


Abstract
Gene silencing by RNA interference (RNAi) has emerged as a powerful treatment strategy across a potentially broad range of diseases. Tailoring siRNAs to silence genes vital for cancer cell growth and function could be an effective treatment, but there are several challenges which must be overcome to enable their use as a therapeutic modality, among which efficient and selective delivery to cancer cells remains paramount. Attempts to use antibodies for siRNA delivery have been reported but these strategies use either nonspecific conjugation resulting in mixtures, or site-specific methods that require multiple steps, introduction of mutations, or use of enzymes. Here, we report a method to generate antibody–siRNA (1:2) conjugates (ARCs) that are structurally defined and easy to assemble. This ARC platform is based on engineered dual variable domain (DVD) antibodies containing a natural uniquely reactive lysine residue for site-specific conjugation to β-lactam linker-functionalized siRNA. The conjugation is efficient, does not compromise the affinity of the parental antibody, and utilizes chemically stabilized siRNA. For proof-of-concept, we generated DVD-ARCs targeting various cell surface antigens on multiple myeloma cells for the selective delivery of siRNA targeting β-catenin (CTNNB1). A set of BCMA-targeting DVD-ARCs at concentrations as low as 10 nM revealed significant CTNNB1 mRNA and protein knockdown.
INTRODUCTION
RNA-mediated post-transcriptional gene silencing, known as RNA interference (RNAi), enables the specific knockdown of any transcribed gene, making it a commonly used technique in basic research. From a therapeutic standpoint, RNAi has the advantage of being able to target any disease-associated RNA-based and RNA-encoded factor, typically a protein translated from mRNA. Notably, many of these RNAi targets are considered ‘undruggable’ by small molecules. Furthermore, the sequence-specific target recognition makes off-target activity and toxicity less of a concern. As one of several RNAi-based strategies, short interfering RNAs (siRNAs) are fully complementary to the target mRNA sequence and are introduced into target cells as a duplex. After entering cells, the siRNA is loaded into an RNA-induced silencing complex (RISC). During the loading process, the passenger (sense) strand is removed and the guide (antisense) strand remains within the RISC where it binds to its complementary site on the target mRNA. The bound mRNA is cleaved by the nuclease activity of RISC and further degraded by intracellular nucleases (1). This process is catalytic, enabling cycles of mRNA binding and degradation resulting in highly potent siRNAs with IC50 values in the single-digit picomolar range (2).
Although siRNAs are highly efficient at gene silencing, there are two key challenges that have to be overcome to enable their use as therapeutics. First, the size and high negative charge prevent passive uptake of siRNAs into cells. Second, unmodified siRNAs have a short half-life under physiological conditions due to rapid degradation by extracellular and intracellular nucleases. For certain tissues, such as the eye and lung, tissue-specific delivery can be achieved by local administration of siRNA via intravitreal injection and inhalation, respectively. For siRNA delivery to the liver via systemic intravenous administration, tremendous progress over the past years has yielded several clinically validated delivery technologies, which have been shown to be safe and effective in humans. One system comprises multi-component lipid nanoparticles (LNPs) in which the siRNAs are encapsulated during most of their journey (3,4). The LNPs are designed to release their siRNA payload into the cytoplasm of hepatocytes, where they can engage with the RISC machinery. The first FDA-approved RNAi-based therapy, patisiran (ONPATTRO®; Alnylam Pharmaceuticals), is an siRNA-loaded LNP for the treatment of the polyneuropathy of hereditary transthyretin amyloidosis (hATTR) (5). Another approach utilizes a trivalent N-acetylgalactosamine (GalNAc) ligand covalently conjugated to the siRNA. The ligand is designed to bind with high affinity and specificity to the asialoglycoprotein receptor (ASGPR), a cell surface lectin expressed on hepatocytes. In addition to the utilization of GalNAc ligands, the development of chemically stabilized siRNA has been critical for the systemic administration of unencapsulated siRNA (6,7). The first RNAi therapeutic based on the GalNAc-siRNA approach, givosiran (GIVLAARI®, Alnylam Pharmaceuticals), was recently approved by the FDA for the treatment of acute hepatic porphyria.
Despite advances in RNAi-based therapies directed towards the liver, the ability to target other tissues is essential for broadening the range of suitable indications including cancer. Monoclonal antibodies (mAbs) are particularly well suited as delivery vehicles due to their high affinity and specificity towards antigens expressed on target cells and their long circulatory half-life. These properties have contributed towards mAbs being a highly successful class of pharmaceuticals with currently >60 FDA-approved antibody-based therapeutics followed by a vast clinical and preclinical pipeline (8,9). Furthermore, mAbs represent an already clinically validated delivery approach for cytotoxic payloads in cancer therapy. This includes seven FDA-approved antibody-drug conjugates (ADCs) which selectively deliver highly toxic small molecules to cancer cells (10). Thus, antibody–siRNA conjugates (ARCs) may offer a promising strategy for the targeted delivery of siRNAs to specific cells. The antibody component of ARCs enables prolonged circulatory half-life and, if properly designed for internalization, intracellular uptake while the siRNA component expands the targetome of antibody-based cancer therapy. Several methods to prepare ARCs have been reported but these strategies use (i) nonspecific conjugation (11–17) resulting in mixtures or (ii) site-specific methods that require either multiple steps and the introduction of mutations (15,18,19) or the use of enzymes such as microbial transglutaminase (20).
We recently reported a site-specific antibody-drug conjugate (ADC) platform that is based on engineering dual-variable-domain (DVD) antibodies composed of an outer variable light (VL) and heavy chain (VH) domain pair (Fv) that selectively targets a cell surface antigen of interest and an inner Fv derived from the anti-hapten mAb h38C2 (Figure 1A) which contains a uniquely reactive lysine (Lys) residue at the bottom of an 11-Å deep hydrophobic pocket (Figure 2) (21). Due to its distinctive environment, this Lys residue (pKa ∼ 6) is unprotonated and highly nucleophilic at physiological pH and reacts specifically with β-lactam functionalized hapten derivatives. When made as DVD-IgG1, there are two binding sites (outer Fv) and two drug attachment sites (inner Fv) within one DVD antibody molecule (Figure 1B). We used the DVD platform to generate highly homogeneous ADCs that potently and selectively killed tumor cells in vitro and in vivo (22). Thus, we envisioned this platform to be highly attractive for siRNA conjugation for the targeted knockdown of mRNA for therapeutic intervention (Figure 1C). For proof-of-concept, we selected multiple myeloma (MM), a hematologic malignancy characterized by aberrant growth of plasma cells in the bone marrow (23). This indication was a suitable place to start as there are several cell surface antigens expressed in MM that have been successfully targeted by ADCs in clinical trials (10).
Figure 1.

ARC assembly platform. (A) Aldolase antibody h38C2 in IgG1 format. Light chains are shown in white, heavy chains in gray. Variable light (VL) and heavy chain domains (VH) are depicted with their three complementarity-determining regions (CDRs). The variable fragment (Fv; VL+VH) of h38C2 contains a uniquely reactive Lys (K) residue (pKa ∼ 6) at the bottom of an 11-Å deep hydrophobic pocket which constitutes the hapten binding and catalytic site (green). (B) DVD-IgG1s in this study graft an outer Fv that binds to a cell surface antigen (such as CD138, BCMA and SLAMF7; Supplementary Figure S1) on top of h38C2 IgG1. (C) Site-specific conjugation of an siRNA to the reactive lysine residue of the DVD-IgG1 yields structurally defined antibody–siRNA conjugates (ARCs) with an siRNA-to-antibody ratio of 2. The siRNA is shown with its sense (passenger) strand in blue and its antisense strand in red. (D) Using a DVD-Fab for site-specific conjugation yields a structurally defined ARC with an siRNA-to-antibody ratio of 1. (E) ARCs are assembled by incubating the DVDs with 2–10 eq of siRNA derivatized with a β-lactam hapten group at the 3′ or 5′ of its sense (passenger) strand. Conjugation to the ϵ-amino group of the reactive Lys residue under mild conditions (PBS pH 7.4) yields a stable amide group and is complete after 2 h at RT.
Figure 2.

Three-dimensional structure of catalytic antibody h38C2 Fab. Based on the recently published three-dimensional structure of the Lys99Arg mutant of h38C2 Fab determined by X-ray crystallography at 2.4-Å resolution (PDB ID: 6U85) (43), a model of the parental catalytic antibody h38C2 was calculated. In the surface rendering model on the left, the Fab's heavy chain (VH-CH1) is shown in teal and the light chain (VL-CL) in gold. The position of the ∼11-Å deep and ∼450-Å3 large hydrophobic pocket between VH and VL is marked by a dotted box and enlarged in the center showing the buried Lys99 residue of VH at the bottom. Shown on the right as ribbon diagram is a top view into the cleft.
MATERIALS AND METHODS
Biological and chemical components
The identification numbers of all biological and chemical components and their conjugates are listed in Supplementary Table S1.
Single-strand siRNA synthesis
We utilized our standard siRNA duplex design (4–7) with 21 nucleosides in the sense (SS) strand and 23 nucleosides in the antisense (AS) strand with a two-nucleoside overhang at the 3′-end of the AS strand (Scheme 1). The 5′- and 3′-termini of AS strands and the 5′- and 3′-termini of SS strands (except for the 3′-conjugates 4, 6 and 7) each contained two phosphorothioate linkages. Four sense (4ss, 5ss, 5ss-bio and 6ss) and two antisense strands (4as targeting CTNNB1 and 6as targeting TTR) (Scheme 1, Supplementary Table S2) were synthesized on an ÄKTA Oligopilot 100 (GE Healthcare) using commercially available 5′-O-(4,4′-dimethoxytrityl)-2′-deoxy-2′-fluoro-, 5′-O-(4,4′-dimethoxytrityl)-2′-O-(tert-butyldimethylsilyl)- and 5′-O-(4,4′-dimethoxytrityl)-2′-O-methyl-3′-O-(2-cyanoethyl-N,N-diisopropyl) phosphoramidite monomers of uridine, 4-N-acetylcytidine, 6-N-benzoyladenosine, and 2-N-isobutyrylguanosine. For introduction of the biotin moiety (5ss-bio), 1-dimethoxytrityloxy-3-O-(N-biotinyl-3-aminopropyl)-triethyleneglycolyl-glyceryl-2-O-(2-cyanoethyl)-(N,N-diisopropyl)-phosphoramidite was used. For oligonucleotides 5ss and 5ss-bio, a N-(aminocaproyl)prolinol-4-phosphate modification was placed at the 5′ end, where the amine was protected with a trifluoroacetic acid (TFA) group. The oligonucleotide was cleaved and deprotected in 50/50 (v/v) solution of AMA [30% (w/v) aqueous ammonia + 40% (w/v) aqueous methylamine] for 3 h at RT. Oligonucleotides 4ss and 6ss had a similar N-(aminocaproyl)-4-hydroxyprolinol modification at the 3′ terminus, which was introduced on controlled pore glass (CPG) support, and were TFA protected and cleaved the same way. Oligonucleotides 4as and 6as have a 5′-(E)-vinylphosophonate-2′-N-methylacetamide-5′-methyluridine monomer, which was introduced and cleaved using previously published protocols (24).
Scheme 1.

Structures of siRNAs and linkers. Shown from top to bottom are the structures of β-lactam (blue) functionalized siRNA at the 3′ (4) and 5′ (5) end targeting human β-catenin (CTNNB1), the structure of control β-lactam functionalized siRNA at the 3′ end (6) targeting human transthyretin (TTR), and the structure of control siRNA targeting CTNNB1 but lacking the β-lactam moiety (7). The siRNAs (4–6) were prepared by reaction of the corresponding sense strands (4-6ss) with symmetrical bis-β-lactam linker (8) followed by annealing with the corresponding antisense strands (4as, 6as). Black circles stand for 2′-OMe and 2′-F-modified nucleosides of the corresponding CTNNB1 and TTR sequences (Supplementary Table S2), orange circles for 2′-O-[2-(methylamino-2-oxoethyl]-5-methyl-uridine containing a 5′-vinylphosphonate (VP) moiety. Yellow bars denote phosphorothioate linkages for exonuclease protection.
After cleavage, all oligonucleotides were filtered through a 0.45-μm filter to remove solid residues, and the support was rinsed with water (1.5 ml/μmol of solid support). The crude ligand-conjugated and unconjugated oligonucleotides were purified by anion-exchange high-performance liquid chromatography (IEX-HPLC) with TSK-Gel Super Q-5PW support (TOSOH) using a linear gradient of 22–42% buffer B over 130 min with 50 ml/min flow rate [buffer A: 0.02 M Na2HPO4 in 10% (v/v) CH3CN, pH 8.5, and buffer B: buffer A plus 1 M NaBr). All oligonucleotides were purified to >85% HPLC (260 nm) purity and then desalted by size exclusion chromatography (SEC) on an ÄKTA Prime instrument using an AP-2 glass column (20 × 300 mm; Waters) custom-packed with Sephadex G25 (GE Healthcare) and sterile nuclease-free water as the mobile phase. The isolated yields for the oligonucleotides were calculated based on the respective ratios of measured to theoretical 260 nm optical density units. The integrities of the purified oligonucleotides were confirmed by LC-MS (Supplementary Table S2) and by analytical IEX-HPLC. Prior to annealing with their respective antisense strands (see below), the four sense strands 4ss, 5ss, 5ss-bio and 6ss were derivatized with a β-lactam linker as described in the following paragraph.
Linker synthesis, conjugation and validation
Among known options of reversible (β-diketone) and irreversible (β-lactam) covalent conjugation of a cargo to the reactive Lys residue (25), we chose the latter to ensure stability of the DVD-ARCs. To better fit in the hydrophobic pocket, the β-lactam linker should contain a phenyl ring which is a structural fragment of the β-diketone-functionalized hapten that had been used to generate a panel of catalytic antibodies with uniquely reactive lysine residues by reactive immunization (26). After initial test experiments with N-p-methoxybenzoyl-β-lactam we found that it reacts instantaneously and quantitatively with benzylamine at ambient conditions in the presence of water in acetonitrile with opening of the β-lactam ring by the primary amine while being relatively inert to water or hydroxyl anions present in the mixture. This result suggested that we can use an excess of a symmetrical linker containing two terminal N-aroyl-β-lactam moieties (bis-β-lactam linker 8) (Scheme 1) for selective functionalization of a sense strand of a siRNA containing a primary amine (4-6ss). The remaining β-lactam moiety thus conjugated to the siRNA would be used for selective conjugation to a highly reactive Lys residue in the hydrophobic pockets of DVDs 1-3.
Bis-β-lactam linker 8 was synthesized as described in detail in Supplementary Scheme S1. Efficient conjugation of the β-lactam linker to sense strand siRNAs was achieved in a liquid-phase reaction using an excess of bis-β-lactam linker 8 with oligonucleotide 5ss and 5ss-bio containing an amino group at the 5′-end and oligonucleotides 4ss and 6ss containing an amino group at the 3′-end (Scheme 1, Supplementary Table S2). In detail, a solution of 5ss (24.4 mg, 3.3 μM) in water (1 ml) was added to a solution of 8 (32.7 mg, 66 μM) in acetonitrile (1 ml) followed by addition of 50 μl of a 10% (v/v) solution of triethylamine (TEA) in acetonitrile. The mixture was kept at RT with occasional swirling for 3 h, and 6 drops of a 10% (v/v) solution of acetic acid in acetonitrile was added. The mixture was diluted with water (7 ml) and dichloromethane (DCM; 4 ml) was added. After extraction, the aqueous phase was separated, washed twice with DCM (5 ml) and stirred in vacuum (up to 10 mbar) for complete evaporation of organic solvents. This afforded the β-lactam linker conjugate of 5ss, termed BL-5ss. The β-lactam linker conjugates of 4ss, 5ss-bio and 6ss, termed 4ss-BL, BL-5ss-bio and 6ss-BL, respectively, were prepared analogously. The β-lactam linker conjugates of the oligonucleotides were analyzed on an Agilent 6130 Quadrupole LC/MS instrument connected to an Agilent 1260 Infinity HPLC system. Standard oligonucleotide LC conditions were used with a column temperature of 60°C and 95 mM of hexafluoro-2-propanol (HFIP) and 16 mM TEA in water as mobile phase A and MeOH as mobile phase B. It was found that in these conditions the β-lactam ring undergoes complete cleavage by all presenting nucleophiles affording water, MeOH, and HFIP adducts correspondingly (Supplementary Figure S2). For reliable analysis of the reaction course and purity of the product, the β-lactam linker conjugates of the oligonucleotides were pre-treated with an excess of butylamine for 5 min followed by LC. The butylamine adduct was found to be stable in standard oligonucleotide LC conditions and gave a single peak of the product, with all impurities existing before the pre-treatment manifesting on the chromatogram. The results suggested a minimum purity of 85% of the product before pre-treatment. The main impurity was the β-lactam ring partial hydrolysis byproduct that apparently formed during the reaction. Finally, BL-5ss, 4ss-BL and 6ss-BL were confirmed by MALDI MS. For this, matrix solutions were prepared as saturated solutions and used within 1 day. A 50 mg/ml solution of diammonium citrate in deionized water and a solution of 10 mg of 2′,4′,6′-trihydroxyacetophenone (THAP) in 1 ml acetonitrile—deionized water (1:1, v/v) were separately prepared, combined in 1:8 ratio, and vortexed to obtain the matrix prior to analysis. One μl of a ∼3 mg/ml solution of the β-lactam linker conjugate of the oligonucleotide was aliquoted into a microcentrifuge tube containing 9 μl of the matrix and mixed well. Of the sample/matrix solution, 1–2 μl was loaded onto a stainless-steel target plate and allowed to dry at ambient temperature and pressure before MALDI MS analysis using a Bruker microflex LRF instrument. The samples were analyzed in the linear positive ion mode, with 500 laser shots collected at random across each sample spot and summed using the automated sample collection mode. The following masses were obtained (Supplementary Figure S3): BL-5ss: MS calc. for [M+H+] 7799.47, observed 7800.79; [M+Na+] calc. 7821.45, observed 7823.56. 4ss-BL: MS calc. for [M+H+] calc. 7768.35, observed 7769.62; [M+Na+] calc. 7790.33, observed 7791.84; [M] as Na salt + Na+ ([M–H+ + 2Na+]) calc. 7812.31, observed 7813.70. 6ss-BL: MS calc. for [M+H+] 7790.41, observed 7792.21.
Double-strand siRNA assembly
Unconjugated siRNA 7 (Scheme 1) was prepared by mixing equimolar amounts of sense (4ss) and antisense strands (4as) and annealed by heating in a water bath at 95°C for 5 min followed by cooling to RT to obtain the duplex. The β-lactam linker-conjugated sense strands were annealed with their complementary antisense strands. For this, aqueous solutions of purified 4ss-BL, 6ss-BL, BL-5ss and BL-5ss-bio were analyzed by UV spectrophotometry to obtain an exact concentration. A subsequent equimolar amount of antisense strand (4as complementary to 4ss-BL, BL-5ss and BL-5ss-bio; 6as complementary to 6ss-BL) was added at a concentration of 20–100 mg/ml in water. The combined strands were vortexed for 30 s and centrifuged to the bottom of a conical tube. For low temperature annealing, the strands were frozen on dry ice and lyophilized to a powder. This afforded siRNAs 4, 6, 5 and 5-bio respectively (Scheme 1, Supplementary Table S2). All siRNAs were dissolved in Ambion nuclease-free water (Thermo Fisher Scientific) to a working concentration of 2.27 or 2.75 mM and stored as aliquots at –80°C. Quality control of the siRNAs included analyses for purity, endotoxins and osmolality.
Cell lines
Human MM cell lines U-266, NCI-H929, and RPMI-8226 were obtained from American Type Culture Collection (ATCC) and cultured in RPMI-1640 medium supplemented with 10% (v/v) FBS, 100 μg/ml streptomycin, and 100 U/ml penicillin (all from Thermo Fisher Scientific) at 37°C in an atmosphere of 5% CO2 and 100% humidity. Expi293F cells were grown in Expi293 Expression Medium (both from Thermo Fisher Scientific) at 37°C in an atmosphere of 8% CO2 and 100% humidity.
Antibody cloning, expression and purification
All variable domain sequences were based on published or patented amino acid sequences. The cloning, expression, and purification of the anti-HER2 DVD-IgG1 was previously described (22). DVD-IgG1s (1-3) were prepared according to the same protocols (27). VH and VL of the targeting antibody (anti-SLAMF7 mAb elotuzumab, anti-BCMA mAb belantamab, or anti-CD138 mAb indatuximab) were linked to VH and VL of mAb h38C2 via a short (ASTKGP; the N-terminal 6 amino acids of human CH1) spacer. The desired sequences were synthesized as gBlocks (Integrated DNA Technologies) and expressed with human IgG1 heavy chain and κ light chain constant domains. The anti-BCMA DVD-Fab was expressed with the same light chain and same VH as the anti-BCMA DVD-IgG1 and an IgG1 heavy chain constant domain CH1 that contained the N-terminal five amino acids (EPKSC) of the IgG1 heavy chain hinge at its C-terminus. The DVD expression cassettes were NheI/BamHI-cloned into a mammalian expression vector (28) and transiently transfected into Expi293F cells using ExpiFectamine according to the manufacturer's instructions (Thermo Fisher Scientific). After 5–7 days, the media were collected, filtered through a 0.22-μm filter, and purified using 1-ml HiTrap Protein A (DVD-IgG1) or Protein L (DVD-Fab) columns (GE Healthcare) in conjunction with an ÄKTA FPLC instrument (GE Healthcare). Yields were typically ∼40 mg/l. The purity of DVD-IgG1s and DVD-Fabs was confirmed by nonreducing and reducing SDS-PAGE followed by Coomassie staining, and the concentration was determined by measuring the absorbance at 280 nm.
Amino acid sequences of DVD IgG1s in the order signal sequence - outer variable domain—spacer—inner variable domain—constant domain(s):
Anti-SLAMF7 DVD-IgG1 (1) light chain:
MPMGSLQPLATLYLLGMLVASVLADIQMTQSPSSLSASVGDRVTITCKASQDVGIAVAWYQQKPGKVPKLLIYWASTRHTGVPDRFSGSGSGTDFTLTISSLQPEDVATYYCQQYSSYPYTFGQGTKVEIKASTKGPELQMTQSPSSLSASVGDRVTITCRSSQSLLHTYGSPYLNWYLQKPGQSPKLLIYKVSNRFSGVPSRFSGSGSGTDFTLTISSLQPEDFAVYFCSQGTHLPYTFGGGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
Anti-SLAMF7 DVD-IgG1 (1) heavy chain:
MPMGSLQPLATLYLLGMLVASVLAEVQLVESGGGLVQPGGSLRLSCAASGFDFSRYWMSWVRQAPGKGLEWIGEINPDSSTINYAPSLKDKFIISRDNAKNSLYLQMNSLRAEDTAVYYCARPDGNYWYFDVWGQGTLVTVSSASTKGPEVQLVESGGGLVQPGGSLRLSCAASGFTFSNYWMSWVRQSPEKGLEWVSEIRLRSDNYATHYAESVKGRFTISRDNSKNTLYLQMNSLRAEDTGIYYCKTYFYSFSYWGQGTLVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGA
Anti-BCMA DVD-IgG1 (2) and DVD-Fab light chain:
MPMGSLQPLATLYLLGMLVASVLADIQMTQSPSSLSASVGDRVTITCSASQDISNYLNWYQQKPGKAPKLLIYYTSNLHSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQYRKLPWTFGQGTKLEIKASTKGPELQMTQSPSSLSASVGDRVTITCRSSQSLLHTYGSPYLNWYLQKPGQSPKLLIYKVSNRFSGVPSRFSGSGSGTDFTLTISSLQPEDFAVYFCSQGTHLPYTFGGGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
Anti-BCMA DVD-IgG1 (2) heavy chain:
MPMGSLQPLATLYLLGMLVASVLAQVQLVQSGAEVKKPGSSVKVSCKASGGTFSNYWMHWVRQAPGQGLEWMGATYRGHSDTYYNQKFKGRVTITADKSTSTAYMELSSLRSEDTAVYYCARGAIYDGYDVLDNWGQGTLVTVSSASTKGPEVQLVESGGGLVQPGGSLRLSCAASGFTFSNYWMSWVRQSPEKGLEWVSEIRLRSDNYATHYAESVKGRFTISRDNSKNTLYLQMNSLRAEDTGIYYCKTYFYSFSYWGQGTLVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGA
Anti-BCMA DVD-Fab heavy chain:
MPMGSLQPLATLYLLGMLVASVLAQVQLVQSGAEVKKPGSSVKVSCKASGGTFSNYWMHWVRQAPGQGLEWMGATYRGHSDTYYNQKFKGRVTITADKSTSTAYMELSSLRSEDTAVYYCARGAIYDGYDVLDNWGQGTLVTVSSASTKGPEVQLVESGGGLVQPGGSLRLSCAASGFTFSNYWMSWVRQSPEKGLEWVSEIRLRSDNYATHYAESVKGRFTISRDNSKNTLYLQMNSLRAEDTGIYYCKTYFYSFSYWGQGTLVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSC
Anti-CD138 DVD-IgG1 (3) light chain:
MPMGSLQPLATLYLLGMLVASVLADIQMTQSTSSLSASLGDRVTISCSASQGINNYLNWYQQKPDGTVELLIYYTSTLQSGVPSRFSGSGSGTDYSLTISNLEPEDIGTYYCQQYSKLPRTFGGGTKLEIKASTKGPELQMTQSPSSLSASVGDRVTITCRSSQSLLHTYGSPYLNWYLQKPGQSPKLLIYKVSNRFSGVPSRFSGSGSGTDFTLTISSLQPEDFAVYFCSQGTHLPYTFGGGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
Anti-CD138 DVD-IgG1 (3) heavy chain:
MPMGSLQPLATLYLLGMLVASVLAQVQLQQSGSELMMPGASVKISCKATGYTFSNYWIEWVKQRPGHGLEWIGEILPGTGRTIYNEKFKGKATFTADISSNTVQMQLSSLTSEDSAVYYCARRDYYGNFYYAMDYWGQGTSVTVSSASTKGPEVQLVESGGGLVQPGGSLRLSCAASGFTFSNYWMSWVRQSPEKGLEWVSEIRLRSDNYATHYAESVKGRFTISRDNSKNTLYLQMNSLRAEDTGIYYCKTYFYSFSYWGQGTLVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGA
Antibody conjugation
Conjugations were performed in PBS (pH 7.4) after the DVD-IgG1s 1, 2 and 3 were diluted to 3.13 mg/ml (15.7 μM). For DVD-ARC 10, 13 and 13-bio assembly, 16.3 μl of 2.75 mM β-lactam linker-functionalized siRNA 4 in H2O (10 eq) or 19.8 μl of 2.27 mM β-lactam linker-functionalized siRNA 5 or 5-bio in H2O (10 eq) was added to 900 μg of each DVD-IgG1. Control anti-BCMA DVD-ARC 15 was prepared by diluting the anti-BCMA DVD-IgG1 2 to 4.08 mg/ml (20.4 μM) and adding 38 μl of 2.59 mM β-lactam linker-functionalized siRNA 6 in H2O (10 eq) to 2 mg of the DVD-IgG1. Anti-HER2 DVD-ARCs 16 and 17 were prepared by diluting the anti-HER2 DVD-IgG1 to 4.47 mg/ml (22.4 μM) and adding 36 μl of 2.75 mM β-lactam linker-functionalized siRNA 4 and 5, respectively, in H2O (10 eq) to 2 mg of the DVD-IgG1. The solutions were incubated for 2 h at room temperature (RT). For the optimized conjugation conditions, anti-BCMA DVD-IgG1 2 was concentrated to 10 mg/ml (50 μM) using a 30-kDa cut-off centrifugal filter device (Millipore). Next, 2 equivalents (eq) of β-lactam linker-functionalized siRNA was added using 2.75 mM 4 or 2.27 mM 5 in H2O and incubated for 4 h at RT. All conjugations were deemed complete by loss of catalytic activity using the methodol assay for which a portion of the crude reaction diluted to 1 μM in PBS was used. Upon completion, unreacted compound was removed by using a PD-10 desalting column (GE Healthcare). Protein containing fractions (Nanodrop A280) were concentrated using a 4-ml 30-kDa cut-off centrifugal filter device (Millipore) and washed with ∼4 ml of PBS three times. During the last wash, the samples were concentrated to a final volume of ∼250 μl. The concentration of the DVD-ARCs was determined using a bicinchoninic acid (BCA) assay kit (Thermo Fisher Scientific) according to the manufacturer's instructions with bovine gamma globulin (Thermo Fisher Scientific) as standard. For DVD-Fab conjugation, 2 mg of the anti-BCMA DVD-Fab was diluted to 7 mg/ml (100 μM) and 21 μl of 2.75 mM β-lactam linker-functionalized siRNA 4 and 5, respectively, in H2O (2 eq) was added. Following incubation for 4 h at RT, the DVD-ARCs were purified as described above. All DVD-ARCs were stored in PBS at 4°C.
IdeS digestion
Five microgram of anti-BCMA DVD-IgG1 or anti-BCMA DVD-ARCs were digested using FabRICATOR (IdeS; Genovis) according to the manufacturer's instructions at 37°C for 1 h. The samples were removed and loaded on a NuPAGE Novex Bis–Tris 4–12% gradient gel in NuPAGE LDS Sample Buffer (both from Thermo Fisher Scientific). Following SDS-PAGE, the gel was stained with SimplyBlue SafeStain (Thermo Fisher Scientific).
SEC
Size-exclusion chromatography (SEC) was performed on an ÄKTA FPLC instrument equipped with a Superdex 200 10/300 GL column (GE Healthcare). For analytical runs, 30-μg samples were analyzed using a flow rate of 0.5 ml/min PBS and a detection wavelength of 280 nm. For purification, 1–2 mg of DVD-ARC were loaded on the column and the desired peaks collected. DVD-ARCs in both formats (IgG1 and Fab) eluted before excess siRNA.
Catalytic activity assay
Catalytic activity was analyzed using methodol as described previously (22,27). IgG1s, DVD-IgG1s, DVD-Fabs and DVD-ARCs were diluted to 1 μM in PBS (pH 7.4) and dispensed in 98-μl aliquots into a 96-well plate in triplicate. Then, 2 μl of 10 mM methodol in ethanol was added and the fluorescence was assessed immediately using a SpectraMax M5 instrument (Molecular Devices) with SoftMax Pro software, a wavelength of excitation (λext) set to 330 nm, a wavelength of emission (λem) set to 452 nm and starting at 0 min using 5-min time points. The signal was determined by normalizing to 98 μl PBS with 2 μl of the methodol solution added.
Flow cytometry
In a V-bottom 96-well plate (Corning), 100,000 cells per well were dispensed. The cells were washed with 200 μl flow cytometry buffer (PBS, 2% (v/v) FBS, 0.01% (w/v) NaN3, pH 7.4), incubated with IgG1s, DVD-IgG1, or DVD-ARCs (50 μl of a 20 nM solution in PBS) for 30 min on ice, washed with 200 μl ice-cold flow cytometry buffer, and stained with Alexa Fluor 647 conjugated polyclonal (Fab’)2 donkey anti-human Fc (Jackson ImmunoResearch Laboratories) for 20 min on ice. After washing twice with 200 μl ice-cold flow cytometry buffer, the cells were analyzed using a Canto II Flow Cytometer (Becton-Dickinson). Data were analyzed using FlowJo software (Tree Star).
SPR
Surface plasmon resonance (SPR) was used to determine the kinetic and thermodynamic parameters of the anti-BCMA DVD-Fab before and after conjugation to β-lactam linker-functionalized siRNA 4 and 5. The ARCs were purified by SEC as described above. A Biacore X100 instrument was used with Biacore reagents and software (GE Healthcare). A mouse anti-human IgG CH2 mAb was immobilized on a CM5 sensor chip using reagents and instructions supplied with the Human Antibody Capture Kit (GE Healthcare). A human BCMA-Fc fusion protein (R&D Systems) was captured at a density not exceeding 1000 RU. Each sensor chip included an empty flow cell for instantaneous background depletion. All binding assays used 1× HBS-EP+ running buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA (pH 7.4) and 0.05% (v/v) Surfactant P20) and a flow rate of 30 μl/min. For affinity measurements, the unconjugated or conjugated anti-BCMA DVD-Fabs were injected at five different concentrations (12.5 (in duplicate), 25, 50, 100 and 200 mM). The sensor chips were regenerated with 3 M MgCl2 from the Human Antibody Capture Kit without any loss of binding capacity. Calculation of association (kon) and dissociation (koff) rate constants was based on a 1:1 Langmuir binding model. The equilibrium dissociation constant (Kd) was calculated from koff/kon.
mRNA knockdown analysis by RT-qPCR
In a 12-well cell culture dish, 500 μl of cells (4 × 105 cells/ml, 200 000 cells per well) were dispensed and 500 μl of DVD-IgG1 or DVD-ARC solution (diluted with RPMI 1640 medium, supplemented with 10% (v/v) FBS, 100 μg/ml streptomycin, and 100 U/ml penicillin) was immediately added at the appropriate concentration. Free siRNA 7 (diluted to 200 nM in 100 μl Opti-MEM medium) was added to a mixture of 6 μl Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific) and 94 μl Opti-MEM following the manufacturer's instructions. After 5 min incubation at RT, 10 μl of this mixture was added to 90 μl of cells (20 000 cells per well in a 96-well cell culture dish) to a final siRNA concentration of 10 nM. After 72 h, extraction of RNA from cell lysates was performed using the RNeasy Mini Kit (Qiagen) followed by cDNA synthesis of 1 μg DNase-digested RNA, using the Maxima First-Strand cDNA Synthesis Kit for RT-qPCR (Invitrogen) according to the manufacturer's instructions. qPCR of the synthesized first-strand cDNA was conducted using the SYBR Green PCR Master Mix (Thermo Fisher Scientific) according to the manufacturer's instructions, performed on an Applied Biosystems’ StepOnePlus Real-Time PCR System, and analyzed using StepOne Software v2.2.2 (both from Thermo Fisher Scientific). All measurements were conducted three times using biological duplicates or triplicates and standardized to the levels of β-actin. Relative changes in gene expression were calculated according to the 2−ΔΔCT algorithm (29). Primer sequences (30): β-catenin forward 5′-AAAATGGCAGTGCGTTTAG-3′; β-catenin reverse 5′-TTTGAAGGCAGTCTGTCGTA-3′ β-actin forward 5′-CCTGTACGCCAACACAGTGC-3′; β-actin reverse 5′-ATACTCCTGCTTGCTGATCC-3′.
Protein knockdown analysis by western blotting
In a 12-well cell culture dish, 500 μl of cells (4 × 105 cells/ml, 200 000 cells per well) were dispensed and 500 μl of DVD-IgG1 or DVD-ARC solution (diluted with RPMI 1640 medium, supplemented with 10% (v/v) FBS, 100 μg/ml streptomycin, and 100 U/ml penicillin) was immediately added at the appropriate concentration. Free siRNA 7 was transfected using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific) according to the manufacturer's instructions to a final siRNA concentration of 10 nM. After 7 days, the cells were washed with PBS and lysed using RIPA Lysis Buffer (Thermo Fisher Scientific) containing a protease inhibitor cocktail (Thermo Fisher Scientific). The samples were diluted with 1× NuPAGE LDS sample buffer (Thermo Fisher Scientific) containing 2% (v/v) β-mercaptoethanol and boiled for 5 min before running on NuPAGE Novex 4–12% Bis–Tris gels (Thermo Fisher Scientific). After transfer to a polyvinylidene difluoride (PVDF) membrane (Millipore) and blocking with 10% (v/v) Western Blocking Reagent (Sigma-Aldrich, cat. no. 11921673001) in Tris-buffered saline containing 0.01% Tween 20 (TBST), the PVDF membrane was incubated with 2 μg/ml mouse anti-human CTNNB1 mAb 12F7 (BioLegend, cat. no. 844602) in 5% (v/v) Western Blocking Reagent in TBST at 4°C overnight. The PVDF membrane was washed with TBST followed by incubation with a 1:5000 dilution (5% (v/v) Western Blocking Reagent in TBST) of peroxidase-conjugated goat anti-mouse IgG polyclonal antibodies (BioLegend, cat. no. 405306) at RT for 3 h before washing with TBST and development using ECL Prime Western Blotting Detection Reagent (GE Healthcare). For β-actin staining, the membrane was incubated with a 1:10 000 dilution (5% (v/v) Western Blocking Reagent in TBST) of peroxidase-conjugated mouse anti-human β-actin mAb AC-15 (Sigma Aldrich, cat. no. A3854) at 4°C overnight, washed with TBST, and then imaged using ECL Prime Western Blotting Detection Reagent. ImageJ software was used for quantification.
Pharmacokinetic (PK) study
Seven-weeks old female CD-1 mice (approximately 25 g each) were purchased from Charles River Laboratories. Three groups of mice (one group in experiment 1 with n = 3; two groups in experiment 2 with n = 5) were injected i.v. (tail vein) with anti-BCMA DVD-ARC 13-bio (experiments 1 and 2) and anti-BCMA DVD-IgG1 2 (experiment 2), respectively, at 6 mg/kg. Using heparinized capillary tubes to prevent clotting, blood was collected from the tip of the tail at 5 min, 30 min, 6 h, 24 h, 48 h, 72 h, 96 h, 144 h, 240 h and 336 h post-injection. Plasma was obtained by centrifuging the samples at 2000 g for 5 min in a microcentrifuge and storing the liquid portion at −80°C until further analysis. ELISA was employed to measure the concentration of intact DVD-ARC and total antibody (conjugated and unconjugated DVD-IgG1) in the first group of mice and DVD-IgG1 in the second group of mice. To do so, individual wells within a 96-well Costar 3690 plate (Corning) were incubated with 100 ng recombinant human BCMA-Fc fusion protein (R&D Systems) in 30 μl PBS buffer (pH 7.4) overnight at 4°C. The wells were subsequently blocked with 150 μl blocking buffer (Pierce Blocker Casein in PBS, Thermo Fisher Scientific) at 37°C for 1 h. Both plasma samples and standards were diluted in PBS and added to the wells for a second round of incubation at 37°C for 1 h. After washing, horseradish peroxidase (HRP)-conjugated avidin (BioLegend) was used to detect the intact DVD-ARC via its biotin moiety on the 3′ end of the sense strand, and Peroxidase-AffiniPure Goat Anti-Human IgG, F(ab')2 Fragment Specific (Jackson ImmunoResearch Laboratories) was used to detect DVD-IgG1. The concentrations of DVD-ARC and DVD-IgG1 in the plasma samples were extrapolated using a minimal four-variable fit standard curve. Circulatory half-lives of DVD-ARC and total antibody (experiments 1 and 2) and DVD-IgG1 (experiment 2) were analyzed with Phoenix WinNonlin PK/PD Modeling and Analysis software (Pharsight).
RESULTS
Generation and characterization of DVD-ARCs
DVD-IgG1s 1, 2 and 3 targeting human SLAMF7 (31), BCMA (32–34) and CD138 (35), respectively, were cloned, expressed and purified. All three cell surface antigens are validated MM targets for antibody-based therapeutics and have been clinically investigated with ADCs (10). The heavy (VH) and light chain variable domain (VL) amino acid sequences used for the outer Fv of the DVD-IgG1s were derived from humanized anti-human SLAMF7 mAb elotuzumab, humanized anti-human BCMA mAb belantamab, and chimeric mouse anti-human CD138 mAb indatuximab, respectively. VH and VL amino acid sequences used for the inner Fv of the DVD-IgG1s were derived from humanized aldolase antibody h38C2 (36) (Figure 1A and B). The VL–VL and VH–VH junction of the outer and inner Fv comprised a short peptide (ASTKGP) derived from the N-terminus of the first constant domain (CH1) of human IgG1. All constant domains were of human κ (CL) and γ1 (CH1, hinge, CH2, CH3) isotypes. The DVD IgG1s were prepared in high purity with retention of specific binding to three MM cell lines as determined by SDS-PAGE and flow cytometry, respectively (Supplementary Figure S1).
Next, β-lactam linker-functionalized siRNAs (4 and 5) targeting human β-catenin (CTNNB1) mRNA were synthesized. CTNNB1 was chosen as the siRNA target because it is overexpressed in MM (37,38). The β-lactam linker functionality served as the reactive handle to conjugate the siRNA to the uniquely reactive Lys residue contained in the inner Fv of the DVD-IgG1s (Figure 1C and E). Two compounds were prepared targeting CTNNB1; siRNA 4 with the β-lactam handle at the 3′ end and siRNA 5 with the β-lactam handle at the 5′ end of the passenger (sense) strand (Scheme 1). In both cases, the passenger strand was modified with the β-lactam handle so RISC complex formation would not be affected. In addition to these compounds, two control siRNAs were synthesized as negative controls; siRNA 6 targeting human transthyretin (TTR), an irrelevant gene in this study, with a β-lactam handle at the 3′ end and siRNA 7 targeting CTNNB1 but lacking the β-lactam linker moiety (Scheme 1). All four siRNAs that were synthesized for this study were chemically stabilized by extensive modification of nucleosides and nucleoside linkages as shown in Scheme 1 and in accordance with the previously reported enhanced stabilization chemistry that does not compromise silencing (7). We utilized vinylphosphonate at the 5′-end of AS strands as a stable phosphate mimic in order to enhance RISC loading. The potential of this modification to substantially improve efficacy, especially in extra-hepatic applications where endogenous phosphorylation of chemically modified strands by kinases may be not as efficient, has been demonstrated previously (39,40). Modification of the 5′- vinylphosphonate containing terminal nucleoside with a N-methylacetamide moiety at the 2′-position can further improve nucleolytic stability and efficacy.
To prepare the desired DVD-ARCs each DVD-IgG1 (1-3) was incubated with 10 eq of β-lactam linker CTNNB1 siRNA 4 or 5 in PBS for 2 h at RT. Thus, a total of six DVD-ARCs were assembled; DVD-ARC 9 = 1 + 4, DVD-ARC 10 = 2 + 4, DVD-ARC 11 = 3 + 4, DVD-ARC 12 = 1 + 5, DVD-ARC 13 = 2 + 5 and DVD-ARC 14 = 3 + 5. Complete conjugation at the reactive Lys residue of the inner Fv of the DVD-IgG1s was verified for all six DVD-ARCs by loss of aldolase activity as previously described (22,27) and shown in Figure 3A. SDS-PAGE analysis of the BCMA-targeting DVD-IgG1 before (2) and after conjugation (10 and 12) revealed an upshift of the protein band. This upshift was also seen after enzymatic digestion of the unconjugated and conjugated DVD-IgG1 with IdeS, a protease from Streptococcus pyogenes that cleaves below the hinge region to generate DVD-F(ab’)2 and Fc fragments (Figure 3B).
Figure 3.

Analysis of DVD-ARC assembly. DVD-IgG1 (1-3) were incubated with 10 eq of β-lactam CTNNB1 siRNA 4 or 5 in PBS for 2 h at RT to yield anti-SLAMF7 DVD-ARCs 9 and 12, anti-BCMA DVD-ARCs 10 and 13, and anti-CD138 DVD-ARCs 11 and 14. (A) The catalytic retro-aldol activity of the reactive Lys residue of the inner Fv of the DVD-IgG1s was measured over 60 min using the conversion of methodol to a fluorescent aldehyde and acetone. The signal is reported in relative fluorescent units (RFU; mean ± SD of triplicates). The assembled DVD-ARCs (9–14) were catalytically inactive due to amide formation at the reactive Lys, indicating complete conjugation. Unconjugated DVD-IgG1s (1–3) were used as positive controls and trastuzumab as a negative control. (B) SDS-PAGE analysis (nonreducing conditions) of the anti-BCMA DVD-IgG1 2 and its DVD-ARCs 10 and 13 before and after digestion with IdeS. The upper band (∼150 kDa when unconjugated) and lower band (∼45 kDa) are the expected bands for DVD-F(ab’)2 and Fc, respectively, following single-site proteolytic cleavage below the IgG1 hinge region by IdeS.
Size exclusion chromatography (SEC), which was used to separate DVD-ARCs from remaining free siRNA, also revealed an increase in size consistent with the expected molecular weight difference between unconjugated and siRNA-conjugated DVD-IgG1. The SEC profiles showed monodisperse DVD-ARC peaks with <10% aggregation (Figure 4A). Furthermore, we found that the conjugation reaction proceeded with equal efficiency when the molar ratio of siRNA to DVD-IgG1 was lowered from 10 eq to 4 eq or even 2 eq and the incubation time increased from 2 to 4 h (Supplementary Figure S4). This result revealed the high efficiency of hapten-driven covalent conjugation as 2 eq correspond to just 1 eq with respect to each of the two reactive Lys residues.
Figure 4.

SEC and flow cytometry analysis of unconjugated and siRNA-conjugated anti-BCMA DVD-IgG1. (A) Shown from top to bottom are the SEC profiles of siRNA 4, anti-BCMA DVD-IgG1 2 alone and after conjugation to 10 eq of siRNA 4 and 5. The separation of the peaks for DVD-ARC (left) and remaining free siRNA (right) facilitated purification of the DVD-ARCs. Note the lower elution volume of the DVD-ARCs (9.7 and 9.8 ml; expected molecular weight ∼230 kDa) compared to the DVD-IgG1 (11.0 ml; ∼200 kDa). (B) Flow cytometry analysis of the binding of anti-BCMA DVD-IgG1 2 (blue line) and anti-BCMA DVD-ARCs 10 (orange line) and 13 (red line) to human MM cell line NCI-H929. Anti-CD138 DVD-IgG1 3 (purple line) and h38C2 IgG1 (black line) were used as a positive and negative control, respectively. The concentration of all primary antibodies was 20 nM. Background staining in the absence of primary antibodies is shown as gray shade.
Due to electrostatic repulsion between the negative charge of the DVD-ARCs introduced by siRNA conjugation and the negative charge of the cell surface, we anticipated weaker cell surface antigen binding of the DVD-ARCs compared to the DVD-IgG1s. This was examined for the BCMA/CTNNB1-targeting DVD-ARCs by flow cytometry. As shown in Figure 4B, DVD-ARCs 10 and 13 indeed revealed diminished binding to H929 cells compared to DVD-IgG1 2. Nonetheless, both DVD-ARCs still bound significantly above background. To further test whether siRNA conjugation to the inner Fv affected binding of the outer Fv to purified BCMA, the kinetic (kon and koff) and thermodynamic (Kd) parameters of binding were analyzed by surface plasmon resonance (SPR). To permit calculation of these parameters based on a 1:1 binding model, a DVD-Fab format that binds BCMA monovalently (Figure 1D) was used. Unconjugated DVD-Fab and siRNA-conjugated DVD-Fab_4 and DVD-Fab_5 revealed single-digit nanomolar affinity with kon and koff values differing by factor 2 or less (Table 1 and Supplementary Figure S5). Thus, conjugation of the CTNNB1-targeting siRNAs to the inner Fv did not compromise the binding of the outer Fv to purified BCMA.
Table 1.
Kinetic and thermodynamic parameters of DVD-Fab binding to BCMA before and after conjugation to siRNAs 4 and 5
| k on (M−1 s−1) | k off (s−1) | K d (nM) | |
|---|---|---|---|
| DVD-Fab | 6.5 × 104 | 3.1 × 10−4 | 4.7 |
| DVD-Fab_siRNA 4 | 8.9 × 104 | 4.2 × 10−4 | 4.7 |
| DVD-Fab_siRNA 5 | 5.5 × 104 | 4.3 × 10−4 | 8.0 |
Functional validation of DVD-ARCs
With the DVD-ARCs (9-14) in hand, knockdown of CTNNB1 mRNA in the MM cell line NCI-H929 was assessed using quantitative reverse transcription polymerase chain reaction (RT-qPCR) and normalized to β-actin as a housekeeping gene for standardization. As expected, significant knockdown (∼80%) was observed for the positive control, free siRNA 7 transfected by Lipofectamine RNAiMAX Transfection Reagent. Significant knockdown (25−75%) was also observed for all six DVD-ARCs following incubation of the NCI-H929 cells with 90 nM for 72 h at 37°C, with the BCMA-targeting DVD-ARCs 10 and 13 eliciting the highest level of silencing (Figure 5A). To further investigate this potency and efficacy, a dose response (1−90 nM) was performed using the BCMA-targeting DVD-ARCs 10 and 13 (Figure 5B). IC50 values were found to be ∼10 nM and ∼7 nM for ARC 10 and 13, respectively. As a negative control, BCMA-targeting DVD-IgG1 2 was conjugated to TTR-targeting siRNA 6 and included as DVD-ARC 15. Significant mRNA knockdown was observed at concentrations as low as 3 nM for both BCMA-targeting DVD-ARCs 10 and 13. By contrast, negative control DVD-ARC 15 did not cause any mRNA knockdown at the highest concentration (90 nM). As an additional set of negative controls, we conjugated CTNNB1-targeting siRNAs 4 and 5 to a previously described HER2-targeting DVD-IgG1, which does not bind to NCI-H929 cells (22), and did not detect silencing at 90 nM by RT-qPCR (Supplementary Figure S6). In addition to MM cell line NCI-H929, free siRNA 7 (transfected by Lipofectamine RNAiMAX Transfection Reagent) caused significant CTNNB1 mRNA knockdown in MM cell lines RPMI-8226 and U266. Anti-BCMA DVD-ARCs 10 and 13 (added at 90 nM for 72 h at 37°C) achieved significant CTNNB1 mRNA knockdown in RPMI-8226 but not U266 cells.
Figure 5.

CTNNB1 mRNA knockdown. (A) CTNNB1 mRNA knockdown in NCI-H929 cells after treatment with 90 nM SLAMF7 (9 and 12), BCMA (10 and 13), or CD138 (11 and 14) targeting DVD-ARCs for 72 h at 37°C. Unconjugated DVD-IgG1s (1–3) and transfected free siRNA (7) were used as negative and positive controls, respectively. Mean ± SD values of independent duplicates (three technical replicates of each of two biological replicates; n = 6) are shown. A Student's t-test was used to determine the significant difference between the unconjugated (blue) and conjugated (orange and red) anti-BCMA DVD-IgG1s (****P < 0.0001). (B) Dose response (1–90 nM) of BCMA-targeting DVD-ARCs (10 and 13) incubated with NCI-H929 cells for 72 h at 37°C. Anti-BCMA DVD-ARC (15) contained an siRNA targeting human TTR (6) and served as negative control. Mean ± SD values of independent duplicates (three technical replicates of each of two biological replicates; n = 6) are shown.
To ensure that CTNNB1 mRNA knockdown correlated with CTNNB1 protein knockdown, western blots were performed after incubation of NCI-H929 cells with the DVD-ARCs at 90 nM for 7 days in three independent experiments (Figure 6). Significant CTNNB1 protein knowdown was observed for both BCMA/CTNNB1-targeting DVD-ARCs but not for the BCMA/TTR-targeting DVD-ARC and the unconjugated anti-BCMA DVD-IgG1. As expected, the HER2/CTNNB1-targeting DVD-ARC was also inactive. Selective CTNNB1 protein knowdown was also observed at two earlier (4 and 6 days) and one later time point (11 days).
Figure 6.

CTNNB1 protein knockdown. CTNNB1 protein knockdown in NCI-H929 cells after treatment with 90 nM BCMA (10 and 13) (orange and red, respectively) targeting DVD-ARCs for 7 days at 37°C. Unconjugated anti-BCMA DVD-IgG1s (2) (blue), anti-BCMA DVD-ARC (15) targeting human TTR, and HER2-targeting DVD-ARCs (16 and 17) served as negative controls. Transfected free siRNA (7) served as positive control. The western blot depicted on the top is a representative example from independent triplicates shown as mean ± SEM values on the bottom.
PK study
Finally, we carried out a PK study to examine the circulatory half-life (t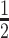 ) of the DVD-ARC in mice. In order to detect the intact DVD-ARC in mouse plasma, we first synthesized a biotin derivative of CTNNB1-targeting siRNA consisting of antisense strand 4as and sense strand BL-5ss with a biotin group at its 3′ end (BL-5ss-bio). We refer to this new siRNA as 5-bio (Supplementary Tables S1 and S2) and to the corresponding DVD-ARC as 13-bio (Supplementary Table S1). An ELISA based on capturing the DVD-ARC and total antibody (conjugated and unconjugated) with immobilized BCMA followed by separately detecting the intact DVD-ARC and total antibody was established. In two independent experiments, three or five immunocompetent mice each were injected with DVD-ARC 13-bio (experiment 1 and 2) or DVD-IgG1 2 (experiment 2) by i.v. (tail vein) injection and peripheral blood was withdrawn at various time points spanning from 5 min to 336 h. Using a calibration curve, plasma concentrations of DVD-ARC and total antibody at the various time points were measured and used to determine t
) of the DVD-ARC in mice. In order to detect the intact DVD-ARC in mouse plasma, we first synthesized a biotin derivative of CTNNB1-targeting siRNA consisting of antisense strand 4as and sense strand BL-5ss with a biotin group at its 3′ end (BL-5ss-bio). We refer to this new siRNA as 5-bio (Supplementary Tables S1 and S2) and to the corresponding DVD-ARC as 13-bio (Supplementary Table S1). An ELISA based on capturing the DVD-ARC and total antibody (conjugated and unconjugated) with immobilized BCMA followed by separately detecting the intact DVD-ARC and total antibody was established. In two independent experiments, three or five immunocompetent mice each were injected with DVD-ARC 13-bio (experiment 1 and 2) or DVD-IgG1 2 (experiment 2) by i.v. (tail vein) injection and peripheral blood was withdrawn at various time points spanning from 5 min to 336 h. Using a calibration curve, plasma concentrations of DVD-ARC and total antibody at the various time points were measured and used to determine t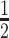 by two-compartment modeling (Table 2). The last time point at which DVD-ARC, total antibody, and DVD-IgG1 were detectable is shown as tlast. The intact DVD-ARC 13-bio revealed a t
by two-compartment modeling (Table 2). The last time point at which DVD-ARC, total antibody, and DVD-IgG1 were detectable is shown as tlast. The intact DVD-ARC 13-bio revealed a t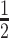 (mean ± SD) of 12.3 ± 1.5 h (experiment 1) and 9.5 ± 2.6 h (experiment 2) compared to 37.2 ± 9.5 h (experiment 1) and 23.6 ± 1.3 h (experiment 2) for total antibody. This observation suggests that the stability of the 5′ or 3′ linker in the siRNA component of the DVD-ARC may be a limiting factor of the circulatory half-life of the intact DVD-ARC 13-bio. The unconjugated DVD-IgG1 had a 2–3 times longer t
(mean ± SD) of 12.3 ± 1.5 h (experiment 1) and 9.5 ± 2.6 h (experiment 2) compared to 37.2 ± 9.5 h (experiment 1) and 23.6 ± 1.3 h (experiment 2) for total antibody. This observation suggests that the stability of the 5′ or 3′ linker in the siRNA component of the DVD-ARC may be a limiting factor of the circulatory half-life of the intact DVD-ARC 13-bio. The unconjugated DVD-IgG1 had a 2–3 times longer t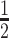 (68.9 ± 15.1 h) compared to the total antibody of the DVD-ARC, confirming reported studies for ADCs (41) and ARCs (19).
(68.9 ± 15.1 h) compared to the total antibody of the DVD-ARC, confirming reported studies for ADCs (41) and ARCs (19).
Table 2.
Circulatory half-lives for DVD-ARC and DVD-IgG1 in mice following i.v. injection
t
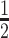 (h) (h) |
t last (h) | |
|---|---|---|
| DVD-ARC 13-bio (experiment 1)a | 12.3 ± 1.5 | 72 |
| Total antibody of DVD-ARC 13-bio (experiment 1)a | 37.2 ± 9.5 | 72 |
| DVD-ARC 13-bio (experiment 2)b | 9.5 ± 2.6 | 54 |
| Total antibody of DVD-ARC 13-bio (experiment 2)b | 23.6 ± 1.3 | 102 |
| DVD-IgG1 2 (experiment 2)b | 68.9 ± 15.1 | 246 |
aMean ± SD values from three mice.
bMean ± SD values from five mice.
DISCUSSION
Here we report a method for generating site-specific ARCs. A primary amino group at the 3′- or 5′-end of the sense strand of a chemically stabilized siRNA is selectively and efficiently functionalized with a symmetrical bis-β-lactam linker. Subsequent low-temperature annealing with the antisense strand yields siRNAs containing the appended β-lactam handle. The biological component is an antibody in DVD-IgG1 format that combines an outer targeting Fv and an inner catalytic Fv and can be expressed, purified, and assembled in good yields. Site-specific drug attachment is achieved by covalent conjugation of the β-lactam-functionalized siRNA to a uniquely reactive Lys residue in the inner catalytic Fv of the DVD-IgG1. Notably, conjugation of antibody and siRNA at low molar ratios proceeds rapidly and efficiently under mild conditions in a single step and can be monitored with a catalytic assay. The generated DVD-ARCs retain binding toward the antigen and successfully induce mRNA and protein knockdown in target cells. Thus, this platform merits further exploration in both oncology and non-oncology indications.
Due to their larger size and their recycling by the neonatal Fc receptor (FcRn), conventional IgG1 (∼150 kDa) have a circulatory half-life of ‘weeks’ in humans whereas chemically stabilized siRNA (∼15 kDa) has a circulatory half-life of ‘hours’. For example, a chemically stabilized siRNA with a trivalent (GalNAc) ligand had a t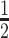 of 1.9 h after i.v. injection into mice (42). Thus, we anticipated that the circulatory half-life of siRNA can be prolonged by attachment to a DVD-IgG1 (∼200 kDa). However, it is known that conjugated antibodies have shorter circulatory half-lives than unconjugated antibodies. For example, in breast cancer patients, trastuzumab vedotin (Kadcyla®; an FDA-approved ADC targeting HER2) has a t
of 1.9 h after i.v. injection into mice (42). Thus, we anticipated that the circulatory half-life of siRNA can be prolonged by attachment to a DVD-IgG1 (∼200 kDa). However, it is known that conjugated antibodies have shorter circulatory half-lives than unconjugated antibodies. For example, in breast cancer patients, trastuzumab vedotin (Kadcyla®; an FDA-approved ADC targeting HER2) has a t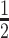 of 3.5 days compared to 12.5 days for trastuzumab (Herceptin®) (41). In addition, our PK study suggested that integrity of the siRNA component was a limiting factor of the overall stability of DVD-ARC 13-bio. Since it was previously reported that the pattern of chemical stabilization of siRNAs that we utilized in this study warrants exceptional stability in vivo (42), we attribute the observed limited stability of the intact DVD-ARC to either the 5′ linker connecting antibody and siRNA or the 3′ linker connecting siRNA and biotin. Both can undergo cleavage at the phosphate moieties in the presence of phosphatases. Despite this shortcoming, the mean t
of 3.5 days compared to 12.5 days for trastuzumab (Herceptin®) (41). In addition, our PK study suggested that integrity of the siRNA component was a limiting factor of the overall stability of DVD-ARC 13-bio. Since it was previously reported that the pattern of chemical stabilization of siRNAs that we utilized in this study warrants exceptional stability in vivo (42), we attribute the observed limited stability of the intact DVD-ARC to either the 5′ linker connecting antibody and siRNA or the 3′ linker connecting siRNA and biotin. Both can undergo cleavage at the phosphate moieties in the presence of phosphatases. Despite this shortcoming, the mean t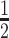 of 10–12 h for the DVD-ARC determined in two independent PK studies reveals a prolonged circulatory half-life of siRNA when attached to the DVD-IgG1, suggesting increased potential for delivery to the target cells.
of 10–12 h for the DVD-ARC determined in two independent PK studies reveals a prolonged circulatory half-life of siRNA when attached to the DVD-IgG1, suggesting increased potential for delivery to the target cells.
The objective of the current study was to establish the DVD-ARC platform biochemically. Building on this proof-of-concept, the next steps will involve a better understanding of the biology, i.e. a qualitative and quantitative assessment of when and where the siRNA payload is released from the ARC, enters the cytoplasm, and engages with the RISC machinery. Analogous to ADCs (10), this information can be used to optimize antibody, linker, and payload components and tailor them to both the antibody and the siRNA target. To translate the DVD-ARC platform to preclinical and, ultimately, clinical investigations, suitable combinations of these two targets enabling functional knockdown need to be identified. Although the BCMA/CTNNB1 combination in our proof-of-concept indication, MM, revealed significant mRNA and protein knockdown, we did not observe cytotoxicity. In ongoing experiments, we are combining antibody-mediated BCMA targeting with siRNA-mediated targeting of intracellular proteins whose downregulation or inhibition is known to lead to MM cell death. As such, comparing the therapeutic index of ARCs to ADCs and small molecules will be a key consideration for clinical translation in MM and beyond.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs Dobeen Hwang and HaJeung Park (The Scripps Research Institute) for preparing Figure 2, Dr Junpeng Qi (The Scripps Research Institute) for supporting data acquisition and analysis for the PK study and Dr Sany Hoxha (The Scripps Research Institute) for help with RT-qPCR. This is manuscript #29915 from The Scripps Research Institute.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Z.X.V. acknowledges a Singapore A*STAR AGS (PDF) scholarship. Funding for open access charge: Alnylam, Inc.
Conflict of interest statement. Alnylam Pharmaceuticals funded this study and is the employer of A.V.K., C.T., J.M.P., A.G., J.K.N., M.A.M. and K.F.
REFERENCES
- 1. Setten R.L., Rossi J.J., Han S.P.. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019; 18:421–446. [DOI] [PubMed] [Google Scholar]
- 2. Zuckerman J.E., Davis M.E.. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat. Rev. Drug Discov. 2015; 14:843–856. [DOI] [PubMed] [Google Scholar]
- 3. Semple S.C., Akinc A., Chen J., Sandhu A.P., Mui B.L., Cho C.K., Sah D.W., Stebbing D., Crosley E.J., Yaworski E. et al.. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010; 28:172–176. [DOI] [PubMed] [Google Scholar]
- 4. Jayaraman M., Ansell S.M., Mui B.L., Tam Y.K., Chen J., Du X., Butler D., Eltepu L., Matsuda S., Narayanannair J.K. et al.. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. 2012; 51:8529–8533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Adams D., Gonzalez-Duarte A., O’Riordan W.D., Yang C.C., Ueda M., Kristen A.V., Tournev I., Schmidt H.H., Coelho T., Berk J.L. et al.. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018; 379:11–21. [DOI] [PubMed] [Google Scholar]
- 6. Nair J.K., Willoughby J.L., Chan A., Charisse K., Alam M.R., Wang Q., Hoekstra M., Kandasamy P., Kel’in A.V., Milstein S. et al.. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014; 136:16958–16961. [DOI] [PubMed] [Google Scholar]
- 7. Foster D.J., Brown C.R., Shaikh S., Trapp C., Schlegel M.K., Qian K., Sehgal A., Rajeev K.G., Jadhav V., Manoharan M. et al.. Advanced siRNA designs further improve in vivo performance of GalNAc-siRNA conjugates. Mol. Ther. 2018; 26:708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carter P.J., Lazar G.A.. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 2017; 17:197–223. [DOI] [PubMed] [Google Scholar]
- 9. Kaplon H., Reichert J.M.. Antibodies to watch in 2019. mAbs. 2019; 11:219–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beck A., Goetsch L., Dumontet C., Corvaia N.. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017; 16:315–337. [DOI] [PubMed] [Google Scholar]
- 11. Kumar P., Ban H.S., Kim S.S., Wu H., Pearson T., Greiner D.L., Laouar A., Yao J., Haridas V., Habiro K. et al.. T cell-specific siRNA delivery suppresses HIV-1 infection in humanized mice. Cell. 2008; 134:577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Song E., Zhu P., Lee S.K., Chowdhury D., Kussman S., Dykxhoorn D.M., Feng Y., Palliser D., Weiner D.B., Shankar P. et al.. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat. Biotechnol. 2005; 23:709–717. [DOI] [PubMed] [Google Scholar]
- 13. Peer D., Zhu P., Carman C.V., Lieberman J., Shimaoka M.. Selective gene silencing in activated leukocytes by targeting siRNAs to the integrin lymphocyte function-associated antigen-1. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:4095–4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xia C.F., Boado R.J., Pardridge W.M.. Antibody-mediated targeting of siRNA via the human insulin receptor using avidin−biotin technology. Mol. Pharm. 2009; 6:747–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lu H., Wang D., Kazane S., Javahishvili T., Tian F., Song F., Sellers A., Barnett B., Schultz P.G.. Site-specific antibody-polymer conjugates for siRNA delivery. J. Am. Chem. Soc. 2013; 135:13885–13891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baumer S., Baumer N., Appel N., Terheyden L., Fremerey J., Schelhaas S., Wardelmann E., Buchholz F., Berdel W.E., Muller-Tidow C.. Antibody-mediated delivery of anti-KRAS-siRNA in vivo overcomes therapy resistance in colon cancer. Clin. Cancer Res. 2015; 21:1383–1394. [DOI] [PubMed] [Google Scholar]
- 17. Baumer N., Appel N., Terheyden L., Buchholz F., Rossig C., Muller-Tidow C., Berdel W.E., Baumer S.. Antibody-coupled siRNA as an efficient method for in vivo mRNA knockdown. Nat. Protoc. 2016; 11:22–36. [DOI] [PubMed] [Google Scholar]
- 18. Sugo T., Terada M., Oikawa T., Miyata K., Nishimura S., Kenjo E., Ogasawara-Shimizu M., Makita Y., Imaichi S., Murata S. et al.. Development of antibody–siRNA conjugate targeted to cardiac and skeletal muscles. J. Control. Release. 2016; 237:1–13. [DOI] [PubMed] [Google Scholar]
- 19. Cuellar T.L., Barnes D., Nelson C., Tanguay J., Yu S.F., Wen X., Scales S.J., Gesch J., Davis D., van Brabant Smith A. et al.. Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB-siRNA conjugates. Nucleic Acids Res. 2015; 43:1189–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huggins I.J., Medina C.A., Springer A.D., van den Berg A., Jadhav S., Cui X., Dowdy S.F.. Site selective antibody-oligonucleotide conjugation via microbial transglutaminase. Molecules. 2019; 24:3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barbas C.F. III, Heine A., Zhong G., Hoffmann T., Gramatikova S., Björnestedt R., List B., Anderson J., Stura E.A., Wilson I.A. et al.. Immune versus natural selection: antibody aldolases with enzymic rates but broader scope. Science. 1997; 278:2085–2092. [DOI] [PubMed] [Google Scholar]
- 22. Nanna A.R., Li X., Walseng E., Pedzisa L., Goydel R.S., Hymel D., Burke T.R. Jr, Roush W.R., Rader C.. Harnessing a catalytic lysine residue for the one-step preparation of homogeneous antibody-drug conjugates. Nat. Commun. 2017; 8:1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kyle R.A., Rajkumar S.V.. Multiple myeloma. N. Engl. J. Med. 2004; 351:1860–1873. [DOI] [PubMed] [Google Scholar]
- 24. Parmar R.G., Brown C.R., Matsuda S., Willoughby J.L.S., Theile C.S., Charisse K., Foster D.J., Zlatev I., Jadhav V., Maier M.A. et al.. Facile synthesis, geometry, and 2′-substituent-dependent in vivo activity of 5′-(E)- and 5′-(Z)-vinylphosphonate-modified siRNA conjugates. J. Med. Chem. 2018; 61:734–744. [DOI] [PubMed] [Google Scholar]
- 25. Wuellner U., Gavrilyuk J.I.;, Barbas C.F. III. Expanding the concept of chemically programmable antibodies to RNA aptamers: chemically programmed biotherapeutics. Angew. Chem. Intl. Ed. 2010; 49:5934–5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Karlstrom A., Zhong G., Rader C., Larsen N.A., Heine A., Fuller R., List B., Tanaka F., Wilson I.A., Barbas C.F. III et al.. Using antibody catalysis to study the outcome of multiple evolutionary trials of a chemical task. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:3878–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nanna A.R., Rader C.. Engineering dual variable domains for the generation of site-specific antibody-drug conjugates. Methods Mol. Biol. 2019; 2033:39–52. [DOI] [PubMed] [Google Scholar]
- 28. Gardner M.R., Fellinger C.H., Prasad N.R., Zhou A.S., Kondur H.R., Joshi V.R., Quinlan B.D., Farzan M.. CD4-induced antibodies promote association of the HIV-1 envelope glycoprotein with CD4-binding site antibodies. J. Virol. 2016; 90:7822–7832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Livak K.J., Schmittgen T.D.. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT. Methods. 2001; 25:402–408. [DOI] [PubMed] [Google Scholar]
- 30. Lv J., Cao X.F., Ji L., Zhu B., Wang D.D., Tao L., Li S.Q.. Association of beta-catenin, Wnt1, Smad4, Hoxa9, and Bmi-1 with the prognosis of esophageal squamous cell carcinoma. Med. Oncol. 2012; 29:151–160. [DOI] [PubMed] [Google Scholar]
- 31. Kumar S.K., Anderson K.C.. Immune therapies in multiple myeloma. Clin. Cancer Res. 2016; 22:5453–5460. [DOI] [PubMed] [Google Scholar]
- 32. Tai Y.T., Mayes P.A., Acharya C., Zhong M.Y., Cea M., Cagnetta A., Craigen J., Yates J., Gliddon L., Fieles W. et al.. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood. 2014; 123:3128–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Trudel S., Lendvai N., Popat R., Voorhees P.M., Reeves B., Libby E.N., Richardson P.G., Anderson L.D. Jr, Sutherland H.J., Yong K. et al.. Targeting B-cell maturation antigen with GSK2857916 antibody-drug conjugate in relapsed or refractory multiple myeloma (BMA117159): a dose escalation and expansion phase 1 trial. Lancet Oncol. 2018; 19:1641–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Trudel S., Lendvai N., Popat R., Voorhees P.M., Reeves B., Libby E.N., Richardson P.G., Hoos A., Gupta I., Bragulat V. et al.. Antibody-drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: an update on safety and efficacy from dose expansion phase I study. Blood Cancer J. 2019; 9:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ikeda H., Hideshima T., Fulciniti M., Lutz R.J., Yasui H., Okawa Y., Kiziltepe T., Vallet S., Pozzi S., Santo L. et al.. The monoclonal antibody nBT062 conjugated to cytotoxic Maytansinoids has selective cytotoxicity against CD138-positive multiple myeloma cells in vitro and in vivo. Clin. Cancer Res. 2009; 15:4028–4037. [DOI] [PubMed] [Google Scholar]
- 36. Rader C., Turner J.M., Heine A., Shabat D., Sinha S.C., Wilson I.A., Lerner R.A., Barbas C.F. III. A humanized aldolase antibody for selective chemotherapy and adaptor immunotherapy. J. Mol. Biol. 2003; 332:889–899. [DOI] [PubMed] [Google Scholar]
- 37. Schmeel L.C., Schmeel F.C., Kim Y., Endo T., Lu D., Schmidt-Wolf I.G.. Targeting the Wnt/beta-catenin pathway in multiple myeloma. Anticancer Res. 2013; 33:4719–4726. [PubMed] [Google Scholar]
- 38. Su N., Wang P., Li Y.. Role of Wnt/β-catenin pathway in inducing autophagy and apoptosis in multiple myeloma cells. Oncol. Lett. 2016; 12:4623–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schirle N.T., Kinberger G.A., Murray H.F., Lima W.F., Prakash T.P., MacRae I.J.. Structural analysis of human argonaute-2 bound to a modified siRNA guide. J. Am. Chem. Soc. 2016; 138:8694–8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Parmar R., Willoughby J.L., Liu J., Foster D.J., Brigham B., Theile C.S., Charisse K., Akinc A., Guidry E., Pei Y. et al.. 5′-(E)-Vinylphosphonate: a stable phosphate mimic can improve the RNAi activity of siRNA-GalNAc conjugates. Chembiochem. 2016; 17:985–989. [DOI] [PubMed] [Google Scholar]
- 41. Krop I.E., Beeram M., Modi S., Jones S.F., Holden S.N., Yu W., Girish S., Tibbitts J., Yi J.H., Sliwkowski M.X. et al.. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J. Clin. Oncol. 2010; 28:2698–2704. [DOI] [PubMed] [Google Scholar]
- 42. Nair J.K., Attarwala H., Sehgal A., Wang Q., Aluri K., Zhang X., Gao M., Liu J., Indrakanti R., Schofield S. et al.. Impact of enhanced metabolic stability on pharmacokinetics and pharmacodynamics of GalNAc-siRNA conjugates. Nucleic Acids Res. 2017; 45:10969–10977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hwang D., Nilchan N., Nanna A.R., Li X., Cameron M.D., Roush W.R., Park H., Rader C.. Site-selective antibody functionalization via orthogonally reactive arginine and lysine residues. Cell Chem. Biol. 2019; 26:1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.