

Abstract
The complement system represents one of the evolutionary oldest arms of our immune system and is commonly recognized as a liver-derived and serum-active system critical for providing protection against invading pathogens. Recent unexpected findings, however, have defined novel and rather ‘uncommon’ locations and activities of complement. Specifically, the discovery of an intracellularly active complement system – the complosome – and its key role in the regulation of cell metabolic pathways that underly normal human T cell responses has taught us that there is still much to be discovered about this system. Here, we summarize the current knowledge about the emerging functions of the complosome in T cell metabolism. We further place complosome activities among the non-canonical roles of other intracellular innate danger sensing systems and argue that a ‘location-centric’ view of complement evolution could logically justify its close connection with the regulation of basic cell physiology.
Keywords: Complement, T cells, metabolism, CD46, complosome
Introduction
Our contemporary immune system has evolved under the constant pressure from a broad range of pathogens that manipulate the host to enhance their own propagation. As diverse as the pathogenic threats are, ranging from intra- and extracellular bacteria, viruses, fungi and complex parasites, as elegant and effective are the defense mechanisms developed and employed by the host to combat this constant onslaught. Particularly, the detection systems that are functioning as the first lines of defense against such invaders are formidable. If a microbe has managed to penetrate the protective physical barriers, such as the skin, lung, etc., a range of innate immune sensors either circulating in the blood, lymph, and interstitial fluids or expressed in and on scavenger and sentinel cells, such as neutrophils and microphages, detect the conserved pathogen-associated molecular patterns (PAMPs). These sensor systems comprise several pattern recognition receptors (PRRs) and include the toll-like receptors (TLRs), the Nod-like receptors (NLRs), the retinoic acid-inducible gene-I (RIG)-like system, several distinct inflammasomes and the complement system-derived receptors(1, 2). Activation of PRRs by a pathogen is most often triggered in several sensor systems in parallel and then initiates the general inflammatory reaction as well as tailored immune cell activation specifically effective towards the identified pathogen. The ability of PRR systems to decisively discriminate between a real threat, that is non-self and dangerous self via recognizing so-called danger-associated molecular patterns (DAMPs), and self and harmless alterations of self is critical for our health. Thus, there are several checks and balances build into the PRR systems that prevent unwanted reactions against sensed innocuous antigens. However, PRRs not only take care to contain tissue injury during their pathogen clearing actions, they also actively contribute to tissue homeostasis via ‘sterile’ and non-inflammatory removal of dying cells (3, 4) and participate in post-inflammatory tissue repair efforts (5). Thus, although all PRRs were initially discovered and defined as pathogen killers, it is now clear that they are also integral parts of maintenance processes that sustain host health on a broader level.
The complement system was the first PRR system to be discovered and is also among the evolutionary oldest sensor systems. Complement was defined by Jules Bordet over a century ago as a liver-derived and serum-circulating system of proteins that is critical for the detection and removal of pathogens (6). Complement ‘idles’ in the host’s body fluids as an inactive pro-enzyme system which becomes activated in a cascade-like fashion when its PRRs domains/proteins are activated through PAMP/DAMP presence. This culminates in the cleavage of the core complement effector molecules C3 and C5 into the anaphylatoxins C3a and C5a and the opsonins C3b and C5b. C5b-tagging of pathogens then initiates formation of the pore-forming membrane attack complex (MAC) and its direct lytic killing, while pathogen-deposited C3b induces its opsonization and destruction by scavenger cells. The anaphylatoxins stimulate their respective G protein-coupled receptors (GPCR), the C3a receptor (C3aR) and the C5a receptor (C5aR1) on innate immune cells and induce their migration and activation to the infection site (7–10). Similar to the other PRR systems, complement activities go well beyond mere pathogen destruction; complement is key to the removal of dangerous self and thus supports tissue maintenance (11, 12). It is also considered a central regulator of adaptive immune responses due to its critical role in delivering co-stimulatory signals via engagement of complement receptors on antigen presenting cells (APCs) or directly on B and T cells (13–16).
Particularly, work on better defining the instructive role of complement on adaptive immune cells led to the unexpected finding that complement activities are strongly dictated by location. While serum-operative complement is undoubtedly indispensable for pathogen removal, the instructive capacity of complement on adaptive immunity is mostly independent of circulating complement but is rather mediated by locally produced and activated complement; for example, by C3 and C5 secreted by APCs and then activated in the extracellular space (17–19). This emerging concept of the compartmentalization of complement activities was then corroborated by the discovery of an intracellularly generated and functioning complement system in human T cells (5, 20). Subsequent efforts to understand the functional roles of intracellular complement, termed ‘the complosome’ to differentiate it from liver-derived and extracellular-active complement (21), then led to the unexpected and exciting realization that the complosome constitutes a key controller of T cell metabolism (5, 15, 22).
Here, we will review our current knowledge about the emerging new roles for the complosome in the regulation of cell metabolic events that underly human T cell homeostasis and effector function. We will also explore how, similar to other PRRs, the roles/functions of complement differ in accordance with its location and make the case that the newly discovered metabolic and cell physiological functions of complement may not be so surprising after all.
Complement and metabolism – early indications
The notion that complement may serve additional roles aside from mere immune protection and participate in more basic cellular pathways is not a recent one. For example, it is known since the 1980 that the des-Arginated form of the anaphylatoxin C3a, C3a desArg stimulates triglyceride (TG) accumulation and glucose transport in adipocytes (23). In consequence, both C3–/– and Factor B (FB)–/– mice have significantly decreased glucose tolerance and delayed clearance of TG when compared with wild type mice (24) and these findings could possibly apply to humans as obese individuals and patients suffering from diabetes type II exhibit increased C3a desArg levels in circulation (9, 25, 26). Furthermore, increased levels of complement activation correlate positively with the onset of insulin resistance, which is frequently caused by severe obesity (27). The underlying mechanisms are currently not clear but C1q may play a central role here. C1q, now recognized as a general sensor for altered self, interacts with adiponectin in such obese patients which in turn augments C3adesArg levels via increased local complement activation and serum carboxypeptidase N-mediated processing of C3a into C3a-desArg (28). Conversely, C1q–/– mice were protected from hepatic insulin resistance when fed a high fat (Western) diet (29). As expected, increased complement activation such as that observed in Cd55–/– (decay acceleration factor, DAF) mice, which lack a key negative regulator of complement activity, results in augmented insulin resistance and adiposity and changes in lipid handling (30). Also, increased complement activation on adipocytes as seen in patients with auto-antibodies called C3 Nephritic Factors, results in systemic complement dysregulation and causes partial lipodystrophy and loss of subcutaneous adipose tissue of the upper body (31). Further, complement has been shown to control energy regulation (32–34) as well as insulin secretion by pancreatic β-beta cells (9, 35). Thus, it has been long recognized in the field that complement clearly partakes in the regulation of metabolic organs and tissues including the pancreas, liver and adipose tissue (35). Furthermore, this early work also already indicated that observations made by these groups may not be exclusively be attributable to the action of systemic, liver-derived complement but that local complement activity could be important as well. Specifically adipocytes produce a range of complement components, including C3, C1q, properdin and Factors B and D (36) and dysregulated synthesis of these factors by adipocytes is associated with their aberrant function and immune cell infiltration (37). What is now needed is the dissection of the exact liver complement and complosome-driven molecular pathways impacting whole body metabolism. Recent discoveries about such pathways in T cells (below) may be of help, and form an anchor point for future studies on this subject. For excellent in-depth overviews on the known roles of systemic complement in metabolism and metabolic disease, please see (35) and (38).
The complosome in human T cell metabolism
Immunologists are currently enthralled by cell metabolism. Although there was always a common understanding that metabolism is the root of life and, thus, sufficient energy provision on a cellular level must be critical to normal immunity, it is fair to say that the extent to which anabolic and catabolic pathways regulate all phases of the life cycle of immune cells was somewhat unexpected. This, in conjunction with strong indication that targeting these pathways in human disease, above all in cancer, may provide a novel and possibly highly effective angle for therapeutic interventions, catapulted cell metabolism studies into the limelight. Particularly work on understanding how metabolism controls T cell immunity has allowed us to gain better insights into key energy pathways governing T cell maintenance and effector functions – but has also revealed how complex the ‘metabolic network’ really is that operates within these and all other cells.
Specific metabolic states underlay the homeostasis of circulating and tissue-resident human CD4+ and CD8+ T cells and dynamic changes in cell metabolism are key drivers in endowing CD8+ T cells with cytotoxic (CTL) activity and allowing CD4+ T cells to develop into various effector cell subtypes, including T helper type (Th) 1, Th2, Th9, Th17 and regulatory T cells (39). Similarly, CTL and CD4+ Th memory cells are characterized by their own specific metabolic needs and profiles (40) with regards to dependences on influx/efflux of nutrients, their subsequent usage and the generation of downstream metabolic products. We currently are witnessing rapid and exciting progress in understanding how these distinct T cell subpopulations utilize nutrients as energy sources and how metabolic break-down products act as cues for effector behavior. An emerging scheme among these new insights is that metabolic pathways and innate PRR systems engage in a strong functional cross-talk within cells (41, 42). Here, we will focus specifically on the newly discovered roles of the intracellular complement system, the complosome, in the regulation of cell metabolic activities underlying normal human Th1 and CTL responses – and we refer the reader to a number of recent in-depth reviews about the role of metabolism and T cell biology on a broader level (43–46).
The complosome in T cell homeostasis.
Liver-derived and serum circulating complement is classically perceived as highly pro-inflammatory in nature and a key mediator of the general inflammatory reaction that accompanies pathogen detection and removal. Thus, the discovery that T cells not only express and intracellularly activate the major complement component C3 but also need intracellular C3 activation fragments to sustain cell homeostasis, was rather unexpected (Figure 1; please note that this figure represents a condensed conceptual summary of the complosome’s known roles in T cell metabolism and more detailed depictions can be found in a recent in-depth review (47)). Specifically, human CD4+ T cells in circulation express low levels of C3 in subcellular compartments, including, but not limited to, the endoplasmatic reticulum (ER), and lysosomes. A portion of these intracellular C3 stores is continuously cleaved by the protease cathepsin L (CTSL) into bio-active C3a and C3b. Intracellular C3a engages the lysosomal-expressed C3aR which results in tonic activation of the nutrient sensor mammalian target of rapamycin (mTOR) and sustained minimal levels of glycolysis required for cell survival (21). In consequence, a stark reduction in intracellular C3 activation, for example by addition of a cell-permeable CTSL inhibitor during CD4+ T cell ex vivo stimulation, induces cell death. Importantly, the provision of exogenous C3a to such CTSL inhibitor treated cells does not rescue their survival (5, 48, 49). This novel role for the complosome in T cell homeostasis was exciting but was initially at odds with the fact that patients with serum-C3 deficiency have normal levels of circulating T cells. Interestingly, closer analysis of T cells isolated from these rare C3-deficient patients revealed that, in all cases analyzed so far, that while patient cells were unable to secrete C3 or C3 activation products, in contrast they contained normal levels of intracellular C3a protein (5, 50). Moreover, despite employing a range of technologies including CRISP-Cas9 we (and our collaborators) have to date been unable to generate a complete C3-deficient human (or mouse) CD4+ T cell (unpublished data). Thus, these data indicate that the complosome may indeed be critical to T cell survival and that complete (intra- and extracellular) C3 deficiency may hence not exist as it would be incompatible with life. Although it has not yet been dissected how exactly the respective C3 gene mutations in affected individuals still allow the generation of the ‘life-saving’ intracellular C3a, specific properties of intracellular C3, not shared by extracellular C3, may be at the heart of this (please see below and Figure 2A).
Figure 1. The Complosome as key driver of T cell metabolism.
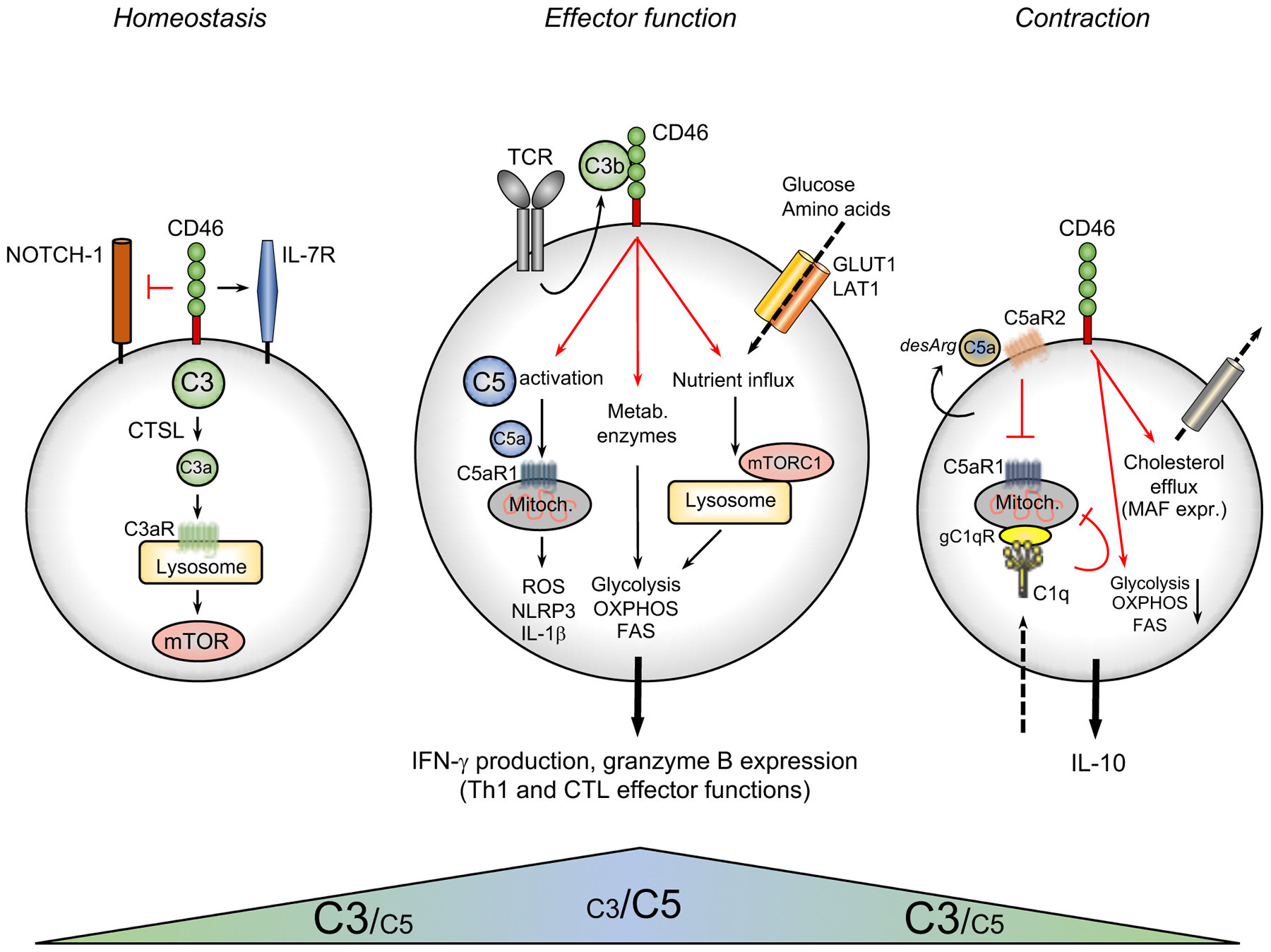
Circulating, non-activated CD4+ and CD8+ T cells generate continuously low levels of intracellular C3a via the cathepsin L-mediated cleavage of intracellular C3 stores (cleavage of C3H2O that can be taken up by in vitro cultured T cells is not depicted here). This C3a generation ensures homeostatic survival of T cells through tonic mTOR activation via C3aR engagement on lysosomes. In addition, CD46 surface expression sustains IL-7R expression also required for homeostatic survival of CD4+ T cells and prevents activating Notch1 stimulation. During TCR activation (and CD28 co-stimulation, not shown here) intracellular C3b translocates rapidly to the cell surface and actively engages CD46. CD46 signaling triggers three key metabolic events: the γ-secretase-processed intracellular domain of CD46 translocates to the nucleus (not shown) and induces gene and protein surface expression of critical nutrient transporters (GLUT1, LAT1) as well as LAMTOR5-driven mTORC1 assembly at the lysosomes. CD46 activation further induces increased expression of metabolic enzymes, including fatty acid synthase (specifically in CD8+ T cells), GAPDH, etc. CD46 also strongly augments activation of intracellular C5 pools with the intracellularly generated C5a engaging intracellular C5aR1 resulting in ROS production and NLRP3 inflammasome activation in CD4+ T cells. Together, these events drive the high levels of glycolysis, OXPHOS and ROS production needed specifically for the induction of IFN-γ production and granzyme B expression. Thus, autocrine complosome activity is an integral part of normal human CD4+ Th1 effector function and CD8+ effector CTL activity. The complosome also contributes to the safe metabolic ‘shut-down’ of human Th1 activity as CD46 (via expression induction of a ‘repressive’ CD46 isoform, not shown)-driven signals reduce glycolysis and OXPHOS while at the same time supporting cholesterol efflux and MAF expression. Autocrine generation of the ‘des-Arginated’ form of C5a (C5a-desArg) engages the repressive C5aR2 on the T cell surface, which reduces C5aR1 activity. Finally, C1q, taken up by the activated T cell hampers normal mitochondrial activity (in CD8+ T cells) via gC1qR via a yet unknown mechanism – together, these events lead to secession of IFN-γ production in T cells. Of note, whilst human CTLs harbor the ‘intracellular C3/C5 systems’ a possible functional role during CTL homeostasis and/or contraction has not yet been explored. Further, although not formerly proven yet, current data strongly suggest that the intracellular C3 system is engaged during all life cycle stages of T cells, while the C5 system is mostly engaged during the Th1 and CTL effector phase. CTSL, cathepsin L; FAS, fatty acid synthase/synthesis; GLUT1; glucose transporter 1; LAT1, large neutral amino acid transporter 1; MAF, cMaf musculoaponeurotic fibrosarcoma oncogene homolog; mTOR, mechanistic target of rapamycin; mTORC1, mechanistic target of rapamycin complex 1; OXPHOS, oxidative phosphorylation; ROS, reactive oxidation species; TCR, T cell receptor.
Figure 2. Location-driven activities of key innate immune sensors.

(A) Suggestion of bi-furcated evolution of our contemporary complement system. Evolutionary older C3 proteins analyzed in precursor organisms such as Porifera and Agnatha contained additional domains with homology to enzymes involved in cell metabolism (126), indicating that C3 may have initially appeared in ancient single cell organisms where it was involved in metabolic activities. With the appearance multi-cellular organisms, complement underwent a bi-furcated evolution into two contemporary principle parts. Liver-derived, and serum-operative complement protecting our vascular space against pathogens through ‘classically’ folded C3 that, upon activation, functions as an opsonin, contributes to C3/C5 convertases formation and initiates the lytic membrane attack complex. Intracellular C3, on the other hand, retained its activity to control cell metabolism. Over time, intracellular C3 lost some its domains and metabolic/cell physiological roles because of the concurrent co-evolution of a ‘dedicated’ cell metabolism protein machinery. We also propose that intracellular C3 forms may not need to acquire the folded structure known for liver-derived C3 but are rather processed by cell-specific proteases to release/activate specific domains needed for cell metabolic activities – for example, C3a (ANA, anaphylatoxin). (B) General scheme for ‘graded’ changes in the focus activities of major innate immune sensor systems, including TLRs, inflammasomes and complement (additional intracellular systems, such as RIG, MAVS etc. are not shown as there is currently only evidence supporting intracellular activity) dependent on their expression location. An intracellular location may come with a strong focus on maintaining normal cell physiology via sensing and rectifying ‘physiological’ stress (such as starvation, non-physiological pH, temperature changes) aside from pathogen sensing, while surface expression allows (micro)environmental sensing and the induction of critical protective pro-inflammatory immune sensors with simultaneous engagement of negative control measures on a cellular level to prevent detrimental tissue pathology. Extracellular activity is mostly concerned with clearance of pathogens that have breached the protective barriers and is highly pro-inflammatory. An intensive cross-talk between the three depicted PRR systems exist intracellularly and at the cell surface and likely between fluid-phase and cellular PRRs. DAMP, Danger/damage-associated molecular pattern; PAMP, pathogen-associated molecular pattern; RAMP, resolution-associated molecular pattern.
A second component of complement, the complement receptor CD46 (also known as membrane cofactor protein, CD46) has also been identified as a key player in the maintenance of T cell homeostasis (5). Of note, CD46 is a human-specific protein that is not expressed in mice and a complement-derived functional homologue of CD46 has so far not been discovered in rodents (51–53). CD46 is intimately connected with the autocrine C3 system/complosome as it is a receptor for the C3b portion of activated C3. CD46 was initially discovered and defined as a complement regulator preventing unwanted complement deposition on host tissue because CD46 also functions as a co-factor for the Factor I-mediated cleavage and rapid inactivation of C3b (54). CD46 is ubiquitously expressed and, over the years, has emerged as a multi-functional protein that orchestrates a wide range of activities on many cell types (53). For example, CD46 is required for normal sperm-egg fusion (51, 55, 56)and is the cell entry receptor for several important human viral and bacterial pathogens such as measles virus, herpesvirus, Streptococci, and Neisseria (57). CD46 is also a clinically ‘attractive’ molecule because single nucleotide polymorphisms (SNPs) in the CD46 gene are either causative or contribute to hemolytic uremic syndrome, age-related macular degeneration, and possibly pre-eclampsia (58–60).
On resting CD4+ T cells, CD46 presence sustains (via signals that have not yet been identified) the expression of the interleukin (IL)-7 receptor (CD127). Incoming IL-7R signals in circulating T cells maintain basic glucose transporter 1 (GLUT-1) surface expression, which, in turn, allows for glucose-mediated protein kinase B (AKT) activation and anti-apoptotic B cell lymphoma (BCL-2) expression, all such events being required for normal T cell survival in the periphery (21, 61–63). Interestingly, CD46 also engages with the evolutionary old Notch-1 system to prevent unwanted T cell activation in the absence of perceived danger (without T cell receptor activation (TCR)). In this case, CD46 directly binds Notch-1 on the resting T cells’ surface and prevents an interaction of Notch-1 with its ‘natural’ ligand Jagged-1, that would normally lead to T cell activation (48) (Figure 1).
Although resting human CD8+ T cells also harbor all complosome components so far detected in CD4+ T cells ((15); Figure 1) and, equally, express Notch-1/2 (64), it remains to be determined whether C3a/complosome activity and the CD46 crosstalk with the Notch system serves a similar homeostatic role in CTLs.
The complosome and cell metabolism regulation during Th1 and CTL effector induction.
Somewhat reminiscent to the serum complement system, where low-level C3 activation via hydrolysis generates a continuously present small pool of active C3b that can immediately latch onto any incoming pathogen, the complosome with its (metabolic) house-keeping functions in resting CD4+ T cells, seems to also function as a ‘spring-loaded’ system that can quickly support T cell activation: TCR- (and CD28-) stimulation induces increased C3 activation and the rapid shuttling of C3a and C3b to the cell surface. The autocrine engagement of CD46 by intrinsic C3b then triggers several events. First, T cell-expressed metalloproteinases (such as a disintegrin and metalloproteinase domain-containing protein 10 (ADAM 10) or matrix metalloprotease 9 (MMP9) release the surface portion of CD46 which then allows Notch-1 to engage in a productive T cell-activating interaction with Jagged-1 (48, 65–68). CD46 stimulation also induces a shift in the isoform expression pattern of CD46 (which can be expressed bearing two distinct cytoplasmic domains, CYT-1 or CYT-2 (53, 54)) by upregulating the ‘inflammatory’ CD46CYT−1 isoform (22, 68). The intracellular domain of CD46 is then processed by the γ-secretase complex and translocates to the nucleus where it mediates the expression of a large number of genes that code for metabolic sensors/regulators, enzymes and/or or nutrient transporters. For example, CD46CYT−1 mediates increased gene transcription of SLC2A1, encoding GLUT-1, of SLC7A5, coding for the large neutral amino acid transporter 1 (LAT-1) and of LAMTOR5 (LAMTOR5 is a scaffolding protein that supports mammalian target of rapamycin complex 1 (mTORC1) assembly at the lysosomes)). Together these events drive the high levels of glycolysis, amino acid influx and lysosomal mTORC1 assembly and activation that are particularly required for metabolically highly demanding IFN-γ and Th1 responses ((22, 69), Figure 1). CD46-mediated signals also support IFN-γ production and granzyme B expression in human CD8+ T cells through engagement of glycolysis in conjunction with a strong augmentation of fatty acid synthesis (Figure 1). This is accomplished through induction of fatty acid synthase and fatty acid binding protein 5, which plays a role on fatty acid uptake and intracellular transport (15). Autocrine CD46CYT−1 signaling also increases the expression of IL-2Ra (CD25) and the subsequent assembly of the high affinity IL-2 receptor on both CD4+ and CD8+ T cell subpopulations, which is necessary for optimal Th1 and CTL lineage induction and responses (47, 70, 71).
Finally, TCR-triggered CD46 co-stimulation on human CD4+ T cells induces the increased activation and mobilization of intracellularly generated C5a via cleavage of C5 storages that are also present in circulating T cells (Figure 1). Of note, whilst we know that CTSL can activate C3 within cells, the protease(s) or enzyme complex(es) that process C5 into active C5a and C5b are currently not defined (72). CD46-mediated amplification of intracellular C5a generation leads to the engagement the C5aR1 expressed on mitochondria and significant production of reactive oxygen species (ROS). How exactly C5aR1 induces mitochondrial ROS is not clear but we have indication that it directly affects activity of the electron transport chain (unpublished data). Increased sensing of ROS production initiates then the assembly of a canonical NLRP3 inflammasome and generation of mature IL-1β. Surprisingly, IL-β production does not, as it often does in other cells, induce pyroptosis in T cells but rather it supports and maintains IFN-γ secretion in an intrinsic fashion ((72), Figure 1).
The importance of CD46CYT−1-mediated regulation of cell metabolism and human Th1 lineage induction is underpinned by the fact that rare patients with CD46 deficiency or with deficiency in C3 (C3b provides the ligand for CD46) have severely reduced Th1 responses – at least early in life – and also suffer from recurrent infections (48, 49, 68). Ex vivo assessments of CD8+ T cells from CD46-deficient patients, on the other hand, demonstrated that CD46 co-stimulation is indeed required to induce their optimal high IFN-γ production, but that CD8+ T cells, in contrast to CD4+ T cells, can still produce ‘basal’ levels of IFN-γ. Moreover, CD8+ T cells, similar to CD4+ T cells, also express all of the complosome components, including stores of C3 and C5 and their cognate complement receptors C3aR1, C5aR1 and C5aR2, and CD46, as well as expressing NLRP3. However, in contrast to CD4+ T cells where canonical NLPR3 inflammasome activity and intrinsic IL-1β production are critical for Th1 induction, they do not seem to be required for normal IFN-γ secretion and CTL activity in CD8+ T cells (15). Thus, while the normal production of their signature cytokine IFN-γ by both Th1 and CTL cells requires complosome-mediated metabolic reprogramming, both T cell subpopulations engage the complosome in a distinct and specific fashion. This observation falls well in line with a wealth of recent publications that clearly demonstrate distinct metabolic requirements, amino acid usages, and dependence on glycolysis and OXPHOS, and fatty acid metabolism, for normal activity of T cell effector subpopulations such as Th1, Th2, Th9 and regulatory T cells (39, 43, 45, 73, 74). Interestingly, CD46 and serum C3-deficient patients have unaltered Th2 responses and we have not been able to observe a defect in their T cell proliferation, at least upon ex vivo assessment (48, 49, 68). The reason for this intimate relationship between the complosome and IFN-γ is currently not clear but may also be rooted in their close co-evolution (see below).
The complosome and Th1 cell contraction.
Successful Th1 immunity is an absolute requirement to prevent pathogen invasion. However, IFN-γ can also cause substantial tissue destruction, thus, the timely resolution of protective Th1 activity is equally important to the hosts’ health because it limits the pathological consequences of a prolonged or chronic response, as observed in non-clearing infections or Th1-driven autoimmune disease states (75–77). The possibly detrimental pro-inflammatory activity of IFN-γ is kept in check via several mechanisms (78–80), one of which is mediated through the immunosuppressive cytokine IL-10, that can inhibit Th1 cell activity directly. The in vivo importance of this regulative concept has been demonstrated in experiments utilizing mice deficient in the Il10 gene. These animals clear certain infections more rapidly through augmented Th1 immunity compared to wild type animals, but then die of severe tissue pathology (76).
Interestingly, CD46 also plays a key role during this vital shut-down program of Th1 responses. Together with signals from the IL-2R, CD46 supports Th1 contraction via the co-induction of IL-10 in Th1 cells once sufficient IFN-γ production and Th1-derived IL-2 levels are established. These cells now also acquire a immunosuppressive and self-regulative phenotype and represent the controlled shut-down phase of Th1 responses. The exact crosstalk between the IL-2R and CD46 that induces the co-secretion of IL-10 in Th1 cells is not understood but requires, at minimum, that the proinflammatory CD46CYT−1 isoform ‘reverts back’ to a predominant CD46CYT−2 form (5, 13, 22). This CD46 isoform switch is then accompanied by a stark change in the effector Th1 cell metabolic signature which is characterized by diminished nutrient influx, the downregulation of GLUT1, LAT1 and LAMTOR5 expression, a reduction in fatty acid synthesis, overall reduced mTORC1 activity and the return of the cell to a metabolically quiescent state (22, 47, 81). Studies to better understand this regulative activity of CD46 have connected this molecule with another key metabolic ‘system’, sterol metabolism. CD46-mediated signals trigger the cholesterol biosynthesis pathway in CD4+ T cells and normal cholesterol flux that is required for the expression of the IL-10 inducing transcription factors proto-oncogen musculoaponeurotic fibrosarcoma oncogene homolog ((c-MAF) (82), (Figure 1)). It is not clear yet though whether the CD46CYT−2 form has an active role in Th1 contraction or if it simply diminishes activity of the CD46CYT−1 isoform via replacement. What is clear though is that perturbations in CD46 signaling during the shutdown phase of Th1 responses contributes to autoimmune disease. For example, increased intracellular C3 activation and CD46 signaling occurs in CD4+ T cells from patients with rheumatoid arthritis and SLE and contributes to their hyperactive Th1 phenotype via increased mTOR activity maintenance. Excitingly, the reduction of such augmented intracellular C3 activation ex vivo through CTSL inhibition normalized IFN-γ production by patients’ T cells (22). A change in the CD46 isoform expression pattern, with a skewed dominance towards the CD46CYT−1 form, has also been connected with reduced Th1 control in multiple sclerosis (83).
Another component of the complosome, the intracellular C5 system, also plays a dual role in the regulation of Th1 responses. Whilst intracellular C5aR1 signals support IFN-γ production via ROS-mediated inflammasome induction, increased production and secretion of the des-Arginated form of C5a, C5a-desArg, engages the inhibitory C5aR2expressed on the T cell surface in an autocrine manner, and leads to a reduction in ROS generation and NLRP3 inflammasome activation (72)(Figure 1). Whether the C5aR2 simply blocks the activity of the C5aR1 (the C5aR2 is mostly recognized in the field as a counter regulator of C5aR1 (84)), or whether it induces a distinct signaling program and actively regulates Th1 contraction is currently not defined.
Similarly, a role for CD46 or other complosome components during CD8+ T cell contraction remains to be investigated. It has been observed that CD46 can induced low levels of IL-10 in CTLs, but the amounts generated are small compared to those observed in CD4+/Th1 cells and a co-induction of suppressive activity in CTLs via CD46 was not investigated (85). Nonetheless, intracellular complement can, analogously to what has been described in human CD4+ T cells, also exert negative control over CD8+ T cells. A recent study by Ling et al. showed that C1q is taken up by activated human CD8+ T cells during activation and then engages the intracellular C1q receptor p32/gC1qR expressed on mitochondria. This C1q–gC1qR interaction reduces mitochondrial activity and reduces CTL killing activity, proliferation, and survival (Figure 1). The biological importance of C1q-mediated negative control was then demonstrated in C1q–/– mice which exhibit augmented CTL responses in models of autoimmunity graft versus host disease (GVHD) and viral infection. Moreover, lack of C1q in humans, which causes systemic lupus erythematosus (SLE), contributes to uncontrolled CD8+ T cell memory responses, which are not causative in the initial phase of SLE in these patients but rather propagate autoimmune pathology in later stages (86). C1q has lately garnered high interest beyond those in the complement field because of its involvement in the pathogenesis of several cancers, with both tumor promoting or tumor-protective activities (87–90). Thus, there is currently a strong effort to understand how exactly C1q enters cells and what exact molecular mechanisms mediate C1q’s effects on mitochondrial activity.
In summary, the complosome emerges as a ‘master regulator’ of the transporter systems that control nutrient (in)flux and of the enzymes that shunt (incoming) nutrients into the correct metabolic adaption pathways required for appropriately catered T cell activities towards sensed antigen and/or incoming environmental cues – and this critical activity spans the complete life cycle of Th1 cells.
Location matters: spatial bias in PRR activities
A picture emerges in which the location of complement activation directs its activity: circulating liver-derived complement protects against invading pathogens via direct tagging and lytic killing of sensed pathogens, while complement generated and activated within (immune) cells is a direct regulator of biochemical reactions, currently mostly of metabolic nature. This intracellular ‘cell physiological and homeostatic’ activity took many by surprise, however, other ancient PRR systems follow a similar spatially-driven and complex activity pattern (Figure 2A). For example, TLRs and inflammasomes were both initially identified and defined as pathogen-sensing and fighting PRR systems but are now recognized players in cell metabolism and cell homeostasis (for comprehensive recent reviews on this subject, please see (91–94)). For the TLRs, it became clear relatively quickly that they occupy both intracellular and cell surface spaces: TLRs 1, 2, 4, 5, and 11 are predominantly found on the cell surface and TLRs 3, and 7–9 are mostly expressed in subcellular compartments such endosomes, the endoplasmatic reticulum (ER), lysosomes, etc. (95) (Figure 2A). All TLRs sense ‘their’ specific PAMP which includes lipopolysaccharides, flagellin, zymosan, and viral single- or double-stranded RNA etc. with high sensitivity and hence protect the host efficiently against extra- and intra-cellular pathogenic microbes. Of note, intracellular complement also serves an immune surveillance activity as C3b that is shunted into the cell by way of C3b-opsonized viruses, triggers mitochondrial anti-viral signaling protein (MAVS)-mediated proinflammatory cytokine secretion in epithelial cells (96). A recent unexpected extracellular function for NLRP3 has been described in which macrophages release oligomeric NLRP3 inflammasome particles that amplify the ongoing inflammatory response (97). Today, it is well accepted that TLRs not only rapidly modify basal cell metabolism upon PAMP sensing by increasing glycolysis-driven ATP production and modulating OXPHOS to allow for a cellular effector response (98–100), but TLRs can also detect cues of perturbed cell metabolism, such as excess lipids in form of fatty acids and oxidized low density lipoprotein (LDL) (101). Moreover, TLR3 has recently emerged as a key intrinsic regulator of glucose metabolic responses (102), while intracellular TLRs 7 and 8 also recognize metabolites generated by increased nucleic acid degradation and convey a ‘stress signal’ to the cell (103). Similarly, NLRP3 inflammasome priming and activation are strongly driven by increased glucose influx, heightened glycolysis and increased ATP production (91), all events generally required for cell activation, proliferation and effector function (99). The NLRP3 inflammasome also integrates signals derived from amino acid and lipid metabolism in cells as well as from ROS production and mTORC1 activation, which are increased by the enhanced mitochondrial activity that is required for cell effector functions. Interestingly, there is now accumulating evidence that the complosome and the NLRP3 inflammasome have a tight physical connection with mitochondria: NLRP3 is transported to the surface of mitochondria where these organelles then create ideal conditions for the exposure/presentation of triggers of NLRP3-ASC assembly, such as cardiolipin externalization (104, 105). Moreover, the contact areas between the ER and mitochondria have recently been identified as metabolic hubs (106) and, thus, could provide energetic support for the assembly of such macromolecular structures. With regards to complement, particularly the C1q receptor gC1qR/gp32, expressed on mitochondria, has an acknowledged history of impacting the activity of these organelles. For example, gC1qR-mediated signals optimize spare respiratory capacity and oxygen consumption by augmenting the transcription of genes coding for proteins involved in mitochondrial biogenesis in human CD8+ T cells (86). We have also found that the C5aR1 is expressed on the mitochondrial outer membrane where it regulates the electron chain flux (unpublished data) and components of the lectin pathways, such as ficolins and mannan-binding lectin (MBL) expressed by cells, can also directly bind mitochondria (107). The impact of lectin pathways proteins on mitochondrial activity, however, remains to be defined.
Importantly, metabolic by-products and particularly those generated during quiescent states or during contracting and tolerogenic response phases, such as AMP, β-hydroxybutyrate (108, 109), and prostaglandin E2 (PGE2) (110) can also function as resolution-associated molecular patterns (RAMPs) (111), inhibit NLRP3 inflammasome activation and contribute to the cessation of cell effector functions. Along this line, TLR2-mediated cell-intrinsic signals are required for the co-induction of IL-10 in Th1 cells and regulatory T cells activity (112). Moreover, all PRR families have dedicated members that are mostly concerned with the negative control of PRR-triggered proinflammatory responses, including TLR10 (113), NLR family CARD domain containing member 3 (NLRC3)(114, 115) and the complement receptor CR3 (CD11b/CD18) (116). A prominent role for intrinsic activity of TLRs and inflammasomes in normal cell (metabolic) homeostasis is supported by the finding that perturbations of these system are mostly connected with metabolic disease and not with recurrent infections (99, 117, 118). Based on the emerging connection between complement and metabolism, it may be worth-while to evaluate patients with idiopathic metabolic disease for potential underlying dysregulated complosome functions.
Thus, it seems that on a single cell level, the intrinsic intracellular activities of the complosome, the TLRs, and the NLRP3 inflammasome are majorly focused on sustaining normal cell homeostasis via constant monitoring for (metabolic) DAMP- and RAMP-derived signals with the help of cellular sub-compartments, and the ability to rapidly integrate incoming PAMP signals to trigger the needed specialized effector responses. Cell surface expression of these PRRs allows the incorporation of incoming environmental cues and moves emphasis towards classic immune surveillance with a balanced protective pro-inflammatory and tissue preserving/repairing anti-inflammatory response. The activity of blood circulating or extracellular PRRs, which are represented largely by liver-derived complement, are currently mostly associated with PAMP sensing and have a strong bias towards a pro-inflammatory nature (Figure 2A). However, this view may change with our increasing understanding of the PRR’s ability to sense ‘whole body metabolism’.
Importantly, complement, TLRs and inflammasome activities heavily engage in functional cross-talks (119–122). For example, TLR- and complement-mediated signals synergize to trigger NLRP3 assembly (123)and can generally amplify each other’s proinflammatory activities to cooperatively fight infections. However, each system can also exert negative control of the other systems, and we hence are faced with a highly complex functional PRR network characterized by intricate positive and negative feedback loops (124). Traditionally, these PRR studies were performed in isolation and the fact that perturbation of one system affects the activity of the other systems makes it clear that the observed effects on cell metabolism (or any other parameter) need to be considered carefully and that clear-cut interpretation of data remains challenging.
A close connection between complement and basic processes of the cell (81, 125, 126) may not only make sense from a location-driven point of view but becomes also apparent upon a closer look at the structural evolution of the key complement component C3 – which is considered the origin of complement (127) and has basically seeded our current knowledge about the complosome in T cells.
A recent more detailed protein domain analysis of ancient C3 forms, for example from tunicates and teleost fish revealed that these older C3 forms often contained distinct additional protein domains that displayed a high homology with metabolically active molecules that are no longer present in ‘contemporary’ C3 (126). For example, C3 in Tunicates harboured a crotonase domain which is commonly present in crotonase/Enoyl-Coenzyme A (CoA) hydratases, enzymes that are key to fatty acid oxidation (128). Similarly, avian C3 has a ferredoxin NADP(H) reductase domain (129), indicating functional involvement in the regulation of the electron transport chain in mitochondria as well as regulation of cholesterol and steroid metabolism (130). Importantly, there are still remnants of such metabolic domains in modern C3. For example, an isoprene C2-like domain (that is common to many proteins regulating cholesterol metabolism (131)) is embedded within the CUB (complement C1r/C1s, Uegf, Bmp1) and TED (thioester containing) domains of mammal C3. Thus, the recently discovered connection between the C3/CD46 axis and cholesterol metabolism during the regulation of Th1 contraction sits well with the metabolic traits of evolutionary older C3.
Based on the intriguing structural composition of old and new C3, the location-driven activity of complement, its strong cross-talk with other PRRs and the emerging key role for basically all PRRs in cell physiology (Figure 2A), our laboratory operates under the notion that complement may have originally appeared in single cell organisms as an intracellular system with the main role to sense and control metabolic pathways/stress within one cell (22, 81, 126, 132). The more restricted size of the genome of such basic organisms is generally reflected in a protein landscape that is characterized by a strong bias towards ‘multitasking’, that is a single protein performs diverse functions (133). The development of life into multi-organ organisms then not only required orchestrated communication between cells within an organism but also new measures to defend the interstitial space, body cavities, and lymph and vascular systems. These needs are met by an increase in the genome and gradual specialization of more proteins with less and rather dedicated functions (134). A bi-furcated evolution of an initially intracellular complement could indeed explain well what we see today: part of the original C3 remained functional mostly intracellularly and continued to serve mostly cell physiological roles but has lost some metabolic domains on its developmental journey due to parallel development of an increasingly complex and dedicated metabolic machinery. In parallel, during evolution, C3 also segregated into a secreted form, mostly produced by hepatocytes and assumed the role of the systemic PRR system as we have known it for decades (Figure 2B).
Based on this compartmentation of function we also suggest that the structure of intra- and extra-cellular C3 is different: C3 generated by the liver and found in circulation needs to be folded into the classically known C3 protein structure (135) to give rise to, upon activation, the opsonin C3b, the backbone of the C3/C5 convertases, and the assembly of the MAC. However, the intracellular C3 form does not require a classically folded C3 structure to perform its functions (Figure 2B) as it is rather processed by cell-specific proteases (for example, furin/CTSL to generate C3a) that release and/or activate the domains mediating its cell metabolic activities. Thus, intracellular C3 may exist mostly as an unfolded or differently folded form. In fact, a C3 form that cannot be secreted due to a second transcription start side that omits expression of the secretory signal peptide has been found in human pancreatic β-cells (136). The structure of intracellular C3 is likely dictated by the specific environment (pH, ion concentration, etc.) of the subcellular compartment that it resides in and a single cell may hence contain several different C3 forms. Moreover, cells can also import the hydrolyzed form of C3 (C3H2O) from the extracellular environment in vitro which can be processed intracellularly by CTSL (137), with the biological significance of this C3 uptake under investigation. In line with the emerging picture that serum-circulating and intracellular C3 have different characteristics, we have found that post-translational modifications of C3 are distinct between monocyte-expressed C3 and liver-derived C3 (unpublished data). Currently, only speculatively, we also expect that there will be additional intracellular C3-processing proteases identified that can release other domains of cellular C3 (aside from C3b/C3a) with novel physiological functions (47).
Current hurdles in dissecting the complosome-metabolism axis in health and disease
Although there is now mounting evidence that the complosome is an integral part of normal cell metabolism, homeostasis and (effector) function in a broad range of cells, and we have further strong indication that perturbations in complosome activity contribute to human disease, we are facing several hurdles to dissect complosome-mediated metabolic pathways in depth.
For example, there is no small animal model available to study the role of CD46 in vivo, as rodents do not express CD46 or a complement-derived protein that recapitulates CD46’s central role in human T cell immunology (51–53). There have been early attempts to generate human CD46-transgenic mice (initially to study infections with pathogens that utilize CD46 as cell entry receptor) but, with very few exceptions, the human CD46 protein did not induce comparable responses in mouse cells, particularly not in any immune cell tested (138–140). Our analysis of T cells isolated from CD46 or C3-deficient patients have allowed us to identify the complosome mechanistically as an unexpected player in cell metabolism. However, even if specific gene mutations have been clearly defined via exome sequencing, one cannot exclude that undetected confounding factors contribute to observed disease pathology in these patients. More importantly, ex vivo analysis of cells may not reflect their in vivo behavior – which is particularly problematic for the analysis of cell metabolism (141) – and ‘sole’ work with patients does not allow one to adress questions surrounding potential roles for CD46 in T cell memory development, tissue residency, TCR repertoire diversity, etc. Thus, to rigorously link CD46 in vitro observations to their in vivo relevance remains a challenge. Nonetheless, our ongoing work on understanding how exactly the intracellular domains of CD46 regulate cell metabolism indicates that CD46 provides a novel, critical and human-specific mode for the direct regulation of the activation state of glycolytic enzymes and the interaction of specific transcription factors with DNA and other proteins (unpublished data). Thus, we are currently taking advantage of the recent advances in sensitive single cell combined ‘(metabol)omic’ analyses (142) of cells isolated from healthy and inflamed human tissues as well as from patients with inborn errors of metabolism (143) to probe for functional changes and/or deviations mediated by CD46 – with an eye on evolutionary conserved CD46 downstream events that can be further analyzed in mice.
Another current barrier in rapidly moving complement-metabolism research forward is the finding that complosome components present in both mice and men, do not always have overlapping locations of expression and may not always serve similar functions. For example, while expression of C3aR and C5aR1 and 2 by human CD4+ T cells is broadly acknowledged (15, 72), their presence in mouse T cells is a matter of ongoing debate with some groups observing C3ar, or C5ar1 or 2 expression on resting or activated T cells (18, 144) and others failing to replicate these findings (145–147). Also, when detected, C5ar2 appears on the mouse T cell surface only upon activation (148), in contrast to human T cells, where C5aR2 is constitutively found on the cell surface in both resting and activated cells. Also, there seem to be large conceptual divides with regards to the autocrine and intrinsic role of C3 in T cells. In human T cells, CTSL drives the intracellular activation of C3, while T cells from Ctsl–/– mice are still able to process C3 into C3b (5). Further, human T cells rely on intrinsic C3a generation and exogenously provided C3/C3a cannot compensate for lack of ‘self-made’ C3a, while provision of C3a by antigen presenting cells seems to restore Th1 induction in C3-deficient mouse T cells (19).
Finally, it is now also understood that almost all human/mouse complement receptors function in a cell-specific fashion. A large majority of published work, however, has been performed using animals with complete deficiency in the complosome/complement components of interest – thus, it is likely that we have to re-evaluate some of the conclusions drawn over the years from this body of work. Moreover, complement components often operate in a bi-phasic mode and partake in both the cell activation (inflammatory) and contraction phases (anti-inflammatory) (72, 149, 150), thus, one ideally wants to employ in vitro and in vivo systems that allow for cell-specific inducible expression or ablation of the complement component of choice. Excitingly, these needed reagents are currently developed in the field and we are hence looking forward to the new insights into the expanding roles of complement in normal cell physiology that these will undoubtedly bring.
Future outlook
Intracellular complement activity, the complosome, emerges as a key player in the regulation of general cell metabolism during all phases of the life cycle of Th1 cells. We therefore expect that the complosome will also impact on the metabolic cues that generate T cell memory and/or establish or sustain tissue residency. These are subjects that we are currently actively pursuing in the laboratory. Importantly, although most of our understanding of this new complement activity has been derived from studying T cells, the complosome (meaning at least the core complement components C3 and C5 and respective activation fragment receptors) has been observed so far in all cells assessed (5). This indicates the broad physiological significance of the complosome and aligns with a new vantage point about the evolutionary roots of complement as an originally ‘single cell’ system. The emergence of complement as a key player in metabolism also conspicuously parallels the rise of other ancient PRRs, such as the TLRs and inflammasome from mere pathogen detectors to orchestrators of normal cell physiology (42). We, thus, not only expect future additional exciting and unexpected news about this ‘old dog’ but would also invite the readers to keep an eye out for a complosome signature in their own (metabolic) studies.
Acknowledgements
Work in the Complement and Inflammation Research Section (Kemper laboratory) was/is financed by the MRC Centre grant MR/J006742/1, an EU-funded Innovative Medicines Initiative BTCURE, a Wellcome Trust Investigator Award, the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London, and by the Division of Intramural Research, National Heart, Lung, and Blood Institute (NHLBI), NIH. We further acknowledge the work of the many scientists, particularly working on T cell metabolism, that we were unable to cite here due to focus on the complosome and T cell metabolism.
Footnotes
Conflict of interest
The authors state that they have no conflict of interest
References
- 1.Lavelle EC, Murphy C, O’Neill LA, Creagh EM. The role of TLRs, NLRs, and RLRs in mucosal innate immunity and homeostasis. Mucosal Immunol.2010;3:17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kohl J. Self, non-self, and danger: a complementary view. Adv Exp Med Biol.2006;586:71–94. [DOI] [PubMed] [Google Scholar]
- 3.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol.2013;13:397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wen H, Miao EA, Ting JP. Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity.2013;39:432–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liszewski MK, Kolev M, Le Friec G, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity.2013;39:1143–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bordet J, Gengou O. Sur l’existence de substances sensibilisatrices dans la plupart des serum antimicrobien. Ann. Inst. Pasteur, 1901:289–302. [Google Scholar]
- 7.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement System Part I - Molecular Mechanisms of Activation and Regulation. Front Immunol.2015;6:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement System Part II: Role in Immunity. Front Immunol.2015;6:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol.2010;11:785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Complement Walport M.. First of two parts. N Engl J Med.2001;344:1058–1066. [DOI] [PubMed] [Google Scholar]
- 11.Botto M, Dell’Agnola C, Bygrave AE, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet.1998;19:56–59. [DOI] [PubMed] [Google Scholar]
- 12.Boackle SA. Complement and autoimmunity. Biomed Pharmacother.2003;57:269–273. [DOI] [PubMed] [Google Scholar]
- 13.Cardone J, Le Friec G, Vantourout P, et al. Complement regulator CD46 temporally regulates cytokine production by conventional and unconventional T cells. Nat Immunol.2010;11:862–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carroll MC, Isenman DE. Regulation of humoral immunity by complement. Immunity.2012;37:199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arbore G, West EE, Rahman J, et al. Complement receptor CD46 co-stimulates optimal human CD8(+) T cell effector function via fatty acid metabolism. Nat Commun.2018;9:4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.West EE, Kolev M, Kemper C. Complement and the Regulation of T Cell Responses. Annu Rev Immunol.2018;36:309–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu J, Miwa T, Hilliard B, et al. The complement inhibitory protein DAF (CD55) suppresses T cell immunity in vivo. J Exp Med.2005;201:567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strainic MG, Liu J, Huang D, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity.2008;28:425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, Heeger PS. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood.2008;112:1759–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liszewski MK, Kolev M, Bertram PM, et al. Cleavage of intracellular C3 into C3a and C3b by cathepsin L is required for human T cell induction. Immunobiology, 2012:1146. [Google Scholar]
- 21.Kolev M, Friec GL, Kemper C. Complement - tapping into new sites and effector systems. Nat Rev Immunol.2014;14:811–820. [DOI] [PubMed] [Google Scholar]
- 22.Kolev M, Dimeloe S, Le Friec G, et al. Complement Regulates Nutrient Influx and Metabolic Reprogramming during Th1 Cell Responses. Immunity.2015;42:1033–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cui W, Paglialunga S, Kalant D, et al. Acylation-stimulating protein/C5L2-neutralizing antibodies alter triglyceride metabolism in vitro and in vivo. Am J Physiol Endocrinol Metab.2007;293:E1482–1491. [DOI] [PubMed] [Google Scholar]
- 24.Paglialunga S, Fisette A, Yan Y, et al. Acylation-stimulating protein deficiency and altered adipose tissue in alternative complement pathway knockout mice. Am J Physiol Endocrinol Metab.2008;294:E521–529. [DOI] [PubMed] [Google Scholar]
- 25.Cianflone K, Xia Z, Chen LY. Critical review of acylation-stimulating protein physiology in humans and rodents. Biochim Biophys Acta.2003;1609:127–143. [DOI] [PubMed] [Google Scholar]
- 26.Barbu A, Hamad OA, Lind L, Ekdahl KN, Nilsson B. The role of complement factor C3 in lipid metabolism. Mol Immunol.2015;67:101–107. [DOI] [PubMed] [Google Scholar]
- 27.Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest.2000;106:473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peake PW, Shen Y, Walther A, Charlesworth JA. Adiponectin binds C1q and activates the classical pathway of complement. Biochem Biophys Res Commun.2008;367:560–565. [DOI] [PubMed] [Google Scholar]
- 29.Hillian AD, McMullen MR, Sebastian BM, et al. Mice lacking C1q are protected from high fat diet-induced hepatic insulin resistance and impaired glucose homeostasis. J Biol Chem.2013;288:22565–22575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lewis RD, Perry MJ, Guschina IA, Jackson CL, Morgan BP, Hughes TR. CD55 deficiency protects against atherosclerosis in ApoE-deficient mice via C3a modulation of lipid metabolism. Am J Pathol.2011;179:1601–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosen BS, Cook KS, Yaglom J, et al. Adipsin and complement factor D activity: an immune-related defect in obesity. Science.1989;244:1483–1487. [DOI] [PubMed] [Google Scholar]
- 32.Faraj M, Sniderman AD, Cianflone K. ASP enhances in situ lipoprotein lipase activity by increasing fatty acid trapping in adipocytes. J Lipid Res.2004;45:657–666. [DOI] [PubMed] [Google Scholar]
- 33.Roy C, Gupta A, Fisette A, et al. C5a receptor deficiency alters energy utilization and fat storage. PLoS One.2013;8:e62531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lim J, Iyer A, Suen JY, et al. C5aR and C3aR antagonists each inhibit diet-induced obesity, metabolic dysfunction, and adipocyte and macrophage signaling. FASEB J.2013;27:822–831. [DOI] [PubMed] [Google Scholar]
- 35.Phieler J, Garcia-Martin R, Lambris JD, Chavakis T. The role of the complement system in metabolic organs and metabolic diseases. Semin Immunol.2013;25:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gauvreau D, Roy C, Tom FQ, et al. A new effector of lipid metabolism: complement factor properdin. Mol Immunol.2012;51:73–81. [DOI] [PubMed] [Google Scholar]
- 37.Vlaicu SI, Tatomir A, Boodhoo D, Vesa S, Mircea PA, Rus H. The role of complement system in adipose tissue-related inflammation. Immunol Res.2016;64:653–664. [DOI] [PubMed] [Google Scholar]
- 38.Saleh J, Al-Maqbali M, Abdel-Hadi D. Role of Complement and Complement-Related Adipokines in Regulation of Energy Metabolism and Fat Storage. Compr Physiol.2019;9:1411–1429. [DOI] [PubMed] [Google Scholar]
- 39.MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol.2013;31:259–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Sullivan D. The metabolic spectrum of memory T cells. Immunol Cell Biol.2019;97:636–646. [DOI] [PubMed] [Google Scholar]
- 41.Könner AC, Brüning JC. Toll-like receptors: linking inflammation to metabolism. Trends Endocrinol Metab.2011;22:16–23. [DOI] [PubMed] [Google Scholar]
- 42.Coll RC, O’Neill L, Schroder K. Questions and controversies in innate immune research: what is the physiological role of NLRP3? Cell Death Discov.2016;2:16019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patel CH, Powell JD. Targeting T cell metabolism to regulate T cell activation, differentiation and function in disease. Curr Opin Immunol.2017;46:82–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Geltink RIK, Kyle RL, Pearce EL. Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu Rev Immunol.2018;36:461–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bantug GR, Galluzzi L, Kroemer G, Hess C. The spectrum of T cell metabolism in health and disease. Nat Rev Immunol.2018;18:19–34. [DOI] [PubMed] [Google Scholar]
- 46.Chapman NM, Boothby MR, Chi H. Metabolic coordination of T cell quiescence and activation. Nat Rev Immunol.2019. [DOI] [PubMed] [Google Scholar]
- 47.West EE, Kemper C. Complement and T Cell Metabolism: Food for Thought. Immunometabolism.2019;1:e190006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Le Friec G, Sheppard D, Whiteman P, et al. The CD46-Jagged1 interaction is critical for human TH1 immunity. Nat Immunol.2012;13:1213–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ghannam A, Fauquert JL, Thomas C, Kemper C, Drouet C. Human complement C3 deficiency: Th1 induction requires T cell-derived complement C3a and CD46 activation. Mol Immunol.2014;58:98–107. [DOI] [PubMed] [Google Scholar]
- 50.de Jorge EG, Yebenes H, Serna M, Tortajada A, Llorca O, de Cordoba SR. How novel structures inform understanding of complement function. Semin Immunopathol.2018;40:3–14. [DOI] [PubMed] [Google Scholar]
- 51.Riley RC, Kemper C, Leung M, Atkinson JP. Characterization of human membrane cofactor protein (MCP; CD46) on spermatozoa. Mol Reprod Dev.2002;62:534–546. [DOI] [PubMed] [Google Scholar]
- 52.Tsujimura A, Shida K, Kitamura M, et al. Molecular cloning of a murine homologue of membrane cofactor protein (CD46): preferential expression in testicular germ cells. Biochem J.1998;330 (Pt 1):163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamamoto H, Fara AF, Dasgupta P, Kemper C. t. Int J Biochem Cell Biol.2013;45:2808–2820. [DOI] [PubMed] [Google Scholar]
- 54.Liszewski M, Post T, Atkinson J. Membrane cofactor protein (MCP or CD46): newest member of the regulators of complement activation gene cluster. Annu Rev Immunol.1991;9:431–455. [DOI] [PubMed] [Google Scholar]
- 55.Frolikova M, Sebkova N, Ded L, Dvorakova-Hortova K. Characterization of CD46 and beta1 integrin dynamics during sperm acrosome reaction. Sci Rep.2016;6:33714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liszewski MK, Kemper C, Price JD, Atkinson JP. Emerging roles and new functions of CD46. Springer Semin Immunopathol.2005;27:345–358. [DOI] [PubMed] [Google Scholar]
- 57.Cattaneo R. Four viruses, two bacteria, and one receptor: membrane cofactor protein (CD46) as pathogens’ magnet. J Virol.2004;78:4385–4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maga TK, Meyer NC, Belsha C, Nishimura CJ, Zhang Y, Smith RJ. A novel deletion in the RCA gene cluster causes atypical hemolytic uremic syndrome. Nephrol Dial Transplant.2011;26:739–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cipriani V, et al. Genetic variation in complement regulators and susceptibility to age-related macular degeneration. Immunobiology.2012;217:158–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Salmon JE, et al. Mutations in complement regulatory proteins predispose to preeclampsia: a genetic analysis of the PROMISSE cohort. PLoS Med.2011;8:e1001013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carrette F, Surh CD. IL-7 signaling and CD127 receptor regulation in the control of T cell homeostasis. Semin Immunol.2012;24:209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jacobs T, Erdmann H, Fleischer B. Molecular interaction of Siglecs (sialic acid-binding Ig-like lectins) with sialylated ligands on Trypanosoma cruzi. Eur J Cell Biol.2010;89:113–116. [DOI] [PubMed] [Google Scholar]
- 63.Wofford JA, Wieman HL, Jacobs SR, Zhao Y, Rathmell JC. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood.2008;111:2101–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duval F, Mathieu M, Labrecque N. Notch controls effector CD8+ T cell differentiation. Oncotarget.2015;6:21787–21788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hakulinen J, Junnikkala S, Sorsa T, Meri S. Complement inhibitor membrane cofactor protein (MCP; CD46) is constitutively shed from cancer cell membranes in vesicles and converted by a metalloproteinase to a functionally active soluble form. Eur J Immunol.2004;34:2620–2629. [DOI] [PubMed] [Google Scholar]
- 66.Ni Choileain S, Astier AL. CD46 processing: a means of expression. Immunobiology.2012;217:169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ellinghaus U, Cortini A, Pinder CL, Le Friec G, Kemper C, Vyse TJ. Dysregulated CD46 shedding interferes with Th1-contraction in systemic lupus erythematosus. Eur J Immunol.2017;47:1200–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ni Choileain S, Weyand NJ, Neumann C, Thomas J, So M, Astier AL. The dynamic processing of CD46 intracellular domains provides a molecular rheostat for T cell activation. PLoS One.2011;6:e16287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chang CH, Curtis JD, Maggi LB, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell.2013;153:1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liao W, Lin JX, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity.2013;38:13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liao W, Lin JX, Leonard WJ. IL-2 family cytokines: new insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr Opin Immunol.2011;23:598–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Arbore G, West EE, Spolski R, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4⁺ T cells. Science 2016;352:aad1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Johnson MO, Wolf MM, Madden MZ, et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell.2018;175:1780–1795 e1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic Instruction of Immunity. Cell.2017;169:570–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stummvoll GH, DiPaolo RJ, Huter EN, et al. Th1, Th2, and Th17 effector T cell-induced autoimmune gastritis differs in pathological pattern and in susceptibility to suppression by regulatory T cells. J Immunol.2008;181:1908–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Trinchieri G. Interleukin-10 production by effector T cells: Th1 cells show self control. J Exp Med.2007;204:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.O’Garra A, Vieira P. T(H)1 cells control themselves by producing interleukin-10. Nat Rev Immunol.2007;7:425–428. [DOI] [PubMed] [Google Scholar]
- 78.Larkin J 3rd, Ahmed CM, Wilson TD, Johnson HM. Regulation of interferon gamma signaling by suppressors of cytokine signaling and regulatory T cells. Front Immunol.2013;4:469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Haring JS, Corbin GA, Harty JT. Dynamic regulation of IFN-gamma signaling in antigen-specific CD8+ T cells responding to infection. J Immunol.2005;174:6791–6802. [DOI] [PubMed] [Google Scholar]
- 80.Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol.2007;96:41–101. [DOI] [PubMed] [Google Scholar]
- 81.Hess C, Kemper C. Complement-Mediated Regulation of Metabolism and Basic Cellular Processes. Immunity.2016;45:240–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Perucha E, Melchiotti R, Bibby JA, et al. The cholesterol biosynthesis pathway regulates IL-10 expression in human Th1 cells. Nat Commun.2019;10:498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Astier AL, Meiffren G, Freeman S, Hafler DA. Alterations in CD46-mediated Tr1 regulatory T cells in patients with multiple sclerosis. J Clin Invest.2006;116:3252–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bamberg CE, Mackay CR, Lee H, et al. The C5a receptor (C5aR) C5L2 is a modulator of C5aR-mediated signal transduction. J Biol Chem.2010;285:7633–7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hansen AS, Slater J, Biltoft M, Bundgaard BB, Moller BK, Hollsberg P. CD46 is a potent co-stimulatory receptor for expansion of human IFN-gamma-producing CD8(+) T cells. Immunol Lett.2018;200:26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ling GS, Crawford G, Buang N, et al. C1q restrains autoimmunity and viral infection by regulating CD8(+) T cell metabolism. Science.2018;360:558–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mangogna A, Agostinis C, Bonazza D, et al. Is the Complement Protein C1q a Pro- or Anti-tumorigenic Factor? Bioinformatics Analysis Involving Human Carcinomas. Front Immunol.2019;10:865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bulla R, Tripodo C, Rami D, et al. C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nat Commun.2016;7:10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kaur A, Sultan SH, Murugaiah V, et al. Human C1q Induces Apoptosis in an Ovarian Cancer Cell Line via Tumor Necrosis Factor Pathway. Front Immunol.2016;7:599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hong Q, Sze CI, Lin SR, et al. Complement C1q activates tumor suppressor WWOX to induce apoptosis in prostate cancer cells. PLoS One.2009;4:e5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Prochnicki T, Latz E. Inflammasomes on the Crossroads of Innate Immune Recognition and Metabolic Control. Cell Metab.2017;26:71–93. [DOI] [PubMed] [Google Scholar]
- 92.Mandrup-Poulsen T. Immunometabolism in 2017: Metabolism and the inflammasome in health and ageing. Nat Rev Endocrinol.2018;14:72–74. [DOI] [PubMed] [Google Scholar]
- 93.Hug H, Mohajeri MH, La Fata G. Toll-Like Receptors: Regulators of the Immune Response in the Human Gut. Nutrients.2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Spiljar M, Merkler D, Trajkovski M. The Immune System Bridges the Gut Microbiota with Systemic Energy Homeostasis: Focus on TLRs, Mucosal Barrier, and SCFAs. Front Immunol.2017;8:1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chaturvedi A, Pierce SK. How location governs toll-like receptor signaling. Traffic.2009;10:621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tam JC, Bidgood SR, McEwan WA, James LC. Intracellular sensing of complement C3 activates cell autonomous immunity. Science.2014;345:1256070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Baroja-Mazo A, Martin-Sanchez F, Gomez AI, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol.2014;15:738–748. [DOI] [PubMed] [Google Scholar]
- 98.McCall CE, El Gazzar M, Liu T, Vachharajani V, Yoza B. Epigenetics, bioenergetics, and microRNA coordinate gene-specific reprogramming during acute systemic inflammation. J Leukoc Biol.2011;90:439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.O’Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med.2016;213:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mills EL, Kelly B, Logan A, et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell.2016;167:457–470 e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Woolard MD, Kevil CG. Paying the Toll for Glucose Regulation: A Central Role for TLR3. Diabetes.2015;64:3345–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Strodthoff D, Ma Z, Wirstrom T, et al. Toll-Like Receptor 3 Influences Glucose Homeostasis and beta-Cell Insulin Secretion. Diabetes.2015;64:3425–3438. [DOI] [PubMed] [Google Scholar]
- 103.Zhang Z, Ohto U, Shibata T, et al. Structural Analysis Reveals that Toll-like Receptor 7 Is a Dual Receptor for Guanosine and Single-Stranded RNA. Immunity.2016;45:737–748. [DOI] [PubMed] [Google Scholar]
- 104.Elliott EI, Miller AN, Banoth B, et al. Cutting Edge: Mitochondrial Assembly of the NLRP3 Inflammasome Complex Is Initiated at Priming. J Immunol.2018;200:3047–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Iyer SS, He Q, Janczy JR, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity.2013;39:311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bantug GR, Fischer M, Grahlert J, et al. Mitochondria-Endoplasmic Reticulum Contact Sites Function as Immunometabolic Hubs that Orchestrate the Rapid Recall Response of Memory CD8(+) T Cells. Immunity.2018;48:542–555 e546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Brinkmann CR, Jensen L, Dagnæs-Hansen F, et al. Mitochondria and the lectin pathway of complement. J Biol Chem.2013;288:8016–8027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Youm YH, Nguyen KY, Grant RW, et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med.2015;21:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bae HR, Kim DH, Park MH, et al. beta-Hydroxybutyrate suppresses inflammasome formation by ameliorating endoplasmic reticulum stress via AMPK activation. Oncotarget.2016;7:66444–66454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol.2001;2:612–619. [DOI] [PubMed] [Google Scholar]
- 111.Shields AM, Panayi GS, Corrigall VM. Resolution-associated molecular patterns (RAMP): RAMParts defending immunological homeostasis? Clin Exp Immunol.2011;165:292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jun JC, Jones MB, Oswald DM, et al. T cell-intrinsic TLR2 stimulation promotes IL-10 expression and suppressive activity by CD45RbHi T cells. PLoS One.2017;12:e0180688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jiang S, Li X, Hess NJ, Guan Y, Tapping RI. TLR10 Is a Negative Regulator of Both MyD88-Dependent and -Independent TLR Signaling. J Immunol.2016;196:3834–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gültekin Y, Eren E, Özören N. Overexpressed NLRC3 Acts as an Anti-Inflammatory Cytosolic Protein. Journal of Innate Immunity.2015;7:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schneider M, Yimmermann AG, Roberts RA, et al. The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-κB. Nature Immunology.2012;13:823–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Reed JH, Jain M, Lee K, et al. Complement receptor 3 influences toll-like receptor 7/8-dependent inflammation: implications for autoimmune diseases characterized by antibody reactivity to ribonucleoproteins. J Biol Chem.2013;288:9077–9083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jialal I, Devaraj S, Rajamani U. Dysregulation of monocyte biology in metabolic syndrome. Expert Rev Endocrinol Metab.2014;9:213–221. [DOI] [PubMed] [Google Scholar]
- 118.Jiang D, Chen S, Sun R, Zhang X, Wang D. The NLRP3 inflammasome: Role in metabolic disorders and regulation by metabolic pathways. Cancer Lett.2018;419:8–19. [DOI] [PubMed] [Google Scholar]
- 119.Arbore G, Kemper C. A novel “complement-metabolism-inflammasome axis” as a key regulator of immune cell effector function. Eur J Immunol.2016;46:1563–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Köhl J. The role of complement in danger sensing and transmission. Immunol Res.2006;34:157–176. [DOI] [PubMed] [Google Scholar]
- 121.Song WC. Crosstalk between complement and toll-like receptors. Toxicol Pathol.2012;40:174–182. [DOI] [PubMed] [Google Scholar]
- 122.Zhang X, Kimura Y, Fang C, et al. Regulation of Toll-like receptor-mediated inflammatory response by complement in vivo. Blood.2007;110:228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Asgari E, Le Friec G, Yamamoto H, et al. C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood.2013;122:3473–3481. [DOI] [PubMed] [Google Scholar]
- 124.Kumar V. The complement system, toll-like receptors and inflammasomes in host defense: three musketeers’ one target. Int Rev Immunol.2019;38:131–156. [DOI] [PubMed] [Google Scholar]
- 125.Janssen BJ, Huizinga EG, Raaijmakers HC, et al. Structures of complement component C3 provide insights into the function and evolution of immunity. Nature.2005;437:505–511. [DOI] [PubMed] [Google Scholar]
- 126.Kolev M, Kemper C. Keeping It All Going-Complement Meets Metabolism. Front Immunol.2017;8:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dodds AW, Law SK. The phylogeny and evolution of the thioester bond-containing proteins C3, C4 and alpha 2-macroglobulin. Immunol Rev.1998;166:15–26. [DOI] [PubMed] [Google Scholar]
- 128.Zhang J, Ibrahim MM, Sun M, Tang J. Enoyl-coenzyme A hydratase in cancer. Clin Chim Acta.2015;448:13–17. [DOI] [PubMed] [Google Scholar]
- 129.Sousa FL, Nelson-Sathi S, Martin WF. One step beyond a ribosome: The ancient anaerobic core. Biochim Biophys Acta.2016;1857:1027–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Aliverti A, Pandini V, Pennati A, de Rosa M, Zanetti G. Structural and functional diversity of ferredoxin-NADP(+) reductases. Arch Biochem Biophys.2008;474:283–291. [DOI] [PubMed] [Google Scholar]
- 131.Bode HB, Zeggel B, Silakowski B, Wenzel SC, Reichenbach H, Müller R. Steroid biosynthesis in prokaryotes: identification of myxobacterial steroids and cloning of the first bacterial 2,3(S)-oxidosqualene cyclase from the myxobacterium Stigmatella aurantiaca. Mol Microbiol.2003;47:471–481. [DOI] [PubMed] [Google Scholar]
- 132.Elvington M, Liszewski MK, Atkinson JP. Evolution of the complement system: from defense of the single cell to guardian of the intravascular space. Immunol Rev.2016;274:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Batut B, Knibbe C, Marais G, Daubin V. Reductive genome evolution at both ends of the bacterial population size spectrum. Nat Rev Microbiol.2014;12:841–850. [DOI] [PubMed] [Google Scholar]
- 134.Streamlining Lynch M. and Simplification of Microbial Genome Architecture. Annu Rev Microbiol, 2006:327–349. [DOI] [PubMed] [Google Scholar]
- 135.Janssen BJ, Gros P. Structural insights into the central complement component C3. Mol Immunol.2007;44:3–10. [DOI] [PubMed] [Google Scholar]
- 136.King BC, Kulak K, Krus U, et al. Complement Component C3 Is Highly Expressed in Human Pancreatic Islets and Prevents beta Cell Death via ATG16L1 Interaction and Autophagy Regulation. Cell Metab.2019;29:202–210 e206. [DOI] [PubMed] [Google Scholar]
- 137.Elvington M, Liszewski MK, Bertram P, Kulkarni HS, Atkinson JP. A C3(H20) recycling pathway is a component of the intracellular complement system. J Clin Invest.2017;127:970–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kemper C, Leung M, Stephensen CB, et al. Membrane cofactor protein (MCP; CD46) expression in transgenic mice. Clin Exp Immunol.2001;124:180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang X, Zhang D, Sjolinder M, Wan Y, Sjolinder H. CD46 accelerates macrophage-mediated host susceptibility to meningococcal sepsis in a murine model. Eur J Immunol.2017;47:119–130. [DOI] [PubMed] [Google Scholar]
- 140.Rall GF, Manchester M, Daniels LR, Callahan EM, Belman AR, Oldstone MB. A transgenic mouse model for measles virus infection of the brain. Proc Natl Acad Sci U S A.1997;94:4659–4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ma EH, Verway MJ, Johnson RM, et al. Metabolic Profiling Using Stable Isotope Tracing Reveals Distinct Patterns of Glucose Utilization by Physiologically Activated CD8(+) T Cells. Immunity.2019;51:856–870 e855. [DOI] [PubMed] [Google Scholar]
- 142.Loft A, Forss I, Mandrup S. Genome-Wide Insights into the Development and Function of Thermogenic Adipocytes. Trends Endocrinol Metab.2017;28:104–120. [DOI] [PubMed] [Google Scholar]
- 143.Ahrens-Nicklas RC, Slap G, Ficicioglu C. Adolescent presentations of inborn errors of metabolism. J Adolesc Health.2015;56:477–482. [DOI] [PubMed] [Google Scholar]
- 144.Strainic MG, Shevach EM, An F, Lin F, Medof ME. Absence of signaling into CD4⁺ cells via C3aR and C5aR enables autoinductive TGF-β1 signaling and induction of Foxp3⁺ regulatory T cells. Nat Immunol.2013;14:162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dunkelberger J, Zhou L, Miwa T, Song WC. C5aR expression in a novel GFP reporter gene knockin mouse: implications for the mechanism of action of C5aR signaling in T cell immunity. J Immunol.2012;188:4032–4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Karsten CM, Wiese AV, Mey F, et al. Monitoring C5aR2 Expression Using a Floxed tdTomato-C5aR2 Knock-In Mouse. J Immunol.2017;199:3234–3248. [DOI] [PubMed] [Google Scholar]
- 147.Quell KM, Karsten CM, Kordowski A, et al. Monitoring C3aR Expression Using a Floxed tdTomato-C3aR Reporter Knock-in Mouse. J Immunol.2017;199:688–706. [DOI] [PubMed] [Google Scholar]
- 148.A Verghese D, Demir M, Chun N, et al. T Cell Expression of C5a Receptor 2 Augments Murine Regulatory T Cell (TREG) Generation and TREG-Dependent Cardiac Allograft Survival. J Immunol.2018;200:2186–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kovtun A, Bergdolt S, Hagele Y, et al. Complement receptors C5aR1 and C5aR2 act differentially during the early immune response after bone fracture but are similarly involved in bone repair. Sci Rep.2017;7:14061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.West EE, Afzali B, Kemper C. Unexpected Roles for Intracellular Complement in the Regulation of Th1 Responses. Adv Immunol.2018;138:35–70. [DOI] [PubMed] [Google Scholar]