

Abstract
The sweet potato weevil, Cylas formicarius (F.) (Coleoptera: Brentidae), is an important pest of sweet potato worldwide. However, there is limited knowledge on the molecular mechanisms underlying growth and differentiation of C. formicarius. The transcriptomes of the eggs, second instar larvae, third instar larvae (L3), pupae, females, and males of C. formicarius were sequenced using Illumina sequencing technology for obtaining global insights into developing transcriptome characteristics and elucidating the relative functional genes. A total of 54,255,544 high-quality reads were produced, trimmed, and de novo assembled into 115,281 contigs. 61,686 unigenes were obtained, with an average length of 1,009 nt. Among these unigenes, 17,348 were annotated into 59 Gene Ontology (GO) terms and 12,660 were assigned to 25 Cluster of Orthologous Groups classes, whereas 24,796 unigenes were mapped to 258 pathways. Differentially expressed unigenes between various developmental stages of C. formicarius were detected. Higher numbers of differentially expressed genes (DEGs) were recorded in the eggs versus L3 and eggs versus male samples (2,141 and 2,058 unigenes, respectively) than the others. Genes preferentially expressed in each stage were also identified. GO and pathway-based enrichment analysis were used to further investigate the functions of the DEGs. In addition, the expression profiles of ten DEGs were validated by quantitative real-time PCR. The transcriptome profiles presented in this study and these DEGs detected by comparative analysis of different developed stages of C. formicarius will facilitate the understanding of the molecular mechanism of various living process and will contribute to further genome-wide research.
Keywords: different stages, differentially expressed genes, sweet potato weevil, transcriptome
Sweet potato (Ipomoea batatas (L.) Lam., Convolvulaceae) possesses remarkable importance in energy production, and the utilization as food, feed, and industrial products in China and worldwide. The sweet potato weevil, Cylas formicarius (F.) (Coleoptera: Brentidae) is one of the most destructive pest of sweet potato in the tropical and temperate growing areas, including China (Kuriwada et al. 2010, Prentice et al. 2015, Reddy and Chi 2015). This pest not only causes huge production losses, but also seriously affects the quality of sweet potato, both in the field and in the storage. C.formicarius infests all plant parts: roots, stems, foliage, and flowers seeds (Edison et al. 2009). Damage caused by the weevils is often worse during dry times (Reddy et al. 2014). In addition to damage caused directly by tunneling, larvae also cause indirect damage by facilitating entry of soil-borne pathogens (Uritani et al. 1975), which renders the product unsuitable for consumption. China accounts for the highest sweet potato production in the world (FAOSTAT 2016). However, its productivity is severely constrained by insect feeding during the production and storage processes, especially by the sweet potato weevil. The yield losses caused by sweet potato weevil damage range from 5 to 50%, and can even lead to a complete loss under heavy infestations (Wang et al. 2014). Reddy and Chi (2015) collected life table data for C. formicarius grown on I. batatas, and showed that the weevil could survive more than 4 mo; the mean of total preoviposition period was 50.1 d and female adults did not contribute to the population growth after 92 d. In southern China, C. formicarius could produce several generations per year, and overwinter in storage or in open fields (Wang et al. 2014). Eggs of C. formicarius are laid in cavities just below the skin of the root, and then larvae feed and pupate in the roots. The peak period of adult occurrence was from the beginning of August to October (Wang et al. 2014, Ye 2015). Control of the pest remains difficult. Efficacy of chemical insecticides to the weevils was limited by the cryptic nature of the larvae, the nocturnal activities of the adults, and increasingly pesticide resistance. Effective crop rotations could reduce the tuber damage compared to monoculture of sweet potato (Pillai et al. 1996, Ehisianya et al. 2013). Sweet potato weevils have the capacity to adapt and develop resistance to active proteins and compounds found or introduced into new sweet potato varieties (Mwanga et al. 2011). Currently, there are few superior varieties with high level of resistance against C. formicarius.
The high throughput sequencing technology is a powerful and cost-efficient tool for advanced research in many areas, including microRNA expression profiling, DNA methylation, especially de novo transcriptome sequencing for non-model organisms (Varshney et al. 2009, Parchman et al. 2010, Wang et al. 2010b, Sun et al. 2012, Zhang et al. 2015c). Illumina sequencing of transcriptome for organisms with completed genomes confirmed that the relatively short reads produced could be effectively assembled and used for gene discovery and gene expression analysis (Marioni et al. 2008, Hegedűs et al. 2009, Wang et al. 2011). The development of next-generation sequencing technologies and the de novo analysis of the transcriptome data could provide a clear insight into the molecular mechanism of different life process. Heyland et al. (2011) analyzed temporal and spatial gene expression changes during embryogenesis, larval development, and metamorphic stages of Aplysia californica (Cooper) using microarrays and in situ hybridization, revealed novel molecular components associated with life history transitions. Daines et al. (2011) provided interesting insights into the extent of alternate splicing in Drosophila melanogaster (Meigen) by the transcriptome of ten developmental stages of D. melanogaster. Zheng et al. (2015) sequenced the transcriptome of the early parasitic second-stage juveniles of Heterodera avenae (Wollenweber) and revealed the genes and molecular mechanisms of the incompatible interaction between H. avenae and the host plant Aegilops variabilis. Prentice et al. (2015) reported the transcriptome analysis of the second instar larvae of Cylas puncticollis (Boheman) and demonstrated that C. puncticollis exhibits a strong and systemic RNAi effect, suggesting the potential of RNAi as a future strategy to control C. formicarius. Though transcriptome sequencing has been broadly applied to illustrate gene expression patterns in many species, genomic, and transcriptomic details about C. formicarius, especially its biosynthetic pathways and gene expression in different stages, have remained largely unknown. Genomic resources available in public databases for C. formicarius were limited to a few sequences. Due to the limitations of information on its genetic basis, it is necessary to carry out an extensive transcriptome study to systematically gauge molecular differences associated with the development of different stages of C. formicarius.
The management of the weevils is vital for sweet potato production. Understanding the gene expression changes at various developmental stages of the sweet potato weevil will provide important insight into mechanisms underlying growth, differentiation, stage structure, and this will be useful for the management programs. In this study, in order to establish a useful database of transcriptome sequences as well as of differentially expressed genes (DEGs) at different developmental stages of C. formicarius, we performed de novo transcriptome sequencing by the Illumina Hiseq2000 sequencing platform. A comprehensive analysis of the transcriptome data for eggs, larvae, pupae, and adults of C. formicarius were present. 61,686 distinct sequences including hundreds of stage specific and metabolism genes were identified. A total of 35,789 unigenes could be annotated with known biological functions; enrichment analysis and pathway analysis were applied to find interesting biological processes. We expect these new dataset will provide genomic resource and new leads for future studies of this species.
Materials and Methods
Insect Rearing and Sample Preparation
A laboratory colony of the C. formicarius was established using the insects collected from Fujian province, China in 2011 and maintained on sweet potato storage roots at 28 ± 1°C, 75 ± 10% relative humidity. For this experiment, 100–150 virgin females and virgin males were collected separately at 2–8 d post eclosion to ensure they were actively feeding and sexually mature. Eggs and pupae were randomly picked out from the sweet potato with a brush. 100–150 second instar larvae (L2) and third instar larvae (L3) were collected ∼11 and 15 d post hatch, respectively. Different instars of C. formicarius were sampled from different generations and the total weight of each life stage (pooled whole body samples) is ∼500 mg. All samples were washed with diethyl pyrocarbonate treated water, then snap-frozen immediately in liquid nitrogen and stored at −80°C until further processing.
RNA Extraction and Library Construction
Total RNA form the five different developmental stages of C. formicarius (mixed sex of eggs, second instar and third instar larvae, pupae; adult females and adult males) were extracted individually using Trizol reagent (Invitrogen, Beijing, China) according to the manufacturer’s protocol. The RNA samples were treated with DNase I (Takara Biotech Co., Dalian, China) for purification from DNA contamination. RNA quality and purity were assessed using Nanodrop2000 spectrophotometer (Thermo Fisher Scientific, MA, USA) based on absorbance ratios at 260/280 and 260/230 nm. The integrity of the RNA preparation was verified using Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) with a minimum integrity number value of 8.0. RNA samples with high purity (OD260/280 between 1.9 and 2.0) and high integrity were selected and used for cDNA library construction. Poly(A) mRNA was purified from total RNA using oligo (dT) magnetic beads. Following purification, the mRNA is fragmented to small pieces using fragmentation buffer under elevated temperature and the cleaved RNA fragments were transcribed into first strand cDNA using reverse transcriptase and random primers. This was followed by second strand cDNA synthesis using DNA polymerase I and RNaseH. Then the high-throughput RNA-sequencing libraries (RNA-seq) were prepared following Illumina’s protocols and were sequenced on the Illumina Hiseq 2000 platform (Illumina, San Diego, USA) at the Beijing Genomics Institute (BGI; Shenzhen, China). The datasets were deposited in NCBI Sequence Read Archive (SRA, http://www.ncbi.nlm.nih.gov/sra) under the accession number SRP067907. This transcriptome shotgun assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GEOI00000000.
De Novo Transcriptome Assembly and Annotation
The raw reads were cleaned by removing adaptor sequence, low-quality sequences (tags with ambiguous sequences ‘N’), empty tags (sequence with only adaptor sequences but no tags), low complexity, and tags with a copy number of 1 (probably sequencing error) ahead of assembly. The quality reads were de novo assembled with Trinity software (Pertea et al. 2003). The TGI clustering tool (Iseli et al. 1999) is used to assemble all the unigenes from different samples to form a single set of non-redundant unigenes. The unigenes were annotated by aligning with the deposited ones in diverse protein databases including NCBI non-redundant protein (NR), UniProt/Swiss-Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG) database, Gene Ontology (GO) and Cluster of Orthologous Groups (COG) database using BLASTX (E-value ≤1e−5). Sequence orientations and the coding sequence (CDS) of unigenes were determined according to the best hit in these databases. ESTScan (Iseli et al. 1999) was used to predict the sequence direction and the CDS when unigenes were unaligned to any of the databases. With NR annotation, Blast2GO program (Conesa et al. 2005) was used to get GO annotation of the unigenes. After getting GO annotation for every unigene, WEGO software (Ye et al. 2006) were used to do functional classification for all unigenes and to decipher the distribution of gene functions at the macro level. Then we used the blast information to extract CDS from unigene sequences and translated them into peptide sequences.
Differentially Expressed Genes
For gene expression analysis, the number of expressed unigenes was calculated and then normalized to TPM (number of transcripts per million tags). A rigorous algorithm was developed to identify DEGs between two samples (Audic and Claverie 1997, Zhang et al. 2015b). The false discovery rate (FDR) was used to determine the threshold of P value in multiple tests (Benjamini and Yekutieli 2001). Fold changes (log2 Ratio) were estimated according to the normalized gene expression level in each sample (Wang et al. 2010a). “FDR ≤0.001 and the absolute value of log2 Ratio ≥1” were used as the threshold to judge the significance of gene expression difference. Pairwise and multi-condition analyses were used to detect DEGs between two samples and among the five development stages of C. formicarius. For the functional and pathway enrichment analysis, the DEGs were then mapped into GO terms and the KEGG databases, significantly enriched GO and KEGG terms were determined by P value ≤0.05. Cluster analysis of gene expression patterns were performed with “cluster” software (Eisen et al. 1998).
Quantitative Real-Time PCR Analysis of Gene Expression
To validate the data obtained by the transcriptome sequencing, the relative expression levels of ten DEGs were analyzed by real-time quantitative PCR (qPCR) with three biological replicates. Specific primers of selected genes were designed by Primer Premier 5.0 (Premier Biosoft International, CA, USA) (Table 1). Total RNA from ∼150 mg of the eggs, L2, L3, pupae, females, and males of C. formicarius was extracted separately as described above. First-strand cDNA was synthesized using AMV first strand cDNA synthesis kit (Sangon, Shanghai, China). The SYBR Green real-time PCR assay was performed on a LightCycler 480 real-time PCR instrument (Roche Diagnostics, Mannheim, Germany) using the SG fast qPCR master mix (BBI Life Science, Ontario, Canada) according to the manufacturer’s protocol in a total volume of 20 µl, containing 2 μl of cDNA, 0.4 μl of 10 μM of each primer, and 10 ml Master mix. The PCR was performed at 95°C for 3 min, followed by 40 cycles of 95°C for 7 s, 57°C for 10 s and 72°C for 20 s. The experiments were repeated three times. The melting curve analysis was conducted from 60 to 95°C. Each of the two primer pairs for the tested genes and endogenous references produced a single peak in melting curve analyses. RPS11 and β-actin were observed to be stable among different stages of C. formicarius and used as an endogenous control. Relative gene expression was calculated by the 2−ΔΔCt method (Pfaffl 2001). Means and standard errors were calculated and analyzed by analysis of variance followed by the Tukey–Kramer test, using SPSS 16.0 (SPSS, Chicago, IL).
Table 1.
Primer sequences for qRT-PCR
| Gene | Forward | Reverse | Length (bp) |
|---|---|---|---|
| PG297 | CGGTGGGTAGTTTTACTGTTCC | CCGTTCCATTCTTTATGAGCC | 112 |
| Lip17338 | CATCCGAGCCGTTCAAAGT | CGACCATTCCACCGACAAA | 125 |
| P450 | TCACCCAAATCATCACCGAA | CGCAGATGTTTCCAAGTCG | 170 |
| OBP3892 | AATCATAATGTTGCGTCTGTGC | GGATGTCTCCGCTAATTGCTT | 188 |
| GST9774 | GGAAAGTAGTATCGGCGAGGTT | GCACTGCCTAACAGCCAAAG | 196 |
| CYP4G27 | ATTGGACCCAGGCTTCTCAT | TCGGTATTCAGGCGATTTGT | 96 |
| H19735 | AGCGAAACCCAAAAGAACG | CTCTTTGCTAAGTTCTTCCCTGTAC | 141 |
| OBPs2 | TGTGCTTCGCAAAATGTCTATC | GGAAAAGTTTACCCATGTCGG | 183 |
| TH14530 | GTTCGCCATCAAGAAGTCGT | GGTCTACCTCGCTAGAGTCGTT | 161 |
| GH48 | CTTCTTGGACTTGTTCGTAGGTG | TTTGGAGGCTTTGGTGAGG | 169 |
Results and Discussion
Sequencing and Assembly of the C. formicarius Transcriptome
In this study, to obtain an overview of gene expression profile at different developmental stages of C. formicarius, total RNA were extracted from the eggs, second and third instar larvae, pupa, females, and males separately. Six cDNA libraries were constructed and sequenced using the Illumina Hiseq 2000 sequencing platform. Each sample yielded more than 3.5 million raw reads, and a total of 60,852,718 raw reads were obtained with cycle Q20 of 97.82% (Table 2). After removing the adaptor sequences and the low-quality reads, a total of 54,255,544 clean reads were obtained with an overall length of more than 4,882 megabase, which accounted for 89.16% of the raw data. The GC content of the transcriptome was 41.41%. Up to 46.55% of the sequences in the reference tag database could be identified by unique tag (Table 2).The distribution of tag expression was used to evaluate the normality of the transcriptome data. In this study, the distribution of total clean tags and distinct clean tags over different tag abundance categories showed similar patterns for all six samples. However, under the distribution of total tags, high-expression tags with copy numbers larger than 100 are in absolute dominance whereas low-expression tags with copy numbers smaller than 10 occupy the majority of distinct tag distributions. A saturation analysis was performed to confirm whether the sequencing depth was sufficient for the transcriptome coverage. The number of detected unigenes increased with the total number of tags. When the number of sequencing tags reaches 2 million, the number of detected genes almost ceases to increase and library capacity reached saturation (Supp Fig. 1 [online only]). Quality reads were de novo assembled into 115,281 contigs, with the average length of 349 nt and the median length value (N50) of 657. The contigs grouped into 61,686 unigenes, with an average length of 1,009 nt and N50 of 1758. Total length of the unigenes was 62,270,490 nt. The size distribution of the contigs and unigenes was shown in Fig. 1. About 46% of all unigenes were smaller than 500 nucleotides, 20% had a size of up to 1,000 nt, 20% were between 1,000 and 2,000 nt long, followed by ∼14% unigenes longer than 2,000 nt.
Table 2.
Statistics of reads in the C.formicarius transcriptomes
| Item | Eggs | L2 | L3 | Pupae | Females | Males |
|---|---|---|---|---|---|---|
| Total raw reads | 3,506,028 | 3,628,506 | 3,691,650 | 3,515,889 | 3,675,722 | 3,655,575 |
| Clean reads | 3,440,924 | 3,575,823 | 3,641,125 | 3,441,973 | 3,606,789 | 3,589,984 |
| Distinct Tag | 51,585 | 42,343 | 41,807 | 57,638 | 53,008 | 52,399 |
| Tags mapping to gene | 3,112,632 | 3,315,991 | 3,395,530 | 3,147,847 | 3,270,395 | 3,259,878 |
| Clean tag mapping to gene (%) | 90.46 | 92.73 | 93.25 | 91.45 | 90.67 | 90.80 |
| Distinct tag mapping to gene | 36,209 | 30,638 | 29,311 | 40,437 | 36,318 | 35,537 |
| All Tag-mapped Genes | 26,822 | 25,104 | 25,125 | 28,715 | 27,477 | 27,469 |
| Tag-mapped Genes (%) | 43.48 | 40.70 | 40.73 | 46.55 | 44.55 | 44.53 |
Fig. 1.

Length distribution of C. formicarius (A) contigs, (B) unigenes, (C, E) CDS and (D, F) predicted proteins. The x-axis indicates the length range of the sequences. The y-axis denotes the sequences number in every range of length.
Unigene Annotation and Classification
For predict proteins and gene annotations, the unigene sequences of sweet potato weevil were searched against protein databases using BLASTX (E-value ≤0.00001) in the following order: NR, SwissProt, KEGG, GO, and COG. Then the blast results were used to extract CDS from unigene sequences and translate them into peptide sequences. CDS of unigenes have no hit in blast were predicted by ESTScan, and 2,131 unigenes obtained significant Blast hits. The size distribution of the CDS and predicted proteins were shown in Fig. 1. Function information of the unigenes could be predicted from annotation of the most similar protein in the above databases. In this study, a total of 35,789 unigenes could be annotated with known biological functions, comprising 58% of all the unigenes in the transcriptome libraries (Table 3). 35,172 (57%) unigenes were annotated against NR database. The characteristics of the homology search of Illumina sequences against the NR database were further analyzed with the E-value distribution, identity distribution, and species distribution (Fig. 2). For the E-value distribution of the predicted proteins, the top hits indicated that 55.5% of the mapped sequences had a significant similarity with a stringent threshold of less than 1e−45, and 44.5% of the similar sequences ranged from 1e−5 to 1e−45 (Fig. 2a). For the similarity distribution of the predicted proteins, 47.6% of the sequences had a similarity higher than 60%, and 14.9% of the sequences had a similarity higher than 80% (Fig. 2b). For the best-matched species distribution (Fig. 2c), 72.4% of the distinct sequences mapped to the sequences of the red flour beetle Tribolium castaneum (Herbst), which is a widely distributed stored-product pest (Richards et al. 2008, Kim et al. 2010); 9.7% mapped to the mountain pine beetle Dendroctonus ponderosae (Hopkins) (Keeling et al. 2012) and 1.3% to the pea aphid Acyrthosiphon pisum (Harris) sequences (IAGC 2010). Searching against Swissprot database found 27,628 unigenes (44.8%) hit deposited genes.
Table 3.
The functional annotations of the C. formicarius transcriptome
| Annotated Databases | Annotated Number | Percentage (%) |
|---|---|---|
| Gene annotation against NR | 35,172 | 57.0 |
| Gene annotation against NT | 13,845 | 22.4 |
| Gene annotation against Swiss-Prot | 27,628 | 44.8 |
| Gene annotation against KEGG | 24,796 | 40.2 |
| Gene annotation against COG | 12,660 | 20.5 |
| GO annotation for NR protein hits | 17,348 | 28.1 |
| All annotated unigenes | 35,789 | 58.02 |
Fig. 2.

Characteristics of the homology search of illumina sequences against the non-redundant (NR) database. (A) E-value distribution of BLAST hits for each sequence with an E-value cut-off of 1e−5. (B) Similarity distribution of the top BLAST hits for each unigenes. (C) Species distribution is shown as a percentage of the total homologous unigenes.
GO is an international standardized gene functional classification system which offers a dynamic-updated controlled vocabulary and a strictly defined concept to comprehensively describe properties of genes and their products in any organism (Botstein et al. 2000). GO functional annotation could be obtained by NR annotation. In this study, according to the GO database, 17,348 sequences were classified into 59 subcategories belonging to 3 main categories, including biological process (27 subcategories), cellular component (15 subcategories), and molecular function (17 subcategories) (Fig. 3). In the category of biological process, ‘cellular process’, ‘metabolic process’, and ‘biological regulation’ comprised the largest proportion of sequences, accounting for 60.7, 46.6, and 28.2% of the total, respectively. Two sequences were categorized into the ‘carbon utilization’ and three sequences were categorized into ‘cell killing’ subgroups. In the category of cellular components, ‘cell part’ and ‘intracellular’ comprised of 7,690 and 6,792 unigenes, and ‘cytoplasm’ comprised of 3,662 unigenes, and these three subgroups were dominant over the others. In the category molecular function, sequences with the functions of ‘binding’, ‘catalytic activity’, and ‘transporter’ comprised 8,566, 8,658, and 1,538 unigenes, respectively.
Fig. 3.

Functional annotation of assembled sequences of C. formicarius based on GO. 17,348 unigenes were annotated into three categories: cellular component, molecular function, and biological process.
To further evaluate the completeness of the transcriptome library and the effectiveness of annotations, all unigenes were aligned to the COG database to predict and classify possible functions. COG is a database where orthologous gene products are classified. Every protein in COG is assumed to be evolved from an ancestor protein, and the whole database is built on coding proteins with complete genome as well as system evolution relationships of bacteria, algae, and eukaryotes (Tatusov et al. 2003). In total, 12,660 unigenes were assigned to 25 functional categories (Fig. 4). The categories of ‘general function prediction only’, ‘replication, recombination and repair’, and ‘translation, ribosomal structure, and biogenesis’ were the top three largest with 4,950, 2,142, and 1,812 unigenes, respectively. The ‘RNA processing and modification’, ‘extracellular structures’, and ‘nuclear structure’ were the smallest COG categories with 124, 17, and 8 unigenes, respectively.
Fig. 4.

Clusters of orthologous group (COG) classification. 12,660 unigenes were functionally classified into 25 COG categories.
Pathway information of unigenes can be obtained from KEGG pathway annotations. The KEGG pathway database records networks of molecular interactions in the cells and helps to further understand genes’ biological functions (Kanehisa and Goto 2000). In this study, KEGG pathway assignment was used to identify the biological pathways that the putative genes are involved in. A total of 24,796 (40.2%) sequences were grouped into 258 pathways (Supp Table 1 [online only]). The most dominant clusters were metabolic pathways (3,619 unigenes), regulation of actin cytoskeleton (942 unigenes), and focal adhesion (824 unigenes). In the metabolism cluster, most abundant pathways involved in purine metabolism, pyrimidine metabolism, glycerolipid metabolism (1.32%), drug metabolism (1.48%) and glycerophospholipid metabolism (1.86%). These functional annotations provide a basis for exploring specific biological processes, functions, subcellular localization, and pathways of gene products in C. formicarius research.
Changes in Gene Expression Profile Among the Different Developmental Stages
The differentially expressed unigenes among the eggs, second instar, and third instar larvae, pupae, female adults, and male adults of C. formicarius were identified (FDR ≤0.001 and log2Ratio ≥1). Differential expression analysis was conducted by comparing multiple samples or two samples (pairwise analysis). A total of 11,258 unigenes showed differential expression across the five developmental stages. To identify genes showing a significant change in expression among the six samples, the DEGs among the five stages were presented by heat-map (Supp Fig. 2 [online only]). The expression profile suggested significant transcriptional complexities during development of sweet potato weevil. The differential expression patterns among libraries revealed that the number of DEGs between eggs and other stages are larger than that of the others (Fig. 5). The largest differences occurred between the egg and L3 libraries with 548 genes up-regulated and 1,593 genes down-regulated, followed by the egg and the male libraries with 983 genes up-regulated and 1,075 genes down-regulated. These results revealed that greater number of unique genes may be required for embryogenesis than for post-embryonic development. The smallest difference was shown between male and female, in which only 351 transcripts were identified with 182 genes up-regulated and 169 genes down-regulated. 388 DEGs were detected between the L2 and L3 libraries with 119 genes up-regulated and 269 genes down-regulated. The unique or shared DEGs in the pairwise comparisons between the different developmental stages are shown in Fig. 6. 165, 274, 197, 184, and 216 genes were exclusively expressed in L2, L3, pupae, females, and males compared with the eggs, respectively. 403 DEGs were commonly expressed across all comparison groups (Supp Table 5 [online only]).
Fig. 5.

Changes in gene expression profile among the eggs, second instar larvae, third instar larvae, pupae, females, and males of C. formicarius. Up-regulated (gray) and down-regulated (white) genes between different stages were quantified.
Fig. 6.

Venn diagrams showing the numbers of unique and shared DEGs differentially expressed unigenes in pairwise analysis.
The differential gene expression levels across different developmental stages of C. formicarius were analyzed. The results showed that a large number of genes were silent or uniquely expressed during the development. Compared with other stages, males, and females had notably higher expression levels for unigenes related to endopolygalacturonase, odorant-binding protein, lipase, CRAL/TRIO domain-containing protein, peritrophic membrane chitin binding protein 2, regucalcin-like isoform 1, and some hypothetical or unknown proteins (Supp Table 4 [online only]). The expression level of endopolygalacturonase in male was the highest with 15,188 TPM, but this value for pupae, eggs, L2, and L3 was only 6.39, 1.45, 1.96, and 0.82, respectively. It has been reported that polygalacturonase plays a key role in the insect feeding (Kirsch et al. 2012, Walker and Allen 2010, Zhang et al. 2015a); odorant-binding proteins are important for odor recognition, which allows insects to find mates and perceive host plant odorants (Li et al. 2016); CRAL/TRIO domain proteins play a conserved role in regulating the protein kinase Ras signaling (Johnson and Kornfeld 2010); peritrophic membrane chitin-binding proteins may play an important role for the protection of the insect midgut epithelium from mechanical damage by food particles, bacterial damage, and parasite invasion (Zhao et al. 2014). Compared with other stages, adults of C. formicarius face more complicated external environment, the high expression levels of these genes in adult may imply their roles in insect adaptability and breeding. The second and third instar larvae stages had high level expression transcripts related to cuticular protein 49Ab, serine protease P40, cathepsin L-like proteinase, filaggrin-2, lysosomal alpha-mannosidase and some hypothetical or unknown proteins (Supp Table 4 [online only]). Serine proteases play important roles during defense response or immune reaction (Yu et al. 2015, He et al. 2016). Cathepsin L proteinase are important in processing the major glutaminerich dietary proteins in cereals (Goptar et al. 2012). However, further studies are still needed to verify the functions of these genes in C. formicarius.
Genes preferentially expressed at each stage were also identified (Supp Fig. 3 [online only]). After filtering low level expression unigenes (TPM <1), 67 unigenes were specifically expressed in males, include unigenes encoding membrane alanyl aminopeptidase, protein kinase c alpha binding protein, calponin-homology and microtubule-associated protein, odorant receptor 89. However, the majority of the specifically expressed unigenes in the males are of unknown function. 51 unigenes specifically expressed in the female library, which annotated as omega subunit family protein, vitellogenin, RNA-binding proteins, synaptotagmin, pdz domain protein arc, serine protease P98, phosphatidylcholine transfer protein and functional unknown proteins; a substantial number of the uniquely expressed genes are related to reproductive and oviposition. 143 unigenes are specifically expressed in pupae, more than half of them have no functional annotation and the others include cuticular protein, cytochrome P450 CYP49a1, hypothetical protein TcasGA2, protein CLEC-199, oxoglutarate (alpha-ketoglutarate) dehydrogenase (lipoamide), etc. 101 unigenes were specifically detected in eggs library, include transcripts of body fluid secretion, cytochrome P450 15A1, serine protease P144, alcohol dehydrogenase, proteolysis proteins, etc.; a large portion of these genes may play roles in embryonic development and hatching. 24 unigenes were specifically expressed in the third instar stage, 18 unigenes were preferentially expressed in the second instar stage, with functional unknown unigenes made up a high proportion. Understanding of the functions of the preferentially expressed genes in further studies will be useful in the development of new pest control strategies.
Functional Annotation of Differentially Expressed Unigenes
An enrichment analysis based on GO terms was performed to investigate the protein function and associated biological process of the DEGs that affect the development process. We compared the gene expression differences occurred between the eggs and other stages of C. formicarius. The results showed an overall similar distribution of genes grouping into the respective terms. In the biological process category, cellular process (71.43–76.42%), metabolic process (60.82–66.79%), primary metabolic process (45.36–50%), cellular metabolic process (44.95–49.47%), and macromolecule metabolic process (31.07–38.48%) were the most significantly enriched GO terms of DEGs. In the cellular component analysis, there were significantly high number of DEGs classified as cell (80.45–84.07%), intracellular part (69.42–73.67%), organelle (60.61–64.82%), cytoplasm (48.21–52.87%), intracellular membrane-bounded organelle (43.94–48.67%), cytoplasmic part (38.84–45.55%), and macromolecular complex (39.12–44.21%). In the molecular function category, the GO terms of DEGs were predominantly enriched in catalytic activity (58.29–60.10%), binding (51.62–55.94%), hydrolase activity (25.11–28.25%), small molecule binding (17.21–17.98%), and nucleotide binding (16.38–17.44%). However, we also detected genes expression specific to a certain developmental library at the molecular function level. The functional DEGs involved in ‘carboxylic acid transmembrane transporter activity’, ‘l-lactate dehydrogenase activity’, and ‘lactate dehydrogenase activity’ were specially present in the eggs versus pupa libraries. When compared the eggs and male libraries, GO analysis revealed specific enrichment of DEGs involved in ‘carbohydrate kinase activity’, ‘carboxypeptidase activity’, and ‘copper ion binding’. Specific enrichment of genes related to ‘cyclin binding’ and ‘enzyme inhibitor activity’ were noticed between the eggs and L2 libraries. Two DEGs in the term of ‘malic enzyme activity’ were specific expressed in eggs versus female libraries. Interestingly, DEGs under the ‘C-acyltransferase activity’ term were enriched in L2 and L3, but not present in the other stages. In contrast, unigenes involved in ‘cargo receptor activity’ were expressed within female, male, and pupa, but not present L2 and L3 libraries. Two genes in the GO term of ‘glyceraldehyde-3-phosphate dehydrogenase (NAD+) (phosphorylating) activity’ were specific expressed in the eggs versus male and eggs versus pupae samples (Supp Table 2 [online only]).
Biological functions of genes could be further understood by pathway-based analysis. To gain a more detailed understanding of the developmental mechanisms, all DEGs were mapped to KEGG database terms and compared with the whole transcriptome data. By comparing all other stages to the egg, metabolic pathways, pancreatic secretion and lysosome were the top three most abundant pathways of the DEGs. Notably, some pathways were only significantly enriched (P value ≤0.05) in certain libraries (Supp Table 3 [online only]). 972 differentially regulated unigenes with KEGG annotation were identified between the egg and female samples, specific significantly enrichment of genes were observed for pathways of phototransduction (0.93%), PPAR signaling pathway (1.44%) and selenocompound metabolism (0.31%). A total of 973 DEGs were found between eggs and L2 samples and specific enrichment of arachidonic acid metabolism (0.72), drug metabolism-cytochrome P450 (0.92%), metabolism of xenobiotics by cytochrome P450 (1.03%), and primary bile acid biosynthesis (0.31%) were noticed. 1,049 differentially regulated unigenes were identified between the eggs and male libraries, transcription of genes associated with pentose phosphate pathway (1.05%) was functionally significantly enriched. 540 differentially regulated unigenes were identified between the eggs and pupa libraries, specific enrichment of unigenes was observed for pathways involved in cardiac muscle contraction (2.41%), gastric acid secretion (2.41%), arginine and proline metabolism (1.67%), and citrate cycle (1.48%). These results indicate that DEGs involved in these pathways may play an important role in the development of different insect stages or instars.
We compared the developmental transcriptomes of C. formicarius and gained a first insight into interesting similarities and differences among different stages. It was reported that the growth of holometabolous insects such as molting, metamorphosis and reproduction is regulated by the molting hormone (20-hydroxyecdysone) and the status quo hormone (juvenile hormone, JH) (Levine et al. 1991). The level of methyl farnesoate (MF), which is a sesquiterpene compound and the crustacean homolog of JH, varied with ovarian maturation; and qPCR results proved the related enzymes in MF biosynthesis exhibited similar variation trends to that of the level of MF in hemolymph (Xie et al. 2015). RNA interference-mediated knockdown of LdTorso of D. melanogaster delayed larval development, impaired pupation and adult emergence, and induced the JH biosynthesis gene expression (Zhu et al. 2015). Tan et al. (2015) demonstrated that the hormonal receptor of ecdysteroids regulates the growth and development in Apolygus lucorum. In this study, we compared the relative expression level of three ecdysone-related genes (E22953–E514) and five JH-related genes (JH13050–JH18635). The result showed that the relative expression of ecdysone and JH related genes differed among the C. formicarius stages (Fig. 7). Further analysis will be needed to address their functions in our subsequent studies.
Fig. 7.

Expression profiles of ecdysone-related genes (E22953–E514) and juvenile hormone-related genes (JH13050–JH18635) in eggs, second instar larvae, third instar larvae, pupae, females and males of C. formicarius.
Functional annotations of DEGs make it possible to know the molecular mechanisms underlying specific biological processes of C. formicarius. However, these DEGs represented only a small proportion of the total transcripts that were found in transcriptome database. In addition to these annotated genes, some of the unigenes could not be matched with any existing ones. Further studies on DEGs associated with the GO terms and pathways are needed. New genes or molecular markers related to different stages formation might be identified from this group in the future.
Validation of DEGs Data by qPCR
Ten DEGs were subjected to RT-qPCR analysis to validate the accuracy and reproducibility of the Illumina RNA-seq results due to the lack of RNA-seq biological replicates. The candidate genes selected for validation were differentially expressed at different developmental stages and associated with growth, metabolism and secondary metabolites biosynthesis. β-actin and RPS11 were used as reference gene for data normalization. No amplification in no-template controls and single specific melt curves confirmed the specificity of the RT-qPCR assays. Fold changes from qPCR were compared with the RNA-seq expression profiles (Fig. 8). In general, genes showed similar expression patterns with the RNA-seq data, confirming the reliable of RNA-seq data analysis.
Fig. 8.
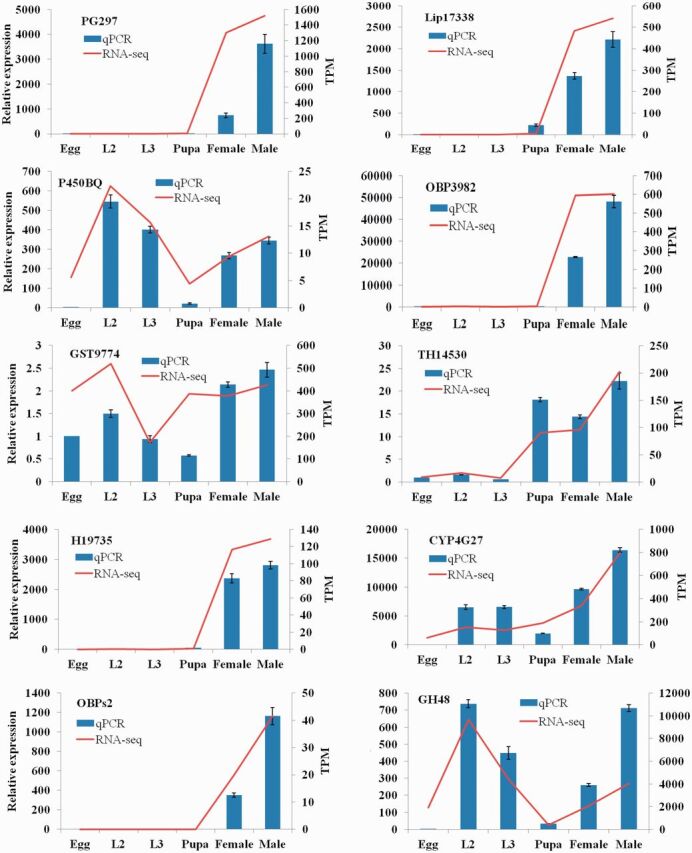
RNA-seq and qPCR analyses of expression levels of development related genes in eggs, second instar larvae, third instar larvae, pupae, females and males of C. formicarius. Gene expression determined by qPCR was provided as mean of three replicates. Error bars represent the standard error of the calculated mean.
The sweet potato weevil C. formicarius is the most important biological threat to productivity and marketability of sweet potato in southern China. The basic molecular biology of the sweet potato weevil is essential for developing new effective control technologies. This study provides an overview of the transcriptome of different developed stages of C. formicarius. More than 61,686 distinct sequences were produced with 35,789 sequences having an above cut-off blast result. Furthermore, we presented a relevant resource for functional analysis of DEGs among different development stages of C. formicarius. These findings provide a substantial contribution to existing sequence resources of C. formicarius and will certainly facilitate our understanding of this and other related species. The annotated transcriptome sequences and gene expression profiles will contribute to explorations of the molecular mechanisms of pest adaptation, formation, development and are benefit to the control of C. formicarius.
Supplementary Data
Supplementary data are available at Journal of Insect Science online.
Acknowledgments
We appreciate Beijing Genomics Institute at Shenzhen, China for help in sequencing. The authors thank Mrs Zhao Limin and Wen Haiyue for preparing insects.
Funding
This study was supported by China Agriculture Research System (CARS-11-B-08), and the National Natural Science Foundation of China (No. 31372265).
References Cited
- Audic S., Claverie J. M. 1997. The significance of digital gene expression profiles. Genome Res. 7: 986–995. [DOI] [PubMed] [Google Scholar]
- Benjamini Y., Yekutieli D. 2001. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 29: 1165–1188. [Google Scholar]
- Botstein D., Cherry J. M., Ashburner M., Ball C. A., Blake J. A., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., et al. 2000. Gene Ontology: tool for the unification of biology. Nat. Genet. 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A., Götz S., García-Gómez J. M., Terol J., Talón M., Robles M. 2005. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21: 3674–3676. [DOI] [PubMed] [Google Scholar]
- Daines B., Wang H., Wang L., Li Y., Han Y., Emmert D., Gelbart W., Wang X., Li W., Gibbs R., et al. 2011. The Drosophila melanogaster transcriptome by paired-end RNA sequencing. Genome Res. 21: 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edison S. V., Hegde, Makeshkumar T., Srinivas T., Padmaja G. 2009. Sweetpotato in the Indian sub-continent pp. 391–414. In:Loebenstein G., Thottappillyeds G. (eds.), The sweetpotato. Springer, Dordrecht. [Google Scholar]
- Ehisianya C. N., Ukeh D. A., Isah M. D., Lale N. E. S., Umeozor O. C. 2013. Field efficacy of neem seed oil and diazinon in the management of sweetpotato weevil, Cylas puncticollis (Boh.) in south-eastern Nigeria. J. Plant Stud. 2: 135–144. [Google Scholar]
- Eisen M. B., Spellman P. T., Brown P. O., Botstein D. 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Natl Acad. Sci. U S A. 95: 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAOSTAT 2016. Available online: http://faostat3.fao.org/browse/Q/QC/E.
- Goptar I. A., Semashko T. A., Danilenko S. A., Lysogorskaya E. N., Oksenoit E. S., Zhuzhikov D. P., Belozerskyd M. A., Dunaevskyd Y. E., Oppert B., Filippova I. Yu., et al. 2012. Cysteine digestive peptidases function as post-glutamine cleaving enzymes in tenebrionid stored product pests. Comp. Biochem. Physiol. 161B: 148–154. [DOI] [PubMed] [Google Scholar]
- He Y. X., Cao, Zhang S., Rogers J., Hartson S., Jiang H. 2016. Changes in the plasma proteome of Manduca sexta larvae in relation to the transcriptome variations after an immune challenge: evidence for high molecular weight immune complex formation. Mol. Cell. Proteomics 15: 1176–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedűs Z., Zakrzewska A., Ágoston V. C., Ordas A., Rácz P., Mink M., Spaink H. P., Meijer A. H. 2009. Deep sequencing of the zebrafish transcriptome response to mycobacterium infection. Mol. Immunol. 46: 2918–2930. [DOI] [PubMed] [Google Scholar]
- Heyland A., Vue Z., Voolstra C. R., Medina M., Moroz L. L. 2011. Developmental transcriptome of Aplysia californica. J. Exp. Zool B. Mol. Dev. E. 15: 113–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Aphid Genomics Consortium. 2010. Genome sequence of the pea aphid Acyrthosiphon pisum. PLoS Biol. 8: e1000313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iseli C., Jongeneel C. V., Bucher P. 1999. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. ISMB 99: 138–148. [PubMed] [Google Scholar]
- Johnson K. G., Kornfeld K. 2010. The CRAL/TRIO and GOLD domain protein TAP-1 regulates RAF-1 activation. Dev. Biol. 341: 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. 2000. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling C., Henderson H., Li M., Yuen M., Clark E. L., Fraser J. D., Huber D. P. W., Liao N. Y., Docking T. R., Birol I., et al. 2012. Transcriptome and full-length cDNA resources for the mountain pine beetle Dendroctonus ponderosae Hopkins, a major insect pest of pine forests. Insect Biochem. Mol. Biol. 42: 525–536. [DOI] [PubMed] [Google Scholar]
- Kim H. S., Murphy T., Xia J., Caragea D., Park Y., Beeman R. W., Lorenzen M. D., Butcher S., Manak J. R., Brown S. J. 2010. BeetleBase in 2010: revisions to provide comprehensive genomic information for Tribolium castaneum. Nucleic Acids Res. 38: D437–D442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch R., Wielsch N., Vogel H., Svatoš A., Heckel D. G., Pauchet Y. 2012. Combining proteomics and transcriptome sequencing to identify active plant-cell-wall-degrading enzymes in a leaf beetle. BMC Genomics 13: 587.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriwada T., Kumano N., Shiromoto K., Haraguchi D. 2010. Effect of mass rearing on life history traits and inbreeding depression in the sweetpotato weevil (Coleoptera: Brentidae). J. Econ. Entomol. 103: 1144–1148. [DOI] [PubMed] [Google Scholar]
- Levine R. B., Fahrback S. E., Weeks J. C. 1991. Steroid hormones and the reorganization of the nervous system during metamorphosis. Sem. Neurosci. 3: 437–447. [Google Scholar]
- Li J., Zhang L., Wang X. 2016. An odorant-binding protein involved in perception of host plant odorant in locust Locusta migratoria. Arch. Insect Biochem. Physiol. 91: 221–229. [DOI] [PubMed] [Google Scholar]
- Marioni J. C., Mason C. E., Mane S. M., Stephens M., Gilad Y. 2008. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 18: 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mwanga R. O. M., Ghislain M., Kreuze J., Ssemakula G. N., Yencho C. 2011. Exploiting the use of biotechnology in sweetpotato for improved nutrition and food security: progress and future outlook. Proceedings of the International Conference On Agbiotech. Biosafety & Seed Systems, pp. 25–31.
- Parchman T. L., Geist K. S., Grahnen J. A., Benkman C. W., Buerkle C. A. 2010. Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery. BMC Genomics 11: 180.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea G., Huang X., Liang F., Antonescu V., Sultana R., Karamycheva S., Lee Y., White J., Cheung F., Parvizi B., et al. 2003. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19: 651–652. [DOI] [PubMed] [Google Scholar]
- Pfaffl M. W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29: e45.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai K. S., Palaniswami M. S., Rajamma P., Ravindran C. S., Premkumar T. 1996. An IPM approach for sweetpotato weevil, pp. 329–339. InKurup G.T., Palaniswami M.S., Potty V.P., Padmaja G., Kabeerathumma, Pillai S.V. (eds.), Tropical tuber crops: problems, prospects and future strategies, Science Publishers, Chennai. [Google Scholar]
- Prentice K., Pertry I., Christiaens O., Bauters L., Bailey A., Niblett C., Ghislain M., Gheysen G., Smagghe G. 2015. Transcriptome analysis and systemic RNAi response in the African sweetpotato weevil (Cylas puncticollis, Coleoptera, Brentidae). PLoS One 10: e0115336.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy G. V., Chi H. 2015. Demographic comparison of sweetpotato weevil reared on a major host, Ipomoea batatas, and an alternative host, I. triloba. Sci. Rep. 5: 11871.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy G. V., Wu S., Mendi R. C., Miller R. H. 2014. Efficacy of pheromone trapping of the sweetpotato weevil (Coleoptera: Brentidae): based on dose, septum age, attractive radius, and mass trapping. Environ. Entomol. 43: 767–773. [DOI] [PubMed] [Google Scholar]
- Richards S., Gibbs R. A., Weinstock G. M., Brown S. J., Denell R., Beeman R. W., Bucher G., Friedrich M., Klingler M., Lorenzen M., et al. 2008. The genome of the model beetle and pest Tribolium castaneum. Nature 452: 949–955. [DOI] [PubMed] [Google Scholar]
- Sun X., Zhou S., Meng F., Liu S. 2012. De novo assembly and characterization of the garlic (Allium sativum) bud transcriptome by Illumina sequencing. Plant Cell Rep. 31: 1823–1828. [DOI] [PubMed] [Google Scholar]
- Tan Y. A., Xiao L. B., Zhao J., Xiao Y. F., Sun Y., Bai L. X. 2015. Ecdysone receptor isoform-B mediates soluble trehalase expression to regulate growth and development in the mirid bug, Apolygus lucorum (Meyer-Dür). Insect Mol. Biol. 24: 611–623. [DOI] [PubMed] [Google Scholar]
- Tatusov R. L., Fedorova N. D., Jackson J. D., Jacobs A. R., Kiryutin B., Koonin E. V., Krylov D. M., Mazumder R., Mekhedov S. L., Nikolskaya A. N., et al. 2003. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4: 41.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uritani I., Saito T., Honda H., Kim W. K. 1975. Induction of furano-terpenoids in sweet potato roots by the larval components of the sweet potato weevils. Agric. Biol. Chem. 39: 1857–1862. [Google Scholar]
- Varshney R. K., Nayak S. N., May G. D., Jackson S. A. 2009. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 27: 522–530. [DOI] [PubMed] [Google Scholar]
- Walker W. B., Allen M. L. 2010. Expression and RNA interference of salivary polygalacturonase genes in the tarnished plant bug, Lygus lineolaris. J. Insect Sci. 10: 173.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Feng Z., Wang X., Wang X., Zhang X. 2010a. DEGseq: an R package for identifying differentially expressed genes from RNA-Seq data. Bioinformatics 26: 136–138. [DOI] [PubMed] [Google Scholar]
- Wang R. Y., Li X. H., Ma J., Gao B., Chen S. L. 2014. Monitoring and control of the sweetpotato weevil Cylas formicarius with pheromone traps in Fujian, China. Plant Protect. 40: 161–165. [Google Scholar]
- Wang X. W., Luan J. B., Li J. M., Bao Y., Zhang C. X., Liu S. S. 2010b. De novo characterization of a whitefly transcriptome and analysis of its gene expression during development. BMC Genomics 11: 326–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Si Y., Dedow L. K., Shao Y., Liu P., Brutnell T. P. 2011. A low-cost library construction protocol and data analysis pipeline for Illumina-based strand specific multiplex RNA-Seq. PLoS One. 6: e26426.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X., Zhu D., Li Y., Qiu X., Cui X., Tang J. 2015. Hemolymph levels of methyl farnesoate during ovarian development of the swimming crab Portunus trituberculatus, and its relation to transcript levels of HMG-CoA reductase and farnesoic acid O-methyltransferase. Biol. Bull. 228: 118–124. [DOI] [PubMed] [Google Scholar]
- Ye M. G. 2015. Investigation and analysis of the occurrence characteristics of Cylas formicarius. Chin. Agric. Sci. Bull. 31: 195–199. [Google Scholar]
- Ye J., Fang L., Zheng H., Zhang Y., Chen J., Zhang Z., Wang J., Li S., Bolund R., Wang J. 2006. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34: 293–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H. Z., Wen D. F., Wang W. L., Geng L., Zhang Y., Xu J. P. 2015. Identification of genes putatively involved in chitin metabolism and insecticide detoxification in the rice leaf folder (Cnaphalocrocis medinalis) larvae through transcriptomic analysis. Int. J. Mol. Sci. 16: 21873–21896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D., Ren L., Yue J. H., Shi Y. B., Zhuo L. H., Wang L., Shen X. H. 2015b. RNA-Seq-based transcriptome analysis of stem development and dwarfing regulation in Agapanthus praecox ssp. orientalis (Leighton) Leighton. Gene 565: 252–267. [DOI] [PubMed] [Google Scholar]
- Zhang L., Xu P., Xiao H., Lu Y., Liang G., Zhang Y., Wu K. 2015a. Molecular characterization and expression profiles of polygalacturonase genes in Apolygus lucorum (Hemiptera: Miridae). PLoS One 10: e0126391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D., Guo W., Li S., Li R., Xu D., Lu X. 2014. Identification of a new peritrophic membrane protein from larval Holotrichia parallela (Coleoptera: Motschulsky). Molecules 19: 17799–17809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M., Long H., Zhao Y., Li L., Xu D., Zhang H., Liu F., Deng G., Pan Z., Yu M. 2015c. RNA-Seq based identification of candidate parasitism genes of cereal cyst nematode (Heterodera avenae) during incompatible infection to Aegilops variabilis. PLoS One 10: e0141095.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu T. T., Meng Q. W., Guo W. C., Li G. Q. 2015. RNA interference suppression of the receptor tyrosine kinase Torso gene impaired pupation and adult emergence in Leptinotarsa decemlineata. J. Insect. Physiol. 83: 53–64. [DOI] [PubMed] [Google Scholar]