

Abstract
Antibacterial-guided fractionation of an extract of a deep-water Topsentia sp. marine sponge led to the isolation of two new indole alkaloids, tulongicin A (1) and dihydrospongotine C (2), along with two known analogues, spongotine C (3) and dibromodeoxytopsentin (4). Their planar structures were determined by NMR spectroscopy. Their absolute configurations were determined through a combination of experimental and computational analyses. Tulongicin (1) is the first natural product to contain a di(6-Br-1H-indol-3-yl)methyl group linked to an imidazole core. The coexistence of tri-indole 1 and bis-indole alcohol 2 suggests a possible route to 1. All of the compounds showed strong antimicrobial activity against Staphylococcus aureus.
Graphical Abstract

The increasing number of drug-resistant bacterial pathogens has become a significant threat to global public health. New classes of antibiotics that are effective toward drug-resistant bacteria or adjuvants that could restore antibiotic sensitivity are urgently needed. Marine organisms have been recognized as a rich source of structurally unique and biologically active metabolites. High-throughput screening of natural products extracts on the Gram-negative bacterium Escherichia coli and Gram-positive bacterium Staphylococcus aureus revealed an organic extract, C031215 (NCI Open Repository), from a Topsentia sp. marine sponge that inhibited the growth of S. aureus and to a lesser degree E. coli. From a natural products perspective, sponges of the genus Topsentia are known for producing unusual sterols,1−6 oxylipins,7 and the bis-indole alkaloids or topsentins.8 Antibacterial-guided fractionation of the extract yielded an unprecedented tri-indole alkaloid, tulongicin (1), three bisindole alkaloids including the new natural product dihydrospongotine C (2), and the known compounds spongotine C (3)9 and dibromodeoxytopsentin (4).10 Here we describe the isolation and structural characterization of compounds 1 and 2 and the integrated approach involving derivatization and comparison of experimental and calculated electronic circular dichroism (ECD) spectra that was necessary to assign the absolute configurations of the new compounds. Antibiotic, anti-HIV, and cytotoxic properties are reported.
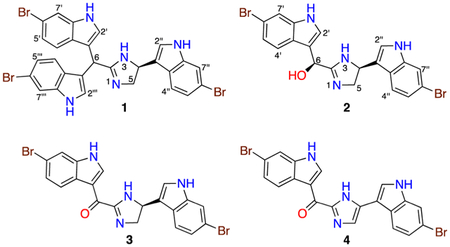
For many years, researchers at the Coral Reef Research Foundation headquartered in Koror, Palau, have collected diverse marine organisms to supply the Natural Products Repository of the National Cancer Institute. In 2008, collections were made near Ulong Channel at a depth of 140 m using the manned submersible Deepworker, providing the sponge used in this study. A portion of a 1:1 MeOH/CH2Cl2 extract (1 g) of the Topsentia sp. marine sponge (Supporting Information) was fractionated over a Sephadex LH-20 column eluting with MeOH to yield a set of fractions that displayed antimicrobial activity (Supporting Information). Active fractions were combined, and solvent was removed in vacuo. The residue was further separated by RP-HPLC to afford compounds 1 (1.6 mg) and 2 (134.9 mg) and the known compounds spongotine C (3, 7.3 mg) and dibromodeoxytopsentin (4, 1.9 mg).
By ESIMS, tulongicin (1) showed a group of positive protonated molecules at m/z 664.0, 666.0, 668.0, and 670.0 with relative intensities of 1:3:3:1, indicating the presence of three bromine atoms in the molecule. HRESIMS analysis of 1 revealed an [M + H]+ ion at m/z 663.9347 consistent with a molecular formula of C28H20Br3N5 that indicated 20 degrees of unsaturation. The 1H NMR and HSQC spectra (Table 1) of 1 revealed 21 resonances assigned to five protons attached to heteroatoms (δH 10.27, s, 1-NH; 10.53, s, 3-NH; 11.35, 2H, d, J = 2.6 Hz, 1′-NH/1″-NH; and 11.43, d, J = 2.6 Hz, 1‴-NH), 12 aromatic protons, one nitrogenous methine (δH 5.67, dd, J = 12.0 and 8.7 Hz, H-4), one methine (5.94, s, H-6), and one methylene group (H-5a: δH 3.84, dd, J = 11.8 and 8.7 Hz; H-5b: δH 4.31, dd, J = 12.0 and 11.8 Hz). The 13C NMR spectrum (Table 1) showed 28 carbon resonances including one imine carbon (δC 170.2, C-2), 12 aromatic nonprotonated carbons, 12 aromatic methine carbons, two sp3 methine carbons, and one methylene carbon. Together the molecular formula and NMR data indicated compound 1 was an indole alkaloid.
Table 1.
1H (500 MHz) and 13C (125 MHz) NMR Data of Compounds 1 and 2 in DMSO-d6
| 1 | 2 | |||
|---|---|---|---|---|
| position | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) |
| 1 NH | 10.27, s | 10.35, s | ||
| 2 | 170.2, C | 171. 5, C | ||
| 3 NH | 10.53, s | 10.53, s | ||
| 4 | 53.7, CH | 5.67, dd (12.0, 8.7) | 53.7, CH | 5.61, dd (12.0, 8.3) |
| 5 Ha | 50.8, CH2 | 3.84, dd (11.8, 8.7) | 50.8, CH2 | 3.80, dd (11.5, 8.3) |
| Hb | 4.31, dd (12.0, 11.8) | 4.31, dd (12.0, 11.5) | ||
| 6 | 32.3, CH | 5.94, s | 61.9, CH | 5.94, d (3.9) |
| 1′ NH | 11.35, d (2.6) | 11.46, d (2.6) | ||
| 2′ | 126.1, CH | 7.22, d (2.6) | 126.2, CH | 7.51, d (2.6) |
| 3′ | 109.9, C | 111.9, C | ||
| 3a′ | 125.0, C | 124.1, C | ||
| 4′ | 120.1, CH | 7.54, d (8.5) | 120.9, CH | 7.66, d (8.6) |
| 5′ | 122.1, CH | 7.20, dd (8.5, 1.8) | 122.1, CH | 7.16, dd (8.6, 1.7) |
| 6′ | 114.5, C | 114.5, C | ||
| 7′ | 114.5, CH | 7.63, d (1.8) | 114.5, CH | 7.64, d (1.7) |
| 7a′ | 137.3, C | 137.3, C | ||
| 1″ NH | 11.35, d (2.6) | 11.40, d (2.6) | ||
| 2″ | 125.6, CH | 7.43, d (2.6) | 125.4, CH | 7.46, d (2.6) |
| 3″ | 112.8, C | 113.0, C | ||
| 3a″ | 123.6, C | 123.6, C | ||
| 4″ | 120.0, CH | 6.83, dd (8.5, 1.7) | 120.1, CH | 7.19, d (8.5) |
| 5″ | 121.7, CH | 7.02, d (8.5) | 121.8, CH | 6.98, dd (8.5, 1.8) |
| 6″ | 114.4, C | 114.5, C | ||
| 7″ | 114.6, CH | 7.57, d (1.7) | 114.6, CH | 7.61, d (1.8) |
| 7a″ | 137.6, C | 137.6, C | ||
| 6 OH | 6.80, d (3.9) | |||
| 1‴ NH | 11.43, d (2.6) | |||
| 2‴ | 126.0, CH | 7.41, d (2.6) | ||
| 3‴ | 109.8, C | |||
| 3a‴ | 124.9, C | |||
| 4‴ | 120.1, CH | 7.46, d (8.6) | ||
| 5‴ | 122.1, CH | 7.17, dd (8.6, 1.8) | ||
| 6‴ | 114.5, C | |||
| 7‴ | 114.5, CH | 7.65, d (1.8) | ||
| 7a‴ | 137.2, C | |||
Analysis of 2D NMR data (Figure 1) led to the assignment of the planar structure of 1. COSY correlations from 1-NH to 3-NH via H-4 and H-5 (indicated in bold) and HMBC correlations from 1-NH, 3-NH, H-4, and H2-5 to C-2 indicated a 2,4-disubstituted-imidazole core. The presence of 1H–1H and2,3JCH couplings from the imidazole NHs implied compound 1 exists in its protonated form from purification in 0.1% trifluoroacetic acid (TFA), and TFA resonances are observed in the 13C spectra. Further examination of the COSY spectrum established substructure A with couplings observed between 1′-NH and H-2′, and H-4′ and H-7′, through the nonprotonated C-6′. Substructures B and C were assigned in the same manner.
Figure 1.
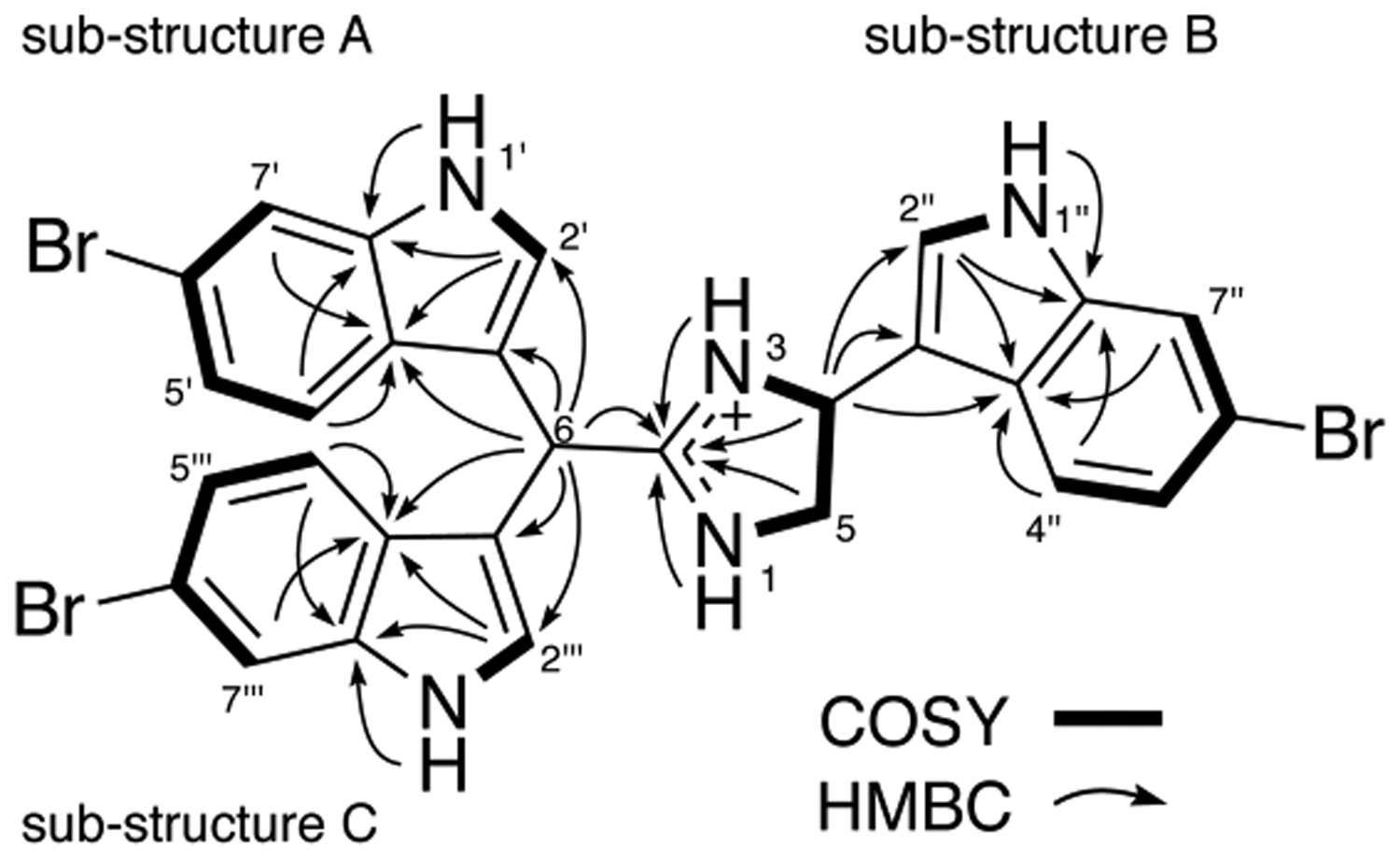
2D NMR correlations for structure elucidation of 1.
To unambiguously assign the 1H and 13C resonances and determine the composition of substructures A–C, HMBC experiments were recorded with spectral widths of 54 ppm in F1 (13C) and 8 ppm in F2 (1H), with 512 and 2048 complex points, respectively. Summarized in Table 1, all indole resonances could be assigned and were not overlapping. The HMBC spectra indicated the presence of three 6-Br-1H-indoles (Table 1, Figure 1). HMBC correlations from H-6 to C-2′, C-3′, C-3a′, C-2‴, C-3‴, C-3a‴, and C-2 and from H-4 to C-2″, C-3″, and C-3a″ unambiguously established that the bis-3-indolemethane was bonded to C-2 and the indole to C-4 of the imizadole group, establishing the structure of 1.
The absolute configuration of 1 was established as 4S by comparing experimental ECD spectra with calculated ones (Figure 2), where the curve of 1 matched closely to that of the (4S) enantiomer.
Figure 2.
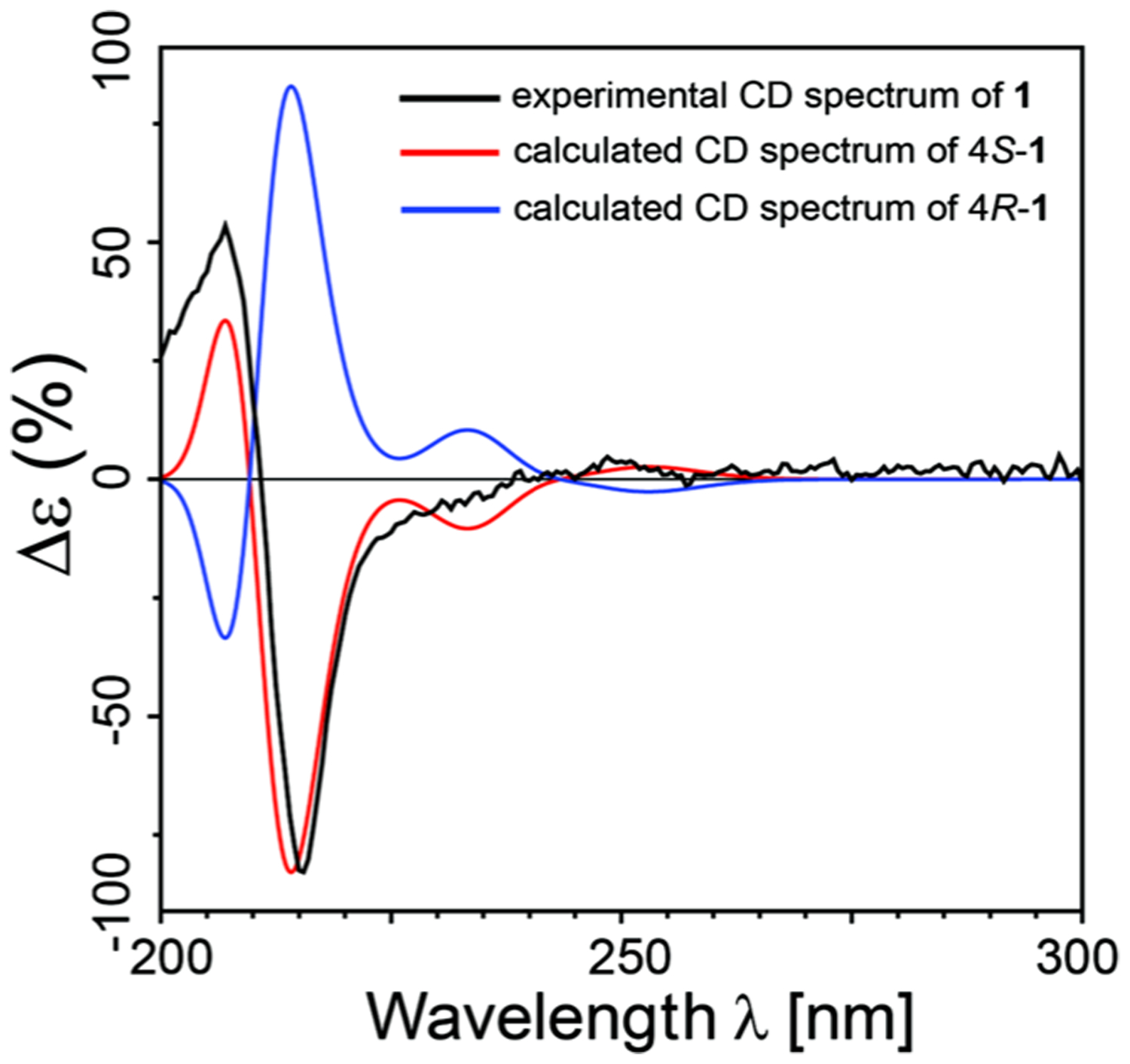
Comparison of the measured ECD spectrum (black line) with the MPW1PW91/6-31G(d,p)-calculated spectra of 4S (red) and 4R (blue) in MeOH.
Dihydrospongotine C (2) was assigned a molecular formula of C20H16Br2N4O by HRESIMS. The NMR data (Table 1) for 2 were similar to those of 1 except for the absence of one 6-Br-1H-indole group and the presence of a hydroxy group at C-6 (δC 61.9). This was confirmed by the COSY correlations between H-6 and 6-OH and HMBC correlations from 6-OH (δH 6.80) to C-6, C-2 (δC 171.5), and C-3′ (δC 111.9). Further analysis of the HMBC spectra showed correlations from H-6 to C-2, C-2′, C-3′, and C-3a′ and from H-4 (δH 5.61) to C-2″ (δC 125.4), C-3″ (δC 113.0), and C-3a″ (δC 123.6), establishing the connections between C-2′/C-6 and C-2″/C-4.
We next sought to determine the absolute configurations of C-4 and C-6 of compound 2. Due to rotation about the C-2/C-6 and C-6/C-3′ bonds, we were unable to determine the relative configurations from NOE data of 2. Attempts to derivatize 2 with α-methoxy-α-trifluoromethylphenylacetic acid to obtain the C-6 Mosher’s ester using a variety of conditions were also unsuccessful. However, while trying to prepare the C-6 methoxy derivative we found that treatment with MeI in the presence of K2CO3 gave a mixture of the N-methylated products 2a–d (Figure 3), whose structures were confirmed by HRESIMS and 1D and 2D NMR data. These included the mono- and di-N-methyl derivatives 2a and 2b and di-N-methyl and tri-N-methyl analogues 2c and 2d, in which respective loss of the C-6 hydroxy or oxidation to the ketone had occurred. With loss of a stereogenic center at C-6, analogue 2c provided a reference compound for determination of the configuration at C-4 by ECD. ECD spectra were recorded and theoretical spectra calculated for each derivative. We first compared the experimental data with the calculated data for the R and S C-4 isomers of 2c and 2d (which lack the stereocenter at C-6). For both compounds, there was excellent agreement with the 4S isomer establishing the S configuration at C-4 of 2 (Supporting Information). ECD spectra were next calculated for the 4S,6R and 4S,6S isomers and compared with experimental curves of 2 and the mono- and dimethylated derivatives 2a and 2b. The experimental and ECD spectra of 2 and the 4S,6S isomer of 2a were nearly identical, while there was very poor agreement between 2 and the 4S,6R isomer. Together the data indicate a 4S,6S configuration for dihydrospongotine C (2). We note that the calculated ECD spectra for the 4S,6R and 4S,6S isomers of dimethyl derivative 2b were similar and were not used to assign the configurations of 2.
Figure 3.

Structures of 2a–d, derivatives of 2 used for ECD spectra and ECD calculations.
The absolute configuration at C-4 of the known compound spongotine C (3) was not assigned in the original report. Comparison of the specific rotations of spongotine C isolated in this study ([α]25D −152) versus the study by Bao et al. ([α]25D −9 (c 0.8, MeOH))9 suggest the same configuration. Because the calculated ECD spectrum of 3 is very similar to the ECD spectra of compounds 1, 2c, and 2d, we propose that spongotine C has the S configuration.
We tested pure compounds 1–4 for antibacterial activity against S. aureus ATCC 29213 and E. coli ATCC 25922 (Table 2). Compounds 1–4 showed strong antibacterial effects toward S. aureus with single-digit μg mL−1 MICs and weak to no inhibition of E. coli at the maximum concentration tested (100 μg/mL). The anti-HIV activity of compounds 1–4 was also evaluated in HIV infectivity assays against the CCR5-tropic primary isolate YU2 and the CXCR4-tropic strain HxB2 (Table 2). Compounds 1–3 inhibited HIV infection at low micro-molar concentrations; surprisingly compound 4 was inactive against HxB2 and showed only weak inhibition toward S. aureus and YU2, implying dehydrogenation of the center core decreases biological activity relative to compounds 1–3. In cytotoxicity assays against a monkey kidney cell line (BSC-1) and a human colorectal tumor cell line (HCT-116) all compounds were inactive (IC50 values >10 μM).
Table 2.
Biological Activities of Compounds 1–4a
| MICs (μg mL−1) | anti-HIV IC50’s (μM) | cytotoxicity (μM) | ||||
|---|---|---|---|---|---|---|
| compound | S. aureus | E. coli | YU2 | HxB2 | BSC-1 | HCT-116 |
| 1 | 1.2 | >100 | 3.9 | 2.7 | 46 | 10 |
| 2 | 3.7 | 100 | 3.5 | 4.5 | 93 | 16 |
| 3 | 1.1 | >100 | 9.5 | 12 | 31 | 27 |
| 4 | 11 | >100 | 57 | NAb | 120 | 180 |
| oxacillin | 0.25 | |||||
| gentamicin | 1 | |||||
See Experimental Section for details of biological assays.
NA, not active.
In summary, two new indole alkaloids have been isolated from a deep-water specimen of Topsentia sp. Tulongicin (1) is the first example of a natural product containing a bisindolemethane linked to an imidazole group. Although a considerable amount is known about the biosynthesis of indole alkaloids,11,12 studies of the chemotypes reported here have yet to be undertaken. Nevertheless, the co-occurrence of compounds 1–4 in a single specimen provides some insight into transformations that may give rise to these compounds. For example tri-indole 1 could derive from a Friedel–Crafts alkylation13 of 2 (or 3) with bromoindole. Oxidation of 2 at C-6 yields spongitine C (3), and dehydrogenation of 3 at C-4/C-5 gives dibromodeoxytopsentin (4). Although tulongicin inhibited the growth of S. aureus with low μg mL−1 MICs, it had no effect on E. coli and was not cytotoxic toward control cell lines.
EXPERIMENTAL SECTION
General Experimental Procedures.
Optical rotations were acquired on a JASCO P-2000 polarimeter. UV–vis spectra were recorded on an Agilent 8453 spectrophotometer, and IR spectra on a PerkinElmer Frontier FT-IR spectrophotometer (Spectrum Two, Real Crystal IR sample card KBr). ECD data were collected on a JASCO J-815 spectrometer. NMR spectra were recorded on a Bruker Avance500 spectrometer equipped with a triple resonance cryoprobe and z gradients. LRESIMS was carried out on an Agilent 1100 LC system and a 6310 MSD. HRESIMS was carried out on a Waters time-of-flight mass spectrometer model LCT Premier. Semipreparative HPLC (Agilent 1100) was performed using a Waters XBridge prep C18 column (10 × 250 mm, S-5 μm, 12 nm). All solvents (HPLC grade) were obtained from Sigma-Aldrich. Sephadex LH-20 was purchased from GE Healthcare.
Biological Material.
The marine sponge used in this study was collected in Palau at a depth of 140 m in August 2008. The sponge, identified as a Topsentia sp., was encrusted by a thin layer (1–2 mm) of Poecillastra incrustans Sollas, 1888. The sponge appeared as a thick, flattened, V-shaped mass, 20 cm long and 5 cm thick, and the surface was slightly fuzzy to the touch, overall undulating and sculpted. Although distinct in its skeletal oxeas, the sponge most closely resembles Topsentia halichondrioides (Dendy, 1905). A full taxonomic description is included in the Supporting Information. Voucher specimens are available at the California Academy of Sciences, specimen number CAS302370.
Extraction and Isolation.
An organic extract of Topsentia sp. was prepared by extracting the sponge material with 1:1 MeOH/CH2Cl2. Extracts were stored at −50 °C until shipment. The extract (1 g) was dissolved in MeOH and chromatographed on a Sephadex LH-20 column eluting with MeOH. Fractions were screened for antimicrobial activity, and 10 active fractions (eluting at 0.3 column volume) were combined (40 mL total) and dried to give fraction F1 (0.5 g). F1 was further separated by semipreparative HPLC (5 mL/min flow rate, eluting with a linear gradient of 20–80%, CH3CN in H2O with 0.1% TFA in 40 min) to yield compounds 1 (1.6 mg, tR 26.5 min), 2 (134.9 mg, tR 16.0 min), spongotine C (3, 7.3 mg, tR 19.5 min), and dibromodeoxytopsentin (4, 1.9 mg, tR 21.5 min).
Tulongicin (1):
yellow powder; [α]25D −32 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 224 (4.71), 285 (4.01) nm; ECD (c 6.0 × 10−5 M, CH3OH) λmax (Δε) 214 (4.27) and 231 (−6.63) nm; IR (KBr) νmax 3419, 1682, 1206, 1142, 803, and 727 cm−1; 1H and 13C NMR data, Table 1 and Table S1; HRESIMS m/z 663.9347 [M + H]+ (calcd for C28H21 79Br3N5, 663.9347).
Dihydrospongotine C (2):
yellow powder; [α]25D −49 (c 0.08, MeOH); UV (MeOH) λmax (log ε) 222 (4.52), 283 (3.78) nm; ECD (c 8.2 × 10−5 M, CH3OH) λmax (Δε) 212 (3.57) and 232 (−4.88) nm; IR (KBr) νmax 3247, 1675, 1611, 1205, 1139, 802, and 723 cm−1; 1H and 13C NMR data, Table 1 and Table S1; HRESIMS m/z 486.9767 [M + H]+ (calcd for C20H17 79Br2N4O, 486.9769).
Methylation of Compound 2.
Compound 2 (18 mg, 37 μmol) was dissolved in acetone (300 μL) in the presence of K2CO3 (1.3 mg) and MeI (7.2 μL, 115 μM) and stirred at rt for 16 h. The solution was dried under a stream of N2 and dissolved in MeOH (1 mL). The CH3OH suspension was separated by semipreparative HPLC (flow rate of 4 mL/min, eluting with a linear gradient of 35–70%, CH3CN in H2O with 0.1% TFA in 30 min) to give compounds 2a (0.5 mg, tR 13.0 min), 2b (3.4 mg, tR 14.5 min), 2c (1.1 mg, tR 16.0 min), and 2d (3.4 mg, tR 21.0 min). The structures of 2a–d were determined by HRMS and 1H and 13C NMR.
2a: yellow powder; [α]25D −23 (c 0.05, MeOH); 1H and 13C NMR data, Table S2; HRESIMS m/z 500.9931 [M + H]+ (calcd for C21H19 79Br2N4O, 500.9926).
2b: yellow powder; [α]25D −34 (c 0.2, MeOH); 1H and 13C NMR data, Table S2; HRESIMS m/z 515.0079 [M + H]+ (calcd for C22H21 79Br2N4O, 515.0082).
2c: yellow powder; [α]25D −37 (c 0.1, MeOH); 1H and 13C NMR data, Table S2; HRESIMS m/z 499.0124 [M + H]+ (calcd forC22H21 79Br2N4, 499.0133).
2d: yellow powder; [α]25D −58 (c 0.2, MeOH); 1H and 13C NMR data, Table S2; HRESIMS m/z 527.0088 [M + H]+ (calcd for C23H21 79Br2N4O, 527.0082).
MIC and Cytotoxicity Determination.
MICs were determined by testing all compounds in duplicate in at least two independent experiments for their antimicrobial activity against bacterial strains listed in Table 2 and as described in the Clinical and Laboratory Standards Institute guidelines.14 All strains were obtained from the American Type Culture Collection. Experimental details for MIC and cytotoxicity data are provided in the Supporting Information.
HIV-1 Neutralization Assays.
HIV envelope pseudotyped viruses were prepared and single-round neutralization assays performed as described previously.15,16 Briefly, 293T cells (American Type Culture Collection) were cotransfected with plasmid SG3 ΔEnv containing an envelope-deficient HIV genome and an HIV-1 envelope-expressing plasmid (NIH AIDS Reagent Program) using X-treme GENE HP DNA (Roche). The media was changed after 24 h, and at 48 h post-transfection the supernatant was collected, passed through a 0.45 μm filter, and stored at −80 °C until further use in neutralization assays. Twofold serial dilutions of the inhibitors were prepared in a 96-well plate after which pseudotyped virus was added followed by TZM-bl cells (NIH AIDS Reagent Program). The plates were incubated at 37 °C in a 5% CO2 atmosphere, and fresh media was added at 24 h. The cells were then lysed, and the plates read for luciferase activity (Bright Glo, Promega) at 48 h postinfection.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. Lloyd for HR-MS data; the NCI Open Repository for organic extracts; and L. Bell, Coral Reef Research Foundation for helpful discussions. This work was supported by the NIH Intramural Research Program (NIDDK).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnatprod.7b00452.
HR-MS; 1H, 13C, and 2D NMR spectra of new compounds; and details for ECD calculations for compounds 1, 2, and 2a–d, and sponge taxonomy (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Mckee TC; Cardellina JH; Tischler M; Snader KM; Boyd MR Tetrahedron Lett. 1993, 34, 389–392. [Google Scholar]
- (2).Gunasekera SP; Sennett SH; Kellyborges M; Bryant RW J. Nat. Prod 1994, 57, 1751–1754. [DOI] [PubMed] [Google Scholar]
- (3).DiGirolamo JA; Li XC; Jacob MR; Clark AM; Ferreira DJ Nat. Prod 2009, 72, 1524–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Fusetani N; Takahashi M; Matsunaga S Tetrahedron 1994, 50, 7765–7770. [Google Scholar]
- (5).Dai JQ; Sorribas A; Yoshida WY; Kelly M; Williams PG J. Nat. Prod 2010, 73, 1597–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ciminiello P; Fattorusso E; Magno S; Mangoni A; Pansini M Steroids 1992, 57, 62–66. [DOI] [PubMed] [Google Scholar]
- (7).Luo X; Li FM; Hong JK; Lee CO; Sim CJ; Im KS; Jung JH J. Nat. Prod 2006, 69, 567–571. [DOI] [PubMed] [Google Scholar]
- (8).Bartik K; Braekman JC; Daloze D; Stoller C; Huysecom J; Vandevyver G; Ottinger R Can. J. Chem 1987, 65, 2118–2121. [Google Scholar]
- (9).Bao B; Sun Q; Yao X; Hong J; Lee C-O; Cho HY; Jung JH J. Nat. Prod 2007, 70, 2–8. [DOI] [PubMed] [Google Scholar]
- (10).Bao B; Sun Q; Yao X; Hong J; Lee C-O; Sim CJ; Im KS; Jung JH J. Nat. Prod 2005, 68, 711–715. [DOI] [PubMed] [Google Scholar]
- (11).Alkhalaf LM; Ryan KS Chem. Biol 2015, 22, 317–328. [DOI] [PubMed] [Google Scholar]
- (12).Ryan KS; Drennan CL Chem. Biol 2009, 16, 351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Li H; Wang Y-Q; Deng L Org. Lett 2006, 8, 4063–4065. [DOI] [PubMed] [Google Scholar]
- (14).Institute, C. a. L. S. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard-ninth ed CLSI document M07-A9. CLSI: Wayne, PA, 2008; Vol. M07–A9. [Google Scholar]
- (15).Gustchina E; Louis JM; Bewley CA; Clore GM J. Mol. Biol 2006, 364, 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Li M; Gao F; Mascola JR; Stamatatos L; Polonis VR; Koutsoukos M; Voss G; Goepfert P; Gilbert P; Greene KM J. Virology 2005, 79, 10108–10125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.