

Abstract
We report the first targeted nuclear medicine application of the lanthanum radionuclides 132/135La. These isotopes represent a matched pair for diagnosis via the positron emissions of 132La and therapy mediated by the Auger electron emissions of 135La. We identify two effective chelators, known as DO3Apic and macropa, for these radionuclides. The 18-membered macrocycle, macropa, bound 132/135La with superior molar activity than DO3Apic under similar conditions. These chelators were conjugated to the prostate-specific membrane antigen (PSMA)-targeting agent DUPA to assess the use of radiolanthanum for in vivo imaging. The 132/135La-labeled targeted constructs showed high uptake in tumor xenografts expressing PSMA. This study validates the use of these radioactive lanthanum isotopes for imaging applications and motivates future work to assess the therapeutic effects of the Auger electron emissions of 135La.
Keywords: lanthanum, radiochemistry, radiopharmaceuticals, positron emission tomography, imaging
Graphical Abstract

Radioactive metal ions, or radiometals, play a critical role in various therapeutic and diagnostic applications in nuclear medicine.[1] For example, the somatostatin receptor 2 agonist derivative DOTA-TATE, comprising the macrocyclic chelator 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetate (DOTA) and the somatostatin receptor-targeting peptide octreotate, has been recently approved for use in conjunction with the diagnostic positron (β+) emitter 68Ga and the therapeutic β– emitter 177Lu. The dual implementation of DOTA-TATE with both a diagnostic and a therapeutic radiometal has enabled exploration of theranostic platforms in the clinic. With DOTA-TATE, imaging and treatment of somatostatin receptor-positive neuroendocrine tumors is achieved with a single molecular construct and these interchangeable radiometals.[2] This example highlights how the identification of chemically similar pairs of radiometals with complementary nuclear properties is of great value for the development of new theranostic agents.[1a, 3]
Because the coordination chemistry of Ga3+ and Lu3+ are very similar, the theranostic use of DOTA-TATE has been successful clinically. The pharmacokinetic differences between the 68Ga and 177Lu conjugates are relatively minor. Moving beyond the 68Ga/177Lu pair, the use of different elements as theranostic pairs can present challenges that arise from different radiolabeling chemistries and pharmacokinetic properties. As such, there has been a large interest in the field to develop matched pairs based on the same elements.
Only a few elements have isotopes with decay properties that are appropriate for both imaging and therapy. Among the candidates, 44/47Sc, 64/67Cu, 86/90Y, and 124/131I have been most thoroughly investigated.[4] Their therapeutic effect results from the emission of β– particles. Although β– particles are effective for treating some forms of cancer and disease, the use of high linear energy transfer (LET) radiation, such as Auger electrons or alpha (α) particles, may be advantageous due to their shorter range in tissue that enables more precise targeting of diseased cells.[5] Furthermore, high LET particles cause more ionization events per distance traveled than β– particles, giving rise to a larger quantity of more lethal DNA double strand breaks. For these reasons, chemically matched theranostic radiometal pairs comprising a high LET therapeutic radionuclide are needed.
In this context, recent studies have identified the 132/135La pair to be highly promising for use in theranostic applications, but these radionuclides have not yet been implemented in targeted nuclear medicine.[6] Produced by proton irradiation of non-isotopically enriched barium targets, 135La decays by electron capture with the subsequent emission of several high LET Auger electrons. It emits 10.9 ± 3.2 electrons per decay, which on average travel less than a micron in tissue.[7] Furthermore, its relatively long half-life of 18.91(2) h makes 135La suitable for distribution to clinical sites without their own cyclotrons.[8] The diagnostic partner of 135La is 132La (t1/2 = 4.59(4) h, 42.1% β+, 76% 464.55 keV γ). This β+-emitting radionuclide is a byproduct of the proton irradiation of barium that can be used for early time-point positron emission tomography (PET) imaging to rapidly screen and quantify in vivo biodistribution of targeted constructs.[6b]
As the successful clinical translation of 68Ga/177Lu-labeled theranostic drugs demonstrates, radiolabels of different elements may also serve as theranostic synthons. La3+ is chemically similar to the therapeutic α emitter 225Ac3+, which has demonstrated clinical utility in the treatment of late-stage cancers that are refractory to traditional chemotherapy and low-LET targeted radiotherapy.[12] Because no isotopes of actinium or of its radioactive daughters possess imageable β+ emissions, 132La may act as a suitable diagnostic congener of 225Ac. Given the promising theranostic properties of the 132/135La and 132La/225Ac pairs, we sought to establish for the first time the radiolabeling conditions for radiolanthanum and its use in targeted imaging studies in vivo.
As noted above, proton irradiation of barium in natural isotopic abundance produces a mixture of the 135La and 132La theranostic matched pair.[6] For this work, irradiations were performed on barium metal pressed into homemade niobium holders that are direct-jet water cooled. The targets tolerate up to 20 μA beam intensities and produce 5.6 ± 1.1 and 0.25 ± 0.05 MBq·μA−1 of 135La and 132La, respectively, at 11.9 MeV. Following irradiation, no-carrier-added lanthanum radioisotopes were isolated from barium and other trace metal contaminants using diglycolamide extraction chromatography resin in nitric acid solution as reported previously.[6b] The final product was obtained in 0.1 M HCl and is suitable for radiolabeling of biologics with in vivo applications (see Section 1.7 in the Supporting Information, SI, for more details).
A key component required for the implementation of 132/135La in targeted imaging and therapy applications is an appropriate chelator that can rapidly bind the La3+ ion and stably retain it in vivo. Because no prior efforts to establish a suitable chelator for radiolanthanum have been undertaken, we first needed to identify a promising candidate for use with 132/135La in biological studies. Towards this aim, we evaluated the previously reported chelators DO3Apic and macropa (Figure 1),[10–11, 13] which were selected based on the high thermodynamic stability of their complexes with La3+ (log KML of 21.17 and 14.99 for DO3Apic and macropa, respectively). 132/135La radiolabeling studies were carried out using different concentrations of these two ligands; the conventional chelator DOTA was also employed for comparison. Upon incubation with the radiolanthanum ions for 30 min, complex formation was achieved for all three chelators, albeit at varying apparent molar activities (Table 1, Figures S1 and S2). At room temperature, DO3Apic and macropa gave rise to complexes with apparent maximum molar activities of 0.37 and 4.34 Ci/μmol, respectively. By contrast, DOTA required heating at 80 °C to achieve comparable apparent molar activity (0.67 Ci/μmol), indicating that this standard chelator is not ideal for use with the large La3+ ion. Our measured apparent molar activity of the La-macropa complex is significantly higher than that of DO3Apic, despite the larger thermodynamic affinity of the latter for La3+. The more effective radiolabeling of macropa in comparison to DO3Apic may be a consequence of their relative selectivities for ions of different sizes. Using the lanthanide series as an example, DO3Apic exhibits only a small change in thermodynamic affinity in traversing this series (logKML ranging from 23.46 (Eu) to 21.17 (La)).[10] By contrast, macropa has a strong thermodynamic preference for the largest lanthanide, La3+, over the smallest lanthanide, Lu3+ (logKLaL = 14.99, logKLuL = 8.25).[11] The greater selectivity of macropa for large ions like La3+ contributes to its more favorable radiolabeling properties because trace impurities of non-radioactive metal ions with smaller ionic radii will not effectively compete for binding. Notably, macropa is also a highly effective chelator for 225Ac3+, for use in targeted α particle therapy.[13]
Figure 1.
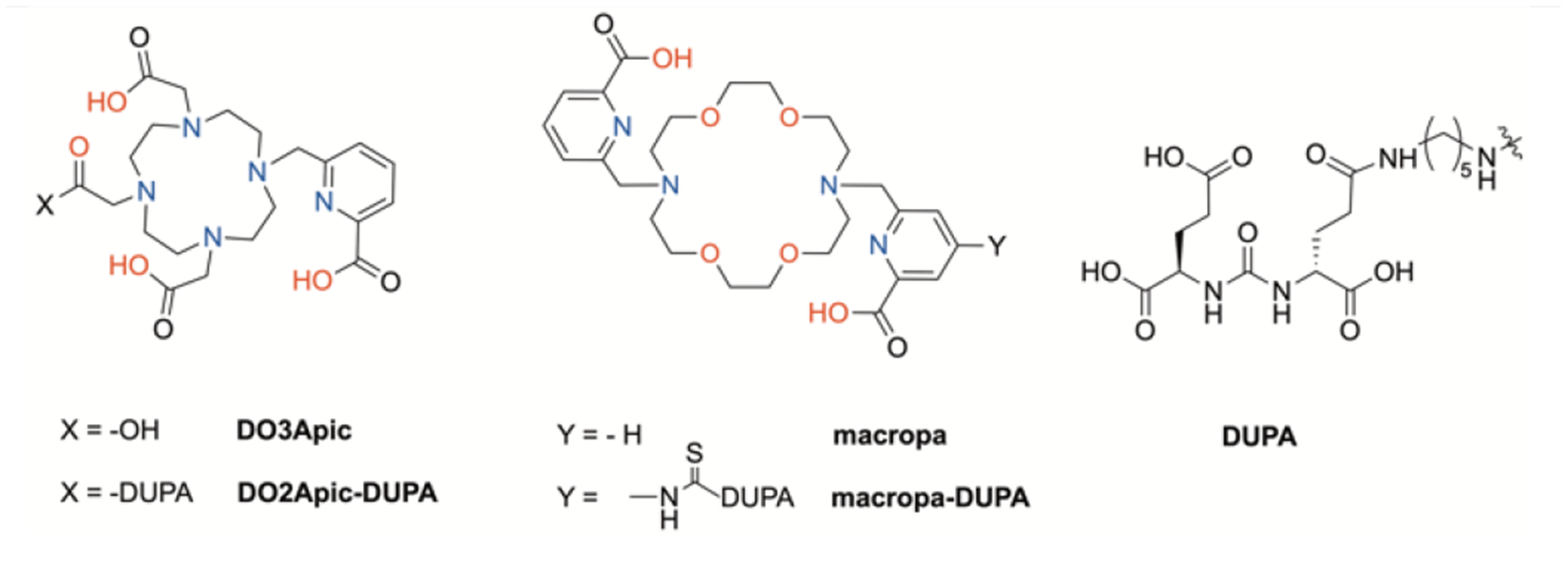
Chemical structures of chelators investigated in this work.
Table 1.
Summary of 132/135La radiolabeling properties, thermodynamic stability, kinetic inertness, and affinity of natLa complexes to PSMA for the ligands investigated in this work.
| compound | Max. apparent molar activity (pH, time, and temperature of radiolabeling conditions) |
Log KLaL | Kinetic inertness (21-day 1000× DTPA challenge) | Ki (PSMA) |
|---|---|---|---|---|
| DOTA | 0.67 Ci/μmol (pH 5.5, 30 min, 80 °C) | 20.7[9] | n/A | n/A |
| DO3Apic | 0.37 Ci/μmol (pH 5.5, 30 min, 25 °C) | 21.17[10] | 100% | n/A |
| DO2Apic-DUPA | 0.35 Ci/μmol (pH 5.5, 30 min, 80 °C) | n/A | ~82% | 12 ± 3 nM |
| macropa | 4.34 Ci/μmol (pH 5.5, 30 min, 25 °C) | 14.99[11] | 100% | n/A |
| macropa-DUPA | 4.04 Ci/μmol (pH 5.5, 30 min, 25 °C) | n/A | ~92% | 17 ± 2 nM |
Having established effective chelators for 132/135La, we next prepared conjugates of these ligands with a prostate-specific membrane antigen- (PSMA)-targeting agent to demonstrate the biological utility of these isotopes. Our choice of PSMA-targeting ligand, the Glu-urea-Glu dipeptides, was motivated by the successful application of the Glu-urea-Glu and Glu-urea-Lys targeting vectors in related radiometal-based diagnostic and therapeutic constructs employing 99mTc, 68Ga, 177Lu, 111In, 225Ac, 44Sc, and other radionuclides.[13–14] Our synthesis (see SI for more details) commenced with the preparation of DUPA, an aliphatic, amino-pentylamine linker version of the Glu-urea-Glu dipeptide that mimics the structural components of the clinically validated 99mTc-based prostate cancer-imaging agent MIP-1427 (Figure 1).[14d] The reactive primary amine of this compound was then conjugated to macropa and DO3Apic. More specifically, the bifunctional isothiocyanate-containing form of macropa, macropa-NCS, was treated with DUPA to form the conjugate macropa-DUPA (Figure 1). By contrast, the conjugate of DO3Apic, DO2Apic-DUPA (Figure 1), was formed via an amide bond coupling reaction employing one carboxylate pendent arm of the DO3Apic scaffold. Both conjugates were purified by reverse-phase HPLC and characterized by conventional methods (Figures S3–S18) prior to use in complexation and biological studies.
Because the formation of a conjugate may significantly alter the coordination properties of the chelator and the receptor affinity of the targeting vector, we next investigated the kinetic stability and PSMA affinity of the complexes of macropa-DUPA and DO2Apic-DUPA with non-radioactive La3+. The complexes were prepared by adjusting the pH of an aqueous mixture of conjugate and La3+ salt to between 7 and 8, purified by reverse-phase HPLC, and then characterized by 1H NMR spectroscopy, analytical HPLC, and UV-vis spectrophotometry (Figures S19–S27). To probe their kinetic inertness, these complexes were challenged with 1000-fold diethylenetriaminepentaacetic acid (DTPA, log KLaL = 19.48)[15] at pH 7.4, conditions under which both La(macropa)-DUPA and La(DO2Apic)-DUPA should thermodynamically lose La3+ to DTPA.[16] Over the course of this experiment, only minimal transchelation was detected for either complex even after 21 days (Table 1, Figures S28 and S29), affirming the exceptional kinetic inertness of these conjugates and validating their potential use in vivo. To confirm that the targeting property of the DUPA moiety was not negatively affected by conjugation to macropa and DO3Apic, the binding affinity (Ki) of both La3+ complexes to the PSMA target was measured using a radioactive displacement assay with the Tc-99m-based, clinically established tracer MIP-1427 in PSMA-expressing PC-3 PiP cells. La(macropa)-DUPA and La(DO2Apic)-DUPA exhibit Ki values of 17 ± 2 and 12 ± 3 nM, respectively (Table 1, Figures S30 and S31). Notably, these values are comparable to those of the 68Ga-based PSMA-targeting tracer PSMA-11 (12 nM),[17] which is currently in clinical trials. Thus, the covalent attachment of macropa or DO3Apic to the DUPA moiety does not significantly diminish its PSMA-binding affinity.
Given the favorable kinetic stability and good PSMA-targeting properties of La(macropa)-DUPA and La(DO2Apic)-DUPA, the conjugates were radiolabeled using 132/135La following the conditions described above for the non-functionalized chelators. For DO2Apic-DUPA, we observed diminished radiolabeling yields at room temperature in comparison to the unfunctionalized DO3Apic ligand. We hypothesize that the poorer radiolabeling ability of DO2Apic-DUPA arises from the conversion of one of the anionic carboxylate donors of DO3Apic to a neutral, more weakly binding carboxamide donor. Despite the poorer radiolabeling at room temperature, quantitative radiolabeling yields could be obtained with this conjugate after 30 min at 80 °C. By contrast, macropa-DUPA retains favorable radiolabeling properties that match those of non-functionalized macropa. However, analysis of the radio-HPLC chromatogram (Figure S32) following purification and reconstitution of 132/135La(macropa)-DUPA reveals the formation of several additional radioactive species. We hypothesize that these decomposition products arise from radiolysis of the conjugate.[18] Notably, this radiolysis effect is not observed for DO2Apic-DUPA (Figure S33), suggesting that it may occur due to the presence of the aromatic thiourea linkage in macropa-DUPA. The radiolysis of 132/135La(macropa)-DUPA could be attenuated via the addition of sodium ascorbate, enabling the intact radiolabeled conjugate to be obtained following solid-phase extraction and formulation (Figure S34).
After confirming suitable radiolabeling conditions for these conjugates, we next evaluated their use as prostate cancer-imaging agents in vivo. To this end, mice were implanted with both PSMA+ (PC-3 PiP) and PSMA– (PC-3 flu) cells to produce palpable, 400–500 mm3 xenografts on the right and left flank, respectively, one week after implantation. Cohorts were injected intravenously with 25–50 μ5–50 e132/135La-radiolabeled macropa-DUPA and DO2Apic-DUPA constructs with effective molar activities of 0.28–0.33 Ci/μmol of 135La. Representative images were obtained with PET-CT at 1 h and 4 h post injection. Additionally, biodistribution analysis was carried out following euthanasia of the mice at the 4 h time point. The results of these studies are shown in Figures 2 and 3, Figure S35, Table, S1, and Table S2. Notably, both radiolabeled conjugates were rapidly taken up by PSMA+ tumors of mice (6.73 ± 0.93 %ID/cc and 5.00 ± 0.50 %ID/cc at 1 h post injection), with the measured activity diminishing only slightly after 4 h. By contrast, negligible activity was observed in PSMA– tumors, even after 4 h (< 0.08 %ID/g for both conjugates). This dramatic difference in uptake between tumor types signifies that the DUPA-based conjugates possess high specificity for the PSMA target.
Figure 2. (.
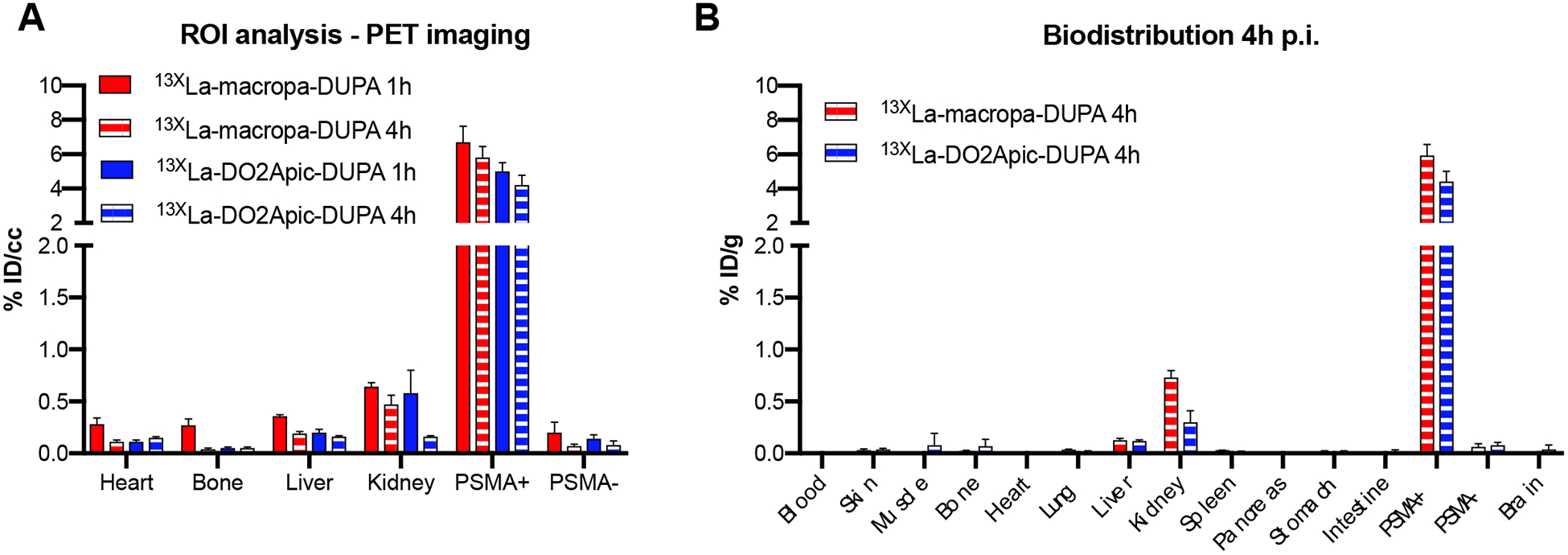
A) Region of interest (ROI) analysis of PET images obtained at 1 and 4 h post injection, expressed in percent injected dose per cubic centimeter (% ID/cc). (B) Biodistribution analysis obtained at 4 h post injection, expressed in percent injected dose per gram (% ID/g). These data indicate that ROI analysis provides reliably quantitative information on distribution in organs of interest.
Figure 3.
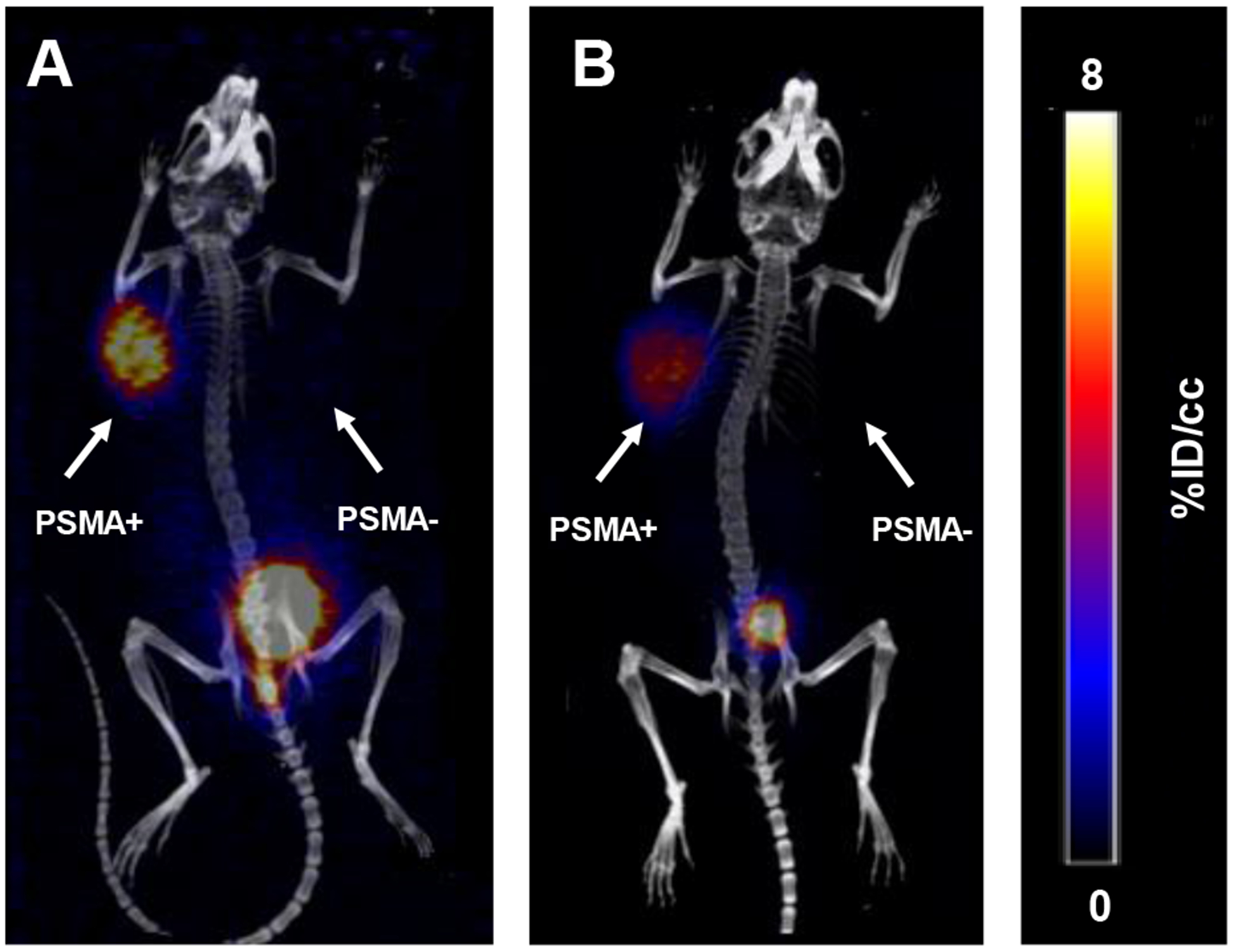
A maximum intensity projection (MIP) image is provided of static PET/CT images of PC3-PiP/Flu tumor-bearing mice 1h after injection of (A) 132/135La(macropa)-DUPA and (B) 132/135La(DO2Apic)-DUPA.
In contrast to the high uptake of the 132/135La-radiolabeled conjugates into PSMA+ tumors, little activity was measured in normal tissues at either time point. Most of the activity was cleared renally, explaining the small amount of uptake by the kidneys (<1 %ID/g). The kidney uptake of both conjugates, however, was significantly lower than that observed previously for other diagnostic and therapeutic PSMA-targeting compounds,[14b, 14d] an important feature that may prevent dose-limiting nephrotoxicity. Because unchelated La3+ rapidly accumulates in the liver and bones,[6b] the presence of activity within these organs would signify instability of the La(macropa)-DUPA and La(DO2Apic)-DUPA complexes. The lack of accumulation of activity in these tissues suggests that the 132/135La complexes remain intact over several hours in vivo. Collectively, the results of these imaging and biodistribution studies demonstrate that both chelators are well suited for targeted in vivo applications with lanthanum radioisotopes.
In summary, our work provides the first account of targeted in vivo imaging with radioactive lanthanum isotopes. We have successfully established the radiochemistry of 132/135La and evaluated two chelators, macropa and DO3Apic, for use in targeted nuclear medicine applications. Our studies identify macropa as an ideal chelator for 132/135La based on its ability to efficiently radiolabel at room temperature and the excellent in vivo stability of the resulting complex. Furthermore, because this chelator is highly effective for 225Ac3+, conjugates containing this ligand can be used for both lanthanum and actinium radionuclides, enabling 132La to be used as a diagnostic partner of 225Ac. Collectively, this work demonstrates a proof-of-principle for the use of radioactive La3+ in targeted nuclear medicine applications. Future efforts are focused on exploring the therapeutic potential of the Auger electron emissions of 135La and the value of 132La as a partner diagnostic for the potent α emitter 225Ac.
Supplementary Material
Acknowledgements
Research reported in this publication was supported in part by the National Cancer Institute of the National Institutes of Health under Award Number T32CA009206 (E.A.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. B.A.V. and K. E. M. acknowledge support through the SBU Chemistry-Biology interface training program (T32GM092714). E.B. acknowledges a Pathway to Independence Award (HL125728) and Stony Brook University for startup funds. J.J.W. thanks the College of Arts and Sciences at Cornell University for financial support of this research.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a Notni J, Wester HJ, J. Labelled Comp. Radiopharm 2018, 61, 141–153; [DOI] [PubMed] [Google Scholar]; b Boros E, Packard AB, Chem. Rev 2019, 119, 870–901; [DOI] [PubMed] [Google Scholar]; c Kostelnik TI, Orvig C, Chem. Rev 2019, 119, 902–956. [DOI] [PubMed] [Google Scholar]
- [2].a Bodei L, Cremonesi M, Grana CM, Fazio N, Iodice S, Baio SM, Bartolomei M, Lombardo D, Ferrari ME, Sansovini M, Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 2125–2135; [DOI] [PubMed] [Google Scholar]; b Kabasakal L, Demirci E, Ocak M, Decristoforo C, Araman A, Ozsoy Y, Uslu I, Kanmaz B, Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 1271–1277. [DOI] [PubMed] [Google Scholar]
- [3].Cutler CS, Hennkens HM, Sisay N, Huclier-Markai S, Jurisson SS, Chem. Rev 2013, 113, 858–883. [DOI] [PubMed] [Google Scholar]
- [4].Qaim SM, Scholten B, Neumaier B, J. Radioanal. Nucl. Chem 2018, 318, 1493–1509. [Google Scholar]
- [5].Aghevlian S, Boyle AJ, Reilly RM, Adv. Drug Deliv. Rev 2017, 109, 102–118. [DOI] [PubMed] [Google Scholar]
- [6].a Fonslet J, Lee BQ, Tran TA, Siragusa M, Jensen M, Kibédi T, Stuchbery AE, Severin GW, Phys. Med. Biol 2017, 63, 015026; [DOI] [PubMed] [Google Scholar]; b Aluicio-Sarduy E, Hernandez R, Olson AP, Barnhart TE, Cai W, Ellison PA, Engle JW, Sci. Rep 2019, 9, 10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Emfietzoglou D, Nikjoo H, Radiat. Res 2007, 167, 110–120. [DOI] [PubMed] [Google Scholar]
- [8].Abel EP, Clause HK, Fonslet J, Nickles RJ, Severin GW, Phys. Rev. C 2018, 97, 034312. [Google Scholar]
- [9].Tóth Ė, Brücher E, Inorg. Chim. Acta 1994, 221, 165–167. [Google Scholar]
- [10].Regueiro-Figueroa M. n, Bensenane B, Ruscsák E, Esteban-Goómez D, Charbonnieère L. c. J, Tircsó G, Tóth I, Blas A. s. d, Rodriíguez-Blas T, Platas-Iglesias C, Inorg. Chem 2011, 50, 4125–4141. [DOI] [PubMed] [Google Scholar]
- [11].Roca-Sabio A, Mato-Iglesias M, Esteban-Gómez D, Tóth É, Blas A. d., Platas-Iglesias C, Rodríguez-Blas T, J. Am. Chem. Soc 2009, 131, 3331–3341. [DOI] [PubMed] [Google Scholar]
- [12].Morgenstern A, Apostolidis C, Kratochwil C, Sathekge M, Krolicki L, Bruchertseifer F, Curr. Radiopharm 2018, 11, 200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Thiele NA, Brown V, Kelly JM, Amor‐Coarasa A, Jermilova U, MacMillan SN, Nikolopoulou A, Ponnala S, Ramogida CF, Robertson AK, Rodríguez-Rodríguez C, Schaffer P, Williams CJ, Babich J, Radchenko V, Wilson J, Angew. Chem. Int. Ed 2017, 56, 14712–14717. [DOI] [PubMed] [Google Scholar]
- [14].a Szabo Z, Mena E, Rowe, Plyku D, Nidal R, Eisenberger MA, Antonarakis ES, Fan H, Dannals RF, Chen Y, Mease RC, Vranesic M, Bhatnagar A, Sgouros G, Cho SY, Pomper MG, Mol. Imaging Biol 2015, 17, 565–574; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Weineisen M, Schottelius M, Simecek J, Baum RP, Yildiz A, Beykan S, Kulkarni HR, Lassmann M, Klette I, Eiber M, J. Nucl. Med 2015, 56, 1169–1176; [DOI] [PubMed] [Google Scholar]; c Umbricht CA, Benešová M, Schmid RM, Türler A, Schibli R, van der Meulen NP, Müller C, EJNMMI res 2017, 7, 9; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hillier SM, Maresca KP, Lu G, Merki RD, Marquis JC, Zimmerman CN, Eckelman WC, Joyal JL, Babich JW, J. Nucl. Med 2013, 54, 1369–1376; [DOI] [PubMed] [Google Scholar]; e Chatalic KL, Heskamp S, Konijnenberg M, Molkenboer-Kuenen JD, Franssen GM, Clahsen-van Groningen MC, Schottelius M, Wester H-J, van Weerden WM, Boerman OC, Theranostics 2016, 6, 849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Martell AE, Smith RM, Critical stability constants, Vol. 1, Springer, 1974. [Google Scholar]
- [16].Alderighi L, Gans P, Ienco A, Peters D, Sabatini A, Vacca A, Coord. Chem. Rev 1999, 184, 311–318. [Google Scholar]
- [17].Eder M, Schäfer M, Bauder-Wüst U, Hull W-E, Wängler C, Mier W, Haberkorn U, Eisenhut M, Bioconjugate Chem. 2012, 23, 688–697. [DOI] [PubMed] [Google Scholar]
- [18].Dey GR, Naik DB, Kishore K, Moorthy PN, J. Chem. Soc. Perkin Trans 1994, 1625–1629. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.