

CONSPECTUS
Cycloaddition reactions are a hallmark in organic synthesis because they provide an efficient way to construct highly substituted carbo- and heterocycles found in natural products and pharmaceutical agents. Most cycloadditions occur under thermal or photochemical conditions, but transition metal complexes can promote reactions that occur beyond these circumstances. Transition metal complexation with alkynes, alkenes, allenes, or dienes often alters the reactivity of those π-systems and facilitates access to diverse cycloaddition products.
This Account describes our efforts toward the design of novel five-carbon synthons for use in rhodium-catalyzed (5+n) cycloadditions, which include 3-acyloxy-1,4-enynes (ACEs) for (5+1) and (5+2) cycloadditions and 3-hydroxy-1,4-enynes (HYEs) for (5+1) cycloadditions. Furthermore, this Account includes relevant computational information, mechanistic insights, and applications of these cycloadditions to synthesize various highly substituted carbo- and heterocycles.
The (5+n) cycloaddition reactions presented herein share the following common mechanistic features: the 1,2-migration of an acyloxy group in propargyl esters or an ionization of a hydroxyl group in propargylic alcohols, oxidative cyclization to form a metallacycle, insertion of the one- or two-carbon component, and reductive elimination to yield the final product.
In conjunction with a cationic rhodium catalyst, we used ACEs for the intramolecular (5+2) cycloaddition with tethered alkynes, alkenes, and allenes. In some cases, an electron-deficient phosphine ligand improved the reaction yields, especially when the ACE featured an internal alkyne. We also demonstrated that chirality could be efficiently transferred from a relatively simple starting material to a more complex bicyclic product. Products derived from ACEs with tethered alkenes and allenes contained one or more stereocenters, and high diastereoselectivity was achieved in most of these cases. For ACEs tethered to an allene, the reaction preferentially occurred at the internal alkene. We also switched the positions of the alkene and the alkyne in the 1,4-enyne of our original ACE to provide an inverted ACE variant, which produced products with complementary functionalities.
After we successfully developed the Rh-catalyzed intramolecular (5+2) cycloaddition, we optimized conditions for the intermolecular version, which required a neutral rhodium catalyst and phosphine ligand. When a terminal alkyne was used as the two-carbon component, high regioselectivity was observed. While investigating the effect of esters on the rate of the intermolecular (5+2) cycloadditions, we determined that an electron-rich ester significantly accelerated the reaction. Subsequently, we demonstrated (5+1) cycloadditions undergo this rate enhancement as well in the presence of an ester.
Aside from ACEs, we synthesized HYEs in four steps from commercially available 2-aminobenzoic acid for use in the (5+1) cycloaddition. Mechanistically, HYEs were designed so that the aniline nitrogen could serve as the nucleophile and the –OH could serve as the leaving group. Using HYEs, we developed a novel method to make substituted carbazoles, dibenzofurans, and tricyclic compounds with a cyclohexadienone moiety.
Although the occurrence of transition-metal catalyzed acyloxy migrations has been known for decades, only recently has their synthetic value been realized. We hope our studies that employ readily available 1,4-enynes as the 5-carbon components in (5+n) cycloadditions can inspire the design of new two-component and multi-component cycloadditions.
Graphical Abstract
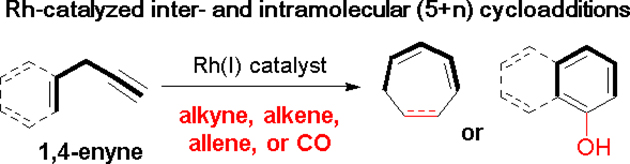
1. Introduction
Cycloaddition reactions are a mainstay in organic synthesis as they provide an efficient method to construct complex carbo- and heterocycles found in natural products and pharmaceutical agents. Forming at least two bonds in a single step, cycloadditions boast high atom-economy, improved step count efficiency, and often occur with a high degree of regio- and stereoselectivity.1
Most cycloadditions occur under thermal or photochemical conditions, but transition metal complexes can also be used to promote reactions that occur beyond these circumstances.2 Transition metal complexation with alkynes, alkenes, allenes, or dienes often alters the reactivity of those π-systems and facilitates access to diverse compounds.
Six-membered rings are easily formed via the Diels-Alder cycloaddition reaction, but a similarly ubiquitous cycloaddition for the formation of seven-membered rings has not been discovered, despite their presence in pharmaceutical agents and natural products (Scheme 1).3 To make seven-membered rings, only three types of two-component cycloadditions are possible: (6+1),4 (5+2)5 and (4+3).6 New methods for the formation of seven-membered rings could be expedited by the development of novel five- and three-carbon synthons.
Scheme 1:
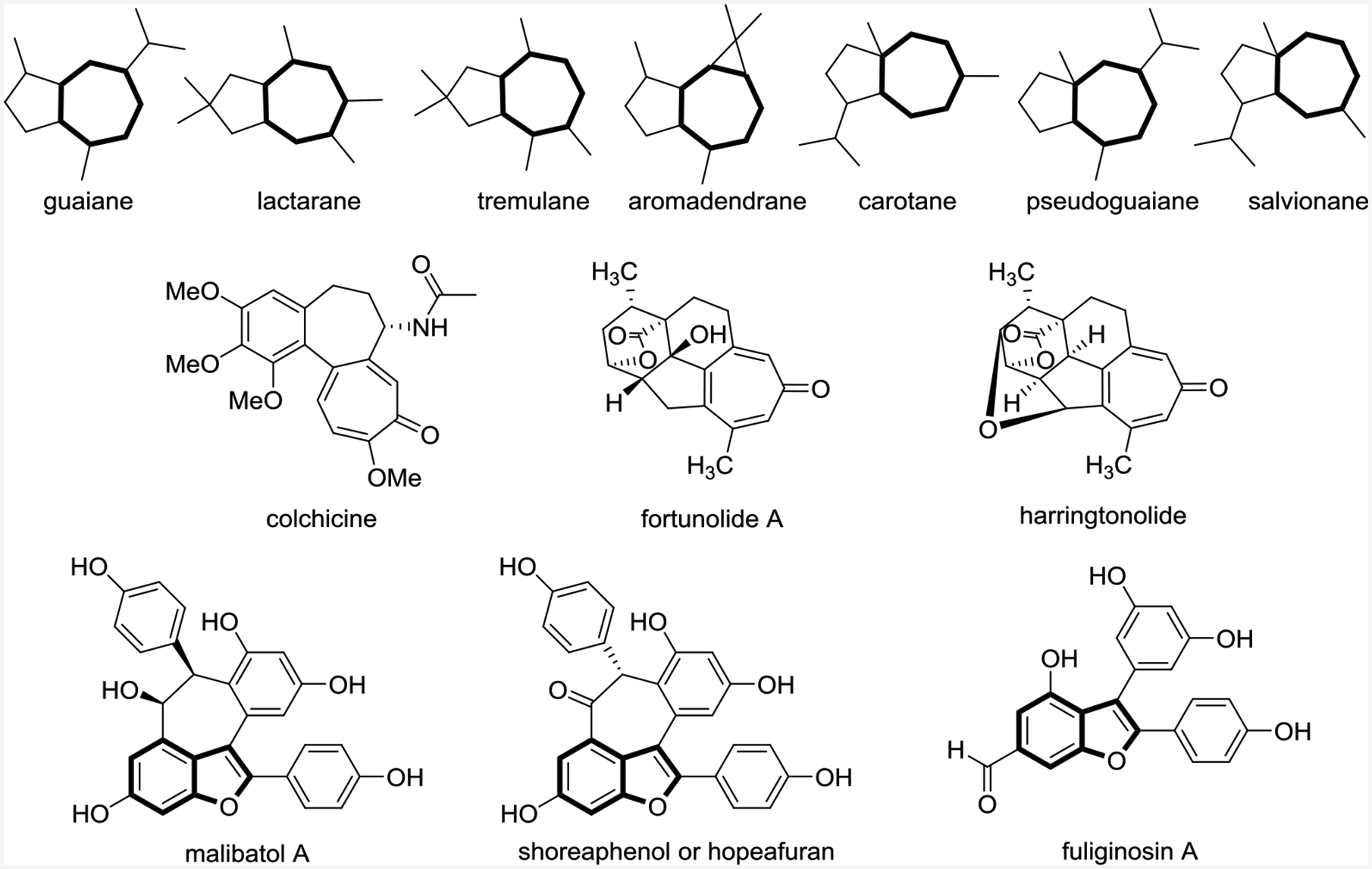
Selected 5–7 fused bicyclic skeletons in sesquiterpenoids and natural products featuring six- and seven-membered rings related to this Account
This Account details our design and use of novel five-carbon components in rhodium-catalyzed (5+n) cycloadditions. These include 3-acyloxy-1,4-enynes (ACEs) for (5+1) and (5+2) cycloadditions and 3-hydroxy-1,4-enynes (HYEs) for (5+1) cycloadditions. Herein, our experimental findings, mechanistic insights, computational rationales, and application-based work will be presented, and an overview of the featured transformations can be found in Scheme 2.
Scheme 2:
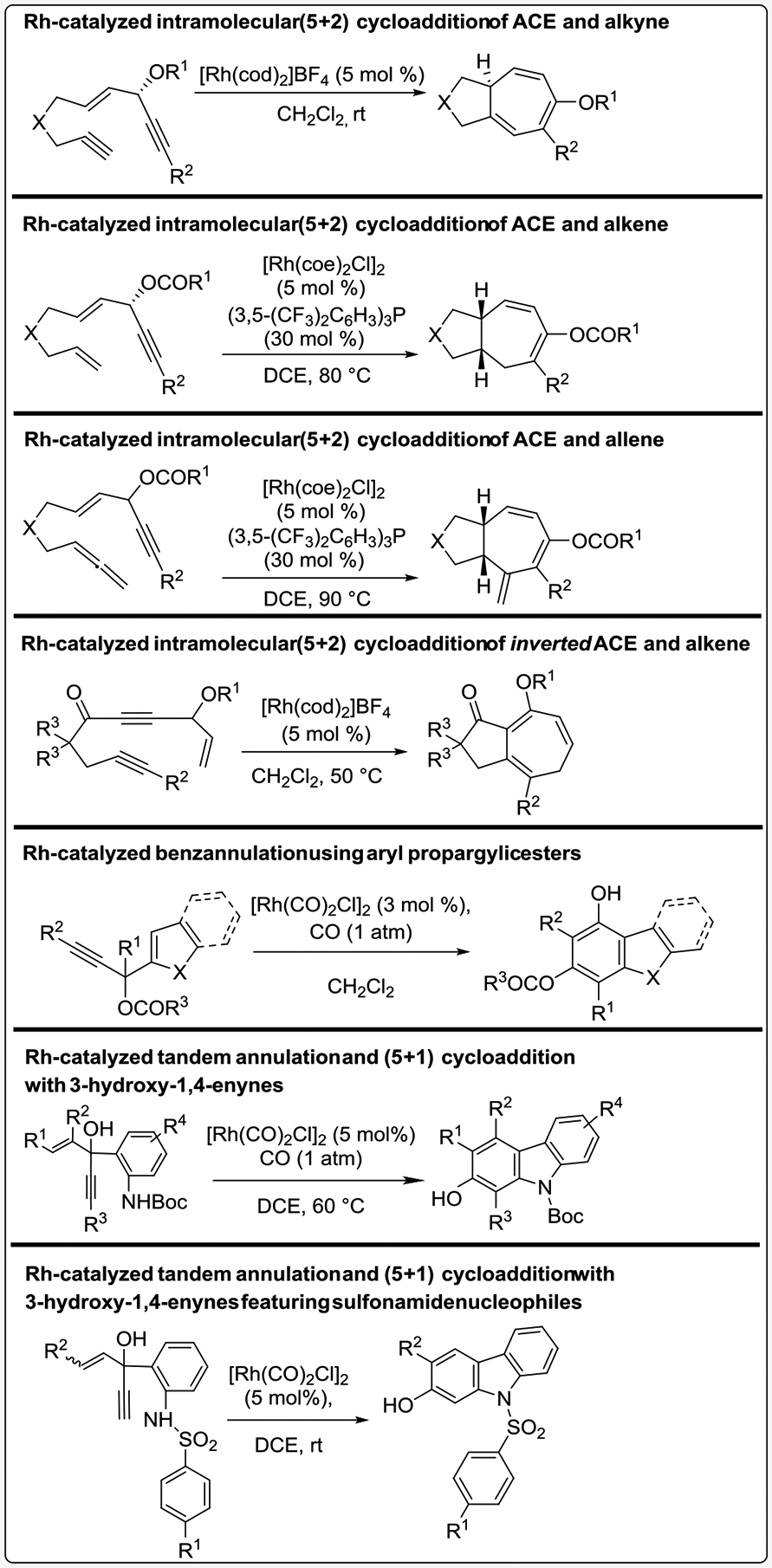
Summary of (5+n) cycloadditions featured in this Account
It should be noted that IUPAC nomenclature defines a cycloaddition as a reaction in which two or more unsaturated molecules (or parts of the same molecule) combine to form a cyclic adduct in which there is a net reduction of the bond multiplicity. While the reactions presented here involve the migration of an acyloxy group to promote ring formation, we have simply referred to them as cycloadditions throughout the text.
2. (5+2) Cycloadditions
2.1. Background
Compared to other transition metal-catalyzed (m+n) cycloadditions, the (5+2) cycloaddition remains largely underdeveloped. Prior to our studies, vinylcyclopropanes (VCPs) were the only known five-carbon synthon for transition metal-catalyzed (5+2) cycloadditions.5,7,8
In 1995 the Wender group reported the first transition-metal catalyzed intramolecular (5+2) cycloaddition between a VCP and a tethered alkyne using a RhCl(PPh3)3 catalyst.7a The same group later used substituted cyclopropanes to investigate the regio- and stereoselectivity of this reaction.7b Not long after, the Trost, Louie, and Fürstner groups determined that Ru-,9 Ni-,10 and Fe-based11 catalysts promoted the intramolecular (5+2) cycloaddition of VCPs with alkynes as well. Intramolecular (5+2) cycloadditions of VCPs and alkenes12 are more difficult reactions to execute because the alkene is less reactive than the previously used alkynes and diastereoselectivity issues can arise. However, the Wender group only observed formation of the cis-fused 5–7 bicyclic compounds. They also discovered that allenes could be tethered to the VCP to promote the (5+2) cycloaddition.13 Additionally, they examined the endo/exo selectivity and chirality transfer in this reaction. Finally, the Wender and Hayashi groups achieved the enantioselective intramolecular (5+2) cycloaddition of VCPs and alkynes or alkenes by using chiral BINAP14 or phosphoramidite ligands, respectively.15
In Diels-Alder [4+2] cycloadditions, a conjugated diene (the 4π system) reacts with an alkyne or alkene (the 2π system), as shown in Scheme 3. For a (5+2) cycloaddition, the 4π system needs to be a five-carbon component. We envisioned that an ester in the middle of a 1,4-enyne might be able to connect the separated alkene and alkyne through a 1,2-acyloxy migration. Inspired by this idea, we proposed a Rh-catalyzed homo-Diels-Alder (5+2) cycloaddition using a 3-acyloxy-1,4-enyne (ACE) as the five-carbon synthon (Scheme 3). The ester in 3.6 could promote this cycloaddition with an accompanying Rautenstrauch-type16 1,2-acyloxy migration. The (5+2) cycloaddition should be thermodynamically favorable because this transformation involves the formation of two C-C σ-bonds and the cleavage of two C-C π-bonds. In addition, depending on the 2-carbon components, a product containing a conjugated triene or diene is formed.
Scheme 3:
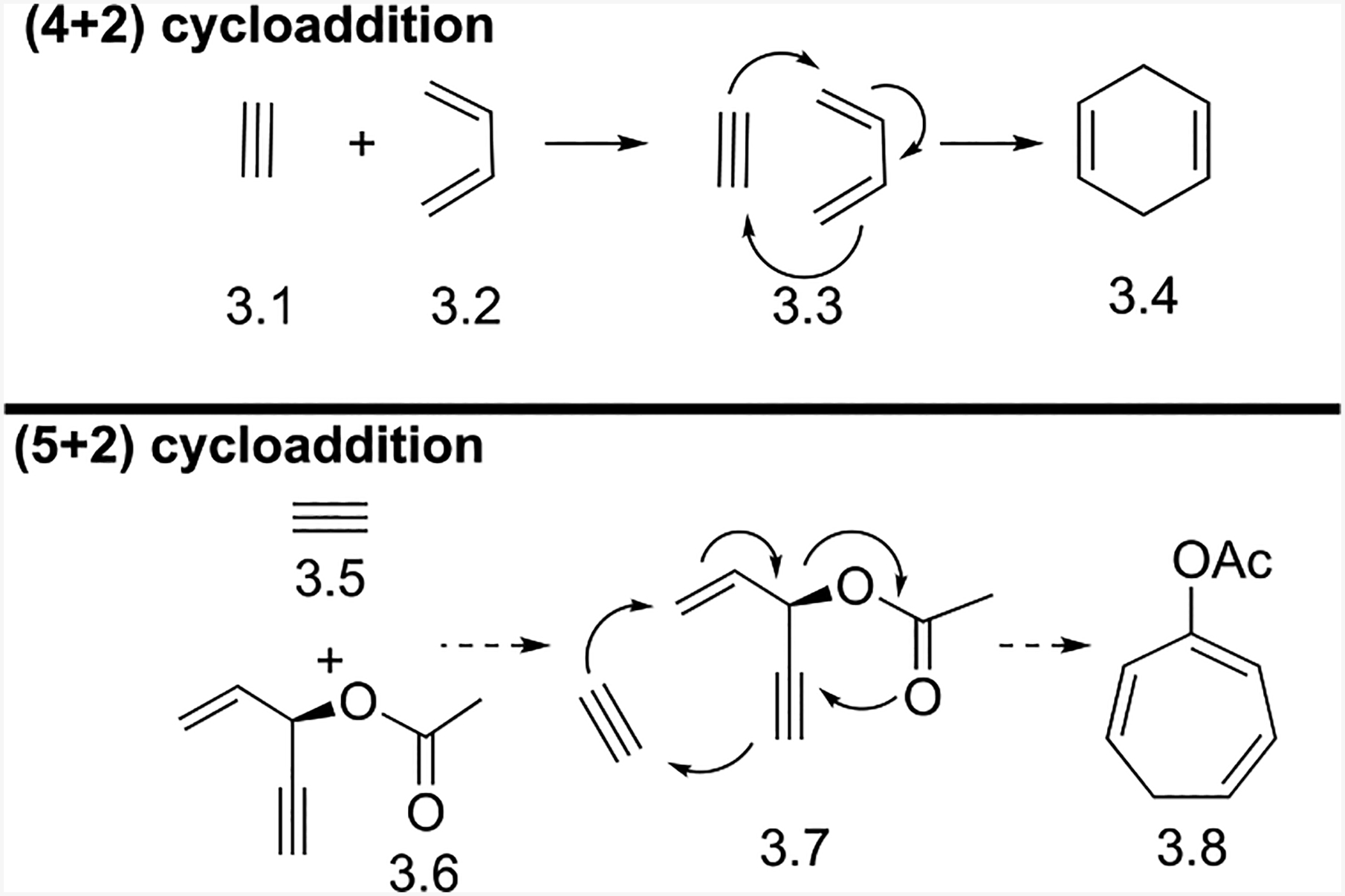
Diels-Alder (4+2) cycloaddition and proposed (5+2) cycloaddition
However, when we ran this reaction with various alkynes and ACEs under thermal conditions, no product formation was observed. We reasoned that a transition metal catalyst might lower the kinetic barrier for the cycloaddition by coordinating to all three π-bonds in 3.7. To further reduce the entropic cost, we decided to tether the five- and two-carbon components together and explore the intramolecular (5+2) cycloaddition first.
2.2. Intramolecular (5+2) Cycloaddition with Alkynes
We were pleased to find that the intramolecular (5+2) cycloaddition with a concomitant 1,2-acyloxy migration proceeded smoothly at room temperature with a variety of Rh(I) catalysts, even though cationic rhodium species [Rh(cod)2]BF4 was superior (Scheme 4).17 The use of platinum or gold complexes did not form the desired products. Successful substrates included those with oxygen, nitrogen, or malonate tethers, esters adjacent to the C1 alkyne (e.g. 5.3), and aryl or alkyl substituents adjacent to the C9 alkyne (e.g. 5.2).
Scheme 4:
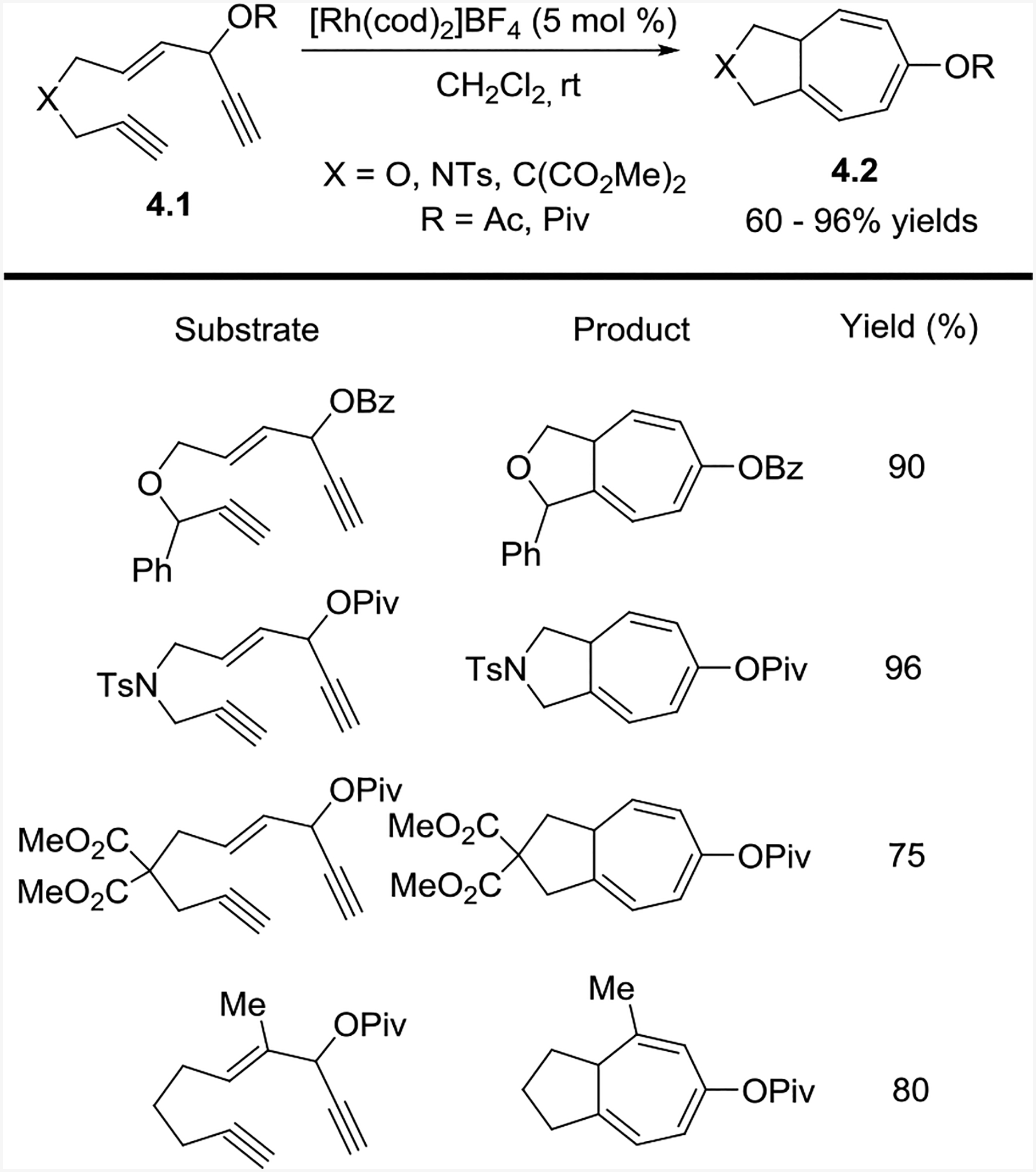
Rh-catalyzed intramolecular (5+2) cycloaddition of ACE and alkyne
When an internal alkyne was the two-carbon component (e.g. 5.5, Scheme 5), cationic rhodium catalysts alone resulted in only trace cycloaddition product formation. However, the addition of an electron-poor phosphite ligand (CF3CH2O)3P significantly increased the reaction yields with these substrates. The alkyne within the 1,4-enyne could be terminal or internal, but internal alkynes required an electron-withdrawing substituent (e.g. 5.7). The limiting factor of this reaction was the tether length; tethers greater than or equal to six atoms did not result in the desired cycloaddition products. This observation was not surprising since π systems separated by more than six atoms are known to be difficult in transition-metal-catalyzed intramolecular cycloisomerization and cycloaddition reactions.2
Scheme 5:
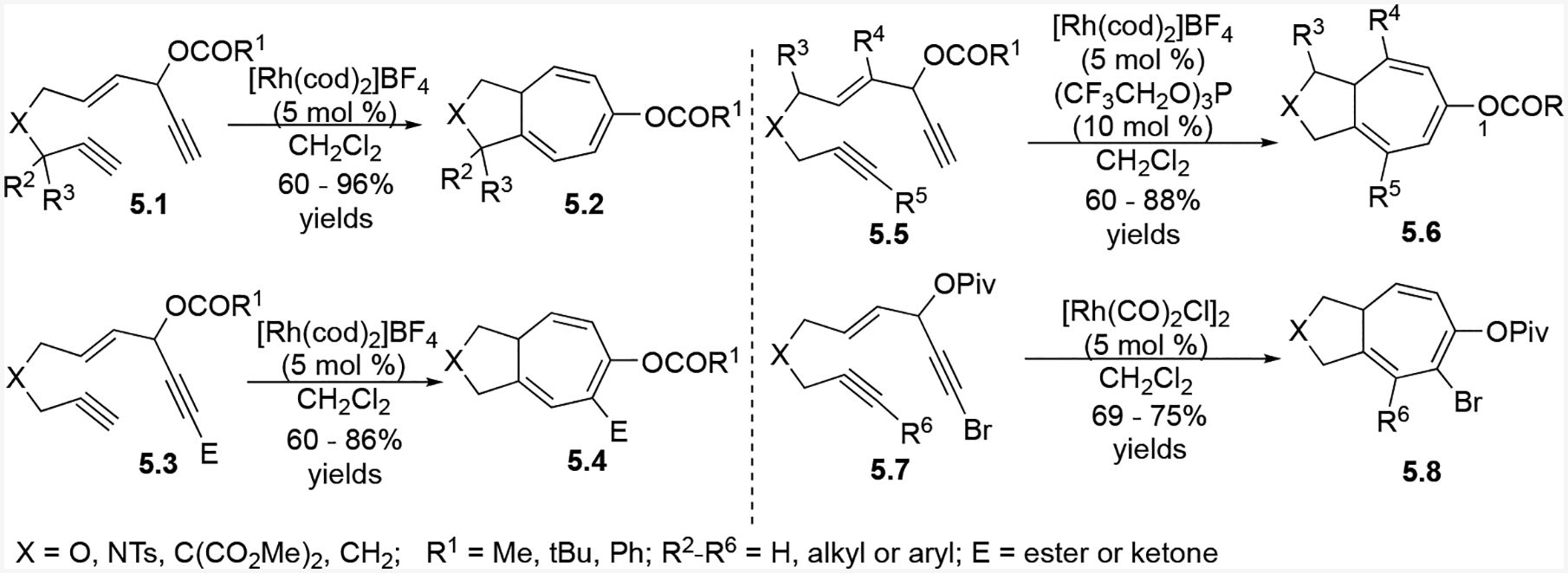
Substrate scope and reaction conditions intramolecular (5+2) cycloaddition between ACEs and tethered alkynes
The resultant bicyclo[5.3.0]decatriene skeletons from this reaction are found in many natural products (Scheme 1),18 and the cycloheptatriene moiety is prevalent in many natural products and pharmaceutical agents.19 Furthermore, the cycloheptatriene has three double bonds that can be easily functionalized. In some cases, we used the cycloheptatriene products to access di- and trisubstituted tropones, which have broad pharmacological activities.20
2.3. Chirality Transfer for the Intramolecular (5+2) Cycloaddition of ACEs and Tethered Alkynes
The Rh-catalyzed intramolecular (5+2) cycloaddition between an ACE and a tethered alkyne provides rapid access to bicyclo[5.3.0]decane skeletons in a stereospecific fashion (Scheme 6).21 In most cases, any erosion of the enantiomeric ratio is minimal. In reactions where an ee loss was observed, the bicyclic products were resubjected to the reaction conditions, which resulted in no significant change in ees. These results suggested that any observed erosion of the enantiomeric ratio occurs before the final cycloaddition product is formed.
Scheme 6:
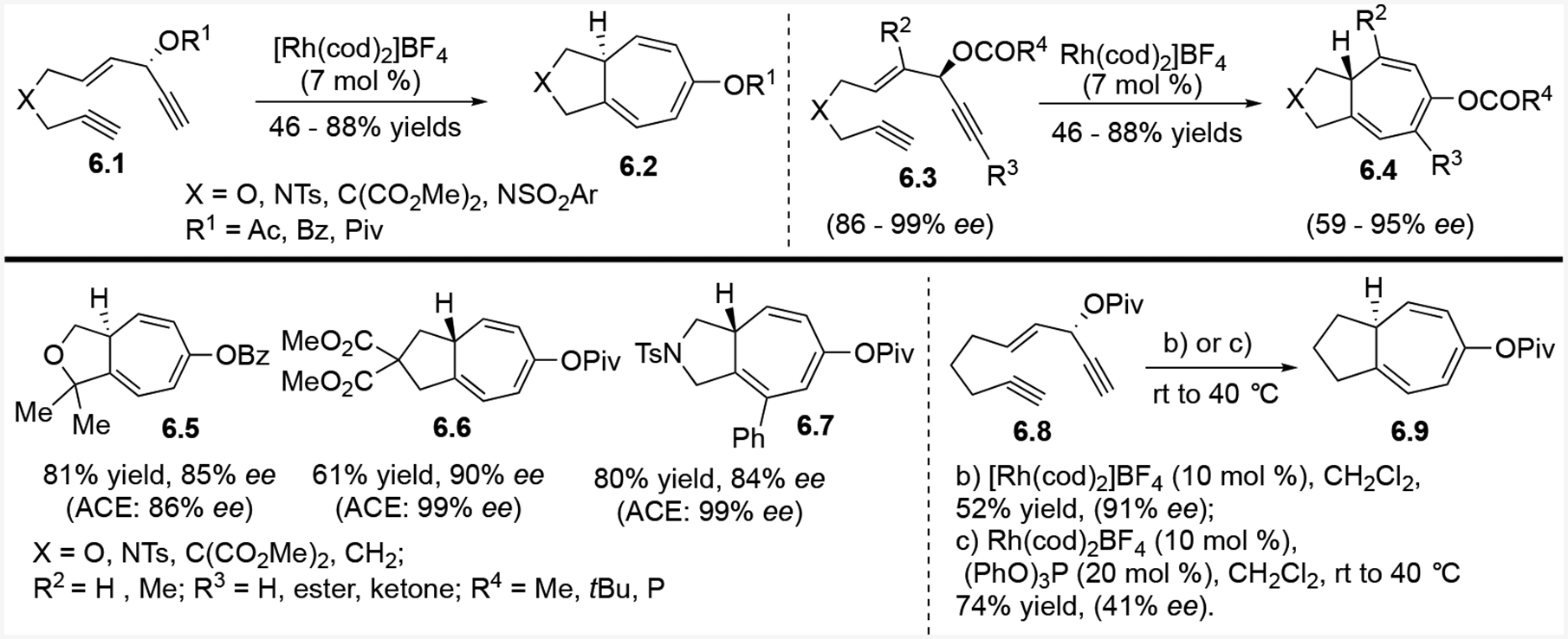
Transfer of chirality in the intramolecular (5+2) cycloaddition
In certain cases, the chirality transfer efficiency is ligand dependent. We knew from previous intramolecular [5+2] cycloaddition research10 that substrates with internal alkynes, such as 6.3, require phosphite ligands to work. However, we observed that phosphite ligands significantly decreased the degree of chirality transfer for most substrates. Nevertheless, we were able to obtain an 84% ee using a phosphite ligand for product 6.7. As seen in the formation of product 6.9, adding (PhO)3P gave a higher yield but a lower ee compared to the Rh catalyst alone. Overall, this work was the first time that bicyclo[5.3.0]decatrienes were prepared in high ees.
Computational studies from the Houk group22 yielded insight into the chirality transfer mechanism. Their DFT calculations suggested the Rh catalyst coordinates to the alkynes and alkene in (S)-7.1 anti to the acyloxy group to yield 7.2 (Scheme 7). The Rh-promoted 1,2-acyloxy migration and cyclization first forms the chiral Rh-allyl intermediate 7.3, which readily interconverts with 7.4. Alkyne insertion can proceed through either 7.3 or 7.4. This syn-addition of the tethered alkyne leads to the formation of the eight-membered rhodacycle 7.5. A final reductive elimination then yields (S)-7.6. Overall, the anti-addition of the Rh catalyst to the ACE and the syn-insertion of the alkyne to the Rh-allyl complex result in the observed stereochemistry for the intramolecular (5+2) cycloaddition.
Scheme 7:
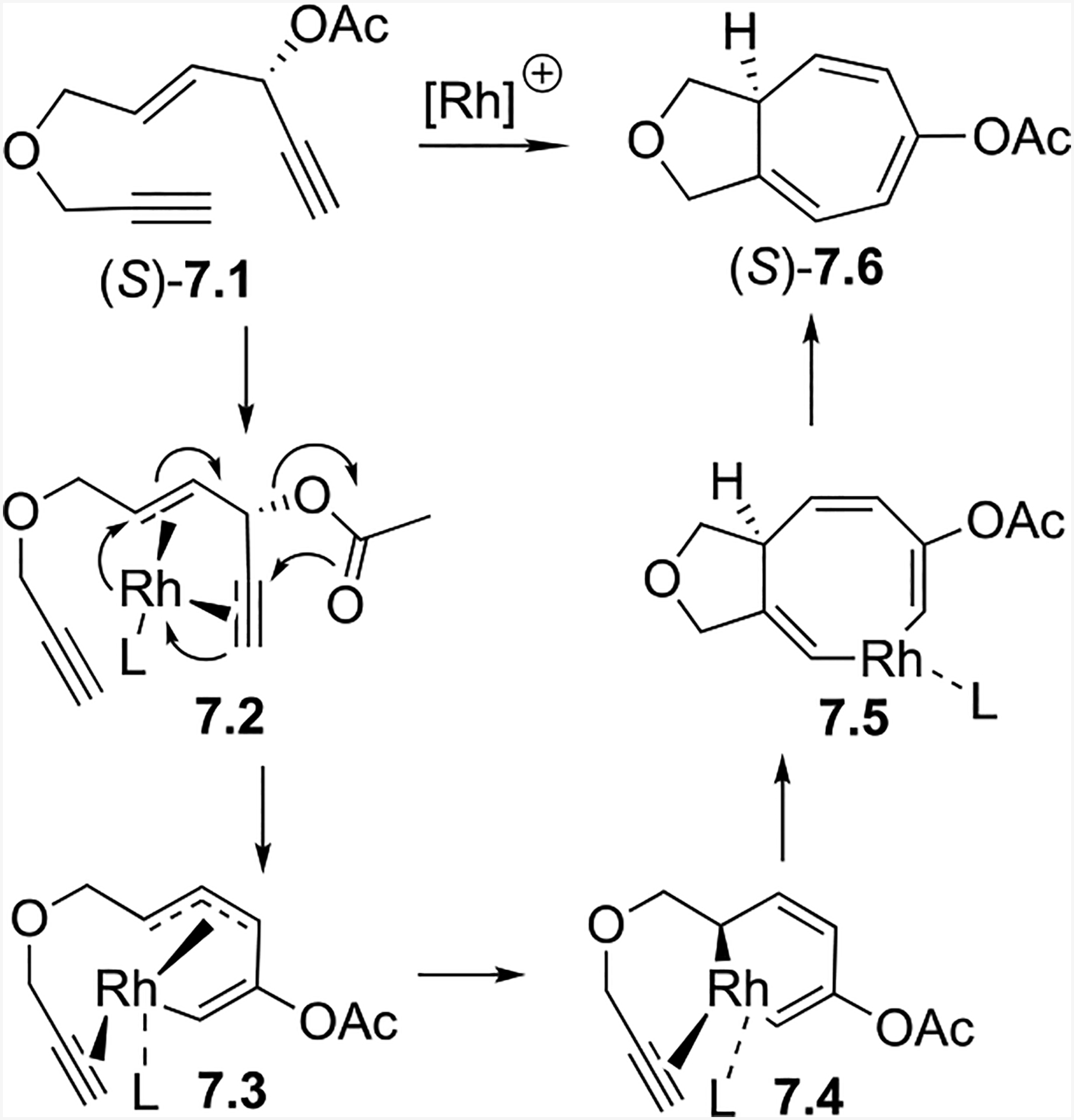
Proposed mechanism for chirality transfer in the intramolecular (5+2) cycloaddition
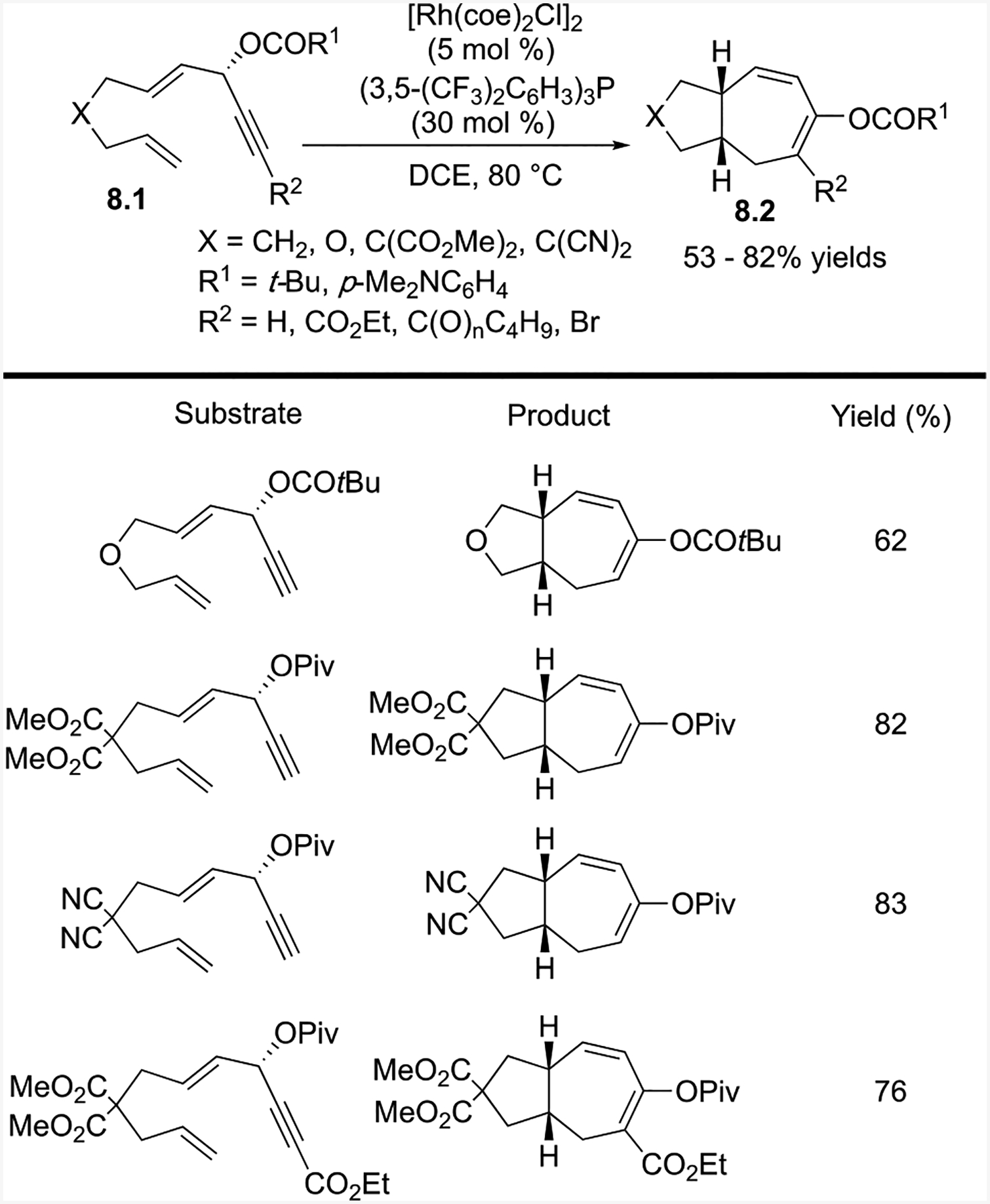
2.4. Intramolecular (5+2) Cycloaddition of ACEs and Tethered Alkenes and Allenes
The intramolecular (5+2) cycloaddition also worked for ACEs with tethered alkenes (Scheme 8 and Scheme 9)23 and allenes (Scheme 10)24 as the two-carbon components. In each case, a neutral rhodium catalyst [Rh(coe)2Cl]2 with an electron-deficient phosphine ligand (3,5-(CF3)2C6H3)3P provided the cycloaddition products, albeit the allene reactions generally resulted in lower yields.
Scheme 8:
Intramolecular (5+2) cycloadditions with alkenes
Scheme 9:
Chirality transfer in the intramolecular (5+2) cycloadditions with alkenes
Scheme 10:
Intramolecular (5+2) cycloadditions with allenes
For tethered alkenes, the two-carbon alkene component could be either terminal or 1,2-disubstituted with an aryl substituent. Chirality transfer from the substrates to the products was possible even for the formation of three contiguous stereogenic centers. Only one diastereomer was observed in each case.
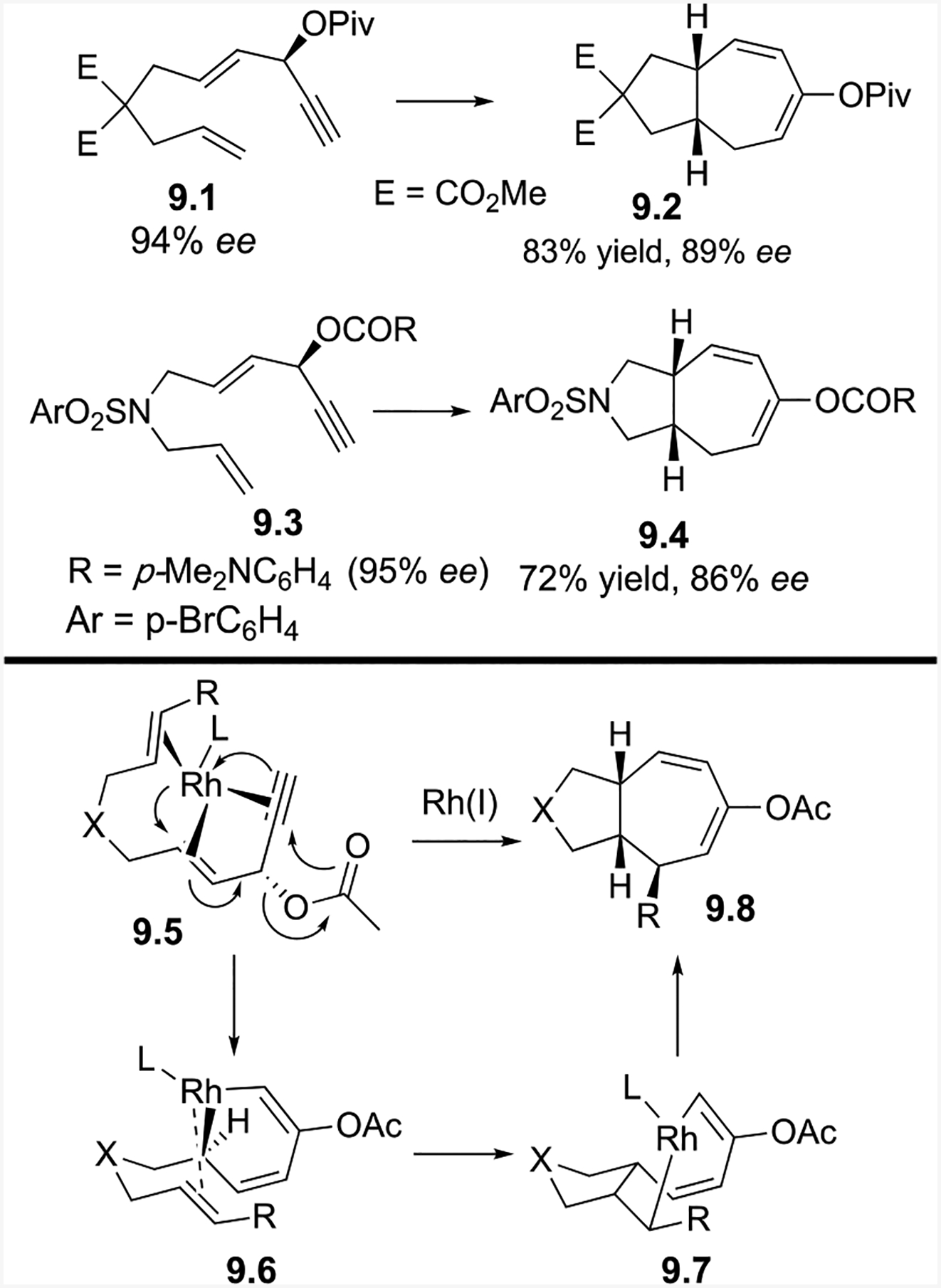
Mechanistically, the Rh catalyst adds anti to the acyloxy group of the ACE to give 9.5 and promotes the 1–2 acyloxy migration and oxidative cyclization, which forms rhodacycle 9.6 (Scheme 9). The tethered alkene then inserts into the rhodacycle to form 9.7. Subsequent reductive elimination of 9.7 forms cycloheptadiene product 9.8. This mechanism is consistent with the observed absolute stereochemistry of propargylic ester 9.3 and bicyclic product 9.4, which was confirmed by X-ray crystallography.
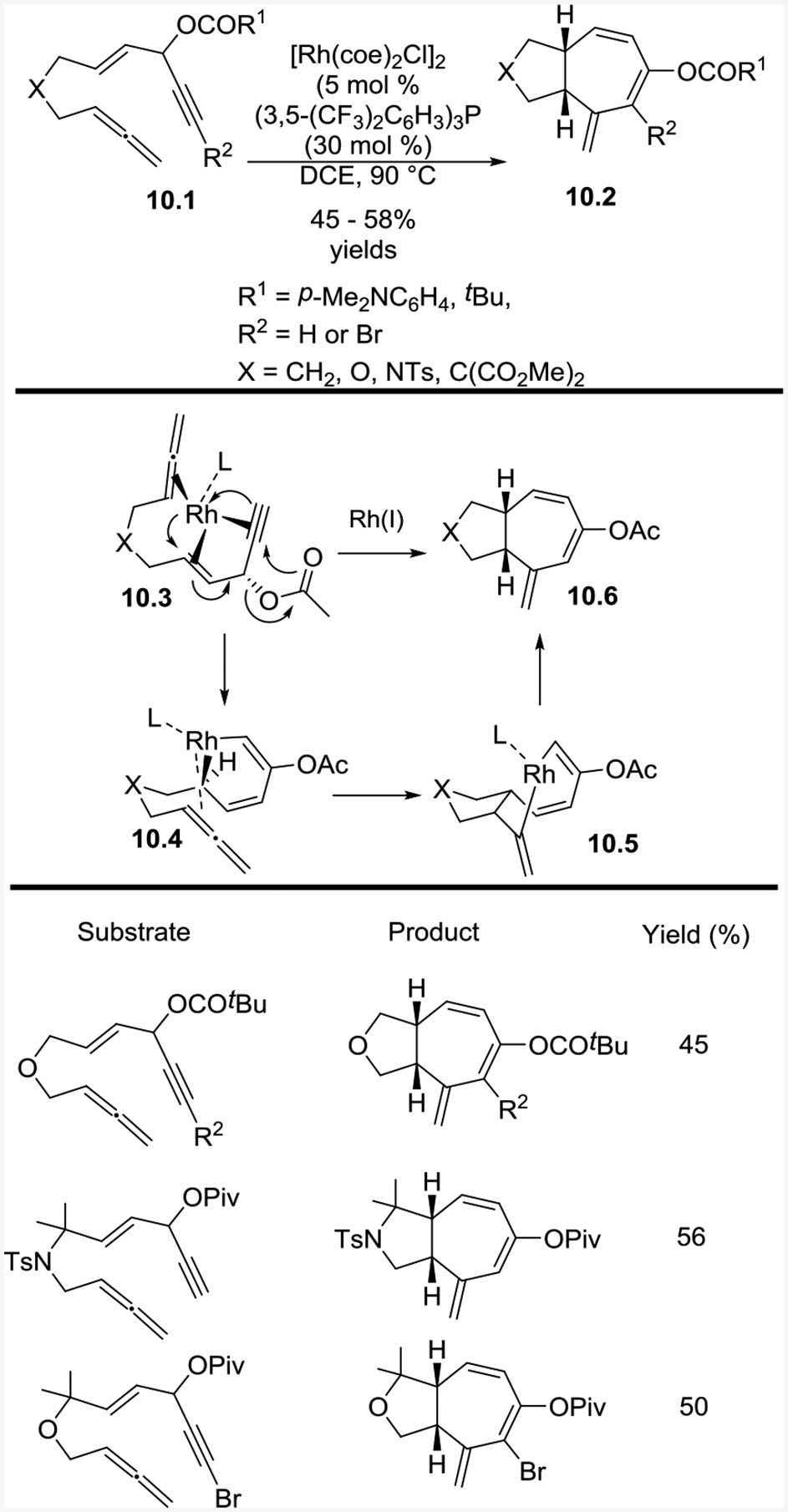
The yields for the (5+2) cycloaddition of allenes were lower than the corresponding reaction with alkenes. Under optimized conditions, the yields of the bicyclic products only ranged from 45–58% percent, due to the isomerization of the allene to dienes under the reaction conditions. The reaction preferentially occurred at the internal alkene.
The reaction begins with Rh addition anti to the acyloxy group to yield 10.3 (Scheme 10). Oxidative cyclization of 10.3, accompanied by a 1,2-acyloxy migration, yields 10.4. The tethered allene then inserts into the rhodium-carbon bond to yield 10.5. Reductive elimination of the eight-membered rhodacycle results in 10.6.
2.5. Inverted Intramolecular (5+2) Cycloaddition with Alkynes
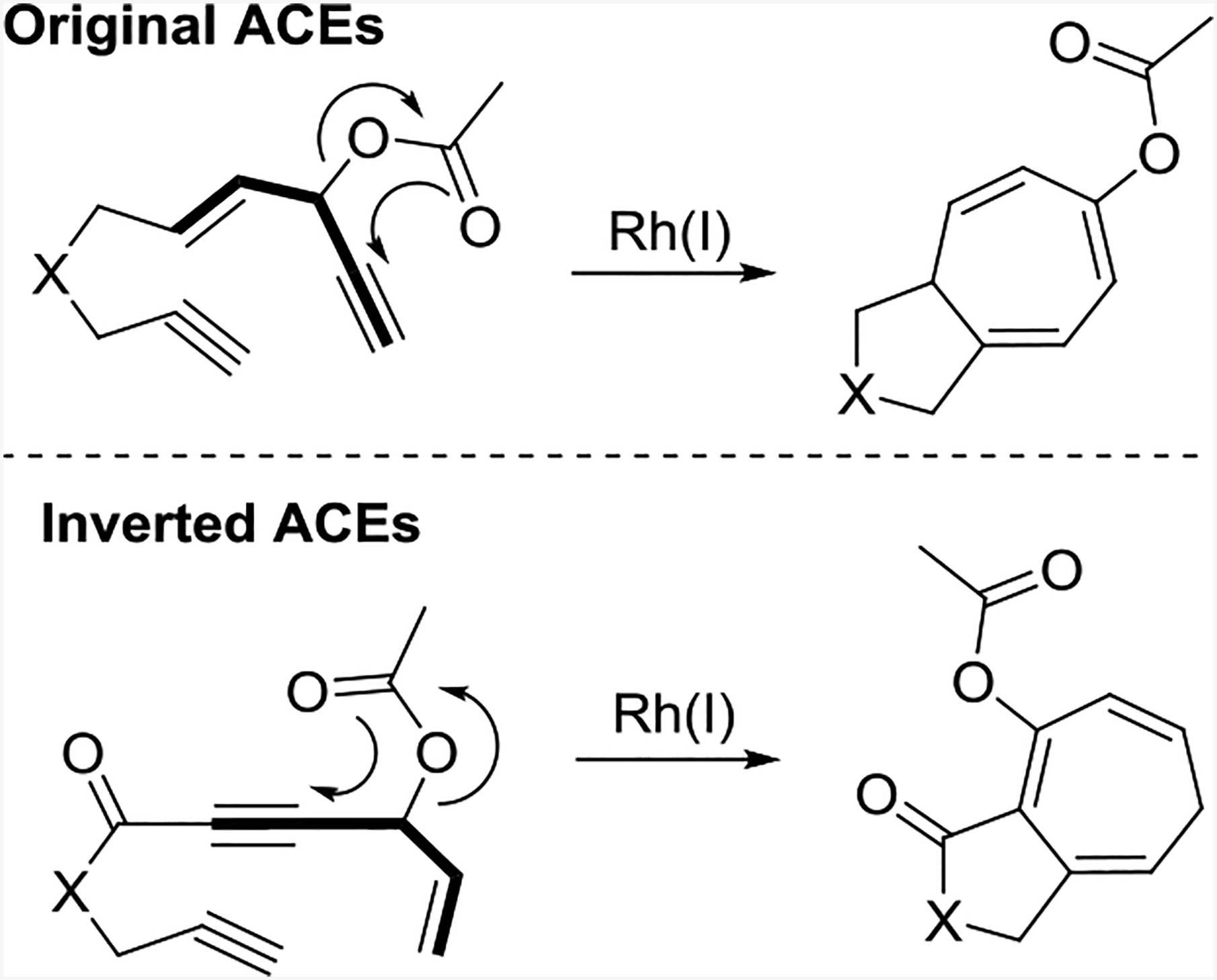
Inspired by Yu’s report25 detailing the design of a new five-carbon building block by swapping the positions of key reaction components, we switched the positions of the alkene and the alkyne in the 1,4-enyne of our original ACE to provide an inverted ACE variant (Scheme 11), which produced products with complementary functionalities.26
Scheme 11:
Original ACEs compared with inverted ACEs
Usually, propargylic esters that contain an internal alkyne favor a 1,3-acyloxy migration to form allenes when used with Rh(I) catalysts.27 However, electron-withdrawing substituents conjugated with an alkyne can alter the selectivity to instead favor a 1,2-acyloxy migration when used in Pt-,28 Rh-,29 and Au-catalyzed reactions because the electron-withdrawing substituents make the C2 position more electrophilic than the C3 position.20a Inverted ACEs 11.1 (Scheme 11) were used with various Rh (I) catalysts to promote the intramolecular (5+2) cycloaddition with tethered alkynes. While neutral Rh catalysts did not give the desired products, cationic Rh catalysts proved efficient for the reaction, and [Rh(COD)2]BF4 was the best. The addition of ligands was ineffective at improving the reaction yields, and neither gold nor platinum catalysts could promote this transformation. The reaction was amenable to substituents on the internal position of the alkene, but no desired product was observed when the external position of the alkene was substituted. A tether length of six atoms decreased the reaction efficiency.
DFT calculations helped elucidate the mechanism of inverted ACE (5+2) cycloadditions. The Rh catalyst coordinates to the internal alkyne and terminal alkene to form 12.4 (Scheme 12). This coordination promotes a 1,2-acyloxy migration and oxidative cyclization that results in intermediates 12.5 and 12.6. Alkyne insertion into the rhodium-carbon bond yields rhodacycle 12.7. Reductive elimination gives 12.8, and Rh catalyst dissociation yields final product 12.9. Based on calculated enthalpies and Gibbs free energies, the 1,2-acyloxy migration is the rate determining step. This inverted ACE cycloaddition provides another way to prepare seven-membered rings, and these rings have complementary functionalities compared to our previous cycloaddition products.
Scheme 12:
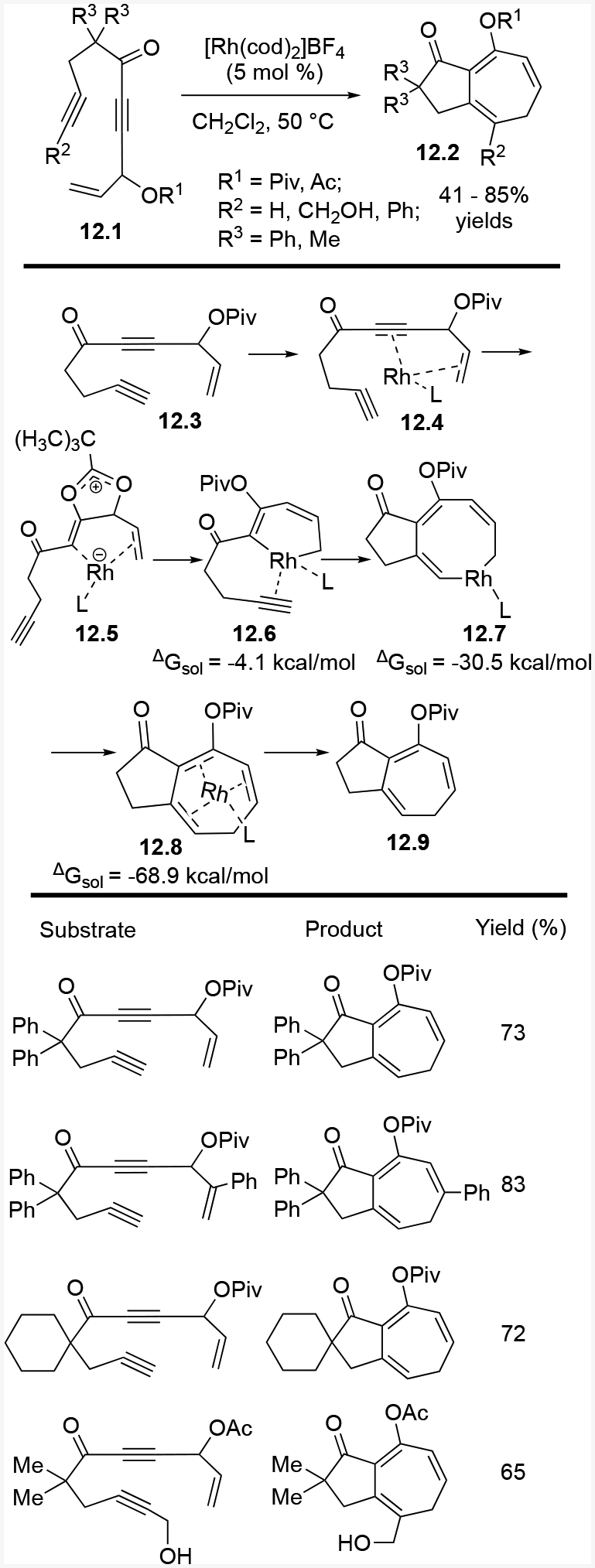
Intramolecular (5+2) cycloaddition with inverted ACEs
2.6. Intermolecular (5+2) Cycloaddition
After developing the Rh-catalyzed intramolecular (5+2) cycloaddition, we turned our attention to the intermolecular version. Unsurprisingly, the intermolecular cycloaddition is more difficult because of potential reactivity, chemo- and regioselectivity issues that result from removing the tether between the two reactants. Conversely, it is easier to obtain the two separate reactants needed to execute the intermolecular version.
When we subjected 13.1 and 13.2 to the [Rh(COD)2]BF4 catalyst used previously in the intramolecular (5+2) cycloaddition, the desired cycloaddition product 13.3 was not observed, regardless of the presence or absence of phosphite ligand (CF3CH2O)3P (Scheme 13).30 Palladium, platinum and gold complexes did not work either. We suspected two molecules of ACE could potentially coordinate to the cationic rhodium catalyst as bidentate ligands as shown in 13.4, which would prevent the two-carbon alkyne component from coordinating to the rhodium. Indeed, only when using a neutral rhodium catalyst with three coordination did we observed the desired product. In most cases, Wilkinson’s catalyst worked well also. The two-carbon alkyne could be either terminal or internal. Polar substituents on the propargylic or homopropargylic positions of the terminal alkynes formed highly regioselective products. Terminal alkynes were generally more reactive than internal alkynes, and while internal alkynes still worked, they required an electron-withdrawing halogen, ketone, or ester as the terminal substituent.
Scheme 13:
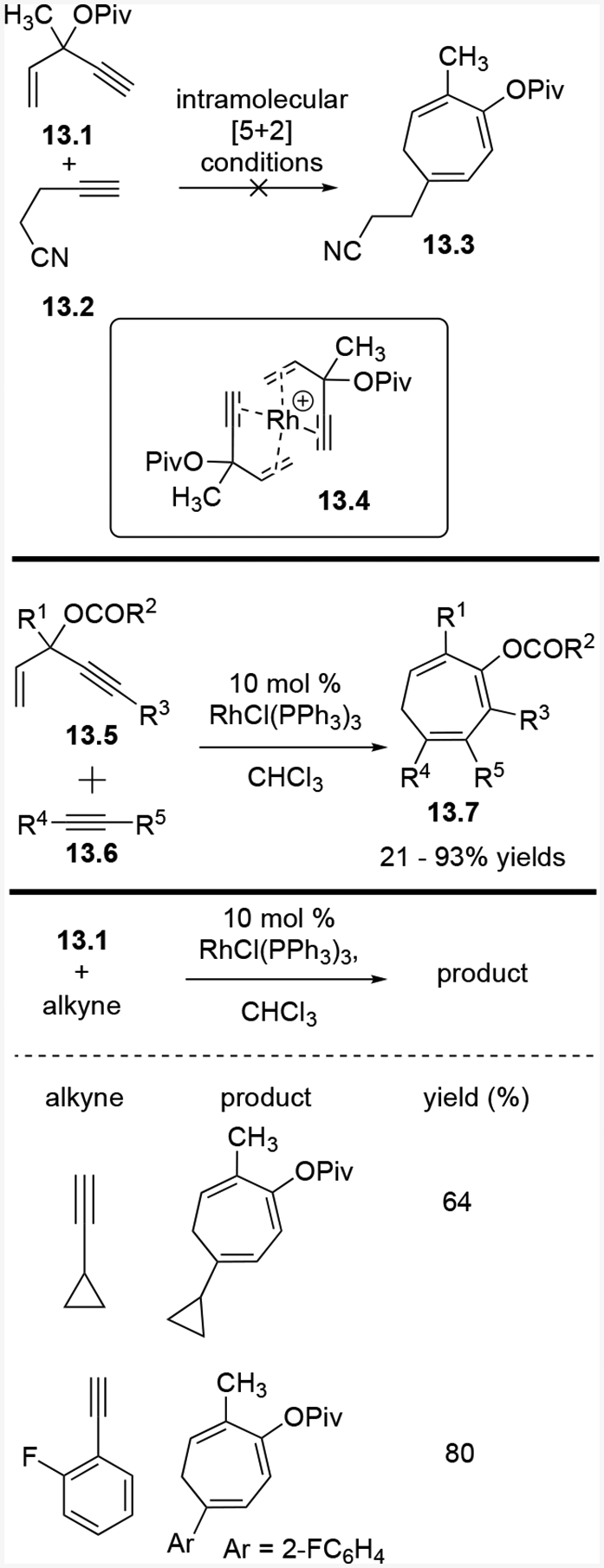
Scope for Rh-catalyzed intermolecular (5+2) cycloaddition of an ACE and an alkyne
Our intermolecular (5+2) cycloaddition method a with concomitant 1,2-acyloxy migration was applicable to a wide variety of substrates, and it was amenable to scale up with high regioselectivity. About 0.76 g of compound 14.3 was obtained with a greater than 20:1 isomeric ratio (Scheme 14). Cycloheptatriene 14.3 could then be functionalized to yield 14.4.
Scheme 14:
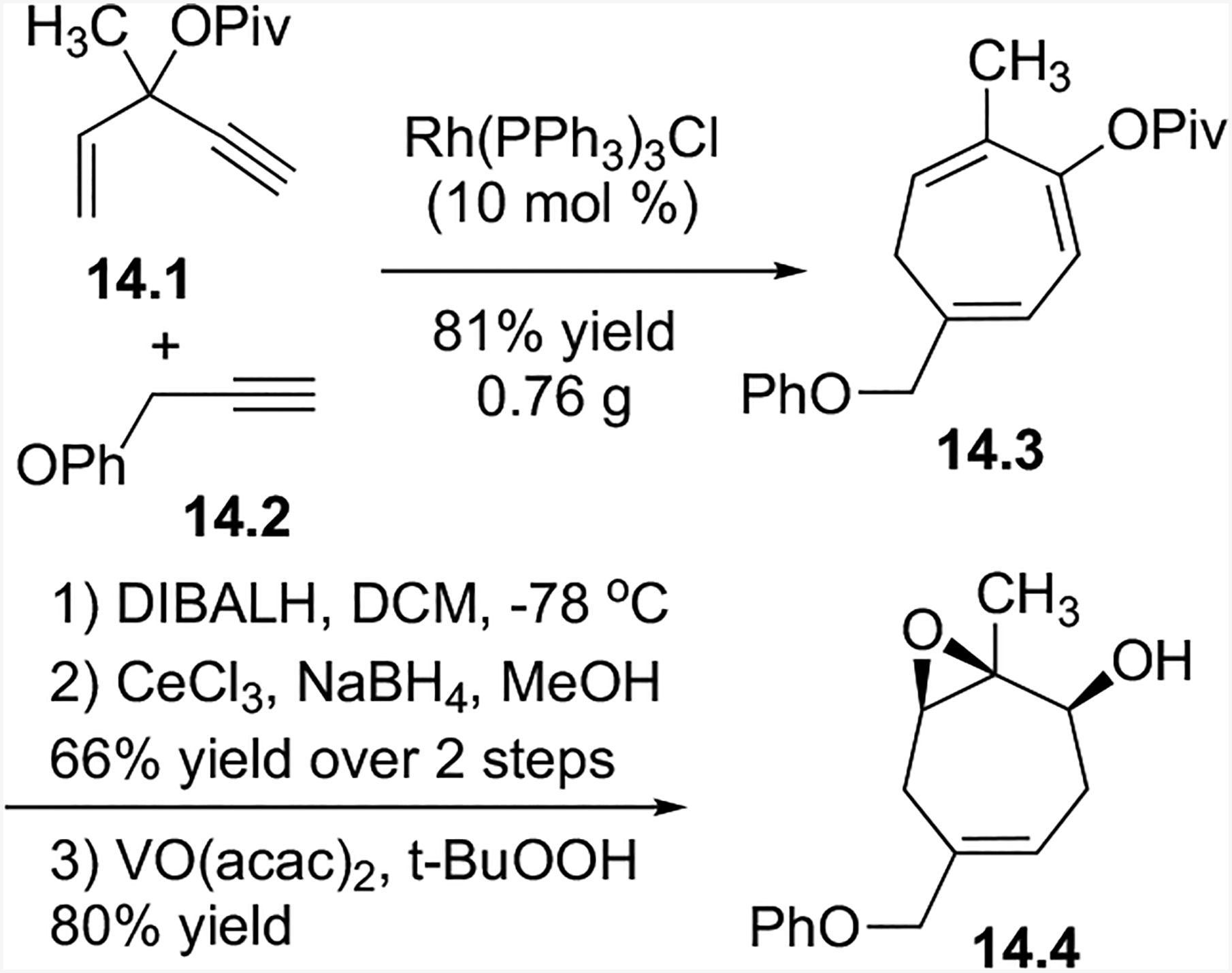
Scale-up and functionalization potential of intermolecular (5+2) cycloaddition product
In collaboration with the Houk group, we performed DFT calculations and our data indicated a concerted cycloaddition mechanism was at work (Scheme 15).15 From ACE 15.1, the rhodium coordinates anti to the acyloxy group, as shown in 15.3. Proceeding through transition state 15.4, rhodacycle intermediate 15.5 is formed. The 1,2-acyloxy migration is believed to be the rate-determining step (RDS). Four rhodacyclooctatriene intermediates can be proposed after the insertion of alkyne 15.2 to rhodacyclohexadiene intermediate 15.5. Via pathway A, the alkyne could insert into either the sp3-carbon-metal bond to form 15.6 or 15.7. Via pathway B, the alkyne could insert into the sp2-carbon-metal bond to form 15.8 or 15.9. For a terminal alkyne, such as 15.2, the R group could be either close (15.6 or 15.9) or distal (15.7 or 15.8) to the forming C-C bond. In Houk’s previous computational studies on Rh-catalyzed reactions involving unsymmetrical alkynes, the bulkier alkyne substituent prefers to be distal to the forming C-C bond.31 From the mostly likely rhodacyclooctatriene intermediate 15.8, final product 15.10 is formed via a reductive elimination step.
Scheme 15:
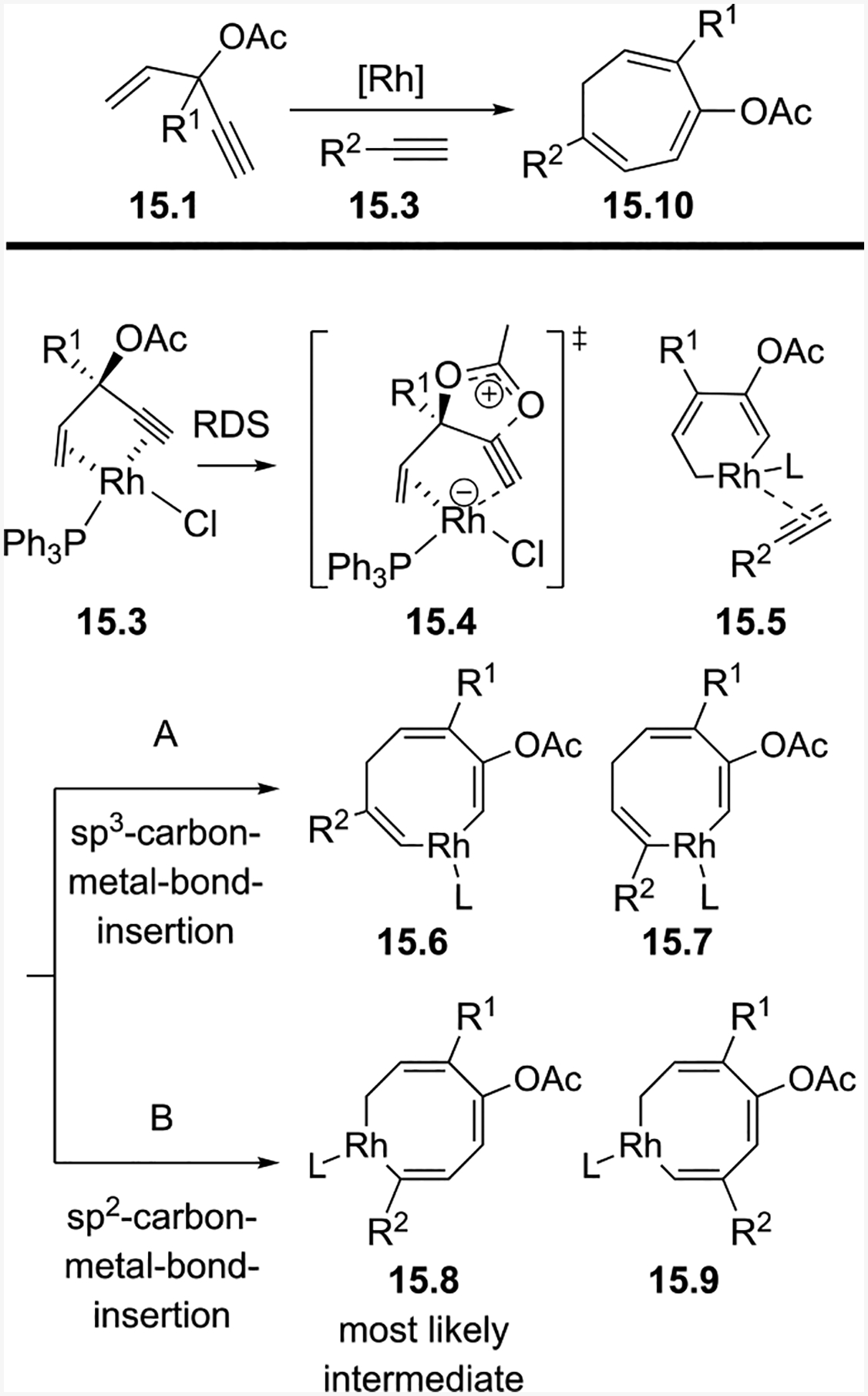
Mechanism of the intermolecular (5+2) cycloaddition
The Rh-catalyzed intermolecular (5+2) cycloaddition between an ACE and an alkyne, which is accompanied by a 1,2-acyloxy migration of a propargyl ester, provides an efficient method to synthesize seven-membered rings in only two steps from commercially available starting materials.
2.7. Effect of Esters on the Intermolecular (5+2) Cycloaddition
In a subsequent study,32 we examined the effect of various esters on the ACE C3-position in Rh-catalyzed intermolecular (5+2) cycloadditions (Scheme 16). By using the electron-donating ester p-dimethylaminobenzoate, the reaction rate was more than 46 times faster than the corresponding reaction with a phenyl ester. We reasoned that the electronic effect of the ester could facilitate the reaction in an early nucleophilic attack step or a late reductive elimination step. After computational calculations,15 we attributed the rate increase to the reductive elimination step.
Scheme 16:
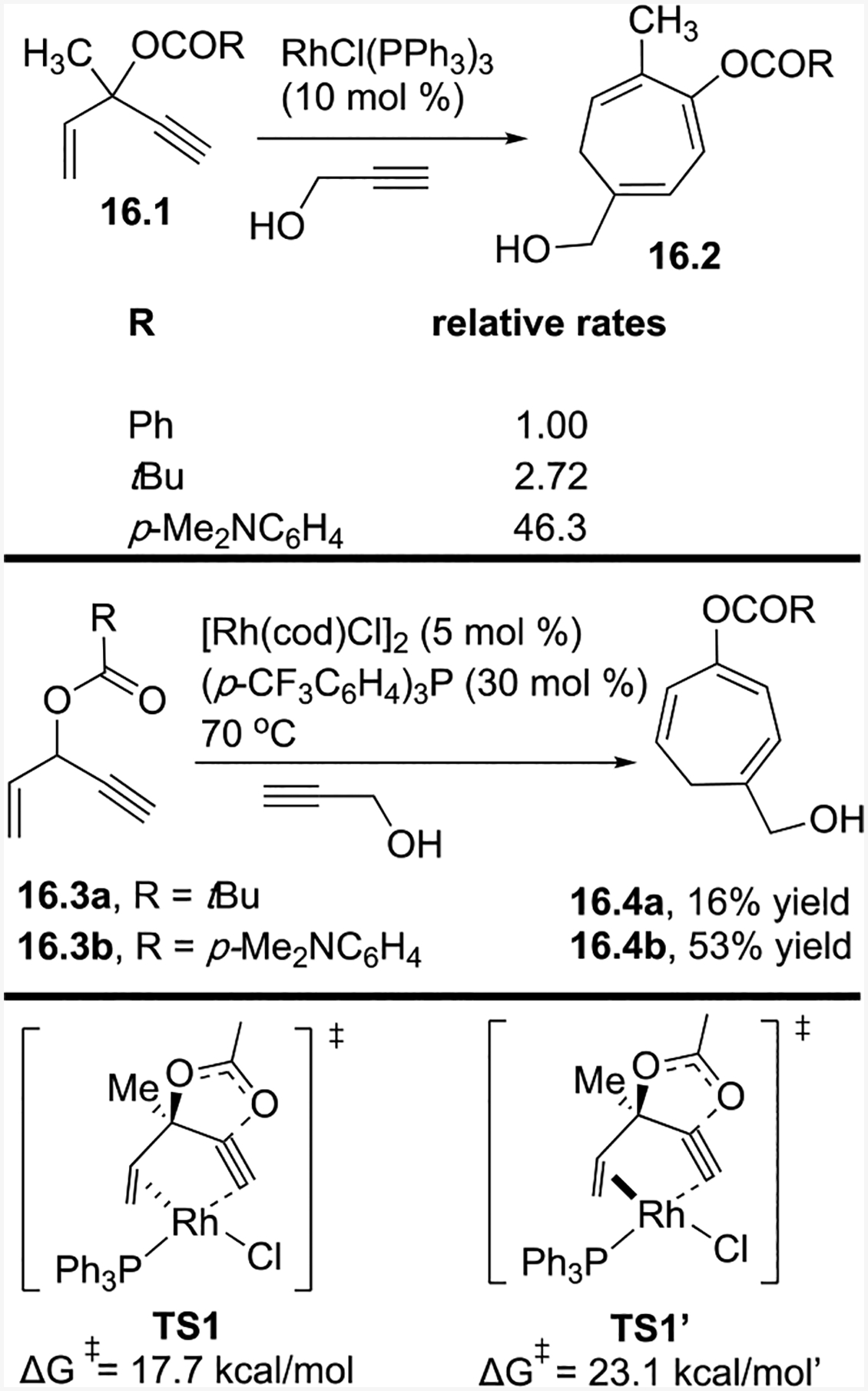
Effect of esters on the rate of Rh-catalyzed intermolecular (5+2) cycloadditions
Since the effect of the electron-donating ester on the rate of the reaction is determined by the barrier of 1,2-acyloxy migration, computational calculations were completed to determine whether the lower transition state energy resulted from the Rh being anti (TS1) or syn (TS1’) to the acyloxy group. We determined that during the concerted process of 1,2-acyloxy migration with the Rh catalyst, the Rh prefers to add in an anti-fashion because the activation free energy is 5.4 kcal/mol less than if the Rh would add in a syn-fashion. The ester’s electron-donating ability also stabilizes the carbonyl carbon’s positive charge (as shown in 15.4) and lowers the transition state’s energy in the rate-determining step. For ACEs with a tertiary ester, the catalyst loading could be reduced to 0.5%. For ACEs with a secondary ester, the yield increased from 16% to 53% (16.4a to 16.4b).
All of the previous research represents the first time that ACEs have served as the five-carbon component in Rh-catalyzed intra- and intermolecular (5+2) cycloadditions with alkynes, and in some cases, intramolecular cycloaddition with alkenes and allenes.
3. (5+1) Cycloadditions
Six-membered carbocycles are among the most prevalent ring compounds. Many two- and three-component cycloadditions have been designed to produce these structures, but difficulties associated with sourcing the five-carbon component hinder further progress of the (5+1) cycloaddition.
3.1. (5+1) Cycloaddition with an ACE and CO
The first Rh-catalyzed (5+1) cycloadditions of ACEs and CO were developed by Fukuyama, Ryu, Fensterbank and Malacria.33 1,3-Dihydroxybenzenes with additional one or two substituents were prepared. However, the reaction requires high pressures of CO (50 atm) and high temperature (80 °C). After discovering that the electron-donating ester can significantly accelerate the rate of the Rh-catalyzed (5+2) cycloaddition,34 we examined the effect of the ester on (5+1) cycloaddition of ACE and CO.35 Indeed, the (5+1) cycloaddition could run under lower temperature (room temperature to 50 °C) and much lower pressure of CO (1 atm) using ACEs that contained an electron-rich dimethylaminobenzoate ester.28,36
3.2. Aryl Propargylic Esters as the Five-carbon Building Block in (5 + 1) Cycloadditions
Besides expanding the scope of (5+2) cycloadditions, we have also developed additional five-carbon synthons for (5+1) cycloadditions to access polycyclic compounds. We envisioned that if the alkene part of an ACE could be embedded in an aromatic system, aryl propargylic esters could then be used in a Rh-catalyzed benzannulation. To our delight, this new type of (5+1) cycloaddition indeed worked and allowed for the synthesis of highly substituted indoles at the C4–C7 positions, complementary to current techniques for indole preparation. We also extended this method to the synthesis of benzofurans, benzothiophenes, carbazoles, and dibenzofurans.37
The DFT calculation supported mechanism is shown in Scheme 17. Starting with 17.1, the Rh(I) catalyst coordinates to the alkyne to form 17.3. This species undergoes the rate-determining stepwise 1,2-acyloxy migration to form 17.4. Then zwitterion intermediate 17.5 forms, isomerizes to a four-membered rhodacycle 17.6, undergoes CO insertion and subsequent reductive elimination to afford ketene intermediate 17.7. Finally, a 6π electrocyclization and aromatization yields benzofuran product 17.2.
Scheme 17:
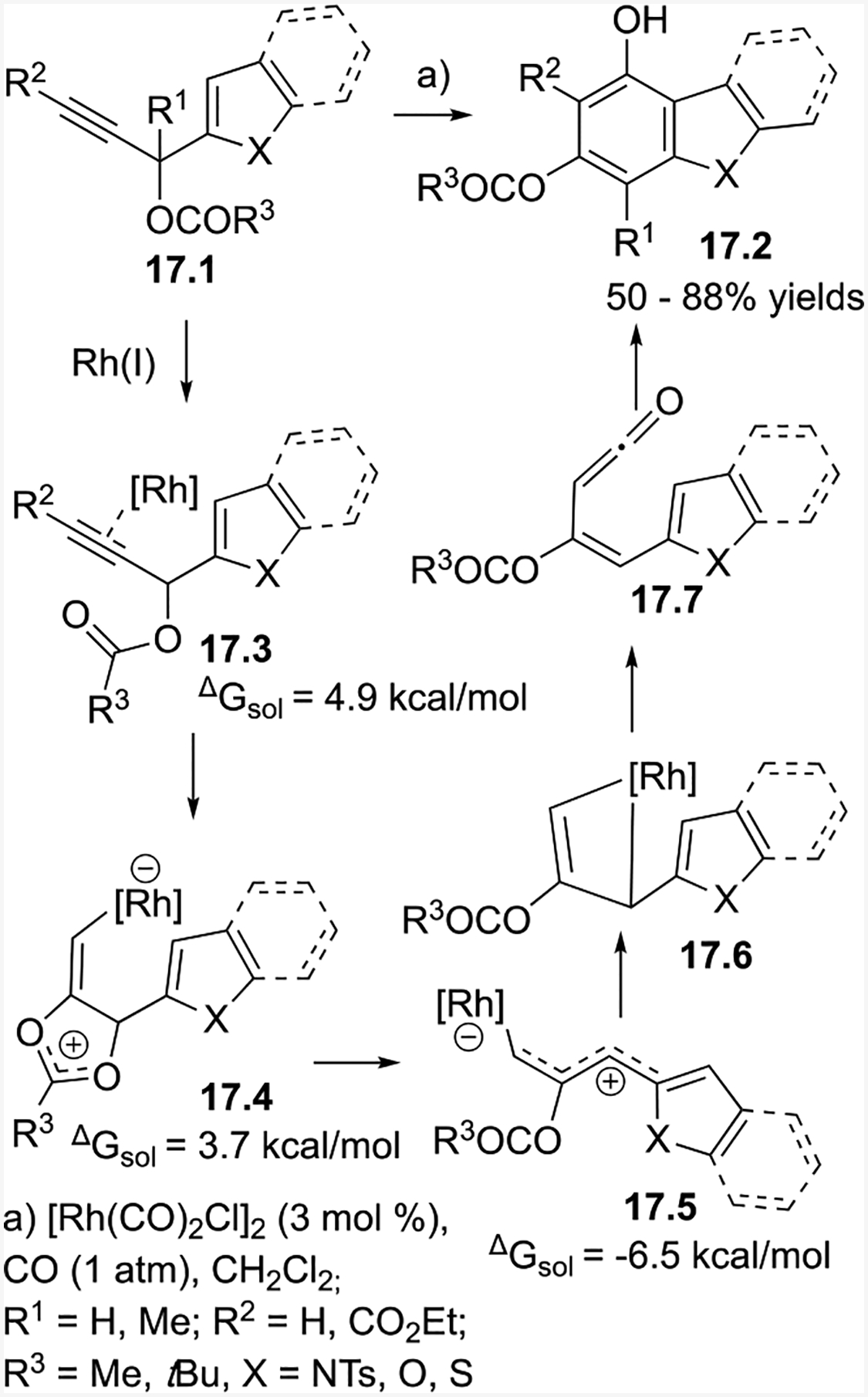
Benzannulation of propargylic esters and heteroaryl propargylic esters
Based on the previous benzannulation of heteroaryl propargylic ester research, we were able to synthesize a variety of highly substituted benzofuran-containing natural products which include: amurensin H (or viniferifuran), malibatol A, shoreaphenol (or hopeafuran), fuliginosin A, and many other related bioactive heterocycles such as diptoindonesin G (Scheme 18).38
Scheme 18:
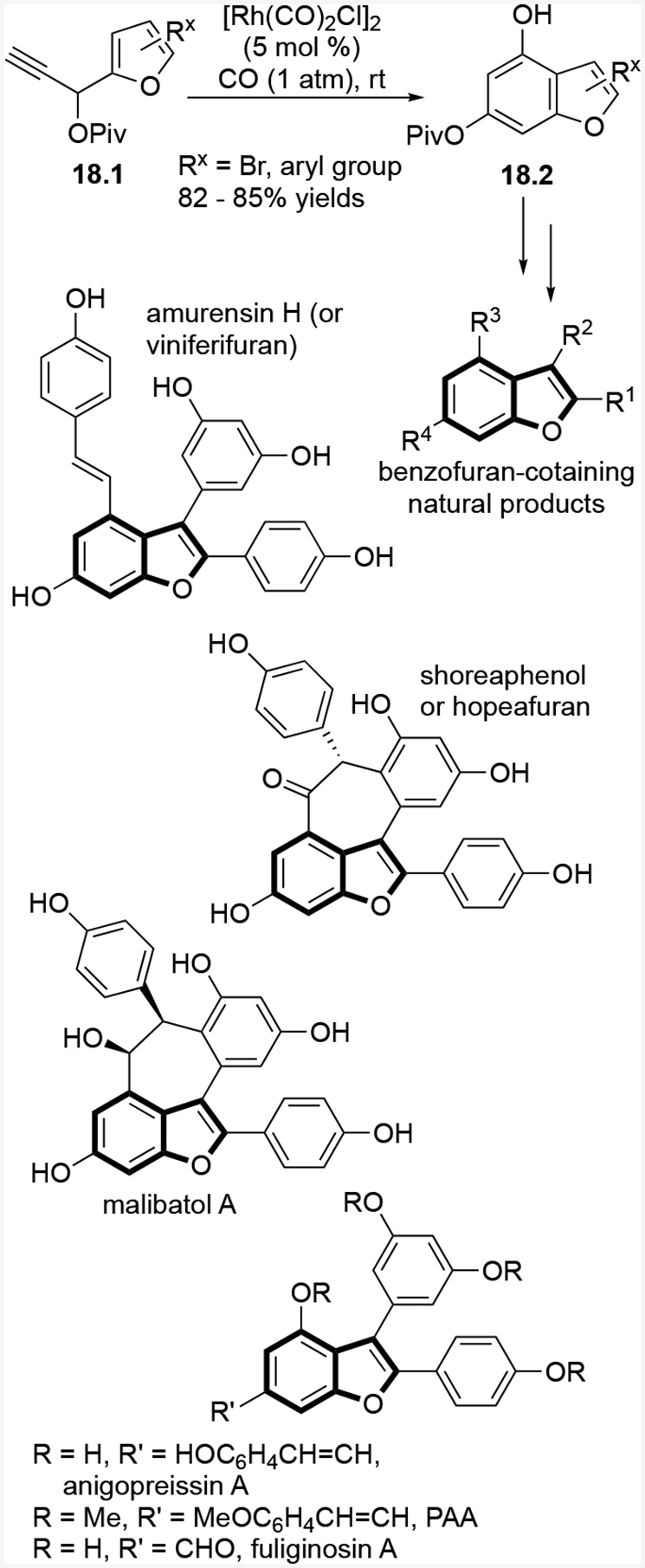
Selected benzofuran-containing natural products synthesized
3.3. 3-Hydroxy-1,4-Enynes and Diaryl Propargylic Alcohols in (5 + 1) Cycloadditions
We then developed a second five-carbon synthon for (5+1) cycloadditions as shown in Scheme 19. We predicted a novel bicyclic rhodacycle intermediate would result if we replaced the acyloxy group in an ACE with a leaving group X and nucleophile Y, as seen in 1,4-enyne 19.4. Through transition state 19.5, the resulting novel rhodacycles 19.6 could result in new cycloadditions.
Scheme 19:
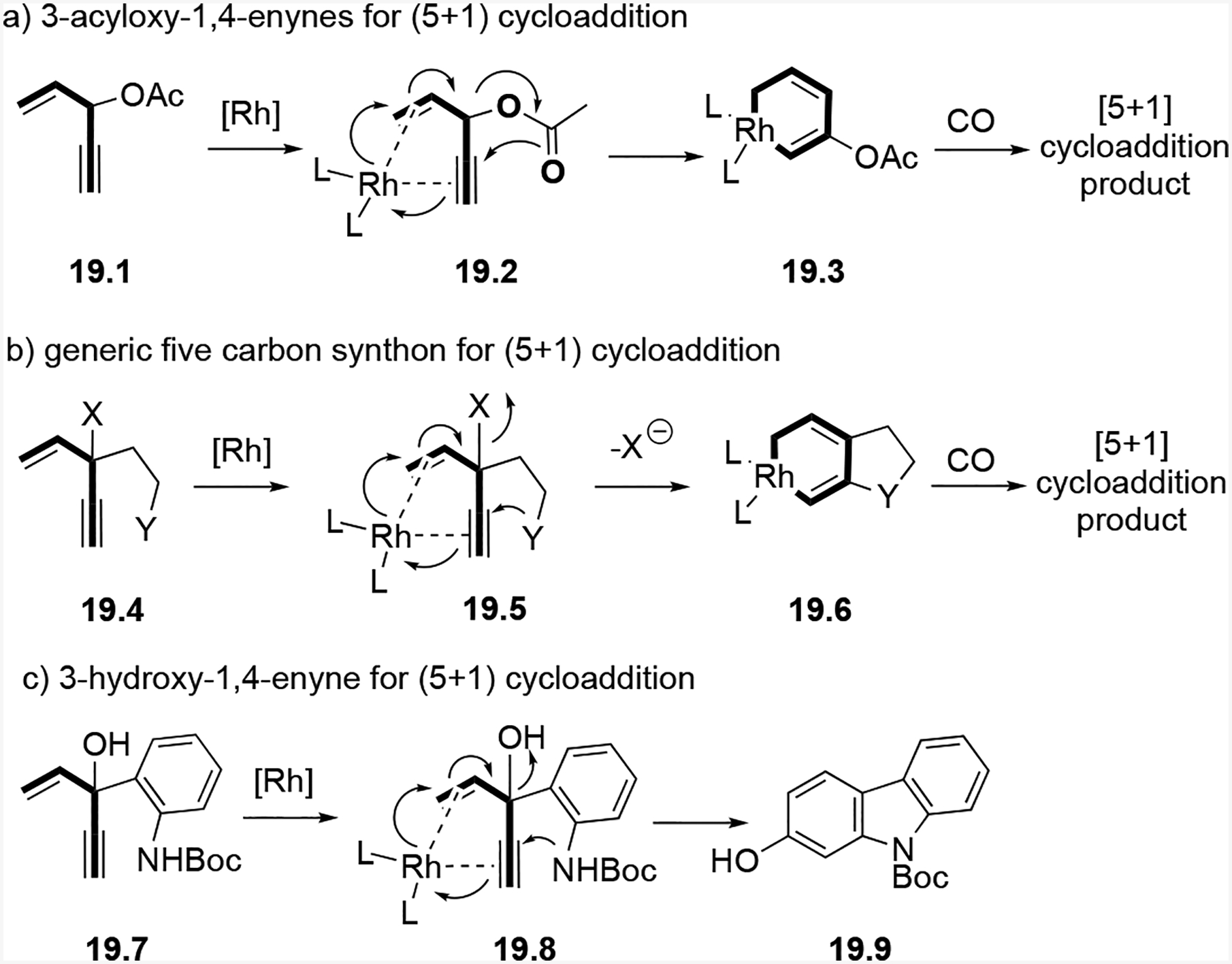
Comparison of five-carbon synthons developed for (5+1) cycloadditions
From commercially available 2-aminobenzoic acid, 3-hydroxy-1,4-enynes (HYEs) 19.7 in Scheme 19 or 20.1 in Scheme 20 were synthesized in four steps to test this hypothesis.39 Mechanistically, HYEs were designed so the aniline nitrogen could serve as the nucleophile and –OH could serve as the leaving group. Using HYEs, we developed a novel method to make substituted carbazoles and carbazoles fused with various heterocycles.
Scheme 20:

Scope of 3-hydroxy-1,4-enynes (HYEs) for the synthesis of polycyclic ring systems
Various Rh(I) catalysts were screened with substrate 19.7 in the presence of CO. The Wilkinson’s catalyst and cationic Rh(I) catalysts provided complex mixtures or no desired product. [Rh(COD)Cl]2 and [Rh(CO)2Cl]2 under one atmosphere of CO led to good yields of 19.9, 81% and 88%, respectively. The CO pressure affected product yields as well, and one atmosphere of CO was optimal. The alkene termini could accommodate alkyl, cyclopropyl, and aryl groups. Bromine, methoxy, and trifluoromethyl substituents on the aromatic ring were all tolerated, and the electronic nature of these substituents did not appear to affect the tandem annulation [5+1] cycloaddition (Scheme 20). However, an internal alkyne did substantially decrease the final product yield. We were pleased to find that phenols could also be used as the nucleophile in these reactions.
We proposed two plausible reaction mechanisms in Scheme 21. From 21.1, via pathway A, a concerted annulation and oxidative cyclization with the loss of water forms 21.3. Insertion of CO and subsequent reductive elimination leads to 21.4. Aromatization then yields carbazole 21.5. Alternatively, the Rh-catalyst could coordinate to the substrate to form 21.6, and then the aniline nitrogen could perform a nucleophilic attack on the alkyne in 21.6 to form 21.7. The loss of water from 21.7 then results in Rh(I) carbene 21.8. From here, a 6π electrocyclization can form 21.3, and CO insertion would go through 21.4 to form 21.5. Otherwise from 21.8, CO insertion could form ketene 21.9, and 6π electrocyclization of this compound would eventually lead to 21.5. The isolation of by-product 21.14 could be explained through ketene 21.11, but this by-product cannot rule out the possibility of either pathway A or pathway B, since the two pathways can be connected by the interconversion between 21.3 and 21.8.
Scheme 21:
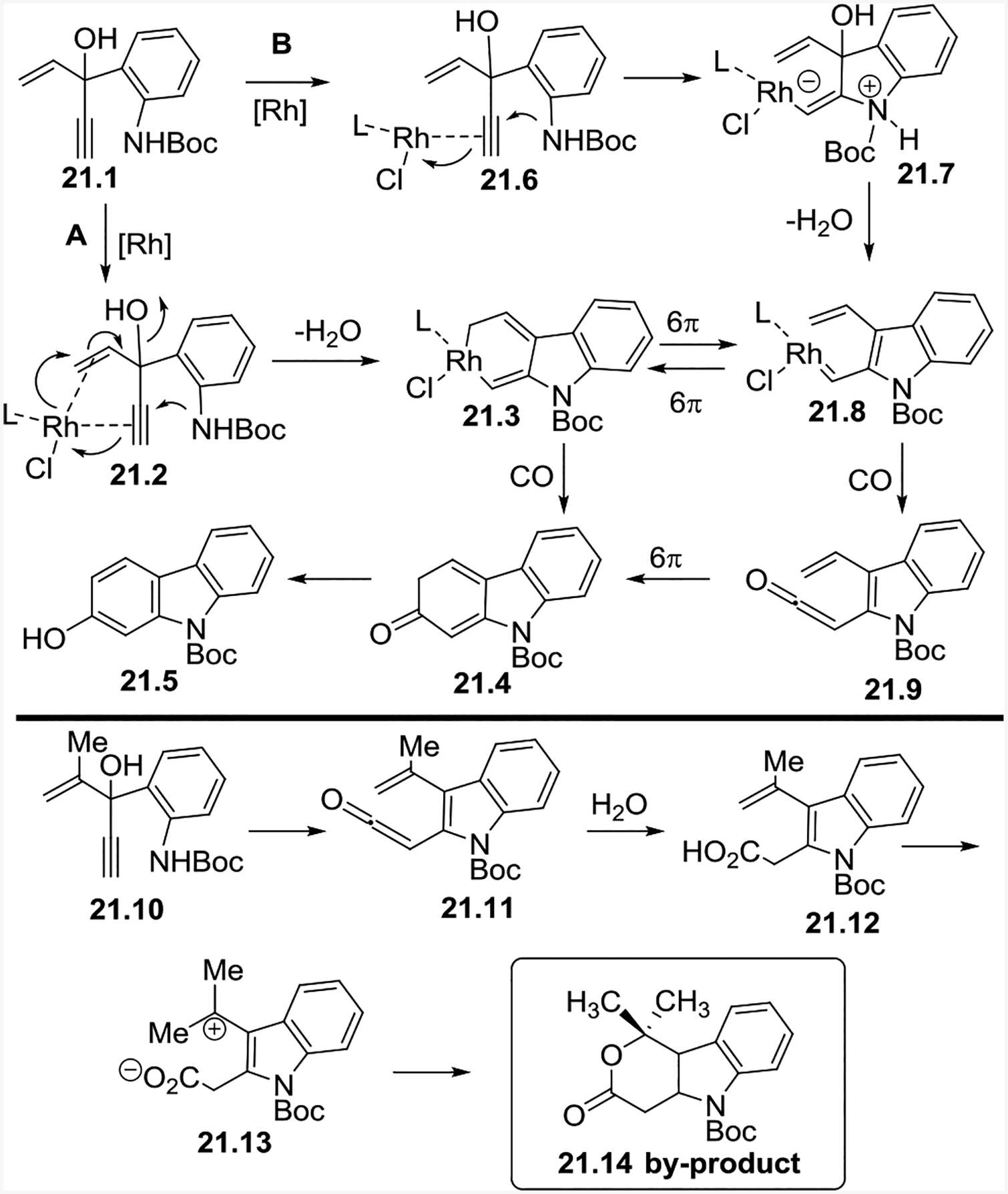
Proposed reaction mechanisms for the tandem annulation (5+1) cycloaddition with HYEs and CO
As opposed to traditional cycloadditions (where two bonds are formed), this reaction formed an additional C-N or C-O bond in conjunction with two other C-C bonds. To demonstrate the power of this method for the synthesis of polycyclic compounds, it was used to synthesize the natural products glycosinine and mahanimbine and achieve the formal synthesis of mukoenine-A and heptaphylline (Scheme 22).32,40
Scheme 22:
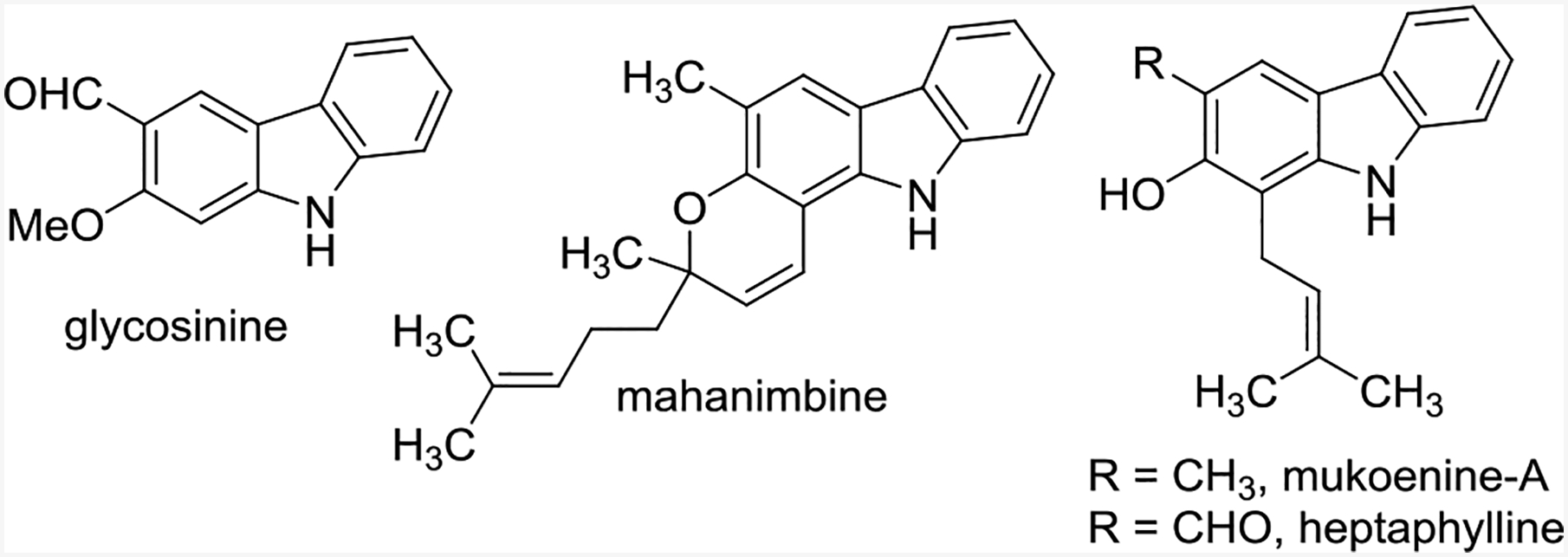
Natural products with carbazole cores synthesized via tandem annulation (5+1) cycloadditions
After employing HYEs with a Boc-activated aniline nucleophile in (5 + 1) cycloadditions, we altered our five-carbon component by replacing the Boc-aniline nucleophile with a sulfonamide (Scheme 23). Boc-substituted compounds don’t react well with certain two-carbon units, such as furans, hence we replaced the aniline nucleophile with a sulfonamide. The reaction tolerated numerous substituents on the alkene termini and aniline ring, and it only appeared to be hampered when substituents were simultaneously present on the alkene and alkyne.
Scheme 23:
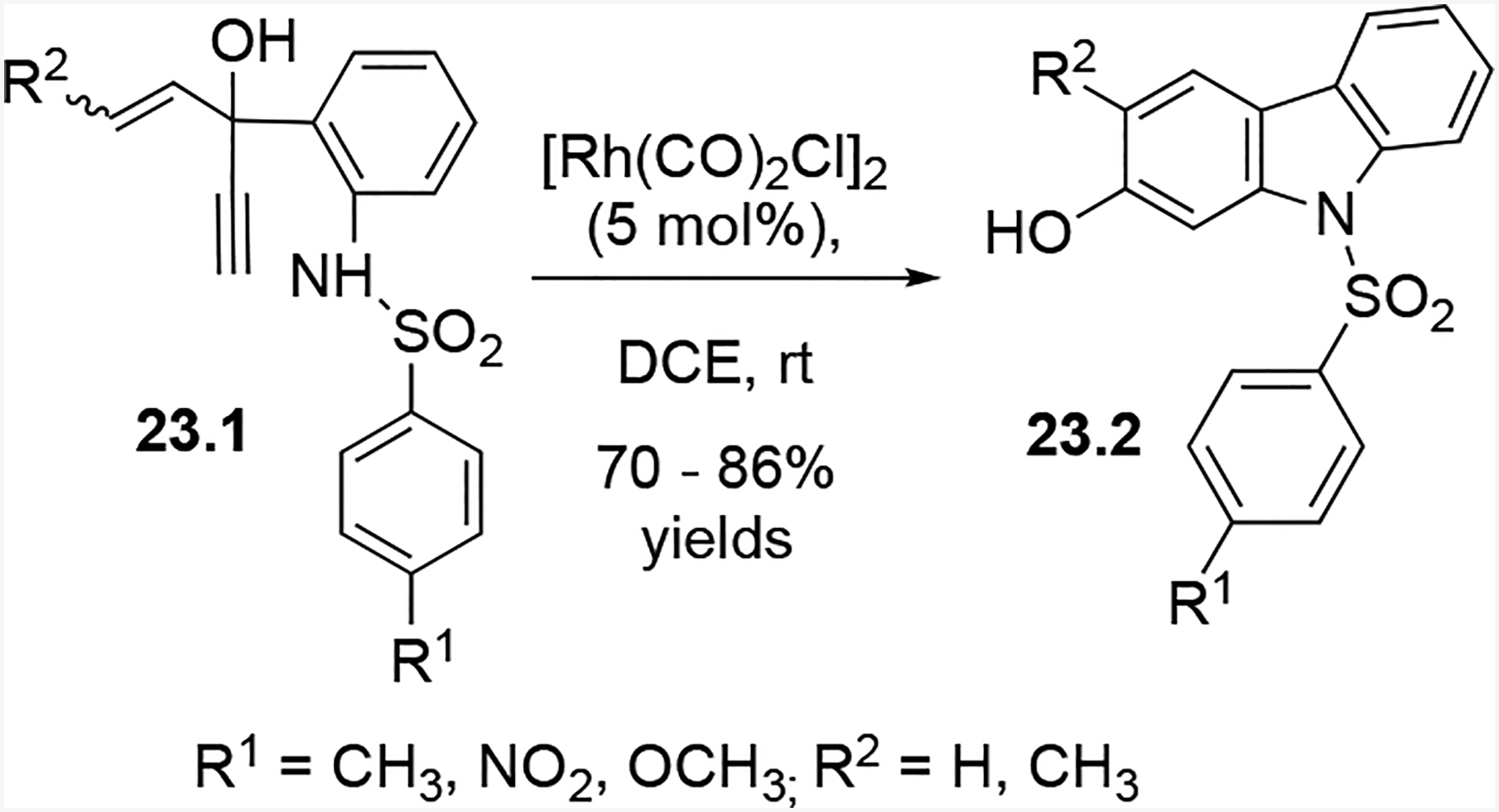
HYEs with sulfonamide nucleophiles
We also prepared a wide variety of substrates to test the ability of the tandem annulation (5+1) cycloaddition to prepare tetracyclic and even pentacyclic heterocycles fused with a carbazole Scheme 24).32,33
Scheme 24:
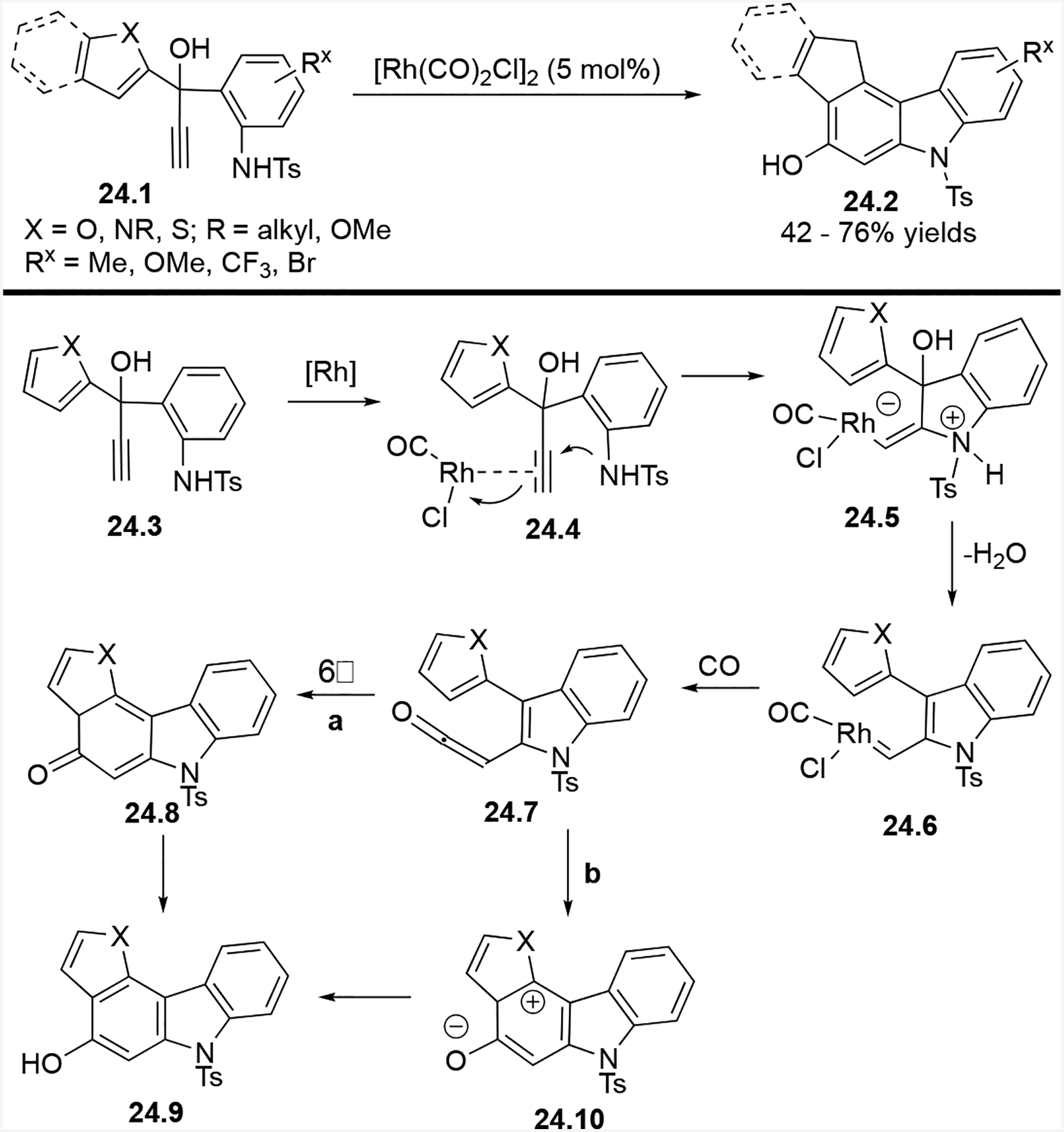
Scope and mechanism of tandem annulation (5+1) cycloaddition for the synthesis of polycyclic heterocycles
To form polycyclic heterocycles, the proposed mechanism begins with Rh(I) coordination to diaryl propargylic alcohol 24.3 to form 24.4. Then nucleophilic addition of the nitrogen to the alkyne, followed by construction of the C–Rh bond in 24.5 occurs. Loss of a water molecule results in Rh(I) carbene 24.6. Insertion of CO and displacement of rhodium forms ketene 24.7. A 6π electrocyclization forms 24.8 (pathway a), and re-aromatization of both five- and six-membered rings yields final product 24.9. Alternatively, Friedel-Crafts acylation followed by re-aromatization event can also provide final product 24.9 through intermediate 24.10 (pathway b).
The de novo synthesis of benzene rings continues to be a challenge for organic chemists. Of the few methods currently available, only a limited number are general enough to be extended to the synthesis of polycyclic heterocycles. The use of diaryl propargylic alcohols is a general, environmentally benign and atom-economical way to synthesize benzene rings that doesn’t rely on stoichiometric amounts of toxic Fischer carbenes used in other methods.36 Using this Rh(I)-catalyzed tandem reaction, the efficient synthesis of highly substituted carbazoles, furocarbazoles, pyrrolocarbazoles, indolocarbazoles, and thiophenocarbazoles can be achieved.
Conclusion
Transition-metal catalysis, specifically rhodium, has been shown to be an invaluable tool for the construction of complex ring systems. In addition, the design of novel five-carbon synthons from relatively simple starting materials has propelled the development of (5+n) cycloadditions. These cycloadditions largely share the common mechanistic features of rhodium coordination, 1,2-migration of an acyl group in propargylic esters or an ionization of hydroxyl group in propargylic alcohols, oxidative cyclization, and reductive elimination to form ring products that are often found in pharmaceutical agents and natural products. Although the occurrence of transition-metal catalyzed acyloxy migrations has been known for decades, only recently has their synthetic value in cycloadditions been realized.
The (5+n) cycloadditions we have developed so far do not have heteroatoms in the 5- or n-atom components. One of the future directions for this work is the introduction of heteroatoms to either of these two components for the preparation of diverse heterocycles. It is our hope that the work previously described, which uses readily available 1,4-enynes as the 5-carbon components in (5+n) cycloadditions, can be extended to other two-component and multi-component cycloadditions.
ACKNOWLEDGMENT
We would like to thank Dr. Peng Wen and Bethany J. McCarty for carefully proofreading the manuscript and thoughtful discussions. We also want to thank all coworkers involved in this research, particularly Dr. Xingzhong Shu, Dr. Xiaoxun Li, Dr. Wangze Song and Ms. Casi M. Schienebeck.
Funding Sources
We would like to thank the University of Wisconsin-Madison and NSF (CHE-1464754) for financial support. S.A.B. was supported in part by the NIH Chemistry-Biology Interface Training Grant (T32GM008505) and the NSF Graduate Research Fellowship.
Biographies
Biographies
Stephanie A. Blaszczyk obtained her B.S. degree in Chemistry from Rockford University after working with Professor Matthew Bork. Following graduation, she worked for Geneva Laboratories and Tate & Lyle. Now a Ph. D. candidate at the University of Wisconsin-Madison, Stephanie develops new synthetic methods under professor Weiping Tang. She is also actively involved in science communication, most recently serving as a 2019 AAAS Mass Media Science & Engineering Fellow at the Milwaukee Journal Sentinel.
Daniel A. Glazier obtained his B.S. in chemistry from the University of Pittsburgh in 2014 after working with Paul Floreancig. He then enrolled as a Ph.D. student at the University of Wisconsin-Madison and joined the Tang Lab. He develops new synthetic methods and also uses DFT calculations to study the mechanism of these reactions. After receiving his PhD, he joined Arrowhead Pharmaceuticals.
Weiping Tang received his B.S. degree from Peking University, M.S. degree from New York University, and Ph.D. degree from Stanford University. He was a HHMI postdoctoral fellow at Harvard University. He is currently a professor in the School of Pharmacy and Department of Chemistry at the University of Wisconsin-Madison. His group is interested in developing new synthetic methods, medicinal chemistry and chemical biology.
Footnotes
The authors declare no competing interest.
REFERENCES
- 1.(a) Trost BM The atom economy--a search for synthetic efficiency. Science 1991, 254, 1471–1477. [DOI] [PubMed] [Google Scholar]; (b) Trost BM Atom Economy—A Challenge for Organic Synthesis: Homogeneous Catalysis Leads the Way. Angew. Chem. Int. Ed 1995, 34, 259–281. [Google Scholar]; (c) Wender PA; Verma VA; Paxton TJ; Pillow TH Function-Oriented Synthesis, Step Economy, and Drug Design. Acc. Chem. Res 2008, 41, 40–29. [DOI] [PubMed] [Google Scholar]; (d) Wender PA; Miller BL Synthesis at the molecule frontier. Nature 2009, 460, 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Lautens M; Klute W; Tam W Transition Metal-Mediated Cycloaddition Reactions. Chem. Rev 1996, 96, 49–92. [DOI] [PubMed] [Google Scholar]; (b) Ojima I; Tzamarioudaki M; Li Z; Donovan RJ Transition Metal-Catalyzed Carbocyclications in Organic Synthesis. Chem Rev. 1996, 96, 635–662. [DOI] [PubMed] [Google Scholar]
- 3.For reviews on seven-membered ring synthesis, see: Battiste MA; Pelphrey PM; Wright DL The Cycloaddition Strategy for the Synthesis of Natural Products Containing Carbocyclic Seven-Membered Rings. Chem. Eur. J 2006, 12, 3438–3447. [DOI] [PubMed] [Google Scholar]; Butenschön H Seven-membered rings by cyclization at transition metals: [4+3], [3+2+2], [5+2]. Angew. Chem. Int. Ed 2008, 47, 5287–5290. [DOI] [PubMed] [Google Scholar]; (c) Nguyen TV; Hartmann JM; Enders D Recent synthetic strategies to access seven-membered carbocycles in natural product synthesis. Synthesis. 2013, 45, 845–873. [Google Scholar]
- 4.Wender PA; Deschamps NM; Sun R Rh(I)-catalyzed C-C bond activation: seven-membered ring synthesis by a [6+1] carbonylative ring-expansion reaction of allenylcyclobutanes. Angew. Chem. Int. Ed 2006, 45, 3957–3960. [DOI] [PubMed] [Google Scholar]
- 5.(a) Selected reviews on (5+2) cycloadditions include:Singh V; Krishna UM; Vikrant; Trivedi GK Cycloaddition of oxidopyrylium species in organic synthesis. Tetrahedron 2008, 64, 405–3428. [Google Scholar]; (b) Pellissier H Recent Developments in the [5+2] Cycloaddition. Adv. Synth. Catal 2011, 353, 189–218. [Google Scholar]; (c) Ylijoki KEO; Stryker JM [5+2] Cycloaddition Reactions in Organic and Natural Product Synthesis. Chem. Rev 2013, 113, 2244–2266. [DOI] [PubMed] [Google Scholar]
- 6.(a) Selected reviews on (4+3) cycloadditions include:Harmata M The [4+3]-cycloaddition reaction: simple allylic cations as dienophiles. Chem. Commun 2010, 46, 8886–8903. [DOI] [PubMed] [Google Scholar]; (b) Harmata M The [4+3]-cycloaddition reaction: heteroatom-substituted allylic cations as dienophiles. Chem. Commun 2010, 46, 8904–8922. [DOI] [PubMed] [Google Scholar]; (c) Lohse AG; Hsung RP [4+3] Cycloaddition Reactions of Nitrogen-Stabilized Oxyallyl Cations. Chem. Eur. J 2011, 17, 3812–3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Wender PA; Takahashi H; Witulski B Transition Metal Catalyzed [5 + 2] Cycloadditions of Vinylcyclopropanes and Alkynes: A Homolog of the Diels-Alder Reaction for the Synthesis of Seven-Membered Rings. J. Am. Chem. Soc 1995, 117, 4720–4721. [Google Scholar]; (b) Wender PA; Husfeld CO; Langkopf E; Love JA First Studies of the Transition Metal-Catalyzed [5+2] Cycloadditions of Alkenes and Vinylcyclopropanes: Scope and Stereochemistry. J. Am. Chem. Soc 1998, 120, 1940–1941. [Google Scholar]
- 8.(a) Wender PA; Rieck H; Fuji M The Transition Metal-Catalyzed Intermolecular [5+2] Cycloaddition: The Homologous Diels−Alder Reaction. J. Am. Chem. Soc 1998, 120, 10976–10977. [Google Scholar]; (b) Yu Z.-x.; Wender PA; Houk KN On the Mechanism of [Rh(CO)2Cl]2-Catalyzed Intermolecular (5 + 2) Reactions between Vinylcyclopropanes and Alkynes. J. Am. Chem. Soc 2004, 126, 9154–9155. [DOI] [PubMed] [Google Scholar]
- 9.(a) Trost BM; Toste FD; Shen H Ruthenium-catalyzed intramolecular (5+2) cycloadditions. J. Am. Chem. Soc 2000, 122 (10), 2379–2380. [Google Scholar]; (b) Trost BM; Shen H Constructing tricyclic compounds containing a seven-membered ring by ruthernium-catalyzed intramolecular (5+2) cycloaddition. Angew. Chem. Int. Ed 2001, 40 (12), 2313–2316. [DOI] [PubMed] [Google Scholar]
- 10.Zuo G; Louie J Selectivity in nickel-catalyzed rearrangements of cyclopropenyl-ynes. J. Am. Chem. Soc 2005, 127 (16), 5798–5799. [DOI] [PubMed] [Google Scholar]
- 11.Fürstner A; Majima K; Martin R; Krause H; Kattnig E; Goddard R; Lehmann CW A cheap metal for a “noble” task: preparative and mechanistic aspects of cycloisomerization and cycloaddition reactions catalyzed by low-valent iron complexes. J. Am. Chem. Soc 2008, 130 (6), 1992–2004. [DOI] [PubMed] [Google Scholar]
- 12.Wender PA; Husfeld CO; Langkopf E; Love JA First studies of the transition metal-catalyzed (5+2) cycloadditions of alkenes and vinylcyclopropanes: scope and stereochemistry. J. Am. Chem. Soc 1998, 120 (8), 1940–1941. [Google Scholar]
- 13.Wender PA; Glorius F; Husfeld CO; Langkopf E; Love JA Transition metal-catalyzed (5+2) cycloadditions of allenes and vinylcyclopropanes: first studies of endo-exo selectivity, chemoselectivity, relative stereochemistry, and chirality transfer. J. Am. Chem. Soc 1999, 121 (22), 5348–5349. [Google Scholar]
- 14.(a) Wender PA; Husfeld CO; Langkopf E; Love JA; Pleuss N The first metal-catalyzed intramolecular (5+2) cycloadditions of alkenes and vinylcyclopropanes: scope, stereochemistry, and asymmetric catalysis. Tetrahedron 1998, 54, 7203–7220.; [Google Scholar]; (b) Wender PA; Haustedt LO; Lim J; Love JA; Williams TJ; Yoon JY Asymmetric Catalysis of the [5 + 2] Cycloaddition Reaction of Vinylcyclopropanes and Pi-Systems. J. Am. Chem. Soc 2006,128, 5354–5355. [DOI] [PubMed] [Google Scholar]
- 15.Shintani R; Nakatsu H; Takatsu K; Hayashi T Rhodium-catalyzed asymmetric (5+2) cycloaddition of alkyne-vinylcyclopropanes. Chem. Eur. J 2009, 15 (35), 8692–8694. [DOI] [PubMed] [Google Scholar]
- 16.Rautenstrauch V 2-Cyclopentenones from 1-ethynyl-2-propenyl acetates. J. Org. Chem 1984, 49, 950–952. [Google Scholar]
- 17.Shu X.-z.; Huang S; Shu D; Guzei IA; Tang W Interception of a Rautenstrauch Intermediate by Alkynes for [5+2] Cycloaddition: Rhodium-Catalyzed Cycloisomerization of 3-Acyloxy-4-ene-1,9-diynes to Bicyclo[5.3.0]decatrienes. Angew. Chem. Int. Ed 2011, 50, 8153–8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.For a recent review on natural products with bicyclo[5.3.0]decane skeletons, see: Foley DA; Maguire AR Synthetic approaches to bicyclo[5.3.0]decane sesquiterpenes. Tetrahedron 2010, 66, 1131. [Google Scholar]
- 19.(a) For recent reviews on cycloheptatriene-containing compounds, see:Zhao J Plant troponoids: chemistry, biological activity, and biosynthesis. Curr. Med. Chem 2007, 14, 2597–262. [DOI] [PubMed] [Google Scholar]; (b) Bentley R A fresh look at natural tropolonoids. Nat. Prod. Rep 2008, 25, 118–138. [DOI] [PubMed] [Google Scholar]
- 20.Song W; Xi B.-m.; Yang K; Tang W Synthesis of naturally occurring tropones and tropolones. Tetrahedron 2015, 71, 5979–5984.26456984 [Google Scholar]
- 21.Shu X-Z; Schienebeck CM; Song W; Guzei IA; Tang W Transfer of Chirality in the Rhodium-Catalyzed Intramolecular [5+2] Cycloaddition of 3-Acyloxy-1,4-Enynes (ACEs) and Alkynes: Synthesis of Enantioenriched Bicyclo[5.3.0]decatrienes. Angew. Chem. Int. Ed 2013, 52, 13601–13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu X; Lui P; Shu X-Z; Tang W; Houk KN Rh-Catalyzed (5+2) Cycloadditions of 3-Acyloxy-1,4-enynes and Alkynes: Computational Study of Mechanism, Reactivity, and Regioselectivity. J. Am. Chem. Soc 2013, 135, 9271–9274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shu X-Z; Schienebeck CM; Li X; Zhou X; Song W; Chen L; Guzei IA; Tang W Rhodium-Catalyzed Stereoselective Intramolecular [5+2] Cycloaddition of 3-Acyloxy 1,4-Enyne and Alkene. Org. Lett 2015, 17, 5128–5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song W; Lynch JC; Shu X.-z.; and Tang W Rhodium-catalyzed [5+2] Cycloaddition of 3-Acyloxy-1,4-enyne and Alkene or Allene. Adv. Syn. Catal 2016, 358, 2007–2011. [Google Scholar]
- 25.(a) Wang Y; Wang J; Su JC; Huang F; Jiao L; Liang Y; Yang D; Zhang S; Wender PA; Yu Z-X A Computationally Design Rh(I)-Catalyzed Two-Component [5 + 2 + 1] Cycloaddition of Ene-Vinylcyclopropanes and CO for the Synthesis of Cyclooctenones. J. Am. Chem. Soc 2007, 129, 10060–10061. [DOI] [PubMed] [Google Scholar]; (b) Jiao L; Yuan C; Yu Z-X Tandem Rh(I)-Catalyzed [(5+2)+1] Cycloaddition/Aldol Reaction for the Construction of Linear Triquinane Skeleton: Total Syntheses of (()-Hirsutene and (()-1-Desoxyhypnophilin. J. Am. Chem. Soc 2008, 130, 4421–4430. [DOI] [PubMed] [Google Scholar]
- 26.Li X; Song W; Zhao X.-l.; Ke X; Xu X; Liu P; Houk KN; Tang W Rhodium(I)-Catalyzed Benzannulation of Heteroaryl Propargylic Esters: Synthesis of Indoles and Related Heterocycles. Chem. Eur. J 2016, 22, 7079–7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.(a) For a recent review, see:Shu X-Z; Shu D; Schienebeck CM; Tang W Rhodium-catalyzed acyloxy migration of propargylic esters in cycloadditions, inspiration from the recent “gold rush”. Chem. Soc. Rev 2012, 41, 7698–7711. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) For selected examples, see:Shu D; Li X; Zhang M; Robichaux PJ; Tang W Synthesis of Highly Functionalized Cyclohexenone Rings: Rhodium-Catalyzed 1,3-Acyloxy Migration and Subsequent [5+1] Cycloaddition. Angew. Chem. Int. Ed 2011, 50, 1346–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li X; Zhang M; Shu D; Robichaux PJ; Huang S; Tang W Rhodium-catalyzed Ring Expansion of Cyclopropanes to Seven-membered Rings by 1,5-C-C Bond Migration. Angew. Chem. Int. Ed 2011, 50, 10421–10424. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Li X; Huang S; Schienebeck CM; Shu D; Tang W Rhodium-Catalyzed Carbonylation of 3-Acyloxy-1,4-enynes for the Synthesis of Cyclopentenones. Org. Lett 2012, 14, 1584–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prasad BAB; Yoshimoto FK; Sarpong R Pt-Catalyzed Pentannulations from In Situ Generated Metallo−Carbenoids Utilizing Propargylic Esters. J. Am. Chem. Soc 2005, 127, 12468–12469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.(a) Shibata Y; Noguchi K; Tanaka K Cationic Rhodium(I) Complex-Catalyzed [3 + 2] and [2 + 1] Cycloadditions of Propargyl Esters with Electron-Deficient Alkynes and Alkenes. J. Am. Chem. Soc 2010, 132, 7896–7898. [DOI] [PubMed] [Google Scholar]; (b) Shibata Y; Noguchi K; Tanaka K Cationic Rhodium(I) Complex-Catalyzed Cotrimerization of Propargyl Esters and Arylacetylenes Leading to Substituted Dihydropentalenes. Org. Lett 2010, 12, 5596–5599. [DOI] [PubMed] [Google Scholar]
- 30.Shu X.-z; Li X; Shu D; Huang S; Schienebeck CM; Zhou X; Robichaux PJ; Tang W J. Am. Chem. Soc 2012, 134, 5211–5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu P; Houk KN Theoretical studies of regioselectivity of Ni-and Rh-catalyzed C–C bond forming reactions with unsymmetrical alkynes. Inorg. Chim. Acta 2011, 369, 2–14. [Google Scholar]
- 32.Schienebeck CM; Robichaux PJ; Li X; Chen L; Tang W Effect of Ester on Rhodium-Catalyzed Intermolecular [5 + 2] Cycloaddition of 3-Acyloxy-1,4-enynes and Alkynes. Chem. Commun 2013, 49, 2616–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.(a) Brancour C; Fukuyama T; Ohta Y; Ryu I; Dhimane AL; Fensterbank L; Malacria M Synthesis of functionalized resorcinols by rhodium-catalyzed [5+1] cycloaddition reaction of 3-acyloxy-1,4-enynes with CO. Chem. Commun 2010, 46, 5470–5472. [DOI] [PubMed] [Google Scholar]; (b) Fukuyama Y; Ohta Y; Brancour C; Miyagawa K; Ryu I; Dhimane A-L; Fensterbank L; Malacria M Rh‐Catalyzed [5+1] and [4+1] Cycloaddition Reactions of 1,4‐Enyne Esters with CO: A Shortcut to Functionalized Resorcinols and Cyclopentenones. Chem. Eur. J 2012, 18, 7243–7247. [DOI] [PubMed] [Google Scholar]
- 34.Schienebeck CM; Robichaux PJ; Li X; Chen L and Tang W Effect of Ester on Rhodium-Catalyzed Intermolecular [5 + 2] Cycloaddition of 3-Acyloxy-1,4-enynes and Alkynes. Chem. Commun, 2013, 49, 2616–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schienebeck CM; Song W; Smits AM; Tang W Rhodium-Catalyzed Intermolecular [5+1] and [5+2] Cycloadditions Using 1,4-Enynes with an Electron-Donating Ester on the 3-Position. Synthesis, 2015, 47, 1076–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song W; Blaszczyk SA; Liu J; Wang S; Tang W Transition metal mediated carbonylative benzannulations. Org. Biomol. Chem 2017, 15, 7490–7504. [DOI] [PubMed] [Google Scholar]
- 37.Li X; Xie H; Fu X; Liu J.-t.; Wang H.-y.; Xi B.-m.; Liu P; Xu X; Tang W Rhodium(I)-Catalyzed Benzannulation of Heteroaryl Propargylic Esters: Synthesis of Indoles and Related Heterocycles. Chem. Eur. J 2016, 22, 10410–10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu JT; Do TJ; Simmons CJ; Lynch JC; Gu W; Ma ZX; Xu W; Tang W Total Synthesis of Diptoindonesin G and Its Analogues as Selective Modulators of Estrogen Receptors. Org. Biomol. Chem 2016, 14, 8927–8930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X; Song W; Tang W Rhodium-Catalyzed Tandem Annulation and (5+1) Cycloaddition: 3-Hydroxy-1,4-enyne as the 5-Carbon Component. J. Am. Chem. Soc 2013, 135, 16797–16800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song W; Li X; Yang K; Zhao X-L; Glazier DA; Xi B-M; Tang W Synthesis of Carbazoles and Carbazole-Containing Heterocycles via Rhodium-Catalyzed Tandem Carbonylative Benzannulations. J. Org. Chem 2016, 81, 2930–2942. [DOI] [PubMed] [Google Scholar]