

Abstract
Hedgehog (Hh) signalling plays conserved roles in controlling embryonic development; its dysregulation causes many diseases including cancers. The G protein-coupled receptor Smoothened (Smo) is the key signal transducer of the Hh pathway, whose posttranslational regulation has been shown to be critical for its accumulation and activation. Ubiquitination has been reported an essential posttranslational regulation of Smo. Here, we identify a novel E3 ligase of Smo, Herc4, which binds to Smo, and regulates Hh signalling by controlling Smo ubiquitination and degradation. Interestingly, our data suggest that Herc4-mediated Smo degradation is regulated by Hh in PKA-primed phosphorylation-dependent and independent manners.
Keywords: Hedgehog, Smoothened, Herc4, ubiquitination
Introduction
Hedgehog (Hh) signalling plays essential roles in embryonic development and adult tissue homeostasis in species ranging from insects to human (Ingham and McMahon, 2001; Lum and Beachy, 2004; Jia and Jiang, 2006; Jiang and Hui, 2008). Aberrant Hh signalling activity is associated with many human disorders including birth defects and cancers (Taipale and Beachy, 2001; Pasca di Magliano and Hebrok, 2003; Jiang and Hui, 2008; Ingham et al., 2011).
Hh transduces signal through binding to its receptor, a 12-transmembrane protein Patched (Ptc), which alleviates Ptc-mediated suppression of Smo, a seven-transmembrane GPCR protein (Hooper and Scott, 1989; Marigo et al., 1996; Stone et al., 1996; Casali and Struhl, 2004; Corbit et al., 2005; Rohatgi et al., 2007; Zhao et al., 2007). Activated Smo recruits the kinesin-like protein Costal2 (Cos2) and the Ser/Thr kinase Fused (Fu) to trigger the activation of latent transcription factor Cubitus interruptus (Ci) (Alcedo et al., 1996; Sisson et al., 1997; Denef et al., 2000; Jia et al., 2003), which enters the nucleus to turn on the expression of Hh target genes including decapentaplegic (dpp), patched (ptc), and engrailed (en) (Ingham and McMahon, 2001; Jia et al., 2005; Smelkinson et al., 2007; Lv et al., 2018a, b).
Protein turnover mediated by ubiquitin modification plays important roles in the regulation of numerous cellular processes during development. Ubiquitination is an enzymatic process by which proteins are modified with ubiquitin chains (Hochstrasser, 1995). This process of delivering ubiquitin to substrates requires the coordinated action of three classes of enzymes: the E1 ubiquitin-activating enzyme, the E2 ubiquitin-conjugating enzyme, and the E3 ubiquitin-protein ligase (Hochstrasser, 1992). While E1 and E2 enzymes are primarily involved in activating and transferring ubiquitin through high-energy thioester bonds, E3 ligases play crucial roles in providing the specificity of substrate recognition (Hochstrasser, 1996). Ubiquitin ligases (E3) have been grouped into two families: the single-subunit forms including single RING-finger, U-box, and HECT-domain ligases (Qiu et al., 2000) and the multi-subunit RING-finger ubiquitin ligases, including the Cullin-Rbx type (Liu and Nussinov, 2011) and the APC E3 ligases (Huang et al., 2005).
The E3 ubiquitin ligases targeting the core components of Hh pathway have been mostly identified. However, the molecular mechanism underlying Smo degradation remains largely unknown. In this study, we identify the HECT E3 ubiquitin ligase Herc4 that compromises Hh signalling by directly controlling Smo protein turnover. Moreover, we find that Herc4 mediates Smo degradation in a manner that is regulated by Hh.
Results
Inactivation of herc4 positively regulates Hh pathway activity
To identify novel Hh signalling regulators, we carried out a genetic modifier screen in the expression of a dominant-negative Smo (Smo–PKA) background with a wing-specific Gal4 driver MS1096 and determined which genes modulated Smo–PKA wing phenotype (Figure 1A and B). From this screen, we identified that knockdown of HECT domain E3 ligase gene herc4 (CG9153) enhanced MS1096 > Smo–PKA-induced wing phenotype (compare Figure 1C with B). To exclude the possible off-target effect of RNAi, we coexpressed HA-Herc4 transgene with the corresponding herc4 RNAi and found that the wing phenotype caused by herc4 RNAi was totally rescued (compare Figure 1D with C). In addition, the mutant of herc4, herc4G5486, also showed herc4 RNAi-like but weak wing phenotype (compare Figure 1F with B). All these results indicate that the wing phenotype caused by downregulation of herc4 is not due to the off-target effect.
Figure 1.

Knockdown of herc4 upregulates Hh signalling activity. (A and B) Comparison of adult wing phenotypes between control (A) and Smo–PKA overexpression flies (B). Arrows indicate the space between veins 3 and 4. (C and D) Overexpression of Herc4 (D) rescued the herc4 knockdown (C)-caused adult wing phenotype under the background of Smo–PKA. (E) Overexpression of Herc4C1030A enhanced the herc4 knockdown-caused adult wing phenotype under the background of Smo–PKA. Arrows indicate the space between veins 3 and 4. (F) The mutant of herc4, herc4G5486, showed herc4 RNAi-like but weak wing phenotypes in expression of Smo–PKA background (compare F with C). (G–H”) Knockdown of herc4 with MS1096-Gal4 under the background of Smo–PKA attenuated the expression of Ptc (compare H–H” with G–G”). Arrows indicate the decrease of Ptc. (I–I”) Overexpression of HA-Herc4 rescued the herc4 knockdown-caused Ptc expression level under the background of Smo–PKA. (J–J”) Overexpression of Herc4C1030A did not rescue the herc4 knockdown-caused Ptc expression level under the background of Smo–PKA. (K–L”) UAS-GFP (green) marks the apG4-mediated gene expression region. apG4 drives UAS transgenes to be specifically expressed in the dorsal region of wing discs. Compared with the control disc (K–K”), knockdown of herc4 by apG4 upregulated the expression of dpp-lacZ (L–L”) (arrow).
To determine whether Herc4 modifies Hh signalling activity, we examined Hh target genes expression in wing discs. Compared with control discs (Figure 1G–G”), knockdown of herc4 with MS1096-Gal4 apparently reduced Ptc level under Smo–PKA background (Figure 1H–H”). However, interestingly, when using a stronger gal4 driver ap-Gal4 (apG4) to knock down herc4 in the dorsal region of wing discs in wild-type background, we found that knockdown of herc4 elevated dpp-lacZ expression (Figure 1K–L”). The explanation of this obvious opposite wing phenotypes is that most possibly the substrate of the E3 ligase Herc4 is Smo. In Smo–PKA background, knockdown of herc4 caused Smo–PKA accumulation, which masked the endogenous smo accumulation effect, at last caused the reduced Ptc level, however, in wild-type background, knockdown of herc4 caused endogenous smo accumulation which is consistent with the increased dpp level. Taken together, these results suggest that Herc4 is a negative regulator of Hh pathway.
In addition, through rescue assays, we found that wild-type Herc4 restored the wing phenotype induced by herc4 RNAi (Figure 1D and I–I”). By contrast, overexpression of a catalytically dead form of Herc4 (Herc4C1030A) (Aerne et al., 2015) deteriorated the phenotype caused by herc4 RNAi (Figure 1E and J–J”), indicating that Herc4 plays the function in the E3 ligase activity dependent manner and Herc4C1030A has a dominant-negative effect.
Herc4 regulates Hh signalling through Smo
To verify our hypothesis that Smo is the target of Herc4, we first checked the subcellular localization of Herc4, found Herc4 was localized in both cytoplasm and nucleus (Supplementary Figure S2). Next, we did the co-IP experiments in S2 cells, and found that Herc4 interacted with Smo (Figure 2A). Except this, we did not observe any interaction between Herc4 and other indicated components of Hh pathway, including Ci, Fu, Cos2, PKA-C1 (Supplementary Figure S1A–D).
Figure 2.

Herc4 binds with Smo. (A) HA-Herc4 interacted with Myc-Smo. (B) Schematic drawings show the domains of Herc4. (C) Both the Herc4 ΔRCC and Herc4 ΔHECT interacted with Smo in S2 cells. (D) Both the Herc4-RCC and Herc4-HECT interacted with Smo in S2 cells. (E) Schematic drawings show the various Smo constructs. (F–H) Herc4 bound with the N-terminus (F), C-terminus (G), and the SAID domain (H) of Smo. (I) Herc4 did not bind with Myc-Smo-CΔSAID.
Herc4 contains two distinct domains: the RCC repeat domain, which is a seven-bladed propeller–like domain, and the HECT domain, which is involved in regulating the E3 ligase activity for ubiquitination (Figure 2B). To map the domains required for Herc4 binding to Smo, we carried out co-IP experiments using truncated fragments of Herc4. Through co-IP experiments, we found either RCC repeat domain or the HECT domain is sufficient to mediate its binding to Smo (Figure 2C–D).
To determine which region of Smo is responsible for binding to Herc4, various Myc-Smo truncated mutants were transfected into S2 cells with HA-Herc4, followed by co-IP assays (Figure 2E). As shown in Figure 2F–G, both N- and C-terminus of Smo (Myc-SmoN and Myc-SmoC) interacted with HA-Herc4. Previous studies have demonstrated that the C-terminally located Smo auto-inhibitory domain (SAID) plays an important role in regulating Smo activity (Zhao et al., 2007; Li et al., 2012). To test whether SAID domain is responsible for SmoC interacting with Herc4, we investigated various SmoC truncations for their ability to bind Herc4. As shown in Figure 2H–I, SAID domain itself could, but SmoC with SAID domain deleted could not bind Herc4, indicating that SAID domain is necessary and sufficient to mediate the interaction between Herc4 and SmoC. Taken together, Herc4 could interact with both N-terminus and C-terminal SAID domain of Smo.
Herc4 regulates Smo stability
Because Herc4 is an E3 ligase and interacts with Smo, we first examined whether Herc4 can regulate Smo level. As shown in Figure 3A, overexpression of Flag-Herc4 (Fg-Herc4) in S2 cells apparently downregulated Myc-Smo protein level, while knockdown of herc4 increased its level. Consistently, overexpression of Fg-Herc4 in S2 cells markedly decreased the cell membrane localized Myc-Smo level (Figure 3B–C”’). Next, we explored the effect of Herc4 on Smo in vivo. Knockdown of herc4 in wing imaginal discs with apGal4 promoted Smo accumulation in anterior-dorsal compartment cells (Figure 3D–D”). Of note, herc4 knockdown did not change the Smo mRNA levels (Figure 3E), suggesting that Smo is regulated by Herc4 through a post-translational mechanism.
Figure 3.

Herc4 affects Smo stability. (A) Overexpression of Fg-Herc4 downregulated Myc-Smo protein level. Knockdown of herc4 upregulated Myc-Smo protein. herc4-dsRNA could effectively knock down herc4 mRNA level in S2 cells (bottom two panels). (B–C”’) S2 cells transfected with indicated constructs were stained by Myc, Flag antibody and DAPI. Of note, Herc4 inhibited Smo cell membrane accumulation (compare C–C”’ with B–B”). The nuclei were showed by DAPI staining. (D–D”) Knockdown of herc4 with apG4 increased the anterior compartment Smo protein level of the wing disc. Arrows indicate the increase of Smo. (E) The relative mRNA level of smo in wing discs. (F and G) Western blots of lysates from S2 cells expressing indicated proteins and treated with CHX for the indicated time intervals. Quantification analyses were shown below. The results were presented as mean ± SD of values from three independent experiments. Of note, Herc4 could promote Smo degradation (F). Herc4C1030A could hamper Smo degradation (G).
To determine whether Herc4 regulates Smo at protein level, we carried out a protein stability assay. S2 cells were transfected with Myc-Smo and Fg-Herc4, then, treated with cycloheximide (CHX) to block protein synthesis for indicated times. The results showed that overexpression of Fg-Herc4 apparently promoted Smo degradation (Figure 3F). While overexpression of HA-Herc4C1030A markedly decreased Smo degradation (Figure 3G). These results suggest that Herc4 negatively regulates Hh signalling by binding and degrading Smo.
Herc4 promotes Smo degradation mainly through SAID domain
From above results we have known that Herc4 regulates Hh signalling through its ubiquitinase activity. Next we examined whether Herc4 ubiquitinates Smo. Via cell-based ubiquitination assay, we found overexpression of the wild-type Herc4 significantly increased Smo ubiquitination, whereas the Herc4C1030A decreased Smo ubiquitination (Figure 4A). Furthermore, as shown in Figure 4B, knockdown of herc4 resulted in a marked reduction of Smo ubiquitination compared to control gfp RNAi treatment. The efficiency of herc4 RNAi was confirmed by semi-quantitative PCR analysis.
Figure 4.

Herc4 promotes Smo degradation mainly through SAID domain. (A) S2 cells transfected with the indicated plasmids were examined by western blotting. Of note, HA-Herc4 increased, but HA-Herc4C1030A decreased Smo ubiquitination. (B) Knockdown of herc4 using herc4-dsRNA attenuated Smo ubiquitination in S2 cells. herc4-dsRNA could effectively knockdown herc4 mRNA level in S2 cells (bottom two panels). (C) S2 cells transfected with indicated plasmids were examined by western blotting. Of note, Fg-Herc4 mainly ubiquitinated SmoC to promote Smo degradation. (D) Schematic drawings show the truncated forms of SmoC. (E) S2 cells were transfected with the indicated plasmids. The western blot result showed that Herc4 mainly regulated the ubiquitination level of SmoSAID domain. (F) The truncated forms of SmoC were transfected alone or with Herc4 into S2 cells. The results showed Herc4 degraded SmoC through its SAID domain. (G) Fg-Herc4 promoted Myc-SmoSAID ubiquitination. (H) Overexpression of Fg-Herc4 apparently downregulated the protein level of Myc-SmoSAID. (I) Overexpression of Fg-Herc4 hardly downregulated the protein level of Myc-SmoΔSAID.
To establish which part of Smo mediates its ubiquitination, we analysed SmoN and SmoC. As shown in Figure 4C, Smo ubiquitination occurred mainly within SmoC. To further analyse which domain of SmoC mediates its ubiquitination, we generated SmoC1, SmoC2, and SmoCΔSAID truncated forms (Figure 4D). As shown in Figure 4E and F, overexpression of Herc4 significantly decreased the protein level and increased the ubiquitination of SmoC1, which comprising SAID domain, but had no effect on SmoC2 and SmoCΔSAID, indicating SAID domain mediated SmoC ubiquitination and degradation. Consistently, as shown in Figure 4G–I, Herc4 obviously promoted the SAID domain ubiquitination and degradation, but failed to degrade SAID-deleted full length Smo (Myc-SmoΔSAID). Taken together, the results support that Herc4 promotes Smo ubiquitination and degradation mainly through its SAID domain.
Herc4 promotes Smo degradation through both lysosome and proteasome
Previous studies have demonstrated that Smo is degradated in both proteasome and lysosome (Molnar et al., 2011; Li et al., 2012; Xia et al., 2012). To evaluate their roles in Herc4-mediated Smo degradation, we overexpressed Myc-Smo with Fg-Herc4 in S2 cells and treated the cells with NH4Cl (lysosome inhibitor) or MG132 (proteasome inhibitor). We found that treating S2 cells with NH4Cl or MG132 stabilized Herc4-mediated Smo, and the combined treatment of cells with MG132 and NH4Cl had an additive effect on Herc4-mediated Smo stabilization. However, MG132 treatment resulted in a marked inhibition in Herc4-mediated Smo degradation compared with treatment with NH4Cl (Figure 5). These results indicate that Smo protein can be degraded through both proteasome and lysosome, but Herc4-mediated Smo degradation is mainly conducted through proteasome.
Figure 5.
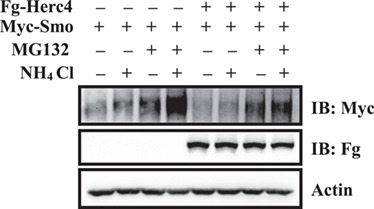
Herc4 degrades Smo mainly through proteasome. S2 cells were transfected with the indicated plasmids, treated with MG132 alone, NH4Cl alone, or together. The results showed Herc4 degraded Smo mainly through proteasome.
Herc4 mediates Smo monoubiquitination and polyubiquitylation
Ubiquitin-mediated protein degradation can occur through monoubiquitination or polyubiquitination (Shabek et al., 2012). To test which type of ubiquitination contributes to Smo degradation, we employed Ub-K0, a mutant form of Ub, in which all seven lysine residues (K6, K11, K27, K29, K33, K48, and K63) are replaced by arginines (Rs), preventing polyubiquitin chain formation (Kulathu and Komander, 2012). Compared with wild-type Ub, when Smo co-expressed with Ub-K0, its ubiquitination level apparently decreased (Figure 6A), suggesting that Smo is subjected to polyubiquitination.
Figure 6.

Herc4 mediates both monoubiquitination and polyubiquitylation of Smo ubiquitination. (A) S2 cells transfected with the indicated plasmids were examined by western blotting. Of note, the ubiquitination level of Smo apparently decreased when co-expressed with Ub-K0 compared with wild-type Ub. (B and C) Co-transfection of Myc-Smo and Flag-Herc4 together with various HA-Ub mutants, the ubiquitination level of Smo was similar to HA-Ub when various HA-Ub mutants were used. (D) Co-expression of HA-UbK0 and Flag-Herc4 together with Myc-SmoK6R or Myc-SmoK7R, Myc-SmoK6R, and Myc-SmoK7R were effectively modified by HA-UbK0. (E) Herc4 ubiquitinated Smo through multiple Lys residues.
We next employed various HA-Ub mutants (Ub-K6R, Ub-K11R, Ub-K27R, Ub-K29R, Ub-K33R, Ub-K48R, and Ub-K63R) to examine the pattern of Smo ubiquitination. Co-transfecting Myc-Smo and Flag-Herc4 together with various HA-Ub mutants, we found the ubiquitination level of Smo was similar to HA-Ub when various HA-Ub mutants were used (Figure 6B and C), indicating that any single type of HA-Ub mutant is not sufficient to block the Smo ubiquitination and Herc4 mediates Smo polyubiquitination with multi-types of ubiquitin chains.
SAID domain harbours a total of 13 Lys residues (K1–K13). We generated two mutant forms of Smo, Myc-SmoK6R and Myc-SmoK7R, in which the K1–K6 and the K7–K13 of SAID domain were mutated to Rs, respectively. Co-expressing HA-UbK0 and Flag-Herc4 together with Myc-SmoK6R or Myc-SmoK7R, we found that Myc-SmoK6R and Myc-SmoK7R were effectively modified by HA-UbK0 (Figure 6D), suggesting that Smo can be monoubiquitinated at multiple sites. Taken together, Herc4 mediates Smo monoubiquitination and polyubiquitylation.
Herc4 mediates Smo ubiquitination at multiple lysine residues
We have known that Herc4 mediates Smo degradation by promoting C-terminal ubiquitination. We speculate that this region might contain Lys residues critical for Smo ubiquitination. Smo C-terminal and intracellular loops contain a total of 49 Lys residues and there are a total of 13 Lys residues in the SAID domain. Using the cell-based ubiquitination assay, we found that both Myc-SmoK13R and Myc-SmoK49R exhibited reduced ubiquitination compared with Myc-Smo by Herc4, but Myc-SmoK49R resulted in a more dramatic reduction in Smo ubiquitination (Figure 6E), suggesting that Smo is ubiquitinated by Herc4 at multiple Lys residues. In addition, the residual ubiquitination of Myc-SmoK13R suggests that Herc4 mediates Smo ubiquitination at one or more Lys residues outside the SAID domain.
Herc4 interacts with USP8, UCHL5, and Hrs to regulate Smo ubiquitination
USP8 and UCHL5 are Smo deubiquitinases that can decrease the Smo ubiquitination level (Li et al., 2012; Xia et al., 2012; Zhou et al., 2018). To test whether USP8, UCHL5 attenuate Herc4-mediated ubiquitination of Smo, we did the western blot experiments. The results showed that USP8, UCHL5 alone could decreased, they together almost abolished Herc4-mediated Smo ubiquitination (Figure 7A). Interestingly, we found overexpression of Fg-UCHL5 and Fg-USP8 alone decreased the interaction between Herc4 and Smo, simultaneous overexpression of Fg-UCHL5 and Fg-USP8 almost blocked the interaction between Herc4 and Smo (Figure 7B), indicating maybe there are competing binding among them.
Figure 7.

Herc4 interacts with Usp8, UCHL5 and Hrs to regulate Smo ubiquitination. (A) Fg-USP8 and Fg-UCHL5 decreased ubiquitination of Smo mediated by Herc4. (B) Fg-USP8 and Fg-UCHL5 attenuated the interaction between Herc4 and Smo. (C) Fg-Hrs interacted with HA-Herc4 in S2 cells. (D) Fg-Hrs did not affect the interaction between HA-Herc4 and Myc-Smo. (E) Knockdown of herc4 decreased the ubiquitin level of Smo mediated by Hrs. herc4-dsRNA could effectively knockdown herc4 mRNA level in S2 cells (bottom two panels).
The previous study has reported that hepatocyte growth factor-regulated tyrosine kinase substrate (Hrs) binds Smo and promotes its ubiquitination (Fan et al., 2013). It is possible that Hrs regulates Smo ubiquitination by facilitating the recruitment of more ubiquitin ligase Herc4 into Hrs/Smo complex. To test this hypothesis, we did a co-IP experiment, and found that Fg-Hrs indeed interacted with HA-Herc4 (Figure 7C), but did not affect the interaction of HA-Herc4 and Myc-Smo (Figure 7D). Furthermore, we found that Hrs-mediated Smo ubiquitination was almost blocked by knockdown of herc4 (Figure 7E). All these results support the idea that Hrs regulates Smo ubiquitination largely through recruiting Herc4 to Smo.
Herc4-mediated Smo ubiquitination is regulated by Hh
The ubiquitination status of Smo is tightly controlled by the Hh signalling. We next want to test whether the interaction between Smo and Herc4 is regulated by Hh. In S2 cells, co-expressing Myc-Smo and Fg-Herc4 with or without Hh treatment, we found that Hh treatment inhibited the interaction between Smo and Herc4 (Figure 8A) and prevented Herc4-mediated Smo ubiquitination (Figure 8B).
Figure 8.
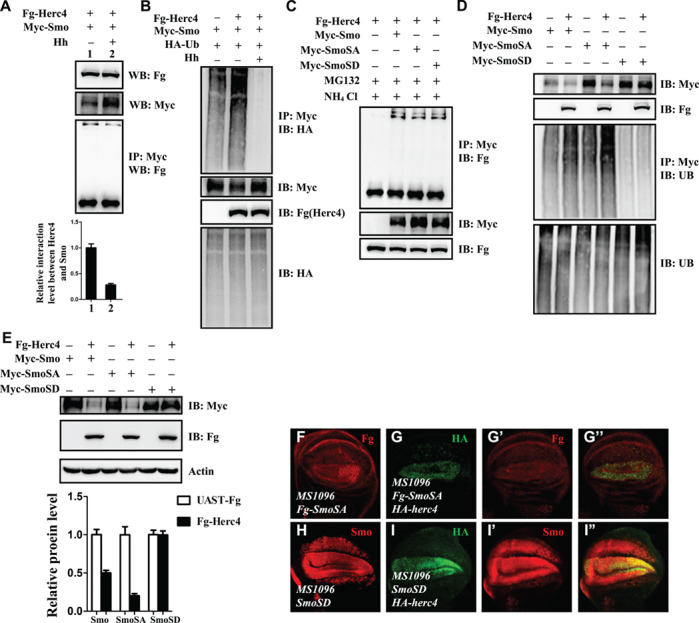
Hh inhibits Herc4-mediated Smo ubiquitination. (A) Hh treatment inhibited the interaction of Herc4 and Smo. (B) Hh decreased Smo ubiquitination mediated by Herc4. (C) S2 cells were transfected with indicated plasmids and treated with the proteasome/lysosome inhibitors MG132 and NH4Cl. Fg-Herc4 interacts equally with Myc-Smo, Myc-SmoSA, and Myc-SmoSD. (D) From cell-based ubiquitin assay, Herc4 upregulated the ubiquitination level of Smo and SmoSA, but did not affect the ubiquitination level of SmoSD. (E) Overexpression of Herc4 in S2 cells apparently decreased the protein level of Smo and SmoSA, but did not affect SmoSD protein level. (F–G”) Overexpression of Herc4 in wing discs with MS1096-gal4 decrease the expression level of Fg-SmoSA (compare G’ with F). (H–I”) Overexpression of Herc4 in wing discs with MS1096-gal4 has no effect on the protein level of SmoSD (compare I’ with H).
Phosphorylation of Smo is tightly controlled by the Hh signal gradient, and is very important for its activity and cell plasma membrane localization (Jia et al., 2004; Zhao et al., 2007). The activated Smo phosphorylation can be mimicked by SmoSD mutants, in which three PKA sites (Ser667, Ser687, and Ser740) and adjacent CKI sites are mutated to aspartic acid. Another Smo mutant, SmoSA, as mentioned before, which has three PKA sites mutated to alanine, can mimic the situation in which the Smo fails to be phosphorylated and no longer responds to the Hh signal. To investigate whether the interaction between Smo and Herc4 is regulated by Smo phosphorylation, we co-expressed HA-Herc4 with Myc-Smo, Myc-SmoSA, or Myc-SmoSD in S2 cells, respectively. In co-IP experiments, S2 cells transfected with the indicated plasmids and treated with the proteasome/lysosome inhibitors MG132/NH4Cl, we found that Herc4 appears to interact equally well with Myc-Smo, Myc-SmoSA, and Myc-SmoSD (Figure 8C), suggesting that the PKA-primed Smo phosphorylation does not affect Smo/Herc4 interaction. Considering Hh treatment inhibited the interaction between Smo and Herc4, we speculate that Hh may regulate the Smo and Herc4 interaction in a manner independent of PKA-primed phosphorylation.
Our data suggested that Hh prevents Herc4-mediated Smo ubiquitination (Figure 8B). This let us investigate the relationship between Smo phosphorylation and Herc4-mediated ubiquitination. As shown in Figure 8D, we found that Herc4 increased the ubiquitin level of Smo, SmoSA, but did not affect the ubiquitin level of SmoSD in S2 cells. Consistently, Herc4 apparently downregulated the protein levels of Smo and SmoSA, but had no effect on the protein level of SmoSD (Figure 8E). To further verify this data in vivo, we co-expressed Herc4 with SmoSA or SmoSD in wing discs using MS1096-gal4 driver. As shown in Figure 8F–I”, overexpression of Herc4 reduced Fg-SmoSA protein level (Figure 8F–G”), but had no apparent effect on SmoSD protein level (Figure 8H–I”).
Taken together, our data suggest that PKA-primed Smo phosphorylation does not affect Herc4 and Smo interaction, instead it affects Smo ubiquitination and protein levels regulated by Herc4.
Discussion
The regulation of Smo intracellular trafficking contributes to transducing the graded Hh signal (Wilson and Chuang, 2010). In the absence of Hh, Smo undergoes ubiquitination in the cytoplasm and fails to accumulate on the cell membrane, culminating in turning off Hh pathway and target genes expression. In the presence of Hh, Ptc relieves its inhibitory effect on Smo by binding Hh, resulting in Smo cell membrane accumulation and Hh pathway activation (Jia et al., 2004; Li et al., 2012; Jiang and Jia, 2015). The ubiquitination of Smo and its localization are tightly linked. In this study, we identified the HECT domain protein Herc4 as a negative regulator of Hh signalling through genetic screening. We further provided both genetic and biochemical evidences that Herc4 binds and ubiquitinates Smo to modulate its stability. We also demonstrated that Herc4-mediated Smo ubiquitination and protein degradation is inhibited by Hh. Our study that unveils a novel E3 ligase of Smo will deepen our understanding of Hh signalling regulation.
Herc4 belongs to the HECT family of ubiquitin ligases featured by the presence of a HECT domain and one or more RCC1-like domains. We found that both HECT and RCC1-like domains of Herc4 could form a complex with Smo. In addition, both N- and C-terminus of Smo interact with Herc4. Multiple sites interaction may facilitate the flexibility of Smo level regulation. Compared with SmoN, SmoC, especially the SAID domain, supplies both the binding and the ubiquitination sites for mediating Smo degradation by Herc4.
Previous study showed that Hrs binds Smo to promote Smo ubiquitination and degradation (Fan et al., 2013). In our study, we found that Hrs could bind Smo, Herc4 and Hrs promoted Smo ubiquitination via Herc4. USP8 and UCHL5 deubiquitinases have been characterized to downregulate Smo ubiquitination (Li et al., 2012; Xia et al., 2012; Zhou et al., 2018). We found that Herc4 promoted Smo ubiquitination, but it was attenuated by USP8 and UCHL5 (Figure 7A). Multiple deubiquitinases involved in regulation of Smo ubiquitination may facilitate Smo efficient recycling, especially in the presence of Hh signalling.
A recent study suggested that Smo is ubiquitylated at multiple lysine residues in the SAID domain (Li et al., 2012). Consistently, our data showed that Herc4 mediated Smo ubiquitination at multiple lysine residues. Different lysine residues ubiquitination and different ubiquitin-types may guide the Smo protein into the proteasome- and lysosome-mediated degradation. Many studies have verified that Smo undergoes both the proteasome- and lysosome-mediated degradation (Molnar et al., 2011; Li et al., 2012; Xia et al., 2012). Our data showed that Herc4 mediated Smo degradation by proteasome and lysosome, but mainly by proteasome.
Hh blocks Smo ubiquitination, which is vital for Smo activity. What is the underlying mechanism? In this study, we found that Hh regulates Smo stability by Herc4 in two ways. On the one hand, Hh inhibited Herc4–Smo association (Figure 8A), interestingly, it is not dependent on the PKA-primed phosphorylation. Based on our published paper (Zhou et al., 2018) and result shown in Figure 7B, it may be caused by the competing binding of UCHL5. On the other hand, Hh inhibited Herc4-mediated Smo ubiquitination, this time, it is dependent on the PKA-primed phosphorylation (Figure 8B and D). It is possible that PKA-primed phosphorylation may cause conformational change of Smo, which somehow hinder the efficient ubiquitination of nearby Smo Ks sites. Together, the results suggest that Hh blocks Smo ubiquitination by Herc4 through inhibiting Herc4–Smo interaction and stimulating PKA-primed phosphorylation.
Materials and methods
Constructs, mutants, and transgenes
The constructs for expression in S2 cells, including Myc-Ci, HA-Fu, HA-Ub, Fg-Smo, Myc-Smo, Myc-SmoN, Myc-SmoC, Myc-SmoSAID, Myc-SmoCΔSAID, and Myc-SmoΔSAID constructs, have been previously described (Doerks et al., 2002; Zhao et al., 2007; Zhou et al., 2015). To construct Fg-Herc4 and HA-Herc4, we amplified the corresponding cDNA fragments using DNA polymerase (Vazyme), and then cloned them into pUAST-3×Fg or pUAST-3×HA vectors. HA-Herc4C1030A was generated using PCR-based site-directed mutagenesis at the background of HA-Herc4. The HA-tagged Herc4-RCC and Herc4-HECT were constructed by inserting the corresponding coding sequences into the pUAST-3×HA vector. The RNAi lines for the screen were obtained from the Vienna Drosophila RNAi Center, including the herc4 RNAi line, v37220, Bloomington Drosophila Stock Center (BDSC), and National Institute of Genetics. The mutant of herc4, herc4G5486, was obtained from BDSC. MS1096, Smo–PKA, ptc-lacZ, apG4, and UAS-GFP have been described (Flybase) (Zhang et al., 2006). The HA-Herc4 and HA-Herc4C1030A transgenic flies were generated by injection of constructs into Drosophila embryos according to the methods described previously. The parental strain for all germline transformations is w1118. All stocks used in this study were maintained and raised under standard conditions.
Immunostaining of wing discs
Immunostaining and in situ hybridization of imaginal discs were performed with standard protocols (Zhou et al., 2015). Images were captured with FV10-ASW 2.0 Olympus confocal microscope. Primary antibodies were used at the following dilutions: mouse anti-Smo (1:100; DSHB); mouse anti-β Gal (1:500; Sigma); mouse anti-En (1:50; DSHB); mouse anti-Ptc (1:200; DSHB); rabbit anti-Fg (1:200; Thermo); mouse anti-HA (F7) (1:200; Santa Cruz); anti-ubiquitin (P4D1) (1:50, Santa Cruz); mouse anti-Myc (9E10) (1:200; Santa Cruz); and DAPI (1:1000; Santa Cruz). Secondary antibodies used in this study were bought from Jackson ImmunoResearch, and were diluted at 1:500.
Cell culture, transfection, immunoprecipitation, western blot, cell immunostaining
S2 cells were maintained at 25°C in Schneider’s Drosophila medium (S9895, Sigma) supplemented with 10% FBS (F0718, Gibco), 100 U/ml penicillin (Life Technologies), and 100 μg/ml streptomycin (Life Technologies). Transfection of S2 cells was performed using calcium phosphate according to the manufacturer’s instructions (Invitrogen). For S2 cell immunostaining, 48 h after transfected, cells were harvested and washed with PBS. Cells were fixed in 4% formaldehyde in PBS buffer for 20 min at room temperature. Then, cells were treated with PBT for 20 min, washed with PBS for 20 min three times. To mark the nucleus, cells were stained with DAPI for 20 min after secondary antibody incubation. Images of cells were acquired under FV10-ASW Olympus confocal microscope. The following antibodies were used for immunoprecipitation and western blot: mouse anti-HA (F-7) (1:2500; Santa Cruz); mouse anti-Myc (9E10) (1:2500; Santa Cruz); mouse anti-Fg (M2) (1:5000; Sigma); LysoTracterRed (1:500; Sigma); MitoTracterRed (1:500; Sigma); ERTracterGreen (1:500; Sigma); mouse anti-Actin (1:5000; ABclonal Technology); goat anti-mouse HRP (1:10000; Abmax). The blots were visualized using a chemiluminescent detection kit (Millipore).
RNA interference
To knock down genes in S2 cells, the double-stranded RNA (dsRNA) was generated by MEGAscript High Yield Transcription Kit (Ambion) according to the manufacturer’s instructions. The primer sequences of herc4 dsRNA are as follows: herc4-RNAi-Fw, 5′-GAATTAATACGACTCACTATAGGGAGAGGGCGGCACCTTGTCTAC-3′, herc4-RNAi-Bw, 5′-GAATTAATACGACTCACTATAGGGAGACTGGGACTGAATGACGAAAC-3′ and herc4-RNAi-5′UTR-Fw, 5′-GAATTAATACGACTCACTATAGGGAGATCCCAACCAGTTGGCATCGCC-3′, herc4-RNAi-5′UTR-Bw, 5′-GAATTAATACGACTCACTTAGGGAGAATTCTTTTGTGTCCACTTGCTCC-3′ were generated by PCR and used for generating dsRNA. dsRNA targeting the GFP full-length coding sequence was used as a control. The dsRNA-mediated gene knockdown was performed as previously described, S2 cells were cultured in serum-free medium containing dsRNA for 12 h at 25°C before transfection with DNA constructs. Then the culture medium was changed to serum medium for transfection and additional culturing for 48 h (Lawrence Lum et al., 2003; Li et al., 2012).
Protein stability assays
S2 cells were plated in 10-cm dishes and transfected with plasmids after 18–24 h. After another 24 h, the cells were split and transferred into 6-well cell culture plates at equivalent densities. Cells were treated with 20 μg/ml CHX (Calbiochem) for the indicated times before harvesting. After western blots, the band intensity was measured by Image J.
Ubiquitination assays
S2 cells were transiently transfected with the indicated combinations of expression vectors. Four hours before cells harvesting, MG132 (Calbiochem) was added to the media at a final concentration of 10 μM. The ubiquitination assays were then carried out based on the previously described protocol. Briefly, cells were lysed with denaturing buffer (1% SDS, 50 mM Tris-base, pH 7.5, 0.5 mM EDTA, and 1 mM DTT) and incubated at 100°C for 5 min. The lysates were then diluted 10-fold with regular lysis buffer and subjected to immunoprecipitation and western blot analysis.
Real-time quantitative PCR
For real-time quantitative PCR, tissues or cells were lysed in TRIzol (Invitrogen) for RNA isolation following standard protocols. All of the cytosol fraction and nuclei pellet RNA were extracted and the reverse transcription was carried out using PrimeScriptRT Reagent Kit with gDNA Eraser (TaKaRa). The primer pairs used were as follows: herc4, 5′-CCAGCCGGAGGAGCTGATGGCCGTG-3′ (forward) and 5′-GTTAAAAAGAGGAGAAAACTCTTC-3′ (reverse); Smo, 5′-CTCACCGACTTCCACACCGAAAAG-3′ (forward) and 5′-GGACGTCCCACAATCGATGGGAAG-3′ (reverse); actin, 5′-CGAAGAAGTTGCTGCTCTGGTTGTCG-3′ (forward) and 5′-GGACGTCCCACAATCGATGGGAAG-3′ (reverse).
Statistical analysis
Imaging data were analysed in the program Image J. The data shown in the figures were representative of three or more independent experiments and were analysed by Student’s t-test, P < 0.05 was considered significant.
Supplementary Material
Acknowledgements
We apologise to colleagues whose works were not cited owing to space limitation. We thank Dr Yun Zhao (Shanghai Institute of Biochemistry and Cell Biology, China) for providing plasmids and reagents. We also appreciate National Institute of Genetics of Japan (NIG), Vienna Drosophila RNAi Center (VDRC), and the Bloomington Stock Center (BSC) for providing fly stocks.
Funding
This work was supported by grants from the National Key Scientific Program of China (2011CB943902), the National Natural Science Foundation of China (30971679, 31071264, 31271531, and 31771615), and the Fundamental Research Funds for the Central Universities (090314380019).
Conflict of interest: none declared.
Author contributions: Q.Z. and W.J. designed the experiments. W.J., X.Y., Z.S., W.L., and Y.G. performed the experiments. W.J., X.Y., and Q.Z. wrote the manuscript.
References
- Aerne B.L., Gailite I., Sims D., et al. (2015). Hippo stabilises its adaptor Salvador by antagonising the HECT ubiquitin ligase Herc4. PLoS One 10, e0131113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcedo J., Ayzenzon M., Von Ohlen T., et al. (1996). The Drosophila Smoothened gene encodes a seven-pass membrane protein, a putative receptor for the Hedgehog signal. Cell 86, 221–232. [DOI] [PubMed] [Google Scholar]
- Casali A., and Struhl G. (2004). Reading the Hedgehog morphogen gradient by measuring the ratio of bound to unbound Patched protein. Nature 431, 76–80. [DOI] [PubMed] [Google Scholar]
- Corbit K.C., Aanstad P., Singla V., et al. (2005). Vertebrate Smoothened functions at the primary cilium. Nature 437, 1018–1021. [DOI] [PubMed] [Google Scholar]
- Denef N., Neubuser D., Perez L., et al. (2000). Hedgehog induces opposite changes in turnover and subcellular localization of Patched and Smoothened. Cell 102, 521–531. [DOI] [PubMed] [Google Scholar]
- Doerks T., Copley R.R., Schultz J., et al. (2002). Systematic identification of novel protein domain families associated with nuclear functions. Genome Res. 12, 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J., Jiang K., Liu Y., et al. (2013). Hrs promotes ubiquitination and mediates endosomal trafficking of Smoothened in Drosophila Hedgehog signaling. PLoS One 8, e79021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser M. (1992). Ubiquitin and intracellular protein degradation. Curr. Opin. Cell Biol. 4, 1024–1031. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M. (1995). Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Curr. Opin. Cell Biol. 7, 215–223. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M. (1996). Ubiquitin-dependent protein degradation. Annu. Rev. Genet. 30, 405–439. [DOI] [PubMed] [Google Scholar]
- Hooper J.E., and Scott M.P. (1989). The Drosophila patched gene encodes a putative membrane protein required for segmental patterning. Cell 59, 751–765. [DOI] [PubMed] [Google Scholar]
- Huang J., Sheung J., Dong G., et al. (2005). High-throughput screening for inhibitors of the e3 ubiquitin ligase APC. Methods Enzymol. 399, 740–754. [DOI] [PubMed] [Google Scholar]
- Ingham P.W., and McMahon A.P. (2001). Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 15, 3059–3087. [DOI] [PubMed] [Google Scholar]
- Ingham P.W., Nakano Y., and Seger C. (2011). Mechanisms and functions of Hedgehog signalling across the metazoa. Nat. Rev. Genet. 12, 393–406. [DOI] [PubMed] [Google Scholar]
- Jia J., and Jiang J. (2006). Decoding the Hedgehog signal in animal development. Cell. Mol. Life Sci. 63, 1249–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J., Tong C., and Jiang J. (2003). Smoothened transduces Hedgehog signal by physically interacting with Costal2/fused complex through its C-terminal tail. Genes Dev. 17, 2709–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J., Tong C., Wang B., et al. (2004). Hedgehog signalling activity of Smoothened requires phosphorylation by protein kinase A and casein kinase I. Nature 432, 1045–1050. [DOI] [PubMed] [Google Scholar]
- Jia J., Zhang L., Zhang Q., et al. (2005). Phosphorylation by double-time/CKIepsilon and CKIα targets cubitus interruptus for Slimb/β-TRCP-mediated proteolytic processing. Dev. Cell 9, 819–830. [DOI] [PubMed] [Google Scholar]
- Jiang J., and Hui C.C. (2008). Hedgehog signaling in development and cancer. Dev. Cell 15, 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang K., and Jia J. (2015). Smoothened regulation in response to Hedgehog stimulation. Front. Biol. 10, 475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulathu Y., and Komander D. (2012). Atypical ubiquitylation—the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat. Rev. Mol. Cell Biol. 13, 508–523. [DOI] [PubMed] [Google Scholar]
- Lum L., Yao S., Mozer B., et al. (2003). Identification of Hedgehog pathway components by RNAi in Drosophila cultured cells. Science 299, 2039–2044. [DOI] [PubMed] [Google Scholar]
- Li S., Chen Y., Shi Q., et al. (2012). Hedgehog-regulated ubiquitination controls Smoothened trafficking and cell surface expression in Drosophila. PLoS Biol. 10, e1001239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., and Nussinov R. (2011). Flexible cullins in cullin-RING E3 ligases allosterically regulate ubiquitination. J. Biol. Chem. 286, 40934–40942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum L., and Beachy P.A. (2004). The Hedgehog response network: sensors, switches, and routers. Science 304, 1755–1759. [DOI] [PubMed] [Google Scholar]
- Lv X., Chen H., Zhang S., et al. (2018a). Distinct functions of polycomb group proteins in regulating Ci transcription in developing Drosophila. J. Mol. Cell Biol. 10, 475–478. [DOI] [PubMed] [Google Scholar]
- Lv X., Chen H., Zhang S., et al. (2018b). Fsh-Pc-Sce complex mediates active transcription of Cubitus interruptus (Ci). J. Mol. Cell Biol. 10, 437–447. [DOI] [PubMed] [Google Scholar]
- Marigo V., Davey R.A., Zuo Y., et al. (1996). Biochemical evidence that Patched is the Hedgehog receptor. Nature 384, 176–179. [DOI] [PubMed] [Google Scholar]
- Molnar C., Ruiz-Gomez A., Martin M., et al. (2011). Role of the Drosophila non-visual ss-arrestin kurtz in Hedgehog signalling. PLoS Genet. 7, e1001335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasca di Magliano M., and Hebrok M. (2003). Hedgehog signalling in cancer formation and maintenance. Nat. Rev. Cancer 3, 903–911. [DOI] [PubMed] [Google Scholar]
- Qiu L., Joazeiro C., Fang N., et al. (2000). Recognition and ubiquitination of Notch by Itch, a hect-type E3 ubiquitin ligase. J. Biol. Chem. 275, 35734–35737. [DOI] [PubMed] [Google Scholar]
- Rohatgi R., Milenkovic L., and Scott M.P. (2007). Patched1 regulates Hedgehog signaling at the primary cilium. Science 317, 372–376. [DOI] [PubMed] [Google Scholar]
- Shabek N., Herman-Bachinsky Y., Buchsbaum S., et al. (2012). The size of the proteasomal substrate determines whether its degradation will be mediated by mono- or polyubiquitylation. Mol. Cell 48, 87–97. [DOI] [PubMed] [Google Scholar]
- Sisson J.C., Ho K.S., Suyama K., et al. (1997). Costal2, a novel kinesin-related protein in the Hedgehog signaling pathway. Cell 90, 235–245. [DOI] [PubMed] [Google Scholar]
- Smelkinson M.G., Zhou Q., and Kalderon D. (2007). Regulation of Ci-SCFSlimb binding, Ci proteolysis, and Hedgehog pathway activity by Ci phosphorylation. Dev. Cell 13, 481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone D.M., Hynes M., Armanini M., et al. (1996). The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 384, 129–134. [DOI] [PubMed] [Google Scholar]
- Taipale J., and Beachy P.A. (2001). The Hedgehog and Wnt signalling pathways in cancer. Nature 411, 349–354. [DOI] [PubMed] [Google Scholar]
- Wilson C.W., and Chuang P.T. (2010). Mechanism and evolution of cytosolic Hedgehog signal transduction. Development 137, 2079–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia R., Jia H., Fan J., et al. (2012). USP8 promotes Smoothened signaling by preventing its ubiquitination and changing its subcellular localization. PLoS Biol. 10, e1001238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Zhang L., Wang B., et al. (2006). A Hedgehog-induced BTB protein modulates Hedgehog signaling by degrading Ci/Gli transcription factor. Dev. Cell 10, 719–729. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Tong C., and Jiang J. (2007). Hedgehog regulates Smoothened activity by inducing a conformational switch. Nature 450, 252–258. [DOI] [PubMed] [Google Scholar]
- Zhou Z., Yao X., Li S., et al. (2015). Deubiquitination of Ci/Gli by Usp7/HAUSP regulates Hedgehog signaling. Dev. Cell 34, 58–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Yao X., Pang S., et al. (2018). The deubiquitinase UCHL5/UCH37 positively regulates Hedgehog signaling by deubiquitinating Smoothened. J. Mol. Cell Biol. 10, 243–257. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.