


Abstract
Antisense oligonucleotides (ASOs) interact with target RNAs via hybridization to modulate gene expression through different mechanisms. ASO therapeutics are chemically modified and include phosphorothioate (PS) backbone modifications and different ribose and base modifications to improve pharmacological properties. Modified PS ASOs display better binding affinity to the target RNAs and increased binding to proteins. Moreover, PS ASO protein interactions can affect many aspects of their performance, including distribution and tissue delivery, cellular uptake, intracellular trafficking, potency and toxicity. In this review, we summarize recent progress in understanding PS ASO protein interactions, highlighting the proteins with which PS ASOs interact, the influence of PS ASO protein interactions on ASO performance, and the structure activity relationships of PS ASO modification and protein interactions. A detailed understanding of these interactions can aid in the design of safer and more potent ASO drugs, as illustrated by recent findings that altering ASO chemical modifications dramatically improves therapeutic index.
INTRODUCTION
Though the concept of designing oligonucleotides to bind via Watson–Crick hybridization to a specific sequence in an RNA target and the term ‘antisense therapeutics’ were introduced in 1978 (1), this novel idea generated little interest till 1989 when several companies were formed to create a new platform for drug discovery focused on targeting RNAs. As proposed, the term ‘antisense technology’ was entirely agnostic as to the structure (single or double stranded oligonucleotides) and the chemistry of the oligonucleotide. Nor did the authors address in any detail what mechanism of action after binding to the RNA might be used to alter the fate or performance of the targeted RNA. Thus, a key to the success of the technology was to develop the medicinal chemistry of oligonucleotides and to define potential mechanism of the binding of the ASO to the RNA that could alter the desired pharmacological effects. Therefore, in this review, the term antisense oligonucleotide (ASO), unless modified, will be used generically to include all RNA targeted ASOs of any chemistry or structure.
Over the past thirty years, the field of RNA targeted therapeutics has advanced sufficiently that it seems likely that the platform will take its place as a broadly enabling drug discovery technology. Today, nine RNA targeted drugs, seven single stranded ASOs and two double stranded ASOs or siRNAs, have been approved for commercial use and scores of RNA targeted agents are in development (for review, see (2–4)). To create this technology, a new conceptual framework that treats RNAs as complex structured ribonucleoproteins that present multiple potential ASO binding sites or receptors had to be established. The medicinal chemistry of oligonucleotides and the molecular pharmacology of these agents also had to be created and understood at progressively more sophisticated levels (for review, see (2,5,6)). Today, ASOs representing multiple chemical classes are available and these agents may be designed to exploit multiple post-RNA binding mechanisms of action. Some commonly used chemical modifications described in this review are shown in Figure 1. The pharmacokinetics of these major chemical classes used therapeutically are also well defined (for review, see (5,7,8)) as are potential toxicities (9–11).
Figure 1.

Schematic prediction of chemical modifications as described. (A) Backbone modification. (B) 2′ modifications. PO, phosphodiester; PS, Phosphorothioate; MOP, methoxypropylphosphonate; Me, methyl; MOE, methoxyethyl; S-cEt, constrained ethyl (cEt); LNA, locked nucleic acid; F, fluoro.
One of the earliest of the modifications, shown in Figure 1, was the substitution of phosphorothioate moieties for the phosphate (PO) at each inter-nucleotide linkage (for review, see (6,12)). This simple chemical change has proven to be a vital component of essentially all of the major chemical and structural classes of ASOs broadly used for therapeutics (2,13). Phosphorothioate (PS) modifications confer increased resistance to the nucleases that degrade ASOs thereby extending their tissue elimination half-lives (for review, see (8,13)). Today, there are numerous chemical designs that can be used to stabilize ASOs to nucleases, but no chemical modification has been identified that provides the optimum in protein binding that the PS moiety confers (7,8,14). In fact, irrespective of the 2′ modification incorporated, the PS moiety is the primary determinant of the distribution of single stranded ASOs after all routes of administration. In plasma, PS containing single stranded ASOs bind to numerous proteins with a wide range of binding affinities (Kd) (14,15). These interactions are critical because, with the exception of the neutrally charged morpholino phosphorodiamidate class, ASOs that lack PS linkages are rapidly cleared by glomerular filtration (8). The extension of the distribution half-life to 1–2 h which is conferred by PS modifications is essential to support exit from the vascular compartment and entry into tissues.
Despite the obvious importance of protein binding by PS ASOs, for the first 20 plus years of effort to create the RNA targeting platform, medicinal chemistry focused almost exclusively on understanding the structure activity relationships of ASO interactions with RNA, both from the perspectives of oligonucleotides and their RNA targets. While substantial progress had been reported over those two decades (for review, see (2)), by 2016, advances in understanding the molecular mechanisms of cell uptake and intracellular distribution, mechanisms of action, mechanisms of toxicities and in in vivo targeted delivery demonstrated that it was essential to understand the structure activity relationships that determine PS ASO interactions with proteins (2,16–18). Put simply, proteins determine the fate of PS ASOs in all biological systems. This means that if a PS ASO is present at a biological site, a protein or proteins are responsible for it being there. Moreover, PS ASOs can alter the fates of many of the proteins with which they interact. The goals, therefore, of this review are to summarize and codify the advances to date and to provide a theoretical framework with which to consider the interactions of these agents with proteins, i.e. to begin to define the language of PS ASO–protein interactions.
Plasma protein binding
That PS ASOs bind to a number of plasma proteins has been known for almost 30 years. At therapeutic doses, and even at doses used in toxicological studies, PS ASOs are extensively protein bound in plasma (19). Though there are slight variations in plasma protein binding as a function of PS ASO sequence, as a general rule, >95% of circulating PS ASO is plasma protein bound (20,21). Slightly greater variation in plasma protein binding is observed as a function of species. Typically, in the mouse, plasma protein binding is slightly less than that in non-human primates (NHP) or humans (for review, see (7)). There are, of course, a large number of proteins in plasma and their plasma concentrations vary widely. For example, in human plasma, albumin is the most abundant and is present at a 600 μM concentration while the 25th most abundant protein, factor V, is present at a 0.02 μM concentration (14).
Given the high concentration of albumin in plasma and in common with most xenobiotics, PS ASOs bind extensively to plasma albumin. The affinity reported has varied widely depending on the method used and whether the albumin is lipidated, with the most recent publication using fluorescence polarization analyses reporting a Kd of 12.7 μM (14,15). Importantly, there is no evidence of competition between PS ASOs and other drugs that bind to albumin, such as warfarin (7,15, 22). However, despite the lower abundance of other plasma proteins, some of these interactions can be important as well. For example, alpha 2 macroglobulin is present in human plasma at a 6 μM concentration and displays high affinity (Kd of 0.05 μM) for PS ASOs (14). In alpha 2 macroglobulin knockout mice, PS ASO plasma protein binding was indistinguishable from normal mice, but a two-fold increase in potency for reducing a target mRNA in the liver was observed (23). Though the mechanism accounting for this increased potency is not defined, it seems likely that it is related to changes in distribution to peripheral tissues. Table 1 lists the major plasma proteins to which PS ASOs bind, their concentrations in plasma and the Kds observed.
Table 1.
Plasma proteins that bind PS ASOs
| Protein | Plasma conc. (μM) | K d (μM) |
|---|---|---|
| Serum albumin | 600 | 12.7 |
| IgG | 75 | 1.6 |
| Apolipoprotein A-I | 40 | 5.3 |
| Apolipoprotein A-II | 24 | >500 |
| Complement factor C3 | 20 | 0.5 |
| Transferrin | 12 | 7.0 |
| α-1 Antitrypsin | 11 | >100 |
| Haptoglobin | 11 | 54.7 |
| Hemopexin | 9.9 | 13.9 |
| Fibrinogen | 9 | 0.87 |
| α-2-Macroglobulin | 6 | 0.05 |
| Prealbumin/TTR | 6 | 132 |
| Antithrombin III | 3.5 | 8.7 |
| α-1-Antichymotrypsin | 3.3 | 21.3 |
| β-2-Glycoprotein | 2.7 | 57.1 |
| Ceruloplasmin | 2 | 22.6 |
| α-1 Acid glycoprotein | 1.7 | >500 |
| Complement component C1q | 1.6 | 3.4 |
| Complement factor C4 | 1.4 | 0.43 |
| Histidine-rich glycoprotein | 1.3 | 0.009 |
| Plasminogen | 1.2 | 2.1 |
| Fibronectin | 0.9 | 0.54 |
| ApoB100 | 0.7 | >10 |
| Factor H | 0.6 | 0.5 |
| Apolipoprotein E | 0.5 | 0.027 |
| Factor V | 0.02 | 0.032 |
Adapted from Gaus et al., Nucleic Acids Research. 2019, 47:1110–1122.
PS ASOs also bind to proteins involved in the intrinsic clotting cascade and the alternative complement pathway. Interactions with the tenase complex have been reported to result in prolongation of the activated partial thromboplastin time (for review, see (24,25)) These effects are peak plasma concentration dependent and therefore are dose dependent. They are transient and decline as PS ASOs exit the vascular compartment. Given the transient nature of the effects, no adverse events associated with this inhibition have been reported. Of substantially more concern have been the effects of PS ASOs on the alternative complement pathway which can result in activation of the complement pathway. Once again, these effects are related to peak plasma concentrations and dose. The NHP appears to be more sensitive than humans to these effects and are often observed at the higher doses administered in toxicological studies in the NHP (for review, see (7,8)).
The binding of PS ASOs to plasma proteins is critical to the ultimate distribution of PS ASOs to tissues. In the absence of plasma protein binding, oligonucleotides are rapidly cleared by glomerular filtration and excreted in urine, resulting in virtually no distribution to peripheral tissues. PS ASO binding to plasma proteins results in a distribution half-life of 1–2 h in all species tested, supporting distribution to peripheral tissues (for review, see (7,8)). Inasmuch as the bulk of plasma protein binding by PS ASOs is albumin, albumin can be considered to determine the fate of PS ASOs in plasma.
Despite broad interest in the interactions of PS ASOs with plasma proteins, structure activity relationships (SAR) for specific interactions and overall binding with these proteins are poorly understood. Slight variations in total plasma protein binding have been observed for different sequences of ASOs within chemical classes (8,15), but essentially nothing is known about the influence of PS ASO sequence on the binding to specific plasma proteins. However, binding to plasma proteins is dependent on PS number, with ∼10–12 PS linkages being necessary for meaningful interactions (7). The effects of PS ASOs containing 2ʹ-methoxyethyl (MOE) modifications have been compared to PS oligodeoxynucleotides, PS oligoribonucleotides and PS 2ʹ-constrained ethyl (cEt) ASOs with regard to total binding and interactions with a variety of specific plasma proteins. While a number of differences in affinity for total plasma protein binding and binding to specific proteins, such as histidine rich glycoprotein were observed, no general principles were evident (14). The effects of conjugation of a few ligands designed to enhance delivery to targeted tissues have also been evaluated. N-Acetyl-galactosamine clusters (GalNAc) are frequently conjugated to PS ASOs to enhance delivery to hepatocytes (for review, see (2)). Despite enabling productive delivery of PS ASOs to hepatocytes, the effects of GalNAc conjugation to PS ASOs on plasma protein binding are minimal (26). In contrast, the conjugation of fatty acids or other lipids to PS ASOs or siRNAs has been shown to substantially alter their binding to total plasma proteins, and in the case of siRNAs, enhance distribution to peripheral tissues (27–32).
Unfortunately, essentially nothing is known about the protein domains that bind PS ASOs although it is known that total plasma protein binding is influenced by ionic strength and pH, suggesting that ionic interactions are important (14). In addition, information regarding the chemical nature of PS ASOs binding to total plasma proteins or to specific plasma proteins is limiting.
Interactions with proteins located in the plasma membranes and the roles of protein binding in cellular uptake and subcellular distribution
Single strand (ss) PS ASOs readily enter mammalian cells both in vitro and in vivo (for review, see (2,7,16,33)). Thanks to advances reported in the past few years, it is known that the bulk of uptake of ss PS ASOs is mainly via endosomes, although the potential presence of a nucleic acids channel in the cell surface has been proposed (34). The major endocytic transport pathways and the relative contribution of each pathway to delivery of pharmacologically effective PS ASO to intracellular sites and the key proteins involved are well defined (Figure 2) (16,35–38). In contrast, and as predicted based on the physical–chemical properties of double strand (ds) ASOs, ds ASOs must either be delivered to cells using cationic lipids or via targeting receptors such as the asialoglycoprotein receptor (ASGPR) by conjugating GalNAc to ds ASOs (for review, see (39–42)). Whether a PS ASO is unconjugated or conjugated with a variety of targeting ligands, the cellular uptake and subcellular distribution process is initiated by interactions of the PS ASO with proteins that are present in the plasma membrane and display binding surfaces at the extra-cellular face of the lipid bilayer. We have shown that this process is best described by a simple adsorption to exposed domains of plasma membrane proteins model (16).
Figure 2.

Schematic prediction of ASO adsorption and intracellular trafficking. PS ASOs enter cells via surface proteins, either through direct binding or mediated by conjugated ligands, tend to be routed to productive uptake pathway. Internalized PS ASOs traffic through the major endocytic process, and are mainly released from LEs, facilitated by ASO binding proteins including STX5, M6PR. Normal cellular transport vesicles such as COPII and M6PR vesicles also mediate productive release of PS ASOs.
Though it has been shown that PS ASOs are able to interact with zwitterionic phosphocholines as determined in liposome systems (43), no direct interactions with the lipid components of the plasma membrane or of intracellular membranes has been demonstrated for any PS ASO, with the possible exception of those compounds conjugated with fatty acids (44). Finally, lipid conjugation results in substantial increases in PS ASO bound to membranes, longer retention of these oligonucleotides in endosomal membranes, and an increase in total activity (44). However, the lipid composition and turnover of lipids in membranes do play important roles in influencing uptake and distribution of PS ASOs. For example, destabilizing membranes by altering the lipid composition such as increasing cholesterol or ceramide content, enhanced release of PS ASOs and increased activity but did not increase total cellular uptake of these agents over time (45). Additionally, ASO activity can be enhanced by palmitic and oleic acid by forming intracellular lipid droplets while ceramide and cholesterol increase membrane ‘leakiness’. Lysobisphosphatidic acid (LBPA) is required for productive uptake and release of PS ASOs because of its role in the activities of late endosomes, especially in the formation of multivesicular bodies (46). Thus, it is clear that membrane lipid composition can influence the uptake and intracellular trafficking of PS ASOs, but much more work is needed to determine the molecular mechanisms that may be involved and how interactions with key proteins as listed in Table 2 may be affected.
Table 2.
Partial list of membrane localized proteins that bind PS ASOs
| A. PS ASO binding proteins localized to plasma membranes | ||||
| Protein | Effects on ASO activity | Ref | ||
| ASGPR | Productive | Tanowitz M, et al., 2018, NAR | ||
| EGFR | Productive | Wang S., et al., 2018, NAR | ||
| LDLR | N/D | Wang S., et al., unpublished | ||
| M6PR | Productive | Liang XH et al., 2020, NAR | ||
| TLR | (May mediate inflammatory effects) | Kandimalla, E.R., et al., 2013, NAR | ||
| Stabilin | Productive | Miller C., et al., 2016, NAR | ||
| SRB | N/D | Wang S., et al., unpublished | ||
| Nucleolin | N/D | Kotula JW., et al., 2012, NAT | ||
| B. Proteins that affect ASO trafficking and endosomal release | ||||
| Protein | Pathway | ASO co-localization | Role in ASO trafficking | Ref. |
| AP2M1 | Clathrin-mediated transport | Not determined | Productive uptake | Koller E., et al., 2011, NAR |
| EEA1 | EE maturation | EE | EE to LE transport | Miller CM et al., 2018, NAT |
| Rab5C | EE maturation | EE | EE to LE transport | |
| Rab7a | LE maturation | LE | EE to LE transport | |
| STX5 | Golgi tethering of COPII | LE | COPII relocalization to LE | Liang XH., et al., 2018, NAR |
| P115 | Golgi tethering of COPII | No | COPII relocalization to LE | |
| COPII | ER-Golgi transport | LE | ASO release from LE | |
| M6PR | Golgi-LE transport | LE / Vesicles | ASO release from LE | Liang XH et al., 2020, NAR |
| GCC2 | Golgi tethering | LE | M6PR LE-Golgi shuttling | |
| ANXA2 | EE – LE transport | LE | ASO release from LE | Liang XH et al., 2015, NAR. Wang S., et al., 2017, NAR. |
| TCP1 | Chaperon | LE | ASO release from LE | Liang XH et al., 2015, NAR |
| ALIX | LE maturation | Not determined | ASO release from LEs | Wang S et al., 2017 NAR |
| TSG101 | Endosomal transport | LE | Inhibit productive uptake | Wagenaar TR et al., 2015, NAR |
| VPS28 | Endosomal transport | LE | Inhibit productive uptake | |
A partial catalog of cell surface localized proteins that may bind PS ASOs is shown in Table 2A. These include stabilin 1/2 (47,48), ASGPR (49), epidermal growth factor receptor (EGFR) (50), mannose-6-phosphate receptor (M6PR) (36), nucleolin (51), toll like receptor (TLR) proteins (52), and the low-density lipoprotein receptor (LDLR) (for review, see (16)). When conjugated with GalNAc, both single and double stranded ASOs bind and are taken up by the ASGPR (for review see (16,42)) and when epidermal growth factor (EGF) is conjugated to an ASO, both the unadorned PS ASO and the EGF-conjugated PS ASO bind to EGFR (50,53). Additionally, conjugation of a glucagon-like peptide 1 (GLP-1) has been reported to dramatically increase productive uptake specifically into the B cells of the pancreas, both in vitro and in vivo (54). In conclusion, PS ASOs may interact with many membrane proteins and it is likely that other membrane localized proteins that interact with PS ASOs will be identified. Interestingly, although some surface receptors like the scavenger receptor class A (SRA) may not directly interact with PS ASOs, receptor mediated ASO uptake can occur through interaction with plasma proteins to which PS ASOs are bound (55). Thus, targeted delivery to specific cells and organs can be achieved and result in important increases in the performance of PS ASOs. Given the extent of efforts to identify novel cell targeting ligands, it seems likely that new ligands that enhance delivery to other cells and organs will be discovered.
The first step in the entry and subcellular distribution of PS ASOs is the binding of these agents to proteins displayed on the external surface of the plasma membrane. Such interactions can lead to subcellular distribution resulting in pharmacological activity (productive delivery), while other interactions can result in non-productive subcellular distribution. While a number of these cell surface proteins have been identified, we suspect that there may be more to which PS ASOs bind, likely in a cell type dependent manner. Interactions can include direct binding to PS ASOs, binding only to a targeting ligand such as GalNAc or some proteins can bind directly to PS ASOs and a targeting ligand. Binding at the cell surface occurs via simple adsorption, does not require energy and is extremely rapid (Figure 2) (for review, see (16,56)). Cellular uptake of PS ASOs is due primarily to endosomal processes with the first step being binding to cell surface proteins that induce endosome formation and trafficking (16). Figure 2 shows the cell surface proteins identified to date that bind PS ASOs directly and mediate ‘productive’ cellular uptake, those that bind only specific targeting ligand and mediate ‘productive uptake’ and that the main non-productive uptake pathway is macropinocytosis (16).
The EGFR is of particular mechanistic interest as PS ASOs bind directly to it and induce cellular internalization of the oligonucleotides without inducing receptor-mediated signaling (50). On the other hand, this receptor can also bind to EGF-conjugated ASOs and induces signaling while delivering PS ASOs to the cell (53). In contrast, ASGPR, a scavenger receptor binds PS ASOs in a fashion that does not support pharmacological activity, but does bind and deliver GalNAc-conjugated PS ASOs resulting in dramatically increased activity (49,57). PS ASOs do not bind GLP1 receptors, but when these ASOs are conjugated with a GLP1 analog, they readily bind this G-protein-coupled receptor on the Beta cells of the pancreas and stimulate signaling while delivering PS ASOs productively to these cells (54). Thus, proteins at the cell surface play a determinative role in defining the extent and the nature of cellular uptake of PS ASOs. There is a growing list of targeting ligands and potential cell surface acceptors/receptors that may be targeted by conjugating PS ASOs to the ligands (for review, see (33,58,59)).
Interactions with intracellular proteins
Several approaches have been taken to identify and characterize the intracellular proteins with which PS ASOs interact. These include protein pull down assays using biotin conjugated PS ASOs coupled to mass spectrometric and western analysis (60–62), a modification of the Nano-Bioluminescence Resonance Energy Transfer (Nano-BRET) assay (63), a modification of the NanoLuc Binary Technology (Nano-Bit) assay (17) and the Proximity-dependent Biotin Identification (BioID) assay (64). Coupling these assays to molecular biological manipulations of target proteins and multiple chemical changes in PS ASOs has supported very rapid advances in understanding the chemical nature of these interactions, the protein domains involved, the impacts on cellular locations of PS ASOs and the impacts of PS ASOs on various proteins and protein complexes. Table 3 presents a list of the major intracellular proteins that interact with PS ASOs, the effects of each protein on the subcellular localization and on the potency and toxicity of these agents.
Table 3.
Intracellular proteins that bind PS ASOs
| Protein | Feature | ASO activity upon protein knockdown | ASO localization | Impact on ASO toxicity | ASO-protein co-localization |
|---|---|---|---|---|---|
| Nucleic acid binding proteins (33) | |||||
| Ago2* | RNA Binding | Reduced (free uptake) | Not characterized | Yes (P-bodies) | |
| CArG binding factor | RNA binding | Not characterized | Not characterized | ||
| DDX21 | RNA binding | Yes (toxic ASO) | |||
| DHX30 | RNA binding | No change | No change | ||
| EIF2S2 | RNA binding | Not characterized | Not characterized | ||
| eIF4H | RNA binding | No change | No change | ||
| GRSF | RNA binding | No change | No change | ||
| HMGB1 | DNA binding | No change | No change | No | |
| hnRNP D1Like | RNA binding | Not characterized | Not characterized | ||
| hnRNPA1$ | RNA binding | Not characterized | Not characterized | ||
| hnRNPA2 | RNA binding | Not characterized | Not characterized | ||
| hnRNPF | RNA binding | Not characterized | Not characterized | ||
| hnRNPH1 | RNA binding | Not characterized | Not characterized | ||
| hnRNPK | RNA binding | Increase | Yes | Yes | |
| hnRNPQ | RNA binding | No change | No change | ||
| hnRNPU | RNA binding | Not characterized | Not characterized | ||
| hnRNPUL | RNA binding | Not characterized | Not characterized | ||
| ILF2 | RNA binding | Not characterized | Not characterized | ||
| ILF3 | RNA binding | No change | No change | ||
| KHSRP | RNA binding | No change | No change | ||
| Ku70 | DNA binding | Increase | No change | No | |
| Ku80 | DNA binding | Increase | No change | No | |
| La/SSB | RNA binding | Reduce | Yes | No | |
| NCL# | RNA binding | No change | No change | Yes (toxic ASO) | |
| NPM1 | RNA binding | Reduce | Yes | Yes (toxic ASO) | |
| P54nrb | RNA/DNA binding | Increase | Yes | Yes | Yes |
| PC4/Sub1 | DNA binding | Reduce | No change | No | |
| PSF | RNA/DNA binding | Increase | Yes | Yes | Yes |
| PSPC1 | RNA binding | Increase | Yes | Yes | |
| RHA | RNA binding | No change | No change | ||
| RNase H1 | DNA/RNA duplex binding | Reduce | No change | Yes | |
| RNF163/ZNF9 | DNA binding | No change | No change | ||
| YBX1 protein | RNA binding | No change | Not characterized | ||
| Chaperone proteins (11) | |||||
| GRP78/Bip | Chaperone protein | No change | No change | ||
| HSC70 | Chaperone protein | Reduce | No change | No | |
| HSP90-AA1 | Chaperone protein | Reduce | No change | Yes | No |
| Hsp90-AB | Chaperone protein | Reduce | No change | Yes | |
| HSPA1L | Chaperone protein | Not characterized | Not characterized | ||
| TCP1-alpha | Chaperone protein | Reduce | Not characterized | Yes (LE) | |
| TCP1-beta | Chaperone protein | Reduce | Not characterized | Yes (PS-body, LE) | |
| TCP1-delta | Chaperone protein | Not characterized | Not characterized | Yes (LE) | |
| TCP1-episilon | Chaperone protein | Reduce | Not characterized | Yes (LE) | |
| TCP1-gamma | Chaperone protein | Not characterized | Not characterized | Yes (LE) | |
| TCP1-Theta | Chaperone protein | Not characterized | Not characterized | Yes (LE) | |
| Other proteins (17) | |||||
| ACLY | Enzyme | No change | No change | ||
| Albumin | Secreted | Not characterized | Not characterized | ||
| Annexin A2 | Membrane binding | Reduce | Yes | Yes (LE) | |
| ATAD3A | Membrane | No change | No change | ||
| FTCD/58K | Enzyme | Reduce | Yes | Yes (LE) | |
| IMP9 | Transport | Reduce | Yes | ||
| JKTPB1 delta 6 | hnNRP-like | Not characterized | Not characterized | ||
| KCTD12 | Membrane receptor | No change | No change | ||
| LRPPRC | Transport/transcription | No change | No change | ||
| NARS | tRNA synthase | No change | No change | ||
| NDKA | Enzyme | Not characterized | Not characterized | ||
| RAN | Transport | Reduce | Yes | ||
| SHMT2 | Enzyme | Not characterized | Not characterized | ||
| Thymidylate kinase | Enzyme | Not characterized | Not characterized | ||
| VARS | tRNA synthase | Reduce | No change | ||
| β-actin (ACTB) | Structure | Reduce | Not characterized | ||
| β-tubulin (TUBB2C) | Structure | No change | Not characterized | ||
Note: Bolded proteins demonstrated to influence ASO activity or localization.
Adapted from Crooke ST et al., Nat Biotechnology. 2017. 35:230–237.
*Castanotto D., et al., 2015, NAR.
# Weidner, D. A. et al., 1995, FEBS Lett.
$ Abdul-Manan et al., 1996, NAR.
Only ∼80 intracellular proteins that bind PS ASOs have been identified (51,61,65). Though it is possible that other intracellular proteins will be identified, it seems likely that most of the key abundant proteins are now known. As expected, PS ASOs interact with numerous proteins with nucleic acid binding domains, but these interactions are not limited to nucleic acid binding domains. A relatively broad range of types of proteins is represented including chaperone proteins, helicases, polymerases, nucleases and other enzymes and other classes of proteins. The proteins that interact with PS ASOs are present in different cellular structures including mitochondrial membranes, the cytosol, nucleoplasm and the nucleolus.
The roles of binding to intracellular proteins in PS ASO subcellular trafficking and distribution
The key first step that has led to the advances in understanding of PS ASO interactions with biologically important proteins was the development of a biotin-PS ASO pull-down assay (60). Using this technique, approximate 55 intracellular proteins were identified in a total cellular homogenate (initially HEK293 cells) and their effects on the behaviors of PS ASOs evaluated (61). The results reported in the publication (61) have since been confirmed and extended using a range of assays, including pull-downs coupled to PAGE separation plus mass-spectrometric analyses. Two new methods, the NanoBRET assay (63) and the NanoBit assay (17), have enabled detailed understanding of PS-ASO-protein binding and visualization of PS ASO–protein interactions in live cells. Table 3 shows the major proteins identified divided into various classes based on known or inferred preferred binding partners and their effects on PS ASO potency in reducing targeted RNAs. As expected, interactions with a number of intracellular proteins have no obvious effect on ASO performance, but a good many interactions alter PS ASO behaviors.
Certainly, one critical process in which proteins play essential roles is cellular uptake and sub-cellular distribution of PS ASOs. A substantial number of proteins that play critical roles in the endosomal uptake, trafficking and release from endosomes have been identified and characterized (Table 2B). Essentially all these proteins are localized to the plasma membrane or early endosome, late endosome, COP2 vesicles or Golgi membranes (35–37,46,66,67). ANXA2 is able to bind PS-ASOs (61), PS ASO interaction is not required for ANXA2 mediated PS ASO transport and release from late endosomes (LEs) (67). On the other hand, although coat proteins of COPII vesicles do not interact with PS ASOs, Golgi-localized STX5 binds PS ASOs, relocates to LEs upon PS ASO uptake, likely in a manner dependent on ASO-protein interactions, and recruits COPII vesicles to LEs to facilitate PS ASO release (35). Similarly, M6PR is able to bind PS ASOs and can mediate PS ASO escape from LEs (36). In this pathway, GCC2 is required for M6PR shuttling between LEs and trans-Golgi network (TGN), and relocate to LEs upon PS ASO incubation, most likely mediated by PS ASO-protein interactions (Figure 2).
The roles of protein binding on the pharmacological activity of PS ASOs after release from membraned organelles
Although some PS ASO binding proteins affect activity by modulating ASO uptake and endosomal trafficking or release, other proteins affect ASO activity at steps after ASOs escape from membraned organelles. It is not unexpected that binding of PS ASOs to cellular proteins may reduce the binding of ASOs to their target RNAs, as supported by earlier observations (68). However, in a cellular environment, PS ASO protein interactions that affect activity are relatively limited in number. In fact, protein interactions may increase or decrease ASO activity. For example, it was found that reduction of La/SSB and NPM1 proteins decreased, and overexpression of these proteins increased PS ASO activity, most likely by modulating PS ASO accumulation in the nucleus (61). On the other hand, nuclear proteins Ku70/Ku80 and paraspeckle proteins P54nrb and PSF inhibit PS ASO activity, by competition with RNase H1 for binding to the RNA/ASO heteroduplex (61,69). In most cases, reduction of an individual PS ASO binding protein caused modest effects on ASO activity (60,61,69). This observation is not surprising, as many cellular proteins bind PS ASOs and each protein interacts with a small fraction of total ASO. Therefore, reducing an individual protein may affect the performance of a small fraction of cellular ASOs.
Interestingly, although ANXA2 facilitates ASO release from endosomes during free uptake, this protein also affects ASO activity upon transfection, an approach that largely bypasses normal endosomal release (61). Thus, ANXA2 may have additional roles in ASO activity after ASOs reach cytosol or nucleus. In addition, some PS ASO binding proteins affect ASO activity via unknown mechanisms, such as PC4, VARS, HSP90 (61,70). It is possible that these proteins may modulate PS ASO distribution or their ability to bind RNA target, facilitate recruitment of RNase H1 to the ASO/RNA duplex, modulate dissociation of RNase H1 or ASO from the RNA substrate, to name a few. Further studies are warranted to understand the underlying mechanisms. Perhaps the best example of proteins enhancing ASO activity is the interaction of siRNAs with the Ago2 loading complex. Once loaded on ASOs, it has been shown that hybridization with the target RNA is facilitated (71).
Proteins involved in the formation of PS ASO-induced aggregates
An important recent advance is the finding that PS ASOs released from membraned organelles can induce the formation of protein–RNA–ASO aggregates. The aggregates induced by PS ASOs vary substantially in the ASO concentrations at which they form, composition, impact on pharmacological activity, subcellular localization, PS ASO associated toxicologic effects and their effects on the cell. In 1998, the first PS ASO-induced cellular aggregate, PS bodies, was reported (72). These small round dot-like structures were observed in the nucleus after transfection of relatively high concentrations of PS ASOs and appeared to have no effect on the pharmacological activities of PS ASOs and no observable effect on cellular function. The protein composition of PS bodies was not defined until 2014 when a single protein, TCP1β, was determined to be bound to PS ASOs in these structures (60). TCP1β is one of the eight subunits of the TCP1 complex. Though all the subunits are able to bind PS ASOs, only TCP1β was found to localize to PS bodies. Depletion of Ras-related nuclear protein (RAN) resulted in the PS bodies relocating to the cytoplasm, thus demonstrating that PS ASO induced aggregates can vary in subcellular localization in response to changes in cellular activities, a characteristic that we now know is common. Table 4 shows the identified PS ASO induced aggregates, the current understanding of their compositions, the minimum concentrations at which they are formed, and their subcellular localizations.
Table 4.
PS ASO induced aggregates
| Aggregates | Protein compositions | Localization | Minimum transfected ASO concentration (nM) | Note |
|---|---|---|---|---|
| PS bodies | TCP1β, Mannosidase II | Nucleus | ∼20 | Upon Ran reduction, form in cytoplasm |
| Paraspeckle-like | P54nrb, PSF et al. | Nucleoplasm | ∼10 | No NEAT1 RNA |
| Nuclear filaments | P54nrb, PSF et al. | Nucleoplasm | ∼30–50 | No TDP43 |
| Loose nucleolar structures | P54nrb, PSF, NPM | Nucleolus | ∼15 | |
| Perinucleolar cap | P54nrb, PSF | Perinucleolar region | ∼40 | Toxic ASO |
| Stress granules | G3BP, mutant FUS, Mutant PSF | Cytoplasm | ∼50 | |
| P-bodies | DDX6, LSM14 et al. | Cytoplasm | ∼1 |
PS ASOs not only form novel aggregates, but also result in the formation of RNP structures that are normally present in cells, with defined compositions and functions. For example, we have shown that under some circumstances, PS ASOs can localize to cytoplasmic stress granules (62). Cytoplasmic stress granules are dynamic RNP complexes with complex compositions that are thought to be formed from stalled ribosomes on mRNAs in response to stress (for review see (73)). Transfected PS ASOs that had 2′modifications including MOE, cEt, locked nucleic acids (LNA) or F at concentrations of 50 nM and greater all are able to localize to cytoplasmic stress granules (62). Further, sodium arsenite-induced stress caused accumulation of PS ASOs in stress granules, and mutation of a nuclear localized PS ASO binding protein, FUS (P525L), or expression of a truncated FUS fused to a nuclear export signal and artificial beta sheet, protein beta 23, resulted in cytoplasmic aggregates, some of which included stress granules. Thus, both in the absence of stress and in response to stress, PS ASOs can accumulate in several cytoplasmic aggregates including stress granules and protein binding is a critical factor in determining the localization of PS ASOs in the cytoplasm. Though stress can increase the potency of PS ASOs in some circumstances (Liang and Crooke, unpublished), it is unclear whether accumulation of PS ASOs in stress granules alters potency per se (62). Similarly, PS ASOs can accumulate in cytoplasmic processing bodies (P-bodies) and actually induce the formation of P-bodies (74,75). P-bodies are cytoplasmic aggregates comprised of mRNAs and multiple proteins involved in inhibition of translation and mRNA degradation (76). Though PS ASOs bind to key P-body scaffold proteins such as the helicase DDX6, theses ASOs can still form P-body-like structures in cells depleted of key P-body proteins. Interestingly, the formation of P-bodies in the cytoplasm does not alter the activity of PS ASOs
PS ASOs can form a number of morphologically distinct nuclear bodies that contain paraspeckle proteins. The interactions of PS ASOs with nuclear paraspeckle proteins are both fascinating and therapeutically important. Paraspeckles are nuclear aggregates that are formed around both the long and short forms of a long non-coding RNA, NEAT1. Paraspeckle proteins include P54nrb, PSF, PSPC-1 and FUS and numerous other ‘outer shell’ proteins (For review see (77,78)). Paraspeckles are thought to play diverse roles in pre-mRNA processing, RNA editing and other RNA biological processes. We showed that PS ASOs containing several different 2′ modifications could displace NEAT1 RNA and form what appeared to be functional paraspeckles that included at least the core complex of proteins (69). Interestingly, in certain cells, PS ASOs can form nuclear filaments that contain paraspeckle proteins P54nrb and PSF at high ASO concentrations. One the other hand, toxic PS ASOs tend to localize to perinucleolar caps and nucleolus, together with these paraspeckle proteins (17). Moreover, 2′ F modified PS ASOs were shown to have higher affinity than other 2′ modifications for P54nrb and PSF and cause rapid cellular degradation of these proteins (63,79,80). In subsequent studies, we also demonstrated that interactions with paraspeckle proteins are a critical step in the induction of nucleolar toxicity by PS ASOs (17,81,82).
The roles of PS ASO–protein interactions in PS ASO induced toxicities
Arguably, the most controversial and most prolonged issue in antisense technology is why some specific PS ASOs are toxic. However, very recent progress has supported a much better understanding of the general mechanisms responsible for most toxic PS ASOs. One possibility considered was that the toxicity of some PS ASOs may be derived from off-target cleavages of essential transcripts by RNase H1, but this has been shown to be relatively less frequent event than once thought (83). Nevertheless, on occasion hybridization-based off-target cleavage can account for the toxicity of specific PS ASOs. Recently, however, we reported a molecular mechanism that appears to be a general solution that applies to nearly all toxic ASOs of all chemical classes studied and all cell lines and organs and species studied to date (17). In brief, toxic PS ASOs bind to paraspeckle proteins with greater affinity than safe PS ASOs and induce RNA-PS ASO–protein complexes that are different (Figure 3). In particular, an interaction with the spacer domain of RNase H1 with toxic PS ASO-induced complexes of the core paraspeckle proteins results in mislocalization of these complexes to the nucleolus, inhibition of pre-rRNA transcription and processing, and nucleolar toxicity that results in apoptosis (17,82). Subsequent studies characterized the various domains of proteins involved, the kinetics of mislocalization of the complex, and found that the major RNA present in toxic PS ASO-induced RNP complexes is 5.8S rRNA (82). Finally, while toxic PS ASOs display higher affinity for many proteins including paraspeckle proteins in general (17,84), there is no strong correlation between the severity of toxicity and affinity of the PS ASOs for a specific protein. Rather, it is the ability to form the toxic complex that appears to determine the severity of the toxic effects (82).
Figure 3.
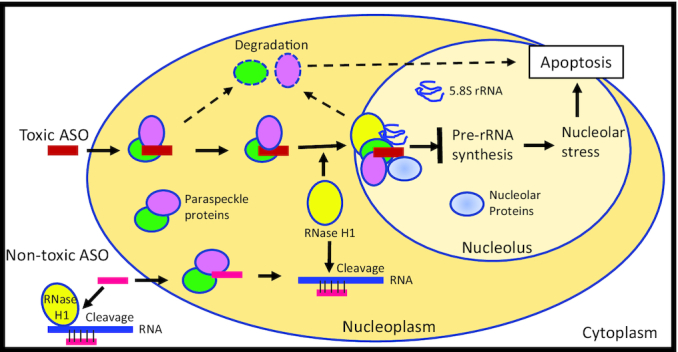
Schematic representation of AOS toxic mechanism mediated by PS ASO protein interactions. ASOs can bind RNA target and trigger RNase H1 cleavage in both the cytoplasm and the nucleus. Toxic ASOs bind tightly to many proteins including paraspeckle proteins, leading to altered interaction and rapid protein degradation. Toxic ASOs also trigger protein mislocalization to the nucleolus mediated by RNase H1, and induced the interactions of paraspeckle proteins with RNase H1 and other components, inhibiting pre-rRNA synthesis and trigger nucleolar stress and apoptotic cell death.
Remarkably, a simple substitution of a 2′-O-methoxy (OMe) in gap position 2 of toxic PS ASOs resulted in ablation or dramatic reduction in toxicity with limited changes in potency (17). This positional effect correlates with previous findings that the 5′ cEt wing and ∼6 DNA nucleotides in the gap region appear to be the binding site for many proteins (70), and substitution of 2′OMe at gap position 2 significantly reduced binding of proteins (17). We have initiated a broad medicinal chemistry effort to fully understand the SAR that support reduction of PS ASO induced toxicities and results published to date are summarized later.
PS ASO binding to model proteins
To develop a more detailed understanding of the basic characteristics of PS ASO interactions, we have selected seven model proteins to study as listed in Table 5. These include the RNA binding proteins nucleolin (NCL), P54nrb, PSF and La, the DNA binding proteins PC4 and SSBP, and DNA/RNA heteroduplex binding protein RNase H1. Of the proteins studied to date, which represent a range of affinities and several types of domains that frequently bind PS ASOs (51,63,82,85), we have shown the presence of one to four PS ASO binding sites with affinities ranging from 0.009 to 83 nM (63). PC4 and SSBP1, for example, have single DNA binding domains, display high affinity and appear to have an effect on the potency of PS ASOs designed to create heteroduplexes that are substrates for RNase H1 (61,63,86). P54nrb and PSF display two binding sites of different domain architectures (Figure 4), are paraspeckle proteins and play critical roles in the molecular mechanisms of toxicity of toxic PS ASOs (17,77,87). La is an important nucleic acid binding protein that has two binding sites and has been shown to enhance ASO activity likely by facilitating ASO accumulation in the nucleus (61). Finally, RNase H1 is one of the key mechanisms by which PS ASOs are designed to work, displays two very different types of binding sites for PS ASOs and is known to be competitively inhibited by PS ASOs (88–90).
Table 5.
PS ASO interaction with model proteins
| ASO binding affinity | ||||||
|---|---|---|---|---|---|---|
| Protein | Feature | DNA/RNA binding domains | ASO binding domain | MOE | cEt | F |
| NCL | RNA binding | 4 | 4 | 0.009 | 1.7 | 0.002 |
| P54nrb | RNA/DNA binding | 2 | 2 | 82.9 | 9.3 | 2.1 |
| PSF | RNA.DNA binding | 2 | 2 | 3.7 | 2.7 | 0.72 |
| La | RNA binding | 2 | 2 | 9.3 | 5.0 | 1.1 |
| PC4 | DNA binding | 1 | 1 | 6.1 | 1.1 | 0.21 |
| SSBP | DNA binding | 1 | 1 | 0.36 | 0.77 | 0.04 |
| RNase H1 | DNA/RNA duplex binding | 2 (duplex) | 2 | 2 | 5 | 2 |
Adapted from Vickers TA., et al., PLoS One. 2016. 11(8):e0161930.
Figure 4.

Schematic prediction of domains of model proteins that interact with PS ASOs. RRM, RNA recognition motif; pQ, poly Q; NOPS, Nona Paraspeckle domain; LaM, La module; SBM, short basic motif; HBD, hybrid-binding domain. ASO binding sites are shown with red lines, with thicker lines indicating higher binding affinity.
These model proteins were selected also because they exemplify a number of general binding properties. Nucleolin is more promiscuous and appears to bind to PS ASOs with different 2′ modifications with approximately equal affinity (63). The binding of nucleolin is also not meaningfully sensitive to PS ASO sequence (51). In contrast, the paraspeckle proteins are sensitive to differences in 2′ modifications and sequence and colocalize with PS ASOs in different structures (17,63,69). PC4 and RNase H1 also have been extensively studied and have structures that are well characterized (86,91,92). In addition, P54nrb, PSF, PC4, and RNase H1 are able to bind ASO/RNA duplex, whereas La and nucleolin mainly bind single stranded PS ASOs (61). Thus, this set of model proteins provides an excellent opportunity for detailed analyses of the chemical nature of the interactions.
PS ASO interactions with PC4
Human positive cofactor 4 (PC4) is an abundant, small, and highly conserved homodimeric single-stranded DNA (ssDNA)-binding protein involved in transcription (93). The interaction between PC4 and ssDNA is mediated by base-stacking interactions that occur through the aromatic amino acids phenylalanine (Phe) and tryptophan (Trp) that are present at positions 77 and 89, respectively (86,94), and by arginine and lysine residues (Arg70, Arg86, Arg100 and Lys101), which form electrostatic and hydrogen bonds with the phosphate backbone (95). PC4 has also been demonstrated to bind PS ASOs, with reduction of PC4 resulting in a decreased ASO potency (61). In collaboration with Nowotny lab, we recently solved the crystal structure of the PC4 DNA binding domain complexed with a 2′-OMe gapmer PS ASO, and identified the specific amino acid interactions involved in ASO binding (96). In addition, the impact of ASO backbone chemistry, 2΄ modifications, and buffer environment on the ASO binding affinity of PC4 was examined in detail (97). Furthermore, using site directed mutagenesis, we characterized the amino acids which are specifically required for ASO binding and by substituting abasic nucleotides, identified the positions on the ASO most important for interaction with the protein.
Binding of the PS ASOs to mutant proteins was highly influenced by the 2′ modification in the wing nucleotides (97). While the R70 mutation reduced the affinity of each of the PS ASOs to a similar extent, the effect of the R100 mutation was much more pronounced for the 2′ F and MOE than for the cEt gapmers. For the cEt PS ASO, contacts at R70 appear to be more important than those at R100, whereas for the MOE ASO R70 and R100 appear to contribute equally. For the 2′ F, loss of R100 had a much more significant impact on binding than loss of R70. Another striking difference was that loss of R86 reduced binding to the 2′ F but did not have a strong effect on the binding of MOE or cEt ASOs. Similarly, loss of the aromatic amino acids at F77 and W89 significantly reduced binding to the 2′ F, but not the MOE or cEt PS ASOs. Taken together these observations suggest that ASOs with 2′F wings may have additional contacts which stabilize protein binding compared to cEt or MOE containing PS ASOs and that the binding interface is differentially modulated by 2′-modifications.
Substitution of the PS ASO with abasic residues resulted in a loss of affinity at positions consistent with the crystal structure. In the crystal structure (Figure 5A) positions 2–5 of the ASO associate with R70, R86, R100 and K101 via electrostatic interactions, while positions 10–13 associate with PC4 via stacking interactions with F77 and W89 as well as electrostatic interactions with R70, R100, and K101. Nucleotides 6–10 do not interact with PC4 in the crystal structure, but instead form a hairpin-like structure between the two DNA-binding interfaces of the PC4 dimer. Abasic substitution at positions 10 and 11 had the most dramatic effect on affinity most likely due to loss of stacking interactions with W89 (97). A second structure that is thought to be an artifact of crystallization is shown in Figure 5B. Of particular interest is that crystal structures showed that the flexibility of PS ASOs support an induced-fit induced by the 2′ modified wings and protein bound, while the PS oligodeoxynucleotide can form a loop that is fully exposed to solvent (96).
Figure 5.
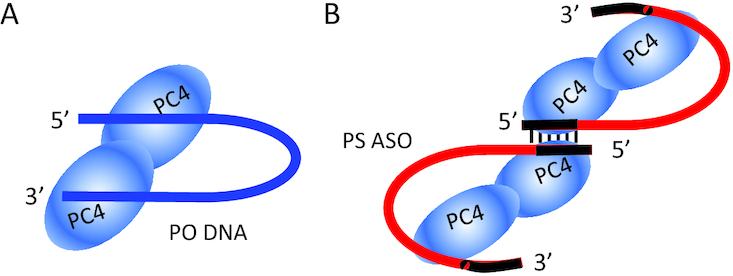
Schematic prediction of PC4 binding to PO DNA or to PS ASO. (A) PC4 complexed with PO DNA. (B) PC4 complexed with 5–10–5 PS OMe ASO. The protein PO DNA complex contains a single DNA molecule (blue) and a PC4 dimer. The protein PS ASO complex contains two ASOs and four protein molecules, with the 5′ portion of ASOs potentially base-paired based on this ASO used. Note that whether such duplex can form in biological systems needs to be investigated. The 2′ modified wings are shown in black, whereas the gap regions are in red.
PS ASO interactions with RNase H1
Human RNase H1 has been shown to play a dominant role in the activity of DNA-like antisense oligonucleotides (98) and interactions of ASOs with a complex that includes P54nrb has been shown to contribute to ASO toxicity (17). The structure of human RNase H1 consists of a DNA/RNA hybrid-binding domain (HBD) that is separated from the catalytic domain by a 62 amino acid spacer region (90,99,100). The catalytic domain is highly conserved with the amino acid sequences of RNase H1 proteins in other species and contains the key catalytic and substrate binding residues required for activity (90,101–103). Although the HBD that is absent in the bacteria enzyme is not required for RNase H activity, this region is responsible for the enhanced binding affinity of the human enzyme for the heteroduplex substrate as well as the strong positional preference for cleavage exhibited by the enzyme (90,104).
PS ASOs interact with the full-length protein similarly with Kd’s ranging between 2 and 4 nM for 2′ F, MOE and cEt gapmers (63). Interestingly, deletion of the spacer domain has no effect on binding of the 2′ F ASOs, but increases affinity for cEt and MOE PS ASOs (82). Binding to the HBD-only is approximately 10-fold weaker than to the full-length protein, whereas PS ASO affinity for the catalytic domain is 4–5-fold less than for the HBD. These data suggest that PS ASOs bind optimally to the full length protein, but that interactions at the HBD contribute more to overall PS ASO affinity than do those at the catalytic domain. The observations also suggest that the binding isotherm for the full-length protein likely requires interactions with two binding sites, one in the HBD and one in the catalytic site.
It has been shown that the conserved Trp43, Lys59 and Lys60 residues constitute the binding surface for the HBD of human RNase H1 (104). While binding affinity of a PO heteroduplex is significantly affected, the K59A and K60A mutations with a nearly 10-fold reduction in affinity relative to the native RNase H1 HBD, and completely ablated by the W43A substitution, only a 2-fold diminution in PS ASO affinity is observed with the Trp43, Lys59 and Lys60 to Ala substitutions (82). Similarly, several amino acids have been identified as important contributors to the interaction of the RNase H1 catalytic domain with the PO heteroduplex (92). Mutations N151A and R179A have the greatest effect on binding of the PO heteroduplex reducing affinity by >7-fold and 2-fold, respectively. A similar effect is observed for binding of the PS ASOs which are strongly reduced by the N151A and R179A mutations, but not other substituted amino acids. Taken together, these data suggest that the PS ASO and PO heteroduplex interact similarly with the catalytic domain, but that the interaction of PS ASOs in the HBD may involve additional interactions relative to the PO heteroduplex.
PS ASO interactions with P54
P54nrb and PSF are essential for multiple cellular processes, including transcription, splicing, polyadenylation, nuclear retention, translation and DNA repair (105). The structure of the human P54nrb/PSF heterodimer encompasses two RNA binding domains: RRM1, RRM2, as well as NOPS, and coiled-coil core domains. P54nrb and PSF form homo- and heterodimers via their core domains (106) and have been shown to interact with nucleic acids through their RRM domains (105). Although the RRM1 domain of P54nrb is canonical, containing four aromatic residues at conserved positions that are typically essential for RNA binding (107), RRM2 is considered noncanonical with three conserved residues substituted to Thr, Lys and Ile, implying that either the RRM2s do not bind RNA, or that they bind in an unexpected manner.
It appears that ASOs must contain PS linkages in order to bind P54nrb and that the affinity of an ASO for P54nrb is directly proportional to the number of PS linkages in the ASO (63,82). Binding interactions occur primarily via the RRM1 and RRM2 domains, but strikingly, the interaction can be influenced by the chemistry of the PS ASO. For example, PS ASOs with the 2′ F modification in the wings bind P54nrb primarily via RRM1, while for MOE or cEt PS ASOs, RRM1 and RRM2 domain interactions contribute nearly equally to overall binding. The strength of PS ASO affinity for RRM2 was also found to be highly influenced by the sodium concentration, while binding to RRM1 was not strongly affected by changes in sodium concentration. The salt-dependence of a bimolecular association is indicative of the contribution of charge–charge interactions to the free energy of binding. Therefore, the greater effect of added sodium chloride on binding to RRM2 as compared to RRM1 suggests that cEt PS ASO interactions with RRM2 are primarily electrostatic in nature, involving contacts between basic residues on the RRM2 domain and the phosphorothioate groups in the PS ASO, while interactions with RRM1 may be mediated by hydrophobic or other forces. This is consistent with the observation that the RRM1 interaction appears to dominate the overall binding to the full-length protein for the more hydrophobic 2′F gapmers, while RRM1 and RRM2 interactions contribute more equally to overall P54nrb binding of the MOE and cEt gapmers.
Chemical characterization of PS ASO–protein interactions
The nature of proteins that interact with PS ASOs
Not surprisingly, among the proteins that bind PS ASOs, proteins with nucleic acid binding domains are well represented. These include domains reported to bind ss or ds RNA or DNA, polymerases, helicases and nucleases. In plasma and in cells, a range of proteins, including albumin and alpha 2 macroglobulin that bind many different types of molecules, heat shock proteins that are involved in protein folding including TCP1 complex proteins, HSP70 and HSP90 proteins that do not normally bind nucleic acids have been shown to bind PS ASOs (60,61,70,108). Interestingly, PS ASOs bind to the mid-domain of HSP90 which is known to bind to client proteins via hydrophobic interactions (109). Though no general conclusions about how and why such a broad range of proteins bind PS ASOs, observations to date show that PS ASOs typically bind to proteins with nucleic acid binding domains with higher affinity than proteins that have no nucleic acid binding domains. For example, PS ASOs bind to chaperone proteins with affinities in the high nanomolar range which is a hundred-fold higher than some proteins that have nucleic acid binding domains (63). Only for albumin have we asked if binding PS ASOs alters binding to types of ligands that typically bind and there is no evidence of competitive interactions. For example, the binding of a range of drugs such as aspirin and penicillin that bind albumin is unaffected by PS ASO binding (110). It will be interesting to determine if PS ASO binding to chaperone proteins affects their interactions with client proteins. So, to date very limited generalization about the types of proteins that bind PS ASOs can be made. Given the obvious ability of PS ASOs to bind a wide variety of proteins, arguably, the better question is why more proteins don’t bind these reagents.
The characteristics of the binding sites in proteins for PS ASOs
As previously discussed, we have identified proteins that contain from one to four binding sites for PS ASOs. In none of the proteins with more than one binding sites have we observed evidence of cooperative or anti-cooperative binding. Only in PC4 have we observed an interaction of a PS ASO binding site and another domain as the ‘flap’ that obscures the nucleic acid binding domain and results in a lower affinity for the entire protein than the binding domain only. A number of proteins display binding sites that differ substantially in affinity. For example, PS ASOs bind to the HBD of RNase H1 with higher affinity than the catalytic site (82). The binding sites studied in depth demonstrate that binding domains may differ significantly in amino acid composition, structure, PI, the presence of clusters of positively charged amino acids and hydrophobic amino acids.
Based on the characteristics of their interactions with PS ASOs, proteins can be divided into several groups (Table 3) (63). A sizeable fraction of proteins appears able to discriminate between PS ASOs that have different 2′ modifications. Many can discriminate between different PS ASO sequences and some can discriminate between differences in sequence and 2′ modifications. Many proteins display a polarity preference with most preferring to bind to the 5′ pole, but a few prefer the 3′ pole of PS ASOs.
The structure activity relationships of PS ASO and protein binding
The roles of phosphorothioates
The replacement of one non-bridging oxygen with a sulfur alters the physicochemical characteristics of the phosphate in important ways as this modification results in the creation of a chiral center at each internucleotide link. The effects of the chiral centers have been extensively studied as have the effects of pure R or S isomers (111–115). Suffice it to say that no reproducible systemic advantages for the use of chirally pure PS ASOs have been identified. Because the sulfur atom is twice as massive as the oxygen atom, the charge distribution, bond angles and stretching of PS links differ substantially from PO linkages (116,117). Put simply, the sulfur substitution spreads the charge and makes the phosphate more ‘lipophilic’ facilitating binding to proteins. As a general rule, for proteins that require PS moieties to bind ASOs, the minimum number of this modification needed to support meaningful protein interactions is 10. In ASOs that contain both PS and PO moieties, the placement of PS units plays an important role, but to date, no general rules about the placement of PS units have emerged, although PS placed at 5′ end of ASOs tend to bind more proteins and have higher affinities for key proteins. An examination of the crystal structure of PC4 bound to a PS ASO versus a PO containing ASO is highly instructive and demonstrates that the PS substitution supports more dispersed interactions with specific sites in the protein and more total binding interactions (96). Consequently, PS ASOs bind to more proteins and higher affinities than PO ASOs profoundly changing behaviors. For example, PS ASOs bind to plasma proteins, resulting in a distribution half-life of 1–2 hours versus rapid clearance via glomerular filtration and urinary excretion of PO ASOs (for review, see (7)). Similarly, PS substituents enhance binding to proteins on the cell surface that facilitate entry into the cell and once in the cell, binding to intracellular proteins determine the intracellular distribution of PS ASOs (16). Despite extensive efforts, no modification that provides the optimal protein binding afforded by PS substitution has been found.
The roles of ASO structure
Though meaningful gaps in our knowledge remain, the conceptual basis to explain the differences between PS containing ss and ds ASOs is straightforward and important. Depending on the length of ASOs, the molecular weight ranges from about 6 to 7.5 kDa. Obviously, the molecular weight of ds oligonucleotides is about double that of ss oligonucleotides. This important difference is often ignored and does have an effect on the level of cellular uptake. In biological systems, ss ASOs contain counterions at most or all PS groups, and waters of hydration are present at most, if not all PS linkages (118). Unfortunately, similar studies on 2′ modified ss ASOs or ds ASOs have not been reported. For example, it would be particularly helpful to understand these characteristics for the currently used designs of siRNAs that are extensively chemically modified.
In solutions, ss PS ASOs are flexible and can adopt a variety of structures (119). Although differences in ASO sequence, length, and chemical modifications affect the physicochemical properties that influence protein binding, the effect of each factor may be amplified by altering the ‘shape’ or topology of ASOs (120), and the surface properties of ASOs may affect the binding of many proteins. The importance of the flexibility of ss PS ASOs is beautifully demonstrated in the crystal structure of a PS 2′OMe gapmer ASO (96). In fact, in this structure, the gap nucleotides are looped into solvent supporting interactions between both poles of the PS ASO to interact with the protein (Figure 5). Though the dimeric structure may be an artifact of crystallization, we prefer the notion that both structures may form in a cell treated with a PS ASO. However, further experimentation is required to understand whether dimeric structure form under biological conditions. If so, the PC4 structures show two other important properties of PS ASOs: they can form homo-and hetero dimeric complexes and intermolecular hybridization driven self-structures (96). In contrast ds oligonucleotides are more rigid and larger, likely resulting in less ability to adopt a conformation that can bind to an individual or a variety of proteins. Of equal importance are two other factors. First, in a duplex, the nucleobases are base-paired and somewhat protected from water. This means that there are fewer hydrogen bonding opportunities and very limited opportunity for the nucleobases to ‘stack’ with aromatic amino acids (121,122). Secondly, there are two charged surfaces which result in a greater ionic character. Clearly, ss PS ASOs can also form intra- and intermolecular structures. However, modern screening methods are effective at avoiding sequences that may form intramolecular structures (for review, see (123)). Additionally, guanosine rich sequences can form G-quartet structures that bind extensively to many proteins (124). Some sequences, particularly in PS ASOs containing high affinity 2′ modifications, may also form intermolecular links via Watson–Crick hybridization. Frequently they form lattice like structures at high concentrations that are easily detected by viscosity measurements. Essentially nothing is known about the potential of such ‘lattice like’ structures to bind proteins, but once again, modern screening techniques make it unlikely that any of these structures would be used to determine the functions of specific RNA or proteins therapeutically. On the other hand, a scientist inexperienced with antisense technology can be misled by accidentally using ASOs that can form such structures.
The roles of 2′ modifications
The 2′ ribose modifications commonly incorporated in PS ASO drugs to enhance pharmacological properties also significantly affect protein binding. More hydrophobic 2′ modifications, e.g., F, LNA or cEt, tend to enhance protein binding relative to more hydrophilic 2′ modifications (MOE and OMe), resulting in a 10-fold difference in binding affinity for many proteins (60,63,70,79). 2′ F modified ASOs appear to have distinct properties with regard to protein binding. For example, PS ASOs with 2′ F modifications tend to trigger rapid degradation of paraspeckle proteins (79,80). In addition, 2′ F PS ASOs also bind differently to P54nrb protein, and preferentially bind RRM1, whereas MOE or cEt PS ASOs bind roughly equally to both RRM1 and RRM2 domains (82). Interestingly, it appears 2′ modification at the 5′ wing has a greater influence on cellular protein binding than at the 3′ wing. Many proteins favor binding to 5′ wing containing more hydrophobic 2′ modifications and ∼6 nucleotides DNA gap sequence (17,70), which constitute the major docking site for many proteins, although this polarity effect may vary for certain ASO sequences and for individual proteins. The polarity effect on protein binding often correlates with ASO toxicity and activity, e.g., higher activity and toxicity might be observed when more hydrophobic 2′ modifications are placed at 5′ wing, as compared with 3′ wing, of ASOs (17,125). This polarity or directional effect may explain why introducing more hydrophilic 2′ OMe, or neutral backbone MOP linkage, at gap position 2–4 tend to reduce protein binding and ASO toxicity.
The roles of PS ASO sequences
Not surprisingly, ASO sequence can substantially affect ASO protein interactions. For example, toxic ASOs tend to bind more proteins more tightly than non-toxic ASOs (17,84), and a 100-fold difference in binding Kd can be observed for different PS ASO sequences (63). Although many proteins demonstrated sensitivity to ASO sequences and many RNA or DNA binding proteins display sequence preference, no ‘consensus’ preferred sequence of pattern has emerged. However, some ASO sequences are frequently found in toxic ASOs, such as TGC and TCC motifs that may be involved in higher protein binding affinity (126). In addition, the CpG motif is known to be involved in immune response (25). It is possible that certain motifs in combination with the type of 2′ modifications incorporated in the PS ASOs may be favored by many proteins. However, no consensus sequence has been identified. In addition, certain sequences, such as G-clusters, A-clusters, as well as polypyrimidine-containing sequences, may also be preferentially bound by proteins that recognize such sequence properties. However, as chemical modifications, especially PS modifications greatly increase the binding affinity to proteins, the discrimination of ASO sequence by binding proteins may be significantly weakened.
The roles of targeting ligands conjugated to PS ASOs
Moieties conjugated to PS ASOs have been used to deliver these drugs into particular tissues or cells, using ligands that display high affinity and specificity for surface proteins that are enriched in target tissues or cells (58,59,127). Compared with formulation with polymers or other transfection reagents, conjugation with specific ligands display several advantages, including tissue or cell type specific targeting, enhanced pharmacokinetics, reduced inflammation, well-defined composition and ease of synthesis. Various peptides or antibodies have been tested that bind specific proteins displayed on cell surface to facilitate adsorption and internalization (for review, see (40,59,127,128)). The surface proteins targeted are normally enriched in target cells and have dynamic cycling between cell surface and endosomes to enhance ASO delivery. Examples include GLP1-analog conjugation to target G-protein coupled receptors on the pancreatic Beta cells (54), ASOs conjugated with antibodies against CD44 and EphA2 in Glioblastoma stem cells (129), and anti-CD71 Fab-conjugation to target siRNAs to cardiac and skeletal muscles (130). Various peptides including those that are cell penetrating have also been conjugated to ASOs to facilitate delivery and cellular release (for review, see (131)).
In addition, other types of ligands have also been conjugated to ASOs to facilitate delivery, including small molecules, aptamers, as well as various lipids. One of the most successful conjugations is the sugar GalNAc that efficiently and specifically delivers the ASOs to liver hepatocytes which express high levels of ASGPR, the high affinity receptor for GalNAc. GalNAc conjugation also enables liver delivery of ds ASOs (42), which otherwise are difficult to enter cells. On the other hand, GalNAc-conjugated ASOs also exhibit 10–30-fold improvement of PS ASO potency in vivo and in the clinic (57,132), and increased ASO binding affinity to ASGPR especially when conjugated with three GalNAc chains (133). Moreover, various lipid conjugations have been evaluated, with enhanced delivery and potency (for review, see (29)). For example, different lipid conjugates have been tested to deliver siRNAs to non-hepatic sites including lung, muscle, heart, fat, adrenal gland, and the CNS (32). Lipid conjugated ss ASOs also demonstrated enhanced potency in muscle (28,134). It has been shown that highly hydrophobic lipids conjugated siRNAs preferentially interact with low density lipoproteins, whereas less lipophilic lipid conjugated siRNAs bind high density lipoproteins, thus lipid-conjugated siRNAs are targeted to tissues enriched in lipoprotein receptors (135). In addition, lipid conjugation can also enhance the interactions of ASOs with membranes, thus facilitating endosomal release (44).
Using medicinal chemical manipulation of protein binding to enhance PS ASO performance
The understanding of ASO protein interactions and the biological consequences provides an opportunity to improve ASO performance through medicinal chemistry. Chemical modifications or conjugations that enhance binding to plasma or cell surface proteins have demonstrated to facilitate delivery to target tissues or cells that dramatically increased ASO activities, as described above. Similarly, altering binding to cellular proteins, especially those proteins involved in ASO release and trafficking, e.g., STX5 and M6PR (35,36), can also facilitate ASO release from endocytic organelles and ASO subcellular distribution to desired compartments. In addition, reducing the binding of ASO/RNA heteroduplex to proteins such as P54nrb, Ku70/Ku80 that compete with RNase H1 should enhance RNase H1 recruitment to target RNAs.
Different approaches have also been explored to modulate protein binding to improve ASO safety by reducing PS ASO protein interactions (17,81). As the PS modification is known to be a major contributor to protein binding, charge-neutral backbone modifications were used to replace PS backbone at different positions of ASOs (81). For example, methylphosphonate (MP) or methoxypropylphosphonate (MOP) linkages were introduced into different positions of toxic ASOs. The toxicity of ASOs was dramatically reduced when these neutral backbones, one or two at a time, were placed at the 5′ portion of the DNA gap region, especially at gap positions 2–4. However, no or little improvement was observed when MOP modifications was placed at the 3′ portion of the DNA gap. This is consistent with the polarity effects of protein binding that favors the 5′ wing/DNA junction region. Indeed, protein binding was significantly reduced when two MOP linkages were placed at gap positions 2–3, 3–4 or 4–5, as compared with parental ASO that is fully modified with PS linkages (81). This modification change resulted in improvement in therapeutic index, with dramatic reduction of toxicity yet no or modest effect on ASO potency.
Similarly, different 2′ modifications were also tested in the DNA gap region for their effects on ASO safety and potency (17). Though introduction of MOE and cEt at positions 2–4 of the DNA gap region may reduce toxicity, such modifications is often accompanied with some meaningful loss of activity. However, full gap positional walks of 2′ modification, one nucleotide at a time, showed that OMe at gap positions 2–3 can significantly reduce toxicity, with no or little loss of potency. OMe at gap position 2 also significantly reduced binding to proteins, as determined using affinity selection assay (17). This positional effect is consistent with the effects of MOP backbone modification, which again align with the polarity effect of ASOs on protein binding. As the 5′ wing/DNA junction region is a preferred protein binding site, introduction of more hydrophilic 2′ modifications at this site may interfere with protein binding. Importantly, large scale studies with more than 450 ASOs showed that introducing 2′OMe at gap position 2 can significantly reduce ASO toxicity for more than 90% of toxic ASO sequences. Improved ASO performance was also observed in different tissues and species tested in vivo, suggesting a general application. Moreover, to further improve ASO performance, additional chemical modifications are also extensively being tested, including modifications at the backbone, ribose, and base of nucleotides. Better understanding the SAR of modification, protein binding as well as PS ASO performance may significantly improve ASO efficacy and improve the therapeutic index of these drugs.
GENERAL PRINCIPLES
Though many questions remain to be answered and there are significant gaps in our understanding of PS ASO-protein interactions, a number of important general principles have emerged.
Proteins determine the fate of PS ASOs
Simply put, in biological systems, if a PS ASO is present at a site, a protein (or proteins) is responsible for being there. Protein binding is critical for distribution from plasma to peripheral tissues, cellular uptake and subcellular distribution, the formation intracellular aggregates and for pharmacological and toxicological effects of PS ASOs.
PS ASOs may affect the fates of the proteins with which they interact
We have identified a number of proteins that are induced by PS ASO binding to re-localize in cells. Certainly, the remarkable interactions between toxic PS ASOs, the spacer domain of RNase H1 and paraspeckle proteins are important and demonstrate how complex such interactions may be. The interaction between STX5 and late endosomes that occurs only in the presence of PS ASOs demonstrates that these ASOs can induce unique interactions between cellular proteins and membraned organelles (35). PS ASOs can induce the formation of novel protein PS ASO aggregates such as PS bodies (60), and the toxic nucleolar complexes as well as aggregates that are normal cellular components, e.g. P bodies or stress granules (17,62,74). Remarkably, PS ASOs can replace a long non-coding RNA, NEAT1, and form paraspeckle-like structures (69). 2′ F containing PS ASOs can even cause rapid degradations of paraspeckle proteins such as P54nrb and PSF (79).
Diverse proteins interact with PS ASOs via multiple domains
Certainly, nucleic acid binding domains are prominently represented in the proteins that bind PS ASOs, but the diversity is impressive with proteins ranging from albumin to chaperone proteins to membrane localized proteins and when targeting ligands are conjugated, additional proteins bind (50,53,61). Moreover, an array of domains is represented. RNase H1 demonstrates that PS ASOs may bind similarly to a natural binding partner RNA as is the case in the catalytic domain, or differently, as is observed in the hybrid-binding domain (82).
We have presented examples of proteins with one to four PS ASO binding sites. Often there are substantial differences in the behaviors of different domains and PS ASO, e.g. P54nrb or RNase H1(82). On the other hand, nucleolin displays four binding sites that behave similarly. Though it seems likely that as more is known, PS ASO binding will be found to alter the conformations of many proteins, to date no evidence of interactions between binding sites has been observed.
Some proteins bind PS ASOs promiscuously while others display a variety of selective binding properties
Nucleolin is particularly interesting as it has four binding sites that are similar and appear to be quite promiscuous. Given that many proteins display meaningful binding preferences, one must ask how can nucleolin be so indifferent to the characteristics of the PS ASOs with which it binds. On the other hand, many proteins display strong binding directionality, the ability to discriminate between 2′modifications, structure and sequence.
The structure activity relationships of PS ASOs and proteins are complex, but understandable
ASO sequence, structure, and chemical modifications substantially affect PS ASO protein interactions. The PS backbone plays a dominant role, whereas 2′ modifications also dramatically affect protein binding. Ionic, hydrophobic, base-stacking with aromatic amino acids all play important roles as well. We have identified a significant numbers of precise contact points between structural elements of the proteins to which they bind and they demonstrate a wide range of potential chemical interactions. The structural flexibility of PS ASOs is likely to prove of general importance supporting an array of ‘induced fits’ with protein binding partners as was demonstrated by PC4 PS ASO binding studies (96,97).
Proteins can affect the pharmacological activities of PS ASOs
Proteins that are bound to RNA in concert with RNA structure are known to influence the number of active sites in RNA and the overall sensitivity to PS ASOs (for review see (5)). We now also know that proteins can compete with RNase H1 for binding to the heteroduplexes formed by ASOs. This is consistent with the observations that RNase H1 is in limiting quantities and that recruitment of RNase H1 to the heteroduplex is slow (136). Proteins can also influence PS ASO activities by affecting release of PS ASOs from endosomes or altering the subcellular distribution of PS ASOs (35,61). Still other proteins, e.g. HSP90, appear to increase PS ASO activities via unknown mechanisms; a topic for future research.
Medicinal chemical approaches designed to alter the protein binding of PS ASOs is feasible and extraordinarily important for future advances in the technology
Arguably, the most surprising and important observation is that a simple gap position 2 insertion of a 2′-methoxy can eliminate or significantly reduce toxicity with only modest effects on potency (17). This observation is remarkable and is altering the screening and design of PS ASOs. As we explore the SAR of these effects, we are learning that other substitutions can provide similar improvements in therapeutic index (81). Similarly, targeted delivery with conjugated ligands has enhanced performance and opened new avenues for medicinal chemistry. Now that the major pathways and the proteins involved are known, it seems likely that medicinal chemical approaches to optimize intracellular distribution should be possible and important.
Conclusions and perspectives
In the past few years, significant progress in understanding the nature and importance of PS ASO-protein interactions has been made. Understanding these interactions has already paid meaningful dividends in the performance of PS ASO and the stage is set for additional advances. As is the case for all scientific inquiry, the work to date has generated more questions than answers. For example, how do PS ASOs that bind to proteins affect the structures of those proteins? Why are some proteins promiscuous in binding to PS ASOs while others display strong preferences? What are the chemical bases for the preferences observed? Can medicinal chemical solutions to enhance subcellular distribution be developed? What new ligands can facilitate targeted delivery to tissues like skeletal muscle and many more. It is exciting to know that the conceptual framework and methods now exist to answer these and other questions.
In this review, we summarized and codified recent important progress in understanding the nature and importance of PS ASOs with proteins. Much remains to be understood. For example, what are the compositions of all PS ASO-induced aggregates and how do the compositions change over time and in response to the environment? Do PS ASO-induced aggregates undergo phase transitions and, if so, what roles do phase transitions play? How generalizable are the basic principles as defined today? Nevertheless, the effort has already yielded important insights into the molecular events induced by PS ASOs and shown that coupling this information with medicinal chemical efforts can result in important gains in therapeutic index.
ACKNOWLEDGEMENTS
We wish to thank Rosanne Crooke for manuscript editing.
FUNDING
Internal funding from Ionis Pharmaceuticals. Funding for open access charge: Internal funding from Ionis Pharmaceuticals
Conflict of interest statement. None declared.
REFERENCES
- 1. Zamecnik P.C., Stephenson M.L.. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. U.S.A. 1978; 75:280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Crooke S.T., Witztum J.L., Bennett C.F., Baker B.F.. RNA-targeted therapeutics. Cell Metab. 2018; 27:714–739. [DOI] [PubMed] [Google Scholar]
- 3. Bennett C.F. Therapeutic antisense oligonucleotides are coming of age. Annu. Rev. Med. 2019; 70:307–321. [DOI] [PubMed] [Google Scholar]
- 4. Bajan S., Hutvagner G.. RNA-based therapeutics: from antisense oligonucleotides to miRNAs. Cells. 2020; 9:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Crooke S.T., Vickers T.A., Lima W.F., Wu H.-J.In: Crooke ST.Antisense Drug Technology - Principles, Strategies, and Applications. 2008; 2nd ednBoca Raton, FL: CRC Press; 3–46. [Google Scholar]
- 6. Swayze E.E., Bhat B.. Crooke ST. Antisense Drug Technology - Principles, Strategies, and Applications. 2008; 2nd ednBoca Raton, FL: CRC Press; 143–182. [Google Scholar]
- 7. Levin A.A., Yu R.Z., Geary R.S.. Crooke ST. Antisense Drug Technology - Principles, Strategies, and Applications. 2008; Boca Raton, FL: CRC Press; 183–216. [Google Scholar]
- 8. Geary R.S., Yu R.Z., Siwkoswki A., Levin A.A.. Crooke ST. Antisense Drug Technology - Principles, Strategies, and Applications. 2008; Boca Raton, FL: CRC Press; 305–326. [Google Scholar]
- 9. Crooke S.T., Baker B.F., Kwoh T.J., Cheng W., Schulz D.J., Xia S., Salgado N., Bui H.H., Hart C.E., Burel S.A. et al.. Integrated safety assessment of 2′-O-Methoxyethyl chimeric antisense oligonucleotides in NonHuman primates and healthy human volunteers. Mol. Ther. 2016; 24:1771–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Crooke S.T., Baker B.F., Witztum J.L., Kwoh T.J., Pham N.C., Salgado N., McEvoy B.W., Cheng W., Hughes S.G., Bhanot S. et al.. The effects of 2′-O-Methoxyethyl containing antisense oligonucleotides on platelets in human clinical trials. Nucleic Acid Ther. 2017; 27:121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Crooke S.T., Baker B.F., Xia S., Yu R.Z., Viney N.J., Wang Y., Tsimikas S., Geary R.S.. Integrated assessment of the clinical performance of GalNAc3-Conjugated 2′-O-methoxyethyl chimeric antisense oligonucleotides: I. human volunteer experience. Nucleic Acid Ther. 2019; 29:16–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eckstein F. Phosphorothioate oligodeoxynucleotides: what is their origin and what is unique about them. Antisense Nucleic Acid Drug Dev. 2000; 10:117–121. [DOI] [PubMed] [Google Scholar]
- 13. Eckstein F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014; 24:374–387. [DOI] [PubMed] [Google Scholar]
- 14. Gaus H.J., Gupta R., Chappell A.E., Ostergaard M.E., Swayze E.E., Seth P.P.. Characterization of the interactions of chemically-modified therapeutic nucleic acids with plasma proteins using a fluorescence polarization assay. Nucleic Acids Res. 2019; 47:1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Watanabe T.A., Geary R.S., Levin A.A.. Plasma protein binding of an antisense oligonucleotide targeting human ICAM-1 (ISIS 2302). Oligonucleotides. 2006; 16:169–180. [DOI] [PubMed] [Google Scholar]
- 16. Crooke S.T., Wang S., Vickers T.A., Shen W., Liang X.H.. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017; 35:230–237. [DOI] [PubMed] [Google Scholar]
- 17. Shen W., De Hoyos C.L., Migawa M.T., Vickers T.A., Sun H., Low A., Bell T.A. 3rd, Rahdar M., Mukhopadhyay S., Hart C.E. et al.. Chemical modification of PS-ASO therapeutics reduces cellular protein-binding and improves the therapeutic index. Nat. Biotechnol. 2019; 37:640–650. [DOI] [PubMed] [Google Scholar]
- 18. Akinc A., Querbes W., De S., Qin J., Frank-Kamenetsky M., Jayaprakash K.N., Jayaraman M., Rajeev K.G., Cantley W.L., Dorkin J.R. et al.. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010; 18:1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Srinivasan S.K., Iversen P.. Review of in vivo pharmacokinetics and toxicology of phosphorothioate oligonucleotides. J. Clin. Lab. Anal. 1995; 9:129–137. [DOI] [PubMed] [Google Scholar]
- 20. Bijsterbosch M.K., Rump E.T., De Vrueh R.L., Dorland R., van Veghel R., Tivel K.L., Biessen E.A., van Berkel T.J., Manoharan M.. Modulation of plasma protein binding and in vivo liver cell uptake of phosphorothioate oligodeoxynucleotides by cholesterol conjugation. Nucleic Acids Res. 2000; 28:2717–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Geary R.S., Norris D., Yu R., Bennett C.F.. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug. Deliv. Rev. 2015; 87:46–51. [DOI] [PubMed] [Google Scholar]
- 22. Agrawal S., Zhang X., Cai Q., Kandimalla E.R., Manning A., Jiang Z., Marcel T., Zhang R.. Effect of aspirin on protein binding and tissue disposition of oligonucleotide phosphorothioate in rats. J. Drug Target. 1998; 5:303–312. [DOI] [PubMed] [Google Scholar]
- 23. Shemesh C.S., Yu R.Z., Gaus H.J., Seth P.P., Swayze E.E., Bennett F.C., Geary R.S., Henry S.P., Wang Y.. Pharmacokinetic and pharmacodynamic investigations of ION-353382, a model antisense oligonucleotide: using alpha-2-macroglobulin and murinoglobulin double-knockout mice. Nucleic Acid Ther. 2016; 26:223–235. [DOI] [PubMed] [Google Scholar]
- 24. Glover J.M., Leeds J.M., Mant T.G., Amin D., Kisner D.L., Zuckerman J.E., Geary R.S., Levin A.A., Shanahan W.R. Jr. Phase I safety and pharmacokinetic profile of an intercellular adhesion molecule-1 antisense oligodeoxynucleotide (ISIS 2302). J. Pharmacol. Exp. Ther. 1997; 282:1173–1180. [PubMed] [Google Scholar]
- 25. Henry S.P., Kim T.-W., Karmer-Stickland K., Zanardi T.A., Fey R.A., Levin A.A.. Crooke ST. Antisense Drug Technology-Principles, Strategies, and Applications. 2008; CRC Press; 327–364. [Google Scholar]
- 26. Crooke S.T., Baker B.F., Pham N.C., Hughes S.G., Kwoh T.J., Cai D., Tsimikas S., Geary R.S., Bhanot S.. The effects of 2′-O-methoxyethyl oligonucleotides on renal function in humans. Nucleic Acid Ther. 2018; 28:10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tseng Y.C., Mozumdar S., Huang L.. Lipid-based systemic delivery of siRNA. Adv. Drug. Deliv. Rev. 2009; 61:721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Prakash T.P., Mullick A.E., Lee R.G., Yu J., Yeh S.T., Low A., Chappell A.E., Ostergaard M.E., Murray S., Gaus H.J. et al.. Fatty acid conjugation enhances potency of antisense oligonucleotides in muscle. Nucleic Acids Res. 2019; 47:6029–6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Osborn M.F., Khvorova A.. Improving siRNA delivery in vivo through lipid conjugation. Nucleic Acid Ther. 2018; 28:128–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haraszti R.A., Miller R., Didiot M.C., Biscans A., Alterman J.F., Hassler M.R., Roux L., Echeverria D., Sapp E., DiFiglia M. et al.. Optimized cholesterol-siRNA chemistry improves productive loading onto extracellular vesicles. Mol. Ther. 2018; 26:1973–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Raouane M., Desmaele D., Urbinati G., Massaad-Massade L., Couvreur P.. Lipid conjugated oligonucleotides: a useful strategy for delivery. Bioconjug. Chem. 2012; 23:1091–1104. [DOI] [PubMed] [Google Scholar]
- 32. Biscans A., Coles A., Haraszti R., Echeverria D., Hassler M., Osborn M., Khvorova A.. Diverse lipid conjugates for functional extra-hepatic siRNA delivery in vivo. Nucleic Acids Res. 2019; 47:1082–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Juliano R.L. Intracellular trafficking and endosomal release of oligonucleotides: what we know and what we don’t. Nucleic Acid Ther. 2018; 28:166–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hanss B., Leal-Pinto E., Bruggeman L.A., Copeland T.D., Klotman P.E.. Identification and characterization of a cell membrane nucleic acid channel. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:1921–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liang X.H., Sun H., Nichols J.G., Allen N., Wang S., Vickers T.A., Shen W., Hsu C.W., Crooke S.T.. COPII vesicles can affect the activity of antisense oligonucleotides by facilitating the release of oligonucleotides from endocytic pathways. Nucleic Acids Res. 2018; 46:10225–10245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liang X.H., Sun H., Hsu C.W., Nichols J.G., Vickers T.A., De Hoyos C.L., Crooke S.T.. Golgi-endosome transport mediated by M6PR facilitates release of antisense oligonucleotides from endosomes. Nucleic Acids Res. 2020; 48:1372–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miller C.M., Wan W.B., Seth P.P., Harris E.N.. Endosomal escape of antisense oligonucleotides internalized by stabilin receptors is regulated by Rab5C and EEA1 during endosomal maturation. Nucleic Acid Ther. 2018; 28:86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Juliano R.L., Wang L., Tavares F., Brown E.G., James L., Ariyarathna Y., Ming X., Mao C., Suto M.. Structure-activity relationships and cellular mechanism of action of small molecules that enhance the delivery of oligonucleotides. Nucleic Acids Res. 2018; 46:1601–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Manoharan M., Rajeev K.G.. Crooke ST. Antisense Drug Technology - Principles, Strategies, and Applications. 2008; Roca Raton, FL: CRC Press; 437–464. [Google Scholar]
- 40. Dowdy S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017; 35:222–229. [DOI] [PubMed] [Google Scholar]
- 41. Manoharan M. RNA interference and chemically modified small interfering RNAs. Curr. Opin. Chem. Biol. 2004; 8:570–579. [DOI] [PubMed] [Google Scholar]
- 42. Springer A.D., Dowdy S.F.. GalNAc-siRNA conjugates: leading the way for delivery of RNAi therapeutics. Nucleic Acid Ther. 2018; 28:109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lu D., Rhodes D.G.. Binding of phosphorothioate oligonucleotides to zwitterionic liposomes. Biochim. Biophys. Acta. 2002; 1563:45–52. [DOI] [PubMed] [Google Scholar]
- 44. Wang S., Allen N., Prakash T.P., Liang X.H., Crooke S.T.. Lipid conjugates enhance endosomal release of antisense oligonucleotides into cells. Nucleic Acid Ther. 2019; 29:245–255. [DOI] [PubMed] [Google Scholar]
- 45. Wang S., Allen N., Liang X.H., Crooke S.T.. Membrane destabilization induced by lipid species increases activity of phosphorothioate-antisense oligonucleotides. Mol Ther Nucleic Acids. 2018; 13:686–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang S., Sun H., Tanowitz M., Liang X.H., Crooke S.T.. Intra-endosomal trafficking mediated by lysobisphosphatidic acid contributes to intracellular release of phosphorothioate-modified antisense oligonucleotides. Nucleic Acids Res. 2017; 45:5309–5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Miller C.M., Donner A.J., Blank E.E., Egger A.W., Kellar B.M., Ostergaard M.E., Seth P.P., Harris E.N.. Stabilin-1 and Stabilin-2 are specific receptors for the cellular internalization of phosphorothioate-modified antisense oligonucleotides (ASOs) in the liver. Nucleic Acids Res. 2016; 44:2782–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gaus H., Miller C.M., Seth P.P., Harris E.N.. Structural determinants for the interactions of chemically modified nucleic acids with the stabilin-2 clearance receptor. Biochemistry. 2018; 57:2061–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tanowitz M., Hettrick L., Revenko A., Kinberger G.A., Prakash T.P., Seth P.P.. Asialoglycoprotein receptor 1 mediates productive uptake of N-acetylgalactosamine-conjugated and unconjugated phosphorothioate antisense oligonucleotides into liver hepatocytes. Nucleic Acids Res. 2017; 45:12388–12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang S., Allen N., Vickers T.A., Revenko A.S., Sun H., Liang X.H., Crooke S.T.. Cellular uptake mediated by epidermal growth factor receptor facilitates the intracellular activity of phosphorothioate-modified antisense oligonucleotides. Nucleic Acids Res. 2018; 46:3579–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weidner D.A., Valdez B.C., Henning D., Greenberg S., Busch H.. Phosphorothioate oligonucleotides bind in a non sequence-specific manner to the nucleolar protein C23/nucleolin. FEBS Lett. 1995; 366:146–150. [DOI] [PubMed] [Google Scholar]
- 52. Kandimalla E.R., Bhagat L., Wang D., Yu D., Sullivan T., La Monica N., Agrawal S.. Design, synthesis and biological evaluation of novel antagonist compounds of Toll-like receptors 7, 8 and 9. Nucleic Acids Res. 2013; 41:3947–3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Deshpande D., Toledo-Velasquez D., Thakkar D., Liang W., Rojanasakul Y.. Enhanced cellular uptake of oligonucleotides by EGF receptor-mediated endocytosis in A549 cells. Pharm. Res. 1996; 13:57–61. [DOI] [PubMed] [Google Scholar]
- 54. Ammala C., Drury W.J. 3rd, Knerr L., Ahlstedt I., Stillemark-Billton P., Wennberg-Huldt C., Andersson E.M., Valeur E., Jansson-Lofmark R., Janzen D. et al.. Targeted delivery of antisense oligonucleotides to pancreatic beta-cells. Sci Adv. 2018; 4:eaat3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Miller C.M., Tanowitz M., Donner A.J., Prakash T.P., Swayze E.E., Harris E.N., Seth P.P.. Receptor-mediated uptake of phosphorothioate antisense oligonucleotides in different cell types of the liver. Nucleic Acid Ther. 2018; 28:119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kang H., Alam M.R., Dixit V., Fisher M., Juliano R.L.. Cellular delivery and biological activity of antisense oligonucleotides conjugated to a targeted protein carrier. Bioconjug. Chem. 2008; 19:2182–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Prakash T.P., Graham M.J., Yu J., Carty R., Low A., Chappell A., Schmidt K., Zhao C., Aghajan M., Murray H.F. et al.. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014; 42:8796–8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Winkler J. Oligonucleotide conjugates for therapeutic applications. Ther Deliv. 2013; 4:791–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Benizri S., Gissot A., Martin A., Vialet B., Grinstaff M.W., Barthelemy P.. Bioconjugated oligonucleotides: recent developments and therapeutic applications. Bioconjug. Chem. 2019; 30:366–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Liang X.H., Shen W., Sun H., Prakash T.P., Crooke S.T.. TCP1 complex proteins interact with phosphorothioate oligonucleotides and can co-localize in oligonucleotide-induced nuclear bodies in mammalian cells. Nucleic Acids Res. 2014; 42:7819–7832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liang X.H., Sun H., Shen W., Crooke S.T.. Identification and characterization of intracellular proteins that bind oligonucleotides with phosphorothioate linkages. Nucleic Acids Res. 2015; 43:2927–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bailey J.K., Shen W., Liang X.H., Crooke S.T.. Nucleic acid binding proteins affect the subcellular distribution of phosphorothioate antisense oligonucleotides. Nucleic Acids Res. 2017; 45:10649–10671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vickers T.A., Crooke S.T.. Development of a quantitative BRET affinity assay for nucleic acid-protein interactions. PLoS One. 2016; 11:e0161930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kim D.I., Jensen S.C., Noble K.A., Kc B., Roux K.H., Motamedchaboki K., Roux K.J.. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell. 2016; 27:1188–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Abdul-Manan N., Williams K.R.. hnRNP A1 binds promiscuously to oligoribonucleotides: utilization of random and homo-oligonucleotides to discriminate sequence from base-specific binding. Nucleic Acids Res. 1996; 24:4063–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Koller E., Vincent T.M., Chappell A., De S., Manoharan M., Bennett C.F.. Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res. 2011; 39:4795–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang S., Sun H., Tanowitz M., Liang X.H., Crooke S.T.. Annexin A2 facilitates endocytic trafficking of antisense oligonucleotides. Nucleic Acids Res. 2016; 44:7314–7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Brukner I., Tremblay G.A.. Cellular proteins prevent antisense phosphorothioate oligonucleotide (SdT18) to target sense RNA (rA18): development of a new in vitro assay. Biochemistry. 2000; 39:11463–11466. [DOI] [PubMed] [Google Scholar]
- 69. Shen W., Liang X.H., Crooke S.T.. Phosphorothioate oligonucleotides can displace NEAT1 RNA and form nuclear paraspeckle-like structures. Nucleic Acids Res. 2014; 42:8648–8662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liang X.H., Shen W., Sun H., Kinberger G.A., Prakash T.P., Nichols J.G., Crooke S.T.. Hsp90 protein interacts with phosphorothioate oligonucleotides containing hydrophobic 2′-modifications and enhances antisense activity. Nucleic Acids Res. 2016; 44:3892–3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Matranga C., Tomari Y., Shin C., Bartel D.P., Zamore P.D.. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell. 2005; 123:607–620. [DOI] [PubMed] [Google Scholar]
- 72. Lorenz P., Baker B.F., Bennett C.F., Spector D.L.. Phosphorothioate antisense oligonucleotides induce the formation of nuclear bodies. Mol. Biol. Cell. 1998; 9:1007–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Protter D.S.W., Parker R.. Principles and properties of stress granules. Trends Cell Biol. 2016; 26:668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wang Y., Shen W., Liang X.H., Crooke S.T.. Phosphorothioate antisense oligonucleotides bind p-body proteins and mediate P-Body assembly. Nucleic Acid Ther. 2019; 29:343–358. [DOI] [PubMed] [Google Scholar]
- 75. Castanotto D., Lin M., Kowolik C., Wang L., Ren X.Q., Soifer H.S., Koch T., Hansen B.R., Oerum H., Armstrong B. et al.. A cytoplasmic pathway for gapmer antisense oligonucleotide-mediated gene silencing in mammalian cells. Nucleic Acids Res. 2015; 43:9350–9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Parker R., Sheth U.. P bodies and the control of mRNA translation and degradation. Mol. Cell. 2007; 25:635–646. [DOI] [PubMed] [Google Scholar]
- 77. Fox A.H., Nakagawa S., Hirose T., Bond C.S.. Paraspeckles: where long noncoding RNA Meets phase separation. Trends Biochem. Sci. 2018; 43:124–135. [DOI] [PubMed] [Google Scholar]
- 78. Nakagawa S., Yamazaki T., Hirose T.. Molecular dissection of nuclear paraspeckles: towards understanding the emerging world of the RNP milieu. Open Biol. 2018; 8:180150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Shen W., Liang X.H., Sun H., Crooke S.T.. 2′-Fluoro-modified phosphorothioate oligonucleotide can cause rapid degradation of P54nrb and PSF. Nucleic Acids Res. 2015; 43:4569–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Shen W., De Hoyos C.L., Sun H., Vickers T.A., Liang X.H., Crooke S.T.. Acute hepatotoxicity of 2′ fluoro-modified 5-10-5 gapmer phosphorothioate oligonucleotides in mice correlates with intracellular protein binding and the loss of DBHS proteins. Nucleic Acids Res. 2018; 46:2204–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Migawa M.T., Shen W., Wan W.B., Vasquez G., Oestergaard M.E., Low A., De Hoyos C.L., Gupta R., Murray S., Tanowitz M. et al.. Site-specific replacement of phosphorothioate with alkyl phosphonate linkages enhances the therapeutic profile of gapmer ASOs by modulating interactions with cellular proteins. Nucleic Acids Res. 2019; 47:5465–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Vickers T.A., Rahdar M., Prakash T.P., Crooke S.T.. Kinetic and subcellular analysis of PS-ASO/protein interactions with P54nrb and RNase H1. Nucleic Acids Res. 2019; 47:10865–10880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lima W.F., Vickers T.A., Nichols J., Li C., Crooke S.T.. Defining the factors that contribute to on-target specificity of antisense oligonucleotides. PLoS One. 2014; 9:e101752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kakiuchi-Kiyota S., Whiteley L.O., Ryan A.M., Mathialagan N.. Development of a method for profiling protein interactions with LNA-Modified antisense oligonucleotides using protein microarrays. Nucleic Acid Ther. 2016; 26:93–101. [DOI] [PubMed] [Google Scholar]
- 85. Lima W.F., Crooke S.T.. Binding affinity and specificity of Escherichia coli RNase H1: impact on the kinetics of catalysis of antisense oligonucleotide-RNA hybrids. Biochemistry. 1997; 36:390–398. [DOI] [PubMed] [Google Scholar]
- 86. Huang J., Zhao Y., Liu H., Huang D., Cheng X., Zhao W., Taylor I.A., Liu J., Peng Y.L.. Substitution of tryptophan 89 with tyrosine switches the DNA binding mode of PC4. Sci. Rep. 2015; 5:8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hirose T., Yamazaki T., Nakagawa S.. Molecular anatomy of the architectural NEAT1 noncoding RNA: The domains, interactors, and biogenesis pathway required to build phase-separated nuclear paraspeckles. Wiley Interdiscip. Rev. RNA. 2019; 10:e1545. [DOI] [PubMed] [Google Scholar]
- 88. Lima W., Wu H., Crooke S.T.. Crooke ST. Antisense Drug Technology - Principles, Strategies, and Applications. 2008; 2nd ednBoca Raton, FL: CRC Press; 47–74. [Google Scholar]
- 89. Wu H., Lima W.F., Crooke S.T.. Properties of cloned and expressed human RNase H1. J. Biol. Chem. 1999; 274:28270–28278. [DOI] [PubMed] [Google Scholar]
- 90. Wu H., Lima W.F., Crooke S.T.. Investigating the structure of human RNase H1 by site-directed mutagenesis. J. Biol. Chem. 2001; 276:23547–23553. [DOI] [PubMed] [Google Scholar]
- 91. Brandsen J., Werten S., van der Vliet P.C., Meisterernst M., Kroon J., Gros P.. C-terminal domain of transcription cofactor PC4 reveals dimeric ssDNA binding site. Nat. Struct. Biol. 1997; 4:900–903. [DOI] [PubMed] [Google Scholar]
- 92. Nowotny M., Gaidamakov S.A., Ghirlando R., Cerritelli S.M., Crouch R.J., Yang W.. Structure of human RNase H1 complexed with an RNA/DNA hybrid: insight into HIV reverse transcription. Mol. Cell. 2007; 28:264–276. [DOI] [PubMed] [Google Scholar]
- 93. Ge H., Roeder R.G.. Purification, cloning, and characterization of a human coactivator, PC4, that mediates transcriptional activation of class II genes. Cell. 1994; 78:513–523. [DOI] [PubMed] [Google Scholar]
- 94. Werten S., Langen F.W., van Schaik R., Timmers H.T., Meisterernst M., van der Vliet P.C.. High-affinity DNA binding by the C-terminal domain of the transcriptional coactivator PC4 requires simultaneous interaction with two opposing unpaired strands and results in helix destabilization. J. Mol. Biol. 1998; 276:367–377. [DOI] [PubMed] [Google Scholar]
- 95. Werten S., Moras D.. A global transcription cofactor bound to juxtaposed strands of unwound DNA. Nat. Struct. Mol. Biol. 2006; 13:181–182. [DOI] [PubMed] [Google Scholar]
- 96. Hyjek-Składanowska M., Vickers T.A., Napiórkowska A., Anderson B., Tanowitz M., Crooke S.T., Liang X.H., Seth P.P., Nowotny M.. Origins of the increased affinity of phosphorothioate-modified therapeutic nucleic acids for proteins. J. Am. Chem. Soc. 2020; https://doi.org/10.1021/jacs.9b13524. [DOI] [PubMed] [Google Scholar]
- 97. Vickers T.A., Migawa M.T., Seth P.P., Crooke S.T.. Interaction of ASOs with PC4 is highly influenced by the cellular environment and ASO chemistry. J. Am. Chem. Soc. 2020; In press. [DOI] [PubMed] [Google Scholar]
- 98. Wu H., Lima W.F., Zhang H., Fan A., Sun H., Crooke S.T.. Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs. J. Biol. Chem. 2004; 279:17181–17189. [DOI] [PubMed] [Google Scholar]
- 99. Cerritelli S.M., Crouch R.J.. The non-RNase H domain of Saccharomyces cerevisiae RNase H1 binds double-stranded RNA: magnesium modulates the switch between double-stranded RNA binding and RNase H activity. RNA. 1995; 1:246–259. [PMC free article] [PubMed] [Google Scholar]
- 100. Evans S.P., Bycroft M.. NMR structure of the N-terminal domain of Saccharomyces cerevisiae RNase HI reveals a fold with a strong resemblance to the N-terminal domain of ribosomal protein L9. J. Mol. Biol. 1999; 291:661–669. [DOI] [PubMed] [Google Scholar]
- 101. Kanaya S., Katayanagi K., Morikawa K., Inoue H., Ohtsuka E., Ikehara M.. Effect of mutagenesis at each of five histidine residues on enzymatic activity and stability of ribonuclease H from Escherichia coli. Eur. J. Biochem. 1991; 198:437–440. [DOI] [PubMed] [Google Scholar]
- 102. Nakamura H., Oda Y., Iwai S., Inoue H., Ohtsuka E., Kanaya S., Kimura S., Katsuda C., Katayanagi K., Morikawa K. et al.. How does RNase H recognize a DNA.RNA hybrid. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:11535–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Yang W., Hendrickson W.A., Crouch R.J., Satow Y.. Structure of ribonuclease H phased at 2 A resolution by MAD analysis of the selenomethionyl protein. Science. 1990; 249:1398–1405. [DOI] [PubMed] [Google Scholar]
- 104. Lima W.F., Wu H., Nichols J.G., Prakash T.P., Ravikumar V., Crooke S.T.. Human RNase H1 uses one tryptophan and two lysines to position the enzyme at the 3′-DNA/5′-RNA terminus of the heteroduplex substrate. J. Biol. Chem. 2003; 278:49860–49867. [DOI] [PubMed] [Google Scholar]
- 105. Shav-Tal Y., Zipori D.. PSF and p54(nrb)/NonO–multi-functional nuclear proteins. FEBS Lett. 2002; 531:109–114. [DOI] [PubMed] [Google Scholar]
- 106. Fox A.H., Bond C.S., Lamond A.I.. P54nrb forms a heterodimer with PSP1 that localizes to paraspeckles in an RNA-dependent manner. Mol. Biol. Cell. 2005; 16:5304–5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Maris C., Dominguez C., Allain F.H.. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005; 272:2118–2131. [DOI] [PubMed] [Google Scholar]
- 108. Vickers T.A., Crooke S.T.. Antisense oligonucleotides capable of promoting specific target mRNA reduction via competing RNase H1-dependent and independent mechanisms. PLoS One. 2014; 9:e108625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Jackson S.E. Hsp90: structure and function. Top. Curr. Chem. 2013; 328:155–240. [DOI] [PubMed] [Google Scholar]
- 110. Crooke S.T., Graham M.J., Zuckerman J.E., Brooks D., Conklin B.S., Cummins L.L., Greig M.J., Guinosso C.J., Kornbrust D., Manoharan M. et al.. Pharmacokinetic properties of several novel oligonucleotide analogs in mice. J. Pharmacol. Exp. Ther. 1996; 277:923–937. [PubMed] [Google Scholar]
- 111. Wan W.B., Migawa M.T., Vasquez G., Murray H.M., Nichols J.G., Gaus H., Berdeja A., Lee S., Hart C.E., Lima W.F. et al.. Synthesis, biophysical properties and biological activity of second generation antisense oligonucleotides containing chiral phosphorothioate linkages. Nucleic Acids Res. 2014; 42:13456–13468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ostergaard M.E., De Hoyos C.L., Wan W.B., Shen W., Low A., Berdeja A., Vasquez G., Murray S., Migawa M.T., Liang X.H. et al.. Understanding the effect of controlling phosphorothioate chirality in the DNA gap on the potency and safety of gapmer antisense oligonucleotides. Nucleic Acids Res. 2020; 48:1691–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Koziolkiewicz M., Krakowiak A., Kwinkowski M., Boczkowska M., Stec W.J.. Stereodifferentiation–the effect of P chirality of oligo(nucleoside phosphorothioates) on the activity of bacterial RNase H. Nucleic Acids Res. 1995; 23:5000–5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Koziolkiewicz M., Wojcik M., Kobylanska A., Karwowski B., Rebowska B., Guga P., Stec W.J.. Stability of stereoregular oligo(nucleoside phosphorothioate)s in human plasma: diastereoselectivity of plasma 3′-exonuclease. Antisense Nucleic Acid Drug Dev. 1997; 7:43–48. [DOI] [PubMed] [Google Scholar]
- 115. Jahns H., Roos M., Imig J., Baumann F., Wang Y., Gilmour R., Hall J.. Stereochemical bias introduced during RNA synthesis modulates the activity of phosphorothioate siRNAs. Nat. Commun. 2015; 6:6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Steinke C.A., Reeves K.K., Powell J.W., Lee S.A., Chen Y.Z., Wyrzykiewicz T., Griffey R.H., Mohan V.. Vibrational analysis of phosphorothioate DNA: II. The POS group in the model compound dimethyl phosphorothioate [(CH3O)2(POS)]. J. Biomol. Struct. Dyn. 1997; 14:509–516. [DOI] [PubMed] [Google Scholar]
- 117. Baraniak J., Frey P.A.. Effect of ion pairing on bond order and charge localization in alkyl phosphorothioates. J. Am. Chem. Soc. 1988; 110:4059–4060. [Google Scholar]
- 118. White A.P., Reeves K.K., Snyder E., Farrell J., Powell J.W., Mohan V., Griffey R.H.. Hydration of single-stranded phosphodiester and phosphorothioate oligodeoxyribonucleotides. Nucleic Acids Res. 1996; 24:3261–3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Plumridge A., Meisburger S.P., Andresen K., Pollack L.. The impact of base stacking on the conformations and electrostatics of single-stranded DNA. Nucleic Acids Res. 2017; 45:3932–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Bohr H.G., Shim I., Stein C., Orum H., Hansen H.F., Koch T.. Electronic structures of LNA phosphorothioate oligonucleotides. Mol Ther Nucleic Acids. 2017; 8:428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Messias A.C., Sattler M.. Structural basis of single-stranded RNA recognition. Acc. Chem. Res. 2004; 37:279–287. [DOI] [PubMed] [Google Scholar]
- 122. Deo R.C., Bonanno J.B., Sonenberg N., Burley S.K.. Recognition of polyadenylate RNA by the poly(A)-binding protein. Cell. 1999; 98:835–845. [DOI] [PubMed] [Google Scholar]
- 123. Freier S.M., Watt A.T.. Crooke ST. Antisense Drug Technology - Principles, Strategies, and Application. 2008; Boca Raton, FL: CRC Press; 117–141. [Google Scholar]
- 124. McRae E.K.S., Booy E.P., Padilla-Meier G.P., McKenna S.A.. On characterizing the interactions between proteins and guanine quadruplex structures of nucleic acids. J. Nucleic Acids. 2017; 2017:9675348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Pandey S.K., Nowak A., Perkins J., Ferng A., Prakash T.P.. Effect of 2′-O-[2-[2-(N,N-dimethylamino)ethoxy]ethyl] modification on activity of gapmer antisense oligonucleotides containing 2′,4′-constrained 2′'-O-ethyl nucleic acid. Bioorg. Med. Chem. Lett. 2015; 25:1688–1691. [DOI] [PubMed] [Google Scholar]
- 126. Burdick A.D., Sciabola S., Mantena S.R., Hollingshead B.D., Stanton R., Warneke J.A., Zeng M., Martsen E., Medvedev A., Makarov S.S. et al.. Sequence motifs associated with hepatotoxicity of locked nucleic acid–modified antisense oligonucleotides. Nucleic Acids Res. 2014; 42:4882–4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Chernikov I.V., Vlassov V.V., Chernolovskaya E.L.. Current development of siRNA bioconjugates: from research to the clinic. Front Pharmacol. 2019; 10:444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Tushir-Singh J. Antibody-siRNA conjugates: drugging the undruggable for anti-leukemic therapy. Expert Opin. Biol. Ther. 2017; 17:325–338. [DOI] [PubMed] [Google Scholar]
- 129. Arnold A.E., Malek-Adamian E., Le P.U., Meng A., Martinez-Montero S., Petrecca K., Damha M.J., Shoichet M.S.. Antibody-antisense oligonucleotide conjugate downregulates a key gene in glioblastoma stem cells. Mol. Ther. Nucleic Acids. 2018; 11:518–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Sugo T., Terada M., Oikawa T., Miyata K., Nishimura S., Kenjo E., Ogasawara-Shimizu M., Makita Y., Imaichi S., Murata S. et al.. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control. Release. 2016; 237:1–13. [DOI] [PubMed] [Google Scholar]
- 131. McClorey G., Banerjee S.. Cell-penetrating peptides to enhance delivery of oligonucleotide-based therapeutics. Biomedicines. 2018; 6:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Viney N.J., van Capelleveen J.C., Geary R.S., Xia S., Tami J.A., Yu R.Z., Marcovina S.M., Hughes S.G., Graham M.J., Crooke R.M. et al.. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet. 2016; 388:2239–2253. [DOI] [PubMed] [Google Scholar]
- 133. Schmidt K., Prakash T.P., Donner A.J., Kinberger G.A., Gaus H.J., Low A., Ostergaard M.E., Bell M., Swayze E.E., Seth P.P.. Characterizing the effect of GalNAc and phosphorothioate backbone on binding of antisense oligonucleotides to the asialoglycoprotein receptor. Nucleic Acids Res. 2017; 45:2294–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ostergaard M.E., Jackson M., Low A., A E.C., R G.L., Peralta R.Q., Yu J., Kinberger G.A., Dan A., Carty R. et al.. Conjugation of hydrophobic moieties enhances potency of antisense oligonucleotides in the muscle of rodents and non-human primates. Nucleic Acids Res. 2019; 47:6045–6058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Osborn M.F., Coles A.H., Biscans A., Haraszti R.A., Roux L., Davis S., Ly S., Echeverria D., Hassler M.R., Godinho B. et al.. Hydrophobicity drives the systemic distribution of lipid-conjugated siRNAs via lipid transport pathways. Nucleic Acids Res. 2018; 47:1070–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Vickers T.A., Crooke S.T.. The rates of the major steps in the molecular mechanism of RNase H1-dependent antisense oligonucleotide induced degradation of RNA. Nucleic Acids Res. 2015; 43:8955–8963. [DOI] [PMC free article] [PubMed] [Google Scholar]