


Abstract
Cohesin SA1 (STAG1) and SA2 (STAG2) are key components of the cohesin complex. Previous studies have highlighted the unique contributions by SA1 and SA2 to 3D chromatin organization, DNA replication fork progression, and DNA double-strand break (DSB) repair. Recently, we discovered that cohesin SA1 and SA2 are DNA binding proteins. Given the recently discovered link between SA2 and RNA-mediated biological pathways, we investigated whether or not SA1 and SA2 directly bind to RNA using a combination of bulk biochemical assays and single-molecule techniques, including atomic force microscopy (AFM) and the DNA tightrope assay. We discovered that both SA1 and SA2 bind to various RNA containing substrates, including ssRNA, dsRNA, RNA:DNA hybrids, and R-loops. Importantly, both SA1 and SA2 localize to regions on dsDNA that contain RNA. We directly compared the SA1/SA2 binding and R-loops sites extracted from Chromatin Immunoprecipitation sequencing (ChIP-seq) and DNA-RNA Immunoprecipitation sequencing (DRIP-Seq) data sets, respectively. This analysis revealed that SA1 and SA2 binding sites overlap significantly with R-loops. The majority of R-loop-localized SA1 and SA2 are also sites where other subunits of the cohesin complex bind. These results provide a new direction for future investigation of the diverse biological functions of SA1 and SA2.
INTRODUCTION
The cohesin complex plays important roles in sister chromatid cohesion, DNA replication, repair and recombination, as well as 3D chromosome organization (1–6). In vertebrates, the core cohesin complex consists of a tripartite ring assembled from SMC1, SMC3 and RAD21 (also known as SCC1), and the stromal antigen subunit (SA) SA1 (STAG1) or SA2 (STAG2). Germline mutations in the core cohesin subunits lead to a wide spectrum of human diseases that are collectively called ‘cohesinopathies’ (2), as well as increased cancer incidence (7,8). Importantly, based on the analysis of somatic point mutations in exome sequences from 4742 human cancers, SA2 has been identified as 1 of only 12 genes that are significantly mutated in four or more cancer types (9–11). SA1 and SA2 were considered to have a supporting role in sister chromatid segregation by stabilizing the ring subunits. However, this notion cannot fully explain the key roles that SA1 and SA2 play in multiple genome maintenance pathways. For example, depletion of SA2 in primary human cells leads to DNA replication fork stalling and activation of DNA damage checkpoint pathways (12). Furthermore, several recent studies demonstrated the synthetic lethality of SA1 and SA2 depletion (13). SA1 depletion does not significantly impact the growth of SA2 proficient cells, whereas SA1 depletion in SA2 deficient cells leads to cell death.
Despite the importance of cohesin SA1 and SA2, their biophysical properties are largely unknown. Recently, we discovered that cohesin SA1 and SA2 are single-stranded (ss) and double-stranded (ds) DNA binding proteins (14,15). SA1 displays similar DNA binding affinities for ds and ssDNA, and binds specifically to double-stranded telomeric sequences mediated through its N-terminal AT-hook domain (14). In contrast, SA2 does not specially recognize either telomeric or centromeric sequences (15). Due to its higher binding affinities for ssDNA than for dsDNA, it recognizes intermediate DNA structures during DNA replication and double-strand break (DSB) repair, such as a dsDNA end, single-stranded overhang, flap, fork and ssDNA gap (15). Furthermore, using the DNA tightrope assay (16,17), we showed that both SA1 and SA2 are capable of switching between the search (1D diffusing) mode on dsDNA and recognition (stable binding) mode at the ssDNA gap (14,15).
Importantly, there is emerging evidence linking SA2 to RNA-mediated pathways, but the underlying mechanism is largely unknown (18,19). For example, depletion of SA2, but not SA1, causes defects in the repression of transcription after induction of DSBs and large-scale genome rearrangements in G1 phase cells (18). SA2 prevents gene translocation when there is strong transcription activity throughout the interphase. Furthermore, studies of the genome-wide distribution of SA2 in embryonic stem cells (ESCs) revealed that most of the SA2 molecules are located in gene promoters that are either in the poised or active transcription state (19). SA1 and SA2 also make specific contributions to genome folding. SA2 promotes the establishment of long-range interaction networks between distant Polycomb-bound promoters, while SA1 helps to maintain topologically associating domain (TAD) borders (19). Strikingly, SA2 is enriched over SA1 along 231 super-enhancer sites, and it is known that enhancers are transcribed into noncoding RNA called enhancer RNAs (eRNAs) (20). However, despite these emerging pieces of evidence for the involvement of SA2 in RNA-mediated pathways, a direct physical association between SA2 and RNA has neither been proved nor disproved.
R-loops are three-stranded nucleic acid structures consisting of an RNA:DNA hybrid and a displaced ssDNA loop (21–23). Using DRIPc-seq (DNA-RNA immunoprecipitation followed by cDNA conversion coupled to high-throughput sequencing), it was shown that R-loops collectively occupy up to 5% of the mammalian genome. R-loop formation occurs at conserved hotspots, including promoters and terminator regions of poly(A) dependent genes (24). R-loops are proposed to be a ‘double-edged sword’. They play critical roles in regulating diverse cellular pathways, including transcription initiation and termination, 3D chromatin architecture formation, immunoglobin class switching, and DNA repair (21,22,25). However, they also tend to induce genome instability when their levels are dysregulated (25–27). In particular, RNA:DNA hybrids form rapidly after DNA DSB induction (28). RNA:DNA hybrid formation and resolution play key roles in the initiation of transcription-associated homologous recombination repair (TA-HRR), for which cohesin function is also critical (5). Despite these established shared pathways between R-loops and cohesin, a direct physical interaction between a cohesin subunit and the R-loop has not been explored.
Our recent observations of cohesin SA1 and SA2 as DNA binding proteins and the emerging links established for SA2 and RNA-mediated pathways raises one important question: Do cohesin SA1 and SA2 directly bind to RNA? To directly address this key question, we probed the interaction between SA1/SA2 and various nucleic acid substrates by applying single-molecule imaging techniques, including Atomic Force Microscopy (AFM) and fluorescence microscopy imaging, as well as bulk fluorescence anisotropy. In addition, to validate RNA binding by SA1 and SA2, we directly compare the binding affinities of SA1 and SA2 to RNA containing substrates with activities of a known RNA binding protein, EWSR1 (29). We discovered that under the same experimental conditions, both cohesin SA1 and SA2 bind tighter to ssRNA than ssDNA of the same length. Furthermore, cohesin SA1 and SA2 bind to various RNA containing nucleic acid substrates, which include ssRNA, dsRNA, dsRNA with an overhang, RNA:DNA hybrids, a model R-loop substrate, and long ssRNA transcripts. Strikingly, cohesin SA1 and SA2 preferentially localize to regions on dsDNA that contain RNA. To address the question of whether or not SA1 and SA2 bind to RNA in vivo, we mined publicly available ChIP-Seq data for SA1, SA2, SMC1, SMC3 and CTCF from four cell lines, and DRIP-Seq data. Our analysis revealed that SA1 and SA2 binding sites overlap significantly with R-loops in vivo. Furthermore, in comparison to SA1/SA2 that do not colocalize with R-loops, SA1/SA2 colocalized with R-loops are positioned at a significantly shorter distance to the nearest transcription start site (TSS). This discovery of previously unknown RNA binding activities of cohesin SA1 and SA2 opens new directions of research to unravel the mechanisms underlying their diverse cellular functions.
MATERIALS AND METHODS
Protein purification
The purification of 6xHis tagged full-length SA1 (1258 AA, 141 KDa) and SA2 (1231 AA, 141 KDa) was described previously (30). Briefly, 6xHis tagged full-length SA1 or SA2 was overexpressed in Sf9 insect cells using the Bac-to-Bac baculovirus expression system (Life Technologies, USA). Recombinant proteins were purified sequentially by affinity chromatography (Ni-NTA agarose, Qiagen), anion exchange (Hitrap Q column, GE Healthcare), and gel filtration chromatography (Superose 6 column, GE Healthcare). The purity of SA1 and SA2 evaluated by SDS-PAGE and coomassie-blue staining was similar to the batches that were shown in our previous studies (14,15). His-tagged EWSR1 was purified in house, and FLAG-tagged EWSR1 was purchased from Origene. His- and FLAG-tagged EWSR1 displayed similar binding affinities for RNA based on the fluorescence anisotropy assay. EWSR1 with an N-terminal 6x-His tag and a C-terminal GFP tag was cloned into baculovirus using the Bac-to-Bac expressions system. Recombinant His-EWSR1 was purified first using Ni-NTA affinity. Afterward, fractions containing the protein were treated with TEV protease to remove the GFP tag, and the mixture was subjected to gel filtration chromatography (Superdex-200 column, GE Healthcare). Purified SA1, SA2, and EWSR1 were free of nuclease and RNase contaminations. This conclusion was supported by the observation of intact DNA and RNA substrates in the presence of these proteins using native gel electrophoresis. T3 RNA polymerase (Promega), as well as bacterial RNase A (Thermofisher) and RNase H (NEB), were purchased from commercial sources.
DNA and RNA substrates
All DNA oligos were purchased from IDT, and RNA oligos were purchased from Dharmacon Inc. (Supplementary Table S1). Linear DNA fragments containing R-loops (R-loop DNA) used for AFM imaging were generated through in vitro transcription. The template DNA, pFC53-Airn plasmid (3991 bp, a gift from the Chedin lab), contains the mouse Airn sequences downstream of a T3 promoter (31). Transcription reactions (50 μL total, 3 μg pFC53 DNA and 4.5 μL T3 RNA polymerase at 18.4 U/μL) were carried out at 37°C for 30 min in 1× Transcription Optimized Buffer (40 mM Tris, pH 7.9, 6 mM MgCl2, 2 mM spermidine and 10 mM NaCl) with additional DTT (20 mM), Tween-20 (0.05%), and rNTP (50 μM). Transcription was terminated by heat inactivation of the enzyme at 65°C for 10 min, and RNA unpaired with DNA was degraded by the addition of RNase A (5 μl of 0.1 mg/mL, Thermofisher) and incubation at 37°C for 30 min. In addition, a negative control DNA without the R-loops (N-R-loop) was prepared by generating R-loop DNA through in vitro transcription under the same conditions, followed by treatment of the sample with both RNase A and RNase H (32). Both the R-loop DNA and negative control DNA samples were purified using phenol/chloroform extraction. The DNA substrates were further digested with ApaLI to generate two fragments, followed by purification using phenol/chloroform extraction.
The long ssRNA was generated using the T7 Megascript kit (Invitrogen) and the linear pTRI-Xef as the template based on the manufacturer's standard protocol. Briefly, the template DNA (1 μg) was incubated with the T7 enzyme mix in 1× Reaction Buffer containing rNTPs (total 20 μL) at 37°C for 4 h. The transcript product was further purified using the MEGAclear Transcription Clean-up Kit (Invitrogen).
The oligo sequences used for the model R-loop substrate are adapted from a previous study (33). The fluorescein-labeled 25-nt RNA oligo was mixed with the 91-nt DNA and 91-nt DNA-com oligos (Supplementary Table S1) followed by heating to 85°C for 5 min and slow cooling to the room temperature in a buffer containing 0.355× PBS and 350 mM LiCl2. Annealed substrates were separated on a 10% native PAGE gel in 1× TBE buffer. The band corresponding to the model R-loop substrate (the upper band) visualized under UV shadowing was excised and eluted overnight in the EB buffer at room temperature. The model R-loop substrate was further concentrated and purified using phenol-chloroform extraction.
For the DNA tightrope assay, control DNA was linearized with BamH1, purified using the QIAquick PCR purification kit (Qiagen), and ligated using the Quick Ligation Kit (NEB). After confirmation of the ligation product by AFM imaging, the ligated control DNA was further purified using phenol/chloroform extraction. To generate long DNA containing R-loops, transcription was carried out using ligated control DNA and T3 RNA polymerases, followed by inactivation of the enzyme at 65°C for 10 min, and degradation of ssRNA by addition of RNase A and incubation at 37°C for 30 min. Finally, the ligated R-loop DNA sample was purified using the Biospin-30 column (BioRad).
Fluorescence anisotropy
His6-tagged full length SA1, SA2 or EWSR1 in DNA Binding Buffer (20 mM HEPES, pH 7.5, 0.1 mM MgCl2, 0.5 mM DTT, 100 mM KCl) was titrated into the binding solution containing substrates (3 nM) until the millipolarization signal (mP) stabilized. Experiments were carried out at 20°C. Each protein titration was repeated in triplicate. The data obtained from fluorescence anisotropy were plotted and analyzed by using the equation P = ((Pbound − Pfree)[protein]/(Kd + [protein])) + Pfree, where P is the polarization measured at a given total protein concentration, Pfree is the initial polarization of fluorescein-labeled DNA without protein binding, Pbound is the maximum polarization of DNA due to binding of proteins, and [protein] is the total protein concentration. A total of three parameters, including Pbound, Pfree and Kd, were fitted by nonlinear least-squares regression analysis.
AFM imaging and image analysis
Purified long RNA transcript alone (5 nM) or in the presence of either SA1 or SA2 (25 nM) were incubated at room temperature in SA2-DNA Reaction Buffer (20 mM HEPES, pH 7.5, 100 mM KCl, and 0.1 mM MgCl2) for 20 min. The reaction mixtures were diluted 15-fold in 1× AFM Imaging Buffer (25 mM NaOAc, 25 mM HEPES–KOH pH 7.5 and 10 mM Mg(OAc)2), and immediately deposited onto a freshly cleaved mica surface (SPI Supply). For imaging of SA1 and SA2 binding to the control DNA substrate or DNA containing R-loops, proteins (60 nM) and DNA (2.3 nM) were incubated in SA2-DNA Reaction Buffer. SA1- and SA2-DNA samples were diluted 10-fold in 1× AFM Imaging Buffer, followed by deposition onto a freshly cleaved mica surface. The samples were then washed with MilliQ water and dried under a stream of nitrogen gas. All images were collected using the AC mode on a MFP-3D-Bio AFM (Asylum Research) and Pointprobe® PPP-FMR probes (Nanosensors, spring constants at ∼2.8 N m−1). All images were captured at a scan size of 1–3 μm × 1–3 μm, a resolution of 512 × 512 pixels, and a scan rate of 1–2 Hz. Positions of SA1 and SA2 proteins on DNA were analyzed using software from Asylum Research. AFM volumes of protein complexes on DNA were determined using Gwyddion software (34). AFM volumes of the ssRNA transcript without or with proteins were measured using the Asylum software. For images of ssRNA transcripts without or with proteins, the threshold for selecting molecules was set at 800 nm2.
Electrophoresis mobility shift assays (EMSAs)
The reactions containing S9.6 antibody (Kerafast Inc., 200 nM) or EWSR1 (200 nM) along with either the model R-loop substrate or control dsDNA (13 nM) were carried out in a buffer containing 25 mM Tris, pH 7.5, 50 mM KCl, 50 μg/ml BSA, 5 mM MgCl2 and 1 mM DTT. The reactions were incubated for 20 min at 25°C. Nucleic acid–protein complexes and substrates alone were resolved by gel electrophoresis on a 6% 29:1 (bisacrylamide:acrylamide) native gel at 150 V for 45 min in 1× TBE buffer at 4°C. The gels were scanned using a Typhoon FLA 7000 Phosphorimager.
DNA tightrope assays
Tracking of quantum dot (QD) labeled proteins on DNA tightropes using oblique angle total internal reflection microscopy was described previously (14,15,17). Briefly, we first immobilized poly-l-lysine (2.5 mg/ml, M.W. > 30 000 kDa, Wako Chemicals) treated silica beads onto a PEGlyated coverslip surface. Then, we introduced ligated DNA substrates into the flow cell using a syringe pump at a flow rate of 300 μl/min to stretch the DNA between poly-l-lysine coated beads. After introducing protein-QDs into the flow cell, all videos were taken on an inverted microscope (Nikon Ti-E) using a 100× objective (APO TIRF, Nikon) and an EMCCD (iXon DU897, Andor Technology) at a time resolution of 50 ms/frame. Biotinylated multivalent chelator tris-nitrilotriacetic acid (BTtris-NTA) was prepared according to protocols published previously (35,36). For QD-labeling of His6-tagged full-length SA1 and SA2, 0.5 μl of red (655 nm) streptavidin-conjugated QDs (Invitrogen, 1 μM) was incubated with 1.5 μL of BTtris-NTA (5 μM) for 20 min. Proteins (1 μL of 1 μM) were then added to the QD-NTA solution and incubated for an additional 20 min. For conjugating FLAG-tagged EWSR1 to secondary antibody-coated QDs, EWSR1 (1 μL of 1 μM) was incubated with FLAG antibody (1 μL of 1 μM) for 20 min at room temperature, followed by the addition of secondary antibody-coated green (565 nm) QDs (1 μL of 1 μM) and additional 20 min of incubation. All conjugated protein samples were diluted to 5 nM using SA2 Imaging Buffer (25 mM HEPES, pH 7.5, 100 mM KCl, 0.5 mM MgCl2, 1 mM DTT and 1 mg/mL BSA) before being introduced into the flow cell. The spacing between adjacent QD-labeled proteins was measured based on the distance between intensity peaks.
Statistical analysis
Data from fluorescence anisotropy, AFM imaging, and DNA tightrope assay were pooled from at least two to three independent experiments. Statistical analysis was carryout out using OriginPro (OriginLab). Unless stated otherwise, the error reported is SEM. The significance of the difference between the AFM height and volume of long RNA transcripts before and after the addition of SA1 and SA2 was evaluated using the paired-sample Wilcoxon signed-rank test. The significance of the difference between AFM volumes of proteins on the linear R-loop and control substrates, between protein densities and position distributions on R-loop and control DNA tightropes, were evaluated using the Mann–Whitney test. The statistically significant level was set at P < 0.05.
RESULTS
Cohesin variants SA1 and SA2 are RNA binding proteins
We recently discovered that cohesin SA1 and SA2 directly bind ssDNA and dsDNA (14,15). To investigate whether nor not SA1 and SA2 directly bind to RNA, we purified full-length His-tagged SA1 and SA2 (Supplementary Figure S1). We then applied fluorescence anisotropy to measure the equilibrium dissociation constants (Kd) of full-length SA1 and SA2 for different RNA-containing nucleic acid substrates. Specifically, we evaluated the binding of SA1 and SA2 to ssRNA, dsRNA, dsRNA with an overhang, and RNA:DNA hybrids (Figures 1 and 2, Table 1, and Supplementary Table S1). Strikingly, at the same substrate length (66 nt), both SA1 and SA2 bound significantly tighter to ssRNA (substrate: 66 nt-1, SA1: Kd = 3.2 ± 0.3 nM; SA2: Kd = 7.2 ± 2.5 nM) than ssDNA (SA1: Kd = 36.5 nM; SA2: Kd = 41.0 nM) (15). Furthermore, the binding affinity of SA1 and SA2 for a second ssRNA (66 nt-2) of the same length was comparable to the 66 nt-1 RNA substrate (Table 1), suggesting that RNA binding is sequence-independent. The binding affinity of SA1 and SA2 for ssRNA was length-dependent. For both SA1 and SA2, the binding affinity for ssRNA dropped significantly as the substrate length decreased from 45 nt (SA1: Kd = 6.0 ± 0.6 nM; SA2: Kd = 11.4 ± 1.2 nM) to 25 nt ssRNA (SA1: Kd = 163.7 ± 37.4 nM; SA2: Kd = 251.1 ± 3.2 nM). In addition, both SA1 and SA2 displayed comparable binding affinities for dsRNA (66 bp, SA1: Kd = 61.5 ± 9.3; SA2: Kd = 31.2 ± 1.8 nM; Figure 2 and Table 1) and dsDNA of the same length (SA1: Kd = 104.0 ± 13.6 nM; SA2: Kd = 76.2 ± 3.9 nM), which were measured under the same conditions as in our previous studies (15). Furthermore, both SA1 and SA2 bound to a dsRNA substrate containing an overhang (45 ds + 21 nt, SA1: Kd = 95.8 ± 6.1 nM; SA2: Kd = 56.8 ± 0.5 nM), RNA:DNA hybrids without an overhang (25 bp, SA1: Kd = 58.6 ± 1.5 nM; SA2: Kd = 43.3 ± 5.6 nM; 45 bp, SA1: Kd = 44.0 ± 5.9 nM; SA2: Kd = 31.7 ± 1.5 nM), and the RNA:DNA hybrid with overhangs (SA1: Kd = 25.8 ± 3.4 nM; SA2: Kd = 23.4 ± 1.3 nM, Figure 2, Table 1).
Figure 1.

Cohesin SA1 and SA2 bind to single-stranded RNA in a length dependence manner. (A and B) Binding of SA1 (A) and SA2 (B) to 25, 45 and 66 nt ssRNA measured by fluorescence anisotropy using fluorescein-labeled RNA. The data were fitted to the law of mass action (R2 > 0.99). The error bars (standard deviations) are from three measurements. The RNA oligo sequences are shown in Supplementary Table S1. The equilibrium dissociation constants (Kd) were calculated from at least two independent experiments (Table 1).
Figure 2.

SA1 and SA2 bind to double-stranded substrates containing RNA. (A) Schematic illustration of double-stranded substrates containing RNA that were used for fluorescence anisotropy experiments. The green star represents the 5′ fluorescein label. The RNA oligo sequences are shown in Supplementary Table S1. (B and C) Concentration-dependent binding of SA1 (B) and SA2 (C) to dsRNA, dsRNA with an overhang, and RNA:DNA hybrids with or without overhangs. The data were fitted to the law of mass action (R2 > 0.96). The error bars (standard deviations) are from three measurements. The equilibrium dissociation constants (Kd) were calculated from at least two independent experiments (Table 1).
Table 1.
The equilibrium dissociation constants (Kd) of SA1, SA2 and EWSR1 for different RNA-containing substrates measured from fluorescence anisotropy experiments
| RNA containing substrates | SA1 Kd [nM] (mean ± SEM) | SA2 Kd [nM] (mean ± SEM) | EWSR1 [nM] (mean ± SEM) | |
|---|---|---|---|---|
| ssRNA | 25 nt (R-loop oligo) | 163.7 ± 37.4 | 251.1 ± 3.2 | 134.2 ± 0.04 |
| 45 nt | 6.0 ± 0.6 | 11.4 ± 1.2 | ||
| 66 nt-1 | 3.2 ± 0.3 | 7.2 ± 2.5 | 35.1 ± 1.8 | |
| 66 nt-2 | 3.0 ± 0.1 | 5.1 ± 0.6 | ||
| dsRNA substrates | dsRNA (66 bp) | 61.5 ± 9.3 | 31.2 ± 1.8 | 34.6 ± 6.9 |
| dsRNA + an overhang | 95.8 ± 6.1 | 56.8 ± 0.5 | 44.0 ± 1.7 | |
| Substrates related to the model R-loop | RNA:DNA hybrid + overhangs | 25.8 ± 3.4 | 23.4 ± 1.3 | 94.7 ± 10.7 |
| RNA:DNA hybrid (25 bp) | 58.6 ±1.5 | 43.3 ± 5.6 | ||
| RNA:DNA hybrid (45 bp) | 44.0 ± 5.9 | 31.7 ± 1.5 | 27.7 ± 4.1 | |
| Model R-loop | 38.4 ± 8.7 | 36.0 ± 4.0 | 28.6 ± 0.8 | |
K d was calculated from two to three independent experiments.
To further validate the RNA binding by SA1 and SA2, we directly compared their nucleic acid binding activities with a previously known RNA binding protein, EWSR1 (Supplementary Figure S2). EWSR1 was reported to bind to G- and U-runs in vitro (29). Fluorescence anisotropy experiments showed that EWSR1 binds to DNA (Supplementary Figure S2A), ssRNA, dsRNA, dsRNA with an overhang (Supplementary Figure S2B), and the RNA:DNA hybrid (Supplementary Figure S2C, Table 1). Overall, RNA binding affinities of SA1 and SA2 were comparable to those of EWSR1 for all RNA containing substrates tested in this study (Table 1). Together, these results from fluorescence anisotropy experiments using various RNA containing substrates demonstrated that SA1 and SA2 are RNA binding proteins.
Cohesin SA1 and SA2 directly bind to long RNA transcripts
Having established that SA1 and SA2 bind ssRNA oligos (Figure 1), we next sought to understand the structure of SA1 and SA2 binding to long ssRNA using AFM imaging. Long ssRNA was transcribed using the linear pTRI-Xef fragment as the template that contains the Xenopus elongation factor 1α gene under the control of the T7 promoter. The full-length transcript using the T7 MegaScript transcription system is expected to be 1.89 kb. Samples containing RNA transcripts alone (5 nM) or in the presence of either cohesin SA1 or SA2 (25 nM) after incubation at room temperature for 20 min were deposited onto a mica surface for AFM imaging. To detect RNA binding by cohesin SA1 and SA2, we measured both the maximum AFM height and volume of molecules from AFM images with either RNA transcripts alone (Figure 3A) or in the presence of proteins (SA1 or SA2, Figure 3B and C). AFM images of the RNA transcripts with Mg2+ present in both the incubation and deposition buffers showed folded structures with maximum AFM heights at 2.0 nm (±0.02 nm) and AFM volumes at 2412 nm3 (±110 nm3, Figure 3). Previously, we showed that cohesin SA1 and SA2 exist as monomers in solution (30), and SA2 displays an average AFM volume of 146 nm3 and AFM height of 1.41 nm under the same experimental conditions (15). On long ssRNA transcripts, both cohesin SA1 and SA2 formed large and heterogeneous clusters on RNA with maximum AFM heights significantly greater than those of proteins or RNA alone (Figure 3B and C). Overall, the addition of cohesin SA1 or SA2 to the RNA sample shifted the distribution of the maximum AFM height to significantly (P < 0.001) greater values (SA1+RNA: 4.3 ± 0.1 nm; SA2 + RNA: 4.5 ± 0.3 nm, Figure 3D). Furthermore, compared to RNA alone, SA1 and SA2 binding to RNA led to significantly (P < 0.001) larger AFM volumes (SA1 + RNA: 4077 ± 568 nm3; SA2+ RNA:5334 ± 531 nm3). These results suggest that each RNA transcript recruited multiple copies of SA1 and SA2 molecules, or SA1 and SA2 protein clusters bridged multiple copies of RNA transcripts (Figure 3B and C). In summary, these results from AFM imaging established that both SA1 and SA2 are capable of binding to long ssRNA transcripts.
Figure 3.

Cohesin SA1 and SA2 bind to long ssRNA transcripts. (A) A representative AFM image of RNA transcripts after purification using the MEGAclear Transcription Clean-up kit. (B and C) AFM images of RNA transcripts in the presence of SA1 (B) or SA2 (C). (D) Histograms of the maximum AFM height (left panel) and AFM volume (right panel) for RNA transcripts alone (black bars, N = 255), and for RNA transcripts in the presence of either SA1 (red bars, N = 135) or SA2 (blue bars, N = 135). Each data set was from two to three independent experiments.
Cohesin SA1 and SA2 bind to a model R-loop substrate
Combining the observations that SA1 and SA2 bind to RNA:DNA hybrids (Figure 2) and ssDNA (15), we hypothesized that SA1 and SA2 might be capable of binding to R-loops. To directly test this hypothesis, we generated a model R-loop substrate that consists of two arms of dsDNA (30 bp), and a three-stranded structure in the middle that contains the RNA:DNA hybrid (25 bp) and the ssDNA loop (31 nt, Figure 4A) (33). The model R-loop substrate was generated by annealing of the three nucleic acid strands, among which the 5′ of the RNA oligo was labeled with fluorescein. We purified the fully annealed R-loop substrate from the native acrylamide gel using phenol/chloroform extraction. This additional purification procedure ensured that in the model R-loop substrate preparation there was no significant contamination from free ssDNA or the RNA:DNA hybrid (Figure 4A). Furthermore, we validated the formation of the RNA:DNA hybrid portion of the R-loop by incubating the purified substrate with the S9.6 antibody that specifically detects RNA:DNA hybrids (37). Considering that EWSR1 showed a strong affinity for the hybrid RNA:DNA substrate in addition to the RNA only substrate, we also evaluated its ability to bind to R-loops. S9.6 antibody and EWSR1 bound to the R-loop substrate and induced a mobility shift under electrophoresis in a native gel (Figure 4B and C). In comparison, under the same conditions, the S9.6 antibody and EWSR1 did not show stable binding to dsDNA, indicating the specificity of their binding. These results using S9.6 antibody validated the integrity of the model R-loop substrate.
Figure 4.

Cohesin SA1 and SA2 bind to the model R-loop substrate. (A) Native gel showing 25-nt RNA, the RNA:DNA hybrid, and the gel-purified three-stranded model R-loop substrate. In the schematic illustrations of the substrates, the star represents the 5′ fluorescein label. The sequences of RNA and DNA oligos for making the model R-loop substrate are shown in Supplementary Table S1. (B) EMSA showing the S9.6 antibody binding to the model R-loop substrate (left panel), and no significant binding of the S9.6 antibody to the control 69-bp dsDNA (right panel). (C) EMSA showing EWSR1 binding to the model R-loop substrate, and no significant stable binding of EWSR1 to the control 66-bp dsDNA. (D) Fluorescence anisotropy showing concentration-dependent binding of SA1 and SA2 to the model R-loop substrate. The data were fitted to the law of mass action (R2 > 0.99). The error bars (standard deviations) are from three measurements. The equilibrium dissociation constants (Kd) were calculated from at least two independent experiments (Table 1).
Furthermore, fluorescence anisotropy experiments demonstrated that both SA1 and SA2 have high binding affinities for the model R-loop substrates (SA1: Kd = 38.4 ± 8.7 nM; SA2: Kd = 36.0 ± 4.0 nM, Table 1, Figure 4D). Their binding affinity for the model R-loop substrate is comparable to that of EWSR1 (Kd = 28.6 ± 0.8 nM, Supplementary Figure S2C). SA2 binds to both ssDNA and dsDNA in a length-dependent manner (15). Based on our previous studies (15), we expect that SA2 does not bind tightly to each 30-bp dsDNA arm. We predict that its binding affinity for the 31-nt ssDNA loop region alone is also relatively weak (for 45-ssDNA: Kd = 117.6 ± 5.3 nM; for 25-ssDNA: Kd = 445.2 ± 11.9 nM) (15). Consequently, we expect that SA1 and SA2 binding to the model R-loop substrate is mediated predominantly through their binding to the 25-bp RNA:DNA hybrid region, along with weaker contributions from their binding to the ssDNA loop and dsDNA arms. In comparison, EWSR1 bound to the model R-loop substrate with affinities (Kd = 28.6 ± 0.8 nM) significantly higher (P < 0.05) than its binding to either the ssRNA component (25 nt RNA, Kd = 134.2 ± 0.04 nM) or the RNA:DNA hybrid with overhangs (Kd = 94.7 ± 10.7 nM, Supplementary Figure S2C, Table 1). Collectively, these results demonstrated that cohesin SA1 and SA2 directly bind to the model R-loop substrate.
Creation of a linear DNA substrate containing R-loops at a defined region
To further establish that cohesin SA1 and SA2 recognize RNA in the context of long dsDNA, we applied AFM imaging to investigate the binding positions of SA1 and SA2 on a linear dsDNA substrate that contains R-loops at a defined region. To make linear dsDNA fragments containing RNA, we first carried out in vitro transcription using T3 RNA polymerase and a template containing the Airn gene cloned downstream of the T3 promoter. Recently, the same pFC53-Airn DNA template was used to demonstrate the R-loop formation after in vitro transcription (31). This previous study established that Airn contains two R-loop forming sites, and the one overlaps the G-stretch form more stable R-loops. We prepared the R-loop DNA substrate by treatment with RNase A after transcription. In parallel, we generated the negative control DNA without R-loops by treatment with both RNase A and RNase H. We first validated the R-loop formation using electrophoresis, which showed that DNA with R-loops after treatment with RNase A displayed slower mobility compared with the negative control DNA. To generate linear DNA substrates for AFM imaging, the circular DNA that contained the R-loop and the negative control DNA substrate were linearized with ApaLI. ApaLI restriction digestion generated a small (1246 bp) and a large linear DNA fragment (2745 bp) that contained the Airn sequences. The distinct lengths of these two fragments (375.1 ± 21.9 nm and 787.6 ± 34.6 nm; mean±SD) enabled us to unambiguously identify them in AFM images. On the large fragment, the G-rich sequences with consecutive Gs (14G and 12G) within the Airn sequences, where R-loops are expected to form, are positioned at 38% and 42%, respectively, from the closest DNA end.
We applied AFM imaging of samples on mica surfaces to characterize the formation of R-loop structures over the Airn template (31). Consistent with that previous study, we observed different R-loop shapes (N = 382), including blobs (40%), spurs (U-shaped, 32%), and loops (28%), formed on DNA after in vitro transcription (Figure 5A and Supplementary Figure S3). Due to the uncertainty in determining positions of R-loop objects with spur and loop shapes along the dsDNA, we focused our analysis on the long linear DNA fragments containing R-loop objects with blob shapes. These DNA fragments showed similar DNA contour lengths (767.1 ± 51.2 nm; mean ± SD), as compared to the negative control without R-loops (787.6 ± 34.6 nm; mean ± SD). To detect R-loops on linear DNA fragments in AFM images, we measured the height of the tallest blob feature (maximum AFM height) along the contour of individual large DNA fragments. In AFM images, dsDNA molecules (N = 200) without R-loops showed maximum AFM heights of 0.34 (±0.09) nm, which were consistent with previous studies (34). AFM imaging revealed that the large linear fragments after transcription and RNase treatment contained features (white arrow, Figure 5A) with heights greater than dsDNA alone. Based on the height of dsDNA only (0.34 ± 0.09 nm), we used 0.5 nm as the cutoff to identify R-loop structures. Based on this selection criterion, ∼43% of the long fragments contained blob features with maximum AFM heights (0.86 ± 0.20 nm) that were significantly greater than dsDNA alone (Supplementary Table S2). These features with greater AFM heights were positioned at 40.0% (±7.5%) from one DNA end (Figure 5D), which were consistent with R-loop formation over the G-rich regions (14G and 12G) on the large linear fragments. It is worth noting that AFM height measurements might underestimate the percentage of DNA fragments containing R-loops. R-loop structures containing shorter stretches of RNA would not be differentiated from dsDNA alone. Importantly, the negative control DNA sample after treatment with both RNase A and RNase H displayed AFM heights (0.49 ± 0.18 nm, N = 100) that were not significantly different from those measured on dsDNA alone. Furthermore, incubation of the R-loop DNA fragments with the S9.6 antibody led to features with heights greater than the R-loop structures (Figure 5B). Based on the height of R-loop structures (0.86 ± 0.20 nm), we used 1.0 nm as the cutoff for identifying R-loops bound by the S9.6 antibody. Based on this selection criterion, after incubating with the S9.6 antibody, ∼29% of the DNA molecules displayed features with maximum AFM heights (2.34 ± 0.85 nm) that were significantly (P < 0.05) greater than R-loop alone (Supplementary Table S2). These features with greater AFM heights were positioned at 37.5% (±7.5%) from one end of the DNA (Figure 5D), consistent with expected locations of the R-loops on the large DNA fragments. In contrast, the S9.6 antibody was randomly distributed on the control dsDNA without R-loops (Figure 5C and E). Collectively, these data from AFM imaging directly validated the formation of R-loops through in vitro transcription over the G-rich regions on the dsDNA template.
Figure 5.

AFM imaging validates the presence of R-loops on the long linear DNA. (A–C) Cartoon drawings of the DNA substrate and S9.6 antibody (top panels), and AFM images of the long linear DNA containing the R-loop (white arrow, R-loop DNA), the short fragment without R-loops (green arrow, A), the R-loop DNA with S9.6 antibody (white arrows, B), and control dsDNA with the S9.6 antibody (white arrow, C). The letter H denotes the maximum AFM height along the linear dsDNA (mean ± SD). (D and E) Position distributions of R-loop structures (N = 200) and R-loop-S9.6 antibody complexes (N = 218) along the linear R-loop DNA (D), and S9.6 antibody along the control dsDNA without R-loops (N = 108, E). Positions were measured from the closest DNA ends. Based on the AFM heights of R-loop (0.86 ± 0.20 nm), structures on the R-loop DNA with AFM heights greater than 1.0 nm were selected as R-loop-S9.6 complexes (Supplementary Table S2). The lines in (D) are Gaussian fits to the data (R2>0.8), with peaks centered at 40.0% (± 7.5%) and 37.5% (±7.5%), respectively, for R-loops and R-loop-S9.6 antiboy complexes.
SA1 and SA2 directly bind to regions containing R-loops on dsDNA substrates
Next, to test whether or not SA1 and SA2 bind to regions containing R-loops on the dsDNA substrate, we applied AFM imaging of the R-loop DNA or negative control dsDNA in the presence of either SA1 or SA2. Similar to what was observed in the presence of the S9.6 antibody, upon incubation of the full-length SA1 or SA2 (170 nM) with the R-loop DNA substrate (7.6 nM), structures with heights greater than the R-loop alone were observed in AFM images (white arrow, Figure 6A and B). For SA1 and SA2, respectively, ∼16% and 24% of the large DNA fragments contained these features (SA1: 1.9 ± 2.7 nm; SA2: 2.9 ± 2.1 nm) with greater AFM heights than R-loop structures alone. Furthermore, for SA1 and SA2, respectively, these complexes were positioned at 39.1% (±8.5%) and 40.4% (±7.8%), from the closest DNA end (Figure 6D). These locations were consistent with SA1 and SA2 binding to regions containing R-loops on the large DNA fragments. In stark contrast, SA1 and SA2 were randomly distributed along the dsDNA without R-loops (Figure 6E). Furthermore, we prepared an additional negative control DNA substrate (N-R-loop DNA) by first generating the R-loop DNA, followed by incubation with both RNase A and RNase H to remove RNA. The distributions of SA1 and SA2 on the N-R-loop DNA were not statistically different from what was observed using the dsDNA control substrate (compare Figure 6E and F).
Figure 6.

Cohesin SA1 and SA2, as well as EWSR1, localize to regions containing R-loops on dsDNA. (A–C) AFM images of the R-loop DNA in the presence of SA1 (A), SA2 (B) or EWSR1 (C) (white arrows). (D–F) The position distributions of SA1, SA2, and EWSR1 on the linear R-loop DNA (D) and control dsDNA (E) and negative control N-R-loop DNA (F). The lines in (D) are Gaussian fits to the data (R2> 0.83), with peaks centered at 39.1% (±8.5%) for SA1, 40.4%(±7.8%) for SA2, and 39.8% (±7.8%) for EWSR1. N = 200 for each data set from at least three independent experiments (Supplementary Table S2). Structures on the R-loop DNA with AFM heights greater than 1.0 nm were selected as R-loop-protein complexes (Supplementary Table S2). (G) AFM volumes of R-loops alone on dsDNA, SA1-R-loop, SA2-R-loop, EWSR1-R-loop, SA1-dsDNA, SA2-dsDNA and EWSR1-dsDNA. N = 50 for each data set. The Box–Whisker plots show 25–75%, the median, and range within 1.5 IQR. *** P < 0.001 based on the Mann–Whitney test.
The AFM volumes of SA1- and SA2-R-loop complexes were heterogeneous (Figure 6G, SA1: 1751 ± 231 nm3; SA2: 1267 ± 173 nm3; N = 50), and approximately 6× greater than the volume of R-loops alone (201 ± 20 nm3, N = 50). Furthermore, the AFM volumes of SA1 on the substrate containing R-loops were significantly (P < 0.001) greater than those on the control dsDNA without R-loops (Figure 6G). Combined with the information on the AFM volume of SA2 alone measured previously (146 nm3) (15), these results from AFM imaging indicated that there were multiple copies of SA1 and SA2 molecules bound at each R-loop region.
To further validate the binding of SA1 and SA2 to R-loop regions on dsDNA, we directly compare their binding to the R-loop DNA to that of EWSR1. Upon incubation of EWSR1 (170 nM) with the R-loop DNA substrate (7.6 nM), structures with maximum AFM heights significantly (P < 0.001) higher than those measured for R-loops alone were observed (Figure 6C). In addition, EWSR1 on dsDNA with R-loops displayed significantly (P < 0.001) higher AFM volumes than that on dsDNA without R-loops (Figure 6G). These results suggested that EWSR1 formed distinct complexes at the R-loop regions on the dsDNA. Furthermore, measurement of the maximum AFM height along each DNA fragment in the presence of EWSR1 revealed that ∼12% of the long DNA fragments contained features with greater AFM heights (1.5 ± 0.5 nm) (Supplementary Table S2). These features unique to the R-loop DNA fragments and EWSR1 incubations were positioned at 39.8% (±7.8%, Figure 6D) from the closest DNA end. In stark contrast, EWSR1 was randomly distributed over the control dsDNA fragment without R-loops and the N-R-loop substrate (Figure 6E and F). In Summary, AFM imaging established that both SA1 and SA2, as well as EWSR1, bind specifically to regions containing R-loops along dsDNA.
SA1 and SA2 localize to regions containing RNA on DNA tightropes
To understand how SA1 and SA2 dynamically bind to RNA on dsDNA in solution, we applied the DNA tightrope assay. In this assay, oblique angle fluorescence microscopy is used to track QD-labeled proteins on DNA anchored between micron-sized silica beads (Figure 7A) (16,17,38,39). In the DNA tightrope assay setup, DNA is stretched under hydrodynamic flow. A DNA tightrope forms when a DNA molecule anchored to one poly-l-lysine coated micro-sized bead stretches out under buffer flow and attaches to a second bead (Figure 7A). For tracking of protein binding dynamics on DNA tightropes, His-tagged proteins can be conjugated to streptavidin-coated QDs through a biotinylated multivalent chelator tris-nitrilotriacetic acid (BTtris-NTA) linker (Figure 7B). Meanwhile, proteins with an epitope tag can be conjugated to antibody-coated QDs (14,40). Specifically, to study sequence- or structure-specific DNA binding, we ligated linear DNA fragments to form DNA tightropes with specific DNA sequences or structures spaced at defined distances. Previously, we also established that under the conditions used in this study, DNA molecules are stretched to ∼90% of their contour lengths. The total lengths of the DNA tightropes formed using ligated DNA are in the range of ∼2.1–22 μm (17). In this study, we created DNA tightropes containing R-loops (Figure 7C) by first ligating linear dsDNA fragments without R-loops, followed by in vitro transcription using T3 polymerase and RNase A treatment. To monitor the recruitment of SA1 and SA2 at R-loops, we conjugated His-tagged SA1 or SA2 to the streptavidin-coated QD (SA1- and SA2-QD, Figure 7B). Previously, we established that SA1 and SA2 conjugated to QDs using this strategy still retain their binding specificities for telomeric DNA and ssDNA gaps, respectively (14,15).
Figure 7.

SA1 and SA2 localize to regions containing RNA on DNA tightropes. (A) Schematics of the DNA tightrope assay showing QD-labeled proteins (red) loading onto DNA (green) anchored between micron-sized beads. (B) QD conjugation strategy: His-NTA-biotin/streptavidin-QD sandwich method for His-tagged SA1 and SA2 proteins. (C) Schematics of ligated DNA tightropes containing R-loops formed through in vitro transcription. (D and E) Images (top panels) and kymographs (bottom panels) of red (655 nm) QD-labeled SA1 (D) and SA2 (E) on the ligated DNA tightropes containing R-loops (left panels) and control DNA tightropes (middle panels), and measurements of the distance between adjacent protein pairs on DNA tightropes (right panels). The dotted white lines mark the contour of the beads. SA1: N = 136 pairs on the control DNA; N = 175 pairs on the R-loop DNA. SA2: N = 116 pairs on the control DNA; N = 257 pairs on the R-loop DNA. Length scale bar: 5 μm; Time scale bar: 1 s.
Previously, we applied the DNA tightrope assay to study sequence-specific binding of telomere proteins, namely TRF1 and TRF2, to telomeric sequences (14,17). In those studies, TRF1 and TRF2 displayed regular spacing between adjacent protein pairs that were consistent with the distance between telomeric regions on DNA tightropes. The distance between adjacent R-loop regions on the ligated R-loop DNA is expected to be 0.6, 1 or 1.4 μm, depending on different end-to-end configurations during ligation of the linear DNA fragments (3.99 kb). These numbers correspond to 0.5, 0.9 and 1.2 μm spacing on the DNA tightropes, taking into consideration that DNA molecules are stretched to ∼90% of their original contour lengths between beads. Based on our previous studies (17), for SA1 and SA2, we reasoned that in comparison to their binding to the negative control DNA tightropes, a higher density of protein binding and defined spacing between adjacent protein pairs on the R-loop DNA tightropes would confirm specific binding of SA1 and SA2 to regions containing RNA. We introduced QD-labeled EWSR1, SA1 or SA2 (5 nM final concentration) into the flow cell that contained either control DNA tightropes or DNA tightropes with R-loops. The FLAG-tagged EWSR1 was conjugated to antibody-coated QDs (Supplementary Figure S4A). EWSR1 bound the R-loop DNA tightropes with high densities (4.0 protein complexes per 10 μm of DNA tightropes, including both static and mobile complexes) and defined spacing (∼1.25 μm) between adjacent pairs (Supplementary Figure S4B and C). EWSR1-QDs on R-loop DNA tightropes were long-lived (Supplementary Figure S4). Greater than 90% of EWSR1 (N = 286) molecules stayed on the R-loop DNA tightropes at the end of the observational time windows (2 min). In comparison, on the DNA tightropes without R-loops, the density of EWSR1 was significantly (P < 0.001) lower (1 protein complex per 10 μm length of DNA tightropes).
SA1 and SA2 on both control and R-loop DNA tightropes were also long-lived (Figure 7D and E). Greater than 90% of SA1 (N = 1326) and 75% of SA2 (N = 2302) molecules stayed on the DNA tightropes at the end of the observational time windows (2 min). The majority of the SA1 and SA2 complexes (>70%) on the R-loop DNA substrates were static. The densities of SA1 and SA2 (including both static and mobile complexes) on the R-loop DNA tightropes were 6.8 and 7.0 complexes per 10 μm of DNA tightropes, respectively. These binding densities were significantly (P < 0.001) higher than the ones observed on control DNA tightropes without R-loops (SA1: 3.9 protein complexes per 10 μm of DNA tightropes; SA2: 2.4 protein complexes per 10 μm of DNA tightropes). The spacing between adjacent QD-labeled SA1/SA2 proteins was measured based on the distance between intensity peaks (Supplementary Figure S5). The histograms of the spacing between adjacent static SA1 or SA2 protein pairs on R-loop DNA tightropes displayed peaks at ∼1.25 μm. In stark contrast, their distributions on the control DNA tightropes without R-loops did not show any defined peak (Figure 7D and E), and significantly (P < 0.001) different from what was observed on R-loop DNA tightropes. Considering an average spacing of 1.25 μm between adjacent R-loops regions on DNA tightropes, if transcription started from every T3 promoter site, eight R-loop regions are expected for every 10 μm of DNA tightropes. An occupation of ∼4 SA1 or SA2 molecules per 10 μm of R-loop DNA tightropes (after deducting the number from nonspecific binding) suggested that these proteins occupied approximately half of the R-loop sites on DNA tightropes. In summary, these results from the DNA tightrope assay directly showed that SA1 and SA2 specifically localize to regions containing RNA on dsDNA.
SA1 and SA2 binding sites and locations of R-loops overlap significantly in vivo
The discovery of high-affinity RNA binding by SA1 and SA2 raised important questions regarding whether or not SA1 and SA2 binding positions colocalize with R-loop sites in cells. To directly address this question, we mined publicly available ChIP-Seq data for SA1, SA2, SMC1, SMC3 and CTCF from HMEC, HCAEC, HeLa and MCF10A cells (Supplementary Table S3), and DRIP-Seq data (110 samples, Supplementary Table S4) as a basis for R-loop locations in the genome. Raw data from these studies were re-processed with a standardized bioinformatics pipeline (Supplementary Materials and Methods). Peak pileup was calculated and visualized around the transcription start site (TSS) for each cell line using the R package ChIPseeker (Figure 8A) (41). Consistent with previous observations, cohesin subunits (SA1/SA2, SMC3) and CTCF were enriched at regions close to promoters (Supplementary Figure S6).
Figure 8.
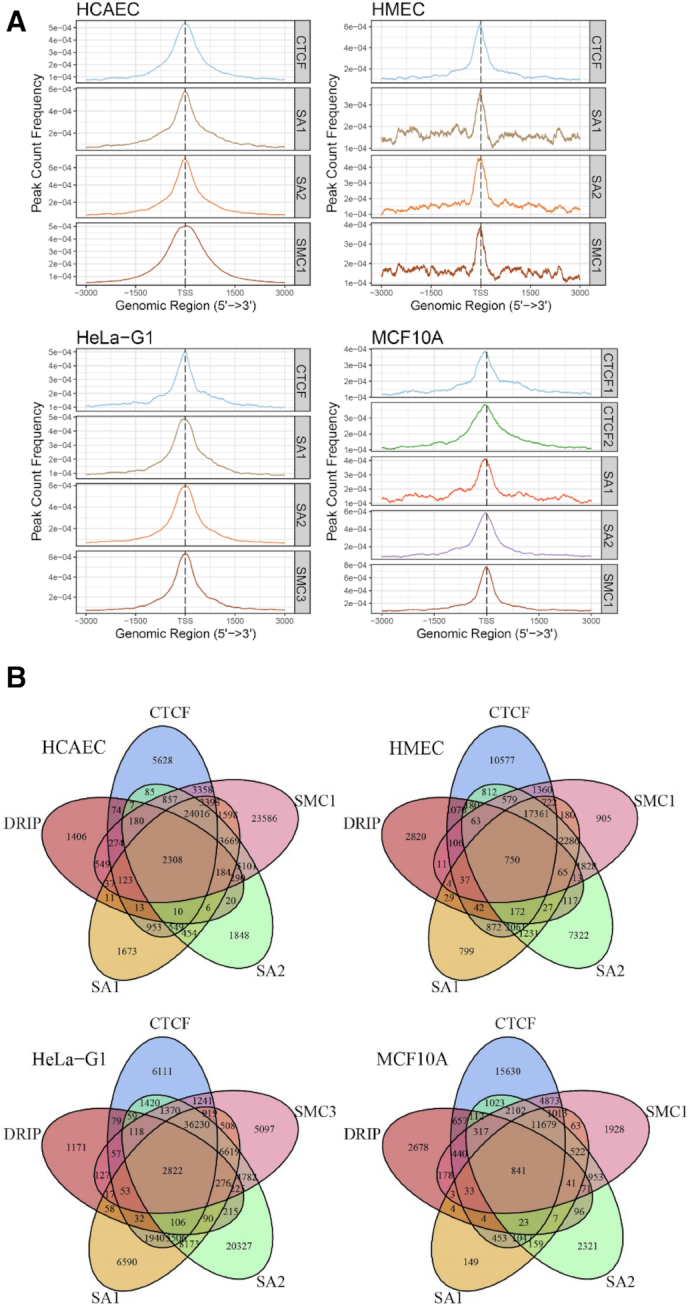
SA1 and SA2 binding sites in vivo overlap significantly with R-loops. (A) ChIP-Seq pileup for CTCF, SA1, SA2, SMC1/SMC3 for four different cell lines (HCAEC, HMEC HeLa, and MCF10A). Peak pileup was calculated and visualized around the TSS for each cell line using the R package ChIPseeker. (B) Venn diagrams showing the overlap of SA1/SA2, cohesin SMC subunit (SMC1 or 3), CTCF and R-loop sites identified through DRIP-seq.
To ensure a high-quality DRIP-Seq analysis, we performed sample pre-filtering using a multi-step principal component analysis (PCA) approach (Supplementary Figure S7). Furthermore, consensus site analysis was performed to determine what proportion of DRIP peaks are conserved across what proportion of samples (Supplementary Figure S8). We then compared SA1 and SA2 binding locations to conserved DRIP sites (5575 peaks). Through this analysis, we discovered that depending on the cell line, ∼20–50% of DRIP peaks overlap with SA1 and SA2 peaks (Figure 8B). We conducted further tests to determine the genomic features where SA1/SA2 colocalize with R-loops. Specifically, SA1 and SA2 peaks that either overlapped with DRIP-peaks or did not overlap with DRIP-peaks were further analyzed for their enrichment with various genomic features (Supplementary Figure S9). From this analysis we noted that SA1 and SA2 peaks that overlap with DRIP peaks tended to be in promoter regions (Supplementary Figure S9) and appeared to be enriched for positions near the TSS. This conclusion was confirmed by analyzing the distance-to-nearest-TSS for every peak in these groups (Figure 9A).
Figure 9.

R-loop-localized SA1 and SA2 are enriched for positions near the TSS and typically in the cohesin complex. (A) The distance to nearest TSS for SA1, SA2, SA1 + SA2 (common), SA1/SA2 overlapping with R-loops (SA1 + SA2 ol w/DRIP), or without overlapping with DRIP-peaks (SA1 + SA2 no ol w/DRIP). **** P ≤ 0.0001 based on Wilcoxon rank sum test. (B) Venn diagrams showing the overlap of the R-loop-localized SA1/SA2 (e.g. SA1_ol_DRIP and SA2_ol_DRIP) with the SMC subunit (SMC1 or SMC3) and CTCF sites.
Considering that our in vitro work examined SA1 or SA2 in isolation, it is possible that their binding to R-loop locations may be independent of the cohesin complex. To address this question, we went on to ask whether R-loop-localized SA1/SA2 also colocalized with other members of the cohesin complex, namely the SMC subunit (SMC1 or SMC3) and CTCF (Figure 9B). The results from this analysis demonstrated that R-loop-localized SA1 and SA2 do colocalize with these other cohesin components in the vast majority of cases suggesting R-loop binding involves the entire cohesin complex. In summary, these data indicate that SA1 and SA2 binding sites overlap significantly with R-loops, and this overlap tends to occur nearer to the promoter region.
DISCUSSION
From recent studies, an emerging view of the cohesin and condensin complexes is that both SMC subunits and non-SMC subunits contribute to DNA binding (4). Supporting this notion, recently, we established that both cohesin SA1 and SA2 directly bind to ssDNA and dsDNA (14,15). In this study, using bulk fluorescence anisotropy, single-molecule AFM and fluorescence imaging, we discovered that both cohesin SA1 and SA2 bind to a variety of RNA containing nucleic acid substrates, which include ssRNA, dsRNA, dsRNA with an overhang, and RNA:DNA hybrids. Furthermore, cohesin SA1 and SA2 are capable of binding to long ssRNA transcripts that form secondary structures. Strikingly, compared to our previous studies of cohesin SA1 and SA2 binding to DNA (14,15), under the same experimental conditions, both cohesin SA1 and SA2 bind to ssRNA with higher affinities than to ssDNA of the same lengths.
To further investigate the binding of cohesin SA1 and SA2 to RNA in the context of long dsDNA that mimic in vivo conditions, we generated dsDNA substrates containing RNA through in vitro transcription using T3 RNA polymerase. Subsequent degradation of unpaired ssRNA with RNase A led to substrates containing R-loops at defined locations along the dsDNA. The presence of R-loops on DNA was validated using the S9.6 antibody. AFM imaging showed that both cohesin SA1 and SA2 specifically localize to the regions containing R-loops. Consistent with results from AFM imaging, on the DNA tightropes containing R-loops, both cohesin SA1 and SA2 stably attached to R-loop regions regularly spaced along the DNA. It is worth noting that for the substrates used for AFM and fluorescence imaging, due to the heterogeneity of the RNA transcripts, it is challenging to quantify the extent of RNA digestion by RNase A. Furthermore, AFM and fluorescence imaging do not provide the resolution to determine which parts of the R-loop region that cohesin SA1 and SA2 directly bind to. However, our in-depth analysis using bulk fluorescence anisotropy and a comprehensive panel of substrates containing RNA provided strong support for cohesin SA1 and SA2 direct binding to the R-loop structures. Fluorescence anisotropy experiments showed that cohesin SA1 and SA2 bind to components of the R-loops (the RNA:DNA hybrid and ssDNA), as well as the model R-loop substrates. It is possible that on the RNA-containing DNA substrates used for AFM imaging and the DNA tightrope assay, cohesin SA1 and SA2 were capable of binding to different nucleic acid structures at the R-loop regions. These structures include RNA:DNA hybrids, displaced ssDNA, and left-over ssRNA that were not degraded by RNase A.
To further validate whether or not SA1 and SA2 bind to RNA in vivo, we analyzed the overlapping frequency between SA1/SA2 binding sites extracted from the ChIP-seq data sets and R-loop sites mined from the DRIP-seq data sets. Our analysis showed that depending on cell lines, ∼20–50% DRIP peaks overlap with SA1 and SA2 peaks. Among the subset of sites where SA1/SA2 co-localize with R-loops, the cohesin SMC subunit (SMC1 or SMC3) and CTCF are also found. One explanation for this result is that the whole complex binds at these R-loop sites rather than just SA1 or SA2. However, current DRIP data do not include information on R-loop dynamics. Separate events of R-loop-dependent recruitment of SA1/SA2 alone and loading of the core cohesin complex to the same site without R-loops could also explain the colocalization of SA1/SA2 with R-loops. Further characterization of cohesin binding in vitro and in vivo are needed to refine the model of cohesin loading at R-loop sites.
Enhancer–promoter interactions via chromatin looping are essential for the regulation of gene expression (20). Since enhancers can be transcribed into RNAs (eRNA), it has been speculated that RNA might mediate looping interactions between enhancers and promoters (20). Consistent with previous results (6), our analysis revealed that there is a substantial subset of promoter sites, where R-loops and SA1/SA2 colocalize. Furthermore, a previous study showed that a high percentage of SA2-only positions (77%) were in enhancers. Our discovery strongly suggests that RNA binding activities of SA1 and SA2 might drive the localization of SA1 or SA2 together with other core cohesin subunits to promoters and perhaps to enhancers. This notion fits with a model in which eRNAs in conjunction with boundary proteins, including CTCF, facilitate the stalling of loop-exclusion by cohesin (6,42). The RNA binding affinity that SA1 and SA2 display in vitro are comparable (Table 1). Higher occupancies of SA2 in comparison to SA1 at enhancer sites observed in vivo might be due to the difference in protein concentrations inside cells; SA2 is more abundant than SA1 (43). Judging by immunoprecipitation data, in HeLa cell extracts, the ratio of SA1 and SA2 is ∼1:3. In addition, SA1 preferentially binds to telomeric DNA sequences through its N-terminal AT-hook domain (14). Binding of SA1 to telomeric and AT-rich sequences could potentially titrate it away from RNA.
Our findings of RNA binding by SA1 and SA2 raise other tantalizing possibilities regarding the mechanisms underlying diverse cellular functions of cohesin SA1 and SA2. The cohesin complex suppresses the joining of distant double-strand DNA ends (44). Postreplicative DSB-recruited cohesin is capable of establishing sister chromatid cohesion (45). It was proposed that binding of cohesin stabilizes DNA ends and limits its mobility to suppress spurious bridging of two distal DNA ends (44). What structure feature secures the cohesin ring at DNA ends is largely unknown. Recent studies demonstrated that transcription can initiate from damage-induced DSBs (46). Our discovery of high-affinity binding of SA1 and SA2 to RNA suggests a new exciting possibility that these proteins could be recruited to DSBs through its RNA binding activities. Future experiments are needed to define the scope of SA1 and SA2 RNA binding activity in a cellular environment.
In summary, our study established that cohesin SA1 and SA2 are versatile nucleic acid binding proteins that bind to both DNA and RNA. We propose that DNA and RNA binding activities of these proteins enable them to efficiently search along the dsDNA and locate regions containing RNA in the genome. Such activities of cohesin SA1 and SA2 likely contribute to their binding specificity for the R-loop regions along the dsDNA. These results open up new directions for investigating the diverse cellular functions of cohesin SA1 and SA2.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank the Weninger and Riehn groups at NCSU for technical support, and Landon Wang at George Washington University for assistance in statistical analysis.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R01GM107559 to H.W., R.R., R01GM123246 to H.W., R.R., Y.J.T., P30 ES025128 through a Pilot Project Grant to H.W. through Center for Human Health and the Environment at NCSU, R01CA152063 and 1R01CA241554 to A.J.R.B., and NCI T32 postdoctoral training grant (T32CA148724) and NCATS TL1 (TL1TR002647) to A.G.]; Caner Prevention & Research Institute of Texas [CPRIT RP150445 to A.J.R.B.]; Welch Foundation [C-1565 to Y.J.T.]; American Association for Cancer Research-AstraZeneca [Stimulating Therapeutic Advancer through Research Training grant (18-40-12-GORT) to A.G.]. Funding for open access charge: National Institutes of Health [R01GM123246].
Conflict of interest statement. None declared.
REFERENCES
- 1. Onn I., Heidinger-Pauli J.M., Guacci V., Unal E., Koshland D.E.. Sister chromatid cohesion: a simple concept with a complex reality. Annu. Rev. Cell Dev. Biol. 2008; 24:105–129. [DOI] [PubMed] [Google Scholar]
- 2. Bose T., Gerton J.L.. Cohesinopathies, gene expression, and chromatin organization. J. Cell Biol. 2010; 189:201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nasmyth K., Haering C.H.. Cohesin: its roles and mechanisms. Annu. Rev. Genet. 2009; 43:525–558. [DOI] [PubMed] [Google Scholar]
- 4. Hassler M., Shaltiel I.A., Haering C.H.. Towards a unified model of SMC complex function. Curr. Biol. 2018; 28:R1266–R1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Litwin I., Pilarczyk E., Wysocki R.. The emerging role of cohesin in the DNA damage response. Genes. 2018; 9:581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kojic A., Cuadrado A., De Koninck M., Gimenez-Llorente D., Rodriguez-Corsino M., Gomez-Lopez G., Le Dily F., Marti-Renom M.A., Losada A.. Distinct roles of cohesin-SA1 and cohesin-SA2 in 3D chromosome organization. Nat. Struct. Mol. Biol. 2018; 25:496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Solomon D.A., Kim J.S., Waldman T.. Cohesin gene mutations in tumorigenesis: from discovery to clinical significance. BMB reports. 2014; 47:299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Watrin E., Kaiser F.J., Wendt K.S.. Gene regulation and chromatin organization: relevance of cohesin mutations to human disease. Curr. Opin. Genet. Dev. 2016; 37:59–66. [DOI] [PubMed] [Google Scholar]
- 9. Lawrence M.S., Stojanov P., Mermel C.H., Robinson J.T., Garraway L.A., Golub T.R., Meyerson M., Gabriel S.B., Lander E.S., Getz G.. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014; 505:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hill V.K., Kim J.S., Waldman T.. Cohesin mutations in human cancer. Biochim. Biophys. Acta. 2016; 1866:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Romero-Perez L., Surdez D., Brunet E., Delattre O., Grunewald T.G.P.. STAG mutations in cancer. Trends Cancer. 2019; 5:506–520. [DOI] [PubMed] [Google Scholar]
- 12. Mondal G., Stevers M., Goode B., Ashworth A., Solomon D.A.. A requirement for STAG2 in replication fork progression creates a targetable synthetic lethality in cohesin-mutant cancers. Nat. Commun. 2019; 10:1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van der Lelij P., Lieb S., Jude J., Wutz G., Santos C.P., Falkenberg K., Schlattl A., Ban J., Schwentner R., Hoffmann T. et al.. Synthetic lethality between the cohesin subunits STAG1 and STAG2 in diverse cancer contexts. Elife. 2017; 6:e26980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin J., Countryman P., Chen H., Pan H., Fan Y., Jiang Y., Kaur P., Miao W., Gurgel G., You C. et al.. Functional interplay between SA1 and TRF1 in telomeric DNA binding and DNA-DNA pairing. Nucleic Acids Res. 2016; 44:6363–6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Countryman P., Fan Y., Gorthi A., Pan H., Strickland J., Kaur P., Wang X., Lin J., Lei X., White C. et al.. Cohesin SA2 is a sequence-independent DNA-binding protein that recognizes DNA replication and repair intermediates. J. Biol. Chem. 2018; 293:1054–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kad N.M., Wang H., Kennedy G.G., Warshaw D.M., Van Houten B.. Collaborative dynamic DNA scanning by nucleotide excision repair proteins investigated by single- molecule imaging of quantum-dot-labeled proteins. Mol. Cell. 2010; 37:702–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin J., Countryman P., Buncher N., Kaur P., E L., Zhang Y., Gibson G., You C., Watkins S.C., Piehler J. et al.. TRF1 and TRF2 use different mechanisms to find telomeric DNA but share a novel mechanism to search for protein partners at telomeres. Nucleic Acids Res. 2014; 42:2493–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meisenberg C., Pinder S.I., Hopkins S.R., Wooller S.K., Benstead-Hume G., Pearl F.M.G., Jeggo P.A., Downs J.A.. Repression of transcription at DNA breaks requires cohesin throughout interphase and prevents genome instability. Mol. Cell. 2019; 73:212–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cuadrado A., Gimenez-Llorente D., Kojic A., Rodriguez-Corsino M., Cuartero Y., Martin-Serrano G., Gomez-Lopez G., Marti-Renom M.A., Losada A.. Specific contributions of cohesin-SA1 and Cohesin-SA2 to TADs and polycomb domains in embryonic stem cells. Cell Rep. 2019; 27:3500–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Lara J.C., Arzate-Mejia R.G., Recillas-Targa F.. Enhancer RNAs: Insights Into Their Biological Role. Epigenet. Insights. 2019; 12:2516865719846093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Santos-Pereira J.M., Aguilera A.. R loops: new modulators of genome dynamics and function. Nat. Rev. Genet. 2015; 16:583–597. [DOI] [PubMed] [Google Scholar]
- 22. Chedin F. Nascent connections: R-loops and chromatin patterning. Trends Genet. 2016; 32:828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pezic D., Weeks S.L., Hadjur S.. More to cohesin than meets the eye: complex diversity for fine-tuning of function. Curr. Opin. Genet. Dev. 2017; 43:93–100. [DOI] [PubMed] [Google Scholar]
- 24. Sanz L.A., Hartono S.R., Lim Y.W., Steyaert S., Rajpurkar A., Ginno P.A., Xu X., Chedin F.. Prevalent, dynamic, and conserved R-loop structures associate with specific epigenomic signatures in mammals. Mol. Cell. 2016; 63:167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aguilera A., Garcia-Muse T.. R loops: from transcription byproducts to threats to genome stability. Mol. Cell. 2012; 46:115–124. [DOI] [PubMed] [Google Scholar]
- 26. Thomas M., White R.L., Davis R.W.. Hybridization of RNA to double-stranded DNA: formation of R-loops. PNAS. 1976; 73:2294–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garcia-Muse T., Aguilera A.. R loops: from physiological to pathological roles. Cell. 2019; 179:604–618. [DOI] [PubMed] [Google Scholar]
- 28. Kato R., Miyagawa K., Yasuhara T.. The role of R-loops in transcription-associated DNA double-strand break repair. Mol Cell Oncol. 2019; 6:1542244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ohno T., Ouchida M., Lee L., Gatalica Z., Rao V.N., Reddy E.S.. The EWS gene, involved in Ewing family of tumors, malignant melanoma of soft parts and desmoplastic small round cell tumors, codes for an RNA binding protein with novel regulatory domains. Oncogene. 1994; 9:3087–3097. [PubMed] [Google Scholar]
- 30. Zhang N., Jiang Y., Mao Q., Demeler B., Tao Y.J., Pati D.. Characterization of the interaction between the cohesin subunits Rad21 and SA1/2. PLoS One. 2013; 8:e69458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Carrasco-Salas Y., Malapert A., Sulthana S., Molcrette B., Chazot-Franguiadakis L., Bernard P., Chedin F., Faivre-Moskalenko C., Vanoosthuyse V.. The extruded non-template strand determines the architecture of R-loops. Nucleic Acids Res. 2019; 47:6783–6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yu K., Roy D., Huang F.T., Lieber M.R.. Detection and structural analysis of R-loops. Methods Enzymol. 2006; 409:316–329. [DOI] [PubMed] [Google Scholar]
- 33. Nguyen H.D., Yadav T., Giri S., Saez B., Graubert T.A., Zou L.. Functions of replication protein a as a sensor of R loops and a regulator of RNaseH1. Mol. Cell. 2017; 65:832–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kaur P., Wu D., Lin J., Countryman P., Bradford K.C., Erie D.A., Riehn R., Opresko P.L., Wang H.. Enhanced electrostatic force microscopy reveals higher-order DNA looping mediated by the telomeric protein TRF2. Sci. Rep. 2016; 6:20513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lata S., Piehler J.. Stable and functional immobilization of histidine-tagged proteins via multivalent chelator headgroups on a molecular poly(ethylene glycol) brush. Anal. Chem. 2005; 77:1096–1105. [DOI] [PubMed] [Google Scholar]
- 36. Reichel A., Schaible D., Al Furoukh N., Cohen M., Schreiber G., Piehler J.. Noncovalent, site-specific biotinylation of histidine-tagged proteins. Anal. Chem. 2007; 79:8590–8600. [DOI] [PubMed] [Google Scholar]
- 37. Boguslawski S.J., Smith D.E., Michalak M.A., Mickelson K.E., Yehle C.O., Patterson W.L., Carrico R.J.. Characterization of monoclonal antibody to DNA.RNA and its application to immunodetection of hybrids. J. Immunol. Methods. 1986; 89:123–130. [DOI] [PubMed] [Google Scholar]
- 38. Dunn A.R., Kad N.M., Nelson S.R., Warshaw D.M., Wallace S.S.. Single Qdot-labeled glycosylase molecules use a wedge amino acid to probe for lesions while scanning along DNA. Nucleic Acids Res. 2011; 39:7487–7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hughes C.D., Wang H., Ghodke H., Simons M., Towheed A., Peng Y., Van Houten B., Kad N.M.. Real-time single-molecule imaging reveals a direct interaction between UvrC and UvrB on DNA tightropes. Nucleic Acids Res. 2013; 41:4901–4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang H., Tessmer I., Croteau D.L., Erie D.A., Van Houten B.. Functional characterization and atomic force microscopy of a DNA repair protein conjugated to a quantum dot. Nano Lett. 2008; 8:1631–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu G., Wang L.G., He Q.Y.. ChIPseeker: an R/bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015; 31:2382–2383. [DOI] [PubMed] [Google Scholar]
- 42. Fudenberg G., Imakaev M., Lu C., Goloborodko A., Abdennur N., Mirny L.A.. Formation of chromosomal domains by loop extrusion. Cell Rep. 2016; 15:2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Losada A., Yokochi T., Kobayashi R., Hirano T.. Identification and characterization of SA/Scc3p subunits in the Xenopus and human cohesin complexes. J. Cell Biol. 2000; 150:405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gelot C., Guirouilh-Barbat J., Le Guen T., Dardillac E., Chailleux C., Canitrot Y., Lopez B.S.. The cohesin complex prevents the end joining of distant DNA double-strand ends. Mol. Cell. 2016; 61:15–26. [DOI] [PubMed] [Google Scholar]
- 45. Strom L., Lindroos H.B., Shirahige K., Sjogren C.. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol. Cell. 2004; 16:1003–1015. [DOI] [PubMed] [Google Scholar]
- 46. Caron P., van der Linden J., van Attikum H.. Bon voyage: a transcriptional journey around DNA breaks. DNA Repair (Amst.). 2019; 82:102686. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.