

Abstract
Aims
Although the effect of the angiotensin receptor blocker neprilysin inhibitor (ARNI) sacubitril/valsartan on heart failure (HF) hospitalizations and cardiovascular death has been evaluated, its effects on functional capacity in patients with HF and ejection fraction (EF) >40% has yet to be determined. In addition, no prior studies have compared sacubitril/valsartan with angiotensin‐converting enzyme inhibitor therapy. We sought to compare the effect of ARNI to background‐medication‐based individualized comparators (BMICs) on N‐terminal pro‐B‐type natriuretic peptide (NT‐proBNP), functional capacity [6 min walk distance (6MWD)], symptoms, and quality of life [Kansas City Cardiomyopathy Questionnaire (KCCQ)] in patients with HF and EF >40% in a randomized clinical trial.
Methods
PARALLAX is a prospective, randomized, controlled, double‐blind multicentre clinical trial in patients with chronic symptomatic HF with EF >40%, New York Heart Association (NYHA) class II–IV symptoms, elevated natriuretic peptides, and evidence of structural heart disease. Eligible patients are randomized to sacubitril/valsartan vs. BMIC for cardiovascular and related co‐morbidities. BMIC includes (i) enalapril, (ii) valsartan, and (iii) placebo depending on the type of medical therapy prior to enrolment. The primary endpoints are the change in plasma NT‐proBNP concentration from baseline to 12 weeks and the change from baseline in 6MWD distance at 24 weeks. The secondary endpoints assess quality of life and symptom burden.
Conclusions
PARALLAX will determine if sacubitril/valsartan compared with standard medical therapy for co‐morbidities improves NT‐proBNP levels, exercise capacity, quality of life, and symptom burden in HF patients with EF >40%.
Keywords: Heart failure with preserved ejection fraction, Heart failure with mid‐range ejection fraction, Functional capacity, Quality of life, Neprilysin inhibition, Randomized controlled trial
1. Background
Heart failure (HF) is associated with high morbidity and mortality and is the most common reason for hospitalization in the Western World. 1 According to current European Society of Cardiology (ESC) Guidelines, HF is classified into three left ventricular ejection fraction (LVEF) categories: (i) HF with reduced ejection fraction ([EF] < 40%, HFrEF), (ii) HF with mid‐range EF (40–49%, HFmrEF) and iii), and HF with preserved EF (≥50%, HFpEF). 2 Approximately up to half of patients with HF have HFpEF, and the proportion of HFpEF in the overall HF population is increasing. 3 While there are pharmacological therapies with mortality benefit in HFrEF, 4 , 5 , 6 , 7 , 8 no therapy has yet shown to convincingly reduce morbidity and mortality in patients with a LVEF of 40% or higher.
Unlike HFrEF patients, HFpEF patients tend to be older and predominantly female. 9 Cardiovascular and metabolic co‐morbidities, such as hypertension, atrial fibrillation, diabetes, renal disease, and obesity, tend to be more common among them than in their HFrEF counterparts. With regards to the burden of the disease, the risk for hospitalization (first and recurrent) is similar in HFpEF as in HFrEF but with a higher incidence of non‐cardiovascular death in HFpEF than in HFrEF. In addition to a substantial morbidity and mortality burden, HFpEF patients suffer from HF symptoms, limitations in exercise capacity, and reduced quality of life comparable with HFrEF patients. International HF guidelines only recommend symptomatic relief for the non‐HFrEF patients, using diuretics in addition to optimizing treatment of their co‐morbidities, given the lack of strong evidence from large outcomes trials in this population. Thus, it is clear that HFpEF is a HF phenotype with a significant unmet medical need for better management options. Specifically, improvements in functional capacity, quality of life, and/or HF related symptoms are important patient‐related outcomes, and a therapy that improves these clinical parameters would be a major advancement in the treatment paradigm of patients with HFpEF.
The angiotensin receptor blocker neprilysin inhibitor (ARNI) sacubitril/valsartan combines the effects of neprilysin inhibition and angiotensin type 1 receptor blockade. Neprilysin degrades biologically active natriuretic peptides, including atrial natriuretic peptide, B‐type natriuretic peptide, and C‐type natriuretic peptide; angiotensin type 1 receptor blockade inhibits the renin angiotensin system. In patients with HFrEF enrolled in the PARADIGM‐HF trial, sacubitril/valsartan reduced HF hospitalizations, and improved survival and symptoms in comparison with enalapril. 7 In the hypothesis‐generating PARAMOUNT trial, sacubitril/valsartan showed, after 12 weeks of treatment, a significant N‐terminal pro‐natriuretic peptide (NT‐proBNP) reduction compared with valsartan in patients with HF and an EF >45%. 10
The PARAGON‐HF trial investigated the effect of sacubitril/valsartan vs. valsartan on HF outcomes in 4822 patients with HF and an EF ≥45%. Treatment with sacubitril/valsartan did not reduce the primary endpoint (total HF hospitalizations and cardiovascular death) in the overall population, but there was a modest, although statistically non‐significant, lower rate of hospitalizations for HF with sacubitril–valsartan than with valsartan. 11
The comparator valsartan in PARAGON‐HF has been criticized as there is no established renin–angiotensin–aldosterone system (RAAS) blockade therapy in HFpEF. A comparator therapy reflecting the RAAS blockade prior to randomization (background‐medication individualized comparators) was warranted. In addition, prior to randomization, patients in PARAGON‐HF had to tolerate valsartan and sacubitril/valsartan in a run‐in‐period. This limits generalizability of the results.
We have designed the PARALLAX trial, a multicentre randomized trial to evaluate the effect of sacubitril/valsartan in comparison with background‐medication‐based individualized comparators (BMIC) for cardiovascular and related co‐morbidities on NT‐proBNP levels and submaximal exercise capacity assessed by 6 min walk test distance (6MWD) in patients with an EF >40%. The study will also evaluate physical function, symptom burden, and social function changes using appropriate quality of life instruments. Here, we describe the rationale and design of the PARALLAX trial and compare it with prior and current trials of HF treatments to improve NT‐proBNP, functional capacity, and quality of life in HFpEF.
2. Methods
PARALLAX is a 24 week, randomized, double‐blind, parallel group, active‐controlled, or placebo‐controlled trial to evaluate sacubitril/valsartan compared wih BMICs on NT‐proBNP, exercise capacity, quality of life, and symptoms in patients with HF and EF >40%. The trial was designed by members of the steering committee in collaboration with the sponsor. The trial has been registered (NCT03066804) and is in compliance with the Declaration of Helsinki. Approval from the responsible local authorities and ethics committees was obtained prior to inclusion of patients.
2.1. Patients
The final key eligibility criteria of the PARALLAX trial are summarized in Table 1 . In summary, patients at an age of 45 years or older, with symptomatic HF requiring the use of diuretics in the last 30 days prior to inclusion, New York Heart Association (NYHA) functional class II to IV, and an EF >40% are to be included. Initially, EF at inclusion was defined as ≥45% but was reduced to >40% in Amendment 2 (dated 12 September 2018) to close the LVEF gap left between the PARADIGM 7 and the PARAGON 11 trial. Elevated NT‐proBNP levels at randomization (cut‐offs >220 pg/mL in patients with no atrial fibrillation or atrial flutter (non‐AF), and > 600 pg/mL in patients with ongoing AF reinforces the diagnosis of HFpEF. Furthermore, objective evidence of structural heart disease (either left atrial enlargement or left ventricular hypertrophy) has to be demonstrated by echocardiography at screening or within 6 months prior to randomization. As this trial specifically evaluates effects of sacubitril/valsartan on quality of life, only patients with a Kansas City Cardiomyopathy Questionnaire (KCCQ) summary score < 75 are included. Such scores reflect the more symptomatic patients and are strongly associated with NYHA class II to IV. Patients on therapy with an angiotensin‐converting enzyme inhibitor (ACEi) or angiotensin II receptor blocker (ARB) [RAAS inhibitor (RAASi) are required to have a history of hypertension.
Table 1.
Key eligibility criteria of the PARALLAX trial
| Inclusion criteria |
|---|
|
• Male or female aged ≥45 years • LVEF >40% and structural heart disease demonstrated by left atrial enlargement or left ventricular hypertrophy on echocardiography at screening or within 6 months prior to screening • Current symptom(s) of HF (NYHA class II–IV) at screening and requiring treatment with diuretics for at least 30 days prior to screening • NT‐proBNP >220 pg/mL for patients with no atrial fibrillation/flutter or >600 pg/mL for those with atrial fibrillation/flutter on electrocardiogram (ECG) at screening • Receiving evidence‐based therapy for relevant co‐morbidities with stable doses for the previous 4 weeks prior to randomization • KCCQ Clinical Summary Score <75 at screening • Patients on ACEi or ARB therapy must have a history of hypertension |
| Exclusion criteria |
|
• Any prior echocardiographic measurement of LVEF ≤40%, under stable conditions • Acute decompensated HF within 30 days prior to screening, or acute coronary syndrome (including myocardial infarction), cardiac surgery, other major CV surgery, or urgent percutaneous coronary intervention within 3 months prior to screening • Any clinical event within the 6 months prior to screening that could have reduced the LVEF, unless an echo measurement was performed after the event confirming the LVEF to be >40% • Walk distance primarily limited by non‐cardiac conditions at screening • Probable alternate diagnosis of the HF symptoms • Prior history of any dilated cardiomyopathy, including peripartum cardiomyopathy, chemotherapy induced cardiomyopathy, or viral myocarditis • Patients with HbA1c > 7.5%, not treated for diabetes • SBP <110 mmHg or ≥ 180 mmHg at screening • SBP >150 to <180 mmHg at screening unless the patient is receiving three or more antihypertensive drugs • Serum potassium >5.2 mmol/L (or equivalent plasma potassium value) at screening • eGFR <30 mL/min/1.73m2 at screening |
ACEi, angiotensin‐converting enzyme inhibitor; ARB, angiotensin II receptor blocker; CV, cardiovascular; ECG electrocardiogram; eGFR, estimated glomerular filtration rate; HbA1c, glycated haemoglobin; HF, heart failure; KCCQ, Kansas City Cardiomyopathy Questionnaire; LVEF, left ventricular ejection fraction; NT‐proBNP, N‐terminal pro BNP; NYHA, New York Heart Association; SBP, systolic blood pressure
2.2. Study design
The study design is summarized in Figure 1 . While all patients in the intervention arm of the trial receive sacubitril/valsartan, the therapy in the control arm is BMICs for cardiovascular and related co‐morbidities. This approach is chosen in order to reflect common medical practice and guideline recommendations for the treatment of co‐morbidities prevalent in HFpEF, such as hypertension, diabetes mellitus, and coronary artery disease. For a vast majority of the HFpEF patients, a RAASi‐based therapy is used. So, if patients receive a RAASi prior to inclusion into the trial, they will be stratified to enalapril (ACEi arm) if they were receiving an ACEi or to valsartan (ARB arm) if they were previously receiving angiotensin type 1 receptor blocker therapy; patients with no prior RAASi will receive placebo.
Figure 1.
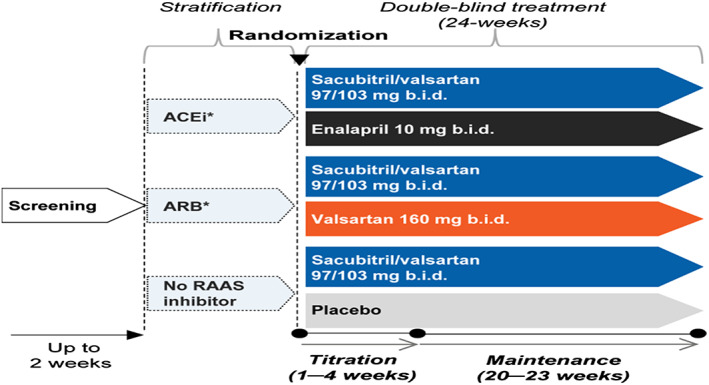
Study design of PARALLAX trial. Patients will be stratified according to therapy prior to study inclusion. Patients with ACEi therapy will be randomized to enalapril or sacubitril/valsartan; patients with ARB therapy will be randomized to valsartan or sacubitril/valsartan, and patients with neither therapy will be randomized to placebo or sacubitril/valsartan. *Patients in the ACEi and ARB strata must have a history of hypertension. ACEi, angiotensin‐converting enzyme inhibitor; ARB, angiotensin II receptor blocker; b.i.d., twice daily; RAAS, renin–angiotensin–aldosterone system; sac/val, sacubitril/valsartan.
2.3. Starting dose and uptitration
The starting dose will depend on the dose of RAASi prior to inclusion (Figure 2 ). Patients on no prior RAASi or a low‐dose RAASi will start on Dose level 1 [2.5 mg enalapril twice daily (b.i.d.), 40 mg valsartan b.i.d., or 24/26 mg sacubitril/valsartan b.i.d]. Patients on a prior high dose of RAASi (definitions of low‐dose and high‐dose RAASi are displayed in Supporting Information Table S1 ) are started on Dose level 2, which is a doubling of dose level 1. Patients should be uptitrated to target doses of 10 mg enalapril b.i.d., 160 mg valsartan b.i.d., and 97/103 mg sacubitril/valsartan b.i.d. within the first 4 weeks after randomization.
Figure 2.
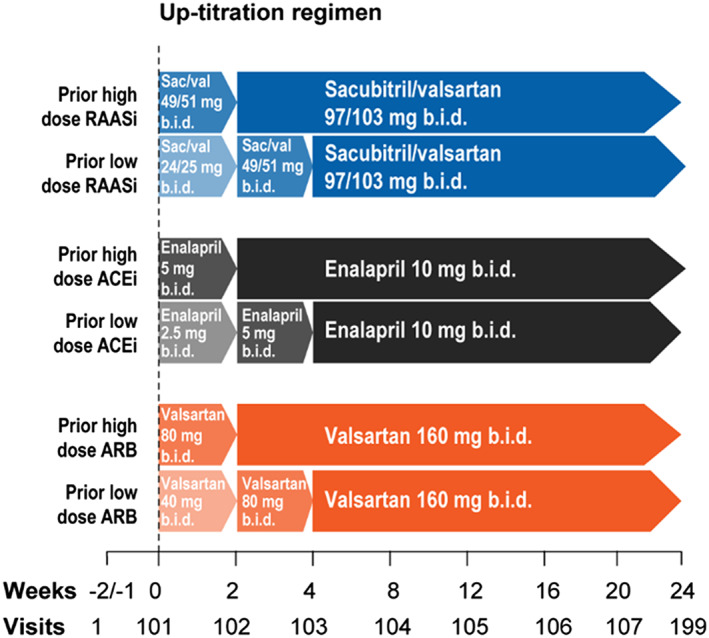
Uptitration in the PARALLAX trial. Baseline doses of study medication will depend on the dosage of medication prior to randomization. Patient with low‐dose RAAS therapy will be randomized to enalapril 2.5 mg b.i.d., valsartan 40 mg b.i.d., or sacubitril/valsartan 24/26 mg b. i. d. (Dose level 1). They will be uptitrated to Dose level 2, which is doubling of Dose level 1, after 2 weeks. Patients with high dose RAAS therapy will directly start with Dose level 2. After 2 weeks on dose level 2, patients will be uptitrated to Dose level 3, which is a doubling of Dose level 2. ACEi, angiotensin‐converting enzyme inhibitor; ARB, angiotensin II receptor blocker; b.i.d., twice daily; RAASi, renin–angiotensin–aldosterone system inhibitor.
Patients are assessed every 4 weeks during this 24 week trial and additionally every 2 weeks after randomization for uptitration of study medication.
2.3.1. Study objectives and endpoints
There are two primary objectives of this trial. The first primary objective is to demonstrate that sacubitril/valsartan is superior to BMIC therapy for cardiovascular or related co‐morbidities in reducing NT‐proBNP levels. This criterion is assessed by the change in NT‐proBNP (on the log scale) from baseline to Week 12. The second primary objective is to demonstrate that sacubitril/valsartan is superior to BMIC for co‐morbidities in improving exercise capacity by change in 6 min walk distance (6MWD) from baseline to Week 24, in a subset of patients with a baseline 6MWD ranging from 100 to 450 m. 6MWD was elevated from a secondary endpoint to a coprimary endpoint to reflect the importance of functional improvement as a therapeutic target in HFpEF in Amendment 2 (dated 12 September 2018). The selected cut‐off was chosen because <450 m indicates exercise impairment and because previous studies have used similar cut‐offs.
Secondary endpoints include comparing sacubitril/valsartan vs. BMIC on (i) the mean change from baseline in KCCQ Clinical Summary Score (CSS) at Week 24, (ii) the proportion of patients with ≥5 points deterioration in KCCQ CSS at Week 24, (iii) the proportion of patients with ≥5 points improvement in KCCQ CSS at Week 24, (iv) change from baseline in NYHA functional class at Week 24 and (v) change from baseline in a Short Form (36) Health Survey–physical component summary score at Week 24.
Safety is assessed based on adverse event (AE) reporting throughout the study.
Suspected cases of angioedema are adjudicated by a blinded and independent angioedema adjudication event committee. A complete list of study objectives including primary, secondary, and exploratory endpoints are shown in Table 2 .
Table 2.
Study objectives and endpoints of the PARALLAX trial
| Objectives | Endpoints | |
|---|---|---|
| Primary | To demonstrate that sacubitril/valsartan is superior to medication‐based individualized therapy for co‐morbidities in | |
| • Reducing NT‐proBNP from baseline after 12 weeks of treatment | • Change from baseline in NT‐proBNP at Week 12 | |
| • Improving exercise capacity after 24 weeks of treatment in a subset of patients | • Change from baseline in 6MWD at Week 24 | |
| Secondary | To compare sacubitril/valsartan to medication‐based individualized therapy for co‐morbidities in/on | |
| • Mean change of KCCQ clinical summary score (CSS) at Week 24 | • Mean change from baseline in KCCQ CSS at Week 24 | |
| • Proportion of patients with a ≥5 points change in KCCQ CSS at Week 24 (separate analyses for ≥5 points improvement and ≥5 points deterioration) | • Proportion of patients with ≥5 points improvement/deterioration in KCCQ CSS at Week 24 | |
| • Improving NYHA functional class at Week 24 | • Change from baseline in NYHA functional class at Week 24 | |
| • Improving symptoms at Week 24 | • Change from baseline in SF‐36 PCS score at Week 24 | |
| Exploratory | To compare sacubitril/valsartan to medication‐based individualized therapy for co‐morbidities in/on | |
| • Reducing NT‐proBNP at Week 24 | • Change from baseline in NT‐proBNP at Week 24 | |
| • Renal function at Week 24 | • Rate of change in eGFR from baseline | |
| • Mean change of KCCQ overall summary score (OSS) at Week 24 | • Mean change from baseline in KCCQ OSS at Week 24 | |
| • Proportion of patients with a mean change ≥5 points change in KCCQ OSS at Week 24 (separate analyses for ≥5 points improvement and ≥5 points deterioration) | • Proportion of patients with ≥5‐points improvement/deterioration in KCCQ OSS at Week 24 | |
| • To evaluate safety of sacubitril/valsartan vs. medication‐based individualized therapy for co‐morbidities | • Frequency of AEs, serious AEs, and laboratory abnormalities |
AE, adverse event; CSS, clinical summary score; KCCQ, Kansas City Cardiomyopathy Questionnaire; NT‐proBNP, N‐terminal pro BNP; NYHA, New York Heart Association; OSS, overall summary score; SF‐36 PCS, The Short Form (36) Health Survey–physical component summary; 6 MWD, 6 min walk distance
2.4. Statistical considerations
2.4.1. Sample size and primary calculation of the primary endpoints
The significance level α α of one‐sided 0.025 (two‐sided 0.05)will be split between the two primary endpoints for the treatment comparisons: 90% for NT‐proBNP and 10% for 6MWD. With a significance level α of one‐sided 0.0225 (two‐sided 0.045) , we estimated the sample size of 2500 randomized patients to provide a power of at least 92% to detect a relative reduction of a minimum 11% in NT‐proBNP change from baseline to Week 12, assuming a standard deviation of 0.81 in log‐transformed NT‐proBNP derived from PARAMOUNT‐HF data for patients with baseline KCCQ CSS <75 and an overall dropout rate of 10%. With a significance level α of one‐sided 0.0025 (two‐sided 0.005), we estimated to a power of at least 90% to detect a mean difference of 22 m in 6MWD change from baseline to Week 24, assuming a standard deviation of 120 m, 12 an overall dropout rate of 10%, and an overall proportion of 88% for patients baseline 6MWD ranging between 100 and 450 m.
2.4.2. Testing strategy
In order to control the overall false positive rate in the multiple treatment comparisons for efficacy, a sequential rejective multiple testing procedure will be employed. 13 The testing strategy is illustrated in Figure 3 . At first, between the two primary endpoints, the overall alpha level will be split in a 9:1 ratio . When either of them is rejected, the corresponding assigned alpha will be propagated and accumulated to test the KCCQ CSS. In case the 6MWD is not rejected at first step but the KCCQ CSS is rejected at the alpha level inherited from the rejection of the NT‐proBNP change, the corresponding alpha level will be propagated to the 6MWD, together with the original assigned alpha, to test the 6MWD again. If both the KCCQ CSS and 6MWD are rejected, then the NYHA class will be tested at the full level of alpha. Otherwise, the testing procedure will be stopped.
Figure 3.
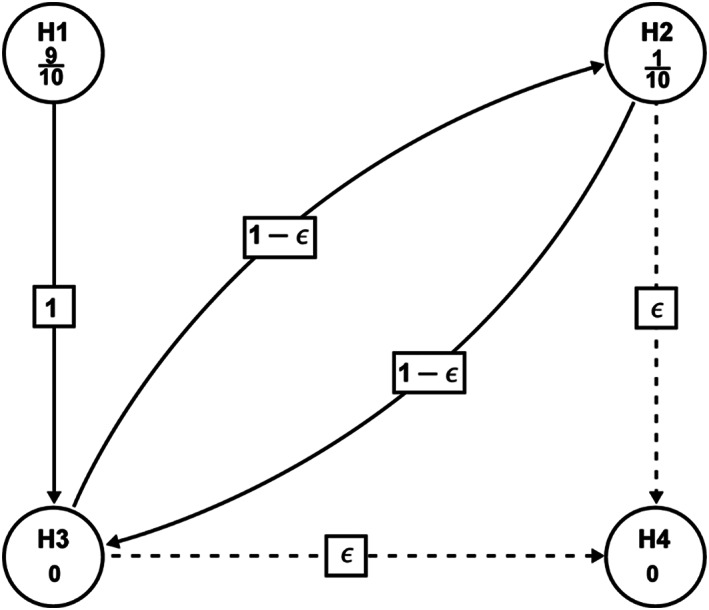
Graphical illustration of the sequentially rejective testing procedure. H1: Sacubitril/valsartan is no better than the comparator in change from baseline in log‐transformed NT‐proBNP at Week 12 (Primary). H2: Sacubitril/Valsartan is no better than the comparator in change from baseline in 6 min walking distance (6MWD) at Week 24. (Primary). H3: Sacubitril/valsartan is no better than the comparator in change from baseline in Kansas City Cardiomyopathy Questionnaire (KCCQ) Clinical Summary Score (CSS) (mean score) at Week 24. H4: Sacubitril/valsartan is no better than the comparator in NYHA class change from baseline at Week 24. In order to control the family‐wise type‐I error rate at the one‐sided 0.025 significance level, a sequentially rejective multiple testing procedure will be employed, whereby H1 and H2 will be tested first at initially assigned level of one‐sided (9/10) × α = 0.0225 and one‐sided (1/10) × α = 0.0025, accordingly. If H1 and/or H2 are rejected, the alpha for the rejected null hypotheses will be propagated to H3, such that, H3 will be tested at the updated alpha level (one‐sided 0.025 if both H1 and H2 are rejected; one‐sided 0.0225 if H1 is rejected but H2 is not rejected; one‐sided 0.0025 if H2 is rejected but H1 is not rejected); if H3 is rejected, the alpha will be propagated to H2 or H4 based on the initial step rejection status
2.4.3. Analyses of the primary endpoints
The primary endpoint of NT‐proBNP will be analysed using a mixed model for repeated measures (MMRM), with the response variable defined as change from baseline in log (NT‐proBNP); stratum (defined by prior ACEi/ARB medication usage), region, study treatment, visits, and treatment‐by‐visit interaction will be included as fixed‐effect factors; baseline log (NT‐proBNP), stratum‐by‐baseline and visit‐by‐baseline interactions will be included as covariates; the within‐patient covariance will be modelled using an unstructured covariance matrix. The analysis will include all post‐baseline scheduled visits up to Week 12 (Week 4, Week 12) and will be performed an assumption of missing at random (MAR) for missing data. 14 , 15 Based on the MMRM model, the estimates and the two‐sided 95% confidence intervals will be provided for the adjusted geometric means for the ratio to baseline in NT‐proBNP at Week 12 in each of the two treatment groups (LCZ696 and individualized medical therapy) and for the ratio of the adjusted geometric means (LCZ696 over individualized medical therapy).
The primary endpoint of 6MWD will be analysed using a similar MMRM modelling approach with the response variable being changes from baseline in 6MWD with the same fixed‐effect factors and baseline 6 min walk distance (B6MWD), baseline systolic blood pressure (BSBP), stratum‐by‐B6MWD, stratum‐by‐baseline systolic blood pressure, and visit‐by‐B6MWD interactions as covariates. The analysis will include all patients with B6MWD ranging from 100 to 450 m, and all post‐baseline scheduled visits up to Week 24 (Week 16 and Week 24), and will be performed based on an assumption of MAR for missing data. To further assess the impact of missing data that are not MAR, additional pattern‐mixture modelling analyses will also be performed.
The secondary endpoints of KCCQ CSS change from baseline at week 24 will be analysed using a similar MMRM model; NYHA class change (improved, unchanged, and worsened) from baseline at Week 24 will be analysed using a longitudinal proportional cumulative odds model with similar fixed‐effect factors and covariates defined above.
3. Discussion
The PARALLAX trial is designed to investigate the effect of sacubitril/valsartan in patients with EF >40% compared with BMIC on NT‐proBNP and 6MWD. PARALLAX aims to address three unexplored areas in the evaluation of sacubitril/valsartan in patients with EF >40% (i) compare ARNI vs. BMIC on NT‐proBNP levels, (ii) assess benefits in functional capacity with ARNI therapy, and (iii) further evaluate improvements in symptoms and quality of life with ARNI.
HFpEF has not been studied as extensively as HFrEF likely because of greater difficulty in diagnosis and more heterogenous patient presentation. To date, no therapy has definitively shown a reduction in morbidity and mortality in HFpEF, and there is medical need to improve physical functioning, exercise capacity, and quality of life in these patients. 2 With the inclusion and exclusion criteria of the PARALLAX trial, a typical HFpEF population with high symptomatic burden is selected. All patients have a KCCQ CSS < 75 reflecting both functional and symptomatic burden. An EF >40% will cover the full spectrum of HFmrEF and HFpEF patients according to current guideline definitions.
PARALLAX will enrol patients irrespective of prior RAASi and dosage. In contrast to PARAGON‐HF and PARADIGM‐HF, there is no run‐in period, possibly rendering data closer to real world exposure and tolerability. The lack of a run‐in period in PARALLAX may also enhance potential effects of sacubitril/valsartan on KCCQ assessment because the randomized treatment will be compared with RAASi and not to run‐in sacubitril/valsartan, as in PARAGON‐HF. There is no established pharmacological treatment for HFpEF, and RAASi are recommended for treatment of co‐morbidities in HFmrEF and HFpEF. 2 Therefore, in HFmrEF and HFpEF, it is appropriate to test individualized background treatment against the intervention, which is reflected in the PARALLAX trial design by using different comparators based on the RAASi prior to study inclusion.
NT‐proBNP was chosen as a primary endpoint in PARALLAX because a reduction in NT‐proBNP has been shown to be a surrogate of structural cardiac changes: In the PARAMOUNT study with 301 HFpEF patients comparing sacubitril/valsartan with valsartan alone, NT‐proBNP was significantly reduced by sacubitril/valsartan after 12 weeks, and this was associated with a reduction in left atrial size. 10 While NT‐proBNP reductions have been associated with a morbidity/mortality benefit in HFrEF, this association is less clear in HFmrEF and HFpEF. 16 In addition to morbidity and mortality, limitation in exercise capacity is a hallmark of HF, and an improvement in exercise capacity (as measured by 6MWD) is also chosen as a primary endpoint. PARALLAX, by trial design, is not powered to assess changes in cardiovascular death or HF hospitalizations. However, tolerability and safety will be monitored throughout the trial.
Table 3 and Supporting Information Table S2 respectively show prior and ongoing randomized multicenter trials in HFpEF with functional capacity such as 6MWD, and VO2 as endpoints. Similar to the morbidity and mortality trials, no trial to date has been able to demonstrate a relevant improvement in exercise capacity in patients with HFpEF. However, these trials were small (not more than 400 patients) and not powered for changes in exercise capacity. PARALLAX has adequate power to show a mean difference of 22 m in 6MWD and is sufficiently powered to detect a minimal clinically important difference that was previously reported to be between 30 m17 and 35 m18.
Table 3.
Comparison of different multicentre HFpEF pharmacological trials with functional capacity endpoints
| Name of trial | N | Investigational drug | Comparator | Duration of follow‐up | 6 min walking distance (m) | Peak VO2 (mL/min/kg) |
|---|---|---|---|---|---|---|
| Aldo‐DHF 19 | 422 | Spironolactone 25 mg/d | Placebo | 12 months | −15 (P = 0.02) | −0.1 (P = 0.81) |
| NEAT‐HFpEF 20 | 110 | Isosorbidmono‐nitrate 120 mg/d | Placebo | 6 weeks | +0.1 (P = 0.91) | NA |
| Anemia in HFpEF 21 | 56/10 | Epoetin alpha | Placebo | 6 months | +11 (P = 0.52) | +2.2 (P < 0.03) |
| RELAX 22 | 216 | Sildenafil 60 mg/d | Placebo | 12 weeks | −10 (P = 0.92) | +0.01 (P = 0.98) |
| EDIFY 23 | 179 | Ivabradine 15 mg/d | Placebo | 8 months | −3.8 (P = 0.88) | NA |
| INDIE‐HFpEF 24 | 105 | Nitrate 138 mg/d | Placebo | 6 weeks | NA | −0.20 (P = 0.27) |
| PANACHE 25 | 305 | Neladenoson 5–30 mg/d | Placebo | 20 weeks | 14–29 (P = n.s.) | NA |
d, day; NA, not available; n.s., non‐significant
Patient‐related outcomes (e.g. KCCQ CSS and KCCQ Overall Summary Score) 26 are considered increasingly important in HF and are included as secondary endpoints. The KCCQ instrument includes a CSS that encompasses the physical limitation score and the total symptom score. KCCQ has been shown to be a valid and reliable tool to measure health status and predict outcomes in patients with HFpEF. 27 Data from HF‐ACTION suggest that a 5 point change in the KCCQ overall score corresponds to clinically significant changes in measures of exercise capacity (peak oxygen consumption and 6MWT). 28 The PARALLAX trial will evaluate not only mean change from baseline but also proportion of patients with ≥5 point change including both improvement and deterioration at the end of the study. The requirement of KCCQ CSS <75 as an inclusion criterion will select HFpEF patients with notable functional impairment. 11 SF‐36 physical component summary and NYHA functional class are investigated as parameters of physical and functional changes.
The PARAGON‐HF trial in a HFpEF population with LVEF >45% did not reach a statistically significant reduction of recurrent HF hospitalizations and cardiovascular mortality with sacubitril/valsartan compared with valsartan in the overall patient population nor a significant improvement in the KCCQ CS at 8 months vs. valsartan; however, efficacy in predefined subgroups and improvements in NYHA class were significant in this trial and warrant further evaluation of sacubitril/valsartan in a HF population with limited treatment options to date.
In summary, PARALLAX will investigate sacubitril/valsartan in comparison with BMICs for co‐morbidities on exercise capacity and symptom benefit in a large population of patients with an EF >40% and improve our understanding of the effects of sacubitril/valsartan on NT‐proBNP, exercise capacity, symptoms, and quality of life.
Conflict of interest
The PARALLAX trial is sponsored by Novartis. R.W. received grants from the Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung, BMBF), Boehringer Ingelheim, German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) and the European Union; personal fees and/or investigator fees from Bayer, Berlin Chemie, Boehringer Ingelheim, Medtronic, Novartis, Servier, Bristol‐Myers Squibb, Pfizer, Sanofi and CVRx, Boston Scientific, Gilead, Johnson & Johnson.
S.J.S. is supported by grants from the National Institutes of Health (NIH; R01 HL107577, R01 HL127028, R01 HL140731, and R01 HL149423); the American Heart Association (AHA; #16SFRN28780016 and #15CVGPSD27260148); and Actelion, AstraZeneca, Corvia, and Novartis and has served as a consultant/advisory board/steering committee member for Abbott, Actelion, AstraZeneca, Amgen, Bayer, Boehringer‐Ingelheim, Cardiora, Coridea, CVRx, Eisai, Ionis, Ironwood, Merck, MyoKardia, Novartis, Pfizer, Sanofi, Tenax, and United Therapeutics.
B.P. is the Principal Investigator of PARALLAX and received personal and institutional honoraria from Novartis for steering committee, advisory board, and speaker activities. BP also received steering committee and/or speaker fees from Bayer Healthcare, Merck, Daiichi‐Sankyo, Servier, BMS, and AstraZeneca.
MRC received grants from Medtronic, Boston Scientific, Abbott, Bayer and ResMed during the conduct of the study and has served as a consultant/advisor/steering committee member for Novartis, Servier, Bayer, Boston Scientific, Abbott, ResMed, AstraZeneca, NovoNordisk, Neurotronik, and FIRE1 Foundry.
Supporting information
Table S1. Low‐dose and high‐dose strata of ACE inhibitors and ARB
Table S2. Other randomized multicenter trials in HFpEF with functional capacity endpoints
Wachter, R. , Shah, S. J. , Cowie, M. R. , Szecsödy, P. , Shi, V. , Ibram, G. , Zhao, Z. , Gong, J. , Klebs, S. , and Pieske, B. (2020) Angiotensin receptor neprilysin inhibition versus individualized RAAS blockade: design and rationale of the PARALLAX trial. ESC Heart Failure, 7: 856–864. 10.1002/ehf2.12694.
Contributor Information
Rolf Wachter, Email: rolf.wachter@medizin.uni-leipzig.de.
Burkert Pieske, Email: burkert.pieske@charite.de.
References
- 1. Christ M, Störk S, Dörr M, Heppner HJ, Müller C, Wachter R, Riemer U, for the Trend HF Germany Project . Heart failure epidemiology 2000‐2013: insights from the German Federal Health Monitoring System. Eur J Heart Fail 2016; 18: 1009–1018. [DOI] [PubMed] [Google Scholar]
- 2. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, Falk V, González‐Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GMC, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P, ESC Scientific Document Group . 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Eur Heart J 2016; 37: 2129–2200. [DOI] [PubMed] [Google Scholar]
- 3. Borlaug BA, Paulus WJ. Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J 2011; 32: 670–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. The SOLVD investigators . Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med 1991; 325: 293–302. [DOI] [PubMed] [Google Scholar]
- 5. No authors listed . Effects of metoprolol CR/XL in chronic heart faliure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT‐HF). Lancet 1999; 353: 2001–2007. [PubMed] [Google Scholar]
- 6. Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe hear faliure. N Engl J Med 1999; 341: 709–717. [DOI] [PubMed] [Google Scholar]
- 7. McMurray JJV, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile MR, PARADIGM‐HF Investigators and Committees . Angiotensin‐neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014; 371: 993–1004. [DOI] [PubMed] [Google Scholar]
- 8. McMurray JJV, Solomon SD, Inzucchi SE, Køber L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Anand IS, Bělohlávek J, Böhm M, Chiang CE, Chopra VK, de Boer RA, Desai AS, Diez M, Drozdz J, Dukát A, Ge J, Howlett JG, Katova T, Kitakaze M, Ljungman CEA, Merkely B, Nicolau JC, O'Meara E, Petrie MC, Vinh PN, Schou M, Tereshchenko S, Verma S, Held C, DeMets D, Docherty KF, Jhund PS, Bengtsson O, Sjöstrand M, Langkilde AM, DAPA‐HF Trial Committees and Investigators . Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med 2019; 381: 1995–2008. [DOI] [PubMed] [Google Scholar]
- 9. Maeder MT, Kaye DM. Heart failure with normal left ventricular ejection fraction. J Am Coll Cardiol 2009; 53: 905–918. [DOI] [PubMed] [Google Scholar]
- 10. Solomon SD, Zile M, Pieske B, Voors A, Shah A, Kraigher‐Krainer E, Shi V, Bransford T, Takeuchi M, Gong J, Lefkowitz M, Packer M, McMurray J, Prospective comparison of ARNI with ARB on Management Of heart failUre with preserved ejectioN fracTion (PARAMOUNT) Investigators . The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: a phase 2 double‐blind randomised controlled trial. Prospective comparison of ARNI with ARB on Management Of heart failUre with preserved ejectioN fracTion (PARAMOUNT) Investigators. Lancet 2012; 380: 1387–1395. [DOI] [PubMed] [Google Scholar]
- 11. Solomon SD, McMurray JJV, Anand IS, Ge J, Lam CSP, Maggioni AP, Martinez F, Packer M, Pfeffer MA, Pieske B, Redfield MM, Rouleau JL, van Veldhuisen D, Zannad F, Zile MR, Desai AS, Claggett B, Jhund PS, Boytsov SA, Comin‐Colet J, Cleland J, Düngen HD, Goncalvesova E, Katova T, Kerr Saraiva JF, Lelonek M, Merkely B, Senni M, Shah SJ, Zhou J, Rizkala AR, Gong J, Shi VC, Lefkowitz MP, PARAGON‐HF Investigators and Committees . Angiotensin‐neprilysin inhibition in heart failure with preserved ejection fraction. N Engl J Med 2019; 381: 1609–1620. [DOI] [PubMed] [Google Scholar]
- 12. Ingle L, Shelton RJ, Rigby AS, Nabb S, Clark AL, Cleland JGF. The reproducibility and sensitivity of the 6‐min walk test in elderly patients with chronic heart failure. Eur Heart J 2005; 26: 1742–1751. [DOI] [PubMed] [Google Scholar]
- 13. Bretz F, Maurer W, Brannath W, Posch M. A graphical approach to sequentially rejective multiple test procedures. Stat Med 2009; 28: 586–604. [DOI] [PubMed] [Google Scholar]
- 14. Siddiqui O, Hung HM, O'Neill R. MMRM vs. LOCF: a comprehensive comparison based on simulation study and 25 NDA datasets. J Biopharm Stat 2009; 19: 227–246. [DOI] [PubMed] [Google Scholar]
- 15. National Research Council . The Prevention and Treatment of Missing Data in Clinical Trials. Panel on Handling Missing Data in Clinical Trials. Committee on National Statistics, Division of Behavioral and Social Sciences and Education. Washington, DC: The National Academies Press; 2010. [Google Scholar]
- 16. Savarese G, Hage C, Orsini N, Dahlström U, Perrone‐Filardi P, Rosano GM, Lund LH. Reductions in N‐terminal pro‐brain natriuretic peptide levels are associated with lower mortality and heart failure hospitalization rates in patients with heart failure with mid‐range and preserved ejection fraction. Circ Heart Fail 2016; 9: e003105. [DOI] [PubMed] [Google Scholar]
- 17. Shoemaker MJ, Curtis AB, Vangsnes E, Dickinson MG. Clinically meaningful change estimates for the six‐minute walk test and daily activity in individuals with chronic heart failure. Cardiopulm Phys Ther J 2013; 24: 21–29. [PMC free article] [PubMed] [Google Scholar]
- 18. Täger T, Hanholz W, Cebola R, Fröhlich H, Franke J, Doesch A, Katus HA, Wians FH Jr, Frankenstein L. Minimal important difference for 6‐minute walk test distances among patients with chronic heart failure. Int J Cardiol 2014; 176: 94–98. [DOI] [PubMed] [Google Scholar]
- 19. Edelmann F, Wachter R, Schmidt AG, Kraigher‐Krainer E, Colantonio C, Kamke W, Duvinage A, Stahrenberg R, Durstewitz K, Löffler M, Düngen HD, Tschöpe C, Herrmann‐Lingen C, Halle M, Hasenfuss G, Gelbrich G, Pieske B, Aldo‐DHF Investigators . Effect of spironolactone on diastolic function and exercise capacity in patients with heart failure with preserved ejection fraction: the Aldo‐DHF randomized controlled trial. JAMA 2013; 309: 781–791. [DOI] [PubMed] [Google Scholar]
- 20. Redfield MM, Anstrom KJ, Levine JA, Koepp GA, Borlaug BA, Chen HH, LeWinter M, Joseph SM, Shah SJ, Semigran MJ, Felker GM, Cole RT, Reeves GR, Tedford RJ, Tang WH, McNulty S, Velazquez EJ, Shah MR, Braunwald E, NHLBI Heart Failure Clinical Research Network . Isosorbide mononitrate in heart failure with preserved ejection fraction. N Engl J Med 2015; 373: 2314–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maurer MS, Teruya S, Chakraborty B, Helmke S, Mancini D. Treating anemia in older adults with heart failure with a preserved ejection fraction with epoetin alfa: single‐blind randomized clinical trial of safety and efficacy. Circ Heart Fail 2013; 6: 254–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, LeWinter M, Rouleau JL, Bull DA, Mann DL, Deswal A, Stevenson LW, Givertz MM, Ofili EO, O'Connor CM, Felker GM, Goldsmith SR, Bart BA, McNulty S, Ibarra JC, Lin G, Oh JK, Patel MR, Kim RJ, Tracy RP, Velazquez EJ, Anstrom KJ, Hernandez AF, Mascette AM, Braunwald E, RELAX Trial . Effect of phosphodiesterase‐5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 2013; 309: 1268–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Komajda M, Isnard R, Cohen‐Solal A, Metra M, Pieske B, Ponikowski P, Voors AA, Dominjon F, Henon‐Goburdhun C, Pannaux M, Böhm M, on behalf of the prEserveD left ventricular ejectIon fraction chronic heart Failure with ivabradine studY (EDIFY) Investigators. Effect of ivabradine in patients with heart failure with preserved ejection fraction: the EDIFY randomized placebo‐controlled trial. Eur J Heart Fail 2017; 19: 1495–1503. [DOI] [PubMed] [Google Scholar]
- 24. Borlaug BA, Anstrom KJ, Lewis GD, Shah SJ, Levine JA, Koepp GA, Givertz MM, Felker GM, LeWinter MM, Mann DL, Margulies KB, Smith AL, Tang WHW, Whellan DJ, Chen HH, Davila‐Roman VG, McNulty S, Desvigne‐Nickens P, Hernandez AF, Braunwald E, Redfield MM, for the National Heart, Lung, and Blood Institute Heart Failure Clinical Research Network . Effect of Inorganic Nitrite vs Placebo on Exercise Capacity Among Patients With Heart Failure With Preserved Ejection Fraction: the INDIE‐HFpEF randomized clinical trial. JAMA 2018. Nov; 320: 1764–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shah SJ, Voors AA, McMurray JJV, Kitzman DW, Viethen T, Bomfim Wirtz A, Huang E, Pap AF, Solomon SD. Effect of Neladenoson Bialanate on Exercise Capacity Among Patients With Heart Failure With Preserved Ejection Fraction: a randomized clinical trial. JAMA 2019; 321: 2101–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kelkar AA, Spertus J, Pang P, Pierson RF, Cody RJ, Pina IL, Hernandez A, Butler J. Utility of patient‐reported outcome instruments in heart failure. JACC Heart Fail 2016; 4: 165–175. [DOI] [PubMed] [Google Scholar]
- 27. Joseph SM, Novak E, Arnold SV, Jones PG, Khattak H, Platts AE, Dávila‐Román VG, Mann DL, Spertus JA. Comparable performance of the Kansas City Cardiomyopathy Questionnaire in patients with heart failure with preserved and reduced ejection fraction. Circ Heart Fail 2013; 6: 1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Flynn KE, Lin L, Moe GW, Howlett JG, Fine LJ, Spertus JA, McConnell TR, Piña IL, Weinfurt KP. Relationships between changes in patient‐reported health status and functional capacity in outpatients with heart failure. Am Heart J 2012; 163: 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Low‐dose and high‐dose strata of ACE inhibitors and ARB
Table S2. Other randomized multicenter trials in HFpEF with functional capacity endpoints