

Abstract
SHOX, the short stature homeobox-containing gene, encodes a critical regulatory protein controlling long bone growth. We examined patients in one family, identified an intronic mutation, and expressed SHOX minigenes in HEK293T cells to characterize the effect on gene splicing. We identified a novel mutation at position −3 (c.−432–3C>A;g.6120C>A) of the intron 1 splice acceptor site; three short (height Z-score −2.4 to −1.7) children were heterozygous and the father (height Z-score −3.4) was homozygous. A wild-type minigene produced alternative transcripts; one utilized the normal splice site between intron 1 and exon 2, the other a cryptic splice site in exon 2. Mutant SHOX minigene generated only the smaller transcript. The exon 2 acceptor splice site is weak; an alternative transcript is normally produced using a downstream cryptic splice site. The c.−432–3C>A mutation causes further weakening, and the cryptic splice site is preferentially utilized, resulting in SHOX deficiency and short stature.
Keywords: short stature, SHOX, splice site mutation
Introduction
SHOX, the short stature homeobox-containing gene, is located in the pseudoautosomal regions on the X and Y chromosomes and encodes a critical regulatory protein that controls bone growth (1). Its location in the pseudoautosomal region allows SHOX to escape X inactivation, and expression of copies of SHOX on both the X and Y chromosomes (2) is required for normal skeletal development. The SHOX gene spans seven exons and includes four exons that have only recently been characterized (3). Two Shox protein isoforms, Shoxa and Shoxb, are generated through the use of alternative 5′-untranslated region (5′-UTR) sequences and final exons (4). Expression of the SHOX gene is regulated by a complex mechanism that utilizes sequential transcriptional and translational control mechanisms (5). Alterations in SHOX expression are associated with several disorders of growth, stature, and skeletal development. Homozygous loss of function of SHOX is the basis for Langer mesomelic dysplasia, characterized by extreme short stature and limb aplasia or hypoplasia of the ulna and fibula (6). Loss of one copy of SHOX or deletions in the pseudoautosomal regions (7) are also the cause of growth failure and skeletal defects in Turner syndrome (1) and Leri Weill dyschondroplasia (8). In both of these conditions, SHOX haploinsufficiency is associated with short stature and the Madelung deformity, a condition in which arrest of epiphyseal growth of the medial and volar portions of the distal radius leads to shortening of the radius and relative overgrowth of the ulna. Deletions and mutations in the SHOX gene have also been identified in patients with idiopathic short stature who lack apparent skeletal deformities (9). By contrast, duplication of Xp, which results in SHOX polyploidy, is associated with tall stature (10) and some cases of type I Mayer-Rokitansky-Kuster-Hauser syndrome (11). Overall, deficiency of SHOX is associated with considerable phenotypic heterogeneity. The variable skeletal phenotypes in patients with SHOX mutations and duplications suggest a quantitative relation of functional protein to bone development and growth.
Most SHOX mutations are due to large deletions that result in haploinsufficiency. However, a variety of point mutations and small deletions have been identified in SHOX that have been implicated in short stature. It is estimated that SHOX mutations are found with a frequency of 1:1000 in the normal population and in up to 2%–15% of children with idiopathic short stature (4).
We report the identification and molecular characterization of a novel SHOX gene intronic mutation at position −3 of the splice acceptor site of exon 2 leading to replacement of the conserved cytosine by adenine (c.−432–3C>A; g.6120C>A). This nucleotide replacement impairs normal splicing and leads to exclusive use of a cryptic splice acceptor site in exon 2 that predicts loss of the homeodomain of the SHOX protein.
Materials and methods
Case reports
The proband (II:2 in pedigree, Figure 1) was born full-term after an un-complicated pregnancy to healthy, non-consanguineous parents of Ashkenazi Jewish descent. Birth weight (3289 g) and length (50.8 cm) were normal. Her developmental milestones were appropriate, and her parents noted declining growth and short stature at the age of 1–2 years. At age 9 years, breast development and pubic hair were first noted, and she was begun on leuprolide and growth hormone by another provider. Breast development regressed on leuprolide but her growth velocity was unchanged (4.4 cm/year) on this combined therapy and her growth remained less than the first percentile. At chronological age 11 years and 5 months, her bone age according to the standards of Greulich and Pyle was 9 years and 5 months and her serum IGF1 level ranged from 55.4 to 74.0 nmol/L. Her physical examination revealed proportionate short stature (−2.4 SDS) with a normal seated height/height SDS of 0.81 (12). Pubertal exam revealed Tanner 1 breasts and Tanner 3 pubic hair. She had a normal female karyotype (46, XX). A skeletal survey was normal and no Madelung deformity was present. At 12 years and 5 months, she was noted to have a mildly elevated serum level of thyroid-stimulating hormone at 8.2 μIU/mL (reference range 0.5–3.8 μIU/mL) and treatment with levothyroxine 50 μg daily was initiated. Leuprolide was discontinued.
Figure 1.

Pedigree of the family described in this report.
The proband (II.2) is indicated with the arrow. The proband and her two brothers (II.1 and II.3) were found to be heterozygous for the SHOX mutation. Her father, (I.1) was homozygous; results of his single nucleotide polymorphism analysis are indicated. Her mother (I.2) had normal SHOX alleles.
The proband’s parents and two brothers were also evaluated (Table 1, Figure 1). The father (I:1) is the smallest member of his extended family, with a height of 152.4 cm (−3.4 SDS) and normal (SDS 1.2) seated height/height ratio (12). He was proportionate without skeletal deformities. Her two brothers (II:1 and II:3), ages 16 and 7 years, had proportionate short stature (height −2.3 and −1.7 SDS, respectively) with normal seated height/height ratios (0.64 and 0.60 SDS, respectively) (12). The mother (I:2) had a normal height (155 cm, −1.3 SDS). The proband’s paternal grandmother was 155 cm tall and the paternal grandfather was 160 cm tall; both were proportionate without skeletal deformities. The maternal grandmother was 152 cm tall and the maternal grandfather was 155 cm tall. No Madelung deformity was present in any family members. Stature is expressed as SDS according to the population standards for age and gender (http://www.magicfoundation.org/). The protocol was approved by the appropriate institutional review boards, and informed consent and/or assent was obtained from all subjects.
Table 1.
Baseline characteristics of family members described in this report.
| Individual | Height, cm | Height SDS | Seated height/height ratio |
|---|---|---|---|
| Proband (age 11 years and 5 months) | 129 | − 2.4 | 0.81 |
| Proband’s brother (age 16 years) | 155 | − 2.3 | 0.64 |
| Proband’s brother (age 7 years) | 113 | − 1.7 | 0.60 |
| Father | 152.4 | − 3.4 | 1.2 |
| Mother | 155 | − 1.3 | a |
| Paternal grandmother | 155 | − 1.3 | a |
| Paternal grandfather | 160 | − 2.3 | a |
| Maternal grandmother | 152 | − 1.7 | a |
| Maternal grandfather | 155 | − 3.0 | a |
Data not obtained.
Mutational analysis of SHOX
Genomic DNA was isolated from peripheral blood mononuclear cells of all subjects and the seven exons and flanking intronic sequences of the SHOX gene were amplified by PCR. The sequences were then screened for point mutations, small deletions and insertions within the SHOX coding sequences, and intron/exon boundaries by denaturing high-performance liquid chromatography (Transgenomic Wave 3500HT, Omaha, NE, USA), and abnormal heteroduplexes were analyzed by direct sequencing of amplicons using previously described techniques (primer sequences and PCR conditions are available upon request) (7). DNA from subject I-1 was also analyzed for larger SHOX gene deletions by multiplex probe ligation amplification using standard techniques (13). DNA mutation numbering is based on cDNA sequence (Accession no. NM_000451.3) and gDNA sequence (NCBI Reference Sequence: NC_000023.10) and expressed using HGVS V2.0 nomenclature.
In silico splice site prediction
The effect of the single base substitution identified in the splice acceptor site of intron 1 was evaluated using several splice site prediction algorithms (14, 15).
Synthesis of hybrid minigenes
Because we were unable to detect SHOX transcripts in peripheral blood mononuclear cells by reverse transcription (RT)-PCR, we synthesized hybrid SHOX minigenes to determine the effect of this mutation on pre-mRNA splicing efficiency. We created wild-type and mutant SHOX minigenes in the pTARGET mammalian expression vector (Promega, Madison, WI, USA) using PCR to amplify and assemble exons 1–3 plus short intronic flanking regions of SHOX from normal human genomic DNA (Figure 2). The PCR products were cloned into pTARGET, and the SHOX sequences were verified by direct sequencing using ABI PRISM Big Dye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Carlsbad, CA, USA).
Figure 2.

SHOX minigene.
Minigenes were created using exons 1 – 3 and short intronic flanking regions. The wild-type sequence is shown above; the arrow indicates the replacement of cytosine by adenine at position −3 of the acceptor splice site in the mutant sequence.
Characterization of SHOX expression
Wild-type or mutant SHOX minigenes were transiently transfected into HEK293T cells using Fugene 6 Transfection Reagent (Roche Applied Science, Indianapolis, IN, USA) according to the manufacturer’s recommendation. Approximately 48 h after transfection, total RNA was extracted from the cells using RNeasy Mini kit (Qiagen Inc., Valencia, CA, USA). The concentration of RNA obtained was measured using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA), and RNA integrity was determined by denaturing agarose gel electrophoresis. Aliquots of total RNA (1.5 μg) were used to generate first-strand cDNA using a cDNA synthesis kit (iScript; BioRad, Hercules, CA), using a blend of oligo(dT) and random primers. After first-strand cDNA synthesis, the reaction was diluted 1/10. For PCR, 5 μL of the diluted cDNA was amplified into a total volume of 20 μL using 10 μM primers located in exon 1 (5′-TCTTTCTACTGCAAACAGAAATGG-3′) and exon 3 (5′-ATTCATAAATCCCTTCTGCCACT-3′) and Amplitaq Gold DNA polymerase (Applied Biosystems) with the following parameters: an initial denaturation at 94°C for 3 min, followed by 40 cycles at 94°C for 30 s, 50°C for 30 s, 72°C for 1 min, and a final extension at 72°C for 5 min. PCR products were analyzed by agarose gel electrophoresis and were sequenced on both strands. Total RNA was extracted from discarded human skull bone fragments and RT-PCR was performed using primers (5′-ATTTCGTTTCCGCGCGTCTCTTTC-3′ and 5′-GCCGGCAATTGGCTTTCTTCTCAT-3′) that were designed to anneal in exons 1 and 2 and conditions described above.
Results
Molecular analysis of SHOX
The proband was found to be heterozygous for a single nucleotide change (C to A) located three nucleotides upstream of the splice acceptor site in intron 1 (c.−432–3C>A). Both of her brothers were also heterozygous for this variant, while their father’s DNA showed only the adenine nucleotide at position c.−432–3. Multiplex ligation-dependent probe amplification (MLPA) analysis of the father’s DNA indicated that he had two copies of the SHOX gene without evidence for an intragenic deletion, thus confirming that he was homozygous for the c.−432–3C>A mutation. The mother carried two wild-type SHOX alleles. Review of a database that contains SHOX gene sequences from >6100 patients with short stature revealed this substitution in only one additional unrelated patient, and no patients with normal stature are reported to carry this variant (personal communication, Frank K Fujimura, PhD., Esoterix Inc., Calabasas Hills, CA). In addition, inspection of the online SHOX gene mutation database (www.hd-lovd.uni-hd.de) similarly failed to disclose this mutation (16, 17).
In silico results
In silico analysis was used to determine the effect of the splice acceptor site mutation on splicing by utilizing splice prediction web interfaces. GeneSplicer (18) (http://www.cbcb.umd.edu/software/GeneSplicer) predicted a reduction in splice site strength from 84% for the wild-type sequence to 14% for the mutant splice site. We used the Cryp Skip web interface (19) (http://cryp-skip.img.cas.cz) to determine whether the C>A mutation might activate a cryptic splice site. This program indicated a 91% probability of cryptic splice site activation.
Splicing efficiency
The SHOX wild-type hybrid minigene produced two alternative transcripts (Figures 3 and 4). The larger and far more abundant transcript, 873 bp, results from the predicted splicing of exon 1 to exon 2 at the native splice acceptor site at position c.−432–3. The smaller transcript, 410 bp, is generated by use of a cryptic splice site in exon 2 at position c.47. However, we were unable to confirm the existence of an alternative transcript that utilizes this cryptic splice site by RT-PCR of normal bone (data not shown). Similarly, a recent comprehensive analysis of SHOX transcripts also failed to identify corresponding transcripts (3). Thus, use of this internal splice site by wild-type alleles in vivo remains speculative; however, utilization appears to require mutation of the normal splice site.
Figure 3.
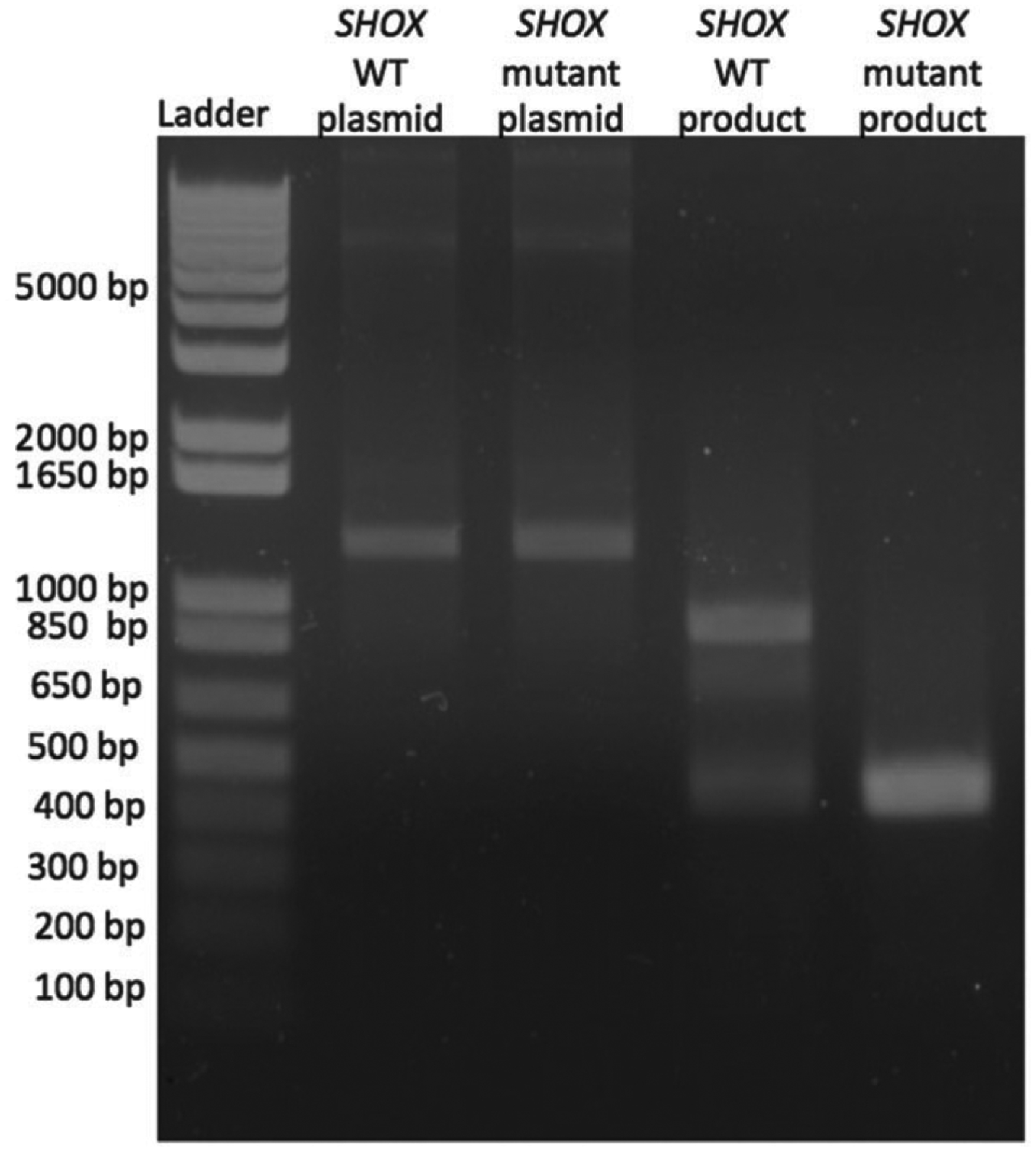
Agarose gel electrophoresis showing RT-PCR products produced by SHOX minigenes.
Lane 1 indicates DNA ladder with molecular sizes shown. Lanes 2 and 3 show PCR products generated from the SHOX wild-type and mutant plasmids, with amplicons of the expected size of 1200 bp. Lane 4 shows the RT-PCR product from transfection of HEK293T cells with the SHOX wild-type plasmid, with the expected amplicons of 893 bp and an additional smaller band of 410 bp. Lane 5 shows the RT-PCR product from HEK293T cells that had been transiently transfected with the SHOX mutant plasmid, which generates only the 410 bp using the cryptic splice site. A water control was negative, and is not shown in this figure.
Figure 4.

Schematic of SHOX transcripts.
The SHOX minigene is indicated in the center of the figure with the exons and truncated flanking intronic sequences. The top product indicates normal splicing, with removal of the introns at the appropriate splice sites, and results in an 893-bp product, as seen on the gel in Figure 4. The dashed lines indicate the use of the cryptic splice site exon 2, which results in the 410-bp product.
The mutant SHOX minigene prevented production of any normal transcript and generated a single, small transcript of 410 bp that was identical to the smaller transcript produced by the wild-type minigene. This transcript contains an in-frame ATG at position c.457 (based on wild-type transcript numbering) in exon 3 that has a strong Kozak sequence. The theoretical use of this alternative initiation codon would produce a truncated Shox protein missing the first 145 amino acids and consequently lacking most of the homeobox domain.
Genotype-phenotype
The father in this family (I-1) was homozygous for the SHOX mutation. Haplotype analysis of the father’s DNA showed homozygosity for microsatellite markers that are linked to SHOX, thereby confirming identity by descent for the SHOX alleles (Figure 1).
Discussion
This case illustrates an example of familial short stature due to a novel acceptor splice site mutation in the SHOX gene. Splice site mutations result in either exon skipping, which utilizes the next available splice site, retention of the intron, or the use of a cryptic splice site (20, 21). Donor splice site mutations occur more frequently than acceptor splice site mutations (20).
Heterozygous loss-of-function mutations in the SHOX gene represent one of the best characterized genetic causes of idiopathic short stature. Although haploinsufficiency of SHOX is frequently associated with skeletal abnormalities and disproportionate seated height (22), there is remarkable phenotypic heterogeneity (23) and many patients with SHOX mutations have proportionate short stature and normal skeletal development. Transmission of SHOX mutations leads to familial short stature, and the father in this family transmitted defective alleles to both his sons and daughter. The unique location of SHOX renders the gene susceptible to pseudoautosomal recombination during paternal meiosis. Genes in the pseudoautosomal region of the X chromosome are believed to escape X inactivation (24), and as a result, active copies of SHOX present on both the X and Y chromosome and both copies are required for normal phenotypic expression. If recombination occurs during meiosis, altered gene expression may result and can be associated with short stature or dysmorphic skeletal features (25). Males have a high frequency of recombination in this region, and several examples of paternal transmission of a heterozygous SHOX mutation to both sons and daughters have been recently described (26), presumably as a result of mutations being transmitted from a paternal X chromosome to a proband’s Y chromosome. However, several lines of evidence indicate that the father of this family was homozygous for the missense mutation, and thereby transmitted mutant X and Y alleles to his affected children. First, haplotype analysis of the father’s DNA using microsatellite markers linked to SHOX (Figure 1) revealed homozygosity across a 7-Mb region that includes SHOX, consistent with identity by descent and a likely previous pseudoautosomal crossover of the mutation. Second, MLPA analysis of the father’s DNA gene failed to identify an intragenic SHOX deletion, indicating that the father had two copies of the SHOX gene and that both alleles carried the same missense mutation. There has been one previous report of a SHOX mutation being transmitted from a paternal X chromosome to a proband Y chromosome during pseudoautosomal recombination during male meiosis (10). Presumed consanguinity likely led to homozygosity of the mutation. This rare recombination event within the pseudoautosomal region allows for male transmission of mutant alleles to sons and a daughter.
We identified a dose effect of SHOX insufficiency in this family, as the homozygous father had more extreme short stature than his heterozygous children. However, despite having two copies of the abnormal SHOX gene, the father lacked features of Langer mesomelic dysplasia. This observation suggests that some functional Shox protein is produced despite the homozygous mutation. Although it is possible that the abnormally spliced SHOX transcript is translated, this is predicted to produce a Shox protein that lacks the homeobox domain, and thus would have very little if any biological activity. Alternatively, some normal transcripts may be produced through the use of the mutated acceptor site, although our in vitro studies did not demonstrate this. A more likely explanation for the phenotypic heterogeneity in this family is the use of alternative transcriptional mechanisms. The principal promoter for SHOX transcription (P1) is located upstream of exon 1 and produces a transcript with a long 5′-UTR. However, SHOX has an alternative (P2) promoter (5) that is located within exon 2 (Figure 5), and which produces a shorter transcript with a truncated 5′-UTR. This nascent SHOX transcript should be unaffected by the splice acceptor site mutation that we have identified in intron 1 and thus should generate a mature transcript that is translated into a fully functional Shox protein. It is unknown how much expression derives from use of the P2 promoter in various cells or at different stages of development; however, given the different translational efficiencies of the two transcripts, it has been suggested that P2 is used in situations with immediate needs of high Shox protein amounts, whereas use of the P1 promoter produces transcripts that facilitate regulation of Shox protein levels by translational control mechanisms (5).
Figure 5.

Organization of SHOX gene.
The SHOX gene is shown above with exons numbered. (Two isoforms differ in exon VI.) Promoter 1 (P1) is within the 5′-UTR. Promoter 2 (P2) is within exon 2 at position c.−298. The normal AUG initiator codon is shown at position c.1. The location of the splice site mutation is indicated at c.−432–3. The cryptic splice site is shown at position c.47.
Finally, although single base pair substitutions at splice sites are thought to be responsible for approximately 10% of all inherited, disease-causing mutations (27), our report emphasizes how uncommon splice site mutations are as a basis of SHOX deficiency. Review of the Human Short Stature Gene Allelic Variant Database (www.hd-lovd.uni-hd.de), which contains a registry of 202 unique disease-related allelic variants in the SHOX gene (16, 17), and the published literature, revealed only one other splice site mutation in SHOX that led to familial short stature (28).
In conclusion, we report a novel SHOX gene splice site mutation in a family with idiopathic short stature. Affected family members demonstrate variable heights with a dose effect that is consistent with a quantitative threshold of functional Shox protein that is necessary for normal development. The affected patients in our studied kindred have normal skeletal development, and therefore is it likely that some residual Shox activity accounts for the mild phenotype in patients carrying one or two mutant SHOX alleles.
Acknowledgments:
We are grateful to Athena Diagnostics Inc. (Worcester, MA 01605, USA) for performing the MLPA analysis of the SHOX gene in subject I-1, and to Mr. Evan Opas for providing excellent technical assistance.
Funding: This work was supported in part by grants from the Cleveland Clinic Lerner Research Institute, the Friedman Family Fund, NIH T32 DK63688-09, Pediatric Endocrine Society, and The Children’s Hospital of Philadelphia.
Footnotes
Conflict of interest statement
Declaration of interest: The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Contributor Information
Jennifer Danzig, Division of Endocrinology and Diabetes, The Children’s Hospital of Philadelphia and Department of Pediatrics, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA.
Michael A. Levine, Division of Endocrinology and Diabetes, The Children’s Hospital of Philadelphia and Department of Pediatrics, Perelman School of Medicine at the University of Pennsylvania, 11 Northwest Tower, Suite 30, 34th and Civic Center Boulevard, Philadelphia, PA 19104, USA.
References
- 1.Rao E, Weiss B, Fukami M, Rump A, Niesler B, et al. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat Genet 1997;16:54–63. [DOI] [PubMed] [Google Scholar]
- 2.Blaschke RJ, Rappold G. The pseudoautosomal regions, SHOX and disease. Curr Opin Genet Dev 2006;16:233–9. [DOI] [PubMed] [Google Scholar]
- 3.Durand C, Roeth R, Dweep H, Vlatkovic I, Decker E, et al. Alternative splicing and nonsense-mediated RNA decay contribute to the regulation of SHOX expression. PLoS One 2011;6:e18115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marchini A, Rappold G, Schneider KU. SHOX at a glance: from gene to protein. Arch Physiol Biochem 2007;113:116–23. [DOI] [PubMed] [Google Scholar]
- 5.Blaschke RJ, Topfer C, Marchini A, Steinbeisser H, Janssen JW, et al. Transcriptional and translational regulation of the Leri-Weill and Turner syndrome homeobox gene SHOX. J Biol Chem 2003;278:47820–6. [DOI] [PubMed] [Google Scholar]
- 6.Zinn AR, Wei F, Zhang L, Elder FF, Scott CI, et al. Complete SHOX deficiency causes Langer mesomelic dysplasia. Am J Med Genet 2002;110:158–63. [DOI] [PubMed] [Google Scholar]
- 7.Benito-Sanz S, Thomas NS, Huber C, Gorbenko del Blanco D, Del Blanco DG, et al. A novel class of pseudoautosomal region 1 deletions downstream of SHOX is associated with Leri-Weill dyschondrosteosis. Am J Hum Genet 2005;77:533–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shears DJ, Vassal HJ, Goodman FR, Palmer RW, Reardon W, et al. Mutation and deletion of the pseudoautosomal gene SHOX cause Leri-Weill dyschondrosteosis. Nat Genet 1998;19:70–3. [DOI] [PubMed] [Google Scholar]
- 9.Morizio E, Stuppia L, Gatta V, Fantasia D, Guanciali Franchi P, et al. Deletion of the SHOX gene in patients with short stature of unknown cause. Am J Med Genet A 2003;119A:293–6. [DOI] [PubMed] [Google Scholar]
- 10.Ogata T, Kosho T, Wakui K, Fukushima Y, Yoshimoto M, et al. Short stature homeobox-containing gene duplication on the der(X) chromosome in a female with 45,X/46,X, der(X), gonadal dysgenesis, and tall stature. J Clin Endocrinol Metab 2000;85:2927–30. [DOI] [PubMed] [Google Scholar]
- 11.Gervasini C, Grati FR, Lalatta F, Tabano S, Gentilin B, et al. SHOX duplications found in some cases with type I Mayer-Rokitansky-Kuster-Hauser syndrome. Genet Med 2010;12:634–40. [DOI] [PubMed] [Google Scholar]
- 12.Fredriks AM, van Buuren S, van Heel WJ, Dijkman-Neerincx RH, Verloove-Vanhorick SP, et al. Nationwide age references for sitting height, leg length, and sitting height/height ratio, and their diagnostic value for disproportionate growth disorders. Arch Dis Child 2005;90:807–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gatta V, Antonucci I, Morizio E, Palka C, Fischetto R, et al. Identification and characterization of different SHOX gene deletions in patients with Leri-Weill dyschondrosteosys by MLPA assay. J Hum Genet 2007;52:21–7. [DOI] [PubMed] [Google Scholar]
- 14.Senapathy P, Shapiro MB, Harris NL. Splice junctions, branch point sites, and exons: sequence statistics, identification, and applications to genome project. Methods Enzymol 1990;183:252–78. [DOI] [PubMed] [Google Scholar]
- 15.Shapiro MB, Senapathy P. RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res 1987;15:7155–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fokkema IF, den Dunnen JT, Taschner PE. LOVD: easy creation of a locus-specific sequence variation database using an “LSDB-in-a-box” approach. Hum Mutat 2005;26:63–8. [DOI] [PubMed] [Google Scholar]
- 17.Niesler B, Roth R, Wilke S, Fujimura F, Fischer C, et al. The novel human SHOX allelic variant database. Hum Mutat 2007;28:933–8. [DOI] [PubMed] [Google Scholar]
- 18.Pertea M, Lin X, Salzberg SL. GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res 2001;29:1185–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Divina P, Kvitkovicova A, Buratti E, Vorechovsky I. Ab initio prediction of mutation-induced cryptic splice-site activation and exon skipping. Eur J Hum Genet 2009;17:759–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krawczak M, Thomas NS, Hundrieser B, Mort M, Wittig M, et al. Single base-pair substitutions in exon-intron junctions of human genes: nature, distribution, and consequences for mRNA splicing. Hum Mutat 2007;28:150–8. [DOI] [PubMed] [Google Scholar]
- 21.Buratti E, Chivers M, Kralovicova J, Romano M, Baralle M, et al. Aberrant 5′ splice sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res 2007;35:4250–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rappold G, Blum WF, Shavrikova EP, Crowe BJ, Roeth R, et al. Genotypes and phenotypes in children with short stature: clinical indicators of SHOX haploinsufficiency. J Med Genet 2007;44:306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jorge AA, Souza SC, Nishi MY, Billerbeck AE, Liborio DC, et al. SHOX mutations in idiopathic short stature and Leri-Weill dyschondrosteosis: frequency and phenotypic variability. Clin Endocrinol (Oxf) 2007;66:130–5. [DOI] [PubMed] [Google Scholar]
- 24.Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005;434:400–4. [DOI] [PubMed] [Google Scholar]
- 25.Day G, Szvetko A, Griffiths L, McPhee IB, Tuffley J, et al. SHOX gene is expressed in vertebral body growth plates in idiopathic and congenital scoliosis: implications for the etiology of scoliosis in Turner syndrome. J Orthop Res 2009;27:807–13. [DOI] [PubMed] [Google Scholar]
- 26.Kant SG, van der Kamp HJ, Kriek M, Bakker E, Bakker B, et al. The jumping SHOX gene – crossover in the pseudoautosomal region resulting in unusual inheritance of Leri-Weill dyschondrosteosis. J Clin Endocrinol Metab 2011;96:E356–9. [DOI] [PubMed] [Google Scholar]
- 27.Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, et al. Human gene mutation database (HGMD): 2003 update. Hum Mutat 2003;21:577–81. [DOI] [PubMed] [Google Scholar]
- 28.Flanagan SF, Munns CF, Hayes M, Williams B, Berry M, et al. Prevalence of mutations in the short stature homeobox containing gene (SHOX) in Madelung deformity of childhood. J Med Genet 2002;39:758–63. [DOI] [PMC free article] [PubMed] [Google Scholar]