 family member was considered affected. In the absence of definitive histological evidence, myocardial inflammation can only be inferred from the constellation of chest pain, elevated serum troponins, and electrocardiographic ST-segment changes.
CONCLUSIONS
The symptoms and clinical phenotype of ACM in the young are highly varied, encompassing a wide disease spectrum that significantly overlaps with other cardiac diagnoses including myocarditis and cardiac failure, which has important implications for accurate diagnosis and management. Disease expression is similar to that seen in the adult population, and while the evolution of different diagnostic features varies with disease subtype, the application of diagnostic criteria appear appropriate for younger patients. Importantly, ARVC was exclusively associated with genetic variants in PKP2, LV disease with variants in DSP and LMNA, and the biventricular phenotype with a more diverse genetic etiology encompassing varying desmosomal genes. The genetic and environmental factors that lead to such early expression require a more comprehensive and detailed understanding.
Supplementary Material
PERSPECTIVES.
COMPETENCY IN MEDICAL KNOWLEDGE:
ACM in the young presents with diverse phenotypes, with right, left, or biventricular subtypes. The genetic basis and phenotypic vari ability of the disease appears similar to that in adults, with n tations primarily in genes encoding cardiac desmosomal proteins.
TRANSLATIONAL OUTLOOK:
The environmental and gens factors that lead to early, severe expression of ACM in young individuals require further investigation.
Acknowledgments
The Inherited Cardiac Arrhythmia Program is funded by the generous philanthropic support of the Mannion and Roberts families. Dr. Lakdawala has acted as a consultant for Array BioPharma and Myokardia. Dr. MacRae is supported by a grant from the Leducq Foundation. Dr. Abrams has acted as a consultant for Audentes Therapeutics.
ABBREVIATIONS AND ACRONYMS
- ACM
arrhythmogenic cardiomyopathy
- ARVC
arrhythmogenic right ventricular cardiomyopathy
- CMR
cardiac magnetic resonance
- DES
desmin
- DSP
desmoplakin
- ECG
electrocardiographic
- EF
ejection fraction
- LD
left dominant
- LGE
late gadolinium enhancement
- LMNA
lamin A/C
- LV
left ventricle
- PKP2
plakophilin-2
- RV
right ventricle
- TFC
International Task Force criteria
- TMEM43
transmembrane protein 43
Footnotes
APPENDIX For a supplemental table, please see the online version of this paper.
All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
REFERENCES
- 1.Sen-Chowdhry S, McKenna WJ. Reconciling the protean manifestations of arrhythmogenic cardiomyopathy. Circ Arrhythm Electrophysiol 2010; 3:566–70. [DOI] [PubMed] [Google Scholar]
- 2.Marcus FI, Fontaine GH, Guiraudon G, et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation 1982;65:384–98. [DOI] [PubMed] [Google Scholar]
- 3.Sen-Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E, McKenna WJ. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation 2007;115:1710–20. [DOI] [PubMed] [Google Scholar]
- 4.van Tintelen JP, Van Gelder IC, Asimaki A, et al. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm 2009;6:1574–83. [DOI] [PubMed] [Google Scholar]
- 5.Quarta G, Syrris P, Ashworth M, et al. Mutations in the lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy.EurHeart J 2012;33:1128–36. [DOI] [PubMed] [Google Scholar]
- 6.Hodgkinson KA, Connors SP, Merner N, et al. The natural history of a genetic subtype of arrhythmogenic right ventricular cardiomyopathy caused by a p.S358L mutation in TMEM43. Clin Genet 2013;83:321–31. [DOI] [PubMed] [Google Scholar]
- 7.Sen-Chowdhry S, Syrris P, Pantazis A, Quarta G, McKenna WJ, Chambers JC. Mutational heterogeneity, modifier genes, and environmental influences contribute to phenotypic diversity of arrhythmogenic cardiomyopathy. Circ Cardiovasc Genet 2010;3:323–30. [DOI] [PubMed] [Google Scholar]
- 8.Te Riele ASJM, James CA, Sawant AC, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy in the pediatric population: clinical characterization and comparison with adult-onset disease. J Am Coll Cardiol EP 2015;1:551–60. [Google Scholar]
- 9.Daliento L, Turrini P, Nava A, et al. Arrhythmogenic right ventricular cardiomyopathy in young versus adult patients: similarities and differences. J Am Coll Cardiol 1995;25:655–64. [DOI] [PubMed] [Google Scholar]
- 10.Bauce B, Rampazzo A, Basso C, et al. Clinical phenotype and diagnosis of arrhythmogenic right ventricular cardiomyopathy in pediatric patients carrying desmosomal gene mutations. Heart Rhythm 2011;8:1686–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation 2010;121: 1533–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sen-Chowdhry S, Syrris P, Prasad SK, et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol 2008;52:2175–87. [DOI] [PubMed] [Google Scholar]
- 13.Richards S, Aziz N, Bale S, et al. , for the ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Basso C, Czarnowska E, Della Barbera M, et al. Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right ventricular cardiomyopathy: an electron microscopy investigation on endomyocardial biopsies. Eur Heart J 2006;27:1847–54. [DOI] [PubMed] [Google Scholar]
- 15.Groeneweg JA, Bhonsale A, James CA, et al. Clinical presentation, long-term follow-up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. Circ Cardiovasc Genet 2015;8:437–46. [DOI] [PubMed] [Google Scholar]
- 16.Martins D, Ovaert C, Khraiche D, Boddaert N, Bonnet D, Raimondi F. Myocardial inflammation detected by cardiac MRI in arrhythmogenic right ventricular cardiomyopathy: a paediatric case series. Int J Cardiol 2018;271:81–6. [DOI] [PubMed] [Google Scholar]
- 17.Blusztein DI, Zentner D, Thompson T, et al. Arrhythmogenic right ventricular cardiomyopathy: a review of living and deceased probands. Heart Lung Circ 2019;28:1034–41. [DOI] [PubMed] [Google Scholar]
- 18.Corrado D, Fontaine G, Marcus FI, et al. , for the Study Group on Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy of the Working Groups on Myocardial and Pericardial Disease and Arrhythmias of the European Society of Cardiology. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: need for an international registry. Circulation 2000;101:E101–6. [DOI] [PubMed] [Google Scholar]
- 19.Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res 2010;107:700–14. [DOI] [PubMed] [Google Scholar]
- 20.Bauce B, Basso C, Rampazzo A, et al. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur Heart J 2005;26: 1666–75. [DOI] [PubMed] [Google Scholar]
- 21.Asimaki A, Tandri H, Duffy ER, et al. Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol 2011;4: 743–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaplan SR, Gard JJ, Protonotarios N, et al. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease). Heart Rhythm 2004;1:3–11. [DOI] [PubMed] [Google Scholar]
- 23.Cerrone M, Noorman M, Lin X, et al. Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc Res 2012;95:460–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noorman M, Hakim S, Kessler E, et al. Remodeling of the cardiac sodium channel, connexin43, and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm 2013;10:412–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cerrone M, Montnach J, Lin X, et al. Plako-philin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat Commun 2017;8:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shanks GW, Tester DJ, Ackerman JP, et al. Importance of variant interpretation in whole-exome molecular autopsy: population-based case series. Circulation 2018;137:2705–15. [DOI] [PubMed] [Google Scholar]
- 27.Grossmann KS, Grund C, Huelsken J, et al. Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J Cell Biol 2004;167:149–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verhagen JMA, van den Born M, Kurul S, et al. Homozygous truncating variant in PKP2 causes hypoplastic left heart syndrome. Circ Genom Precis Med 2018;11:e002397. [DOI] [PubMed] [Google Scholar]
- 29.Jin SC, Homsy J, Zaidi S, et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet 2017;49:1593–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.James CA, Bhonsale A, Tichnell C, et al. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/ cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol 2013;62:1290–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruwald AC, Marcus F, Estes NA 3rd., et al. Association of competitive and recreational sport participation with cardiac events in patients with arrhythmogenic right ventricular cardiomyopathy: results from the North American multidisciplinary study of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J 2015;36:1735–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nannenberg EA, van Rijsingen IAW, van der Zwaag PA, et al. Effect of ascertainment bias on estimates of patient mortality in inherited cardiac diseases. Circ Genom Precis Med 2018;11: e001797. [DOI] [PubMed] [Google Scholar]
- 33.Heuser A, Plovie ER, Ellinor PT, et al. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 2006; 79:1081–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.