

Abstract
Chinese hamster ovary (CHO) cells are the preferred workhorse for the biopharmaceutical industry, and CRISPR/Cas9 has proven powerful for generating targeted gene perturbations in CHO cells. Here we expand the CRISPR engineering toolbox with CRISPR activation (CRISPRa) to increase transcription of endogenous genes. We successfully increased transcription of Mgat3 and St6gal1, and verified on a functional level by subsequently detecting that the appropriate glycan structures were produced. This work demonstrates CRISPRa can make targeted alterations of CHO cells for desired phenotype.
Keywords: CRISPRa, CHO, glycosylation, Mgat3, St6gal1
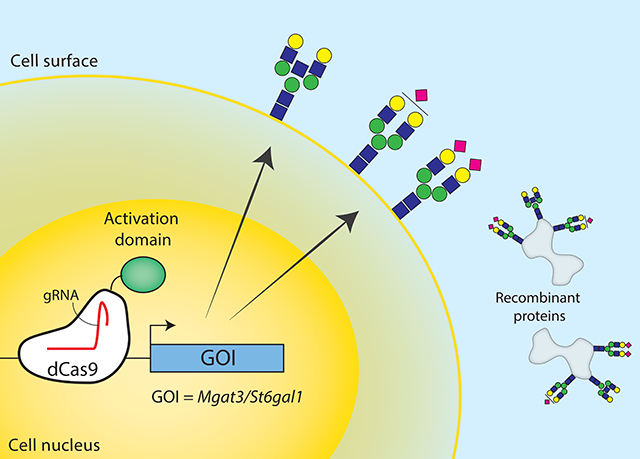
Graphical Abstract
Mammalian genomes encode over 20,000 genes but only a fraction of these are expressed in any given tissue (Uhlén et al., 2015). This is true for Chinese hamster ovary (CHO) cells as well; across the course of culture, nearly half of all genes are never expressed in CHO-S (Hefzi et al., 2016). For recombinant therapeutic protein productions, CHO cells are the most widely used expression host. Despite the ubiquity of CHO cells, the resulting recombinant proteins are not perfect matches to their native human form, in part due to post-translational modifications, such as differences in glycosylation (Amann et al., 2018). For example, the bisecting N-acetylglucosamine attached by Mgat3 (Beta-1,4-mannosyl-glycoprotein 4-beta-N-acetylglucosaminyltransferase) and the alpha 2,6 sialic acid attached by St6gal1 (Beta-galactoside alpha-2,6-sialyltransferase 1) are common on many human glycoproteins (Amann et al., 2018; Popp et al., 2018), but missing from proteins expressed in CHO cells. While the lack of these epitopes has not led to immunogenic responses (Butler & Spearman, 2014), these human epitopes may impact drug activity (Chiang et al., 2016; Reusch & Tejada, 2015; Tian et al., 2019). Fortunately, recent advances in gene editing tools and genomic understanding have increased our ability to manipulate CHO cells to overcome some of these shortcomings.
The CRISPR/Cas9 system enabled rapid and cost-effective genome editing and works efficiently for generating CHO knockout cell lines (Amann et al., 2018; Grav, la Cour Karottki, Lee, & Kildegaard, 2017; Grav et al., 2015). It is not limited to gene disruption; by mutating the nuclease domains of the native Cas9 endonuclease to a catalytically inactive Cas9--referred to as dead Cas9 (dCas9)--and fusing it with various domains, the system has been repurposed to deliver effector molecules to sites of interest. For example, dCas9 can be fused to transcription activators, a tool known as CRISPR activation (CRISPRa), and guided to a region upstream of the transcription start site (TSS). This can increase transcription of endogenous genes across diverse eukaryotic organisms (Chavez et al., 2016; Gilbert et al., 2013; Lin, Ewen-Campen, Ni, Housden, & Perrimon, 2015). Similarly, fusion to TET can be used to demethylate promoter regions, reversing silencing of genes (Marx et al., 2018). Here we demonstrate that the CRISPRa system can be implemented in CHO cells. To demonstrate this, we targeted two silenced glycosyltransferases, Mgat3 and St6gal1, which are not expressed in these lines and their expression would enable the synthesis of more human-like glycan structures.
A central factor for successful activation using CRISPRa is the gRNA design. As comprehensive experimental determination of TSSs in CHO has not been published (e.g., with GRO-cap or CAGE methods), each TSS was defined to be the first base pair of the target gene using the CHO-K1 annotation available on NCBI. The gRNAs were designed to target close to the NCBI TSS locations, consistent with suggested distances from the TSS that previously lead to the highest CRISPRa activity (25 to 550 bp upstream) in other mammalian cells (Gilbert et al., 2014; Horlbeck et al., 2016) (Figure 1A). We selected two genes relevant to achieving human-like glycosylation (Mgat3 and St6gal1) that are not expressed in CHO-S transcriptomic data (Hefzi et al., 2016) (Supplementary Table S1). VPR was used as the activator construct--consisting of three activation domains VP64, p65 and rta--since it is the simplest construct that showed high levels of activation in previous studies (Chavez et al., 2016) (Figure 1B). Finally, Mgat3 and St6gal1 activation was carried out in an engineered cell line designed to produce primarily bianntenary, nonsialylated, nonfucosylated glycans to simplify the detection of altered glycans (see Methods).
Figure 1. Components for CRISPRa.

A) gRNAs were designed using the CHO-K1 annotation from NCBI. Since experimentally reported TSSs have not been reported for CHO cells, the TSS was defined as the starting base pair of the annotated target genes. Three gRNAs (red rectangles) were designed between 25 bp and 550 bp upstream of the TSS. gRNA positions for each gene targeted in this study are given in Supplementary Table S2. B) A plasmid encoding VPR-dCas9 was transfected along with either one of the gRNA plasmids or a mix of all gRNA plasmids. The gRNAs guide the VPR-dCas9 to a region upstream of the TSS where VPR increases expression of the target gene.
Using gRNAs designed to target these TSSs increased the transcription of both target genes (Figure 2). RNA expression levels for Mgat3 and St6gal1 were increased noticeably. There were clear differences between the individual gRNAs on expression level. For example, gRNA 1 against Mgat3 and gRNA 2 against St6gal1 failed to increase RNA expression. In both cases, transfecting with a mixture of the three gRNAs showed the most potent activation, corroborating findings in literature (Chavez et al., 2016; Cheng et al., 2013).
Figure 2. CRISPR activation of endogenous genes in CHO cells using gRNAs designed against the NCBI annotation.

Activation of Mgat3 and St6gal1 via transient expression of gRNA and VPR-dCas9 was measured by qRT-PCR. CHO cells were transfected with individual gRNAs 1–3, or a mix of the 3 gRNAs in three biological replicates. mRNA was harvested 48h after transfection. For controls, cells were transfected with non-targeting gRNA and VPR-dCas9 (NT-VPR-dCas9) or non-targeting gRNA (NT). Relative expression is calculated with respect to the NT samples. Error bars represent the 95% percentile confidence interval as calculated by the Relative Quantification module of Thermo Fisher Connect (see Methods). Relative expression of Mgat3 and St6gal1 with respect to only the housekeeping genes is shown in Supplementary Figure S2.
To confirm the transcriptional activation has a functional impact, we assayed and quantified the glycans on the secretome from cells targeted with the gRNA mix and the non-targeting (NT) gRNAs for Mgat3 and St6gal1 (see Methods). In the NT gRNA samples, we did not detect glycans with the epitopes associated with Mgat3 (bisecting GlcNAc) or St6gal1 (alpha-2,6 sialic acid) activity, whereas in gRNA mix samples, we found modest amounts of glycans containing bisecting GlcNAcs and alpha-2,6 sialylation (Figure 3). Thus, transcriptional activation with the CRISPRa system can revive silenced gene functions in CHO cells.
Figure 3. Glycan analysis of secreted proteins during CRISPR induced activation of Mgat3 and St6gal1.

Fraction of the total N-glycans on secreted proteins that contain bisecting GlcNAc (increased Mgat3 activity) or any number (mono- or bi-) or alpha-2,6 linked sialic acids (increased St6gal1 activity). Glycoform(s) of interest displayed above each bar. NT: non-targeting gRNA treated samples; ND: not detected. Error bars represent standard deviation (N=3).
Here we successfully implemented CRISPRa to expand the CRISPR toolbox for CHO cell line engineering increased expression of two silenced glycosyltransferases that are important for human-like glycosylation. This was achieved by transiently transfecting dCas9 fused with activator domain VPR and gRNAs targeting the region directly upstream of reported TSSs in the NCBI annotation. Consistent with reports when CRISPRa was applied to other species, the increase in transcription varied across individual gRNAs and transfecting all gRNAs showed the highest activation (e.g., up to 1500-fold for St6gal1). The activation of Mgat3 and St6gal1 was confirmed on a functional level by observing the more human-like glycan structures with LC-MS. Thus, CRISPRa provides another tool that will enable the study and engineering of improved CHO cell hosts for recombinant protein production. Future work will be essential to identify more effective CRISPRa gRNA designs, but this tool can be of immediate value by identifying genes that can be overexpressed for desired traits in CHO cells.
Among the various CRISPR-based tools, it has been more difficult to unravel the rules governing the efficacy of gRNAs for CRISPRa. Our experience was consistent with this, in that each gRNA showed vast differences in activation. Indeed, a third unexpressed gene targeted, Aldh18a1, showed limited to no activation by any of the gRNAs we tested (Supplementary Figure S1). It is expected that many factors can impact gRNA efficacy, including chromatin state (for dCas9 and gRNA accessibility), proximity to TSSs, or local sequence (Horlbeck et al., 2016; Sanson et al., 2018). There is a need for work wherein large panels of gRNAs are designed and tested to learn the rules governing effective gRNAs for CRISPRa in mammals in general. Until then, it is recommended that multiple gRNAs are designed for each gene of interest. This will be particularly important for CHO cells, since all TSSs are currently computationally predicted. Thus, it remains unclear where exactly gRNAs should be placed for efficient recruitment of the transcriptional machinery. Despite these challenges, we showed here that gRNAs can be effectively designed for CRISPRa.
Our work demonstrated that even silenced genes can be activated in CHO cells with CRISPRa. Thus, this tool will be particularly powerful as an inexpensive and rapid tool to identify which genes should be stably overexpressed to facilitate the production of difficult-to-produce proteins or to maintain desired phenotypes in the cells. For example, while it can be expensive to synthesize and overexpress a large number of genes, one can instead conduct pooled CRISPRa screens, wherein gRNAs targeting thousands of different endogenous genes can be implemented at once (Gilbert et al., 2014; Horlbeck et al., 2016). From these screens, one can identify genes whose activation confers a phenotype of interest, such as increased cell density, increased viability, decreased cell aggregation, etc., and then efforts can be made to stably transfect more highly expressing copies of such genes in a platform cell line.
In summary, the CRISPR toolbox has enabled rapid and extensive modifications of gene expression levels in diverse cell types. Here we add CRISPRa to the list of tools that can be employed to better understand how gene expression can be modulated in CHO cells and guide further efforts to engineer enhanced platform cell lines that produce a variety of recombinant proteins of interest.
Materials and Methods
gRNA Design
gRNAs were designed using the NCBI CHO-K1 annotation of Mgat3 (GeneID: 100689076), St6gal1 (GeneID: 100689389), and Aldh18a1 (GeneID: 100755485). A 1000 bp region upstream of the first basepair of the gene was obtained from the CHO-K1 assembly on NCBI. This region was analyzed using an inhouse gRNA prediction tool (available at https://github.com/laeblab/crispy) to rank all possible gRNA sequences in the region based on the number of off-targets across the whole genome. The gRNAs were further filtered for a proximity of 550 bp-25 bp upstream of the predicted TSS. Finally, the most 5’ nucleotide was replaced with a G for compatibility with the U6 promoter. Target sequences and gRNA oligos are listed in Supplementary Table S2 and Table S3.
Vector Construction
gRNA vectors were constructed using Uracil-Specific Excision Reagent (USER) friendly cloning as previously described (Ronda et al., 2014). The CHO codon-optimized dCas9 cassette was generated from a synthesized CHO codon-optimized wild-type Cas9 (GeneArt, Thermo Fisher Scientific) as previously described (Xiong et al., 2019). The CHO codon-optimized dCas9 was then fused to a VPR domain cloned from a AAV_NLS-SaCas9-NLS-VPR vector (a gift from George Church (Addgene plasmid #68496)). The construct is here referred to as VPR_dCas9 and available at Addgene (Addgene Plasmid #134601). The plasmid construct and sequence are listed in Supplementary Figure S3. All plasmids were purified using NucleoBond Xtra Midi EF (Macherey-Nagel) according to manufacturer’s protocol and verified by Sanger sequencing.
Cell Culture and Transfection
CHO-S cells (Thermo Fisher Scientific) and a CHO-S mutant with knockouts of Mgat4a,4b and 5, St3gal3,4 and 6, B3gnt2, Sppl3, and Fut8 (referred to here as CHO-C1) were maintained in CD-CHO medium supplemented with 8mM L-glutamine (Thermo Fisher Scientific) and 1 μL/mL Anti-Clumping (AC) reagent (Life Technologies), unless otherwise specified. The cells were cultivated in 125mL Erlenmeyer shake flasks (Corning) in a humidified incubator at 37˚C, 5% CO2 at 120 RPM and passaged every 2–3 days. Viable cell density (VCD) and viability were monitored using the Nucleocounter NC-200 Cell Counter (ChemoMetec). One day prior to transfection, 0.6 × 106 cells/mL cells were spun down at 200x g and resuspended in 6-well plates (BD Biosciences) in 3 mL culture medium without AC per well. Cells were transfected using a total of 3.75 μg DNA and 3.75 μL FreeStyle™ Max transfection reagent (Thermo Fisher Scientific) together with OptiPRO SFM (Life Technologies) according to manufacturer’s protocol. The ratio of DNA used from respective plasmids is listed in Supplementary Table S4. Plasmids for gRNAs targeting Aldh18a1 were transfected into CHO-S cells; those targeting Mgat3 and St6gal1 were transfected into CHO-C1 cells. In addition, non-targeting gRNA with and without dCas9 were transfected for each cell line as controls. All transfections were done in three biological replicates.
RNA Isolation, cDNA Generation and Quantitative Reverse-Transcription PCR
qRT-PCR was carried out to determine the relative expression of target genes after CRISPRa using pre-designed TaqMan Gene Expression Assays (St6gal1: Cg04423241_g1, Aldh18a1: Cg04498748_m1, and Hprt: Cg04448435_m1) (Thermo Fisher Scientific) or custom assays (Mgat3 and Gnb1--see Supplementary Table S5). Two days after transfection, 500 μL of cells were spun at 200 x g. The supernatant was removed and the pellet was resuspended in 300 μL TRIzol™ Reagent (Thermo Fisher Scientific). RNA was isolated using 96 well Direct-zol RNA kits (Zymo Research) following manufacturer’s protocol, followed by quality and quantity measurement on the NanoDrop spectrophotometer 2000 (Thermo Fisher Scientific). Isolated RNA was reverse transcribed into cDNA using using the qScript Flex cDNA synthesis kit (QuantaBio), followed by qRT-PCR with Hprt and Gnb1 as housekeeping genes following manufacturer’s protocol using the QuantStudio 5 instrument (Applied Biosystems). Amplification was performed under the following conditions: 50°C for 2 min, 95°C for 10 min; 45x: 95°C for 15 s, 60°C for 1 min. Three technical replicates were performed for each sample. Transcript levels were normalized to the mean of Hprt and Gnb1 and relative expression levels were calculated using using the 2−ΔΔCt method (Livak & Schmittgen, 2001). For St6gal1 no amplification was detected in the NT samples so the Ct value was arbitrarily set to 46 (1 cycle after the run was terminated). Analysis was done using the Relative Quantification module of Thermo Fisher Connect (https://www.thermofisher.com/dk/en/home/digital-science/thermo-fisher-connect/all-analysis-modules.html).
Glycan Analysis
After centrifugation for cell and debris removal, the supernatant (~3 mL) was concentrated using Amicon® Ultra-4 Centrifugal Filter Units per manufacturer’ instructions. Secretome proteins were fluorescently labeled with GlycoWorks RapiFluor-MS N-Glycan Kit (Waters, Milford, MA) per manufacturer’s protocol. N-linked glycan analysis was performed by LC-MS using a Thermo Ultimate 3000 HPLC with the fluorescence detector coupled on-line to a Thermo Velos Pro Iontrap MS (run in positive mode). Separation gradient was 30% to 43% buffer. The amount of N-glycan was measured by integrating the areas under the normalized fluorescence spectrum peaks with Thermo Xcalibur software (Thermo Fisher Scientific, Waltham, MA) giving a normalized, relative glycan quantities.
Supplementary Material
Acknowledgements
The authors thank Helle Munck Petersen and Sanne Schoffelen for help with glycosylation labeling.
The authors acknowledge support from the Novo Nordisk Foundation (NNF10CC1016517 and NNF16OC0021638) and NIGMS (R35 GM119850).
References
- Amann T, Hansen AH, Kol S, Hansen HG, Arnsdorf J, Nallapareddy S, … Kildegaard HF (2018). Glyco-engineered CHO cell lines producing alpha-1-antitrypsin and C1 esterase inhibitor with fully humanized N-glycosylation profiles. Metabolic Engineering, 52, 143–152. [DOI] [PubMed] [Google Scholar]
- Butler M, & Spearman M (2014). The choice of mammalian cell host and possibilities for glycosylation engineering. Current Opinion in Biotechnology, 30, 107–112. [DOI] [PubMed] [Google Scholar]
- Chavez A, Tuttle M, Pruitt BW, Ewen-Campen B, Chari R, Ter-Ovanesyan D, … Church G (2016). Comparison of Cas9 activators in multiple species. Nature Methods, 13(7), 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AW, Wang H, Yang H, Shi L, Katz Y, Theunissen TW, … Jaenisch R (2013). Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Research, 23(10), 1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang AW, Li S, Spahn PN, Richelle A, Kuo C-C, Samoudi M, & Lewis NE (2016). Modulating carbohydrate-protein interactions through glycoengineering of monoclonal antibodies to impact cancer physiology. Current Opinion in Structural Biology, 40, 104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, … Weissman JS (2014). Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell, 159(3), 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, … Qi LS (2013). CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell, 154(2), 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grav LM, la Cour Karottki KJ, Lee JS, & Kildegaard HF (2017). Application of CRISPR/Cas9 Genome Editing to Improve Recombinant Protein Production in CHO Cells. In Methods in Molecular Biology (pp. 101–118). [DOI] [PubMed] [Google Scholar]
- Grav LM, Lee JS, Gerling S, Kallehauge TB, Hansen AH, Kol S, … Kildegaard HF (2015). One-step generation of triple knockout CHO cell lines using CRISPR/Cas9 and fluorescent enrichment. Biotechnology Journal, 10(9), 1446–1456. [DOI] [PubMed] [Google Scholar]
- Hefzi H, Ang KS, Hanscho M, Bordbar A, Ruckerbauer D, Lakshmanan M, … Lewis NE (2016). A Consensus Genome-scale Reconstruction of Chinese Hamster Ovary Cell Metabolism. Cell Systems, 3(5), 434–443.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, … Weissman JS (2016). Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife, 5 10.7554/eLife.19760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Ewen-Campen B, Ni X, Housden BE, & Perrimon N (2015). In Vivo Transcriptional Activation Using CRISPR/Cas9 in Drosophila. Genetics, 201(2), 433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, & Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 25(4), 402–408. [DOI] [PubMed] [Google Scholar]
- Marx N, Grünwald-Gruber C, Bydlinski N, Dhiman H, Ngoc Nguyen L, Klanert G, & Borth N (2018). CRISPR-Based Targeted Epigenetic Editing Enables Gene Expression Modulation of the Silenced Beta-Galactoside Alpha-2,6-Sialyltransferase 1 in CHO Cells. Biotechnology Journal, 13(10), e1700217. [DOI] [PubMed] [Google Scholar]
- Popp O, Moser S, Zielonka J, Rüger P, Hansen S, & Plöttner O (2018). Development of a pre-glycoengineered CHO-K1 host cell line for the expression of antibodies with enhanced Fc mediated effector function. mAbs, 10(2), 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reusch D, & Tejada ML (2015). Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology, 25(12), 1325–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronda C, Pedersen LE, Hansen HG, Kallehauge TB, Betenbaugh MJ, Nielsen AT, & Kildegaard HF (2014). Accelerating genome editing in CHO cells using CRISPR Cas9 and CRISPy, a web-based target finding tool. Biotechnology and Bioengineering, 111(8), 1604–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanson KR, Hanna RE, Hegde M, Donovan KF, Strand C, Sullender ME, … Doench JG (2018). Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nature Communications, 9(1), 5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W, Ye Z, Wang S, Schulz MA, Van Coillie J, Sun L, … Yang Z (2019). The glycosylation design space for recombinant lysosomal replacement enzymes produced in CHO cells. Nature Communications, Vol. 10 10.1038/s41467-019-09809-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, … Pontén F (2015). Proteomics. Tissue-based map of the human proteome. Science, 347(6220), 1260419. [DOI] [PubMed] [Google Scholar]
- Xiong K, Marquart KF, la Cour Karottki KJ, Li S, Shamie I, Lee JS, … Kildegaard HF (2019). Reduced apoptosis in Chinese hamster ovary cells via optimized CRISPR interference. Biotechnology and Bioengineering. 10.1002/bit.26969 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.