

Abstract
Background:
Short stature can be caused by mutations in a multitude of different genes. 3-M syndrome is a rare growth disorder marked by severe pre- and postnatal growth retardation along with subtle dysmorphic features. There have only been 2 prior reports of mutations in CCDC8 causing 3-M syndrome.
Methods:
Two patients presenting with mild short stature underwent whole exome sequencing. The mutation was confirmed via Sanger sequencing. We compare the clinical characteristics of our 2 patients to patients previously reported with mutations in the same gene.
Results:
Exome sequencing identified a homozygous frameshift mutation in CCDC8 in both patients. They presented with a much milder phenotype than previously described patients with the same mutation.
Conclusion:
In this study, we report a case of 2 sisters with relatively mild short stature who were found via exome sequencing to carry a previously reported homozygous mutation in CCDC8. These patients expand the anthropometric phenotype of 3-M syndrome and demonstrate the power of exome sequencing in the diagnosis of children with short stature. 3-M syndrome should be considered in children with mild skeletal abnormalities, normal/high growth hormone-IGF axis parameters, and normal intelligence.
Keywords: 3-M syndrome, CCDC8, Whole exome sequencing, Short stature
Introduction
3-M syndrome (MIM 273750, 612921, and 614205) was first recognized by Miller et al. [1] in 1975, who described a rare autosomal recessive primordial dwarfism characterized by severe pre- and postnatal growth retardation but with normal mental development. Approximately 100 cases have been reported in the literature since 1975 [2]. Children are characteristically born extremely small for gestational age and achieve a final height 5–6 SDS below the mean. Clinical characteristics include distinctive facial dysmorphism such as a triangular face, frontal bossing, mid-facial hypoplasia, fleshy tipped nose, and full fleshy lips. Characteristic radiological findings include slender long bones with diaphyseal constriction and flared metaphyses, tall vertebral bodies, anterior wedging of thoracic vertebral bodies, irregular upper and lower endplates, thoracic kyphoscoliosis, spina bifida occulta, small pelvic bones, small iliac wings, broad thorax with slender and horizontal ribs, and slightly delayed bone age. Prominent heels are most easily recognized in affected younger children. While the gonadal status of female patients with 3-M syndrome appears to be normal, male patients can develop gonadal failure with small testicular volumes, elevated gonadotropin concentrations, and sub- or infertility [3, 4]. From an endocrine perspective, serum growth hormone concentrations are usually normal and IGF-I concentrations are normal or low, while response to growth hormone therapy is variable but typically poor [5].
3-M syndrome is a genetically heterogeneous condition with 3 causal genes currently identified. In 2005, au-tozygosity mapping revealed a 3-M syndrome locus on chromosome 6p21.1, and pathogenic mutations in Cullin 7 (CUL7 [MIM609577]) were subsequently identified as the primary cause of 3-M syndrome [6]. A previous study showed that mutations in CUL7 account for approximately 84% of cases of 3-M syndrome (52/62) [7]. Cullin 7 is a major structural component of E3 ubiquitin ligase, which is involved in the proteasomal degradation path- way targeting p53 and the IGF-1/insulin signalling molecule IRS-1 [8]. In 2009, mutations in Obscurin-like 1 (OBSL1 [MIM 610991]) were found as the second most common cause of 3-M syndrome [9]. OBSL1 encodes a cytoskeletal adaptor protein which has been shown to physically interact with CUL7, and loss-of-function mutations in OBSL1 lead to decreased levels of CUL7 [9]. A third 3-M causal gene was identified in 2011, in which 6 patients from 5 families carried truncating mutations in CCDC8, 4 of whom carried the same frameshift mutation p.K205EfsX59 [10]. In co-expression studies, CCDC8 was shown to directly interact with OBSL1 but not CUL7 [10]. However, similar to CUL7, CCDC8 acts as a co-factor for p53-induced apoptotic processes [8]. To date, there has only been 1 additional report of a single family with a mutation in CCDC8 [11], and thus, the full clinical spectrum of patients with mutations in this gene is yet to be elucidated.
There are a multitude of genetic causes of short stature [12], many of which can be difficult to diagnose due to the subtlety of the associated clinical features. Next generation sequencing provides the opportunity to rapidly assess many genes and even the entire exome, facilitating the diagnosis of rare genetic conditions [13]. Herein, we report a case of 2 sisters who lacked an obvious syndromic cause for their relatively mild short stature, and, via exome sequencing, were found to carry the previously reported homozygous p.K205EfsX59 mutation in CCDC8. These patients have the mildest degree of short stature associated with 3-M syndrome to date and demonstrate the power of exome sequencing in the diagnosis of children with short stature.
Case Report
Subject 1
Subject 1 was born at 37 weeks’ gestation with a pregnancy complicated by intrauterine growth restriction, with a birthweight of 2.72 kg (–1.4 SDS). Her antenatal and postnatal course was otherwise unremarkable, and she was completely weaned from breastfeeding at 1 year of age. Her growth remained below the 3rd percentile throughout childhood (Fig. 1a). Further investigations performed indicated a normal karyotype, coeliac screen, thyroid function tests, plasma amino acids, immunoglobulins, and faecal elastase. Her appetite appeared normal throughout this period, and there were no developmental concerns.
Fig. 1.
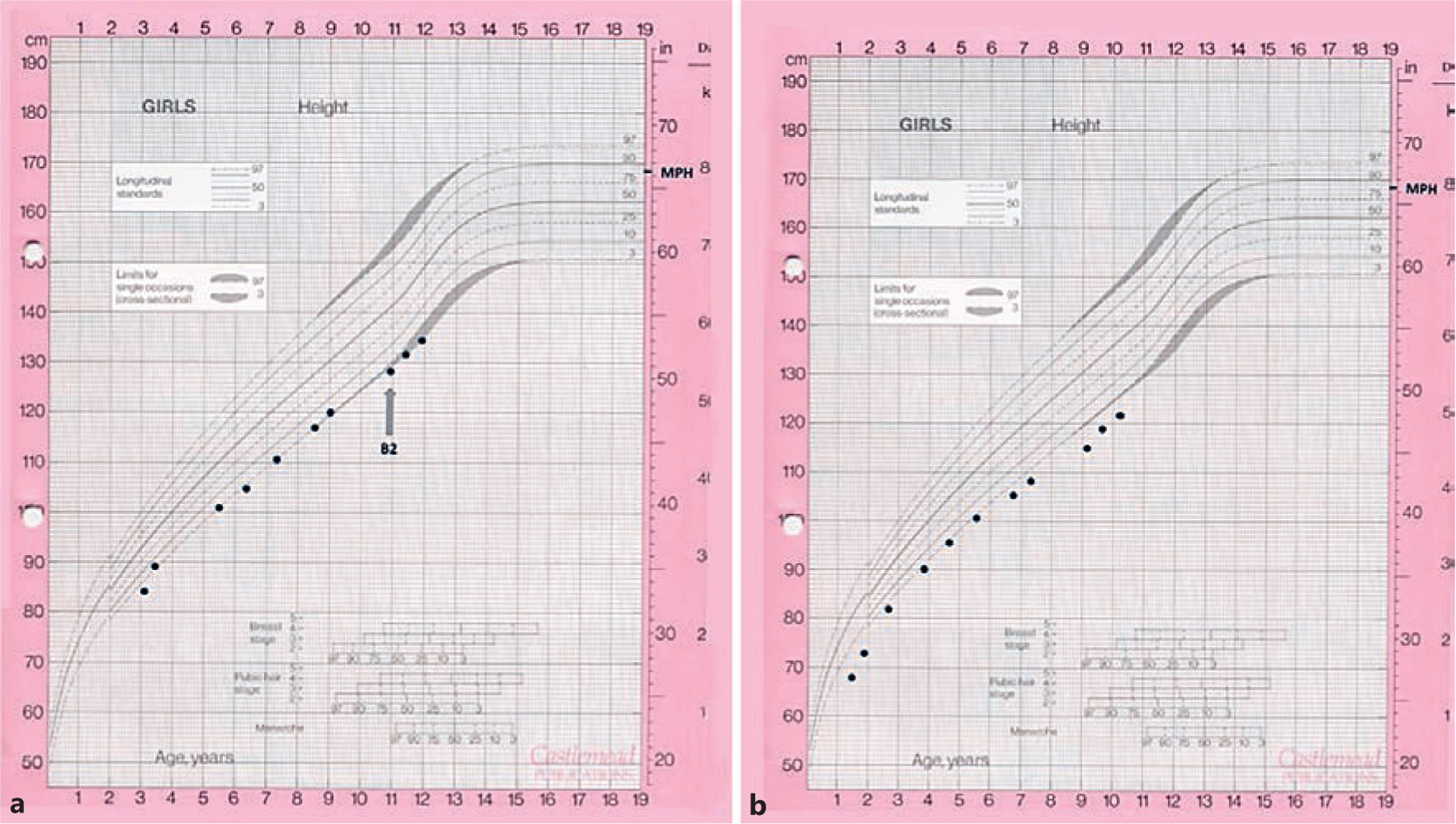
Growth charts of the 2 subjects. a Subject 1. b Subject 2.
The subject is of Pakistani descent. Family history was significant for parental consanguinity. Her maternal great-grandfather and paternal grandfather were brothers. Her younger sister was also referred to the paediatric endocrinology team for concerns regarding her growth, but she has 2 older brothers of normal height (188 and 175 cm). Her father is 180 cm and her mother 170 cm, giving her a mid-parental target height of 168 cm (75th–90th percentile).
Examination revealed proportionate short stature, mild clinodactyly, mild frontal bossing, and depression of her nasal bridge. After the molecular diagnosis was made via exome sequencing (see below), a closer examination indicated a triangular face, exaggerated lumbar lordosis, upturned nares, fleshy nose and lips, and prominent heels, which, although mild in presentation, could, together with her skeletal survey findings and family history, be suggestive of 3-M syndrome. IGF-1 and IGFBP-3 concentrations were normal: 119 ng/ml (−0.8 SDS, normal range 49–283) and 2.3 mg/L (−0.6 SDS, normal range 1.0–4.7), respectively. Height velocity remained normal throughout childhood. A skeletal survey revealed gracile long bones and ribs, pectus excavatum, and spina bifida occulta with a vertebral ossification defect in L4–5 but normal vertebral height (Fig. 2).
Fig. 2.
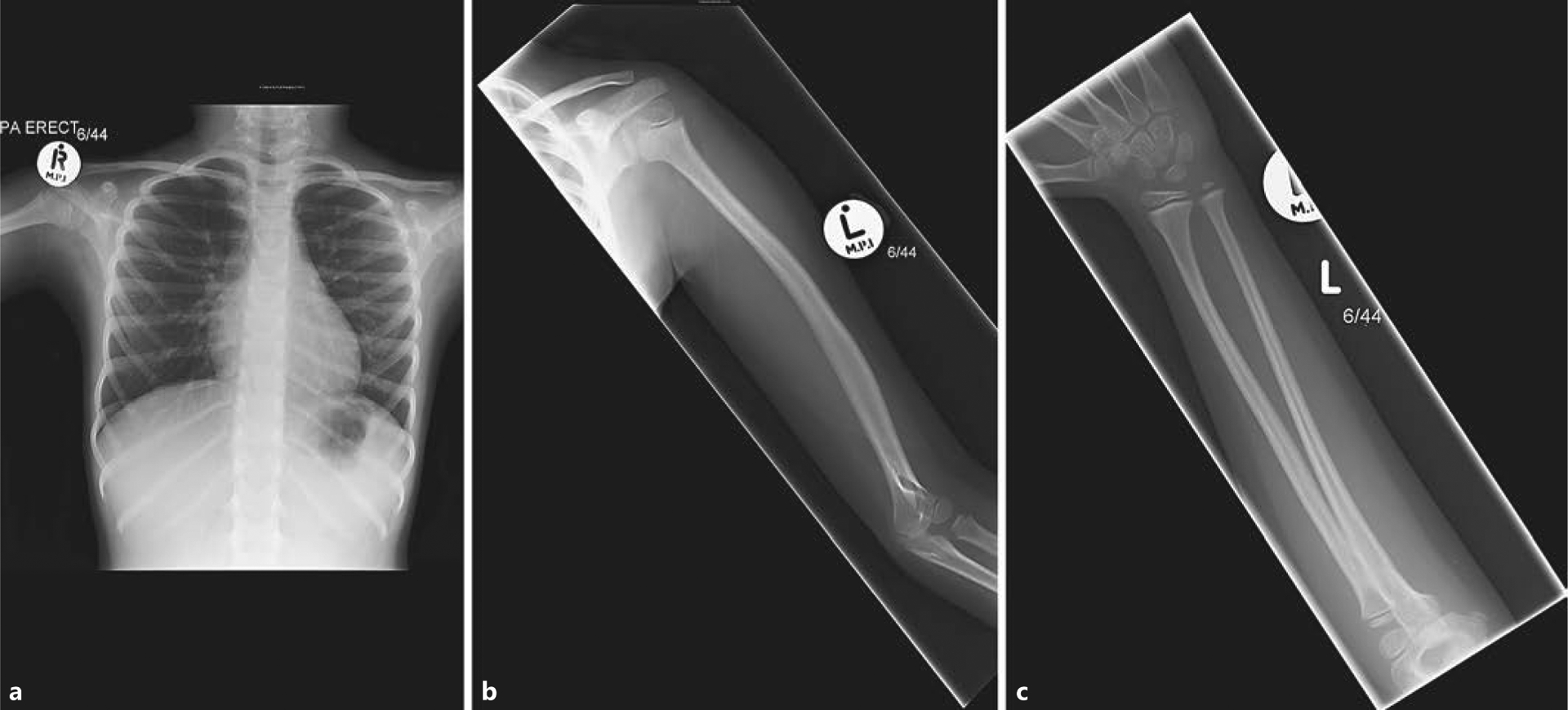
Skeletal survey images of subject 1 demonstrating gracile ribs with evidence of pectus excavatum (oblique anterior rib angles and relatively horizontal posterior ribs) (a) and gracile long bones in the upper limb (b, c). The left humerus (b) is also bowed with slight expansion of the diaphysis. Similar long bone appearances were observed in the lower limbs (not shown).
At the age of 11 years, subject 1 started puberty with Tanner stage 2 breast development. A glucagon stimulation test was performed at this point due to her slightly suboptimal height velocity of 4.2 cm/year, but this revealed a normal growth hormone peak of 15.2 ng/mL and a normal cortisol peak of 1,070 nmol/L. The rest of her pituitary function screen was normal: free T4 15.3 pmol/L (normal range 10.8–19.0), TSH 1.4 mU/L (normal range <6.0), LH <0.2 U/L, FSH 0.3 U/L, oestradiol <44 pmol/L, and PRL 115 mU/L. Height at last follow-up (11.9 years) was 134.6 cm (–2.2 SDS), weight 34.5 kg (–1.0 SDS), BMI 19.0 (+0.4 SDS), and head circumference 53.2 cm (–0.35 SDS).
Subject 2
Subject 2, the younger sister of subject 1, was born at 36 weeks’ gestation with a pregnancy complicated by intrauterine growth restriction, with a birthweight of 2.27 kg (−2.6 SDS). She was noted to feed poorly and exhibited poor weight gain, only reaching 6.6 kg (−4.9 SDS) at 1.3 years of life when she was referred to paediatric endocrinology. Early clinical examination revealed frontal bossing and mid-facial hypoplasia. Investigations indicated a normal karyotype, coeliac screen, and thyroid function tests. IGF-1 and IGFBP-3 concentrations were normal: 111 ng/mL (−1.0 SDS, normal range 51–303) and 2.6 mg/L (+0.3 SDS, normal range 0.8–3.9), respectively. Height velocity remained normal throughout childhood (Fig. 1b).
Further examination performed at 5.7 years of age revealed similar features to her sister, with upturned nares, fleshy nose and lips, long palpebral fissures, and prominent heels. Repeat endocrine biochemistry was performed at 9.1 years due to a deceleration in height velocity but these investigations were all normal: IGF-1 concentrations 111 ng/mL (−1.7 SDS, normal range 88–452), IGFBP-3 concentrations 3.9 mg/L (−0.7 SDS, normal range 2.1–7.7), free T4 15.6 pmol/L (normal range 10.8–19.0), TSH 2.6 mU/L (normal <6.0), LH <0.2 U/L, FSH 0.3 U/L, and PRL 92 mU/L. A glucagon stimulation test demonstrated a normal growth hormone peak of 29 ng/mL and a cortisol peak of 1,286 nmol/L. Height at last follow-up (10.2 years) was 121.8 cm (–2.7 SDS), weight 21.3 kg (–2.9 SDS), BMI 14.4 (–1.5 SDS), and head circumference 51.2 cm (–1.0 SDS).
Molecular Analysis
Ethical approval for the protocol was granted by the Cincinnati Children’s Hospital Medical Center Institutional Review Board. Written informed consent was obtained from the subjects and their parents. Both subjects underwent whole exome sequencing at Cincinnati Children’s Hospital Medical Center using previously described methods [14]. Given the known consanguinity and 2 affected siblings with unaffected parents, we hypothesized that the causal variant would be a rare homozygous non-synonymous variant present in both subjects. We excluded all variants present with a minor allele frequency greater than 0.001 in the 1,000 Genomes database (www.1000genomes.org), the Exome Aggregation Consortium browser (http://exac.broadinstitute.org), or our internal variant database. Only a single variant met these criteria, the homozygous p.K205EfsX59 mutation in CCDC8. We then confirmed this mutation via Sanger sequencing (Fig. 3). PCR primers are available on request.
Fig. 3.
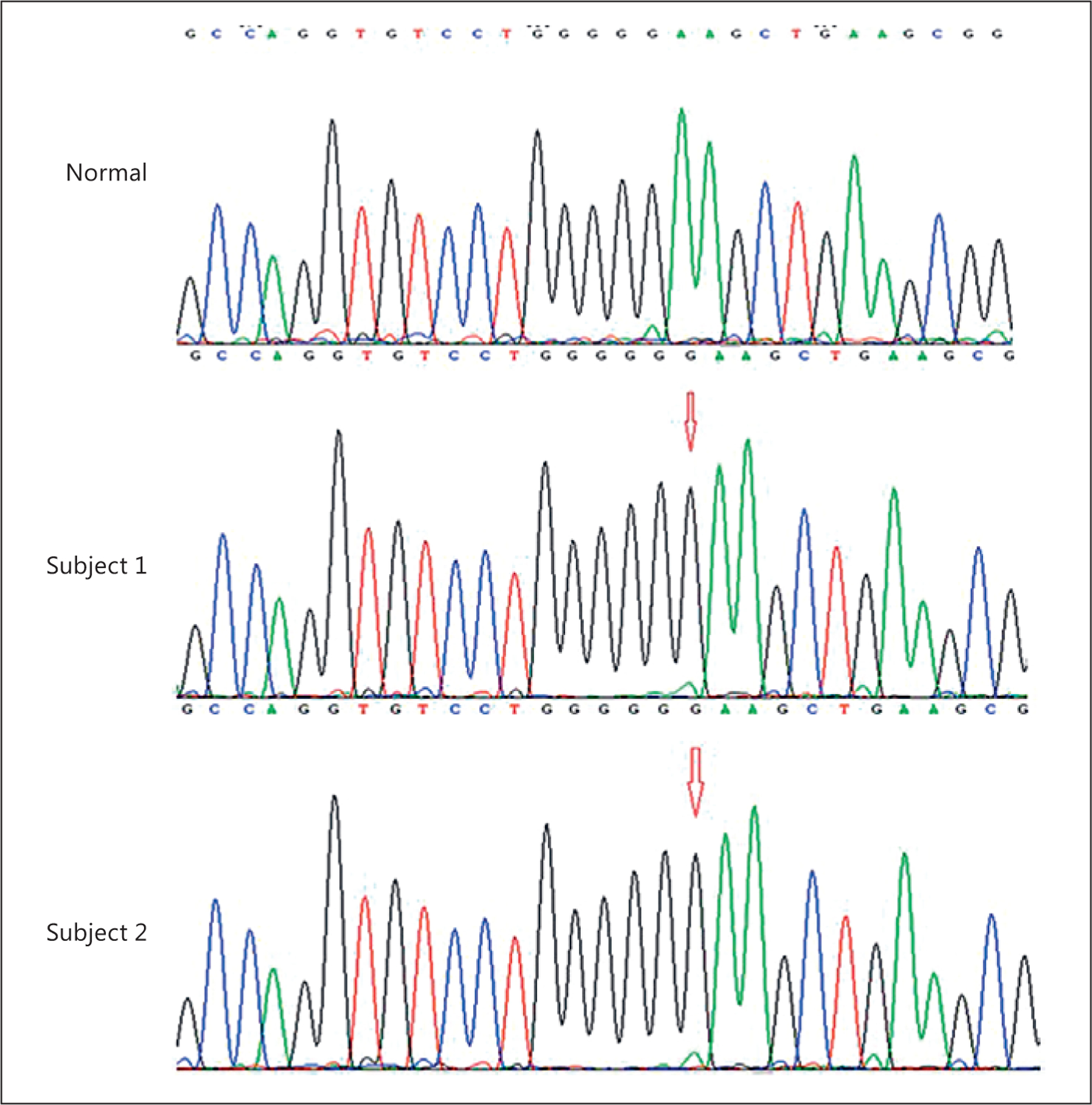
Chromatograms showing a homozygous insertion of a guanine nucleotide in the 2 subjects.
Discussion
To date, there have only been 2 reports describing a total of 9 patients in 6 families carrying CCDC8 mutations causing 3-M syndrome, and only 3 mutations were identified [10, 11]. The typical description of 3-M syndrome includes severe prenatal (usual birth weight SD scores <−3 SD) and postnatal growth retardation (adult height in the range of −8 to −4 SD), although the birth weight can fall within the lower part of the normal range [8]. Interestingly, patients with mutations in CCDC8 (i.e., 3-M syndrome type 3, MIM 614205) tend to have a milder phenotype with a mean height at presentation of –4.4 SD (Table 1). In this report, we describe 2 patients with a previously described homozygous frameshift mutation in CCDC8 who present with a considerably milder phenotype than previously described (Table 1). Their current height SDS scores of −2.2 and −2.7 fall within the relatively mild-moderate short stature range and, notably, subject 1 had a normal birth weight, thus lacking prenatal growth retardation. Unfortunately, birth length data were unavailable for these patients so it is possible that subject 1 was small for gestational age based on length criteria. Notably, their parents were rather tall with a mid-parental target height of +1 SD; thus, their actual degree of growth failure is more significant when this is taken into account. This highlights the importance in considering the deviation from target height when assessing a patient’s degree of short stature. Both subjects had normal head circumferences as is observed in 3-M syndrome. In retrospect, both subjects do exhibit mild features seen in 3-M syndrome including a slightly exaggerated lumbar lordosis, pectus excavatum, and hypermobile finger joints, but these are relatively non-specific and did not raise suspicion for this rare diagnosis. The fleshy heels, a distinct finding in 3-M syndrome, became less prominent with time. Similarly, the radiological features were quite subtle and were not fully appreciated until after the molecular diagnosis was obtained.
Table 1.
Clinical characteristics of the 2 affected subjects compared to patients previously reported with CCDC8 mutations
| Characteristic | Previous reported patients with CCDC8 mutation (n = 6) [10,11] | Subject 1 | Subject 2 |
|---|---|---|---|
| Birth weight | |||
| Mean −3.4 SDS | (−1.4 SDS) | (−2.6 SDS) | |
| Postnatal growth, height | |||
| Mean −4.4 SDS | (−2.2 SDS, 11.9 years) | (−2.7 SDS, 10.2 years) | |
| Postnatal growth, weight | |||
| Mean −3.8 SDS | (−1.0 SDS, 11.9 years) | (−2.9 SDS, 10.2 years) | |
| Head circumference | 4 out of 6 subjects with recorded head circumference were normal | ||
| (−0.35 SDS, 11.9 years) | (−1.0 SDS, 10.2 years) | ||
| Skeletal survey | Tall vertebral bodies (5/9), slender long bones (5/9), short thorax (6/6) | Gracile long bones and ribs, pectus excavatum and spina bifida occulta with a vertebral ossification defect in L4−−5 but normal vertebral height | Slightly exaggerated lumbar lordosis, pectus excavatum |
| Facial features | Triangular face (7/9), fleshy tipped nose (9/9), frontal bossing (8/9), pointed chin (7/9), midface hypoplasia (6/9), long philtrum (1/6) | Triangular face, mild clinodactyly, upturned nares, fleshy nose and lips, mild frontal bossing, depression of the nasal bridge | Frontal bossing and midfacial hypoplasia, upturned nares, fleshy nose and lips, long palpebral fissures |
| Other distinguishing features | Prominent fleshy heels (6/9), hypermobility of joints (2/9) | Fleshy heels, hypermobile finger joints | Fleshy heels, hypermobile finger joints |
| Puberty | NA | Tanner stage 2 breast development (11.9 years) | Prepubertal (10.2 years) |
It is interesting to note that both subjects had elevated peak growth hormone and normal cortisol concentrations during a glucagon stimulation test. Additionally, they had normal IGF-1 levels. Taken together, the relatively robust growth hormone levels with normal IGF-1 levels may suggest a degree of growth hormone and possibly IGF-1 resistance, as has been previously described in 3-M syndrome [5]. In other cases reported in the literature, 2 patients with 3-M syndrome have been shown to have complete or partial growth hormone deficiency, although the majority have a normal growth hormone axis on testing [5, 15–17]. The rest of their pituitary function (TSH, LH, FSH, and PRL) screening was normal.
These 2 girls are older than the CCDC8-deficient patients reported in the previous study, and the elder sister has started puberty with no evidence of gonadal failure. Gonadal status of female patients with 3-M syndrome has previously been reported to be normal, while males may have hypergonadotropic hypogonadism. It is not clear whether mutations in CCDC8 affect gonadal status or if this is only the case in individuals with CUL7 mutations. Further follow-up of these patients is needed.
In conclusion, we report a case of 2 siblings carrying a previously reported homozygous frameshift mutation in CCDC8 with mild short stature, one of whom had a normal birth weight, thus representing the mildest presentation of 3-M syndrome to date. An explanation for the same mutation causing highly variable clinical presentations remains to be fully elucidated, although it is important to consider the distance from the mid-parental target height when quantifying the degree of growth failure. 3-M syndrome should be considered in children with mild skeletal abnormalities, normal/high growth hormone-IGF axis parameters and normal intelligence. We highlight the power of exome sequencing in children with short stature, which provides comprehensive genetic evaluation for patients who lack obvious specific molecular aetiologies.
Established Facts.
3-M syndrome is a growth disorder presenting with severe pre- and postnatal growth retardation and is caused by mutations in 3 genes (CUL7, OBSL1, and CCDC8).
Novel Insights.
We describe 2 siblings with quite mild short stature who carry a homozygous pathogenic mutation in CCDC8 causative of 3-M syndrome. This is the mildest case of 3-M syndrome reported to date and demonstrates the utility of genomic screening in children with undiagnosed growth disorders, especially from consanguineous families.
Acknowledgements
This work was supported by grant K23HD07335 (to A.D.) from the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Disclosure Statement
The authors have no conflicts of interest to declare.
References
- 1.Miller JD, McKusick VA, Malvaux P, Temtamy S, Salinas C: The 3-M syndrome: a heritable low birthweight dwarfism. Birth Defects Orig Artic Ser 1975;11:39–47. [PubMed] [Google Scholar]
- 2.Holder-Espinasse M, Irving M, Cormier-Daire V: Clinical utility gene card for: 3-M syndrome – update 2013. Eur J Hum Genet 2014;2014:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Wal G, Otten BJ, Brunner HG, van der Burgt l: 3-M syndrome: description of six new patients with review of the literature. Clin Dysmorphol 2001;10:241–252. [DOI] [PubMed] [Google Scholar]
- 4.Temtamy SA, Aglan MS, Ashour AM, Ramzy MI, Hosny LA, Mostafa MI: 3-M syndrome: a report of three Egyptian cases with review of the literature. Clin Dysmorphol 2006;15:55–64. [DOI] [PubMed] [Google Scholar]
- 5.Hanson D, Murray PG, Coulson T, Sud A, Omokanye A, Stratta E, Sakhinia F, Bonshek C, Wilson LC, Wakeling E, Temtamy SA, Aglan M, Rosser EM, Mansour S, Carcavilla A, Nampoothiri S, Khan WI, Banerjee I, Chandler KE, Black GC, Clayton PE: Mutations in CUL7, OBSL1, and CCDC8 in 3-M syndrome lead to disordered growth factor signaling. J Mol Endocrinol 2012;49:261–275. [DOI] [PubMed] [Google Scholar]
- 6.Huber C, Dias-Santagata D, Glaser A, O’Sullivan J, Brauner R, Wu K, Xu X, Pearce K, Wang R, Uzielli ML, Dagoneau N, Chemaitilly W, Superti-Furga A, Dos Santos H, Mégarbané A, Morin G, Gillessen-Kaesbach G, Hennekam R, van der Burgt I, Black GC, Clayton PE, Read A, Le Merrer M, Scambler PJ, Munnich A, Pan ZQ, Winter R, Cormier-Daire V: Identification of mutations in CUL7 in 3-M syndrome. Nat Genet 2005;37:1119–1124. [DOI] [PubMed] [Google Scholar]
- 7.Huber C, Delezoide AL, Guimiot F, Baumann C, Malan V, Le Merrer M, Da Silva DB, Bon-neau D, Chatelain P, Chu C, Clark R, Cox H, Edery P, Edouard T, Fano V, Gibson K, Gil-lessen-Kaesbach G, Giovannucci-Uzielli ML, Graul-Neumann LM, van Hagen JM, van Hest L, Horovitz D, Melki J, Partsch CJ, Plau-chu H, Rajab A, Rossi M, Sillence D, Steichen-Gersdorf E, Stewart H, Unger S, Zenker M, Munnich A, Cormier-Daire V: A large-scale mutation search reveals genetic heterogeneity in 3M syndrome. Eur J Hum Genet 2009;17: 395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clayton PE, Hanson D, Magee L, Murray PG, Saunders E, Abu-Amero SN, Moore GE, Black GC: Exploring the spectrum of 3-M syndrome, a primordial short stature disorder of disrupted ubiquitination. Clin Endocrinol (Oxf) 2012;77:335–342. [DOI] [PubMed] [Google Scholar]
- 9.Hanson D, Murray PG, Sud A, Temtamy SA, Aglan M, Superti-Furga A, Holder SE, Urqu-hart J, Hilton E, Manson FD, Scambler P, Black GC, Clayton PE: The primordial growth disorder 3-M syndrome connects ubiquitination to the cytoskeletal adaptor OBSL1. Am J Hum Genet 2009;84:801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanson D, Murray PG, O’Sullivan J, Urqu-hart J, Daly S, Bhaskar SS, Biesecker LG, Skae M, Smith C, Cole T, Kirk J, Chandler K, Kingston H, Donnai D, Clayton PE, Black GC: Exome sequencing identifies CCDC8 mutations in 3-M syndrome, suggesting that CCDC8 contributes in a pathway with CUL7 and OBSL1 to control human growth. Am J Hum Genet 2011;89:148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Al-Dosari MS, Al-Shammari M, Shaheen R, Faqeih E, AlGhofely MA, Boukai A, Alkuraya FS: 3M Syndrome: an easily recognizable yet underdiagnosed cause of proportionate short stature. J Pediatr 2012;161:139–145. [DOI] [PubMed] [Google Scholar]
- 12.Dauber A, Rosenfeld RG, Hirschhorn JN: Genetic evaluation of short stature. J Clin Endocrinol Metab 2014;99:3080–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rabbani B, Tekin M, Mahdieh N: The promise of whole-exome sequencing in medical genetics. J Hum Genet 2014;59:5–15. [DOI] [PubMed] [Google Scholar]
- 14.de Bruin C, Mericq V, Andrew SF, van Duyvenvoorde HA, Verkaik NS, Losekoot M, Porollo A, Garcia H, Kuang Y, Hanson D, Clayton P, van Gent DC, Wit JM, Hwa V, Dauber A: An XRCC4 splice mutation associated with severe short stature, gonadal failure, and early-onset metabolic syndrome. J Clin Endocrinol Metab 2015;100:E789–E798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meazza C, Lausch E, Pagani S, Bozzola E, Calcaterra V, Superti-Furga A, Silengo M, Bozzola M: 3-M syndrome associated with growth hormone deficiency: 18-year follow-up of a patient. Ital J Pediatr 2013;39:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller JD, McKusick VA, Malvaux P, Temtamy S, Salinas C: The 3-M syndrome: a heritable low birthweight dwarfism. Birth Defects Orig Artic Ser 1975;11:39–47. [PubMed] [Google Scholar]
- 17.Dauber A, Stoler J, Hechter E, Safer J, Hirschhorn JN: Whole exome sequencing reveals a novel mutation in CUL7 in a patient with an undiagnosed growth disorder. J Pediatr 2013;162:202–204.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]