

Abstract
Cardiovascular disease (CVD) and cancer are leading causes of morbidity and mortality worldwide. While conventionally managed as separate disease processes, recent research has lent insight into compelling commonalities between CVD and cancer, including shared mechanisms for disease development and progression. In this review, we discuss several pathophysiologic processes common to both CVD and cancer, such as inflammation, resistance to cell death, cellular proliferation, neurohormonal stress, angiogenesis, and genomic instability, in an effort to understand common mechanisms of both disease states. In particular, we highlight key circulating and genomic biomarkers associated with each of these processes, as well as their associations with risk and prognosis in both cancer and CVD. The purpose of this state-of-the-art review is to further our understanding of the potential mechanisms underlying cancer and CVD by contextualizing biomarkers common to both diseases.
Keywords: Biomarkers, Cancer, Cardiovascular Disease
CONDENSED ABSTRACT
Although conventionally viewed as distinct disease processes, cardiovascular disease (CVD) and cancer share common pathophysiologic mechanisms for disease development and progression. Importantly, blood- and tissue-based biomarkers may provide insight into the mechanistic overlap underlying these two diseases. Cellular stress, inflammation, cellular proliferation, neurohormonal stress, angiogenesis, and genomic instability each represent shared mechanisms for which candidate biomarkers may improve our understanding of the fundamental commonalities, risk, and prognosis of CVD and cancer.
Introduction
Cardiovascular disease (CVD) and cancer are leading causes of morbidity and mortality in the United States and represent a significant public health burden.(1, 2) Although traditionally viewed as distinct disease processes, recent attention has focused on the potential interplay between these two major health priorities.(3) Indeed, the potential cardiovascular (CV) toxicities of cancer therapy are now widely recognized.(4) However, beyond therapy-related CV clinical toxicities, much interest remains in the fundamental epidemiologic and biologic overlap between CVD and cancer, including the shared risk factors and pathophysiologic processes associated with both CV and oncologic disease incidence and progression.
Nearly two decades ago, Weinberg and Hanahan characterized the “hallmarks of cancer” by outlining the central biologic principles upon which the complexities of human cancer tumorigenesis could be organized.(5) These fundamental processes comprise several acquired capabilities that are common to neoplastic disease, including sustained proliferation, resistance of cell death, and angiogenesis, as well as two enabling characteristics common to cancer, including genomic instability and inflammation (Central Illustration) (5,6). Importantly, several of these oncogenic biologic processes are also central to other pathologic disease states, including CVD. Building on the biologic overlap between CVD and cancer, recent research has focused on the potential mechanistic links between CVD and cancer, and understanding how circulating biomarkers can inform diagnosis, prognosis, and therapy across both diseases. The purpose of this state-of-the-art review is to further our understanding of the potential shared mechanisms between cancer and CVD, as exemplified by biomarkers common to both diseases. We highlight key candidate biomarkers representative of several fundamental biologic processes that are common to both CVD and cancer, including inflammation, cell proliferation, resistance to cell death, neurohormonal stress, angiogenesis, and genomic instability, and describe biomarker associations with respective CV and cancer disease outcomes. While many of these biomarkers appear to provide prognostic information across both CVD and cancer, future research is necessary to clarify their mechanistic roles and to inform their potential utility in clinical practice.
Central Illustration. Shared Pathophysiologic Mechanisms Between Cardiovascular Disease and Cancer.
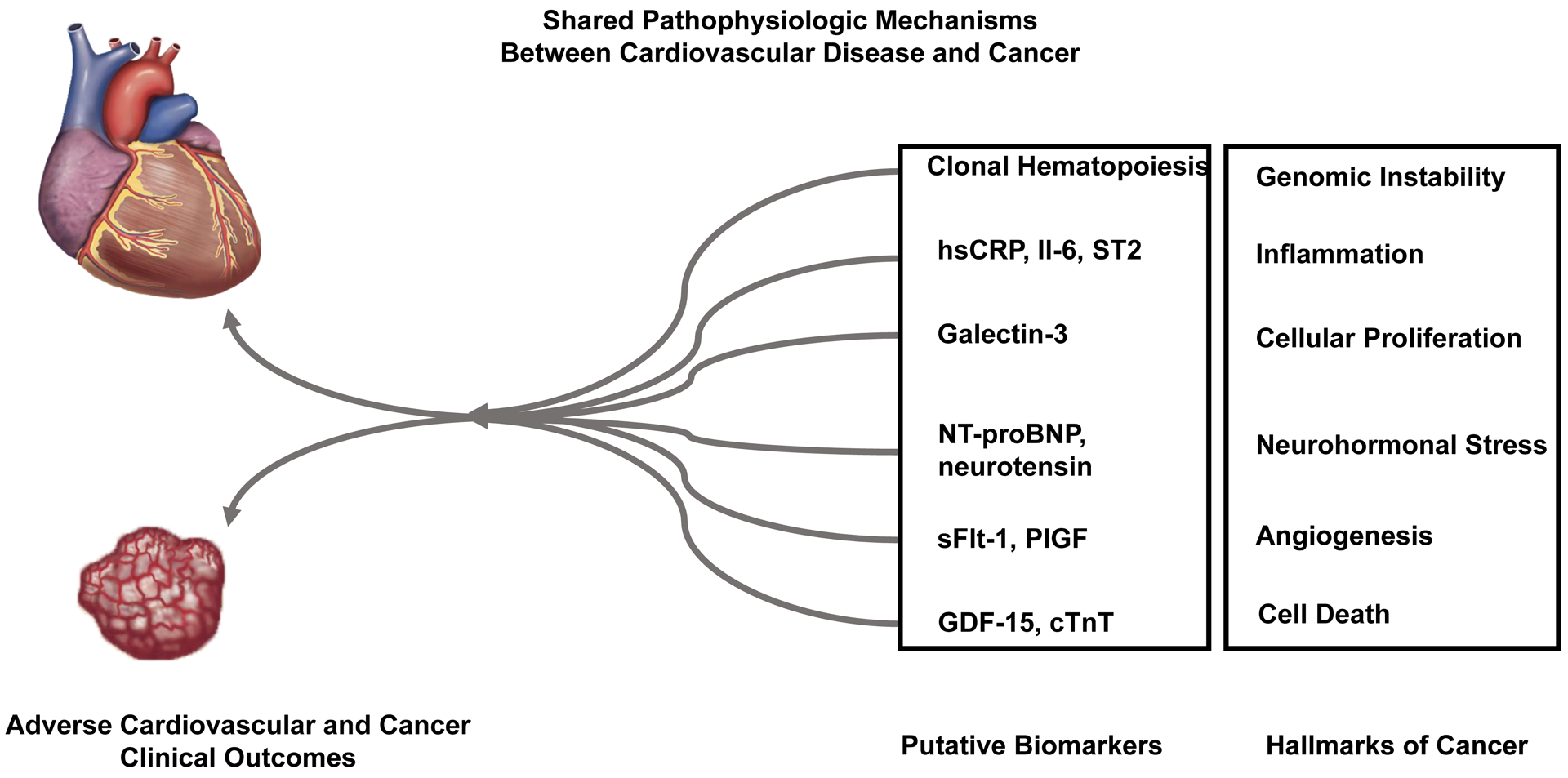
Several classic “hallmarks of cancer” share biologic overlap with cardiovascular disease. Recent research has focused on the potential mechanistic links between cardiovascular disease and cancer, and how circulating biomarkers may inform diagnosis, prognosis, and therapy across both diseases.
INFLAMMATION – High sensitivity C-reactive protein, Interleukin-6, and Suppression of tumorigenicity 2 (ST2)
Immune dysregulation, often characterized by increased levels of pro-inflammatory markers in blood and tissues in the absence of a clear precipitating factor, is associated with several age-associated chronic diseases, including CVD and cancer.(7) Indeed, inflammation is often considered a characteristic of biologic aging and functional disability.(7) An increase in inflammation may be attributed to a variety of underlying mechanisms, including primary dysregulation of immune cells, cell senescence, or genetic susceptibility.(7) In addition, conditions such as obesity are associated with inflammation, and abdominal and visceral adipocytes produce proinflammatory factors.(7)
Vascular inflammation is a primary factor in atherosclerotic pathogenesis and progression.(7) Although the causal role remains debated, increased circulating levels of proinflammatory factors, including high sensitivity C-reactive protein (hsCRP) and interleukin-6 (IL-6), are associated with CVD independent of traditional risk factors (Table 1).(8–11) Moreover, hsCRP is used to predict CVD risk and is often used to enrich patient populations for CV risk-reducing interventions (12). For example, rosuvastatin therapy is a risk-reducing strategy for the prevention of major CV events in patients without hyperlipidemia but with elevated hsCRP levels.(13) Consequently, anti-inflammatory therapeutic interventions, particularly targeting the CRP/IL-6/IL-1 inflammatory axis, may lead to improved CV outcomes. In a randomized trial of over 10,000 patients, the monoclonal anti-IL1β antibody, canakinumab, improved the secondary prevention of major CV events in patients with residual inflammation (elevated hsCRP) following myocardial infarction (MI) (Hazard Ratio (HR) 0.83, 95% CI 0.73 – 0.95).(14–16) Interestingly, in this trial, an exploratory analysis also indicated a reduction in incident lung cancer with canakinumab therapy (HR 0.61, 95% CI 0.39 – 0.97), therefore suggesting potential oncologic benefit through such an anti-inflammatory therapeutic intervention.(17) This observation has motivated a number of trials investigating the safety and potential benefit of canakinumab on overall survival in metastatic non-small cell lung cancer (e.g. NCT03447769, NCT03626545). Therefore, understanding the clinical effect of reducing inflammation, as quantified by hsCRP, in CVD has helped to stimulate investigations into novel therapies for cancer.
Table 1:
Biomarker Associations with Cancer and Cardiovascular Outcomes
| Hallmark of Cancer | Example Biomarkers | Mechanistic Links | Associations with Cancer Outcome | Associations with CV Outcome | Potential Therapeutic Implications |
|---|---|---|---|---|---|
| Inflammation | hsCRP IL-6 |
Enabling characteristic resulting in tissue damage and stress Implicated in atherosclerotic pathogenesis and progression, tissue invasion, and metastasis |
All-cause cancer incidence (28) Cancer-related mortality (29) |
MI, ischemic CVA (8) Composite CV Events (death from CHD, nonfatal MI or stroke, need for coronary revascularization) (9) MI or coronary death (10) |
Anti-inflammatory therapeutic interventions. (e.g. targeting CRP/IL-6/IL-1 inflammatory axis with canakinumab, an anti-IL-1β monoclonal antibody) Statin therapy |
| ST2 | Implicated in myocardial hypertrophy and fibrosis (particularly the soluble isoform) Involved in the modulation of the tumor microenvironment |
Overall survival (41, 42) | All-cause mortality, CV mortality, HF rehospitalization in patients with HF (33, 34) All-cause and CV mortality in patients with stable CAD (35) Major adverse cardiac events following acute MI (36) Incident HF, major CV events and CV mortality in the general population (38, 39) |
||
| Cellular Proliferation | Galectin-3 | Mitogenic functions stimulating cellular proliferation Implicated in CV tissue remodeling, cardiac fibrosis |
Overall survival (55) Disease or Progression-free survival (55) |
Composite CV events (death, sudden death with resuscitation, hospitalization for HF, or administration of an intravenous inotropic or vasodilator drug for ≥4 hours without hospitalization) (46) All-cause mortality and rehospitalization due to HF (48) |
Galectin-3 inhibitors (e.g. GR-MD-02) |
| Resistance to Cell Death | GDF-15 | Reflective of cellular stress response, apoptosis | Cancer Mortality (62) Incident Cancer or Cancer Death (63, 64) |
MI, thromboembolic stroke, or CV death (61) Composite CV events (MI, coronary insufficiency, CHD death, HF, and stroke) (39) Overall Survival (39) |
|
| cTnT | Reflective of cardiac injury and cell death | Overall survival with AL amyloidosis (68) Key factor in AL amyloidosis clinical staging (68) |
Major adverse cardiac events following acute MI (68, 111) | Risk stratification for therapy for AL amyloidosis | |
| Neurohormonal Stress | NT-proBNPMR-proANP MR-proADM CT-proET1 Neurotensin Sortilin | Incident Cancer (79) | All-cause mortality (among patients with suspected or confirmed cancer) (72) Incident CVD (MI and stroke) (79) CV mortality (79) |
SR48692 (an NTSR1 antagonist) | |
| Angiogenesis | sFlt-1 PlGF | Endothelial cell survival Implicated in tumorigenesis, invasion, and metastasis |
Overall survival (97) | Acute HF (92) All-cause mortality (93) Composite adverse HF outcomes (Death, OHT, or LVAD) (94) |
VEGF and VEGFreceptor targeting agents. (e.g. sunitinib, pazopanib, axitinib) |
| Genomic Instability | CHIP | Hematologic malignant precursor lesion Associated with vascular inflammation and accelerated atherogenesis |
0.5 – 1.0% annual risk for progression to hematologic malignancy (100) | Incident CHD (103) CV mortality (102, 103) |
Abbreviations: CAD, coronary artery disease; CHD, coronary heart disease; CHIP, clonal hematopoiesis of indeterminate potential; CT-proET1, C-terminal pro-endothelin-1; cTnT, cardiac troponin T; CVA, cerebrovascular accident; HF, heart failure; hsCRP, high-sensitivity C-reactive protein; IL-1, interleukin-1; IL-6, interleukin-6; LVAD, left ventricular assist device; MI, myocardial infarction; MR-proADM, mid-regional pro-adrenomedullin; MR-proANP, mid-regional pro-atrial natriuretic peptide; NT-proBNP, N-terminal pro-B natriuretic peptide; NTSR1, neurotensin receptor 1; OHT, orthotopic heart transplant, VEGF, vascular endothelial growth factor
Admittedly, however, the relationship between cancer and inflammation is complex, with inflammatory factors serving both adaptive and maladaptive functions.(18) Induction of IL-6 expression is associated with tissue damage or stress, and IL-6 dysregulation in the tumor microenvironment promotes cancer growth by enabling a variety of oncologic hallmarks, including tissue invasion and metastasis, angiogenesis, DNA damage repair, and anti-apoptotic effects.(19) Inflammatory factors within the tumor microenvironment may promote tumor growth and facilitate resistance to cancer therapies (19).
Elevated levels of serum or tumor tissue IL-6 are present in a large variety of malignancies, such as breast, prostate, renal, and lung carcinomas,(20–23) and have been associated with aggressive tumor growth, impaired response to cancer therapy, and inferior clinical outcomes (24–26). The prognostic significance of inflammatory biomarkers has been described in multiple oncologic settings. For example, elevated baseline levels of CRP or IL-6 have each been associated with worse overall survival in metastatic colorectal cancer patients, and elevated inflammatory biomarkers were predictive of a new-onset cancer diagnosis among >8,000 apparently healthy individuals enrolled in a prospective observational cohort study (e.g. hsCRP adjusted HR 1.08, 95% CI 1.04 – 1.13).(27, 28) This same study also suggested that heart failure (HF) contributed to tumor formation and progression, perhaps through activation of these inflammatory pathways.(28) Furthermore, in a national cohort study of over 6,000 participants, elevated levels of IL-6 independently predicted both all-cause mortality (HR 1.22, 95% CI 1.12 – 1.33) and cancer-related mortality (HR 1.13, 95% CI 1.00 – 1.29).(29)
Suppression of tumorigenicity 2 (ST2) is a member of the IL-1 superfamily that serves as a receptor for IL-33, a predominantly proinflammatory cytokine. The transmembrane isoform of ST2 (ST2L) is expressed in cardiomyocytes, fibroblasts and multiple immune cells including macrophages, T cells, basophils, eosinophils and mast cells.(30) The binding of IL-33 to ST2L initiates a cascade of downstream inflammatory processes. Similarly, soluble ST2 (sST2) acts as a decoy receptor sequestering free IL-33, thereby promoting anti-inflammatory effects.(30, 31)
IL-33/ST2 inflammatory signaling appears to be upregulated in response to myocardial stress and promotes cardioprotective effects by reducing apoptosis, myocardial hypertrophy, and fibrosis, while sST2 appears to block these beneficial effects.(31) As a result, there has been great interest in the negative prognostic value of sST2 in patients with HF. Elevations in sST2 levels are reported in both acute and chronic HF, and higher sST2 levels are associated with adverse myocardial remodeling, disease severity, and poor prognosis.(32) A meta-analysis including ten studies with 4,835 patients hospitalized for acute HF showed that levels of sST2 at hospital admission were predictive of all-cause death (HR per doubling 2.46, 95% CI 1.80 – 3.37) and cardiovascular death (HR per doubling 2.29, 95% CI 1.41 – 3.73). Pre-discharge sST2 levels were also predictive of all-cause death (HR per doubling 2.06, 95% CI 1.37 – 3.11), cardiovascular death (HR per doubling 2.20, 95% CI 1.48 – 3.25), and HF rehospitalization (HR per doubling 1.54, 95%CI is 1.03 to 2.32).(33) Similar adverse outcomes have been observed in patients with chronic HF, stable coronary artery disease, MI, and acute aortic dissection.(34–39)
IL-33/ST2 signaling is also a potent modulator of the tumor microenvironment through the recruitment of immune cells affecting tumor phenotype, and this pathway has been shown to be involved in the pathogenesis and progression of multiple malignancies.(30) Although opposing effects have been reported in some malignancies, increased levels of sST2 have been associated with more severe disease and poor prognosis. For instance, in patients with ER-positive breast cancer, circulating levels of sST2 were significantly elevated as compared to healthy controls, and higher levels correlated with markers of angiogenesis and prognostic factors such as age, disease stage, histologic type and tumor size.(40) Similarly, high plasma levels of sST2 were independently associated with worse overall survival in patients with pancreatic adenocarcinoma undergoing systemic chemotherapy (HR for all-cause mortality 2.10, 95% CI 1.33 – 3.41).(41) Conversely, in hepatocellular cancer, low levels of sST2 were associated with improved overall survival independent of established prognostic variables (HR for all-cause mortality 0.09, 95% CI 0.01 – 0.69), thus supporting the negative prognostic association for clinical cancer outcomes with elevated sST2 (42).
CELLULAR PROLIFERATION – Galectin-3
Galectins represent a family of carbohydrate-binding proteins with a high affinity for β-galactoside, and are widely expressed in human tissues, including epithelial and endothelial cells, sensory neurons, and immune cells (Table 1).(43, 44) Though predominantly located in the cell cytoplasm, galectins can localize to the nucleus, cell surface, or extracellular compartment, and may be detected in the serum circulation (43). As a result, galectins play a dynamic role in numerous biologic functions, including proliferation, differentiation, angiogenesis, cell adhesion, and regulation of the immune system (43,44). In particular, galectin-3 is implicated as a mitogen that is capable of stimulating cell proliferation through paracrine interactions.(45)
Through these proliferative functions, galectin-3 is believed to play a role in the pathogenesis of cardiovascular tissue remodeling and to represent a biomarker reflective of cardiac fibrosis and inflammation.(44) Accordingly, the prognostic utility of galectin-3 has been most extensively studied in patients with HF. Increased expression levels of galectin-3 are detected in a large proportion of chronic HF patients, and increases over time are associated with multiple biomarkers of HF severity (high sensitivity troponin, N-terminal pro-B-type natriuretic peptide (NT-proBNP), left ventricular ejection fraction, estimated glomerular filtration rate, and hsCRP), as well as adverse CV outcomes (defined as death, sudden death with resuscitation, hospitalization for HF, or administration of an intravenous inotropic or vasodilator drug for ≥4 hours without hospitalization; HR 1.021, 95% CI 1.002 – 1.040).(46) Similarly, in a prospective cohort study of coronary heart disease patients with chronic HF, increased concentrations of galectin-3 were independently associated with mortality and rehospitalization, particularly among patients with HF with preserved ejection fraction (HFpEF; relative risk [RR] 1.231, 95% CI 1.066 – 1.422) as compared to those with HF with reduced ejection fraction (HFrEF; RR 1.024, 95% CI 1.000 – 1.049).(47–49) The prognostic utility of galectin-3 does not appear to be influenced by common HF therapies, including angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, or mineralocorticoid receptor antagonists.(49–51) However, as it is ultimately somewhat less prognostic than other CV biomarkers such as NT-proBNP, the primary incremental value of galectin-3 may be in combination with other established HF biomarkers.(44) For example, the combination of elevated galectin-3 and BNP was a potent predictor of CV events in patients following hospital discharge after an episode of acute decompensated HF.(52)
As galectin-3 serves mitogenic functions, thereby promoting proliferation and differentiation, its value as a cancer biomarker has also been explored. Serum levels of galectin-3 are increased in patients with cancer relative to healthy individuals, and further increases are observed during progression from a localized malignancy to metastatic disease.(43) However, cancer tissue expression is complex and may vary by malignancy, with increased galectin-3 levels observed in thyroid, gastrointestinal, and hepatobiliary or pancreatic malignancies, and down-regulated expression in breast and prostate malignancies.(43) Such findings may be partially explained by varying nuclear to cytoplasmic translocation of galectin-3 during cancer progression across these various malignancies.(53, 54)
Similar to CVD, galectin-3 provides prognostic value in patients with cancer. In a meta-analysis of 36 studies evaluating cancer tissue and/or serum expression of galectin-3 and clinical outcomes, an elevated galectin-3 level was associated with worse overall and disease-free survival (HR 1.79, 95% CI 1.42 – 2.27; HR 1.57, 95% CI 1.04 – 2.37, respectively).(55) Many hypotheses underlying the inferior clinical cancer outcomes observed with increased galectin-3 levels have been explored. Circulating galectin-3 may enable metastatic dissemination of cancer by the facilitation of endothelial cell adherence and avoidance of immune surveillance.(56) Galectin-3 may also promote cancer cell survival by contributing to the immunosuppressive tumor microenvironment and attenuating T cell activation by binding to the T cell receptor (57). Given these observed roles of galectin-3 in promoting tumor growth, metastasis, and immune suppression, early phase clinical trials are ongoing to evaluate novel galectin-3 inhibitors (e.g. GR-MD-02) in combination with immune checkpoint inhibitors for the treatment of advanced cancers (e.g. NCT02575404, NCT02117362). In these studies, it would also be interesting to understand the potential impact of modulation of this pathway on cardiac remodeling.
RESISTANCE TO CELL DEATH – Growth Differentiation Factor-15 and Cardiac Troponin T
The cellular stress response is a universal defense mechanism that occurs in response to damaging environmental pressures that are associated with biologic aging and disease and that trigger cellular apoptosis (Table 1).(58) These external pressures include hypoxia, inflammation, telomere erosion, or oxidative stress.(58) Growth differentiation factor-15 (GDF-15), a member of the transforming growth factor β cytokine superfamily, is a biomarker reflective of this fundamental response to cellular stress.(59) The GDF-15 gene promoter contains two binding sites for the p53 transcription factor, which is itself activated through a variety of cellular stress mechanisms.(59) As a result, GDF-15 expression increases in relation to p53-mediated responses to the acute or chronic cellular stressors that often underlie biologic aging and disease (59).
Given its associations with cellular stress, aging, and disease, multiple studies have evaluated the prognostic value of circulating levels of GDF-15 in CV and non-CV disease settings. In community-dwelling patients with CVD or known CVD risk factors (defined as male sex, tobacco use, hypertension, diabetes, elevated low density lipoprotein cholesterol, and decreased high density lipoprotein cholesterol), concentrations of GDF-15 reflected underlying CVD burden independent of conventional measures.(60) Similarly, in a prospective nested case-control study of over 500 women without CVD history from the Women’s Health Study, increased GDF-15 levels (>856 pg/mL) were independently associated with a 2.7-fold increase in the risk of fatal or non-fatal CV events (95% CI 1.6 – 4.9).(61) In the Framingham Offspring Cohort Study, increased GDF-15 concentrations were associated with incident HF (HR 1.52, 95% CI 1.29 – 1.78), major CV events (defined as MI, coronary insufficiency, coronary heart disease death, HF, and stroke; HR 1.26, 95% CI 1.12 – 1.41), and all-cause mortality (HR 1.66, 95% CI 1.51 – 1.81).(39).
Comparable prognostic associations have also been identified between GDF-15 and oncologic outcomes. In the Rancho Bernardo study of mostly elderly adults, increased log GDF-15 was associated with increased mortality from any cancer (HR per standard deviation [SD] increase 1.8, 95% CI 1.3 – 2.4).(62) Similarly, in addition to an association with fatal or nonfatal CV events, increased GDF-15 independently predicted incident cancer diagnosis or cancer mortality in the Uppsala Longitudinal Study of Adult Men (HR 1.24, 95% CI 1.05 – 1.47), and was associated with incident colorectal cancer diagnosis in men and women enrolled in the Nurses’ Health Study and the Health Professionals Follow-up Study (adjusted RR 1.93, 95% CI 1.27 to 2.94) (63,64).
Although such associations demonstrate the potential CV and oncologic prognostic utility of GDF-15, the lack of tissue or disease specificity of the biomarker has limited its ability to serve as an effective screening or diagnostic tool. Rather, we propose that GDF-15 may ultimately have its most important role in refining patient risk stratification for an enriched evaluation of interventions that may be effective for reducing both CV and cancer morbidity and mortality.
Cardiac troponins serve as a sensitive and specific marker of cardiac injury and cell death, and the diagnostic and prognostic value of troponins for assessing cardiac injury and dysfunction is well established. Moreover, cardiac troponins have been extensively investigated as complementary approaches to imaging modalities for the early identification of cancer therapy-related cardiotoxicity. However, while several studies have demonstrated an association between elevated troponin levels and the development of subsequent cancer therapy-related cardiotoxicity,(65, 66) it remains unknown whether routine assessment of cardiac troponins improves long-term clinical outcomes.(67) Furthermore, the test characteristics of cardiac troponins in oncologic populations, often with comorbid medical conditions, remain uncertain.
However, one oncologic condition in which cardiac troponin T (cTnT) has clear prognostic value is primary systemic or light chain (AL) amyloidosis, in which immunoglobulin light chain-derived amyloid deposits from an underlying plasma cell dyscrasia may create multi-organ dysfunction. In particular, the majority of patients with AL amyloidosis have cardiac amyloid infiltration, and clinical outcomes are highly dependent on the extent of cardiac involvement. Furthermore, light chain deposits may have a direct cardiotoxic effect. As such, as a direct biomarker of cardiac injury and dysfunction, a cTnT level ≥ 0.025 ng/ml is a primary prognostic marker in AL amyloidosis,(68) thus representing a readily available biomarker reflective of injury and prognosis in both primary cardiac and oncologic conditions.
NEUROHORMONAL STRESS – Vasoactive Peptides and Diuretic Hormones
CV neurohormones, such as NT-proBNP, are routinely used as prognostic biomarkers in patients with acute or chronic HF (Table 1). However, elevated levels of functional CV neurohormones have also been identified in patients with cancer.(69, 70) These neurohormones may be produced by malignant cells of the tumor vascular endothelium, and are often abnormally elevated prior to the initiation of potential cardiotoxic anti-cancer therapy. For example, in a prospective study of patients with metastatic renal cell carcinoma, approximately 16% of patients had elevated baseline BNP concentrations >100 pg/ml.(71) However, the functional significance of these elevations and the potential implications for cardiac- and/or cancer-related risk remain unclear.
A recent prospective study of consecutive cancer patients sought to evaluate the association between baseline circulating CV neurohormones, including NT-proBNP, mid-regional pro-atrial natriuretic peptide (MR-proANP), mid-regional pro-adrenomedullin (MR-proADM), and C-terminal pro-endothelin-1 (CT-proET1), and all-cause mortality in an unselected, newly-diagnosed oncology patient population.(72) The majority of included cancer patients had a lack of CVD history, no significant electrocardiographic or echocardiographic abnormalities, and an NT-proBNP<400 pg/mL.(72) Elevated concentrations (up to 100-fold the reference limit) of NT-proBNP, MR-proANP, MR-proADM, and CT-proET1 were found in patients with cancer, and biomarker levels increased with advancing tumor stage. Furthermore, increased neurohormone levels were independently associated with reduced overall survival (HR 1.54, 95% CI 1.24 – 1.90 for NT-proBNP; HR 1.40, 95% CI 1.10 – 1.79 for MR-proANP; HR 1.31, 95% CI 1.19 – 1.44 for MR-proADM; HR 1.21, 95% CI 1.14 – 1.30 for CT-proET1).(72) Interestingly, elevations in these neurohormones also correlated with elevations in pro-inflammatory markers, including IL-6 and CRP.
Neurotensin (NTS) is an additional neurohormone with broad CV physiologic implications, including heart rate regulation and myocardial and vascular function (73–75). NTS is stored alongside norepinephrine in cardiac sympathetic nerves and is spatially associated with the coronary vasculature (75,76). Dysregulation of NTS, along with its intracellular receptor sortilin, has been associated with the development of CAD and adverse myocardial remodeling (75,77,78)
In the Malmo Diet and Cancer Study, proneurotensin (pNTS), the stable pro-fragment of NTS, was associated with incident CVD (HR 1.17, 95% CI 1.07 – 1.27) and CV mortality (HR 1.29, 95% CI 1.12 – 1.49),(79) and in participants from the Framingham Heart Study, the logpNTS plasma concentration predicted incident CVD (HR per SD increase 1.24, 95% CI 1.11 – 1.39).(80) Serum sortilin levels have also been associated with higher risk of major adverse cerebrovascular and CV events (HR 1.70 per SD, 95% CI 1.30 – 2.20) and abdominal aortic calcification (odds ratio [OR] 1.43 per SD, 95% CI 1.10 – 1.85).(81)
NTS and its receptors are upregulated in many cancers, including breast, ovarian, prostate, colorectal, and pancreatic.(82–86) In particular, NTS seems to have a link specifically with hormone-responsive cancers. In prostate cancer, androgen deprivation therapy stimulates NTS production, and in breast cancer, estrogen receptor (ER) positive cell proliferation is linked to NTS expression (while similar associations are not seen in triple negative breast cancer) (82, 84, 87). In the aforementioned Malmo Diet and Cancer Study, elevated pNTS was associated with incident breast cancer in women (HR 1.44, 95% CI 1.21 – 1.71), a finding subsequently validated in participants from the Malmo Preventive Project (OR 2.09, 95% CI 1.79 – 2.44).(79, 88) Interestingly, receptor antagonists such as SR48692 (an NTSR1 antagonist) have shown therapeutic promise in the treatment of some of these diseases, with multiple ongoing investigations.(89, 90)
These findings shed light on the potential cross-talk and functional significance of CV neurohormonal biomarker elevations in oncology populations. Vasoactive peptides play a role in the incidence and progression of CVD, and some can be effectively therapeutically modulated with CV medications. However, many of these hormones have also been implicated in tumor progression. Therefore, the worse prognosis that is associated with elevations of these neurohormones in oncologic populations may pathophysiologically result from either tumor proliferative effects or potentially via effects on CV function.(72)
ANGIOGENESIS – Soluble Fms-like Tyrosine Kinase-1 and Placental Growth Factor
Vascular changes are of interest in both CVD and cancer, as the CV system and the growing tumor each seek to moderate and maintain blood flow. Placental growth factor (PlGF) is a member of the vascular endothelial growth factor (VEGF) family that plays a key role in angiogenesis, and soluble fms-like tyrosine kinase 1 (sFlt-1) is a splice variant of Flt-1 (also known as VEGF receptor 1) that circulates in the blood and is able to sequester and inhibit PlGF activity.(91) These angiogenic biomarkers have been associated with clinical outcomes across both CVD and cancer.
In patients with acute MI, sFlt-1 concentrations were increased compared to controls, correlated with duration of hospitalization (r=0.23, p=0.002), and predicted the development of acute severe HF (OR per 100 pg/mL increase in sFlt-1 of 1.14, 95% CI 1.02 – 1.27).(92) Additionally, in patients with suspected acute MI, sFlt-1 >84 ng/L (HR 2.6, 95% CI 1.2 – 5.4) and PlGF >20 ng/L (HR 3.6, 95% CI 1.3 – 10.4) both predicted mortality during one-year of follow-up.(93) An investigation of chronic HF patients demonstrated that patients in the highest quartile of sFlt-1 (>379 pg/mL) had a 6.17-fold increased risk of adverse events, as defined by all-cause mortality, cardiac transplantation, and ventricular assist device placement (95% CI 4.30 – 8.86).(94) The PlGF to sFlt-1 ratio (PlGF/sFlt-1) has also been studied. In patients with stable CAD, a higher PlGF/sFlt-1 ratio was associated with increased risk of all-cause death (HR 3.32, 95% CI 1.43 – 7.72) and CV events (CV death, nonfatal MI, nonfatal cerebral infarction, hospitalization for HF, and the introduction of maintenance hemodialysis; HR 2.23, 95% CI 1.23 – 4.03) (95).
The VEGF family facilitates endothelial cell survival and is heavily implicated in tumorigenesis, invasion, and metastasis.(96) Although sFlt-1 itself does not seem to correlate directly with tumor prognosis, the imbalance of sFlt-1 and PlGF has been associated with outcomes in certain cancers.(97) In addition, preclinical studies have shown that PlGF inhibition results in decreased tumor growth and metastasis and that PlGF is involved in promoting tumor immune escape.(98) Since 2005, several VEGF and VEGF receptor targeting agents have been commonly used for the treatment of many solid tumor malignancies, including renal cell, thyroid, and hepatocellular carcinomas.(97) While these anti-VEGF therapies have yielded significant improvements in oncologic outcomes, they have also been associated with clinically important CV toxicity, including incident hypertension and cardiomyopathy. However, the clinical importance of the VEGF biomarkers in defining CV toxicity risk specifically in cancer patients treated with these therapies still remains to be defined. Nonetheless, these findings support a shared dependence of carcinogenesis and normal cardiovascular function on VEGF biology.
GENOME INSTABILITY – Clonal Hematopoeisis
Clonal hematopoiesis (CH), an expansion of a population of blood cells derived from a single hematopoietic stem cell precursor, is a defining feature of hematologic malignancies, as well as a frequent occurrence in normal human aging (Table 1) (99). This recognition of CH in normally aging individuals has led to the characterization of clonal hematopoiesis of indeterminate potential (CHIP), which is most commonly defined as CH occurring due to a somatic driver mutation often associated with hematologic malignancy (such as DNMT3A, TET2, ASXL1, JAK2) with a variant allele frequency of at least 2%, but not yet meeting standard clinicopathologic criteria for a hematologic malignancy (100,101). Indeed, up to 15% of persons aged >70 years and 30% of persons aged >85 years may have underlying CHIP.(100) As the presence of CHIP confers a 0.5 – 1.0% annual risk for developing a hematologic cancer, CHIP may be considered a pre-malignant state (100), and has been associated with worse overall survival. In addition to age, apparent risk factors for CHIP include male sex, cigarette smoking, and a germline polymorphism in TERT, a gene encoding a component of the telomerase complex.(99, 100)
Notably, in addition to representing a potential malignant precursor lesion, CHIP has also been associated with increased CV risk, including MI, stroke, and CV-related mortality.(102, 103) In comparison to patients without CH, patients with CHIP demonstrate a near doubling of the relative risk for CVD after adjusting for traditional risk factors (RR 1.9, 95% CI 1.4 – 2.7).(103) Notably, while increasing age is associated with both CVD and CHIP, the strong association between CHIP and CV risk is retained among patients <50 years of age, with a near 4-fold increased risk of CVD when compared to non-CHIP carriers. In addition, adverse CV outcomes can occur in carriers of CHIP regardless of whether the underlying CH ultimately progresses to an overt hematologic malignancy.(104) Moreover, while a larger clone size has been associated with an increased risk for malignant transformation, it is not yet known whether the burden of the circulating mutant clone in the peripheral blood correlates with CV risk.(104, 105)
The biologic underpinnings of this association with CVD are under active investigation, however, a suggested mechanism is the pathologic interaction between clonally-derived cells (monocytes, macrophages) and the vascular endothelium, resulting in vascular inflammation and accelerated atherogenesis.(103) In LDL receptor-deficient murine models prone to atherosclerosis, macrophages that are TET2 deficient resulted in increased secretion of inflammatory cytokines, including IL-6 and IL-1β, which are known to contribute to inflammation-associated atherosclerotic plaque progression.(106)
Practically, testing for CHIP is not routinely performed in all current clinical settings, as such testing requires peripheral blood sequencing. Additionally, the clinical utility of such testing to inform appropriate CVD risk mitigation strategies in CHIP carriers currently remains unclear. Future research is needed to understand the role of CVD risk-reducing strategies in patients harboring this novel CV biomarker, as well as how this might impact screening and treatment strategies in cancer patients.
CLINICAL AND THERAPEUTIC IMPLICATIONS
Biomarkers are used across cardiovascular and oncologic medicine to inform diagnosis and prognosis, and to select patients for risk-reducing interventions or disease therapy. For example, elevated LDL is used to identify patients who will benefit from statin therapy, and prostate-specific antigen (PSA) levels are used to screen for prostate cancer and to longitudinally monitor disease burden. As we look towards an era of personalized medicine, it is hopeful that novel mechanistic biomarkers will be used to inform and improve disease prognosis across both CVD and cancer. Understanding the role of these biomarkers can additionally inform the application of existing therapies and the discovery of new strategies that can target both CVD and cancer. As noted above, canakinumab is one such example.
There are also other commonly used CV medications that have demonstrated an impact on the aforementioned biomarkers, as well as potential beneficial effects on both CV and oncologic outcomes. Statins, classically known for their roles in lowering cholesterol and reducing cardiac events, also have been shown to play a role in cancer by modifying cell signaling, leading to increased apoptosis and decreased proliferation of cancer cells.(107) Additionally, statins decrease hsCRP levels and may suppress angiogenesis.(108, 109) A 2016 meta-analysis indicated that treatment with statins appeared superior to observation alone in the treatment of hepatocellular carcinoma,(110) and in breast cancer, statins are associated with reduced mortality and recurrence (108).
While many of the biomarkers discussed in this review demonstrate compelling prognostic associations with cancer and CVD clinical outcomes, future research will be required to clarify their mechanistic roles and their potential clinical utility with risk prediction, screening, and therapeutic monitoring. As the fields of cardiology and oncology move forward, it is hopeful that biomarkers can be used to guide cross-cutting therapeutic strategies, and that new drug development will be able to serve multiple indications and improve both CV and oncologic outcomes.
CONCLUSION
CVD and cancer represent the most prevalent and deadly diseases in the United States. While typically viewed as distinct disease processes, recent research has unveiled remarkable overlap between these fields. In particular, many of the “hallmarks of cancer”, including the resistance of cell death, inflammation, cellular proliferation, angiogenesis, and genomic instability, represent shared pathophysiologic processes with CVD. Research into the linking mechanistic biomarkers between CVD and cancer provide the important opportunity to better understand these shared fundamental biologic processes, and to facilitate risk prediction, screening, therapeutic monitoring, and new drug discovery for these diseases. Indeed, much progress has already been made in evaluating standard CV lifestyle interventions and medications as potential cancer therapy. As the field of cardio-oncology expands its scope to include a focus on the shared epidemiology and mechanisms underlying both CVD and cancer, the study of shared biomarkers will undoubtedly play a key role in improving our fundamental understanding and successful translation to the clinics. This review highlights the critical need for research clarifying the role of diagnostic, prognostic, and predictive biomarkers, as well as new therapies that can effectively treat cancer while mitigating cardiotoxicity risk.
KEY POINTS.
Cardiovascular disease (CVD) and cancer share several common pathophysiologic mechanisms for disease incidence and progression.
Several circulating and genomic biomarkers are reflective of this fundamental biologic overlap and shared mechanisms.
Before such biomarkers can be utilized in practice, their clinical role in enhancing risk prediction, screening, and therapeutic monitoring of both CVD and cancer should be further defined.
Acknowledgments
Author Disclosures:
Dr. Narayan has received research grant support from Pfizer, Bristol-Myers Squibb, Glaxo-Smith Kline, and Peloton Therapeutics. He has served as a consultant for Astra-Zeneca, Merck, and Exilixis.
Ms. Thompson is supported by the American Heart Association Student Scholarship in Cardiovascular Disease, the Perelman School of Medicine Center for Clinical Epidemiology and Biostatistics Summer Research Fellowship, and the Okun Family Cardiovascular Scholarship. Dr. Ho is supported in part by NIH R01-HL134893 and R01-HL140224. She has received research support from Gilead Sciences, Bayer, and research supplies from EcoNugenics, Inc. Dr. Januzzi is supported in part by the Hutter Family Professorship, is a Trustee of the American College of Cardiology, has received grant support from Novartis Pharmaceuticals, Roche Diagnostics, Abbott, Singulex and Prevencio, consulting income from Abbott, Janssen, Novartis, Pfizer, Merck, and Roche Diagnostics, and participates in clinical endpoint committees/data safety monitoring boards for Abbott, AbbVie, Amgen, Boehringer-Ingelheim, Janssen, and Takeda.
Dr. Ky is supported by NHLBI R01-HL118018 and R21HL141802. She has previously received research support from Roche Diagnostics, Inc and served as a consultant for Bristol Myers Squibb.
The remaining authors have nothing to disclose.
Abbreviations:
- CHIP
clonal hematopoiesis of indeterminate potential
- ER
estrogen receptor
- GDF-15
growth differentiation factor 15
- hsCRP
high sensitivity C-reactive protein
- IL-6
interleukin 6
- NTS
neurotensin
- PlGF
placental growth factor
- sFlt-1
soluble fms-like tyrosine kinase 1
- sST2
soluble Suppression of Tumorigenicity 2
- VEGF
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation. 2019. March 05,;139(10):e56–e528. [DOI] [PubMed] [Google Scholar]
- 2.Cancer of Any Site - Cancer Stat Facts [Internet]. [] Available from: https://seer.cancer.gov/statfacts/html/all.html.
- 3.Koene RJ, Prizment AE, Blaes A, Konety SH. Shared Risk Factors in Cardiovascular Disease and Cancer. Circulation. 2016. March 15,;133(11):1104–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Narayan V, Ky B. Common Cardiovascular Complications of Cancer Therapy: Epidemiology, Risk Prediction, and Prevention. Annu Rev Med. 2018. January 29,;69:97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000. January 07,;100(1):57–70. [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011. March 04,;144(5):646–74. [DOI] [PubMed] [Google Scholar]
- 7.Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018. September;15(9):505–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997. April 03,;336(14):973–9. [DOI] [PubMed] [Google Scholar]
- 9.Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000. March 23,;342(12):836–43. [DOI] [PubMed] [Google Scholar]
- 10.Cushman M, Arnold AM, Psaty BM, Manolio TA, Kuller LH, Burke GL, et al. C-reactive protein and the 10-year incidence of coronary heart disease in older men and women: the cardiovascular health study. Circulation. 2005. July 05,;112(1):25–31. [DOI] [PubMed] [Google Scholar]
- 11.Cesari M, Penninx, Brenda WJH, Newman AB, Kritchevsky SB, Nicklas BJ, Sutton-Tyrrell K, et al. Inflammatory markers and onset of cardiovascular events: results from the Health ABC study. Circulation. 2003. November 11,;108(19):2317–22. [DOI] [PubMed] [Google Scholar]
- 12.Ridker PM. From C-Reactive Protein to Interleukin-6 to Interleukin-1: Moving Upstream To Identify Novel Targets for Atheroprotection. Circ Res. 2016. January 08,;118(1):145–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM, Kastelein JJ, et al. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet. 2009. April 04,;373(9670):1175–82. [DOI] [PubMed] [Google Scholar]
- 14.Ridker PM, Howard CP, Walter V, Everett B, Libby P, Hensen J, et al. Effects of interleukin-1β inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo-controlled trial. Circulation. 2012. December 04,;126(23):2739–48. [DOI] [PubMed] [Google Scholar]
- 15.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017. September 21,;377(12):1119–31. [DOI] [PubMed] [Google Scholar]
- 16.Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet. 2018. January 27,;391(10118):319–28. [DOI] [PubMed] [Google Scholar]
- 17.Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. 2017. October 21,;390(10105):1833–42. [DOI] [PubMed] [Google Scholar]
- 18.Chabner BA, Nabel CS. Canakinumab and Lung Cancer: Intriguing, but Is It Real? Oncologist. 2018. June;23(6):637–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumari N, Dwarakanath BS, Das A, Bhatt AN. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016. September;37(9):11553–72. [DOI] [PubMed] [Google Scholar]
- 20.Dethlefsen C, Højfeldt G, Hojman P. The role of intratumoral and systemic IL-6 in breast cancer. Breast Cancer Res Treat. 2013. April;138(3):657–64. [DOI] [PubMed] [Google Scholar]
- 21.Culig Z, Puhr M. Interleukin-6: a multifunctional targetable cytokine in human prostate cancer. Mol Cell Endocrinol. 2012. September 05,;360(1–2):52–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang CH, Hsiao CF, Yeh YM, Chang GC, Tsai YH, Chen YM, et al. Circulating interleukin-6 level is a prognostic marker for survival in advanced nonsmall cell lung cancer patients treated with chemotherapy. Int J Cancer. 2013. May 01,;132(9):1977–85. [DOI] [PubMed] [Google Scholar]
- 23.Altundag O, Altundag K, Gunduz E. Interleukin-6 and C-reactive protein in metastatic renal cell carcinoma. J Clin Oncol. 2005. February 10,;23(5):1044; author reply 1044–1045. [DOI] [PubMed] [Google Scholar]
- 24.Wu C, Chen M, Chen W, Hsieh C. The role of IL-6 in the radiation response of prostate cancer. Radiat Oncol. 2013. June 27,;8:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev. 2012. November;38(7):904–10. [DOI] [PubMed] [Google Scholar]
- 26.Nolen BM, Marks JR, Ta’san S, Rand A, Luong TM, Wang Y, et al. Serum biomarker profiles and response to neoadjuvant chemotherapy for locally advanced breast cancer. Breast Cancer Res. 2008;10(3):R45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomsen M, Kersten C, Sorbye H, Skovlund E, Glimelius B, Pfeiffer P, et al. Interleukin-6 and C-reactive protein as prognostic biomarkers in metastatic colorectal cancer. Oncotarget. 2016. November 15,;7(46):75013–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meijers WC, Maglione M, Bakker SJL, Oberhuber R, Kieneker LM, de Jong S, et al. Heart Failure Stimulates Tumor Growth by Circulating Factors. Circulation. 2018. August 14,;138(7):678–91. [DOI] [PubMed] [Google Scholar]
- 29.Singh-Manoux A, Shipley MJ, Bell JA, Canonico M, Elbaz A, Kivimäki M. Association between inflammatory biomarkers and all-cause, cardiovascular and cancer-related mortality. CMAJ. 2017. Mar 13,;189(10):E384–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larsen KM, Minaya MK, Vaish V, Pena MMO. The Role of IL-33/ST2 Pathway in Tumorigenesis. Int J Mol Sci. 2018. September 09;19(9): 10.3390/ijms19092676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pascual-Figal DA, Januzzi JL. The biology of ST2: the International ST2 Consensus Panel. Am J Cardiol. 2015. April 02;115(7 Suppl):3B–7B. [DOI] [PubMed] [Google Scholar]
- 32.McCarthy CP, Januzzi JL. Soluble ST2 in Heart Failure. Heart Fail Clin. 2018. January 01;14(1):41–8. [DOI] [PubMed] [Google Scholar]
- 33.Aimo A, Vergaro G, Ripoli A, Bayes-Genis A, Pascual Figal DA, de Boer RA, et al. Meta-Analysis of Soluble Suppression of Tumorigenicity-2 and Prognosis in Acute Heart Failure. JACC Heart Fail. 2017. April 01;5(4):287–96. [DOI] [PubMed] [Google Scholar]
- 34.Aimo A, Vergaro G, Passino C, Ripoli A, Ky B, Miller WL, et al. Prognostic Value of Soluble Suppression of Tumorigenicity-2 in Chronic Heart Failure: A Meta-Analysis. JACC Heart Fail. 2017. April 01;5(4):280–6. [DOI] [PubMed] [Google Scholar]
- 35.Dieplinger B, Egger M, Haltmayer M, Kleber ME, Scharnagl H, Silbernagel G, et al. Increased soluble ST2 predicts long-term mortality in patients with stable coronary artery disease: results from the Ludwigshafen risk and cardiovascular health study. Clin Chem. 2014. March 01;60(3):530–40. [DOI] [PubMed] [Google Scholar]
- 36.Wang YP, Wang JH, Wang XL, Liu JY, Jiang FY, Huang XL, et al. Roles of ST2, IL-33 and BNP in predicting major adverse cardiovascular events in acute myocardial infarction after percutaneous coronary intervention. J Cell Mol Med. 2017. November 01;21(11):2677–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Tan X, Gao H, Yuan H, Hu R, Jia L, et al. Magnitude of Soluble ST2 as a Novel Biomarker for Acute Aortic Dissection. Circulation. 2018. January 16;137(3):259–69. [DOI] [PubMed] [Google Scholar]
- 38.Chen LQ, de Lemos JA, Das SR, Ayers CR, Rohatgi A. Soluble ST2 is associated with all-cause and cardiovascular mortality in a population-based cohort: the Dallas Heart Study. Clin Chem. 2013. March 01;59(3):536–46. [DOI] [PubMed] [Google Scholar]
- 39.Wang TJ, Wollert KC, Larson MG, Coglianese E, McCabe EL, Cheng S, et al. Prognostic utility of novel biomarkers of cardiovascular stress: the Framingham Heart Study. Circulation. 2012. September 25,;126(13):1596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu DP, Zhou XY, Yao LT, Liu CG, Ma W, Jin F, et al. Serum soluble ST2 is associated with ER-positive breast cancer. BMC Cancer. 2014. March 18;14:198–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kieler M, Unseld M, Wojta J, Kaider A, Bianconi D, Demyanets S, et al. Plasma levels of interleukin-33 and soluble suppression of tumorigenicity 2 in patients with advanced pancreatic ductal adenocarcinoma undergoing systemic chemotherapy. Med Oncol. 2018. November 13;36(1):1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bergis D, Kassis V, Ranglack A, Koeberle V, Piiper A, Kronenberger B, et al. High Serum Levels of the Interleukin-33 Receptor Soluble ST2 as a Negative Prognostic Factor in Hepatocellular Carcinoma. Transl Oncol. 2013. June 01;6(3):311–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang L, Guo X. Molecular regulation of galectin-3 expression and therapeutic implication in cancer progression. Biomed Pharmacother. 2016. March;78:165–71. [DOI] [PubMed] [Google Scholar]
- 44.Dong R, Zhang M, Hu Q, Zheng S, Soh A, Zheng Y, et al. Galectin-3 as a novel biomarker for disease diagnosis and a target for therapy (Review). Int J Mol Med. 2018. February;41(2):599–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Inohara H, Akahani S, Raz A. Galectin-3 stimulates cell proliferation. Exp Cell Res. 1998. December 15;245(2):294–302. [DOI] [PubMed] [Google Scholar]
- 46.Anand IS, Rector TS, Kuskowski M, Adourian A, Muntendam P, Cohn JN. Baseline and serial measurements of galectin-3 in patients with heart failure: relationship to prognosis and effect of treatment with valsartan in the Val-HeFT. Eur J Heart Fail. 2013. May;15(5):511–8. [DOI] [PubMed] [Google Scholar]
- 47.Medvedeva EA, Berezin II, Surkova EA, Yaranov DM, Shchukin YV. Galectin-3 in patients with chronic heart failure: association with oxidative stress, inflammation, renal dysfunction and prognosis. Minerva Cardioangiol. 2016. December;64(6):595–602. [PubMed] [Google Scholar]
- 48.Yu X, Sun Y, Zhao Y, Zhang W, Yang Z, Gao Y, et al. Prognostic value of plasma galectin-3 levels in patients with coronary heart disease and chronic heart failure. Int Heart J. 2015. May 13,;56(3):314–8. [DOI] [PubMed] [Google Scholar]
- 49.Edelmann F, Holzendorf V, Wachter R, Nolte K, Schmidt AG, Kraigher-Krainer E, et al. Galectin-3 in patients with heart failure with preserved ejection fraction: results from the Aldo-DHF trial. Eur J Heart Fail. 2015. February;17(2):214–23. [DOI] [PubMed] [Google Scholar]
- 50.Motiwala SR, Szymonifka J, Belcher A, Weiner RB, Baggish AL, Sluss P, et al. Serial measurement of galectin-3 in patients with chronic heart failure: results from the ProBNP Outpatient Tailored Chronic Heart Failure Therapy (PROTECT) study. Eur J Heart Fail. 2013. October;15(10):1157–63. [DOI] [PubMed] [Google Scholar]
- 51.Koukoui F, Desmoulin F, Galinier M, Barutaut M, Caubère C, Evaristi MF, et al. The prognostic value of plasma galectin-3 in chronic heart failure patients is maintained when treated with mineralocorticoid receptor antagonists. PLoS ONE. 2015;10(3):e0119160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feola M, Testa M, Leto L, Cardone M, Sola M, Rosso GL. Role of galectin-3 and plasma B type-natriuretic peptide in predicting prognosis in discharged chronic heart failure patients. Medicine (Baltimore). 2016. June;95(26):e4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lotz MM, Andrews CW, Korzelius CA, Lee EC, Steele GD, Clarke A, et al. Decreased expression of Mac-2 (carbohydrate binding protein 35) and loss of its nuclear localization are associated with the neoplastic progression of colon carcinoma. Proc Natl Acad Sci U S A. 1993. April 15,;90(8):3466–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van den Brûle FA, Waltregny D, Liu FT, Castronovo V. Alteration of the cytoplasmic/nuclear expression pattern of galectin-3 correlates with prostate carcinoma progression. Int J Cancer. 2000. July 20,;89(4):361–7. [DOI] [PubMed] [Google Scholar]
- 55.Wang Y, Liu S, Tian Y, Wang Y, Zhang Q, Zhou X, et al. Prognostic role of galectin-3 expression in patients with solid tumors: a meta-analysis of 36 eligible studies. Cancer Cell Int. 2018;18:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu L Circulating galectin-3 in the bloodstream: An emerging promoter of cancer metastasis. World J Gastrointest Oncol. 2010. April 15,;2(4):177–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Farhad M, Rolig AS, Redmond WL. The role of Galectin-3 in modulating tumor growth and immunosuppression within the tumor microenvironment. Oncoimmunology. 2018;7(6):e1434467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emmerson PJ, Duffin KL, Chintharlapalli S, Wu X. GDF15 and Growth Control. Front Physiol. 2018;9:1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wollert KC, Kempf T, Wallentin L. Growth Differentiation Factor 15 as a Biomarker in Cardiovascular Disease. Clin Chem. 2017. January;63(1):140–51. [DOI] [PubMed] [Google Scholar]
- 60.Lind L, Wallentin L, Kempf T, Tapken H, Quint A, Lindahl B, et al. Growth-differentiation factor-15 is an independent marker of cardiovascular dysfunction and disease in the elderly: results from the Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS) Study. Eur Heart J. 2009. October;30(19):2346–53. [DOI] [PubMed] [Google Scholar]
- 61.Brown DA, Breit SN, Buring J, Fairlie WD, Bauskin AR, Liu T, et al. Concentration in plasma of macrophage inhibitory cytokine-1 and risk of cardiovascular events in women: a nested case-control study. Lancet. 2002. June 22,;359(9324):2159–63. [DOI] [PubMed] [Google Scholar]
- 62.Daniels LB, Clopton P, Laughlin GA, Maisel AS, Barrett-Connor E. Growth-differentiation factor-15 is a robust, independent predictor of 11-year mortality risk in community-dwelling older adults: the Rancho Bernardo Study. Circulation. 2011. May 17,;123(19):2101–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wallentin L, Zethelius B, Berglund L, Eggers KM, Lind L, Lindahl B, et al. GDF-15 for prognostication of cardiovascular and cancer morbidity and mortality in men. PLoS ONE. 2013;8(12):e78797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Raaj S Mehta Mingyang Song, Bezawada Navya, Wu Kana, Xabier Garcia-Albeniz Teppei Morikawa, et al. A Prospective Study of Macrophage Inhibitory Cytokine-1 (MIC-1/GDF15) and Risk of Colorectal Cancer. Journal of the National Cancer Institute. 2014. April 1,;106(4):dju016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cardinale D, Sandri MT, Colombo A, Colombo N, Boeri M, Lamantia G, et al. Prognostic value of troponin I in cardiac risk stratification of cancer patients undergoing high-dose chemotherapy. Circulation. 2004. June 08;109(22):2749–54. [DOI] [PubMed] [Google Scholar]
- 66.Demissei BG, Hubbard RA, Zhang L, Smith AM, Sheline K, McDonald C, et al. Changes in Cardiovascular Biomarkers With Breast Cancer Therapy and Associations With Cardiac Dysfunction. J Am Heart Assoc. 2020. January 21;9(2):e014708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu AF, Ky B. Roadmap for biomarkers of cancer therapy cardiotoxicity. Heart. 2016. March 01;102(6):425–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012. March 20;30(9):989–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Burjonroppa SC, Tong AT, Xiao L, Johnson MM, Yusuf SW, Lenihan DJ. Cancer patients with markedly elevated B-type natriuretic peptide may not have volume overload. Am J Clin Oncol. 2007. June;30(3):287–93. [DOI] [PubMed] [Google Scholar]
- 70.Popat J, Rivero A, Pratap P, Guglin M. What is causing extremely elevated amino terminal brain natriuretic peptide in cancer patients? Congest Heart Fail. 2013. May-Jun;19(3):143–8. [DOI] [PubMed] [Google Scholar]
- 71.Narayan V, Keefe S, Haas N, Wang L, Puzanov I, Putt M, et al. Prospective Evaluation of Sunitinib-Induced Cardiotoxicity in Patients with Metastatic Renal Cell Carcinoma. Clin Cancer Res. 2017. July 15,;23(14):3601–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pavo N, Raderer M, Hülsmann M, Neuhold S, Adlbrecht C, Strunk G, et al. Cardiovascular biomarkers in patients with cancer and their association with all-cause mortality. Heart. 2015. December;101(23):1874–80. [DOI] [PubMed] [Google Scholar]
- 73.Kitabgi P Prohormone convertases differentially process pro-neurotensin/neuromedin N in tissues and cell lines. J Mol Med (Berl). 2006. August 01;84(8):628–34. [DOI] [PubMed] [Google Scholar]
- 74.Osadchii OE. Emerging role of neurotensin in regulation of the cardiovascular system. Eur J Pharmacol. 2015. September 05;762:184–92. [DOI] [PubMed] [Google Scholar]
- 75.Widiapradja A, Chunduri P, Levick SP. The role of neuropeptides in adverse myocardial remodeling and heart failure. Cell Mol Life Sci. 2017. June 01;74(11):2019–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reinecke M, Weihe E, Carraway RE, Leeman SE, Forssmann WG. Localization of neurotensin immunoreactive nerve fibers in the guinea-pig heart: evidence derived by immunohistochemistry, radioimmunoassay and chromatography. Neuroscience. 1982. July 01;7(7):1785–95. [DOI] [PubMed] [Google Scholar]
- 77.Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010. August 05;466(7307):714–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Redegeld FA, Yu Y, Kumari S, Charles N, Blank U. Non-IgE mediated mast cell activation. Immunological Reviews. 2018. March;282(1):87–113. [DOI] [PubMed] [Google Scholar]
- 79.Melander O, Maisel AS, Almgren P, Manjer J, Belting M, Hedblad B, et al. Plasma proneurotensin and incidence of diabetes, cardiovascular disease, breast cancer, and mortality. JAMA. 2012. October 10;308(14):1469–75. [DOI] [PubMed] [Google Scholar]
- 80.Januzzi JL, Lyass A, Liu Y, Gaggin H, Trebnick A, Maisel AS, et al. Circulating Proneurotensin Concentrations and Cardiovascular Disease Events in the Community: The Framingham Heart Study. Arterioscler Thromb Vasc Biol. 2016. August 01;36(8):1692–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Goettsch C, Iwata H, Hutcheson JD, O’Donnell CJ, Chapurlat R, Cook NR, et al. Serum Sortilin Associates With Aortic Calcification and Cardiovascular Risk in Men. Arterioscler Thromb Vasc Biol. 2017. May;37(5):1005–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dupouy S, Viardot-Foucault V, Alifano M, Souaze F, Plu-Bureau G, Chaouat M, et al. The neurotensin receptor-1 pathway contributes to human ductal breast cancer progression. PLoS One. 2009;4(1):e4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Norris EJ, Zhang Q, Jones WD, DeStephanis D, Sutker AP, Livasy CA, et al. Increased expression of neurotensin in high grade serous ovarian carcinoma with evidence of serous tubal intraepithelial carcinoma. J Pathol. 2019. July;248(3):352–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhu S, Tian H, Niu X, Wang J, Li X, Jiang N, et al. Neurotensin and its receptors mediate neuroendocrine transdifferentiation in prostate cancer. Oncogene. 2019. June 01;38(24):4875–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kontovounisios C, Qiu S, Rasheed S, Darzi A, Tekkis P. The role of neurotensin as a novel biomarker in the endoscopic screening of high-risk population for developing colorectal neoplasia. Updates Surg. 2017. September;69(3):397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Körner M, Waser B, Strobel O, Büchler M, Reubi JC. Neurotensin receptors in pancreatic ductal carcinomas. EJNMMI Res. 2015;5:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Johnson J, Li J, Evers BM. Abstract 3458: Neurotensin increases breast cancer proliferation through activation of AMPK. Cancer Res. 2018. -July-01 00:00:00;78(13 Supplement):3458.29716915 [Google Scholar]
- 88.Melander O, Belting M, Manjer J, Maisel AS, Hedblad B, Engstrom G, et al. Validation of plasma proneurotensin as a novel biomarker for the prediction of incident breast cancer. Cancer Epidemiol Biomarkers Prev. 2014. August 01;23(8):1672–6. [DOI] [PubMed] [Google Scholar]
- 89.Schulz J, Rohracker M, Stiebler M, Goldschmidt J, Stöber F, Noriega M, et al. Proof of Therapeutic Efficacy of a 177Lu-Labeled Neurotensin Receptor 1 Antagonist in a Colon Carcinoma Xenograft Model. J Nucl Med. 2017. June;58(6):936–41. [DOI] [PubMed] [Google Scholar]
- 90.Dong Z, Lei Q, Yang R, Zhu S, Ke X, Yang L, et al. Inhibition of neurotensin receptor 1 induces intrinsic apoptosis via let-7a-3p/Bcl-w axis in glioblastoma. Br J Cancer. 2017. June 06,;116(12):1572–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lecarpentier E, Tsatsaris V. Angiogenic balance (sFlt-1/PlGF) and preeclampsia. Ann Endocrinol (Paris). 2016. June;77(2):97–100. [DOI] [PubMed] [Google Scholar]
- 92.Onoue K, Uemura S, Takeda Y, Somekawa S, Iwama H, Nishida T, et al. Usefulness of soluble Fms-like tyrosine kinase-1 as a biomarker of acute severe heart failure in patients with acute myocardial infarction. Am J Cardiol. 2009. December 01,;104(11):1478–83. [DOI] [PubMed] [Google Scholar]
- 93.Hochholzer W, Reichlin T, Stelzig C, Hochholzer K, Meissner J, Breidthardt T, et al. Impact of soluble fms-like tyrosine kinase-1 and placental growth factor serum levels for risk stratification and early diagnosis in patients with suspected acute myocardial infarction. Eur Heart J. 2011. February 01;32(3):326–35. [DOI] [PubMed] [Google Scholar]
- 94.Ky B, French B, Ruparel K, Sweitzer NK, Fang JC, Levy WC, et al. The vascular marker soluble fms-like tyrosine kinase 1 is associated with disease severity and adverse outcomes in chronic heart failure. J Am Coll Cardiol. 2011. July 19,;58(4):386–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Matsumoto T, Uemura S, Takeda Y, Matsui M, Okada S, Nishida T, et al. An elevated ratio of placental growth factor to soluble fms-like tyrosine kinase-1 predicts adverse outcomes in patients with stable coronary artery disease. Intern Med. 2013;52(10):1019–27. [DOI] [PubMed] [Google Scholar]
- 96.Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005. February 10;23(5):1011–27. [DOI] [PubMed] [Google Scholar]
- 97.Yang F, Jin C, Jiang YJ, Li J, Di Y, Fu DL. Potential role of soluble VEGFR-1 in antiangiogenesis therapy for cancer. Expert Rev Anticancer Ther. 2011. April 01;11(4):541–9. [DOI] [PubMed] [Google Scholar]
- 98.Albonici L, Giganti MG, Modesti A, Manzari V, Bei R. Multifaceted Role of the Placental Growth Factor (PlGF) in the Antitumor Immune Response and Cancer Progression. Int J Mol Sci. 2019. June 18;20(12):1. 10.3390/ijms20122970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014. December 25,;371(26):2477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Steensma DP. Clinical Implications of Clonal Hematopoiesis. Mayo Clin Proc. 2018. August;93(8):1122–30. [DOI] [PubMed] [Google Scholar]
- 101.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014. December;20(12):1472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014. December 25,;371(26):2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med. 2017. July 13,;377(2):111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Libby P, Ebert BL. CHIP (Clonal Hematopoiesis of Indeterminate Potential). Circulation. 2018. August 14,;138(7):666–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Boettcher S, Ebert BL. Clonal Hematopoiesis of Indeterminate Potential. J Clin Oncol. 2019. February 10,;37(5):419–22. [DOI] [PubMed] [Google Scholar]
- 106.Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017. February 24,;355(6327):842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Masoudkabir F, Sarrafzadegan N, Gotay C, Ignaszewski A, Krahn AD, Davis MK, et al. Cardiovascular disease and cancer: Evidence for shared disease pathways and pharmacologic prevention. Atherosclerosis. 2017. August 01;263:343–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Beckwitt CH, Brufsky A, Oltvai ZN, Wells A. Statin drugs to reduce breast cancer recurrence and mortality. Breast Cancer Res. 2018. November 20,;20(1):144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Asher J, Houston M. Statins and C-reactive protein levels. J Clin Hypertens (Greenwich). 2007. August;9(8):622–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhou Y, Zhu G, Wang Y, Zheng J, Ruan L, Cheng Z, et al. Systematic review with network meta-analysis: statins and risk of hepatocellular carcinoma. Oncotarget. 2016. April 19,;7(16):21753–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vohra A, Asnani A. Biomarker Discovery in Cardio-Oncology. Curr Cardiol Rep. 2018. May 25;20(7):52–y. [DOI] [PubMed] [Google Scholar]