

Abstract
Desmin‐related myopathy (DRM) is a rare heritable cardiac and skeletal muscle disease caused by mutations in the desmin gene (DES). DRM is generally characterized by skeletal muscle weakness, conduction disturbance, and dilated cardiomyopathy. However, the clinical cardiac phenotypes of DRM are not yet fully understood. Herein, we report the first case of DRM with the de novo missense DES mutation, R454W, that is characterized by left ventricular non‐compaction cardiomyopathy, progressive cardiac conduction defect, spontaneous coronary artery dissection, and no skeletal muscle weakness. Our case findings suggest that clinicians should genetically test patients who have cardiomyopathy, progressive cardiac conduction defect, and coronary artery dissection, even if the patient has neither family history of DRM nor skeletal muscle symptoms.
Keywords: Cardiomyopathy, coronary artery dissection, heart failure
1. Introduction
Desmin is the main component of intermediate filament protein expressed in skeletal, cardiac, and smooth muscle.1 Desmin‐related myopathy (DRM) is a rare heritable cardiac and skeletal muscle disease caused by mutations in the desmin gene (DES).2 DRM is typically characterized by skeletal muscle weakness, conduction disturbance, and dilated cardiomyopathy (DCM)1, 2; however, the clinical cardiac phenotypes of DRM are not yet fully understood. Herein, we present an individual case of left ventricular non‐compaction cardiomyopathy (LVNC), progressive cardiac conduction defect, and spontaneous coronary artery dissection (SCAD) associated with a de novo missense mutation of the DES, R454W.
2. Case report
A previously healthy Japanese 6‐year‐old female was referred to our hospital for left‐axis deviation of electrocardiogram by school heart screening. During follow‐up, she developed symptomatic complete atrioventricular block, and she underwent implantation of permanent dual‐chamber pacemaker when she was 11 years old (Figure 1 ). Atrial flutter (common type) occurred 3 years later, and so catheter ablation was performed. The coronary angiography showed normal coronary arteries, and the left ventriculogram showed normal left ventricular wall motion.
Figure 1.

Electrocardiogram changes. Electrocardiogram at 11 years old demonstrates atrioventricular block.
At 17 years old, the patient experienced several months of progressive dyspnoea. Transthoracic echocardiography revealed a dilated left ventricle (LV) and reduced LV wall motion with an LV ejection fraction of 36%. Colour Doppler showed blood flowing within the deep intertrabecular recesses of the prominent trabecular meshwork of the LV apex. Figure 2 shows the increased non‐compacted (NC) endomyocardial layer depth compared with the compacted epicardial (C) layer (NC/C ratio at end‐systole >2.0) at the apex. Based on these findings and previously reported diagnostic criteria,3 the patient was diagnosed with LVNC and treated with carvedilol 1.25 mg/day, enalapril 2.5 mg/day, furosemide 20 mg/day, and spironolactone 25 mg/day.
Figure 2.

Transthoracic echocardiogram shows prominent trabeculation with deep intertrabecular recesses on the lateral and anterior apical walls. (A) The image shows the end‐diastolic apical short‐axis view. (B) The image shows the end‐systolic apical short‐axis view. (C) Colour Doppler image shows blood flow in the recess between trabeculae. C, compacted epicardial layer; NC, non‐compacted endomyocardial layer.
At 20 years old, the patient was admitted to our hospital with chest pain. She was haemodynamically stable with no abnormal findings upon physical examination. The electrocardiogram showed no significant changes. Blood tests revealed elevated creatine kinase levels (987 U/L) and troponin I levels (38.7 ng/mL, normal range 0.0–0.1 ng/mL). Because the patient's symptoms persisted, she underwent coronary angiography, which revealed 99% stenosis with defective contrast filling in the middle left anterior descending artery (LAD). Intravascular ultrasound revealed a continuity disruption in the internal elastic lamia around the lesion of middle LAD with thrombus and localized dissection with false lumen and intramural hematoma in the left main trunk (LMT) (Figure 3 ). Thus, we diagnosed the patient with SCAD.
Figure 3.

(A) Coronary angiogram shows the lesion in the middle of the left anterior descending coronary artery (white arrow). (B) Intravascular ultrasound image shows the dissection with false lumen in the left main trunk with a haematoma (yellow arrows). (C) Intravascular ultrasound image shows the intramural thrombus and haematoma in the middle of the left anterior descending coronary artery (yellow arrows). FL, false lumen; TL, true lumen.
After the intravascular ultrasound imaging was performed, the patient's coronary blood flow rapidly decreased from Grade 2 to Grade 0 on the Thrombolysis in Myocardial Infarction Trial scale; the patient went into cardiac shock. We inserted an intra‐aortic balloon pump and proceeded with percutaneous coronary intervention to LAD and LMT. After cautious wiring and confirmation of the guidewire placement in the true lumen with selective coronary contrast injection, we deployed a 4.0 × 16 mm bare metal stent (Liberte, Boston Scientific) in LMT and a 2.25 × 16 mm drug eluting stent (SYNERGY, Boston Scientific) in mid LAD. Then, the coronary blood flow improved to Thrombolysis in Myocardial Infarction Trial 3. The patient was discharged after 17 days of hospital stay. Three months later, the patient was readmitted with exacerbation of heart failure, and cardiac resynchronization therapy was implemented.
Approximately 1 year later, the 22‐year‐old patient went to the emergency room with continuous chest pain. Although no marked changes were identified from the results of either the ECG or the chest radiogram, creatine kinase levels (537 U/L) and troponin I levels (15.67 ng/mL) were elevated. We performed coronary CT that revealed two stenotic lesions in the distal LAD and the distal right coronary artery (Figure 4 ). There were no stent abnormalities. Based on the patient's medical history and multiple new lesions, the diagnosis was a recurrence of SCAD. We treated conservatively because both lesions were found in the distal coronary arteries.
Figure 4.

(A, C) Coronary CT angiogram shows a new stenotic lesion in the distal right coronary artery (arrows). (B, D) Coronary CT angiogram shows a new stenotic lesion in the distal left anterior descending artery (LAD) (arrows). RCA, right coronary artery.
The patient's heart failure worsened with gradual damage to both liver and kidney functions. While undergoing treatment with dobutamine and milrinone, her heart failure deteriorated to the New York Heart Association's classification of a functional class IV, the LV ejection fraction was only 13%, and maximum oxygen uptake measured 6.7 mL/min/kg with decreased exercise tolerance. The right‐cardiac catheter test results determined that the cardiac index was 1.6 L/min/m2 and the pulmonary capillary wedge pressure was 37.6 mmHg. Therefore, the patient underwent the implantation of an LV assist device (EVAHEART 2: SUN MEDICAL, Inc, Nagano, Japan) using a double tipless cannula, as a bridge to heart transplantation. When we inserted the cannula into the LV, we obtained myocardium that included both compaction and non‐compaction areas from the LV apical wall; the biopsy was submitted to pathology for pathological examination. Pathological findings showed signs of fibrosis located primarily in the sub‐endocardium of the LV, but no evidence of secondary cardiomyopathy was observed (Figure 5 ). After surgery, the patient was placed on the heart transplant waiting list and discharged from the hospital. Currently, 1 year after the implantation of the LV assist device, the patient regularly visits the outpatient clinic while waiting for her heart transplantation.
Figure 5.
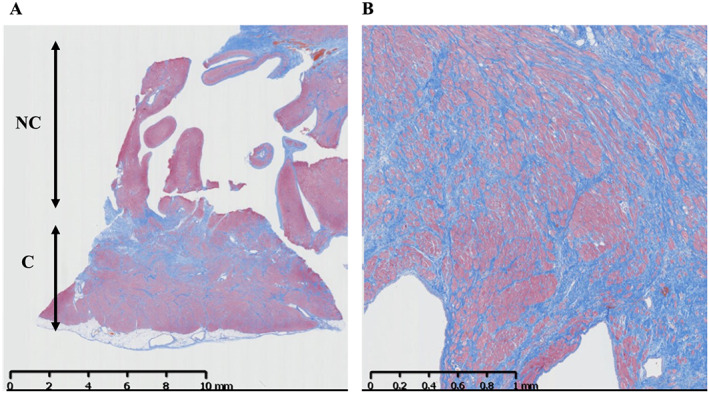
The sample of myocardium includes both compaction and non‐compaction areas, which was obtained from the left ventricle apical wall. Masson trichrome histological staining of the left ventricle shows fibrosis replacement of myocardial tissue with no evidence of secondary cardiomyopathy. (A) 100× magnification. (B) 400× magnification. C, compacted epicardial layer; NC, non‐compacted endomyocardial layer.
In addition, we performed targeted exon sequencing using methods, which we previously reported, for a custom HaloPlex™‐designed capture panel of 90 genes linked to either cardiac arrhythmias or sudden cardiac death.4 After mapping, variant calling, filtering out rare variants, and validating with the Sanger sequencing method, we found a heterozygous missense mutation (c.C1360T, p.R454W) in the proband but not in the her parents, which indicates a de novo mutation (Figure 6 ). The in silico prediction tools, SIFT and PolyPhen‐2, suggest that this mutation leads to a deleterious protein function. These data strongly suggest that DES‐R454W is the pathogenic mutation that causes progressive cardiac conduction defect with LVNC. To evaluate the skeletal muscle symptoms, a detailed physical examination by expert neurologists confirmed that there were no abnormalities in the skeletal muscular strength of the patient. Creatine kinase levels were also within the normal range (70–100 U/L).
Figure 6.

(A) Sequencing electropherogram shows the heterozygous DES mutation R454W in the proband. Arrow and bar indicate the missense mutation; R (CGG) to W (TGG). (B) Diagram shows the family tree where both the parents and brother of the proband do not carry the DES gene mutation. WT, wild type.
3. Discussion
We identified three key clinical characteristics in this case of DRM: the clinical phenotype of DRM may be associated with LVNC, SCAD may aggravate heart failure in DRM patients, and DRM may develop in patients that have neither skeletal muscle symptoms nor family history of DRM.
DES is expressed in cardiac, skeletal, and smooth muscle cells. Even though most patients with DES mutations present a combined skeletal and cardiac myopathy,1, 2 the clinical phenotypes associated with DES mutations are heterogeneous. Among the previously reported DES mutations, R454W, which was found in the case, exhibited a severer clinical course than other DES mutations.5, 6, 7 It is reported that this type of mutation was associated with an accelerated and severe cardiac phenotype with a high incidence of ventricular arrhythmia, progressive cardiomyopathy, and sudden death.5, 6, 7 Therefore, R454W mutation is a ‘red flag’ for clinicians.7 However, the clinical phenotypes of DRM, especially those associated with R454W, are not fully understood.8 Our findings in this case suggest that LVNC may be associated with clinical phenotype of DRM. Currently, more than 50 DES mutations have been identified; most are associated with DCM. Only three DES mutations (L69P, R212Q, and A360S) were previously known to be associated with LVNC.1 The association between DES‐R454W mutation and LVNC has not been reported.
In this case, SCAD aggravated heart failure. SCAD was recently identified as an important cause of acute coronary syndrome and sudden cardiac death.9, 10 Our patient did not have any of the known SCAD‐associated risk factors.11 Although there are no previous case reports that link SCAD and DES mutations, in vitro studies suggest that there is a relationship between DES mutations and the disruption of the cardiac smooth muscle layers.12 SCAD is characterized by a deep coronary arterial wall dissection that usually occurs in the area that comprises vascular smooth muscle and elastic fibres13; therefore, the recurring SCAD complications in this DRM patient may not be just coincidental. This case suggests that coronary arterial dissections may lead to sudden heart failure in patients of DRM.
Typically, DRM causes skeletal muscle weakness, and most DRM patients have a family history of DRM.1, 14 However, our patient has neither family history nor skeletal muscle weakness. Our case suggests that genetic testing for potential DES gene mutations should not be excluded from patient assessments, even if the patient has neither skeletal muscle weakness symptoms nor DRM family history; this is an especially important method for identifying the DES‐R454W mutation because it is known as a ‘red flag’ mutation. Therefore, an early genetic workup is essential in patients that have the triple combination of cardiomyopathy, cardiac conduction defect, and coronary artery disease.
To the best of our knowledge, we report the first de novo case of a DRM patient with no skeletal muscle weakness that also has LVNC, a progressive cardiac conduction defect, and SCAD. Therefore, clinicians should genetically test patients that have the triple combination of cardiomyopathy, cardiac conduction defect, and coronary artery dissection, even if the patient has neither family history of DRM nor skeletal muscle weakness symptoms.
Conflict of interest
None declared.
Tamiya, R. , Saito, Y. , Fukamachi, D. , Nagashima, K. , Aizawa, Y. , Ohkubo, K. , Hatta, T. , Sezai, A. , Tanaka, M. , Ishikawa, T. , Makita, N. , Sumitomo, N. , and Okumura, Y. (2020) Desmin‐related myopathy characterized by non‐compaction cardiomyopathy, cardiac conduction defect, and coronary artery dissection. ESC Heart Failure, 7: 1338–1343. 10.1002/ehf2.12667.
References
- 1. Brodehl A, Gaertner‐Rommel A, Milting H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys Rev 2018; 10: 983–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Taylor MR, Slavov D, Ku L, Di Lenarda A, Sinagra G, Carniel E, Haubold K, Boucek MM, Ferguson D, Graw SL, Zhu X, Cavanaugh J, Sucharov CC, Long CS, Bristow MR, Lavori P, Mestroni L. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation 2007; 115: 1244–1251. [DOI] [PubMed] [Google Scholar]
- 3. Jenni R, Oechslin E, Schneider J, Attenhofer Jost C, Kaufmann PA. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non‐compaction: a step towards classification as a distinct cardiomyopathy. Heart 2001; 86: 666–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Daumy X, Amarouch MY, Lindenbaum P, Bonnaud S, Charpentier E, Bianchi B, Nafzger S, Baron E, Fouchard S, Thollet A, Kyndt F, Barc J, Le Scouarnec S, Makita N, Le Marec H, Dina C, Gourraud JB, Probst V, Abriel H, Redon R, Schott JJ. Targeted resequencing identifies TRPM4 as a major gene predisposing to progressive familial heart block type I. Int J Cardiol 2016; 207: 349–358. [DOI] [PubMed] [Google Scholar]
- 5. Ackerman JP, Bartos DC, Kapplinger JD, Tester DJ, Delisle BP, Ackerman MJ. The promise and peril of precision medicine: phenotyping still matters most. Mayo Clin Proc 2016; 91: 1606–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gearhart AS, Batra AS. Isolated left bundle branch block progressing to complete heart block and asystole: a novel presentation of a desmin mutation. HeartRhythm Case Rep 2018; 4: 184–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Oomen A, Jones K, Yeates L, Semsarian C, Ingles J, Sy RW. Rare desmin variant causing penetrant life‐threatening arrhythmic cardiomyopathy. HeartRhythm Case Rep 2018; 4: 318–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Otten E, Asimaki A, Maass A, van Langen IM, van der Wal A, de Jonge N, van den Berg MP, Saffitz JE, Wilde AA, Jongbloed JD, van Tintelen JP. Desmin mutations as a cause of right ventricular heart failure affect the intercalated disks. Heart Rhythm 2010; 7: 1058–1064. [DOI] [PubMed] [Google Scholar]
- 9. Almeda FQ, Barkatullah S, Kavinsky CJ. Spontaneous coronary artery dissection. Clin Cardiol 2004; 27: 377–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saw J, Humphries K, Aymong E, Sedlak T, Prakash R, Starovoytov A, Mancini GBJ. Spontaneous coronary artery dissection: clinical outcomes and risk of recurrence. J Am Coll Cardiol 2017; 70: 1148–1158. [DOI] [PubMed] [Google Scholar]
- 11. Alfonso F, Bastante T, Rivero F, Cuesta J, Benedicto A, Saw J, Gulati R. Spontaneous coronary artery dissection. Circ J 2014; 78: 2099–2110. [DOI] [PubMed] [Google Scholar]
- 12. Weisleder N, Soumaka E, Abbasi S, Taegtmeyer H, Capetanaki Y. Cardiomyocyte‐specific desmin rescue of desmin null cardiomyopathy excludes vascular involvement. J Mol Cell Cardiol 2004; 36: 121–128. [DOI] [PubMed] [Google Scholar]
- 13. Parry R, MacConnell T, Wilde P. Case report: spontaneous coronary artery dissection. Clin Radiol 1994; 49: 142–143. [DOI] [PubMed] [Google Scholar]
- 14. Wahbi K, Behin A, Charron P, Dunand M, Richard P, Meune C, Vicart P, Laforêt P, Stojkovic T, Bécane HM, Kuntzer T, Duboc D. High cardiovascular morbidity and mortality in myofibrillar myopathies due to DES gene mutations: a 10‐year longitudinal study. Neuromuscul Disord 2012; 22: 211–218. [DOI] [PubMed] [Google Scholar]