

Abstract
The clinical steroidal selective estrogen receptor (ER) degrader (SERD), fUlvestrant, is effective in metastatic breast cancer, but limited by poor pharmacokinetics, prompting the development of orally bioavailable, nonsteroidal SERDs, currently in clinical trials. These trials address local breast cancer as well as peripheral metastases, but patients with brain metastases are generally excluded because of the lack of blood–brain barrier penetration. A novel family of benzothiophene SERDs with a basic amino side arm (B-SERDs) was synthesized. Proteasomal degradation of ERα was induced by B-SERDs that achieved the objectives of oral and brain bioavailability, while maintaining high affinity binding to ERα and both potency and efficacy comparable to fulvestrant in cell lines resistant to endocrine therapy or bearing ESR1 mutations. A novel 3-oxyazetidine side chain was designed, leading to 37d, a B-SERD that caused endocrine-resistant ER+ tumors to regress in a mouse orthotopic xenograft model.
Graphical Abstract
INTRODUCTION
One in eight women will develop invasive breast cancer during their lifetime and, in Europe and the United States, approximately 190 000 women are expected to die from breast cancer in 2019.1 The majority of breast cancers express estrogen receptor α (ERα), which drives proliferation and survival of these tumors.2,3 Endocrine therapy of ER positive (ER+) breast cancer has had a remarkable effect on long-term survival. In premenopausal and postmenopausal women, treatment with tamoxifen (TAM, 1a, Figure 1) and aromatase inhibitors (AIs), respectively, provides effective first line and adjuvant therapy.4,5 AIs prevent the synthesis of estrogens by inhibiting the aromatase enzyme; whereas, the selective ER modulator (SERM), TAM, binds to ER, stabilizing an antagonist conformation in breast cancer cells, resulting in antiproliferative signaling.6,7 Notwithstanding these successes, more women die annually of ER+ breast cancer than triple-negative breast cancer (TNBC), despite TNBC having no safe, targeted therapy.8 This is explained by the high prevalence of ER+ breast cancer and the high rate of resistance to endocrine therapy.9 The battlefield has therefore shifted to endocrine-resistant, metastatic breast cancer (MBC), in which TAM and AIs have lost efficacy.
Figure 1.
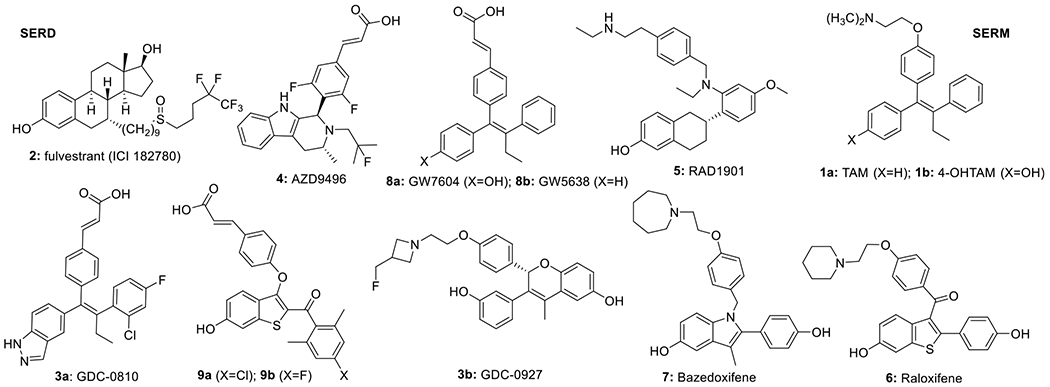
Structures of the SERD fulvestrant (2), oral SERDs (3, 4, 8, 9), together with “SERM/SERDs” (5, 7), and SERMs (1, 6). Several of these SERDs are currently being studied in clinical trials: NCT01823835, NCT02316509, NCT03332797, NCT03236974, NCT03616587, NCT02734615, NCT03284957, NCT02338349, NCT03455270.
In the resistance setting, up to 90% of tumors remain ER+ (i.e., express ERα), wherein ER provides prosurvival signaling even in the absence of estrogens and/or the presence of TAM.10 ER, therefore, remains a target for therapeutics in endocrine-resistant breast cancer, amply demonstrated by the clinical efficacy of the SERD, fulvestrant, 2, which has moved to first line therapy for metastatic ER+ breast cancer.11 Importantly, resistance to one endocrine therapy does not lead to “cross-resistance” to all ER-targeted therapeutics.12 For example, patients who have progressed on SERM treatment will commonly be treated with AIs or fulvestrant.5,13 Similarly, patients with breast cancer tumors resistant to AIs will generally respond to fulvestrant.14 Resistance to endocrine therapy is multifactorial, including upregulation of growth factor signaling, modifications in prosurvival pathways, ER functioning as a ligand-independent transcription factor, and mutations in ER.15–18 Hence, Cdk4/6 inhibitors that block an alternate growth pathway in ER-expressing breast cancer, used in combination with AIs and fulvestrant, have rapidly changed standard of care.19–22
While effective in ER+ breast cancer, fulvestrant (ICI182, 780, 2) has significant pharmaceutical liabilities including poor solubility and pharmacokinetics (PK), which require intramuscular injection. The combination of these issues leads to significant clinical problems in establishing stable and efficacious drug levels.23,24 This has been a strong impetus for the recent entry into clinical trials of orally bioavailable SERDs, including GDC-0810(3a)/GDC-0927(3b)/GDC-9545 (Genentech), AZD9496(4)/AZD9833 (AstraZeneca), LSZ-102 (Novartis), SAR439859 (Sanofi), G1T48 (G1, 9b), and RAD1901 (Radius, 5) (Figure 1). The classification of a drug as a SERD requires the demonstration of enhanced proteosomal degradation of ERα, in addition to antagonist actions on binding to ER.25 In the clinical setting, SERMs are the standard-of-care for premenopausal women because SERMs do not act as antiestrogens in all tissues, in contrast to AIs and fulvestrant; and indeed, the SERM, raloxifene (6), is used clinically to treat postmenopausal osteoporosis.26 The SERM, bazedoxifene (7), approved for postmenopausal osteoporosis in Europe, has been described as a SERM/SERD hybrid;27 and RAD1901 (5), labeled as a SERD, preserves bone mass,28 thereby reclassifying it as a SERM/SERD hybrid (Figure 1).28,29
A majority of SERDs in clinical trials are nonsteroidal acrylates. The acrylate side chain engages in a hydrogen bonding network with helix 12 (H12), as observed in the co-crystal structure of 8a with ERα (pdb 1R5K; Figure 1), causing displacement and destabilization of H12, a conformational trigger to expose a hydrophobic surface, leading to proteasomal degradation of ER. The acrylate side chain can limit brain bioavailability, and in some cases clinical trials of oral SERDs exclude patients with brain metastases. Patients with brain metastases have extremely poor prognosis; therefore, our motivation in developing a SERD with a basic amino side arm (B-SERD) was to ensure good brain bioavailability to allow treatment of this population.30–32 The replacement of an acrylate anion with a basic amino group would be expected to improve blood–brain barrier penetration. We recently developed and optimized a unique benzothiophene chemical scaffold as the basis for a family of potent acrylate benzothiophene SERDs (e.g., 9) with oral bioavailability and in vivo efficacy.33 We therefore used this scaffold to explore a variety of basic side arms, with the objective of maintaining the excellent potency and efficacy of 2, whilst gaining the oral and brain bioavailability lacking in 2.
STRUCTURE DESIGN
We have made numerous modifications to benzothiophene scaffolds to diversify the biological activity of ER ligands.34–41 To successfully generate the potent, oral SERD, 9, we designed a unique scaffold substituted with an acrylate containing side chain.33 Co-crystal structures of SERMs, containing the archetypical SERM 2-phenoxyethylamino side chain, bound to ERα reveal the key salt–bridge interaction between the SERM side chain amino group and Asp-351, and we hypothesized that retaining this interaction and extending the aliphatic side chain would displace H12, expose its hydrophobic surface and result in ERα degradation.42–45 Molecular modeling of putative B-SERDs using the co-crystal structure of 4 bound to ERα (pdb 5ACC) supported this hypothesis; and suggested that favorable interactions targeted for 9 with the two hydrophobic cavities formed by Leu 384 and Leu 428 (pdb 1R5K) could be maintained in a B-SERD (Figure 2).
Figure 2.
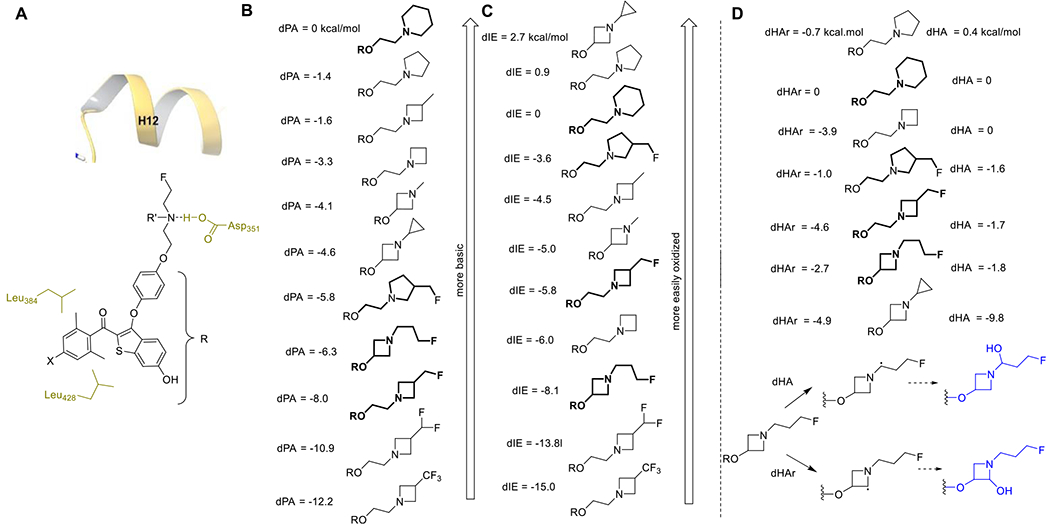
Structure design. H-bonding of the amine side chain to Asp-351 in the ERα ligand-binding pocket should allow engagement of ring substituents with hydrophobic pockets formed in the region of Leu-384 and Leu-428 (increasing affinity), whilst displacing H12 (causing ERα degradation). The design of the amine side chain using a conformationally restricted heterocycle considered the ability to interact with H12 (A); the amine basicity (B); and susceptibility to oxidation (C,D). DFT molecular orbital calculations of proton affinity (dPA) (B), ionization energy (dIE) (C), and H-atom abstraction (dHA) (D) were normalized relative to the calculated free energy for the piperidine side chain: dHAr corresponds to heterocyclic ring-C oxidation; dHA refers to oxidation of the alternate carbon. The R group in (A) is modeled by H in calculations.
The choice of constrained basic side arm for a B-SERD ranges from the pyrrolidine, piperidine, and azepane rings found in SERMs and SERDs, to the azetidine ring found in a SERD reported in 2019, after completion of our lead optimization campaign (3b).46 Effective side arms would presumably need to maintain a salt bridge or H-bond with Asp-351, with the strength of this interaction influenced by amine basicity. In addition, SERMs are well known to undergo oxidative metabolism leading to metabolites formed from N-desalkylation. Because both characteristics are expected to be strongly influenced by ring size (hybridization and ring strain) and electron-withdrawing substituents, calculations were performed on candidate side arms using density functional theory (DFT) molecular orbital calculations at RI-MP2/6– 311++G**//B3LYP/6-31+G** (Figure 2 and Supporting Information). The influence of ring size is to decrease nitrogen basicity as the ring size contracts, and to further decrease basicity with electron-withdrawing group substitution. In general, a similar trend is seen for ionization energy, indicating that azetidine side arms are less susceptible to oxidation, although a cyclopropyl substituent on the azetidine ring stabilizes the radical cation formed on oxidation. The relative energy for H atom abstraction (dHA) α to N is an indicator of susceptibility to phase 1 oxidation, potentially leading to N-desalkylation (Figure 2D). Again, the azetidine ring carbons are predicted to be less readily oxidized, especially in the fluoro-substituted derivatives. Although calculated electronic contributions can be overwhelmed by ligand binding site interactions and the microenvironment, the calculated trends encouraged exploration of azetidine side arms.
CHEMISTRY
To construct the 2-keto-benozothiophene core required by our structure design, commercially available 4-methoxycinnamic acid was used, followed by cyclization to afford 3-chloro-6-methoxybenzo[b]thiophene-2-carbonyl chloride (10) (Scheme 1). The acyl chloride was converted to a Weinreb amide, which was subsequently reduced to the corresponding aldehyde (12) with DIBAL-H at −40 °C. A variety of Grignard reagents were shown to react with the aldehyde to afford diverse secondary alcohols 13a–d that were oxidized to generate phenyl ketones 14b–e. Meanwhile, ketones 14a, 14f, 14h, and 14i were obtained from the reaction of Weinreb amide with Grignard reagents at 0 °C. Compound 14g was obtained from the Grignard reagent reacting with acyl chloride 10. The deprotection of the methoxy group of compounds 14 was performed with Lewis acid BF3·Me2S in an ice bath.33 The resulting phenol was protected with tetrahydropyran (THP) to afford the primary precursor synthons 16a–i. To obtain the azetidine synthon required by our structure design, we started with t-butyl 3-(hydroxymethyl)azetidine-1-carboxylate 17, installing the fluoro substitution using tetrabutylammonium fluoride under reflux to generate compound 19. The azetidine synthon 20 was obtained by deprotection using 4 M HCl; and the pyrrolidine synthon 24 was obtained in a similar way from 21 (Scheme 1). For the construction of the side chain, 2-(4-(2-bromoethoxy)phenoxy)tetrahydro-2H-pyran 26 was obtained from 4-((tetrahydro-2H-pyran-2-yl)oxy)phenol 25. Coupling of 26 to the appropriate amine under basic conditions (NaH) gave the THP-protected side chain: 27a–f (Scheme 2). Deprotection of the THP protecting group afforded compounds 28a–f that underwent the SNAr reaction with synthons 16a–i to produce 29a–o, which gave final products 30a–o after deprotection.
Scheme 1. Synthetic Routes for Precursor Synthonsa.
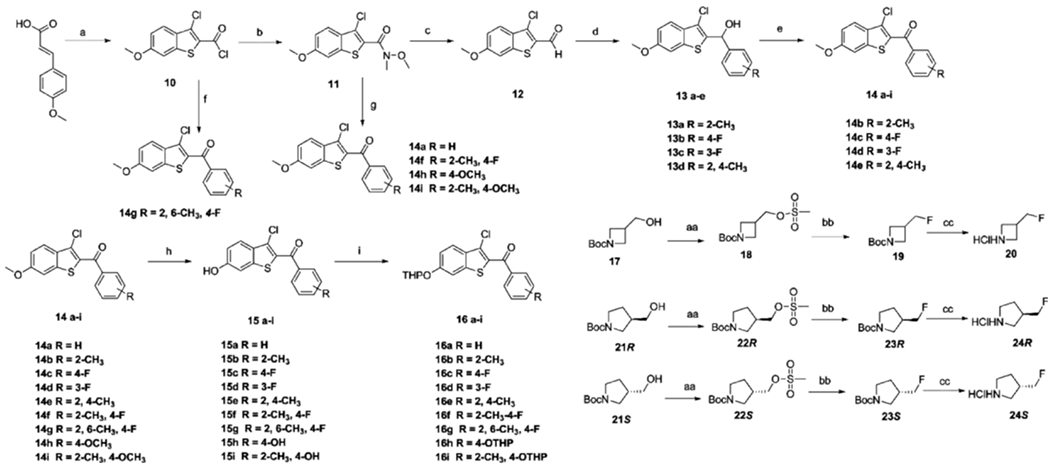
aReagents and conditions: (a) SOCI2, pyridine, chlorobenzene, reflux, 50%. (b) N-Methoxymethylamine, Et3N, DCM, rt, 90%. (c) DIBAL-H, THF, −40 °C, 60%. (d) Grignard reagent, THF, 0 °C to rt, 75–85%. (e) PCC, DCM, rt, 55–65%. (f) (4-Fluoro-2,6-dimethylphenyl)magnesium bromide, CuCN-2LiCl, THF, 0 °C to rt, 90%. (g) Grignard reagent, THF, 0 °C to rt, 70–80%. (h) BBr3, DCM, −78 °C to rt, 40–60%. (i) 3,4-Dihydro-2H-pyran, PPTS, DCM, rt, 70–80%. (aa) Methanesulfonyl chloride, Et3N, DCM, 0 °C to rt, 95%. (bb) TBAF, 80 °C, 70%. (cc) 6 M HCI MeOH, rt, 60%.
Scheme 2. Synthetic Routes for Candidate Piperidine, Azetidine, and Pyrrolidine B-SERDsa.
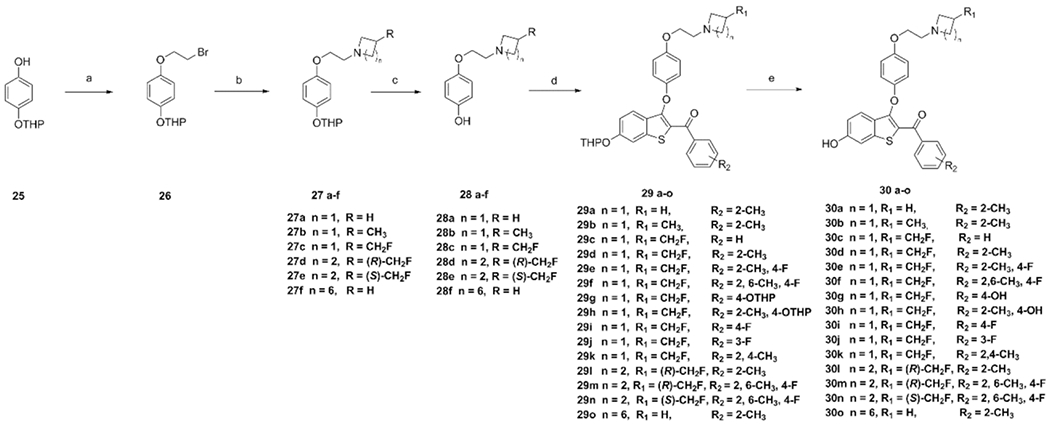
aReagents and conditions: (a) 1,2-dibromoethane, KOH, THF, reflux, 50%. (b) Azetidine hydrochloride, 3-methylazetidine hydrochloride, 20, 24, piperidine, NaH, THF, 0–60 °C, 55%, (c) p-TsOH, MeOH, rt, 60%. (d) 16a-i, Cs2CO3, DMF, 90 °C, 55–75%. (e) p-TsOH, MeOH, rt, 60–80%.
The 3-oxy-azetidine, or “reverse azetidine”, side arm was derived from the reaction of t-butyl 3-iodoazetidine-1-carboxylate with 4-(benzyloxy)phenol 31 to afford t-butyl 3-(4-(benzyloxy)phenoxy)azetidine-1-carboxylate 32 (Scheme 3). Deprotection in trifluoroacetic acid (TFA) to give 33 was followed by alkylation with 1-bromo-3-fluoropropane or (1-ethoxycyclopropoxy) trimethylsilane, to give 34a and 34b, respectively. Deprotection was performed under Pd/C and H2 to afford compounds 35a,b, which were coupled with the appropriate precursor synthon (l6a,b,d,f,g) to yield 36a–f using the procedure shown in Scheme 2. Deprotection of THP under mild acidic conditions gave 37a–f.
Scheme 3. Synthetic Routes for Candidate Reverse-Azetidine B-SERDsa.
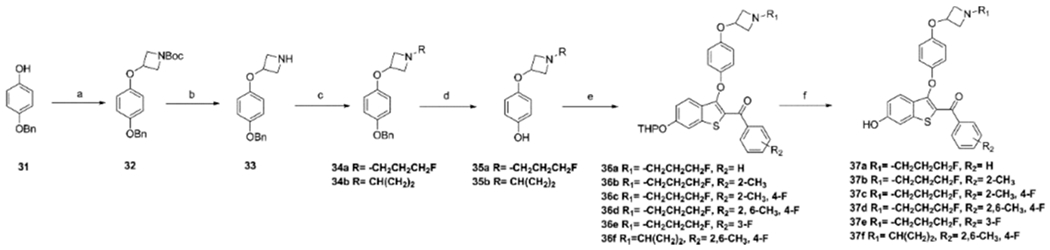
aReagents and conditions: (a) 11-boc-3-iodoazetidine, Cs2CO3, DMF, 140 °C, 50%. (b) CF3COOH, DCM, rt, 70%. (c) 1-Bromo-3-fluoropropane, NaH, 0–60 °C, 55%; or (1-methoxycyclopropoxy)trimethylsilane, AcOH, NaBH3CH, 80% (d) H2, Pd/C, rt, 70%. (e) 16a, 16b, 16d, 16f, 16g, Cs2CO3, DMF, 90 °C, 75–85%. (f) p-TsOH, MeOH, rt, 70–80%.
BIOLOGICAL TESTING
Since the ultimate goal of this study was to discover an oral SERD suitable for treatment of brain metastases in endocrine-resistant ER+ breast cancer, lead optimization was driven by efficacy in endocrine-resistant, ER+ breast cancer cell lines. A development lead would need to demonstrate efficacy in breast cancer cells bearing mutations in ESR1, brain penetration, and cause regression of endocrine-resistant tumors in a mouse xenograft study.
Optimization of the B-SERD scaffold was driven by in vitro assays in 2D and 3D breast cancer cell cultures. The primary objective, to develop new oral SERDs for treatment of MBC resistant to endocrine therapy, required the use of breast cancer cell lines modeling TAM and AI resistance. The MCF7:TAM1 cell line models resistance to TAM and AIs, having been developed by long-term exposure of parental, endocrine-dependent MCF7:WS8 cell cultures to the active metabolite of TAM, 4OH-TAM (1b, Figure 1) with concurrent long-term estrogen deprivation (LTED); whereas the MCF7:5C cell line was developed from LTED of MCF7:WS8 cell cultures.34,47 Both cell lines undergo estrogen-independent growth and are insensitive to treatment with 1b, in contrast to the parent MCF7:WS8 cell line, growth of which is dependent on estrogens and inhibited by 1b. In addition to these cell lines, we evaluated the T47D:Y537S and T47D:D538G cell lines obtained from CRISPR-Cas9 manipulation of T47D-WT cells as described previously.48 ESR1 mutations are prevalent in ER+ MBC after AI therapy and the Y537S and D538G mutations are associated with AI resistance and a more aggressive disease.49 Not only do the T47D ESR1 mutant cell lines provide an additional model of resistance, the T47D cell line itself provides a different ER+ tumor background.
In our previous development of oral SERDs, exemplified by 9, we used oral SERD 3a as a benchmark; however, this oral SERD failed in clinical trials; thus, we now use fulvestrant, 2, as a benchmark, because it is currently the only clinical SERD. It should be noted that: 2 demonstrates high efficacy and potency in cell cultures and cell-derived xenografts (CDX); and the severe pharmacokinetic limitations experienced in the clinic, which limit efficacy, are not recapitulated in preclinical models. Therefore, the desired B-SERD will match the high potency and efficacy of 2 in cell lines and CDX, but in contrast to 2, will demonstrate oral bioavailability and brain exposure. All cell lines used for B-SERD optimization are sensitive to growth inhibition by 2. In both cell cultures and in vivo measurements of target engagement and side effects, we have also compared against the SERDs: 3b and 9; and the SERM/SERDs, 5 and 7.
Cell viability assays were performed in 2D monolayer cell and 3D spheroidal cell cultures to measure the ability of B-SERDs to inhibit growth of both endocrine-resistant and parent cell lines. Full concentration–response curves were obtained for B-SERDs and for 2 to derive potency for inhibition of cell growth in 2D monolayer cultures of MCF7:WS8 and MCF7:5C cell lines, measuring cell DNA content. The maximum efficacy for inhibition is reported relative to vehicle (0%) and cell medium only (100%), such that the maximum efficacy of 2 was measured as 66 and 48% inhibition of cell growth in parental and endocrine-resistant breast cancer cell lines, respectively. Inhibition of estrogenic activity, required for SERD activity, was measured in the endocrine-dependent parental cell line in competition with E2, using a transient ER response element (ERE)-luciferase transcriptional reporter and compared to relative binding affinity (RBA) to ERα using a radioligand binding assay. The effect of treatments on the ER protein level was measured using in-cell westerns (ICW) in MCF7:WS8 cells and confirmed by western blots in the presence and absence of a proteasome inhibitor to show proteasomal degradation. While the monolayer cell culture allows a higher throughput and allows the multiplate measurement of ER content using ICW, 3D spheroids provide a more physiologically relevant cell model, more closely mimicking the increased cell–cell signaling and hypoxic core, observed in solid tumors.50 Cell viability for 3D spheroid cultures was measured in parent MCF7:WS8 and endocrine-resistant MCF7:TAM1 cell cultures as well as in three T47D cell lines, including those with Y537S and D538G mutations in ESR1. The novel B-SERD showing a superior PK profile was: (1) compared with 2 in an MCF7:TAM1 orthotopic CDX mouse model; (2) compared with 2 and 7 in the assessment of unwanted uterotrophic effects in juvenile rats; and (3) compared with 9a in assessment of target engagement (ERα immunoassay) in the uterus and ovaries of intact female mice.
RESULTS
The benchmark SERD, 2, inhibits growth of endocrine-resistant and parental cell lines in 2D cultures with high potency (pIC50 = 8.8–9.2) and with observed reduced maximal efficacy in the endocrine-resistant MCF7:5C cell line (Table 1). Of the B-SERDs with a pyrrolidine side arm, 30l showed superior potency and efficacy to 2 in endocrine-dependent and -independent cell cultures (Table 1). The enantiomer, 30m was marginally inferior to 30n in both cell lines. As also observed for 2, all the pyrrolidines studied (30l–n) lost significant efficacy in endocrine-resistant MCF7:5C cells. In contrast to 2 and to the pyrrolidines, the piperidine 30o did not inhibit growth of MCF7:5C cells. MCF7:5C cells are TAM resistant and are cross-resistant to the SERM raloxifene 6 (Table 1). The very similar cellular phenotype induced by 6 and 30o strongly suggests that 30o is a SERM and that cross-resistance in MCF7:5C cells also extend to this SERM.
Table 1.
Antiproliferative Activity of Pyrrolidine and Piperidine Ligands in Breast Cancer Cellsa
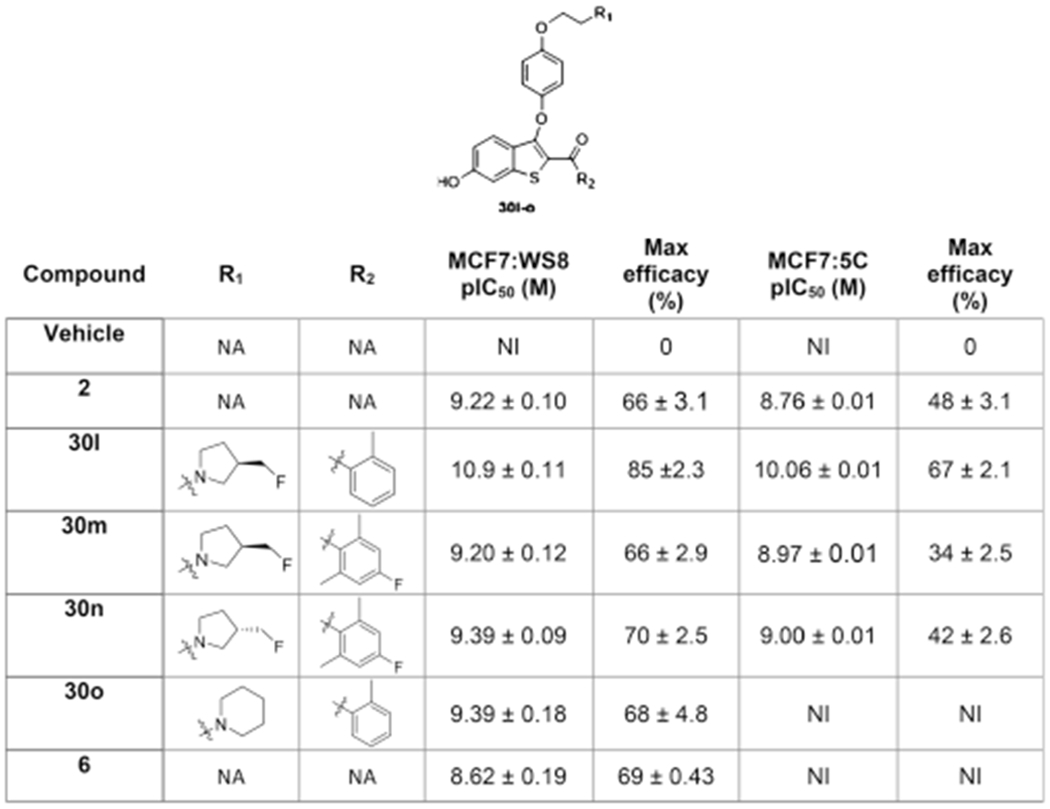 |
Potency was determined by DNA content after 4 or 6 days of incubation in MCF:WS8 or MCF7:5C respectively, following treatment with B-SERDs. The signal was normalized to vehicle control. Data shown as mean ± SEM from at least three biological and analytical replicates. Maximum efficacy was normalized to vehicle (0%) and no cells (100%). NA = not applicable; NI = no inhibition.
Moving from a pyrrolidine to an azetidine side arm led to no loss of efficacy and potency in growth inhibition of MCF7:WS8 cells (9.0 < pIC50 < 10.4; Table 2). Comparison of the 2-Me-phenyl-substituted series of azetidine ligands bearing different substitutions on the azetidine ring, 30a, 30b, and 30d, showed identical efficacy, with 30b having the higher growth inhibition potency of the parental cell line. As we saw for 30o (and 6), high potency in the parental cell line can translate to a total loss of efficacy in the MCF7:5C cell line for a SERM (Table 1). The unadorned 4-membered azetidine ring of 30a confers potent SERD activity in inhibiting growth of both cell lines, but significantly reduced efficacy in MCF7:5C cells. Structurally, 30a resembles a SERM; however, the observed activity shows that TAM-resistant MCF7:5C cells that are cross-resistant to the piperidine SERMs, 6 and 30o, are not cross-resistant to 30a, leading to speculation that 30a may fall in the SERM/SERD classification. Regardless, the lack of cross-resistance supports the selection of the azetidine side arm for further exploration.
Table 2.
Antiproliferative Activity of Azetidine Ligands in Breast Cancer Cellsa
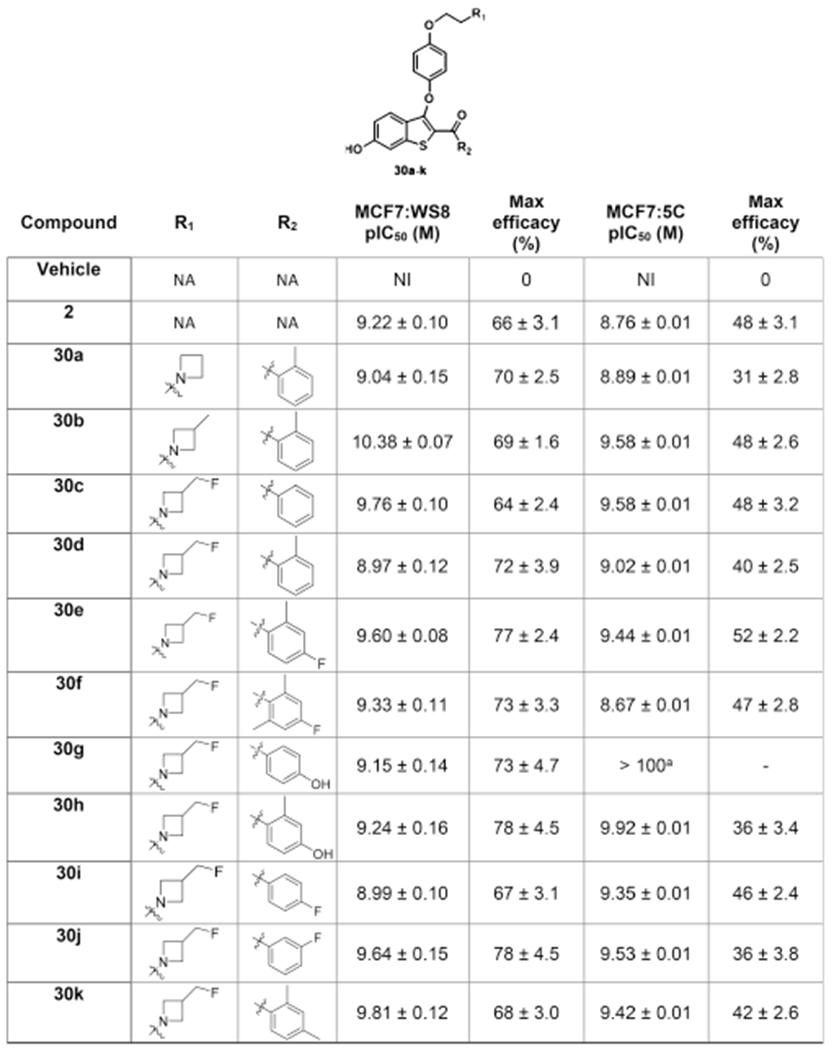 |
Potency was determined by DNA content after 4 or 6 days of incubation in MCF7:WS8 or MCF7:5C respectively, following treatment with B-SERDs. Signal was normalized to vehicle control.
IC50 > 100 nM; 45% inhibition at 1 μM. Data shown as mean ± SEM from at least three biological and analytical replicates. Maximum efficacy was normalized to vehicle (0%) and no cells (100%). NA = not applicable; NI = no inhibition.
Comparison of the nine 3-fluoromethyl azetidine derivatives, 30c–k, allows comparison of the effect of different substitution patterns of the benzoyl ring; however, all derivatives potently inhibited growth in MCF7:WS8 cells (8.9 < pIC50 < 9.8). In endocrine-resistant cells, many derivatives showed a small loss of potency, and a loss of efficacy; with the exception of 30g that lost efficacy in MCF7:5C cells. We speculate that the provision of a second phenolic group in 30g and 30h, in addition to the benzothiophene phenol, facilitates an alternative binding mode that is more akin to a SERM, and MC7:5C cells are, in general, resistant to SERMs. The metasubstitution in 30j is also not a preferred substitution as indicated by efficacy in MCF7:5C cells. These observations support the importance of the o-methyl group in exploiting the small hydrophobic pockets created by Leu-384 and Leu-428, explored in the optimization of the SERD 9, in maintaining potency in the endocrine-resistant cell line.
We next explored derivatives with a “reverse azetidine”, or 3-oxy-azetidine, side arm. This modification moves the conformational lock of the azo-ring closer to the more rigid benzothiophene core. To induce the displacement of H12, which underlies the high potency and efficacy observed with azetidine ligands (Table 2), a longer and flexible fluoropropyl chain was conjugated to the azetidine nitrogen. Given the potential for the N-cyclopropyl substituent to increase stability to phase 1 metabolism (Figure 2), compound 37f was prepared; although this ligand was potent and effective in MCF7:WS8 cells, disappointingly, it showed a loss of efficacy in TAM-resistant cells (Table 3). All reverse azetidines, 37a–f, were potent (9.3 < pIC50 < 10.2) and efficacious inhibitors of MCF7:WS8 cell growth.
Table 3.
Antiproliferative Activity of Reverse Azetidine Ligands in Breast Cancer Cellsa
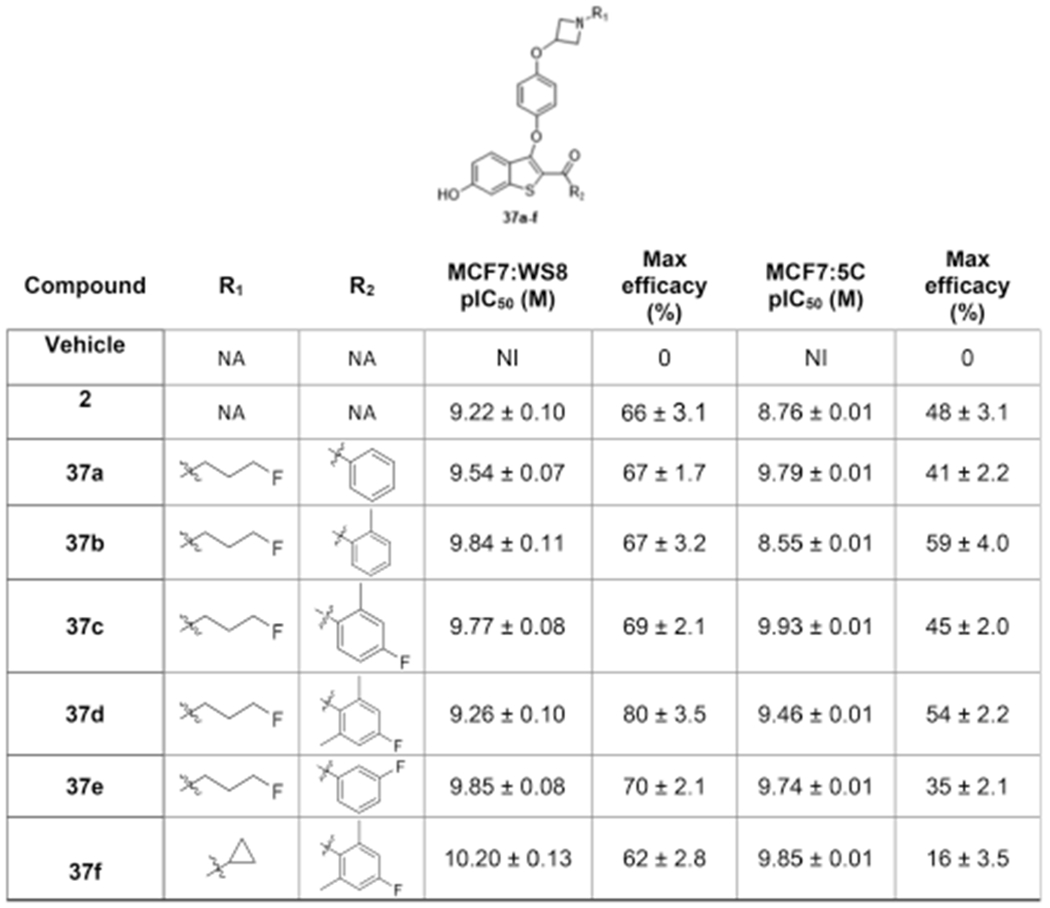 |
Potency was determined by DNA content after 4 or 6 days of incubation in MCF7:WS8 or MCF7:5C respectively, following treatment with B-SERDs. Signal was normalized to vehicle control. Data shown as mean ± SEM from at least three biological and analytical replicates. Maximum efficacy was normalized to vehicle (0%) and no cells (100%). NA = not applicable; NI = no inhibition.
As with the 3-fluoromethyl azetidine series, the predicted reduced basicity of the N-fluoropropyl azetidine nitrogen did not influence potency or efficacy. Having established the efficacy of the putative B-SERDs in inhibiting growth of endocrine-resistant and parental cell lines, it was essential to measure ERα levels to confirm the functional identity of these compounds as SERDs. Potency toward degradation of ERα was studied in MCF7:WS8 cells measured by ICW. All B-SERDs that induced growth inhibition also caused the loss of ERα with potency comparable to 2 (Table 4). The SERM, 30o, did not induce ER degradation (Figure S1). The cyclopropylidene derivatized reverse-azetidine, 37f, showed low efficacy in the ICW assay. Despite predicted stability against metabolic N-desalkylation (see Figure 2) this compound was not considered as a development candidate. Interestingly, the unadorned azetidine (30a) and fluoromethyl pyrrolidine (30n) derivatives also manifested reduced efficacy, as did the bisphenolic derivative, 30h. These compounds also showed lower efficacy in growth inhibition of MCF7:5C cells.
Table 4.
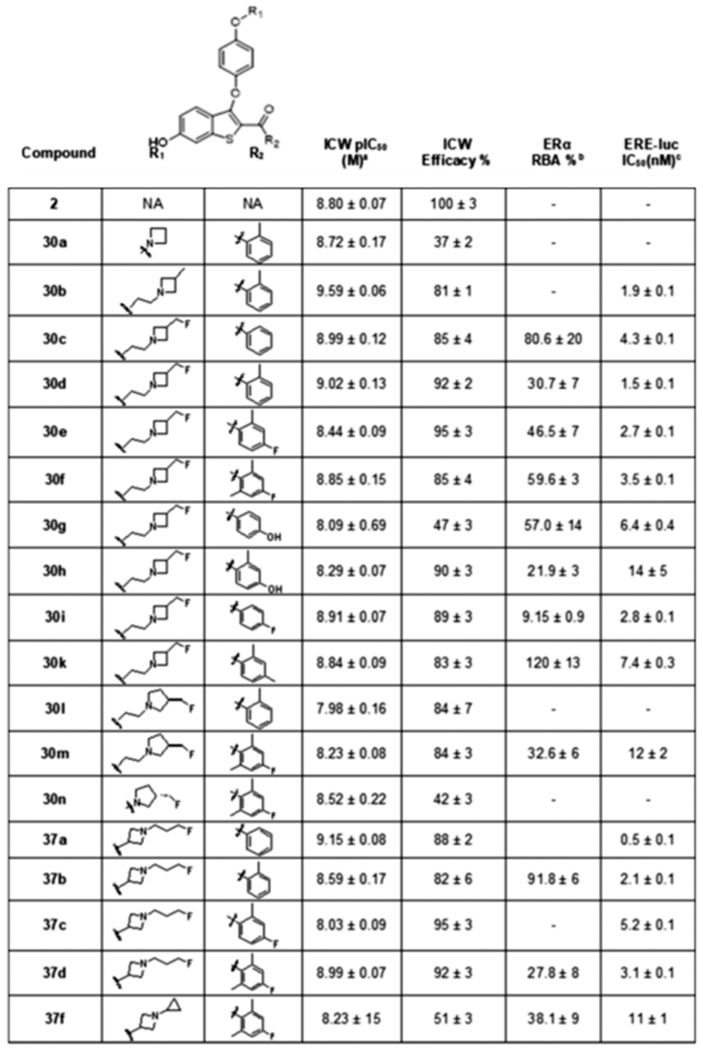 |
ER degradation potency and efficacy normalized to vehicle (100%) and 1 μM 2 (0%).
RBA values, determined by radioligand displacement assays expressed as IC50 estradiol/IC50 compound × 100 (RBA, estradiol = 100%).
Estrogenic activity (ERE IC50), as determined by ERE dual luciferase in competition with 1 nM E2, normalized to 1 nM E2 alone (100%) and vehicle (0%).
Data shown as mean ± SEM from at least three biological and analytical replicates.
Data shown as mean ± SEM from three analytical replicates. NA = not applicable; NI = no inhibition.
SERDs and SERMs, by definition, antagonize the actions of E2 in breast cancer cells, by inducing the binding of a repressed ER transcriptional complex to the ERE causing inhibition of selective gene transcription. All B-SERDs tested antagonized ERE-luciferase induction by E2 with low nanomolar potency, with 30h, 30m, and 37f excluded from further consideration, based on lower potency (Table 4). For selected compounds, RBA was measured to confirm sub-nanomolar affinity for ERα (Table 4). The selectivity for ERα/ERβ given by RBA measurements was less than fourfold for all B-SERDs tested (Table S1).
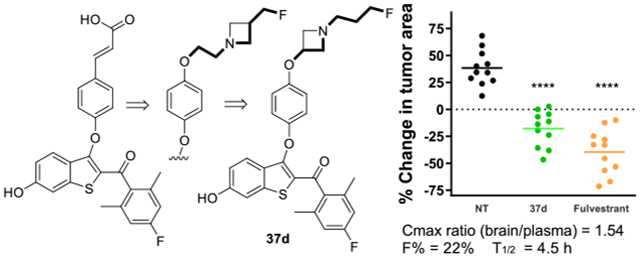
Based upon the foregoing data collected in 2D cell cultures, several B-SERDs were excluded from further study; however, many examples remained with high potency and efficacy, and clear evidence of target engagement. The success of the 2,6-dimethyl,4-halo acrylate SERDs (9) in our previous preclinical studies33 biased us toward these derivatives; therefore, 30f, 30m, and 37d, were prioritized for a preliminary study of oral brain bioavailability. The three B-SERDs tested were delivered orally at 100 mg/kg to C57/BL6 mice with plasma and brain concentrations measured by LC-MS/MS at 30 and 120 min and compared to fulvestrant delivered by the standard s.c. route (Table 5). B-SERDs showed the desired brain bioavailability; for example, the concentration of 30f in the brain at 0.5 and 2 h was 600–750 nM and that of 37d exceeded 1 μM, whereas the concentration of 2 in the brain was below the limit of quantification at 30 min. The 3-oxyazetidine side-armed B-SERD, 37d, gave substantially higher plasma and brain concentrations compared to the analogues bearing either azetidine (30f) or pyrrolidine side-arms (30m). The pyrrolidine, 30m, was not studied further.
Table 5.
Preliminary Pharmacokinetic Screen of 2,6-Dimethyl-4-fluoro B-SERDs Compared to 2a
| plasma concentration (nM) |
brain concentration (nM) |
|||
|---|---|---|---|---|
| compound | 0.5 h | 2 h | 0.5 h | 2h |
| 2 | 31.2 ± 16.5 | 301 ± 268 | 0.7 ± 0.8b | 47.4 ± 61.1 |
| 30f | 786 ± 179 | 659 ± 329 | 750 ± 673 | 588 ± 274 |
| 30m | 211 ± 45 | 163 ± 141 | 121 ± 61 | 276 ± 93 |
| 37d | 2490 ± 797 | 2150 ± 201 | 1630 ± 1110 | 1250 ± 406 |
Plasma and brain concentration of 30f, 30m and 37d (100 mg/kg p.o.), and 2 (5 mg s.c.) showing mean and s.d. (N = 3).
Below LOQbrain = 5nM.
B-SERDs 30f and 37d were compared with congeners, 30i and 37b, respectively, and with SERD 2 in 3D cultures following treatment for 14 days (Figure 3A). In the parental, MCF7:WS8 cell line, all SERDs (10 nM) were equally effective in inhibiting growth of spheroids as shown by spheroid size and viability measured by ATP content (Figure 3A,B). However, in the TAM-resistant MCF7:TAM1 spheroids, significant differences were observed between treatments, with 2, 30f, and 37d having greater efficacy. Growth of MCF7:TAM1 cells is endocrine-independent and TAM-resistant, and because this is an LTED cell line, it models resistance to AI; however, it does not harbor mutations in ESR1, which are known to be associated with acquired AI resistance. The efficacy of B-SERD 37d was compared to SERDs (2, 3b) and SERM (1b) in T47D cell lines expressing only mutant ERα (TYS = Y537S; TDG = D538G) or WT ERα.48 B-SERD 37d was equally as effective as the SERDs, fulvestrant (2) and GDC-0927 (3b), and as the SERM 1b (Figure 3C).
Figure 3.
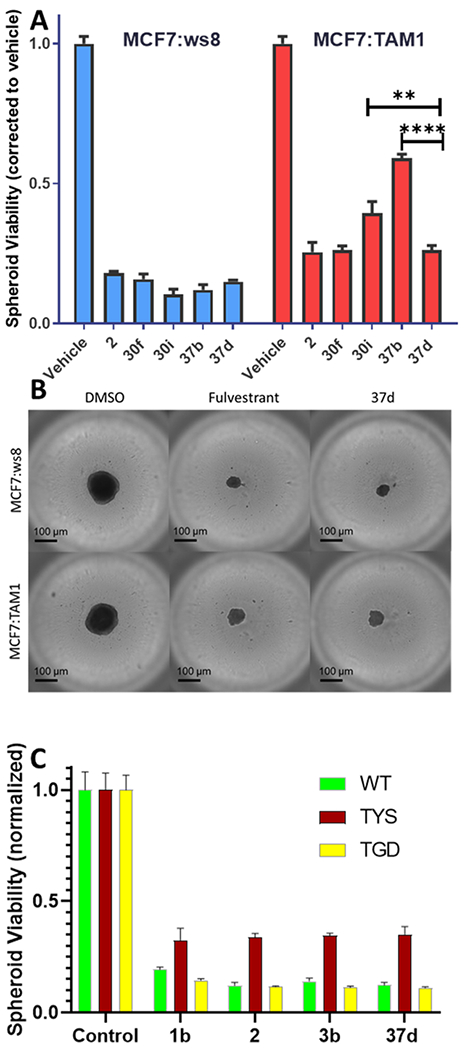
Spheroid viability. (A) MCF7:WS8 and MCF7:TAM1 spheroid viability following treatment with B-SERDs (30f, 30i, 27b, and 37d, 10 nM) compared to 2 (10 nM). (B) Representative images on day 10 of treatment with DMSO (0.01%), 2 (10 nM), and 37d (10 nM) in MCF7:WS8 and MCF7:TAM1 spheroids. (C) T47D:WT, T47D:TYS, and T47D:TGD cells grown for 3 days were treated for a further 11 days with test compounds (10 nM). (A,C) Luminescence normalized to vehicle/control (1.0) and background (0.0). Data shown as mean ± SEM from three biological and analytical replicates. Significance compared to vehicle/control by one-way ANOVA: p <0.0001.
Western blots supported the observations on B-SERD-induced ER degradation in cell cultures made using ICW, and using co-treatment with the proteasomal inhibitor, MG-132, western blots also confirmed the role of the proteasome in degradation (Figure 4A). Representative western blots demonstrating ERα degradation from treatment of the parent cell line are shown in Figure 4B (and Figure S2), providing a comparison of 37d with the structurally related acrylate, 9a, clinical SERD, 2, and SERM/SERD, 5. We extended these in vitro observations to examine the effects of oral administration of the B-SERD, 37d, compared to the congenic acrylate SERD, 9a, in intact female mice (Figure 4C). In these experiments, animals were sacrificed and tissues flash frozen, at either 5 or 7 h after oral drug administration. In the acute treatment paradigm, the mice received only one dose before sacrifice; whereas, in chronic treatment, the mice were treated with the drug for 3 days. Treatment with 37d or 9a (50 mg/kg) under all conditions led to significant (p < 0.01 control vs treated groups) loss of ERα, as quantified by the western blot of tissue homogenates (Figure 4C). After single dose administration, degradation of ERα in ovaries was significantly greater with 37d versus acrylate SERD 9a. After multiple doses, degradation of ERα in the uterus was significantly greater with 37d versus acrylate SERD 9a.
Figure 4.
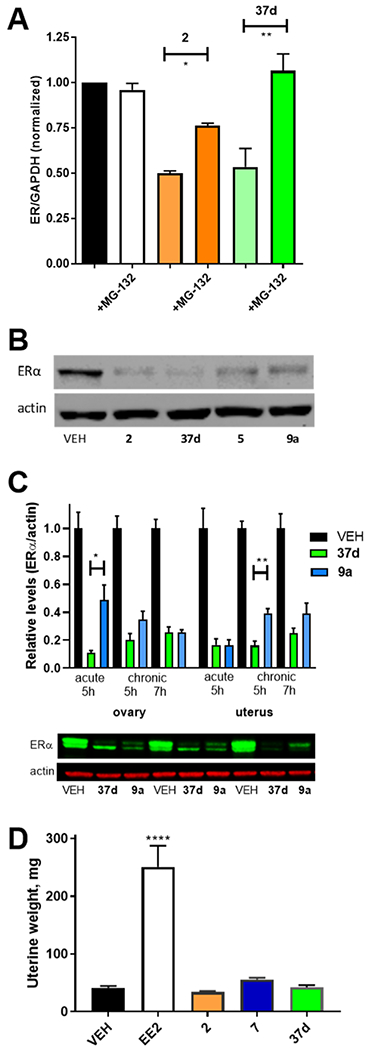
B-SERD vs SERD comparisons. (A). ERα degradation after 24 h treatment ofMCF7:WS8 cells with 10 nM 37d or 2 measured by western blot and inhibited by proteasomal inhibitor MG-132 (1 μM) normalized to vehicle (1.0). (B) ERα western blots after 24 h treatment of MCF7:WS8 cells with 100 nM 2, 37d, 5, and 9a measured by the western blot. (C) ERα degradation after oral dosing of female mice with vehicle, 37d or 9a, measued by western blot analysis of tissues, with representative immunoblots shown from individual mouse uterus. (D) Uterine weight from juvenile female rats dosed with 2, 7, and 37d, compared to EE2 as a positive control. Cell culture data shown as mean ± SEM from three biological and analytical replicates. Statistical analysis by one-way ANOVA with multiple comparisons (p* < 0.01; *** > 0.001; **** < 0.0001).
The juvenile rat model is widely used for assessment of uterotrophic activity of ER ligands because the young female rat is highly sensitive to ER ligands, but without a background of high levels of circulating endogenous estrogens. Ethinylestradiol (EE2) was administered as a positive control, yielding a significant increase in uterine weight (Figure 4D). There was no significant uterotrophic effect of the SERM/SERD, 7, nor the SERDs, 2 and 37d, versus the vehicle control.
Full PK analysis for 37d was conducted in CD-1 mice to determine rigorous oral bioavailability parameters, demonstrating absolute bioavailability of 22%, a half-life of 4.5 h, and a brain/plasma ratio for Cmax and AUClast of 1.54 and 1.26, respectively (Table 6 and Figure S3).
Table 6.
Pharmacokinetics of 37da
| dose (mg/kg) | T1/2 (h) | Tmax (m) | Cmax (ng/mL) | AUClast (h ng/mL) | AUCInf (h ng/mL) | AUCExtrapobs % | MRTInfobs (h) | AUClast/D (ng/mL) | F (%) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Plasma | 50 p.o. | 4.48 | 1.00 | 413 | 1678 | 2292 | 26.8 | 6.05 | 33.6 | 21.9 |
| Brain | 50 p.o. | 1.00 | 635 | 2106 | 3450 | |||||
| Plasma | 5 i.v. | 1.68 | 1592b | 766 | 778 | 1.52 | 1.07 | 153 |
PK parameters from measurements at 0.25, 0.5, 1, 2, 4, 8 and 24 h (N = 5).
C0.
Efficacy in MCF7:TAM1 spheroids is predictive of efficacy in xenografts and therefore 37d was tested in breast cancer tumors that had been established by injecting MCF7:TAM1 cells into the mammary fat pads of nude mice. Tumors in mice treated with the vehicle or 1a continued to grow over the treatment period, whereas in mice treated with 2 (s.c.) or 37d (p.o.) tumor regression was observed (Figure 5A). The observations with 1a reinforced that these breast cancer tumors are TAM resistant. Analysis of individual tumors is shown for SERD treatment compared to the no treatment (NT) group, to show statistical significance in tumor regression (Figure 5B).
Figure 5.
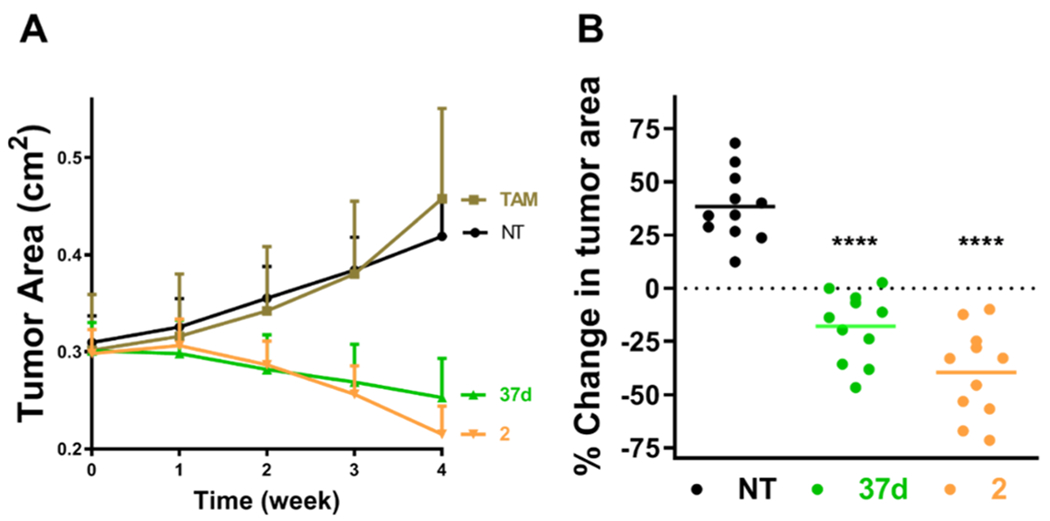
Effect on tumor growth. (A) Tumor growth or regression of MCF7:TAM1 CDX grown to 0.3 cm2 before initiating daily treatment with TAM (1a), 37d (100 mg/kg oral gavage) or 2 Individual tumor % area change after 4 weeks treatment: ****p < 0.0001 vs NT group, by one-way ANOVA with Dunnett’s test.
DISCUSSION
The search for an orally, brain bioavailable B-SERD, started with our unique benzothiophene scaffold developed for the acrylate SERD (9b), currently in clinical trials. Modification of this scaffold on the benzoyl ring, and with a basic side chain yielded a number of interesting ERα ligands. The piperidine derivative, 30o, demonstrated SERM-like characteristics; whereas most derivatives that substituted the piperidine side arm with a pyrrolidine, azetidine, or reverse azetidine, demonstrated SERD activity. The azetidine 30a, a direct analogue of 30o, inhibited growth of a TAM-resistant cell line and lowered ERα levels; however, the observed reduced efficacy indicates a mixed SERM/SERD activity. Several other derivatives, such as 30g, and the cyclopropyl azetidine 37f, showed activity profiles worthy of further study; however, our objective was to select a SERD with comparable potency and efficacy to the clinical SERD 2, but with oral and brain bioavailability. The compound selected, 37d, bears a novel reverse azetidine side arm.
The success of 2 in treating advanced, metastatic ER+ breast cancer provides the rationale for oral SERD development; therefore, efficacy in endocrine-resistant breast cancer cells is crucial. In parent cell lines, pyrrolidine, azetidine, and reverse azetidine derivatives with potent and high efficacy SERD activity demonstrated high potency and high efficacy inhibition of cell growth. However, almost all compounds demonstrated reduced efficacy in growth inhibition of the endocrine-resistant cell line, including 2. In this cell line, derivatives containing fluoromethyl pyrrolidine, fluoromethyl azetidine, or N-fluoropropyl azetidine, bearing benzoyl rings substituted o- or p- with fluoro or methyl groups, demonstrated a higher potency (pIC50 = 9.3 ± 0.4) and identical efficacy to 2 (Emax = 48 ± 9%). With no clear bias for benzoyl substitution, analogues with the 2,5-dimethyl-4-fluoro substitution, contained in clinical candidate 9b, were selected for progression. Of these, 37d demonstrated markedly superior oral bioavailability and excellent brain bioavailability.
Compound 37d, fulfilled the requirements for a brain bioavailable B-SERD with efficacy in multiple ER+ breast cancer cell lines, including two endocrine-resistant cell lines, and two ESR1 mutant cell lines, in 2D and 3D cultures. This B-SERD showed equivalent potency and efficacy to the clinical SERD 2 both in vitro and in vivo (Figures 3–6), but importantly was both orally bioavailable and brain penetrant, in contrast to 2. As measured by western blotting for ERα, coadministration of a proteasome inhibitor blocked the actions of both 2 and 37d, demonstrating that 37d is indeed a SERD, inducing proteasomal degradation of ERα. An almost complete degradation of ERα in gynecological tissues was observed on oral administration of 37d to female mice and 37d was devoid of uterotrophic effects.
Figure 6.
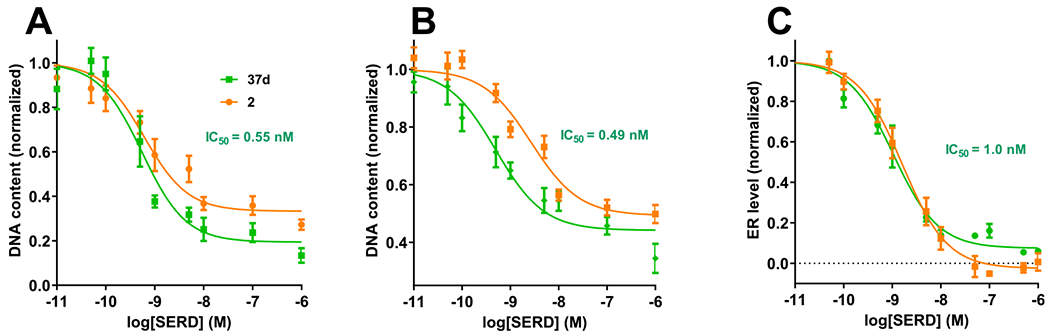
Summary of B-SERD in vitro activity. 2D growth inhibition in MCF7:WS8 (A) and MCF7:5C (B) cells as a result of treatment with 37d (green) or 2 (orange). Data normalized to vehicle, dimethyl sulfoxide (DMSO) (1.0) and no cells (0.0) shown as mean ± SEM, from three biological and analytical replicates. (C) ER level following treatment for 24 h with 37d or 2 evaluated by ICW. Data corrected to vehicle (1) and 1 μM 2 shown as mean ± SEM, from three biological and analytical replicates.
During completion of this work, a paper was published reporting the substitution of the piperidine ring of the SERM acolbifene (EM-652) with a pyrrolidine ring (38),45 which can be seen as a similar substitution to that of the piperidine ring in 30o with pyrrolidine in 30l–n. Interestingly, R-38 demonstrated SERD-like properties, whereas S-38 did not lower ERα and showed SERM-like properties, which was explained by stereospecific interactions in 38/ERα co-crystal structures causing H12 destabilization only for the R-isomer. In our benzothiophene series, reported herein, 30o showed SERM-like properties; however both 3-fluoromethyl-pyrrolidine isomers (30m,n) showed SERM/SERD or SERD-like properties. The R-38/ERα structure (pdb 5UFX) was used to model the potential interactions of 30f and 37d with ERα: the H-bond between Asp-351 and the azetidine N forces the fluoropropyl arm to interact with and destabilize H12 (Figure 7).
Figure 7.
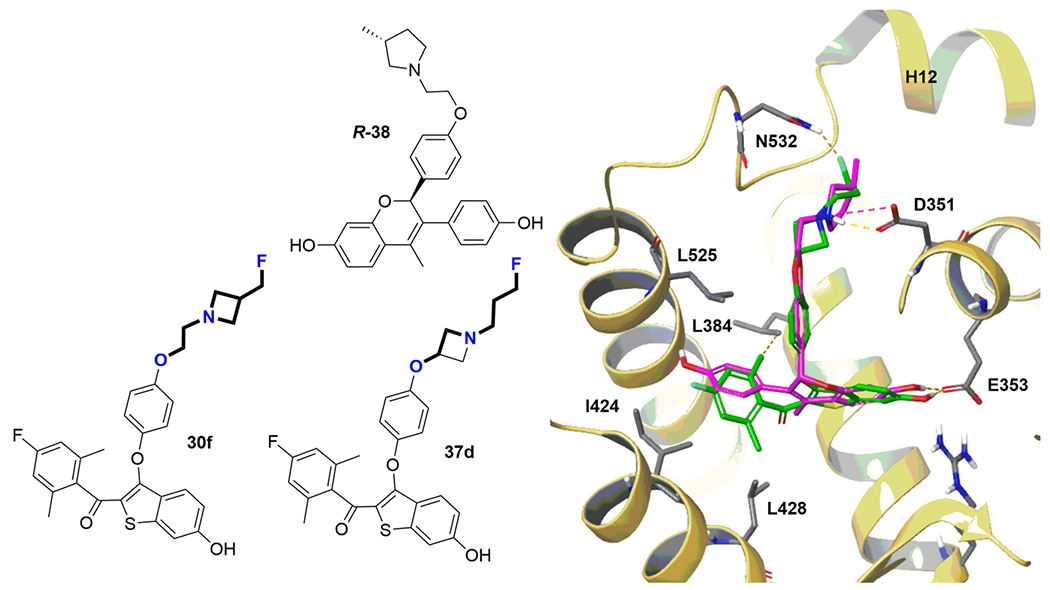
B-SERD docking to ER. B-SERDs 30f and 37d have a common basic side-arm motif: 3-fluoro-N-(2-oxyethyl)propan-1-amine. The structure design envisaged the occupation of the hydrophobic pockets formed by leucines 384, 428, 525, and isoleucine 424 by the substituted benzoyl ring, allowing a salt bridge interaction between the amine side arm amine and Asp-351 in the ERα ligand binding site. B-SERD 37d (green) was docked to the ligand binding domain of ERα obtained from the co-crystal structure with the SERD R-38 (magenta) (pdb 5UFX) confirming the proposed binding site interactions leading to destabilization of H12.
In various assays, we compared the B-SERD, 37d, with the analogous acrylate SERD 9a; SERD 2; the SERM/SERDs 5 and 7; and the fluoromethyl azetidine SERD 3b. The search for an oral SERD was driven initially by 8b, a derivative of the SERM, TAM, which appends an acrylate side arm in place of the SERM amine side arm. This led to the acrylate SERD 3a that includes a modification of the TAM scaffold. Similarly, the amine side arm of the SERM acolbifene, was replaced with a methyl pyrrolidine in R-38; whereas SERD 3b contains a modification of the acolbifene scaffold and a fluoromethyl azetidine side arm. Recent reports on R-38 and 3b posit that ER degradation is subservient to the silencing of the ER transcriptional complex in growth inhibition of breast cancer cells and xenografts by SERDs.45,51 In particular, in a detailed ChIP-seq, ATAC-seq, and RNA-seq analysis, the acrylate SERD 3a was shown to be inferior to 3b in this respect. Similar sequencing assays to compare the B-SERD 37d with the directly analogous acrylate SERD 9b could provide further mechanistic insight and direction for drug development.
CONCLUSIONS
B-SERDs were identified with very high potency and efficacy in models of treatment-resistant breast cancer, building out from the unique benzothiophene scaffold optimized in our previous pursuit of acrylate SERDs.33 The novel N-fluoropropyl 3-oxyazetidine side-armed B-SERD, 37d, was selected as a development candidate, based on the combination of sub-nanomolar potency and superior PK characteristics. The preclinical data support the development of B-SERDs for treatment of patients with metastatic ER+ breast cancer, including those with brain metastases.
EXPERIMENTAL SECTION
Cell Lines and Culture Conditions.
The parental, endocrine-sensitive cell line, MCF7:WS8, was derived from a single-cell clone by the Jordan group. This cell line was maintained in phenol red-containing RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), 1% antibiotic–antimycotic, 1% glutamax, and 10 ng/mL insulin at 37 °C and 5% CO2. The MCF7:5C cell line was also derived from a single cell clone by the Jordan group.47 These cells were maintained in phenol red-free RPMI-1640 medium supplemented with 10% charcoal:dextran stripped FBS, 1% antibiotic–antimycotic, and 1% glutamax at 37 °C and 5% CO2. The MCF7:TAM1 cell line was generated through long-term exposure to increasing concentrations of 1b until resistance was established (>25 passages).34 MCF7:TAM1 cells are maintained in 1b (1 μM) and phenol red-free, stripped RPMI-1640. The T47D:TYS, T47D:TDG, and parent WT T-47D cell lines were a kind gift from David Shapiro (UIUC) and were cultured and maintained as previously described.48
DNA Content Assay.
MCF7:WS8 cells were stripped of estrogens for 2 days prior to plating each experiment by changing media to phenol red-free RPMI-1640 and stripped FBS. Cells were seeded in a 96-well, clear, flat bottom microplate at a density of 5000 cells/well and treated with either 0.01% (v/v) DMSO, 1 nM E2, or the compound of interest. All compounds stored were dissolved in DMSO and diluted to the specific treatment concentration through serial dilution. On day 5 (MCF7:WS8) or day 7 (MCF7:5C), the media was removed and cells were lysed through hydrolysis at −80 °C overnight. DNA content was determined by Hoechst 33258 dye in TNE buffer (1 mg/mL Hoechst in TE buffer + 2 M NaCl). The fluorescence signal was measured using a Synergy Neo (BioTek).
ER ICW Assay.
MCF7:WS8 cells were stripped using phenol red-free media and stripped FBS for 2 days prior to plating at 2.5 × 104 cells/well in black, clear bottom 96-well microplate. Cells were incubated for 48 h prior to treatment for 24 h. Fixation, detection of ERα (H10, Santa Cruz Biotechnologies), and imaging were performed per LI-COR manufacturer’s protocol using the In-Cell Western Assay Kits and LI-COR Odyssey SA imaging system. The IRDye 800CW (anti-rabbit) signal was normalized to CellTag 700 stain.
ER Western Assay.
Cells were stripped of estrogens for 3 days prior to plating. Cells were grown to 80% confluency in a 6-well plate and treated with vehicle or MG-132 (1 μM) for 30 min followed by 2 h of SERD treatment (10 nM). For comparison of SERDs and B-SERDs, similar experiments were performed without prior treatment with MG-132. ER protein content was quantified by the western blot where ERα (antibody Cell Signaling 8644) was normalized to actin or GAPDH. Blots were quantified using the LI-COR Odyssey SA imaging system.
Spheroid Growth Assay.
The cells were plated at a density of 1000–1500 cells/well in Corning 96-well black, clear round bottom, ultra-low attachment spheroid microplates and grown in the absence of treatment for 1–3 days. Spheroids were then treated with 2× treatment media following the removal of half of the media (100 μL) from each well. Treatment at a 1× concentration was repeated every 2–3 days for 14 days. The CellTiter-Glo 3D Cell Viability Assay protocol was used to determine growth inhibition of the spheroids, as per manufacturer’s instructions. A luminescence signal was read using a Synergy Neo (BioTek). Data were normalized to blank (media with CellTiter Glo 3D reagent).
Binding Affinity Studies.
Binding affinities were determined by a competitive radiometric binding assay using 2 nM [3H]estradiol as a tracer (PerkinElmer, Waltham, MA) and full-length purified human ERα (Pan Vera/Invitrogen, Carlsbad, CA), as reported previously.52,53 The RBA values were calculated using the following equation: IC50 estradiol/IC50 compound × 100.
Estrogenicity Assay.
MCF7:WS8 cells were grown in phenol red-free RPMI-1640 and stripped FBS for 3 days prior to plating. Cells were then plated at a density of 1 × 105 cells/well in clear, flat bottom, 48-well plates and incubated for 24 h. The cells were co-transfected with 5 μg of the pERE-luciferase plasmid per plate, which contains three copies of the Xenopus laevis vitellogenin A2 ERE upstream of firefly luciferase and 0.5 μg of the pRL-TK plasmid (Promega, Madison, WI) containing a cDNA encoding Renilla luciferase. Transfection was performed for 6 h using a 2 μL/well Lipofectamine 2000 transfection reagent (Invitrogen) in Opti-MEM media. Cells were then treated with test compounds. Luciferase activity was measured after 18 h of treatment using the Dual Luciferase assay system (Promega) with Synergy Neo (BioTek).
Animal Experiments.
The Animal Care and Use Committee of the University of Illinois at Chicago approved all animal procedures. Animal care adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Juvenile Rat Study.
Female Sprague Dawley rats (12 days old) were purchased from Envigo, USA. Sucklings were housed with foster nursing dams (6 per dam). After one week of acclimation period, at age 19 days, treatment was initiated by administering: 37d or 7 (Sigma, USA) (p.o. 10 mg/kg suspended in 2% Tween 80, 0.5% methylcellulose); EE2 (Sigma, USA) (p.o. 0.1 mg/kg in peanut oil); 2 (s.c. 2 mg/kg in peanut oil). Twenty-four h after the last of 3 daily doses, animals were euthanized with CO2 followed by exsanguination from the abdominal aorta. Uteri, including uterine horn were carefully excised and weighed after removing fat and mesentery. Uteri were cut in the sagittal plane into two equal halves: one half was fixed in phosphate-buffered saline (pH 7.4) buffered 4% paraformaldehyde for 24 h, rinsed with water, washed twice with 70% ethanol, paraffin embedded, and processed for hematoxylin eosin staining and imaging.
Mouse Gynecological Tissue Immunoassay.
Female C57BL/6J mice (8 W) were purchased from the Jackson Laboratory (Bar Harbor, ME) and treated at 9–12 W. The mice were housed in groups of 3–5 with food and water available ad libitum. Dosing: 9a (p.o. 50 mg/kg in polyethylene glycol 400 (PEG 400), 0.5% carboxymethyl cellulose, polyvinylpyrrolidone, and Tween-80; 9:90:0.5:0.5 v/v); 37d (p.o. 50 mg/kg in PEG 400, 10% (2-hydroxypropyl)-β-cyclodextrin (HPCD); 1:9 v/v); the vehicle control group received the PEG/HPCD vehicle. In the acute experiment, mice (n = 5) were given a single administration of drug and euthanized 5 h later. In the chronic experiments, the mice were administered the drug once daily for 3 days and euthanized 5 h (n = 5) or 7 h (n = 6) after the last drug administration. The mice were euthanized by CO2 inhalation followed by decapitation. Ovaries and uterus were immediately removed and frozen on dry ice and stored at −80 °C until being processed for western blots. Tissue samples were homogenized in 200 μL of lysis buffer (Cell Signaling Technology, Danvers, MA; 20 mM Tris-HCl, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid, 1 mM ethylene glycol tetraacetic acid, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/mL leupeptin, and 1 μg/ mL aprotinin) containing the complete protease inhibitor cocktail (Roche, Indianapolis, IN) and clarified by centrifugation for 15 min at 10 000 rpm at 4 °C. Protein concentrations were determined using the BCA Protein Assay Kit (Thermo Fisher Scientific). Equal amounts of protein (20 μg) were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis on Novex 10% Tris-glycine gels (Thermo Fisher Scientific) and transferred to polyvinylidene fluoride membranes. Membranes were blocked with 5% bovine serum albumin in TBST (25 mM Tris-HCl, 137 mM NaCl and 0.1% Tween-20) and then incubated with primary antibodies overnight at 4 °C (anti-ERα, 1:1000, Millipore Sigma, #06–935; anti-β-actin, 1:10 000; Millipore Sigma, #A5441). Membranes were incubated with IRDye donkey antirabbit and donkey antimouse secondary antibodies (LI-COR Biosciences, Lincoln, NE, #925–32213 and #925–68072) at room temperature. Blots were imaged using the Odyssey Fc Imaging System (LI-COR Biosciences). Band intensities were determined using with LI-COR Image Studio software and ERα normalized to β-actin.
Mouse Xenograft Experiments.
MCF7:TAM1 tumors were established in the 4–6 week old ovariectomized athymic female nude mice (Harlan Laboratories) and E2 was administered via silastic capsules (1.0 cm) implanted subcutaneously between the scapulae as previously described.54,55 SERD 37d and SERM 1a were administered (p.o. 100 mg/kg/day) in 10% HPCD/PEG-400 (9:1 v/v) solution, whereas 2 was administered (s.c. 5 mg/week) in sesame oil. The tumor cross-sectional area was determined weekly using Vernier calipers and calculated using the formula (length/2) × (width/2) × π as described previously.54,55 The mean tumor area was plotted against time (in weeks) to monitor tumor growth.
Pharmacokinetic Experiments.
PK screening was conducted in female 3M C57BL/6 mice at two time points (N = 3), collecting plasma and brain tissues from perfused mice postmortem, after gavage administration of 30f, 30m, or 37d in HPβD/PEG-400, or s.c. administration of 2. Standard curves were established in the corresponding biological matrix using a common internal standard and optimization of analyte measurement by LC-MS/MS in the MRM mode. Further PK measurements were performed by Pharmaron Inc. at 10 time points, administering 37d in solution (p.o. 50 mg/kg in water and 10% SBE-β-CD/PEG-400 9:1 v/v). Working solutions were made by serial dilution of analyte in 50% acetonitrile in water. Plasma samples were diluted in 50% acetonitrile to achieve a range of dilutions for analysis and quantitation by LC-MS/MS.
General Synthetic Procedures.
All chemicals and solvents were purchased from Sigma-Aldrich, Fisher Scientific, Matrix Scientific, or Oakwood Chemical and were used without further purification. Synthetic intermediates were purified using a Biotage flash chromatography system on 230–400 mesh silica gel. 1H and 13C NMR spectra were obtained using a Bruker DPX-400 or AVANCE-400 spectrometer at 400 and 100 MHz, respectively. NMR chemical shifts are described in δ (ppm) using residual solvent peaks as the standard (CDCl3, 7.26 ppm (1H), 77.16 ppm (13C); CD3OD, 3.31 ppm (1H), 49.00 ppm (13C); DMSO-d6, 2.50 ppm (1H), 39.52 ppm (13C); acetone-d6, 2.05 ppm (1H), 29.84 ppm, and 206.26 ppm (13C)). Data are reported in the following format: chemical shift, multiplicity (s = singlet, d = doublet, dd = doublet of doublet, t = triplet, q = quartet, br = broad, and m = multiplet), coupling constants, and number of protons. High-resolution mass spectral data were measured using a Shimadzu IT-TOF LC/MS for all final compounds. All compounds submitted for biological testing were confirmed to be ≥96% pure by analytical high-performance liquid chromatography (HPLC), supported by 1H analysis, unless otherwise stated. The purity of final compounds were determined by HPLC using an Agilent Eclipse XDB-C18 column (4.6 × 250 mm, 5 μm) with UV absorbance detection at 254 nm, eluting with a linear gradient from 10% aqueous MeCN to 90% MeCN over 18 min, holding at 90% MeCN for a further 5 min. For the synthesis of Grignard reagents, the following procedure was used: to a dried round-bottomed flask were added aryl bromide (1 equiv) in anhydrous tetrahydrofuran (THF) and magnesium turnings (1.1 equiv) under an argon atmosphere. One granule of iodine was added to initiate the reaction along with a hot fan. The solution turned pale white and then a brownish color along with strong heat release. The Grignard reagent was ready for use without further purification when magnesium was consumed. For full experimental details of all compounds, see Supporting Information. Representative synthetic methods, spectral data, and HRMS for novel compounds are described in detail below. Full spectra and chromatograms are supplied in Supporting Information.
3-Chloro-6-methoxybenzo[b]thiophene-2-carbonyl Chloride (10).
To a solution of chlorobenzene was added (10 g, 56.18 mmol) (E)-3-(4-methoxyphenyl)acrylic acid, (40 mL, 561.8 mmol) SOCl2, (0.45 mL, 5.618 mmol) pyridine, and molecular sieves. The reaction was heated at reflux for 3 days. Excess thionyl chloride was removed under reduced pressure and the remaining material was suspended in hot hexane and then filtered. The filtrate collected and evaporated under reduced pressure to give compound 10 as a pale solid (yield: 7 g, 40%).
3-Chloro-N,6-dimethoxy-N-methylbenzo[b]thiophene-2-carboxamide (11).
To an oven-dried round-bottom flask was dissolved compound 10 (2 g, 7.7 mmol) in (15 mL) of anhydrous dichloromethane under an argon atmosphere. N,O-Dimethylhydroxylamine hydrochloride (0.83 g, 8.5 mmol) was added in one portion followed by Et3N (5.4 mL, 38.8 mmol) dropwise to the mixture. The reaction was stirred at room temperature and monitored by thin layer chromatography (TLC). Upon completion, the reaction was quenched by water and extracted with ethyl acetate, washed by water and brine, and dried over Na2SO4. The organic extracts were evaporated under reduced pressure and purified by flash chromatography (10–30% ethyl acetate in hexanes) to give a light yellow solid (yield: 1.7 g, 76%). 1H NMR (400 MHz, CDCl3,): δ 7.80 (d, J = 8.9 Hz, 1H), 7.22 (d, J = 2.2 Hz, 1H), 7.08 (dd, J = 8.9, 2.3 Hz, 1H), 3.88 (s, 3H), 3.72 (s, 3H), 3.38 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 161.45, 159.31, 139.78, 129.61, 124.47, 123.58, 122.66, 115.53, 103.71, 61.47, 55.29, 33.16.
3-Chloro-6-methoxybenzo[b]thiophene-2-carbaldehyde (12).
To a solution of anhydrous THF (10 mL) was dissolved compound 11 (1 g, 3.9 mmol). The reaction mixture was stirred at −78 °C for 0.5 h. Diisobutylaluminium hydride (3.85 mL, 4.29 mmol) was added dropwise slowly to the reaction mixture and then stirred at room temperature until the starting material was consumed completely. Upon completion, the reaction was quenched by potassium sodium tartrate solution at 0 °C and stirred at room temperature until most of the amorphous precipitation was dissolved. The reaction was extracted by ethyl acetate, washed by water and brine and dried over Na2SO4. The organic extracts were evaporated under reduced pressure and purified by flash chromatography (10–30% ethyl acetate in hexanes) to give a white solid (yield: 0.5 g, 64%). 1H NMR (400 MHz, CDCl3): δ 10.27 (s, 1H), 7.90 (d, J = 9.0 Hz, 1H), 7.47 (d, J = 2.2 Hz, 1H), 7.15 (dd, J = 9.0, 2.2 Hz, 1H), 3.95 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 182.88, 161.39, 141.85, 132.85, 131.05, 130.45, 124.79, 117.07, 104.83, 55.83.
(3-Chloro-6-methoxybenzo[b]thiophen-2-yl)(o-tolyl)methanol (13a).
To a solution of THF (8 mL) was added compound 12 (0.5 g, 2.2 mmol) and stirred at 0 °C for 0.5 h. o-Tolylmagnesium bromide solution (1.3 mL, 2.6 mmol, 2 M in diethyl ether) was dropwise to the reaction mixture slowly at 0 °C. The reaction was then stirred at room temperature for 2 h and monitored by TLC. Upon completion, the reaction was quenched by water and extracted by ethyl acetate, washed by water and brine and dried over Na2SO4. The organic extracts were evaporated under reduced pressure and purified by flash chromatography (10–40% ethyl acetate in hexanes) to give a white solid (yield: 0.5 g, 71%). 1H NMR (400 MHz, acetone-d6): δ 7.71–7.69 (m, 2H), 7.46 (d, J = 2.3 Hz, 1H), 7.29–7.19 (m, 2H), 7.17 (d, J = 7.1 Hz, 1H), 7.11 (dd, J = 8.8, 2.3 Hz, 1H), 6.46 (d, J = 4.0 Hz, 1H), 5.31 (d, J = 4.0 Hz, 1H), 3.88 (s, 3H), 2.32 (s, 3H). 13C NMR (101 MHz, acetone-d6): δ 158.51, 141.11, 139.77, 138.78, 135.17, 130.24, 130.17, 127.57, 125.90, 125.82, 122.02, 116.23, 115.03, 105.38, 66.31, 55.15, 18.37.
(3-Chloro-6-methoxybenzo[b]thiophen-2-yl)(phenyl)methanol (13c).
This compound was prepared using a procedure similar to that of 13a (yield: 0.52 g, 85%). 1H NMR (400 MHz, acetone-d6): δ 7.63 (d, J = 8.8 Hz, 1H), 7.59–7.52 (m, 2H), 7.47 (d, J = 2.3 Hz, 1H), 7.12 (t, J = 7.8 Hz, 2H), 7.09 (dd, J = 8.9, 2.2 Hz, 1H), 6.33 (d, J = 3.0 Hz, 1H), 5.63 (d, J = 3.5 Hz, 1H), 3.87 (s, 3H). 13C NMR (101 MHz, acetone-d6): δ 162.14 (d, J = 243.9 Hz), 158.49, 140.63, 139.27, 139.24, 138.55, 130.26, 128.23 (d, J = 8.2 Hz), 121.93, 115.04, 114.93 (d, J = 21.8 Hz), 105.46, 69.02, 55.17.
(2,4-Dimethylphenyl)(6-methoxybenzo[b]thiophen-2-yl)-methanol (13e).
This compound was prepared using a procedure similar to that of 13a (yield: 0.55 g, 85%). 1H NMR (400 MHz, acetone-d6): δ 7.66 (d, J = 8.8 Hz, 1H), 7.56 (d, J = 7.9 Hz, 1H), 7.45 (d, J = 2.2 Hz, 1H), 7.10 (dd, J = 8.9, 2.3 Hz, 1H), 7.07 (d, J = 7.9 Hz, 1H), 6.99 (s, 1H), 6.42 (d, J = 4.0 Hz, 1H), 5.24 (d, J = 4.0 Hz, 1H), 3.87 (s, 3H), 2.29 (s, 6H). 13C NMR (101 MHz, acetone-d6): δ 158.46, 140.15, 138.74, 138.20, 136.99, 135.01, 130.91, 130.31, 126.50, 125.91, 121.98, 115.99, 114.98, 105.39, 66.29, 55.15, 20.17, 18.35.
(3-Chloro-6-methoxybenzo[b]thiophen-2-yl)(phenyl)methanone (14a).
This compound was prepared using a procedure similar to that of 13a (yield: 0.5 g, 55%). 1H NMR (400 MHz, acetone-d6): δ 7.95–7.85 (m, 3H), 7.71–7.69 (m, 1H), 7.64 (d, J = 2.1 Hz, 1H), 7.60 (t, J = 7.7 Hz, 2H), 7.24 (dd, J = 9.0, 2.3 Hz, 1H), 3.97 (s, 3H). 13C NMR (101 MHz, acetone-d6): δ 188.19, 160.71, 140.85, 138.19, 133.01, 131.79, 130.60, 129.26, 128.53, 124.35, 123.75, 116.92, 104.78, 55.42.
(3-Chloro-6-methoxybenzo[b]thiophen-2-yl)(o-tolyl)methanone (14b).
To a solution of dichloromethane (5 mL) was added compound 13b (0.3 g, 0.94 mmol), pyridinium chlorochromate (PCC; 0.24 g, 1.1 mmol) and stirred at room temperature for 3 h. The reaction was monitored by TLC. Upon completion, the reaction mixture was extracted by dichloromethane and washed by water and brine, and dried over Na2SO4. The organic extracts were evaporated under reduced pressure and purified by flash chromatography (1020% ethyl acetate in hexanes) to give a yellow solid (yield: 0.14 g, 70%). 1H NMR (400 MHz, CDCl3): δ 7.83 (d, J = 9.0 Hz, 1H), 7.47–7.39 (m, 2H), 7.33–7.29 (m, 2H), 7.27 (d, J = 2.2 Hz, 1H), 7.12, (dd, J = 9.0, 2.3 Hz, 1H), 3.94 (s, 3H), 2.42 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 190.94, 160.77, 141.69, 139.28, 136.09, 133.74, 131.47, 130.90, 130.60, 127.80, 126.33, 125.64, 125.20, 116.74, 104.32, 55.79, 19.58.
(3-Chloro-6-methoxybenzo[b]thiophen-2-yl)(4-fluorophenyl)-methanone (14c).
This compound was prepared using a procedure similar to that of 14b (yield: 0.35 g, 60%). 1H NMR (400 MHz, acetone-d6): δ 8.02–7.94 (m, 2H), 7.85 (d, J = 9.0 Hz, 1H), 7.61 (d, J = 2.1 Hz, 1H), 7.35 (t, J = 8.8 Hz, 2H), 7.22 (dd, J = 9.0, 2.2 Hz, 1H), 3.96 (s, 3H). 13C NMR (101 MHz, acetone-d6): δ 186.79, 165.62 (d, J = 252.5 Hz), 160.72, 140.84, 134.57 (d, J = 2.5 Hz), 132.28 (d, J = 9.4 Hz), 131.61, 130.54, 124.35, 123.75, 116.92, 115.56 (d, J = 22.3 Hz), 104.76, 55.43.
(3-Chloro-6-methoxybenzo[b]thiophen-2-yl)(3-fluorophenyl)-methanone (14d).
This compound was prepared using a procedure similar to that of 14b (yield: 0.42 g, 62%). 1H NMR (400 MHz, acetone-d6): δ 7.89 (d, J = 9.0 Hz, 1H), 7.78–7.69 (m, 1H), 7.697.60 (m, 3H), 7.55–7.46 (m, 1H), 7.24 (dd, J = 9.0, 2.3 Hz, 1H), 3.97 (s, 3H). 13C NMR (101 MHz, acetone-d6): δ 186.93, 162.53 (d, J = 246.2 Hz), 160.92, 141.11, 140.45 (d, J = 6.5 Hz), 131.37, 130.69, 130.68 (d, J = 8.3 Hz), 125.28 (d, J = 2.5 Hz), 124.52, 119.64 (d, J = 21.6 Hz), 117.06, 115.59 (d, J = 23.1 Hz), 104.76, 55.45.
(2,4-Dimethylphenyl)(6-methoxybenzo[b]thiophen-2-yl)-methanone (14e).
This compound was prepared using a procedure similar to that of 14b (yield: 0.38 g, 65%). 1H NMR (400 MHz, acetone-d6): δ 7.83 (d, J = 9.0 Hz, 1H), 7.58 (d, J = 2.2 Hz, 1H), 7.38 (d, J = 7.7 Hz, 1H), 7.23–7.10 (m, 3H), 3.95 (s, 3H), 2.39 (s, 3H), 2.34 (s, 3H). 13C NMR (101 MHz, acetone-d6): δ 189.83, 160.95, 141.32 141.10, 136.49, 136.17, 133.87, 131.64, 130.96, 128.41, 126.28, 125.03, 124.63, 116.91, 104.78, 55.43, 20.55, 18.80.
(3-Chloro-6-methoxybenzo[b]thiophen-2-yl)(4-fluoro-2-methylphenyl)methanone (14f).
This compound was prepared using a procedure similar to that of 13a (yield: 0.5 g, 60%). 1H NMR (400 MHz, CDCl3): δ 7.84–7.77 (m, 1H), 7.43 (dd, J = 8.4, 5.8 Hz, 1H), 7.25 (d, J = 2.2 Hz, 1H), 7.10 (dd, J = 9.0, 2.3 Hz, 1H), 7.03–6.92 (m, 2H), 3.91 (s, 3H) 2.21 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 189.77, 163.89 (d, J = 250.8 Hz), 160.83, 141.66, 139.85 (d, J = 8.6 Hz), 135.26, 133.62, 131.37, 130.48 (d, J = 9.2 Hz), 126.23, 125.19, 117.87 (d, J = 21.5 Hz), 116.83, 112.70 (d, J = 21.7 Hz), 104.32, 55.80, 19.81.
(3-Chloro-6-methoxybenzo[b]thiophen-2-yl)(4-fluoro-2,6-dimethylphenyl)methanone (14g).
The preparation of Grignard reagent (4-fluoro-2,6-dimethylphenyl)magnesium bromide is as follows, 2-bromo-5-fluoro-1,3-dimethylbenzene (0.57 g, 2.85 mmol) was dissolved in anhydrous THF, magnesium (0.14 g, 5.7 mmol), and iodine (0.07 g, 0.285 mmol) were then added to the reaction mixture, the reaction was stirred at room temperature for 2 h and then ready to use. To a solution of anhydrous THF was added compound 10 (0.5 g, 1.9 mmol), followed by dropwise addition of (4-fluoro-2,6-dimethylphenyl)magnesium bromide at 0 °C, then the reaction was stirred at room temperature for 5 h, and monitored by TLC. Upon completion, the reaction mixture was quenched by water in an ice bath, extracted by EtOAc and washed by water and brine, and dried over Na2SO4. The organic extracts were combined, evaporated under reduced pressure, and purified by flash chromatography (10–30% ethyl acetate in hexanes) to give a white solid (yield: 0.4 g, 60%). 1H NMR (400 MHz, CDCl3): δ 7.79 (d, J = 9.0 Hz, 1H), 7.25 (d, J = 2.0 Hz, 1H), 7.09 (dd, J = 9.0, 2.1 Hz, 1H), 6.80 (d, J = 9.5 Hz, 2H), 3.91 (s, 3H), 2.21 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 191.75, 162.97 (d, J = 247.5 Hz), 161.25, 142.26, 137.08 (d, J = 8.6 Hz), 136.28 (d, J = 3.0 Hz), 134.27, 131.68, 126.98, 125.60, 117.02, 114.67 (d, J = 21.3 Hz), 104.51, 55.92, 19.43, 19.42.
(3-Chloro-6-methoxybenzo[b]thiophen-2-yl)(4-methoxyphenyl)-methanone (14h).
This compound was prepared using a procedure similar to that of 13a (yield: 0.35 g, 60%). 1H NMR (400 MHz, acetone-d6): δ 7.95–7.89 (m, 2H), 7.88 (d, J = 8.9 Hz, 1H), 7.65 (d, J = 2.2 Hz, 1H), 7.24 (dd, J = 9.0, 2.3 Hz, 1H), 7.12 (d, J = 8.9 Hz, 2H). 3.97 (s, 3H), 3.95 (s, 3H).
(3-Chloro-6-methoxybenzo[b]thiophen-2-yl)(4-methoxyphenyl)-methanone (14i).
This compound was prepared using a procedure similar to that of 13a (yield: 0.4 g, 45%). 1H NMR (400 MHz, CDCl3): δ 7.83 (d, J = 9.0 Hz, 1H), 7.49 (d, J = 8.5 Hz, 1H), 7.27 (d, J = 2.2 Hz, 1H), 7.12 (dd, J = 9.0, 2.3 Hz, 1H), 6.84 (d, J = 2.2 Hz, 1H), 6.79 (dd, J = 8.5, 2.4 Hz, 1H), 3.94 (s, 3H), 3.89 (s, 3H). 2.21 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 189.76, 161.76, 160.43, 141.20, 140.08, 134.08, 131.71, 131.31, 131.22, 125.00, 124.91, 116.70, 116.54, 110.60, 104.34, 55.77, 55.33, 20.44.
(3-Chloro-6-hydroxybenzo[b]thiophen-2-yl)(phenyl)methanone (15a).
To a solution of dichloromethane (5 mL) was added compound 14a (0.2 g, 0.63 mmol) and stirred at −78 °C for 0.5 h. BBr3 (0.29 mL, 3.1 mmol) was added dropwise slowly to the reaction mixture. The reaction was stirred at room temperature and monitored by TLC. Upon completion, the reaction mixture was quenched by water at 0 °C and extracted by dichloromethane, washed by water and brine, and dried over Na2SO4. The organic extracts were evaporated under reduced pressure and purified by flash chromatography (10–40% ethyl acetate in hexanes) (yield: 0.4 g, 60%). 1H NMR (400 MHz, acetone-d6): δ 7.95–7.79 (m, 3H), 7.71 (t, J = 7.4 Hz, 1H), 7.59, (t, J = 7.6 Hz, 2H), 7.47 (d, J = 1.7 Hz, 1H), 7.20 (dd, J = 8.8, 1.9 Hz, 1H). 13C NMR (101 MHz, acetone-d6): δ 188.22, 158.58, 140,82, 138.30, 132.91, 131.13, 130.01, 129.21, 128.50, 124.72, 123.95, 116.82, 107.43.
(3-Chloro-6-hydroxybenzo[b]thiophen-2-yl)(o-tolyl)methanone (15b).
This compound was prepared using a procedure similar to that of 15a (yield: 0.12 g, 68%). 1H NMR (400 MHz, CDCl3): δ 7.80 (d, J = 8.9 Hz, 1H), 7.44 (t, J = 7.5 Hz, 1H), 7.43 (d, J = 7.5 Hz, 1H), 7.32 (d, J = 7.8 Hz, 1H), 7.23 (s, 1H), 7.07 (dd, J = 8.9, 0.9 Hz, 1H), 5.32 (s, 1H), 2.41 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 191.90, 157.45, 141.74, 139.13, 136.03, 133.28, 131.44, 130.98, 130.81, 127.76, 127.21, 125.74, 125.71, 116.62, 107.66, 19.57.
(3-Chloro-6-hydroxybenzo[b]thiophen-2-yl)(4-fluorophenyl)-methanone (15c).
This compound was prepared using a procedure similar to that of 15a (yield 0.25 g, 57%). 1H NMR (400 MHz, acetone-d6): δ 8.01–7.94 (m, 2H), 7.84 (d, J = 8.8 Hz, 1H), 7.46 (d, J = 2.1 Hz, 1H), 7.39–7.31 (m, 2H), 7.20 (dd, J = 8.8, 2.2 Hz, 1H). 13C NMR (101 MHz, acetone-d6): δ 186.83, 165.57 (d, J = 252.3 Hz), 158.64, 140.82, 134.69 (d, J = 2.6 Hz), 132.23 (d, J = 9.4 Hz), 130.95, 129.94, 124.73, 123.93, 116.86, 115.53 (d, J = 22.3 Hz), 107.44.
(3-Chloro-6-hydroxybenzo[b]thiophen-2-yl)(3-fluorophenyl)-methanone (15d).
This compound was prepared using a procedure similar to that of 15a (yield: 0.38 g, 58%). 1H NMR (400 MHz, acetone-d6): δ 7.83 (d, J = 8.9 Hz, 1H), 7.70–7.65 (m, 1H), 7.67–7.56 (m, 2H), 7.52–7.38 (m, 2H), 7.19 (dd, J = 8.8, 2.2 Hz, 1H). 13C NMR (101 MHz, acetone-d6): δ 186.90, 162.50 (d, J = 246.2 Hz), 158.79, 141.07, 140.55 (d, J = 6.5 Hz), 131.37, 130.67, 130.63 (d, J = 7.9 Hz), 125.23 (d, J = 2.2 Hz), 124.90, 124.77, 119.52 (d, J = 21.5 Hz), 116.94, 115.55 (d, J = 23.0 Hz), 107.42.
(3-Chloro-6-hydroxybenzo[b]thiophen-2-yl)(2,4-dimethylphenyl)methanone (15e).
This compound was prepared using a procedure similar to that of 15a (yield: 0.2 g, 55%). 1H NMR (400 MHz, acetone-d6): δ 9.32 (s, 1H), 7.81 (d, J = 8.9 Hz, 1H), 7.43 (d, J = 2.2 Hz, 1H), 7.37 (d, J = 7.7 Hz, 1H), 7.22–7.06 (m, 3H), 2.39 (s, 3H), 2.34 (s, 3H). 13C NMR (101 MHz, acetone-d6): δ 189.86 158.86, 141.29, 140.97, 136.64, 136.04, 133.21, 131.58, 130.37, 128.28, 126.25, 125.03, 124.85, 116.82, 107.46, 20.52, 18.75.
(3-Chloro-6-hydroxybenzo[b]thiophen-2-yl)(4-fluoro-2-methylphenyl)methanone (15f).
This compound was prepared using a procedure similar to that of 15a (yield: 0.38 g, 56%). 1H NMR (400 MHz, acetone-d6): δ 9.34 (s, 1H), 7.77 (d, J = 8.9 Hz, 1H), 7.52 (dd, J = 8.4, 5.9 Hz, 1H), 7.41 (d, J = 2.0 Hz, 1H), 7.23–6.91 (m, 3H), 2.36 (s, 3H). 13C NMR (101 MHz, acetone-d6): δ 188.95, 163.72 (d, J = 248.7 Hz), 159.07, 141.51, 139.51 (d, J = 8.6 Hz), 135.83, 132.98, 130.46, 130.37, 125.48, 125.18, 117.50 (d, J = 21.7 Hz), 116.95, 112.58 (d, J = 21.8 Hz), 107.50, 18.78.
(3-Chloro-6-hydroxybenzo[b]thiophen-2-yl)(4-fluoro-2,6-dimethylphenyl)methanone (15g).
This compound was prepared using a procedure similar to that of 15a (yield: 0.4 g, 55%). 1H NMR (400 MHz, acetone-d6): δ 9.40 (s, 1H), 7.83 (d, J = 8.9 Hz, 1H), 7.45 (d, J = 2.1 Hz, 1H), 7.17 (dd, J = 8.9, 2.2 Hz, 1H), 6.97 (d, J = 9.7 Hz, 2H), 2.22 (s, 6H). 13C NMR (101 MHz, acetone-d6): δ 190.54, 162.78 (d, J = 245.6 Hz), 159.41, 141.93, 137.03 (d, J = 8.7 Hz), 136.72 (d, J = 2.7 Hz), 133.47, 130.57, 126.02, 125.54, 117.05, 114.28 (d, J = 21.7 Hz), 107.62, 18.34.
(3-Chloro-6-hydroxybenzo[b]thiophen-2-yl)(4-hydroxyphenyl)-methanone (15h).
This compound was prepared using a procedure similar to that of 15a (yield: 0.28 g, 55%). 1H NMR (400 MHz, MeOD): δ 7.84–7.74 (m, 3H), 7.27 (d, J = 2.1 Hz, 1H), 7.08 (dd, J = 8.8, 2.1 Hz, 1H), 6.90 (d, J = 8.7 Hz, 2H). 13C NMR (101 MHz, MeOD): δ 187.90, 162.94, 158.34, 140.46, 132.32, 130.71, 129.62, 129.0, 124.03, 123.01, 116.29, 114.90, 106.76.
(3-Chloro-6-hydroxybenzo[b]thiophen-2-yl)(4-hydroxy-2-methylphenyl)methanone (15i).
This compound was prepared using a procedure similar to that of 15a (yield: 0.32 g, 55%). 1H NMR (400 MHz, MeOD): δ 7.75 (d, J = 8.9 Hz, 1H), 7.35 (d, J = 8.4 Hz, 1H), 7.25 (d, J = 1.8 Hz, 1H), 7.05 (dd, J = 8.9, 2.1 Hz, 1H), 6.75 (d, J = 1.4 Hz, 1H), 6.69 (dd, J = 8.4, 2.0 Hz, 1H), 2.36 (s, 3H). 13C NMR (101 MHz, MeOD): δ 190.34, 160.43, 158.81, 141.17, 139.87,132.86, 131.82, 130.01, 129.84, 124.62, 124.49, 117.57, 116.33, 112.06, 106.78, 19.02.
(3-Chloro-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]thiophen-2-yl)(phenyl)methanone (16a).
To a solution of dichloromethane (5 mL) was added compound 15a (0.2 g, 0.66 mmol), 3,4-dihydropyran (0.3 mL, 3.3 mmol), and pyridinium p-toluenesulfonate (0.01 g, 0.07 mmol). The reaction mixture was stirred at room temperature and monitored by TLC. Upon completion, the reaction was extracted by dichloromethane, washed by water and brine, and dried over Na2SO4. The organic extracts were evaporated under reduced pressure and purified by flash chromatography (10–40% ethyl acetate in hexanes) (yield: 0.3 g, 77%). 1H NMR (400 MHz, acetone-d6): δ 7.91 (d, J = 8.4 Hz, 3H), 7.75–7.69 (m, 2H), 7.60 (t, J = 7.7 Hz, 2H), 7.37–7.32 (m, 1H), 5.66 (t, J = 3.1 Hz, 1H), 3.96–3.80 (m, 1H), 3.70–3.60 (m, 1H), 1.78–1.60 (m, 3H), 1.60–1.36 (m, 3H). 13C NMR (101 MHz, acetone-d6): δ 188.23, 157.86, 140.34, 138.12, 133.08, 132.20, 131.18, 129.30 128.55, 124.29, 123.62, 117.83, 108.45, 96.54, 61.70, 29.99, 25.42 18.50.
(3-Chloro-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]thiophen-2-yl)(o-tolyl)methanone (16b).
This compound was prepared using a procedure similar to that of 16a (yield: 0.21 g, 84%). 1H NMR (400 MHz, acetone-d6): δ 7.88 (d, J = 9.0 Hz, 1H), 7.71 (d, J = 2.1 Hz, 1H), 7.50–7.46 (m, 2H), 7.42–7.38 (m, 2H), 7.31 (dd, J = 9.0, 2.2 Hz, 1H), 5.66 (t, J = 3.1 Hz, 1H), 3.88–3.85 (m, 1H), 3.69–3.59 (m, 1H), 2.36 (s, 3H), 2.02–1.81 (m, 3H), 1.79–1.47 (m, 3H). 13C NMR (101 MHz, acetone-d6): δ 190.06, 158.28, 141.01, 139.46, 135.69, 134.11, 131.60, 130.84, 130.65, 127.68, 125.77, 125.11, 124.74, 117.88, 108.44, 96.50, 61.71, 29.95, 24.92, 18.65, 18.47.
(3-Chloro-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]thiophen-2-yl)(4-fluorophenyl)methanone (16c).
This compound was prepared using a procedure similar to that of 16a (yield 0.2 g, 68%). 1H NMR (400 MHz, acetone-d6): δ 7.99–7.97 (m, 2H), 7.89 (d, J = 8.9 Hz, 1H), 7.72 (d, J = 2.0 Hz, 1H), 7.35 (t, J = 6.6 Hz, 2H), 7.32 (dd, J = 6.7, 2.2 Hz, 1H), 5.64 (t, J = 3.0 Hz, 1H), 3.89–3.75 (m, 1H), 3.45–3.42 (m, 1H), 1.94–1.77 (m, 2H), 1.77–1.40 (m, 4H). 13C NMR (101 MHz, acetone-d6): δ 186.83, 165.68 (d, J = 252.7 Hz), 157.88, 140.33, 134.52 (d, J = 2.7 Hz), 132.34 (d, J = 9.4 Hz), 131.12, 129.51, 124.30, 123.57, 117.86, 115.59 (d, J = 22.3 Hz), 108.45, 96.53, 61.70, 30.31, 24.93, 18.49.
(3-Chloro-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]thiophen-2-yl)(3-fluorophenyl)methanone (16d).
This compound was prepared using a procedure similar to that of 16a (yield: 0.3 g, 69%). 1H NMR (400 MHz, acetone-d6): δ 7.89 (d, J = 8.9 Hz, 1H), 7.75–7.72 (m, 2H), 7.68–7.60 (m, 2H), 7.53–7.44 (m, 1H), 7.32 (dd, J = 8.9, 2.2 Hz, 1H), 5.64 (t, J = 3.0 Hz, 1H), 3.83–3.81 (m, 1H), 3.64–3.61 (m, 1H), 2.05–1.78 (m, 3H), 1.78–1.41 (m, 3H). 13C NMR (101 MHz, acetone-d6): δ 186.93, 162.53 (d, J = 246.3 Hz), 158.04, 140.58, 140.36 (d, J = 6.7 Hz), 131.74, 131.18, 130.69 (d, J = 7.9 Hz), 125.34 (d, J = 2.8 Hz), 124.46, 124.41, 119.71 (d, J = 21.5 Hz), 117.92, 115.63 (d, J = 23.0 Hz), 108.38, 96.52, 61.70, 29.97, 24.94, 18.49.
(3-Chloro-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]thiophen-2-yl)(2,4-dimethylphenyl)methanone (16e).
This compound was prepared using a procedure similar to that of 16a (yield: 0.18 g, 73%). 1H NMR (400 MHz, acetone-d6): δ 7.87 (d, J = 8.9 Hz, 1H), 7.69 (d, J = 1.9 Hz, 1H), 7.39 (d, J = 7.8 Hz, 1H), 7.30 (dd, J = 8.9, 2.2 Hz, 1H), 7.20 (s, 1H), 7.16 (d, J = 7.8 Hz, 1H), 5.64 (s, 1H), 3.91–3.73 (m, 1H), 3.73–3.58 (m, 1H), 2.40 (s, 3H), 2.35 (s, 3H), 2.02–1.77 (m, 3H), 1.77–1.44 (m, 3H). 13C NMR (101 MHz, acetone-d6): δ 189.85, 158.12, 141.18, 140.80, 136.43, 136.23, 134.28, 131.67, 131.54, 128.49, 126.29, 124.60, 124.52, 117.80, 108.43, 96.50, 61.70, 29.97, 24.93, 20.54, 18.82, 18.48.
(3-Chloro-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]thiophen-2-yl)(4-fluoro-2-methylphenyl)methanone (16f).
This compound was prepared using a procedure similar to that of 16a (yield: 0.35 g, 70%). 1H NMR (400 MHz, acetone-d6): δ 7.88 (t, J = 10.0 Hz, 1H), 7.73 (d, J = 2.0 Hz, 1H), 7.59 (dd, J = 8.5, 5.9 Hz, 1H), 7.37–7.30 (m, 1H), 7.25–7.17 (m, 1H), 7.17–7.06 (m, 1H), 5.67 (s, 1H), 3.97–3.73 (m, 1H), 3.66–3.64 (m, 1H), 2.21 (s, 3H) 2.17–1.80 (m, 2H), 1.80–1.28 (m, 4H). 13C NMR (101 MHz, acetone-d6): δ 188.98, 163.82 (d, J = 249.0 Hz), 158.31, 141.03, 139.69 (d, J = 8.8 Hz), 135.67, 134.07, 131.54, 130.62 (d, J = 9.2 Hz), 125.12, 124.77, 117.93, 117.58 (d, J = 21.7 Hz), 112.63 (d, J = 21.7 Hz), 108.44,96.51, 93.23, 61.72, 29.96, 24.92, 18.48.
(3-Chloro-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]thiophen-2-yl)(4-fluoro-2,6-dimethylphenyl)-methanone (16g).
This compound was prepared using a procedure similar to that of 16a (yield: 0.45 g, 75%). 1H NMR (400 MHz, acetone-d6): δ 7.89 (d, J = 9.0 Hz, 1H), 7.73 (d, J = 2.1 Hz, 1H), 7.31 (dd, J = 9.0, 2.2 Hz, 1H), 6.98 (d, J = 9.8 Hz, 2H), 5.67 (t, J = 3.1 Hz, 1H), 3.88–3.78 (m, 1H), 3.70–3.57 (m, 1H), 2.23 (s, 6H), 1.91–1.89 (m, 2H), 1.68–1.64 (m, 4H). 13C NMR (101 MHz, acetone-d6): δ 190.66, 161.47 (d, J = 242.4 Hz), 158.63, 141.48, 137.07 (d, J = 8.8 Hz), 136.12, 133.19, 131.75, 125.12, 118.02, 116.59, 114.32 (d, J = 21.6 Hz), 108.54, 96.46, 61.69,29.91, 24.90, 18.42, 18.34.
(3-Chloro-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]thiophen-2-yl)(4-((tetrahydro-2H-pyran-2-yl)oxy)phenyl)methanone (16h).
This compound was prepared using a procedure similar to that of 16a (yield: 0.35 g, 70%). 1H NMR (400 MHz, acetone-d6): δ 7.97–7.83 (m, 3H), 7.72 (d, J = 1.8 Hz, 1H), 7.33 (dd, J = 8.9, 2.0 Hz, 1H), 7.21 (d, J = 8.8 Hz, 2H), 5.64 (s, 1H), 4.82 (s, 1H), 4.00–3.90 (m, 1H), 3.83–3.81 (m, 1H), 3.64–3.62 (m, 1H), 3.47–3.45 (m, 1H), 1.94–1.92 (m, 6H), 1.78–1.33 (m, 6H). 13C NMR (101 MHz, acetone-d6): δ 186.60, 161.56, 157.55, 139.92, 132.09, 123.99, 117.77, 116.1, 108.49, 108.34, 97.51, 96.54, 96.08, 62.00, 61.75, 61.68, 30.72, 30.02, 29.93, 25.40, 24.96, 19.26, 18.49.
(3-Chloro-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]thiophen-2-yl)(2-methyl-4-((tetrahydro-2H-pyran-2-yl)oxy)phenyl)-methanone (16i).
This compound was prepared using a procedure similar to that of 16a (yield: 0.3 g, 72%). 1H NMR (400 MHz, acetone-d6): δ 7.88 (d, J = 8.9 Hz, 1H), 7.71 (d, J = 2.1 Hz, 1H), 7.51 (d, J = 8.5 Hz, 1H), 7.32 (dd, J = 8.9, 2.2 Hz, 1H), 7.04 (d, J = 2.1 Hz, 1H), 7.00 (dd, J = 8.5, 2.3 Hz, 1H), 5.62–5.60 (m, 2H), 3.85–3.83 (m, 2H), 3.71–3.56 (m, 2H), 3.56–3.38 (m, 1H), 2.41 (s, 3H), 2.04–1.80 (m, 5H), 1.77–1.41 (m, 6H). 13C NMR (101 MHz, acetone-d6): δ 188.93, 159.47, 157.95, 140.56, 139.29, 134.44, 131.89, 131.26, 124.43, 118.81, 117.74, 113.07, 108.47, 96.52, 95.99, 61.69, 61.67, 30.04, 29.98, 24.97, 24.93, 19.42, 19.24, 19.13, 18.56, 18.50.
t-Butyl 3-(((Methylsulfonyl)oxy)methyl)azetidine-1-carboxylate (18).
To a solution of t-butyl 3-(hydroxymethyl)azetidine-1-carboxylate (5 g, 26.7 mmol), triethylamine (7.4 mL, 53.4 mmol), and dichloromethane (50 mL), methanesulfonyl chloride (32 mL, 401 mmol) was added dropwise over 15 min at 0 °C. The resulting cloudy orange mixture was stirred at 0 °C for 1 h and then diluted with 10% aqueous citric acid (20 mL). The layers were separated, and the organic phase was washed by 10% aqueous citric acid, saturated NaHCO3, and water. The organic phase was dried over Na2SO4 and concentrated to give the title compound as dark orange oil (yield: 6 g, 85%). 1H NMR (400 MHz, DMSO-d6): δ 4.33 (d, J = 5.3 Hz, 2H), 3.91 (m, 2H), 3.61 (m, 2H), 3.21 (s, 3H), 2.89 (m, 1H), 1.37 (s, 9H).
t-Butyl 3-(Fluoromethyl)azetidine-l-carboxylate (19).
A solution of compound 18 (7 g, 26.7 mmol) and tetrabutylammonium fluoride (50 mL, 50 mmol, 1 M in THF) was refluxed for 1 h and monitored by TLC. Upon completion, the reaction mixture was evaporated under reduced pressure to remove the solvent THF. The resulting thick oil was diluted with ethyl acetate and then washed with water and brine, and dried over Na2SO4. The organic extracts were evaporated under reduced pressure and purified by flash chromatography (10–40% ethyl acetate in hexanes) to give yellow oil (yield: 4.2 g, 85% over two steps). 1H NMR (400 MHz, DMSO-d6): δ 4.52 (dd, J = 47.3, 5.3 Hz, 2H), 3.94–3.83 (m, 2H), 3.66–3.52 (m, 2H), 2.94–2.77 (m, 1H), 1.37 (s, 9H).
3-(Fluoromethyl)azetidine Hydrochloride (20).
To a solution of methanol (45 mL) was added compound 19 (4.2 g, 22.2 mmol), and aqueous HCI (6 M, 11.1 mL, 66.6 mmol) was dropwise added slowly to the reaction at 0 °C. The reaction was stirred at room temperature and monitored by TLC. Upon completion, the reaction was evaporated to become solidified under high vacuum to give the title compound (yield: 2.7 g, 97%) as a hygroscopic white solid. 1H NMR (400 MHz, DMSO-d6): δ 9.18 (br s, 2H), 4.56 (dd, J = 47.6, 5.3 Hz, 2H), 4.03–3.92 (m, 2H), 3.78–3.68 (m, 2H), 3.19–3.00 (m, 1H).
(R)-t-Butyl-3-(((methylsulfonyl)oxy)methyl)pyrrolidine-1-carboxylate (22R).
This compound was prepared using a procedure similar to that of 18 (yield: 0.8 g, 88%). 1H NMR (400 MHz, DMSO-d6): δ 4.26–4.11 (m, 2H), 3.44–3.28 (m, 2H), 3.26–3.14 (m, 1H), 3.18 (s, 3H), 3.05–2.93 (m, 1H), 2.62–2.49 (m, 1H), 2.00–1.87 (m, 1H), 1.72–1.56 (m, 1H), 1.40 (s, 9H).
(R)-t-Butyl 3-(Fluoromethyl)pyrrolidine-1-carboxylate (23R).
This compound was prepared using a procedure similar to that of 19 (yield: 0.2 g, 60%). 1H NMR (400 MHz, DMSO-d6): δ 4.49–4.41 (m, 1H), 4.37–4.29 (m, 1H), 3.40–3.28 (m, 2H), 3.24–3.18 (m, 1H), 3.02–2.98 (m, 1H), 2.58–2.52 (m, 1H), 1.95–1.88 (m, 1H), 1.67–1.54 (m, 1H), 1.38 (s, 9H).
(R)-3-(Fluoromethyl)pyrrolidine Hydrochloride (24R).
This compound was prepared using a procedure similar to that of 20 (yield: 0.1 g, 70%). 1H NMR (400 MHz, DMSO-d6, HCI salt): δ 9.35 (br s, 1H), 4.57–4.47 (m, 1H), 4.44–4.33 (m, 1H), 3.33–3.10 (m, 3H), 2.95–2.87 (m, 1H), 2.69–2.57 (m, 1H), 2.05–1.97 (m, 1H), 1.70–1.61 (m, 1H).
2-(4-(2-Bromoethoxy)phenoxy)tetrahydro-2H-pyran (26).
To a solution of THF (30 mL) was added deoxyarbutin, (2 g, 10.2 mmol), 1,2-dibromoethane (1.2 mL, 0.11 mol), NaOH (1.23 g, 31 mmol). The reaction mixture was reflux for 24 h and monitored by TLC. The reaction mixture was evaporated under reduced pressure and diluted by ethyl acetate, washed by water, brine and dried over Na2SO4. The organic extracts were evaporated under reduced pressure and purified by flash chromatography (10–25% ethyl acetate in hexanes) to give a white solid (yield: 1.4 g, 46%). 1H NMR (400 MHz, acetone-d6): δ 7.05–6.97 (m, 2H), 6.95–6.86 (m, 2H), 5.33 (t, J = 3.2 Hz, 1H), 4.31 (t, J = 5.7 Hz, 2H), 3.95–3.82 (m, 1H), 3.75 (t, J = 5.5 Hz, 2H), 3.56–3.52 (m, 1H), 1.98–1.96 (m, 1H), 1.90–1.73 (m, 2H), 1.71–1.50 (m, 3H). 13C NMR (101 MHz, acetone-d6): δ 153.18, 151.77, 117.74, 115.55, 96.95, 68.58, 61.50, 30.33, 30.29, 25.13, 18.79.
1-(2-(4-((Tetrahydro-2H-pyran-2-yl)oxy)phenoxy)ethyl)azetidine (27a).
To acetonitrile (5 mL) was added compound 26 (0.5 g, 1.6 mmol), azetidine hydrochloride (0.3 g, 3.2 mmol), and potassium carbonate (0.66 g, 4.8 mmol). The reaction mixture was stirred at 60 °C and monitored by TLC. Upon completion, the reaction was extracted by ethyl acetate, washed by water and brine, and dried over Na2SO4. The organic extracts were evaporated under reduced pressure and purified by flash chromatography (1–10%) of methanol in dichloromethane (yield: 0.3 g, 70%). 1H NMR (400 MHz, acetone-d6): δ 6.98 (d, J = 9.1 Hz, 2H), 6.84 (d, J = 9.1 Hz, 2H), 5.30 (t, J = 2.2 Hz, 1H), 3.90 (t, J = 5.8 Hz, 2H), 3.89–3.87 (m, 1H), 3.55–3.53 (m, 1H), 3.24 (t, J = 6.9 Hz, 4H), 2.72 (t, J = 5.8 Hz, 2H), 2.02 (t, J = 6.9 Hz, 2H), 1.97–1.95 (m, 1H), 1.88–1.75 (m, 2H), 1.62–1.59 (m, 3H). 13C NMR (101 MHz, acetone-d6): δ 153.92, 151.27, 117.70, 115.00, 97.03, 67.08, 61.48, 58.13, 55.63, 30.39, 25.18, 18.84, 17.91.
3-Methyl-1-(2-(4-((tetrahydro-2H-pyran-2-yl)oxy)phenoxy)-ethyl)azetidine (27b).
This compound was prepared using a procedure similar to that of 27a (yield: 0.3 g, 56%). 1H NMR (400 MHz, acetone-d6): δ 6.98 (d, J = 9.0 Hz, 2H), 6.84 (d, J = 9.1 Hz, 2H), 5.30 (t, J = 3.3 Hz, 1H), 3.92 (t, J = 5.8 Hz, 2H), 3.51 (t, J = 7.4 Hz, 2H), 2.85 (t, J = 7.0 Hz, 2H), 2.77 (t, J = 5.8 Hz, 2H), 2.51 (dq, J = 13.8, 6.9 Hz, 1H), 2.03–1.89 (m, 1H), 1.89–1.73 (m, 2H), 1.71–1.52 (m, 3H), 1.52–1.36 (m, 2H), 1.15 (d, J = 6.8 Hz, 3H). 13C NMR (101 MHz, acetone-d6): δ 153.87, 151.28, 117.70, 115.03, 97.03, 66.98, 62.39, 61.49, 57.85, 30.37, 26.23, 25.16, 18.82, 18.48.
2-(Fluoromethyl)-1-(2-(4-((tetrahydro-2H-pyran-2-yl)oxy)-phenoxy)ethyl)azetidine (27c).
This compound was prepared using a procedure similar to that of 27a (yield: 0.45 g, 88%). 1H NMR (400 MHz, CDCl3): δ 7.04–6.96 (m, 2H), 6.87–6.79 (m, 2H), 5.31 (t, J = 3.3 Hz, 1H), 4.56 (dd, J = 47.6, 5.3 Hz, 2H), 3.95 (t, J = 5.5 Hz, 2H), 3.72–3.48 (m, 2H), 3.20 (t, J = 7.0 Hz, 2H), 2.99–2.88 (m, 2H), 2.86 (t, J = 5.5 Hz, 2H), 2.08–1.93 (m, 1H), 1.86–1.83 (m, 2H), 1.75–1.56 (m, 4H). 13C NMR (101 MHz, CDCl3): δ 153.67, 151.25, 117.73, 115.28, 97.31, 84.21 (d, J = 167.0 Hz), 66.89, 62.10, 57.91, 56.56 (d, J = 6.9 Hz), 31.50, 31.40 (d, J = 20.3 Hz), 25.26, 18.94.
(3R)-3-(Fluoromethyl)-1-(2-(4-((tetrahydro-2H-pyran-2-yl)oxy)-phenoxy)ethyl)pyrrolidine (27d).
This compound was prepared using a procedure similar to that of 27a (yield: 0.5 g, 70%). 1H NMR (400 MHz, CDCl3): δ 7.00 (d, J = 8.9 Hz, 2H), 6.85 (d, J = 8.9 Hz, 2H), 5.31 (t, J = 3.1 Hz, 1H), 4.36 (dd, J = 47.4, 6.6 Hz, 2H), 4.9 (t, J = 5.7 Hz, 2H), 3.99–3.91 (m, 1H), 3.68–3.55 (m, 1H), 3.03–2.81 (m, 3H), 2.81–2.46 (m, 4H), 2.13–1.93 (m, 2H), 1.91–1.78 (m, 2H), 1.77–1.48 (m, 4H). 13C NMR (101 MHz, CDCl3): δ 153.63, 151.29, 117.76, 115.40, 97.30, 85.80 (d, J = 168.8 Hz), 67.27, 62.10, 56.65 (d, J = 5.2 Hz), 54.86, 54.32, 37.71 (d, J = 18.7 Hz), 30.50, 26.12 (d, J = 6.8 Hz), 25.26, 18.94.
(3S)-3-(Fluoromethyl)-1-(2-(4-(( tetrahydro-2H-pyran-2-yl)oxy)-phenoxy)ethyl)pyrrolidine (27e).
This compound was prepared using a procedure similar to that of 27a (yield: 0.4 g, 77%). 1H NMR (400 MHz, CDCl3): δ 6.99 (d, J = 9.0 Hz, 2H), 6.84 (d, J = 9.0 Hz, 2H), 5.29 (t, J = 3.3 Hz, 1H), 4.33 (dd, J = 47.4, 6.7 Hz, 2H), 4.04 (t, J = 5.9 Hz, 2H), 3.99–3.89 (m, 1H), 3.59–3.55 (m, 1H), 2.94–2.73 (m, 3H), 2.73–2.48 (m, 4H), 2.09–1.89 (m, 2H), 1.84–1.78 (m, 2H), 1.75– 1.43 (m, 4H). 13C NMR (101 MHz, CDCl3): δ 153.72, 151.22, 117.72, 115.35, 97.26, 85.93 (d, J = 168.7 Hz), 67.46, 62.05, 56.72 (d, J = 5.2 Hz), 54.90, 54.31, 37.72 (d, J = 18.7 Hz), 30.49, 26.14 (d, J = 6.8 Hz), 25.26, 18.93.
1-(2-(4-((Tetrahydro-2H-pyran-2-yl)oxy)phenoxy)ethyl)-piperidine (27f).
This compound was prepared using a procedure similar to that of 27a (yield: 0.3 g, 60%). 1H NMR (400 MHz, acetone-d6): δ 6.98 (d, J = 9.1 Hz, 2H), 6.90 (d, J = 9.1 Hz, 2H), 5.31 (t, J = 3.3 Hz, 1H), 4.04 (t, J = 6.1 Hz, 2H), 3.93–3.81 (m, 1H), 3.63–3.47 (m, 1H), 2.67 (t, J = 6.0 Hz, 3H), 2.48 (br s, 4H), 2.12–1.86 (m, 2H), 1.86–1.72 (m, 2H), 1.72–1.47 (m, 5H), 1.43–1.41 (m, 2H). 13C NMR (101 MHz, acetone-d6): δ 153.92, 151.26, 117.69, 115.10, 97.02, 66.54, 61.47, 57.91, 54.87, 30.35, 25.98, 25.14, 24.19, 18.81.
4-(2-(Azetidin-1-yl)ethoxy)phenol (28a).
To a solution of methanol (5 mL) was added compound 27a (0.18 g, 0.65 mmol) and p-toluenesulfonic acid (0.33 g, 1.95 mmol). The reaction was stirred at room temperature and monitored by TLC. Upon completion, the reaction was extracted by ethyl acetate, washed by saturated NaHCO3, water, and brine, and dried over Na2SO4. The organic extracts were evaporated under reduced pressure and purified by flash chromatography (1–10%) methanol in dichloromethane (yield: 0.15 g, 60%). 1H NMR (400 MHz, acetone-d6): δ 6.96 (s, 4H), 3.86 (t, J = 5.8 Hz, 2H), 3.24 (t, J = 6.9 Hz, 4H), 2.71 (t, J = 5.8 Hz, 2H), 2.04–1.96 (m, 2H). 13C NMR (101 MHz, acetone-d6): δ 152.30 151.34, 115.73, 115.33, 67.12, 58.19, 55.58, 17.85.
4-(2-(3-Methylazetidin-1-yl)ethoxy)phenol (28b).
This compound was prepared using a procedure similar to that of 28a (yield: 0.15 g, 70%). 1H NMR (400 MHz, MeOD): δ 6.77 (d, J = 9.1 Hz, 2H), 6.71 (d, J = 9.1 Hz, 2H), 3.93 (t, J = 5.3 Hz, 2H), 3.68 (t, J = 8.1 Hz, 2H), 3.03 (t, J = 7.9 Hz, 2H), 2.91 (t, J = 5.3 Hz, 2H), 2.66 (dq, J = 14.4, 7.2 Hz, 1H), 1.18 (d, J = 6.8 Hz, 3H). 13C NMR (101 MHz, MeOD): δ 152.00, 151.19, 115.47, 115.19, 66.16, 62.09, 57.32, 26.04, 17.71.
4-(2-(3-(Fluoromethyl)azetidin-1-yl)ethoxy)phenol (28c).
This compound was prepared using a procedure similar to that of 28a (yield: 0.1 g, 71%). 1H NMR (400 MHz, acetone-d6): δ 6.76 (s, 4H), 4.55 (dd, J = 47.7, 6.3 Hz, 2H), 3.89 (t, J = 5.7 Hz, 2H), 3.41 (t, J = 7.6 Hz, 2H), 3.13 (d, J = 7.6 Hz, 2H), 2.78–2.76 (m, J = 5.7 Hz, 3H). 13C NMR (101 MHz, acetone-d6): δ 152.28, 151.34, 115.72, 115.34, 84.57 (d, J = 164.9 Hz), 67.23, 57.74, 56.39 (d, J = 7.7 Hz), 31.37 (d, J = 20.2 Hz).
(R)-4-(2-(3-(Fluoromethyl)pyrrolidin-1-yl)ethoxy)phenol (28d).
This compound was prepared using a procedure similar to that of 28a (yield: 0.2 g, 65%). 1H NMR (400 MHz, CDCl3): δ 6.72–6.63 (m, 4H), 4.45–4.32 (m, 2H), 4.02 (t, J = 5.7 Hz, 2H), 2.99–2.86 (m, 3H), 2.86–2.76 (m, 1H), 2.76–2.47 (m, 3H), 2.09–1.95 (m, 1H), 1.59 (dq, J = 8.0, 6.0 Hz, 1H). 13C NMR (101 MHz, CDCl3): δ 152.40, 150.22, 116.46, 115.56, 85.67 (d, J = 168.8 Hz), 67.25, 56.82 (d, J = 5.0 Hz), 55.01, 54.51, 37.61 (d, J = 18.8 Hz), 26.04 (d, J = 6.7 Hz).
4-(2-(Piperidin-1-yl)ethoxy)phenol (28f).
This compound was prepared using a procedure similar to that of 28a (yield: 0.15 g, 58%). 1H NMR (400 MHz, MeOD): δ 6.79 (d, J = 9.0 Hz, 2H), 6.71 (d, J = 9.0 Hz, 2H), 4.90 (s, 1H), 4.05 (t, J = 5.7 Hz, 2H), 2.75 (t, J = 5.7 Hz, 2H), 2.57 (br s, 4H), 1.65–1.64 (m, 4H), 1.50–1.48 (m, 2H). 13C NMR (101 MHz, MeOD): δ 152.09, 151.10, 115.43, 115.24, 65.64, 57.73, 54.52, 25.02, 23.61.
(3-(4-(2-(Azetidin-1-yl)ethoxy)phenoxy)-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]thiophen-2-yl)(o-tolyl)methanone (29a).
To a solution of dimethylformamide (DMF) (10 mL) was added 16b (0.5 g, 1.3 mmol), 28a (0.31 g, 1.6 mmol), and cesium carbonate (1.26 g, 3.9 mmol). The reaction mixture was stirred at 110 °C for 5 h and monitored by TLC. Upon completion, the reaction mixture was extracted by ethyl acetate, washed by water and brine, and dried over Na2SO4. The organic extracts were evaporated under reduced pressure and purified by flash chromatography (1–10%) of methanol in DCM (yield: 0.13 g, 63%). 1H NMR (400 MHz, acetone-d6): δ 7.67 (d, J = 2.1 Hz, 1H), 7.44 (d, J = 8.9 Hz, 1H), 7.41–7.35 (m, 1H), 7.29 (td, J = 7.6, 1.2 Hz, 1H), 7.15 (d, J = 6.1 Hz, 2H), 7.10 (dd, J = 8.9, 2.1 Hz, 1H), 6.69 (d, J = 9.1 Hz, 2H), 6.46 (d, J = 9.1 Hz, 2H), 5.63 (t, J = 3.0 Hz, 1H), 3.85–3.82 (m, 3H), 3.73–3.58 (m, 1H), 3.25–3.15 (m, 4H), 2.71–2.62 (m, 2H), 2.16 (s, 3H), 2.00–1.96 (m, 2H), 1.92–1.80 (m, 2H), 1.77–1.51 (m, 4H). 13C NMR (101 MHz, acetone-d6): δ 189.92, 158.08, 154.54, 152.23, 149.27, 141.39, 139.63, 135.42, 130.32, 129.91, 127.51, 127.47, 125.06, 124.52, 116.94, 116.01, 115.50, 115.04, 108.95, 96.42, 67.18, 61.71, 57.96, 55.56, 29.98, 24.94, 18.52, 18.50, 17.87.
(3-(4-(2-(3-(Fluoromethyl)azetidin-1-yl)ethoxy)phenoxy)-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo-[b]thiophen-2-yl)(2-methyl-4-((tetrahydro-2H-pyran-2-yl)oxy)phenyl)methanone (29b).
This compound was prepared using a procedure similar to that of 29a (yield: 0.18 g, 65%). 1H NMR (400 MHz, acetone-d6): δ 7.67 (d, J = 2.0 Hz, 1H), 7.44 (d, J = 8.9 Hz, 1H), 7.37 (d, J = 7.5 Hz, 1H), 7.29 (m, 1H), 7.15 (d, J = 5.8 Hz, 2H), 7.10 (dd, J = 8.9, 2.1 Hz, 1H), 6.69 (d, J = 9.1 Hz, 2H), 6.46 (d, J = 9.1 Hz, 2H), 5.71–5.58 (m, 1H), 3.85 (t, J = 5.8 Hz, 3H), 3.67–3.58 (m, 1H), 3.43 (d, J = 7.2 Hz, 2H), 2.78 (t, J = 4.6 Hz, 2H), 2.70 (t, J = 5.8 Hz, 2H), 2.57–2.41 (m, 2H), 2.16 (s, 3H), 1.88–1.86 (m, 2H), 1.77–1.56 (m, 3H), 1.13 (d, J = 6.8 Hz, 3H). 13C NMR (101 MHz, acetone-d6): δ 189.92, 158.07, 154.55, 152.22, 149.27, 141.39, 139.63, 135.43, 130.32, 129.90, 127.52, 127.47 125.06, 124.52, 116.94, 116.01, 115.03, 108.94, 96.42, 67.26, 62.50, 61.70, 58.02, 46.12, 29.98, 26.27, 24.95, 18.54, 18.51.
(3-(4-(2-(3-(Fluoromethyl)azetidin-1-yl)ethoxy)phenoxy)-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]thiophen-2-yl)(phenyl)-methanone (29c).
This compound was prepared using a procedure similar to that of 29a (yield: 0.3 g, 73%). 1H NMR (400 MHz, acetone-d6): δ 7.75 (d, J = 8.3 Hz, 1H), 7.69 (d, J = 2.0 Hz, 1H), 7.57 (t, J = 7.5 Hz, 2H), 7.50–7.38 (m, 3H), 7.13 (dd, J = 8.9, 2.1 Hz, 1H), 6.74 (d, J = 9.1 Hz, 2H), 6.62 (d, J = 9.1 Hz, 2H), 5.65 (s, 1H), 4.54 (dd, J = 47.7, 6.2 Hz, 2H), 3.89–3.76 (m, 3H), 3.65–3.62 (m, 2H), 3.49–3.33 (m, 3H), 3.12–3.10 (m, 2H), 2.78–2.67 (m, 1H), 1.88– 1.86 (m, 2H), 1.75–1.41 (m, 4H). 13C NMR (101 MHz, acetone-d6): δ 188.26, 157.81, 154.57, 152.11, 148.30, 140.99, 138.66, 132.15, 128.69, 127.93, 127.26, 125.68, 124.36, 116.89, 116.56, 115.15, 108.87, 96.45, 85.40 (d, J = 164.8 Hz), 67.24, 61.71, 57.55, 56.36 (d, J = 6.3 Hz), 31.39 (d, J = 20.2 Hz), 30.00, 24.95, 18.52.
(3-(4-(2-(3-(Fluoromethyl)azetidin-1-yl)ethoxy)phenoxy)-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]thiophen-2-yl)(o-tolyl) Methanone (29d).
This compound was prepared using a procedure similar to that of 29a (yield: 0.44 g, 62%). 1H NMR (400 MHz, CDCl3): δ 7.51 (d, J = 1.9 Hz, 1H), 7.42 (d, J = 8.9 Hz, 1H), 7.29 (d, J = 7.3 Hz, 1H), 7.22 (t, J = 7.4 Hz, 1H), 7.07 (t, J = 6.9 Hz, 2H), 7.01 (dd, J = 8.8 2.0 Hz, 1H), 6.58 (d, J = 9.0 Hz, 2H), 6.35 (d, J = 9.0 Hz, 2H), 5.51 (t, J = 2.7 Hz, 1H), 4.50 (dd, J = 47.4, 5.6 Hz, 2H), 3.91–3.81 (m, 3H), 3.64 (m, 2H), 3.50 (br s, 2H), 3.16 (br s, 1H), 2.94–2.76 (m, 3H), 2.15 (s, 3H), 2.06–1.95 (m, 1H), 1.90–1.88 (m, 2H),1.78– 1.49 (m, 3H). 13C NMR (101 MHz, CDCl3): δ 190.88, 157.96, 154.04, 152.50, 149.48, 141.70, 139.38, 135.77, 130.39, 129.94, 127.72 127.62, 127.56, 125.03, 124.69, 116.85, 115.85, 115.04, 108.79 96.66, 84.13 (d, J = 167.2 Hz), 66.98, 62.10, 57.87, 56.55 (d, J = 6.5 Hz), 31.39 (d, J = 20.4 Hz), 30.21, 25.09, 19.28, 18.54.
(4-Fluoro-2-methylphenyl)(3-(4-(2-(3-(fluoromethyl)azetidin-1-yl)ethoxy)phenoxy)-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]-thiophen-2-yl)methanone (29e).
This compound was prepared using a procedure similar to that of 29a (yield: 0.32 g, 70%). 1H NMR (400 MHz, acetone-d6): δ 7.67 (d, J = 2.0 Hz, 1H), 7.49–7.41 (m, 2H), 7.11 (dd, J = 7.4, 3.7 Hz, 1H), 6.91 (d, J = 13.7 Hz, 2H), 6.73 (d, J = 6.9 Hz, 2H), 6.53–6.46 (d, J = 6.9 Hz, 2H), 5.63 (t, J = 3.0 Hz, 1H), 4.54 (dd, J = 47.7, 6.3 Hz, 2H), 3.97–3.77 (m, 3H), 3.71–3.57 (m, 1H), 3.47–3.28 (m, 2H), 3.14–3.12 (m, 2H), 2.80–2.70 (m, 3H), 2.17 (s, 3H), 2.07–1.79 (m, 3H), 1.78–1.48 (m, 3H). 13C NMR (101 MHz, acetone-d6): δ 188.79, 163.34 (d, J = 247.9 Hz), 158.15, 154.58, 152.25, 149.21, 141.44, 139.27 (d, J = 8.6 Hz), 135.83, 130.20 (d, J = 9.1 Hz), 127.45, 127.32, 124.51, 117.02, 116.92 (d, J = 19.2 Hz), 115.22, 115.13, 111.84 (d, J = 21.7 Hz), 108.94, 96.42, 84.58 (d, J = 165.0 Hz), 67.33, 61.71, 57.56, 56.39 (d, J = 7.2 Hz), 31.39 (d, J = 20.1 Hz), 29.98, 24.94, 18.61, 18.50.
(4-Fluoro-2,6-dimethylphenyl)(3-(4-(2-(3-(fluoromethyl)azetidin-1-yl)ethoxy)phenoxy)-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]-thiophen-2-yl)methanone (29f).
This compound was prepared using a procedure similar to that of 29a (yield: 0.4 g, 58%). 1H NMR (400 MHz, acetone-d6): δ 7.69 (d, J = 2.1 Hz, 1H), 7.37 (d, J = 8.9 Hz, 1H), 7.09 (dd, J = 8.9, 2.2 Hz, 1H), 6.74 (d, J = 9.1 Hz, 2H), 6.70 (d, J = 9.8 Hz, 2H), 6.50 (d, J = 9.0 Hz, 2H), 5.65 (t, J = 3.2 Hz, 1H), 4.54 (dd, J = 47.7, 6.1 Hz, 2H), 3.90 (t, J = 4.8 Hz, 2H), 3.88–3.76 (m, 2H), 3.64–3.62 (m, 1H), 3.55–3.40 (m, 1H), 2.98–2.95 (m, 2H), 2.87–2.65 (m, 2H), 2.13 (s, 6H), 1.97–1.94 (m, 1H), 1.92–1.79 (m, 2H), 1.78–1.65 (m, 2H), 1.65–1.43 (m, 2H). 13C NMR (101 MHz, acetone-d6): δ 190.57, 162.26 (d, J = 244.6 Hz), 158.28, 154.60, 151.79, 149.44, 141.73, 136.98, 136.64 (d, J = 8.6 Hz), 128.40, 127.26, 124.82, 117.08, 115.57, 114.98, 113.68 (d, J = 21.5 Hz), 109.09, 96.39, 93.23, 84.58 (d, J = 165.0 Hz), 67.37, 61.70,61.54, 57.49, 56.21, 29.94 (d, J = 19.7 Hz), 24.92, 18.46.
(3-(4-(2-(3-(Fluoromethyl)azetidin-1-yl)ethoxy)phenoxy)-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo-[b]thiophen-2-yl)(4-((tetrahy-dro-2H-pyran-2-yl)oxy)phenyl)methanone (29g).
This compound was prepared using a procedure similar to that of 29a (yield: 0.2 g, 70%). 1H NMR (400 MHz, acetone-d6): δ 7.83–7.73 (m, 2H), 7.67 (d, J = 2.1 Hz, 1H), 7.51 (d, J = 8.9 Hz, 1H), 7.13 (dd, J = 8.9, 2.2 Hz, 1H), 7.10–7.03 (m, 2H), 6.76–6.70 (m, 2H), 6.69–6.60 (m, 2H), 5.62 (m, 2H), 4.53 (dd, J = 47.7, 6.3 Hz, 2H), 3.86–3.84 (m, 2H), 3.68–3.57 (m, 2H), 3.37 (t, J = 7.0 Hz, 2H), 3.08 (t, J = 6.5 Hz, 2H), 2.75–2.74 (m, 1H), 2.73 (t, J = 5.6 Hz, 2H), 1.99 (m, 2h ), 1.88 (m, 6H), 1.77–1.51 (m, 6H). 13C NMR (101 MHz, acetone-d6): δ 186.73, 160.81, 157.57, 154.51, 152.23, 147.40, 140.53, 131.56, 131.16, 127.40, 125.69, 124.07, 116.81, 116.60, 115.57, 115.15, 108.85, 96.46, 95.96, 84.59 (d, J = 164.9 Hz), 67.22, 61.66, 61.62, 57.58, 56.39 (d, J = 7.7 Hz), 31.37 (d, J = 20.1 Hz), 30.02, 29.92, 24.95, 18.53, 18.47.
(3-(4-(2-(3-(Fluoromethyl)azetidin-1-yl)ethoxy)phenoxy)-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]thiophen-2-yl)(2-methyl-4-((tetrahydro-2H-pyran-2-yl)oxy)phenyl)methanone (29h).
This compound was prepared using a procedure similar to that of 29a (yield: 0.2 g, 58%). 1H NMR (400 MHz, acetone-d6): δ 7.67 (d, J = 2.0 Hz, 1H), 7.53 (d, J = 8.9 Hz, 1H), 7.43 (d, J = 8.5 Hz, 1H), 7.14 (dd, J = 8.9, 2.1 Hz, 1H), 6.85 (dd, J = 8.5, 2.3 Hz, 1H), 6.81 (d, J = 2.0 Hz, 1H), 6.72–6.67 (m, 2H), 6.50–6.45 (m, 2H). 5.64 (t, J = 3.0 Hz, 1H), 5.53 (t, J = 2.9 Hz, 1H), 4.54 (dd, J = 47.7, 6.3 Hz, 2H), 3.89– 3.86 (m, 4H), 3.69–3.56 (m, 2H), 3.39 (t, J = 7.1 Hz, 2H), 3.10 (t, J = 6.4 Hz, 2H), 2.78–2.76 (m, 1H), 2.75 (t, J = 5.4 Hz, 2H), 2.13 (s, 3H), 2.02–1.93 (m, 2H), 1.93–1.79 (m, 4H), 1.75–1.54 (m, 6H). 13C NMR (101 MHz, acetone-d6): δ 188.91, 158.80, 157.92, 154.43, 152.58, 148.50, 140.92, 138.59, 132.32, 130.61, 127.83, 127.42, 124.18, 118.43, 116.90, 115.97, 115.09, 112.72, 108.90, 96.45, 95.83, 84.57 (d, J = 164.9 Hz), 67.31, 61.70, 61.48, 57.54, 56.38 (d, J = 7.7 Hz), 31.38 (d, J = 20.1 Hz), 30.01 25.03, 24.95, 19.03, 18.52, 18.49.
(3-(4-(2-(3-(Fluoromethyl)azetidin-1-yl)ethoxy)phenoxy)-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]-thiophen-2-yl)(3-fluorophenyl)methanone (29j).
This compound was prepared using a procedure similar to that of 29a (yield: 0.2 g, 70%). 1H NMR (400 MHz, acetone-d6): δ 7.68 (d, J = 1.9 Hz, 1H), 7.57 (d, J = 7.6 Hz, 1H), 7.49–7.41 (m, 3H), 7.32 (td, J = 8.6, 2.5 Hz, 1H), 7.12 (dd, J = 8.9, 2.1 Hz, 1H), 6.75 (d, J = 9.1 Hz, 2H), 6.64 (d, J = 9.1 Hz, 2H), 5.63 (t, J = 2.8 Hz, 1H), 4.53 (dd, J = 47.7, 6.3 Hz, 2H), 3.88–3.83 (m, 3H), 3.65–3.62 (m, 1H), 3.37 (t, J = 7.2 Hz, 2H), 3.08 (t, J = 6.4 Hz, 2H), 2.73–2.69 (m, 3H), 2.01–1.80 (m, 3H), 1.76–1.52 (m, 3H). 13C NMR (101 MHz, acetone-d6): δ 186.94, 162.22 (d, J = 245.4 Hz), 157.99, 154.67, 152.00, 148.86, 141.30, 140.88 (d, J = 6.6 Hz), 129.99 (d, J = 7.8 Hz), 127.11, 125.23, 124.58, 124.55, 118.72 (d, J = 21.4 Hz), 116.98, 116.54, 115.25, 115.02 (d, J = 23.0 Hz), 108.87, 96.44, 84.60 (d, J = 165.0 Hz), 67.27, 61.71, 57.59, 56.40 (d, J = 7.7 Hz), 31.38 (d, J = 20.1 Hz), 29.98, 24.94, 18.50.
(2,4-Dimethylphenyl)(3-(4-(2-(3-(fluoromethyl)azetidin-1-yl)-ethoxy)phenoxy)-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]-thiophen-2-yl)methanone (29k).
This compound was prepared using a procedure similar to that of 29a (yield: 0.2 g, 72%). 1H NMR (400 MHz, acetone-d6): δ 7.67 (d, J = 2.1 Hz, 1H), 7.48 (d, J = 8.9 Hz, 1H), 7.31 (d, J = 7.5 Hz, 1H), 7.12 (dd, J = 8.9, 2.2 Hz, 1H), 6.98–6.92 (m, 2H), 6.71–6.69 (m, 2H), 6.52–6.42 (m, 2H), 5.64 (d, J = 3.0 Hz, 1H), 4.54 (dd, J = 47.7, 6.3 Hz, 2H), 3.95–3.82 (m, 3H), 3.70–3.60 (m, 1H), 3.39–3.37 (m, 2H), 3.11–3.09 (m, 2H), 2.79–2.72 (m, 3H), 2.32 (s, 3H), 2.13 (s, 3H), 2.03–1.82 (m, 3H), 1.80–1.54 (m, 3H). 13C NMR (101 MHz, acetone-d6): δ 189.76, 157.99, 154.49, 152.40, 148.86, 141.14, 140.21, 136.64, 135.80, 131.10, 128.12, 127.67, 127.51, 125.60, 124.36, 116.91, 116.03, 115.00, 108.91, 96.43, 84.57 (d, J = 164.9 Hz), 67.31, 61.70, 57.55, 56.38 (d, J = 7.6 Hz), 31.38 (d, J = 19.8 Hz), 29.99, 24.94, 20.44, 18.56, 18.50.
(3-(4-(2-((R)-3-(Fluoromethyl)pyrrolidin-1-yl)ethoxy)phenoxy)-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]thiophen-2-yl)(o-tolyl)-methanone (29l).
This compound was prepared using a procedure similar to that of 29a (yield 0.15 g, 80%). 1H NMR (400 MHz, acetone-d6): δ 7.68 (d, J = 2.1 Hz, 1H), 7.45 (d, J = 8.9 Hz, 1H), 7.39 (d, J = 5.6, 1H), 7.30 (td, J = 7.6, 1.2 Hz, 1H), 7.16 (dd, J = 7.2, 3.4 Hz, 2H), 7.11 (dd, J = 9.0, 2.2 Hz, 1H), 6.77–6.69 (m, 2H), 6.52–6.43 (m, 2H), 5.64 (t, J = 3.1 Hz, 1H), 4.31 (dd, J = 47.7, 6.8 Hz, 2H), 4.03 (t, J = 5.9 Hz, 2H), 3.91–3.75 (m, 1H), 3.68–3.65 (m, 1H), 2.83–2.81 (m, 2H), 2.69–2.67 (m, 2H), 2.55–2.52 (m, 3H), 2.16 (s, 3H), 2.02–1.77 (m, 4H), 1.72–1.66 (m, 4H). 13C NMR (101 MHz, acetone): δ 189.93, 158.09, 154.50, 152.29, 149.26, 141.38, 139.63, 135.42, 130.32, 129.91, 127.52, 127.47, 125.07, 124.51, 116.95, 116.03, 115.15, 108.95, 96.42, 85.68 (d, J = 167.2 Hz), 67.56, 61.71, 56.41 (d, J = 5.2 Hz), 54.33, 53.82, 37.76 (d, J = 18.6 Hz), 29.97, 25.84 (d, J = 7.0 Hz), 24.94, 18.50.
(4-Fluoro-2,6-dimethylphenyl)(3-(4-(2-((R)-3-(fluoromethyl)-pyrrolidin-1-yl)ethoxy)phenoxy)-6-(tetrahydro-2H-pyran-2-yl)-benzo[b]thiophen-2-yl)methanone (29m).
This compound was prepared using a procedure similar to that of 29a (yield: 0.15 g, 72%). 1H NMR (400 MHz, acetone-d6): δ 7.69 (d, J = 2.0 Hz, 1H), 7.37 (d, J = 8.9 Hz, 1H), 7.09 (dd, J = 8.9, 2.1 Hz, 1H), 6.80–6.73 (m, 2H), 6.70 (d, J = 9.7 Hz, 2H), 6.55–6.44 (m, 2H), 5.64 (t, J = 2.9 Hz, 1H), 4.31 (dd, J = 47.7, 6.7 Hz, 2H), 4.04 (t, J = 5.9 Hz, 2H), 3.89– 3.76 (m, 1H), 3.70–3.54 (m, 1H), 2.82 (d, J = 5.8 Hz, 2H), 2.72–2.62 (m, 2H), 2.59–2.45 (m, 2H), 2.13 (s, 6H), 1.90–1.83 (m, 4H), 1.78–1.52 (m, 4H), 1.52–1.35 (m, 1H). 13C NMR (101 MHz, acetone-d6): δ 190.55, 162.26 (d, J = 244.5 Hz), 158.28, 154.58, 151.82, 149.44, 141.73, 136.95, 136.65 (d, J = 8.6 Hz), 128.41, 127.28, 124.82, 124.10, 117.08, 115.58, 115.08, 113.69 (d, J = 21.5 Hz), 109.10, 96.40, 85.68 (d, J = 167.2 Hz), 67.58, 61.70, 56.42 (d, J = 5.2 Hz), 54.33, 53.83, 37.76 (d, J = 18.6 Hz), 29.94, 25.85 (d, J = 6.9 Hz), 24.92, 18.47.
(4-Fluoro-2,6-dimethylphenyl)(3-(4-(2-((R)-3-(fluoromethyl)-pyrrolidin-1-yl)ethoxy)phenoxy)-6-((tetrahydro-2H-pyran-2-yl)-oxy)benzo[b]thiophen-2-yl)methanone (29n).
This compound was prepared using a procedure similar to that of 29a (yield: 0.3 g, 64%). 1H NMR (400 MHz, acetone-d6): δ 7.69 (d, J = 2.0 Hz, 1H), 7.37 (d, J = 8.9 Hz, 1H), 7.09 (dd, J = 8.9, 2.2 Hz, 1H), 6.77 (d, J = 9.2 Hz, 2H), 6.71 (s, 1H), 6.69 (s, 1H), 6.51 (d, J = 9.1 Hz, 2H), 5.64 (s, 1H), 4.31 (dd, J = 47.7, 6.8 Hz, 2H), 4.04 (t, J = 5.9 Hz, 2H), 3.84–3.82 (m, 1H), 3.73–3.58 (m, 1H), 2.81–2.79 (m, 2H), 2.68–2.65 (m, 2H), 2.53–2.51 (m, 2H), 2.12 (s, 6H), 2.03–1.83 (m, 4H), 1.66–1.64 (m, 4H), 1.54–1.40 (m, 1H). 13C NMR (101 MHz, acetone-d6): δ 190.57, 162.26 (d, J = 244.6 Hz), 158.28, 154.59, 151.82, 149.45, 141.73, 136.97, 136.65 (d, J = 8.6 Hz), 128.41, 127.28, 124.83, 117.09, 115.58, 115.07, 113.69 (d, J = 21.5 Hz), 109.10, 96.39, 85.70 (d, J = 167.2 Hz), 67.60, 61.71, 56.44 (d, J = 5.2 Hz), 54.35, 53.85, 37.76 (d, J = 18.6 Hz), 29.95, 25.86 (d, J = 7.0 Hz), 24.93, 18.47.
(3-(4-(2-(Piperidin-1-yl)ethoxy)phenoxy)-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]thiophen-2-yl)(o-tolyl)methanone (29o).
This compound was prepared using a procedure similar to that of 29a (yield: 0.1 g, 65%). 1H NMR (400 MHz, acetone-d6): δ 7.67 (d, J = 1.9 Hz, 1H), 7.44 (d, J = 8.9 Hz, 1H), 7.38 (d, J = 8.0 Hz, 1H), 7.29 (t, J = 7.5 Hz, 1H), 7.17–7.11 (m, 2H), 7.09 (dd, J = 8.9, 2.0 Hz, 1H), 6.71 (d, J = 9.1 Hz, 2H), 6.46 (d, J = 9.1 Hz, 2H), 5.62 (t, J = 3.0 Hz, 1H), 3.99 (t, J = 6.0 Hz, 2H), 3.88–3.79 (m, 1H), 3.74–3.57 (m, 1H), 2.65 (t, J = 6.0 Hz, 2H), 2.45–2.43 (m, 4H), 2.16 (s, 3H), 1.97–1.80 (m, 2H), 1.70–1.68 (m, 2H), 1.65–1.50 (m, 6H), 1.41–1.39 (m, 2H). 13C NMR (101 MHz, acetone-d6): δ 189.91, 158.08, 154.54, 152.25, 149.27, 141.40, 139.63, 135.44, 130.33, 129.92, 127.49, 125.07, 124.67, 124.53, 118.16, 116.95, 116.02, 115.16, 108.95, 96.42, 66.63, 61.71, 57.81, 54.87, 29.99, 25.99, 24.96, 24.20, 18.57, 18.51.
(3-(4-(2-(Azetidin-1-yl)ethoxy)phenoxy)-6-hydroxybenzo[b]-thiophen-2-yl)(o-tolyl)methanone (30a).
To a solution of methanol (5 mL) was added compound 29a (0.2 g, 0.37 mmol) and p-toluenesulfonic acid (0.2 g, 1.2 mmol). The reaction was stirred at room temperature and monitored by TLC. Upon completion, the reaction was extracted by ethyl acetate, washed by saturated NaHCO3, water, and brine, and dried over Na2SO4. The organic extracts were evaporated under reduced pressure and purified by flash chromatography (1–10% methanol in dichloromethane) Further purification was by RP-HPLC (phase A 0.05% TFA in water, phase B 0.05% TFA in MeOH) running gradients from 40 to 100% B (yield: 0.02 g, 11%). 1H NMR (400 MHz, acetone-d6): δ 7.43–7.32 (m, 3H), 7.26–7.23 (m, 1H), 7.15–7.07 (m, 2H), 6.96 (dd, J = 8.8, 2.1 Hz, 1H), 6.67 (d, J = 9.1 Hz, 2H), 6.43 (d, J = 9.1 Hz, 2H), 3.89 (t, J = 5.6 Hz, 2H), 3.33 (t, J = 7.0 Hz, 4H), 2.79 (t, J = 5.6 Hz, 2H), 2.14 (s, 3H), 2.05–2.02 (m, 2H). 13C NMR (101 MHz, acetone-d6): δ 189.85, 159.23, 154.35, 152.31, 149.60, 141.94, 139.84, 135.29, 130.25, 129.74, 127.34, 126.27, 125.97, 125.03, 124.87, 116.19, 115.95, 115.02, 108.05, 66.80, 57.66, 55.52, 18.49, 17.75. HRMS (m/ z): [M + H]+ calcd for C27H26NO4S, 460.1504; observed, 460.1509.
(6-Hydroxy-3-(4-(2-(3-methylazetidin-1-l)ethoxy)phenoxy)-benzo[b]thiophen-2-yl)(o-tolyl) Methanone (30b).
This compound was prepared using a procedure similar to that of 30a (yield: 0.047 g, 15%). 1H NMR (400 MHz, acetone-d6): δ 7.42 (d, J = 2.1 Hz, 1H), 7.37 (t, J = 8.4 Hz, 2H), 7.28 (t, J = 7.5 Hz, 1H), 7.14–7.12 (m, 2H), 6.98 (dd, J = 8.8, 2.1 Hz, 1H), 6.72–6.66 (m, 2H), 6.47–6.41 (m, 2H), 3.89 (t, J = 5.7 Hz, 2H), 3.51 (t, J = 7.3 Hz, 2H), 2.85 (t, J = 6.8 Hz, 2H), 2.75–2.73 (m, 5H), 2.52–2.50 (m, 1H) 2.15 (s, 3H). 13C NMR (101 MHz, acetone-d6): δ 189.84, 158.99, 154.42, 152.28, 149.56 141.88, 139.82, 135.29, 130.25, 129.74, 127.34, 126.36, 126.08, 125.02, 124.87, 116.05, 115.95, 115.03, 108.00, 67.02, 62.38, 57.74, 45.96, 26.20, 18.46, 18.43. HRMS (m/z): [M + H]+ calcd for C28H28NO4S, 474.1661; observed, 474.1660.
(3-(4-(2-(3-(Fluoromethyl)azetidin-1-yl)ethoxy)phenoxy)-6-hydroxybenzo[b]thiophen-2-yl)(phenyl)methanone (30c).
This compound was prepared using a procedure similar to that of 30a (yield: 0.004 g, 10%). 1H NMR (400 MHz, acetone-d6): δ 7.72–7.66 (m, 2H), 7.51 (t, J = 7.4 Hz, 1H), 7.37–7.34 (m, 2H), 7.33 (d, J = 2.0 Hz, 2H), 6.93 (dd, J = 8.8, 2.1 Hz, 1H), 6.73–6.67 (m, 2H), 6.62–6.52 (m, 2H), 4.52 (dd, J = 47.7, 6.3 Hz, 2H), 3.85 (t, J = 5.6 Hz, 2H), 3.34 (t, J = 7.6 Hz, 2H), 3.09–3.02 (m, 2H), 2.71–2.69 (m, 3H). 13C NMR (101 MHz, acetone-d6): δ 188.10, 154.42, 152.19, 148.90, 141.88, 139.09, 134.04, 131.73, 128.55, 127.79, 124.73, 124.57, 123.72, 116.76, 116.45, 115.07, 107.98, 84.62 (d, J = 165.0 Hz), 67.21, 57.64, 56.39 (d, J = 7.7 Hz), 31.37 (d, J = 20.1 Hz). HRMS (m/z): [M + H]+ calcd for C27H25FNO4S, 478.1483; observed, 478.1408.
(3-(4-(2-(3-(Fluoromethyl)azetidin-1-yl)ethoxy)phenoxy)-6-hydroxybenzo[b]thiophen-2-yl)(o-tolyl)methanone (30d).
(Yield: 0.1 g, 20%). 1H NMR (400 MHz, CDCl3): δ 7.37–7.15 (m, 4H), 7.08–7.06 (m, 2H), 6.81 (dd, J = 8.8, 1.9 Hz, 1H), 6.50 (d, J = 9.1 Hz, 2H), 6.32 (d, J = 9.0 Hz, 2H), 4.46 (dd, J = 47.3, 4.7 Hz, 2H), 3.93 (d, J = 4.7 Hz, 2H), 3.72 (d, J = 7.7 Hz, 2H), 3.39 (t, J = 7.2 Hz, 2H), 2.99 (br s, 3H), 2.16 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 191.2, 158.46, 153.55, 152.61, 149.90, 142.16, 139.48, 135.63, 130.35, 129.88, 127.44, 126.54, 126.29, 125.06, 125.05, 116.22, 115.87, 114.88, 108.36, 83.85 (d, J = 168.2 Hz), 65.97, 57.25, 56.15 (d, J = 6.2 Hz), 50.87, 31.30 (d, J = 20.6 Hz), 19.25. HRMS (m/z): [M + H]+ calcd for C28H27FNO4S, 492.1645; observed, 492.1626.
(4-Fluoro-2-methylphenyl)(3-(4-(2-(3-(fluoromethyl)azetidin-1-yl)ethoxy)phenoxy)-6-hydroxybenzo[b]thiophen-2-yl)methanone (30e).
This compound was prepared using a procedure similar to that of 30a (yield: 0.0147 g, 23%). 1H NMR (400 MHz, acetone-d6): δ 7.48–7.42 (m, 1H), 7.40 (d, J = 9.2 Hz, 2H), 6.98 (dd, J = 8.8, 2.1 Hz, 1H), 6.92 (d, J = 9.2 Hz, 2H), 6.72 (d, J = 9.1 Hz, 2H), 6.49 (d, J = 9.1 Hz, 2H), 4.54 (dd, J = 47.7, 6.3 Hz, 2H), 3.90 (t, J = 5.6 Hz, 2H), 3.40 (t, J = 7.2 Hz, 2H), 3.15 (t, J = 6.4 Hz, 2H), 2.76–2.74 (m, 3H), 2.17 (s, 3H). 13C NMR (101 MHz, acetone-d6): δ 188.72, 163.26 (d, J = 247.5 Hz), 159.11, 154.47, 152.30, 149.52, 141.96, 139.11 (d, J = 8.6 Hz), 136.03, 130.05 (d, J = 9.1 Hz), 126.13 (d, J = 18.6 Hz), 124.88, 116.96, 116.85 (d, J = 21.6 Hz), 115.82, 115.10, 111.79 (d, J = 21.6 Hz), 108.02, 84.45 (d, J = 165.0 Hz), 67.16, 57.48, 56.35 (d, J = 7.6 Hz), 31.34 (d, J = 20.2 Hz), 18.55. HRMS (m/z): [M + H]+ calcd for C28H26F2NO4S, 510.1472; observed, 510.1478.
(4-Fluoro-2,6-dimethylphenyl)(3-(4-(2-(3-(fluoromethyl)azetidin-1-yl)ethoxy)phenoxy)-6-hydroxybenzo[b]thiophen-2-yl)-methanone (30f).
This compound was prepared using a procedure similar to that of 30a (yield: 0.4 g, 20%). 1H NMR (400 MHz, acetone-d6): δ 7.42 (d, J = 2.0 Hz, 1H), 7.31 (d, J = 8.8 Hz, 1H), 6.96 (dd, J = 8.8, 2.1 Hz, 1H), 6.75–6.66 (m, 4H), 6.52–6.45 (m, 2H), 4.54 (dd, J = 47.7, 6.3 Hz, 2H), 3.90 (d, J = 5.6 Hz, 2H), 3.41 (t, J = 7.5 Hz, 2H), 3.12 (t, J = 6.5 Hz, 2H), 2.77–2.75 (m, 3H), 2.12 (s, 6H). 13C NMR (101 MHz, acetone-d6): δ 190.40, 162.21 (d, J = 244.4 Hz), 159.13, 154.52, 151.82, 149.70, 142.20, 137.06, 136.61 (d, J = 8.6 Hz), 127.30, 125.91, 125.20, 116.17, 115.52, 114.94, 113.64 (d, J = 21.5 Hz), 108.18, 84.52 (d, J = 165.1 Hz), 67.25, 57.56, 56.38 (d, J = 7.7 Hz), 31.36 (d, J = 20.2 Hz), 18.45. HRMS m/z: (M + H)+ calcd for C29H28F2NO4S, 524.1702; observed, 524.1694.
(3-(4-(2-(3-(Fluoromethyl)azetidin-1-yl)ethoxy)phenoxy)-6-hydroxybenzo[b]thiophen-2-yl)(4-hydroxyphenyl)methanone (30g).
This compound was prepared using a procedure similar to that of 30a (yield: 0.017 g, 15%). 1H NMR (400 MHz, acetone-d6): δ 7.78– 7.66 (m, 2H), 7.43 (d, J = 8.8 Hz, 1H), 7.39 (d, J = 2.1 Hz, 1H), 6.98 (dd, J = 8.8, 2.2 Hz, 1H), 6.90–6.83 (m, 2H), 6.79–6.69 (m, 2H), 6.69–6.60 (m, 2H), 4.53 (dd, J = 47.7, 6.3 Hz, 2H), 3.88 (t, J = 5.6 Hz, 2H), 3.39 (d, J = 4.4 Hz, 2H), 3.10 (t, J = 6.6 Hz, 2H), 2.88–2.61 (m, 3H). 13C NMR (101 MHz, acetone-d6): δ 186.50, 161.77, 158.18, 154.43, 152.24, 147.16, 140.87, 131.75, 129.97, 126.01, 124.86, 124.34, 116.56, 115.77, 115.10, 114.72, 107.81, 84.51 (d, J = 165.0 Hz), 67.10, 57.57, 56.34 (d, J = 7.7 Hz), 31.34 (d, J = 20.2 Hz). HRMS (m/z): [M + H]+ calcd for C27H25FNO5S, 494.1359; observed, 494.1357.
(3-(4-(2-(3-(Fluoromethyl)azetidin-1-yl)ethoxy)phenoxy)-6-hydroxybenzo[b]thiophen-2-yl)(4-hydroxy-2-methylphenyl)-methanone (30h).
This compound was prepared using a procedure similar to that of 30a (yield: 0.013 g, 18%). 1H NMR (400 MHz, acetone-d6): δ 7.32 (d, J = 8.5 Hz, 2H), 7.22 (d, J = 1.9 Hz, 1H), 6.90 (dd, J = 8.9, 2.0 Hz, 1H), 6.67 (d, J = 9.1 Hz, 2H), 6.61 (dd, J = 8.4, 2.4 Hz, 1H), 6.57 (d, J = 2.1 Hz, 1H), 6.49 (d, J = 9.1 Hz, 2H), 4.53 (dd, J = 47.7, 6.4 Hz, 2H), 3.86 (t, J = 5.6 Hz, 2H), 3.37 (dd, J = 7.6, 6.3 Hz, 2H), 3.13–3.04 (t, J = 6.2 Hz, 2H), 2.72–2.69 (m, 3H), 2.09 (s, 3H). 13C NMR (101 MHz, acetone-d6): δ 188.39, 163.61, 160.93, 154.18, 152.73, 148.74, 141.88, 138.99, 131.47, 130.01, 124.31, 123.98, 123.77, 117.79, 117.71, 116.09, 114.92, 112.00, 108.22, 84.64 (d, J = 164.9 Hz), 67.25, 57.63, 56.37 (d, J = 7.7 Hz), 31.37 (d, J = 20.1 Hz), 19.24. HRMS (m/z): [M + H]+ calcd for C28H27FNO5S, 508.1516; observed, 508.1511.
(3-(4-(2-(3-(Fluoromethyl)azetidin-1-yl)ethoxy)phenoxy)-6-hydroxybenzo[b]thiophen-2-yl)(4-fluorophenyl)methanone (30i).
This compound was prepared using a procedure similar to that of 30a (yield: 0.015 g, 18%). 1H NMR (400 MHz, acetone-d6): δ 7.91–7.71 (m, 2H), 7.42–7.39 (m, 2H), 7.18 (t, J = 8.8 Hz, 2H), 7.00 (dd, J = 8.9, 2.0 Hz, 1H), 6.74 (d, J = 9.1 Hz, 2H), 6.64 (d, J = 9.1 Hz, 2H), 4.54 (dd, J = 47.7, 6.3 Hz, 2H), 3.89 (t, J = 5.6 Hz, 2H), 3.39 (t, J = 7.2 Hz, 2H), 3.10 (t, J = 6.5 Hz, 2H), 2.76–2.74 (m, 1H), 2.75 (t, J = 5.5 Hz, 2H). 13C NMR (101 MHz, acetone-d6): δ 186.79, 165.02 (d, J = 250.6 Hz), 158.73, 154.54, 152.09, 148.53, 141.55, 135.19 (d, J = 2.0 Hz), 131.47 (d, J = 9.2 Hz), 125.78, 124.71, 124.39, 116.39, 115.98, 115.18, 114.83 (d, J = 22.1 Hz), 107.82, 84.53 (d, J = 165.0 Hz), 67.18, 57.53, 56.37 (d, J = 7.6 Hz), 31.35 (d, J = 20.2 Hz). HRMS (m/z): [M + H]+ calcd for C27H24F2NO4S, 496.1316; observed, 496.1322.
(3-(4-(2-(3-(Fluoromethyl)azetidin-1-yl)ethoxy)phenoxy)-6-hydroxybenzo[b]thiophen-2-yl)(3-fluorophenyl)methanone (30j).
This compound was prepared using a procedure similar to that of 30a (yield: 0.034 g, 15%). 1H NMR (400 MHz, acetone-d6): δ 7.55 (dt, J = 7.6, 1.1 Hz, 1H), 7.49–7.38 (m, 4H), 7.36–7.26 (m, 1H), 6.98 (dd, J = 8.9, 2.1 Hz, 1H), 6.74 (d, J = 9.2 Hz, 2H), 6.63 (d, J = 9.2 Hz, 2H), 4.54 (dd, J = 47.7, 6.3 Hz, 2H), 3.89 (t, J = 5.6 Hz, 2H), 3.39 (td, J = 7.7, 1.3 Hz, 2H), 3.13–3.05 (m, 2H), 2.84–2.68 (m, 3H). 13C NMR (101 MHz, acetone-d6): δ 186.90, 162.20 (d, J = 245.3 Hz), 158.82, 154.60, 152.04, 149.15, 141.81, 141.06 (d, J = 6.7 Hz), 129.94 (d, J = 7.8 Hz), 125.79, 124.93, 124.51 (d, J = 2.8 Hz), 124.24, 118.53 (d, J = 21.5 Hz), 116.46, 116.06, 115.22, 114.95 (d, J = 23.1 Hz), 107.92, 84.54 (d, J = 164.9 Hz), 67.19, 57.57, 56.36 (d, J = 7.6 Hz), 31.35 (d, J = 20.1 Hz). HRMS (m/z): [M + H]+ calcd for C27H24F2NO4S, 496.1316; observed, 496.1322. HPLC purity 95.04%.
(2,4-Dimethylphenyl)(3-(4-(2-(3-(fluoromethyl)azetidin-1-yl)-ethoxy)phenoxy)-6-hydroxybenzo[b]thiophen-2-yl)methanone (30k).
This compound was prepared using a procedure similar to that of 30a (yield: 0.067 g, 18%). 1H NMR (400 MHz, acetone-d6): δ 7.42–7.38 (m, 2H), 7.28 (d, J = 7.5 Hz, 1H), 6.98 (dd, J = 8.8, 2.1 Hz, 1H), 6.95 (s, 2H), 6.70 (d, J = 9.1 Hz, 2H), 6.46 (d, J = 9.1 Hz, 2H), 4.54 (dd, J = 47.7, 6.3 Hz, 2H), 3.89 (t, J = 5.6 Hz, 2H), 3.40 (t, J = 7.6 Hz, 2H), 3.11 (t, J = 6.5 Hz, 2H), 2.85–2.68 (m, 3H), 2.31 (s, 3H), (2.10, s, 3H). 13C NMR (101 MHz, acetone-d6): δ 189.71, 158.79, 154.41, 152.42, 149.17, 141.66, 140.02, 136.82, 135.67, 131.3, 127.99, 126.44, 126.31, 125.56, 124.74, 115.97, 115.24, 114.95, 107.94, 84.54 (d, J = 165.1 Hz), 67.26, 57.57, 56.37 (d, J = 7.6 Hz), 31.37 (d, J = 20.2 Hz), 20.44, 18.54. HRMS (m/z): [M + H]+ calcd for C29H29FNO4S, 506.1723; observed, 506.1713.
(R)-(3-(4-(2-(3-(Fluoromethyl)pyrrolidin-1-yl)ethoxy)phenoxy)-6-hydroxybenzo[b]thiophen-2-yl)(o-tolyl)methanone (30l).
This compound was prepared using a procedure similar to that of 30a (yield: 0.028 g, 16%). 1H NMR (400 MHz, acetone-d6): δ 7.44–7.32 (m, 3H), 7.31–7.22 (m, 1H), 7.17–7.09 (m, 2H), 6.98 (dd, J = 8.8, 2.0 Hz, 1H), 6.73 (d, J = 6.9 Hz, 2H), 6.45 (d J = 6.9 Hz, 2H), 4.34 (dd, J = 47.6, 6.8 Hz, 2H), 4.12–4.07 (m, 2H), 2.92–2.90 (m, 2H), 2.81–2.78 (m, 2H), 2.72–2.43 (m, 3H), 2.15 (s, 3H), 2.00–1.85 (m, 1H), 1.56–1.52 (m, 1H). 13C NMR (101 MHz, acetone-d6): δ 189.85 158.89, 154.30, 152.37, 149.53, 141.87, 139.79, 135.31, 130.26, 129.77, 127.36, 126.41, 126.16, 125.03, 124.88, 116.03, 115.98, 115.16, 108.01, 85.46 (d, J = 167.1 Hz), 67.12, 56.28 (d, J = 5.1 Hz), 54.24, 53.83, 37.68 (d, J = 18.7 Hz), 25.75 (d, J = 6.9 Hz), 18.48 HRMS (m/z): [M + H]+ calcd for C29H29FNO4S, 506.1796; observed, 506.1727.
(R)-(4-Fluoro-2,6-dimethylphenyl)(3-(4-(2-(3-(fluoromethyl)-pyrrolidin-1-yl)ethoxy)phenoxy)-6-hydroxybenzo[b]thiophen-2-yl)-methanone (30m).
This compound was prepared using a procedure similar to that of 30a (yield: 0.04 g, 20%). 1H NMR (400 MHz, acetone-d6): δ 7.45 (d, J = 1.8 Hz, 1H), 7.30 (d, J = 8.8 Hz, 1H), 6.98 (dd, J = 8.8, 2.0 Hz, 1H), 6.78–6.73 (m, 2H), 6.68 (d, J = 9.8 Hz, 2H), 6.48 (d, J = 9.0 Hz, 2H), 4.34 (dd, J = 47.6, 6.8 Hz, 2H), 4.08 (t, J = 5.7 Hz, 2H), 2.92 (t, J = 5.7 Hz, 2H), 2.79–2.76 (m, 2H), 2.75–2.65 (m, 1H), 2.65–2.47 (m, 2H), 2.12 (s, 6H), 2.01–1.89 (m, 1H), 1.52–1.49 (m, 1H). 13C NMR (101 MHz, acetone-d6): δ 190.39, 162.21 (d, J = 244.4 Hz), 159.47, 154.37, 151.93, 149.75, 142.19, 137.07, 136.62 (d, J = 8.6 Hz), 127.15, 125.74, 125.10, 116.33, 115.54, 115.07, 113.64 (d, J = 21.5 Hz), 108.23, 85.47 (d, J = 167.2 Hz), 67.14, 56.30 (d, J = 5.3 Hz), 54.27, 53.86, 37.69 (d, J = 18.7 Hz), 25.78 (d, J = 6.9 Hz), 18.48. HRMS (m/z): [M + H]+ calcd for C30H30F2NO4S, 538.1785; observed, 538.1798.
(S)-(4-Fluoro-2,6-dimethylphenyl)(3-(4-(2-(3-(fluoromethyl)-pyrrolidin-1-yl)ethoxy)phenoxy)-6-hydroxybenzo[b]thiophen-2-yl)-methanone (30n).
This compound was prepared using a procedure similar to that of 30a (yield: 0.10 g, 25%). 1H NMR (400 MHz, acetone-d6): δ 7.41 (d, J = 1.9 Hz, 1H), 7.31 (d, J = 8.8 Hz, 1H), 6.95 (dd, J = 8.8, 2.1 Hz, 1H), 6.75 (d, J = 9.1 Hz, 2H), 6.70 (s, 1H), 6.67 (s, 1H), 6.48 (d, J = 9.1 Hz, 2H), 4.32 (dd, J = 47.7, 6.8 Hz, 2H), 4.05 (t, J = 5.7 Hz, 2H), 2.84 (t, J = 5.7 Hz, 2H), 2.76–2.66 (m, 2H), 2.62–2.46 (m, 3H), 2.12 (s, 6H), 1.92–1.85 (m, 1H), 1.48–1.45 (m, 1H). 13C NMR (101 MHz, acetone-d6): δ 190.41, 162.22 (d, J = 244.4 Hz), 159.14, 154.49, 151.86, 149.71, 142.20, 137.07 (d, J = 2.8 Hz) 136.62 (d, J = 8.7 Hz), 127.31, 125.92, 125.20, 116.19, 115.53, 115.3, 113.64 (d, J = 21.4 Hz), 108.20, 85.65 (d, J = 167.2 Hz), 67.48, 56.41 (d, J = 5.4 Hz), 54.36, 53.84, 37.74 (d, J = 18.6 Hz), 25.84 (d, J = 6.9 Hz), 18.47. HRMS (m/z): [M + H]+ calcd for C30H30F2NO4S, 538.1785; observed, 538.1798.
(6-Hydroxy-3-(4-(2-(piperidin-1-yl)ethoxy)phenoxy)benzo[b]-thiophen-2-yl)(o-tolyl) Methanone (30o).
This compound was prepared using a procedure similar to that of 30a (yield: 0.04 g, 17%). 1H NMR (400 MHz, acetone-d6): δ 7.43 (d, J = 2.1 Hz, 1H), 7.37 (t, J = 8.3 Hz, 2H), 7.28 (td, J = 7.5, 1.3 Hz, 1H), 7.14–7.12 (m, 2H), 6.98 (dd, J = 8.8, 2.2 Hz, 1H), 6.71 (d, J = 9.1 Hz, 2H), 6.45 (d, J = 9.1 Hz, 2H), 4.02 (t, J = 6.0 Hz, 2H), 2.69 (t, J = 6.0 Hz, 2H), 2.50–2.49 (m, 4H), 2.15 (s, 3H), 1.55–1.53 (m, 4H), 1.46–1.36 (m, 2H). 13C NMR (101 MHz, acetone-d6): δ 189.84, 159.07, 154.44, 152.29, 149.60, 141.89, 139.83, 135.30, 130.25, 129.75, 127.36, 126.31, 126.04, 125.03, 124.86, 116.11, 115.96, 115.13, 108.02, 66.45, 57.73, 54.79, 25.83, 24.08, 18.49. HRMS (m/z): [M + H]+ calcd for C29H30NO4S, 488.1817; observed, 488.1819.
t-Butyl 3-(4-(Benzyloxy)phenoxy)azetidine-1-carboxylate (32).
To a solution of DMF (5 mL) was added 4-(benzyloxy)phenol (0.3 g, 1.5 mmol), t-butyl 3-(4-(benzyloxy)phenoxy)azetidine-1-carboxylate (0.5 g, 1.8 mmol), and cesium carbonate (1.4 g, 4.5 mmol). The reaction mixture was stirred at 140 °C for 3 h and monitored by TLC. Upon completion, the reaction mixture was extracted by ethyl acetate, washed by water and brine, and dried over Na2SO4. The organic extracts were evaporated under reduced pressure and purified by flash chromatography (10–50%) ethyl acetate in hexanes to give a white solid (yield: 0.33 g, 63%). 1H NMR (400 MHz, CDCl3): δ 7.46–7.30 (m, 5H), 6.92 (d, J = 9.0 Hz, 2H), 6.70 (d, J = 9.0 Hz, 2H), 5.04 (s, 2H), 4.89–4.83 (m, 1H), 4.28 (dd, J = 9.6, 6.4 Hz, 2H), 4.01 (dd, J = 9.7, 4.1 Hz, 2H), 1.47 (s, 9H). 13C NMR (101 MHz, CDCl3): δ 156.19, 153.62, 150.87, 137.11, 128.59, 127.96, 127.47, 116.04, 115.51, 79.78, 70.65, 66.05, 56.48, 28.38.
3-(4-(Benzyloxy)phenoxy)azetidine (33).
To a solution of dichloromethane (5 mL) was added compound 32 (0.5 g, 1.9 mmol), followed by dropwise addition of TFA (0.72 mL, 9.5 mmol) at 0 °C. The reaction mixture then was stirred at room temperature and monitored by TLC. Upon completion, the reaction mixture was extracted by dichloromethane, washed by water and brine, and dried over Na2SO4. The organic extracts were evaporated under reduced pressure and purified by flash chromatography (1–10% methanol in dichloromethane) to give a white solid (yield: 0.3 g, 85%). 1H NMR (400 MHz, acetone-d6): δ 7.47 (d, J = 7.6 Hz, 2H), 7.40 (dd, J = 8.0, 7.0 Hz, 2H), 7.33 (t, J = 6.7 Hz, 1H), 6.96 (d, J = 8.9 Hz, 2H), 6.81 (d, J = 8.9 Hz, 2H), 5.08 (s, 2H), 4.75 (br s, 1H), 3.99–3.69 (m, 2H), 3.54–3.44 (m, 2H). 13C NMR (101 MHz, acetone-d6): δ 153.47, 151.28, 137.75, 128.37, 127.66, 127.46, 115.86, 115.58, 70.04, 65.29, 53.41.
3-(4-(Benzyloxy)phenoxy)-1-(3-fluoropropyl)azetidine (34a).
To a solution of anhydrous DMF was added compound 33 (0.5 g, 1.9 mmol), followed by addition of NaH (0.15 g, 3.8 mmol) at 0 °C. The reaction mixture was stirred at room temperature for 0.5 h. 1-Bromo-3-fluoropropane (0.53 g, 3.8 mmol) was added slowly to the reaction mixture in an ice bath. The reaction was stirred at 40 °C and monitored by TLC. Upon completion, the reaction was quenched by water and extracted by EtOAc, washed by water and brine, and dried over Na2SO4. The organic phase was evaporated under reduced pressure and purified by flash chromatography (1–100% EtOAc in hexanes) to give a white powder (yield: 0.37 g, 50%). 1H NMR (400 MHz, acetone-d6): δ 7.39–7.36 (m, 5H), 7.00–6.88 (m, 3H), 6.82–6.69 (m, 1H), 5.07 (s, 2H), 4.71 (p, J = 5.7 Hz, 1H), 4.49 (dt, J = 47.5, 6.1 Hz, 2H), 3.82–3.72 (m, 1H), 3.02–2.94 (m, 1H), 2.90–2.64 (m, 2H), 2.65–2.54 (m, 2H), 1.94–1.61 (m, 2H). 13C NMR (101 MHz, acetone-d6): δ 153.32, 151.56, 137.78, 128.36, 127.65, 127.47, 115.81, 115.43, 81.90 (d, J = 145.1 Hz), 70.05, 66.79, 61.21, 55.16 (d, J = 5.6 Hz), 27.62.
4-((1-(3-Fluoropropyl)azetidin-3-yl)oxy)phenol (35a).
To a solution of methanol (10 mL) was added compound 34a (0.5 g, 1.5 mmol) and palladium carbon (0.15 g, 0.15 mmol, 10 wt %). The reaction was degassed by hydrogen for at least three times and stirred at room temperature for 5 h monitored by TLC. Upon completion, the reaction mixture was extracted by ethyl acetate, washed by water and brine, and dried over Na2SO4. The organic extracts were evaporated under reduced pressure and purified by flash chromatography (1–10% methanol in dichloromethane) to give a white solid (yield: 0.3 g, 85%). 1H NMR (400 MHz, acetone-d6): δ 6.79–6.71 (m, 2H), 6.71–6.62 (m, 2H), 4.70–4.60 (m, 1H), 4.49 (dt, J = 47.5, 6.1 Hz, 2H), 3.78 (td, J = 6.1, 1.9 Hz, 1H), 2.98 (td, J = 5.7, 1.9 Hz, 2H), 2.88–2.64 (m, 1H), 2.55 (t, J = 6.9 Hz, 2H), 1.91–1.64 (m, 2H). 13C NMR (101 MHz, acetone-d6): δ 151.63, 150.53, 115.85, 115.51 81.80 (d, J = 162.4 Hz), 70.05, 66.80, 61.28, 55.18 (d, J = 5.5 Hz).
4-((1-Cyclopropylazetidin-3-yl)oxy)phenol (35b).
This compound was prepared using a procedure similar to that of 35a (yield: 0.14 g, 55%). 1H NMR (400 MHz, DMSO-d6): 6.80 (d, J = 9.0 Hz, 2H), 6.60 (d, J = 9.0 Hz, 2H), 5.75 (br s, 1H), 4.95 (m, 1H), 4.09 (dd, J2 = 11.6 Hz, J2 = 6.6 Hz, 2H), 3.54 (dd, J1 = 11.5, J2 = 5.8 Hz, 2H), 2.89 (m, 1H), 0.95 (m, 2H), 0.86 (m, 2H).
(3-(4-((1-(3-Fluoropropyl)azetidin-3-yl)oxy)phenoxy)-6-((tetrahy-dro-2H-pyran-2-yl)oxy)benzo[b]thiophen-2-yl)(phenyl)methanone (36a).
This compound was prepared using a procedure similar to that of 29a (yield: 0.13 g, 77%). 1H NMR (400 MHz, acetone-d6): δ 7.78– 7.65 (m, 3H), 7.60–7.52 (m, 1H), 7.52–7.37 (m, 3H), 7.12 (dd, J = 8.9, 2.2 Hz, 1H), 6.68–6.55 (m, 4H), 5.65–5.61 (m, 1H), 4.75–4.62 (m, 1H), 4.49 (dt, J = 47.5, 5.3 Hz, 2H), 3.78–3.69 (m, 4H), 2.97–2.95 (m 2H), 2.57–2.55 (m, 2H), 1.94–1.79 (m, 3H), 1.77–1.62 (m, 5H). 13C NMR (101 MHz, acetone): δ 188.22,157.83, 152.74, 152.46, 148.21, 140.98, 138.62, 132.19, 128.71, 127.96, 127.29, 125.67, 124.31, 116.94, 116.67, 115.38, 108.88, 96.46, 81.81 (d, J = 162.6 Hz), 66.80, 61.72, 61.05, 61.04, 55.13 (d, J = 5.0 Hz), 30.01, 24.97, 18.53.
(4-Fluoro-2-methylphenyl)(3-(4-((1-(3-fluoropropyl)azetidin-3-yl)oxy)phenoxy)-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]-thiophen-2-yl)methanone (36c).
This compound was prepared using a procedure similar to that of 29a (yield: 0.2 g, 72%). 1H NMR (400 MHz, acetone-d6): δ 7.68 (d, J = 2.1 Hz, 1H), 7.48 (d, J = 8.9 Hz, 1H), 7.12 (dd, J = 8.9, 2.2 Hz, 1H), 7.03 (d, J = 9.1 Hz, 1H), 6.92 (d, J = 9.3 Hz, 1H), 6.79 (d, J = 9.1 Hz, 1H), 6.64 (d, J = 9.1 Hz, 2H), 6.49 (d, J = 9.2 Hz, 2H), 5.64 (s, 1H), 4.72–4.64 (m, 1H), 4.48 (dt, J = 47.5, 6.1 Hz, 2H), 3.93–3.71 (m, 4H), 3.02 (br s, 4H), 2.58 (t, J = 6.8 Hz, 2H), 2.15 (s, 3H), 2.01–1.83 (m, 3H), 1.80–1.54 (m, 3H). 13C NMR (101 MHz, acetone-d6): δ 188.76, 163.38 (d, J = 247.9 Hz), 161.92, 158.19, 152.64, 149.09, 141.42, 139.32 (d, J = 8.6 Hz), 135.90, 130.25 (d, J = 9.2 Hz), 127.51, 124.43, 117.08, 117.07 (d, J = 23.0 Hz), 115.95, 115.42, 115.33, 111.86 (d, J = 21.7 Hz), 108.95, 96.44, 81.82 (d, J = 162.5 Hz), 66.83, 61.73, 61.14, 61.02, 55.12 (d, J = 5.4 Hz), 35.29, 29.97, 24.94, 18.50.
(4-Fluoro-2,6-dimethylphenyl)(3-(4-((1-(3-fluoropropyl)azetidin-3-yl)oxy)phenoxy)-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]-thiophen-2-yl)methanone (36d).
This compound was prepared using a procedure similar to that of 29a (yield: 0.16 g, 62%). 1H NMR (400 MHz, acetone-d6): δ 7.69 (d, J = 2.1 Hz, 1H), 7.39 (d, J = 8.9 Hz, 1H), 7.10 (dd, J = 8.9, 2.1 Hz, 1H), 6.70 (s, 1H), 6.67 (s, 1H),6.65 (d, J = 9.1 Hz, 2H), 6.48 (d, J = 9.1 Hz, 2H). 5.63–5.59 (m, 1H), 4.69–4.65 (m, 1H), 4.55 (t, J = 6.0 Hz, 1H), 4.43 (t, J = 6.0 Hz, 1H), 3.89–3.70 (m, 3H), 3.70–3.52 (m, 1H), 2.98–2.96 (m, 2H), 2.61–2.58 (m, 2H), 2.11 (s, 6H), 1.94–1.92 (m, 3H), 1.67–1.65 (m, 5H). 13C NMR (101 MHz, acetone-d6): δ 190.51, 162.28 (d, J = 244.4 Hz), 158.32, 152.74, 152.19, 149.26, 141.70, 136.86, 136.67 (d, J = 8.7 Hz), 128.52, 127.35, 124.73, 117.15, 115.60, 115.17, 113.70 (d, J = 21.5 Hz), 109.09, 96.39, 81.78 (d, J = 162.6 Hz), 66.85, 61.70, 60.98, 55.05 (d, J = 5.2 Hz), 29.94, 24.93, 18.47.
(3-Fluorophenyl)(3-(4-((1-(3-fluoropropyl)azetidin-3-yl)oxy)-phenoxy)-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]thiophen-2-yl)methanone (36e).
This compound was prepared using a procedure similar to that of 29a (yield: 0.1 g, 73%). 1H NMR (400 MHz, acetone-d6): δ 7.68 (d, J = 1.9 Hz, 1H), 7.56 (d, J = 7.7 Hz, 1H), 7.52–7.39 (m, 3H), 7.32–7.28 (m, 1H), 7.13 (dd, J = 8.9, 2.1 Hz, 1H), 6.72–6.59 (m, 4H), 5.64 (t, J = 2.9 Hz, 1H), 4.71–4.68 (m, 1H), 4.49 (dt, J = 47.5, 6.0 Hz, 2H), 3.89–3.75 (m, 4H), 3.03 (d, J = 7.9 Hz, 2H), 2.61 (t, J = 7.0 Hz, 2H), 1.91–1.89 (m, 3H), 1.83–1.65 (m, 5H). 13C NMR (101 MHz, acetone-d6): δ 186.91, 162.23 (d, J = 245.4 Hz), 158.03, 152.78, 152.42, 148.73, 141.27, 140.85 (d, J = 6.6 Hz), 130.02 (d, J = 7.9 Hz), 127.14, 125.21, 124.59 (d, J = 2.7 Hz), 124.46, 118.75 (d, J = 21.4 Hz), 117.03, 116.63, 115.00 (d, J = 23.0 Hz), 114.89, 108.87, 96.44, 81.74 (d, J = 162.7 Hz), 66.73, 61.71,60.94, 60.93, 54.95 (d, J = 5.5 Hz), 29.98, 24.94, 18.50.
(3-(4-((1-Cyclopropylazetidin-3-yl)oxy)phenoxy)-6-((tetrahydro-2H-pyran-2-yl)oxy)benzo[b]thiophen-2-yl)(4-fl uoro-2,6-dimethylphenyl)methanone (36f).
This compound was prepared using a procedure similar to that of 29a (yield: 0.11 g, 75%). 1H NMR (400 MHz, acetone-d6): δ 7.70 (d, J = 2.0 Hz, 1H), 7.40 (d, J = 8.9 Hz, 1H), 7.11 (dd, J = 8.9, 2.1 Hz, 1H), 6.70 (s, 1H), 6.67 (d, J = 6.6 Hz, 1H), 6.64 (d, J = 9.1 Hz, 2H), 6.48 (d, J = 9.1 Hz, 2H). 5.65 (t, J = 2.8 Hz, 1H), 4.65–4.62 (m, 1H), 3.92–3.81 (m, 1H), 3.78 (t, J = 7.2 Hz, 2H), 3.71–3.58 (m, 1H), 3.20–3.18 (m, 2H), 2.12 (s, 6H), 2.01–1.82 (m, 3H), 1.77–1.55 (m, 3H). 0.41–0.35 (m, 3H), 0.30–0.28 (m, 2H).
(3-(4-((1-(3-Fluoropropyl)azetidin-3-yl)oxy)phenoxy)-6-hydroxybenzo[b]thiophen-2-yl)(phenyl)methanone (37a).
This compound was prepared using a procedure similar to that of 30a (yield: 0.03 g, 18%). 1H NMR (400 MHz, acetone-d6): δ 7.73–7.69 (m, 2H), 7.57–7.51 (m, 1H), 7.45–7.37 (m, 4H), 6.99 (dd, J = 8.9, 2.1 Hz, 1H), 6.66–6.53 (m, 4H), 4.67–4.63 (m, 1H), 4.48 (dt, J = 47.5, 6.1 Hz, 2H), 3.80–3.68 (m, 2H), 3.05–2.89 (m, 2H), 2.58 (t, J = 7.0 Hz, 2H), 1.82–1.63 (m, 2H). 13C NMR (101 MHz, acetone-d6): δ 188.19, 158.85, 152.65, 152.51, 148.54, 141.50, 138.82, 131.98, 128.60, 127.88, 125.82, 124.64, 124.54, 116.59, 116.08, 115.35, 107.91, 81.78 (d, J = 162.6 Hz), 66.78, 61.01, 55.10 (d, J = 5.7 Hz). HRMS (m/z): [M + H]+ calcd for C27H25FNO4S, 478.1410; observed, 478.1472.
(3-(4-((1-(3-Fluoropropyl)azetidin-3-yl)oxy)phenoxy)-6-hydroxybenzo[b]thiophen-2-yl)(o-tolyl)methanone (37b).
This compound was prepared using a procedure similar to that of 30a (yield: 0.006 g, 17%). 1H NMR (400 MHz, acetone-d6): δ 7.43–7.41 (m, 2H), 7.36 (d, J = 7.6 Hz, 1H), 7.32–7.26 (m, 1H), 7.14 (t, J = 7.7 Hz, 2H), 6.98 (dd, J = 8.8, 1.9 Hz, 1H), 6.61 (d, J = 9.0 Hz, 2H), 6.44 (d, J = 9.0 Hz, 2H), 4.68 (dt, J = 11.3, 5.7 Hz, 1H), 4.49 (dt, J = 47.5, 6.1 Hz, 2H), 3.78–3.73 (m, 2H), 2.98–2.95 (m, 2H), 2.57 (t, J = 6.9 Hz, 2H), 2.13 (s, 3H), 1.82–1.65 (m, 2H). 13C NMR (101 MHz, acetone-d6): δ 189.79, 158.84, 152.67, 152.63, 149.41, 141.85, 139.71, 135.36, 130.27, 129.83, 127.41, 126.44, 126.26, 125.04, 124.82, 116.3, 115.22, 115.21, 107.97, 81.79 (d, J = 162.5 Hz), 66.81, 61.05, 55.13 (d, J = 5.6 Hz), 18.44. HRMS (m/z): [M + H]+ calcd for C28H27FNO4S, 492.1639; observed, 492.1627.
(4-Fluoro-2-methylphenyl)(3-(4-((1-(3-fluoropropyl)azetidin-3-yl)oxy) phenoxy)-6-hydroxybenzo[b]thiophen-2-yl)methanone (37c).
This compound was prepared using a procedure similar to that of 30a (yield: 0.015 g, 19%). 1H NMR (400 MHz, acetone-d6): δ 7.47–7.39 (m, 3H), 6.98 (dd, J = 8.8, 2.2 Hz, 1H), 6.94–6.86 (m, 2H), 6.63 (d, J = 9.1 Hz, 2H), 6.47 (d, J = 9.1 Hz, 2H), 4.77–4.64 (m, 1H), 4.49 (dt, J = 47.5, 6.1 Hz, 2H), 3.77 (t, J = 7.0 Hz, 2H), 3.06–2.96 (m, 2H), 2.59 (t, J = 6.9 Hz, 2H), 2.14 (s, 3H), 1.83–1.65 (m, 2H). 13C NMR (101 MHz, acetone-d6): δ 188.67, 163.30 (d, J = 247.6 Hz), 159.05, 149.38, 141.93, 139.17 (d, J = 8.6 Hz), 135.90, 130.11 (d, J = 9.1 Hz), 126.25, 126.15, 124.81, 117.80, 116.87 (d, J = 21.7 Hz), 116.17, 115.89, 115.42, 115.31, 111.80 (d, J = 21.8 Hz), 108.01, 81.79 (d, J = 162.7 Hz), 66.83, 61.00, 55.12 (d, J = 5.6 Hz), 18.54. HRMS (m/z): [M + H]+ calcd for C28H26F2NO4S, 510.1545; observed, 510.1537.
(4-Fluoro-2,6-dimethylphenyl)(3-(4-((1-(3-fluoropropyl)azetidin-3-yl)oxy)phenoxy)-6-hydroxybenzo[b]thiophen-2-yl)methanone (37d).
This compound was prepared using a procedure similar to that of 30a (yield: 2.0 g, 25%). 1H NMR (400 MHz, acetone): δ 7.42 (d, J = 2.0 Hz, 1H), 7.34 (d, J = 8.8 Hz, 1H), 6.97 (dd, J = 8.8, 2.1 Hz, 1H), 6.75–6.59 (m, 4H), 6.54–6.39 (m, 2H), 4.77–4.65 (m, 1H), 4.55 (t, J = 6.0 Hz, 1H), 4.43 (t, J = 6.1 Hz, 1H), 3.77–3.75 (m, 2H), 3.10–2.92 (m, 2H), 2.60 (t, J = 6.9 Hz, 2H), 2.11 (s, 6H), 1.75–1.65 (m, 2H). 13C NMR (101 MHz, acetone-d6): δ 190.36, 162.25 (d, J = 244.6 Hz), 159.15, 152.69, 152.22, 149.54, 142.18, 136.98, 136.64 (d, J = 8.6 Hz), 127.44, 126.03, 125.12, 116.23, 115.57, 115.15, 113.66 (d, J = 21.5 Hz), 108.19, 81.80 (d, J = 162.6 Hz), 66.86, 61.01, 60.98, 55.12 (d, J = 5.6 Hz), 18.46. HRMS (m/z): [m + H]+ calcd for C29H28F2NO4S, 524.1629; observed, 524.1627.
(3-Fluorophenyl)(3-(4-((1-(3-fluoropropyl)azetidin-3-yl)oxy)-phenoxy)-6-hydroxybenzo[b]thiophen-2-yl)methanone (37e).
This compound was prepared using a procedure similar to that of 30a (yield: 0.03 g, 22%). 1H NMR (400 MHz, acetone-d6): δ 7.53 (d, J = 7.5 Hz, 1H), 7.50–7.35 (m, 4H), 7.30 (td, J = 8.4, 1.8 Hz, 1H), 7.00 (dd, J = 8.8, 2.0 Hz, 1H), 6.63 (q, J = 9.3 Hz, 4H), 4.68 (p, J = 5.7 Hz, 1H), 4.48 (dt, J = 47.5, 6.1 Hz, 2H), 3.75 (t, J = 7.2 Hz, 2H), 3.05–2.91 (m, 2H), 2.58 (t, J = 6.9 Hz, 2H), 1.83–1.63 (m, 2H). 13C NMR (101 MHz, acetone-d6): δ 186.85, 162.20 (d, J = 245.2 Hz), 159.30, 152.75, 152.42, 149.13, 141.84, 141.08 (d, J = 6.7 Hz), 129.94 (d, J = 7.8 Hz), 125.56, 124.78, 124.51, 124.01, 118.51 (d, J = 21.5 Hz), 116.54, 116.27, 115.44, 114.92 (d, J = 23.0 Hz), 107.94, 81.79 (d, J = 162.5 Hz), 66.80, 60.99, 55.11 (d, J = 5.6 Hz). HRMS (m/z): [M + H]+ calcd for C27H24F2NO4S, 496.1389; observed, 496.1402.
(3-(4-((1-Cyclopropylazetidin-3-yl)oxy)phenoxy)-6-hydroxybenzo[b]thiophen-2-yl)(4-fluoro-2,6-dimethylphenyl)-methanone (37f).
This compound was prepared using a procedure similar to that of 30a (yield: 0.03 g, 19%). 1H NMR (400 MHz, acetone-d6): δ 7.44 (d, J = 2.1 Hz, 1H), 7.32 (d, J = 8.8 Hz, 1H), 6.98 (dd, J = 8.8, 2.1 Hz, 1H), 6.69 (s, 1H), 6.66 (s, 1H), 6.64 (d, J = 9.1 Hz, 2H), 6.46 (d, J = 9.1 Hz, 2H). 4.64–4.61 (m, 1H), 3.79–3.69 (m, 2H), 3.20–3.10 (m, 2H), 2.12 (s, 6H), 1.90–1.88 (m, 1H), 0.36–0.34 (m, 2H), 0.30–0.22 (m, 2H). 13C NMR (101 MHz, acetone-d6): δ 190.31, 162.23 (d, J = 244.4 Hz) 159.53, 152.72, 152.20, 149.59, 142.19, 137.03, 136.63 (d, J = 8.7 Hz), 126.82, 125.80, 125.02, 116.35, 115.54, 115.09, 113.63 (d, J = 21.5 Hz), 108.18, 66.91, 60.34, 37.94, 18.44, 4.52. HRMS (m/z): [M + H]+ calcd for C29H27FNO4S, 504.1567; observed, 504.1572.
Supplementary Material
ACKNOWLEDGMENTS
For financial support: Chancellor’s Proof-of-Concept Fund, UICentre (Drug Discovery @ UIC), UIC Center for Clinical and Translational Science grant UL1RR029879 and UL1TR002003. We would also like to thank John A. Katzenellenbogen, Kathryn Carlson and Teresa Martin (Department of Chemistry, University of Illinois at Urbana-Champaign), for carrying out RBA assays and scientific insight. We thank David Shapiro (Department of Biochemistry, University of Illinois at Urbana-Champaign) for providing ESR1 mutant cell lines.
ABBREVIATIONS
- AIs
aromatase inhibitors
- B-SERD
basic selective estrogen receptor degrader
- Cdk
cyclin-dependent kinases
- ER+
estrogen receptor positive
- ERα
estrogen receptor α
- ERβ
estrogen receptor β
- E2
estradiol
- ERE
estrogen response element
- HPCD
2-hydroxypropyl-β-cyclodextrin
- ICW
in-cell western
- MBC
metastatic breast cancer
- PEG-400
polyethylene glycol 400
- PCC
pyridinium chlorochromate
- PK
pharmacokinetics
- RBA
relative binding affinity
- RT
room temperature
- SBE-β-CD
sulfobutylether-β-cyclodextrin
- SERM
selective estrogen receptor modulator
- SERD
selective estrogen receptor degrader
- TAM
tamoxifen
- 4OH-TAM
4-hydroxy tamoxifen
- TNBC
triple-negative breast cancer
- TLC
thin layer chromatography
- THP
tetrahydropyran
- TFA
trifluoroacetic acid
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01580.
Molecular formula strings, effect of 30o treatment on the ER level. , representative western blot, PK profiles for 37d., ER isoform binding specificity, molecular orbital calculations, final compound spectra, final compound purity, and PDB: 1R5K, 5ACC, 5UFX (PDF)
Molecular formula strings (CSV)
The authors declare no competing financial interest.
REFERENCES
- (1).Bray F; Ferlay J; Soerjomataram I; Siegel RL; Torre LA; Jemal A Global Cancer Statistics 2018: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. Ca-Cancer J. Clin. 2018, 68, 394–424. [DOI] [PubMed] [Google Scholar]
- (2).Lemieux P; Fuqua S The Role of the Estrogen Receptor in Tumor Progression. J. Steroid Biochem. Mol. Biol. 1996, 56, 87–91. [DOI] [PubMed] [Google Scholar]
- (3).Sommer S; Fuqua SAW Estrogen Receptor and Breast Cancer. Semin. Cancer Biol. 2001, 11, 339–352. [DOI] [PubMed] [Google Scholar]
- (4).Nicholson RI; Johnston SR Endocrine Therapy—Current Benefits and Limitations. Breast Cancer Res. Treat. 2005, 93, S3–S10. [DOI] [PubMed] [Google Scholar]
- (5).Smith IE; Dowsett M Aromatase Inhibitors in Breast Cancer. N. Engl. J. Med. 2003, 348, 2431–2442. [DOI] [PubMed] [Google Scholar]
- (6).Wu Y-L; Yang X; Ren Z; McDonnell DP; Norris JD; Willson TM; Greene GL Structural Basis for an Unexpected Mode of Serm-Mediated Er Antagonism. Mol. Cell 2005, 18, 413–424. [DOI] [PubMed] [Google Scholar]
- (7).Jordan VC Tamoxifen: Catalyst for the Change to Targeted Therapy. Eur. J. Cancer 2008, 44, 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Haque R; Ahmed SA; Inzhakova G; Shi J; Avila C; Polikoff J; Bernstein L; Enger SM; Press MF Impact of Breast Cancer Subtypes and Treatment on Survival: An Analysis Spanning Two Decades. Cancer Epidemiol., Biomarkers Prev. 2012, 21, 1848–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sweeney EE; McDaniel RE; Maximov PY; Fan P; Jordan VC Models and Mechanisms of Acquired Antihormone Resistance in Breast Cancer: Significant Clinical Progress Despite Limitations. Horm. Mol. Biol. Clin. Invest. 2012, 9, 143–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kuukasjarvi T; Kononen J; Helin H; Holli K; Isola J Loss of Estrogen Receptor in Recurrent Breast Cancer Is Associated with Poor Response to Endocrine Therapy. J. Clin. Oncol. 1996, 14, 2584–2589. [DOI] [PubMed] [Google Scholar]
- (11).Boér K Fulvestrant in Advanced Breast Cancer: Evidence to Date and Place in Therapy. Ther. Adv. Med. Oncol. 2017, 9, 465–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Clarke R; Skaar TC; Bouker KB; Davis N; Lee YR; Welch JN; Leonessa F Molecular and Pharmacological Aspects of Antiestrogen Resistance. J. Steroid Biochem. Mol. Biol. 2001, 76, 71–84. [DOI] [PubMed] [Google Scholar]
- (13).Ciruelos E; Pascual T; Arroyo Vozmediano ML; Blanco M; Manso L; Parrilla L; Munoz C; Vega E; Calderon MJ; Sancho B; Cortes-Funes H The Therapeutic Role of Fulvestrant in the Management of Patients with Hormone Receptor-Positive Breast Cancer. Breast 2014, 23, 201–208. [DOI] [PubMed] [Google Scholar]
- (14).Perey L; Paridaens R; Hawle H; Zaman K; Nole F; Wildiers H; Fiche M; Dietrich D; Clement P; Koberle D; Goldhirsch A; Thurlimann B Clinical benefit of fulvestrant in postmenopausal women with advanced breast cancer and primary or acquired resistance to aromatase inhibitors: final results of phase II Swiss Group for Clinical Cancer Research Trial (SAKK 21/00). Ann. Oncol. 2007, 18, 64–69. [DOI] [PubMed] [Google Scholar]
- (15).Katzenellenbogen BS; Montano MM; Ekena K; Herman ME; McInerney EM Antiestrogens: Mechanisms of action and resistance in breast cancer. Breast Cancer Res. Treat. 1997, 44, 23–38. [DOI] [PubMed] [Google Scholar]
- (16).Massarweh S; Schiff R Unraveling the Mechanisms of Endocrine Resistance in Breast Cancer: New Therapeutic Oppor-tunities. Clin. Cancer Res. 2007, 13, 1950–1954. [DOI] [PubMed] [Google Scholar]
- (17).Osborne CK; Schiff R Mechanisms of Endocrine Resistance in Breast Cancer. Annu. Rev. Med. 2011, 62, 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mills JN; Rutkovsky AC; Giordano A Mechanisms of Resistance in Estrogen Receptor Positive Breast Cancer: Overcoming Resistance to Tamoxifen/Aromatase Inhibitors. Curr. Opin. Pharmacol. 2018, 41, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Finn RS; Aleshin A; Slamon DJ Targeting the Cyclin-Dependent Kinases (Cdk) 4/6 in Estrogen Receptor-Positive Breast Cancers. Breast Cancer Res. 2016, 18, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Pernas S; Tolaney SM; Winer EP; Goel S Cdk4/6 Inhibition in Breast Cancer: Current Practice and Future Directions. Ther. Adv. Med. Oncol. 2018, 10, 1758835918786451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).O’Leary B; Finn RS; Turner NC Treating Cancer with Selective Cdk4/6 Inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [DOI] [PubMed] [Google Scholar]
- (22).O’Sullivan CC Overcoming Endocrine Resistance in Hormone-Receptor Positive Advanced Breast Cancer-the Emerging Role of Cdk4/6 Inhibitors. Int. J. Cancer Clin. Res. 2015, 2, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).van Kruchten M; de Vries EG; Glaudemans AW; van Lanschot MC; van Faassen M; Kema IP; Brown M; Schröder CP; de Vries EF; Hospers GA Measuring Residual Estrogen Receptor Availability During Fulvestrant Therapy in Patients with Metastatic Breast Cancer. Cancer Discovery 2015, 5, 72–81. [DOI] [PubMed] [Google Scholar]
- (24).Young OE; Renshaw L; Macaskill EJ; White S; Faratian D ; Thomas JSJ; Dixon JM Effects of Fulvestrant 750mg in Premenopausal Women with Oestrogen-Receptor-Positive Primary Breast Cancer. Eur. J. Cancer 2008, 44, 391–399. [DOI] [PubMed] [Google Scholar]
- (25).Osborne CK; Wakeling A; Nicholson RI Fulvestrant: An Oestrogen Receptor Antagonist with a Novel Mechanism of Action. Br. J. Cancer 2004, 90, S2–S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ettinger B; Black DM; Mitlak BH; Knickerbocker RK; Nickelsen T; Genant HK; Christiansen C; Delmas PD; Zanchetta JR; Stakkestad J; Gluer CC; Krueger K; Cohen FJ; Eckert S; Ensrud KE; Avioli LV; Lips P; Cummings SR Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene: results from a 3-year randomized clinical trial. Multiple Outcomes of Raloxifene Evaluation (MORE) Investigators. JAMA 1999, 282, 637–645. [DOI] [PubMed] [Google Scholar]
- (27).Wardell SE; Nelson ER; Chao CA; McDonnell DP Bazedoxifene exhibits antiestrogenic activity in animal models of tamoxifen-resistant breast cancer: implications for treatment of advanced disease. Clin Cancer Res 2013, 19, 2420–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Garner F; Shomali M; Paquin D; Lyttle CR; Hattersley G Rad1901: A Novel, Orally Bioavailable Selective Estrogen Receptor Degrader That Demonstrates Antitumor Activity in Breast Cancer Xenograft Models. Anticancer Drugs 2015, 26, 948–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wardell SE; Nelson ER; Chao CA; Alley HM; McDonnell DP Evaluation of the Pharmacological Activities of Rad1901, a Selective Estrogen Receptor Degrader. Endocr.-Relat. Cancer 2015, 22, 713–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lim E; Lin NU Updates on the management of breast cancer brain metastases. Oncology 2014, 28, 572–578. [PubMed] [Google Scholar]
- (31).Martin AM; Cagney DN; Catalano PJ; Warren LE; Bellon JR; Punglia RS; Claus EB; Lee EQ; Wen PY; Haas-Kogan DA; Alexander BM; Lin NU; Aizer AA Brain Metastases in Newly Diagnosed Breast Cancer. JAMA Oncol. 2017, 3, 1069–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lin NU; Amiri-Kordestani L; Palmieri D; Liewehr DJ; Steeg PS Cns Metastases in Breast Cancer: Old Challenge, New Frontiers. Clin. Cancer Res. 2013, 19, 6404–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Xiong R; Zhao J; Gutgesell LM; Wang Y; Lee S; Karumudi B; Zhao H; Lu Y; Tonetti DA; Thatcher GRJ Novel Selective Estrogen Receptor Downregulators (Serds) Developed against Treatment-Resistant Breast Cancer. J. Med. Chem. 2017, 60, 1325–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Xiong R; Patel HK; Gutgesell LM; Zhao J; Delgado-Rivera L; Pham TND; Zhao H; Carlson K; Martin T; Katzenellenbogen JA; Moore TW; Tonetti DA; Thatcher GRJ Selective Human Estrogen Receptor Partial Agonists (Sherpas) for Tamoxifen-Resistant Breast Cancer. J. Med. Chem. 2016, 59, 219–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kastrati I; Edirisinghe PD; Hemachandra L-P-MP; Chandrasena ER; Choi J; Wang Y-T; Bolton JL; Thatcher GRJ Raloxifene and Desmethylarzoxifene Block Estrogen-Induced Malignant Transformation of Human Breast Epithelial Cells. PLoS One 2011, 6, No. e27876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Abdelhamid R; Luo J; Vandevrede L; Kundu I; Michalsen B ; Litosh VA; Schiefer IT; Gherezghiher T; Yao P; Qin Z; Thatcher GR J. Benzothiophene Selective Estrogen Receptor Modulators Provide Neuroprotection by a Novel Gpr30-Dependent Mechanism. ACS Chem. Neurosci. 2011, 2, 256–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Qin Z; Kastrati I; Ashgodom RT; Lantvit DD; Overk CR; Choi Y; van Breemen RB; Bolton JL; Thatcher GRJ Structural Modulation of Oxidative Metabolism in Design of Improved Benzothiophene Selective Estrogen Receptor Modulators. Drug Metab. Dispos. 2009, 37, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Yu B; Dietz BM; Dunlap T; Kastrati I; Lantvit DD; Overk CR; Yao P; Qin Z; Bolton JL; Thatcher GRJ Structural Modulation of Reactivity/Activity in Design of Improved Benzothiophene Selective Estrogen Receptor Modulators: Induction of Chemopreventive Mechanisms. Mol. Cancer Ther. 2007, 6, 2418–2428. [DOI] [PubMed] [Google Scholar]
- (39).Overk CR; Peng K-W; Asghodom RT; Kastrati I; Lantvit DD; Qin Z; Frasor J; Bolton JL; Thatcher GR J. Structure-Activity Relationships for a Family of Benzothiophene Selective Estrogen Receptor Modulators Including Raloxifene and Arzoxifene. ChemMedChem 2007, 2, 1520–1526. [DOI] [PubMed] [Google Scholar]
- (40).Qin Z; Kastrati I; Chandrasena REP; Liu H; Yao P; Petukhov PA; Bolton JL; Thatcher GRJ Benzothiophene Selective Estrogen Receptor Modulators with Modulated Oxidative Activity and Receptor Affinity. J. Med. Chem. 2007, 50, 2682–2692. [DOI] [PubMed] [Google Scholar]
- (41).Liu H; Bolton JL; Thatcher GRJ Chemical Modification Modulates Estrogenic Activity, Oxidative Reactivity, and Metabolic Stability in 4’f-Dma, a New Benzothiophene Selective Estrogen Receptor Modulator. Chem. Res. Toxicol. 2006, 19, 779–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Brzozowski AM; Pike ACW; Dauter Z; Hubbard RE; Bonn T; Engstrom O; Ohman L; Greene GL; Gustafsson J-A; Carlquist M Molecular Basis of Agonism and Antagonism in the Oestrogen Receptor. Nature 1997, 389, 753–758. [DOI] [PubMed] [Google Scholar]
- (43).Shiau AK; Barstad D; Loria PM; Cheng L; Kushner PJ; Agard DA; Greene GL The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell 1998, 95, 927–937. [DOI] [PubMed] [Google Scholar]
- (44).Nagasawa J; Govek S; Kahraman M; Lai A; Bonnefous C; Douglas K; Sensintaffar J; Lu N; Lee K; Aparicio A; Kaufman J; Qian J; Shao G; Prudente R; Joseph JD; Darimont B; Brigham D; Maheu K; Heyman R; Rix PJ; Hager JH; Smith ND Identification of an Orally Bioavailable Chromene-Based Selective Estrogen Receptor Degrader (Serd) That Demonstrates Robust Activity in a Model of Tamoxifen-Resistant Breast Cancer. J. Med. Chem. 2018, 61, 7917–7928. [DOI] [PubMed] [Google Scholar]
- (45).Fanning SW; Hodges-Gallagher L; Myles DC; Sun R; Fowler CE; Plant IN; Green BD; Harmon CL; Greene GL; Kushner PJ Specific Stereochemistry of Op-1074 Disrupts Estrogen Receptor Alpha Helix 12 and Confers Pure Antiestrogenic Activity. Nat. Commun 2018, 9, 2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Kahraman M; Govek SP; Nagasawa JY; Lai A; Bonnefous C; Douglas K; Sensintaffar J; Liu N; Lee K; Aparicio A; Kaufman J; Qian J; Shao G; Prudente R; Joseph JD; Darimont B; Brigham D; Heyman R; Rix PJ; Hager JH; Smith ND Maximizing ER-α Degradation Maximizes Activity in a Tamoxifen-Resistant Breast Cancer Model: Identification of GDC-0927. ACS Med. Chem. Lett. 2019, 10, 50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Jiang S.-y.; Wolf DM; Yingling JM; Chang C; Jordan VB An Estrogen Receptor Positive Mcf-7 Clone That Is Resistant to Antiestrogens and Estradiol. Mol. Cell. Endocrinol. 1992, 90, 77–86. [DOI] [PubMed] [Google Scholar]
- (48).Mao C; Livezey M; Kim JE; Shapiro DJ Antiestrogen Resistant Cell Lines Expressing Estrogen Receptor a Mutations Upregulate the Unfolded Protein Response and Are Killed by Bhpi. Sci Rep. 2016, 6, 34753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Chandarlapaty S; Chen D; He W; Sung P; Samoila A; You D; Bhatt T; Patel P; Voi M; Gnant M; Hortobagyi G; Baselga J; Moynahan ME Prevalence of Esr1 Mutations in Cell-Free DNA and Outcomes in Metastatic Breast Cancer: A Secondary Analysis of the Bolero-2 Clinical Trial. JAMA Oncol. 2016, 2, 1310–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Breslin S; O’Driscoll L Three-Dimensional Cell Culture: The Missing Link in Drug Discovery. Drug Discovery Today 2013, 18, 240–249. [DOI] [PubMed] [Google Scholar]
- (51).Guan J; Zhou W; Hafner M; Blake RA; Chalouni C; Chen IP; De Bruyn T; Giltnane JM; Hartman SJ; Heidersbach A; Houtman R; Ingalla E; Kategaya L; Kleinheinz T; Li J; Martin SE; Modrusan Z; Nannini M; Oeh J; Ubhayakar S; Wang X; Wertz IE; Young A; Yu M; Sampath B ; Hager JH; Friedman LS; Daemen A; Metcalfe C Therapeutic Ligands Antagonize Estrogen Receptor Function by Impairing Its Mobility. Cell 2019, 178, 949–963. [DOI] [PubMed] [Google Scholar]
- (52).Katzenellenbogen JA; Johnson HJ Jr.; Myers HN Photoaffinity Labels for Estrogen Binding Proteins of Rat Uterus. Biochemistry 1973, 12, 4085–4092. [DOI] [PubMed] [Google Scholar]
- (53).Carlson KE; Choi I; Gee A; Katzenellenbogen BS; Katzenellenbogen JA Altered Ligand Binding Properties and Enhanced Stability of a Constitutively Active Estrogen Receptor: Evidence That an Open Pocket Conformation Is Required for Ligand Interaction. Biochemistry 1997, 36, 14897–14905. [DOI] [PubMed] [Google Scholar]
- (54).Chisamore MJ; Ahmed Y; Bentrem DJ; Jordan VC; Tonetti DA Novel antitumor effect of estradiol in athymic mice injected with a T47D breast cancer cell line overexpressing protein kinase C alpha. Clin. Cancer Res. 2001, 7, 3156–3165. [PubMed] [Google Scholar]
- (55).Molloy ME; White BEP; Gherezghiher T; Michalsen BT; Xiong R; Patel H; Zhao H; Maximov PY; Jordan VC; Thatcher GRJ; Tonetti DA Novel Selective Estrogen Mimics for the Treatment of Tamoxifen-Resistant Breast Cancer. Mol. Cancer Ther. 2014, 13, 2515–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.