

Abstract
The ULK (UNC51-like) enzymes are a family of mammalian kinases that have critical roles in autophagy and development. While ULK1, ULK2, and ULK3 have been characterized, very little is known about ULK4. However, recently, deletions in ULK4 have been genetically linked to increased susceptibility to developing schizophrenia, a devastating neuropsychiatric disease with high heritability but few genes identified. Interestingly, ULK4 is a pseudokinase with some unusual mutations in the kinase catalytic motifs. Here, we report the first structure of the human ULK4 kinase at high resolution and show that although ULK4 has no apparent phosphotransfer activity, it can strongly bind ATP. We find an unusual mechanism for binding ATP in a Mg2+-independent manner including a rare hydrophobic bridge in the active site. In addition, we develop two assays for ATP binding to ULK4, perform a virtual and experimental screen to identify small molecule binders of ULK4 and identify several novel scaffolds that bind ULK4 and can lead the way to more selective small molecules that may help shed light on the function of this enigmatic protein.
Graphical Abstract

The ULK family of kinases comprises 5 genes in mammals: ULK1 through ULK4 and STK36. These enzymes share a conserved N-terminal kinase domain that is homologous to the C. elegans UNC-51 and the yeast Atg1, the original kinase identified in the autophagy pathway1. All the enzymes are thought to be important for development, in particular neurological development. Indeed, the unc-51 (uncoordinated movement) gene was originally identified as causing paralysis and other defects, consistent with axonal formation deficiencies2, before it was known as the autophagy kinase (atg1) orthologue. In mammals, ULK1 and ULK2, seemingly redundant3–4, have been shown to be required for autophagy, but also have important roles in development, as their double knockout causes neurological defects5, axonal pathfinding defects, in a likely autophagy-independent manner6. However, the role of ULK4 has been elusive. ULK4 is a large neuron-specific 142 kDa polypeptide consisting of an N-terminal pseudokinase domain and 5 predicted C-terminal HEAT repeats, which are a series of repeats found in other large proteins, including mTOR, thought to be involved in scaffolding and interacting with one or more partner proteins7–8. However, it is unknown what interacts with the HEAT repeats of ULK4. Recently, it was reported that ULK4 is genetically linked to schizophrenia,9 a highly genetic debilitating disease with few genetic causes identified. ULK4 was discovered as a rare risk factor for schizophrenia, as well as autism and depression. It was shown that knockdown of ULK4 leads to errors in cortical development in mice, consistent with the ideas that the ULK kinases are important for neurological development.10 However, the mechanism by which ULK4 can affect brain development, as a pseudokinase with no known catalytic activity or interaction partners is unclear.
Pseudokinases are members of the protein kinase family that have divergent amino acids from the conserved catalytic core of conventional kinases and were therefore predicted to be catalytically inactive11. Nevertheless, some pseudokinases do in fact have phosphotransfer activity using different amino acids to perform the catalytic functions. Many at least have the ability to bind ATP. It is thought for this latter class of pseudokinases that they might act as sensors for ATP and undergo conformational changes upon ATP binding that allows them to respond to ATP binding without phosphotransfer and act as scaffolds for signaling pathways12–14. For example, if they interact with another kinase they could activate the active kinase, as many pseudokinases have active kinase partners. Having selective inhibitors of ATP binding of a pseudokinase could therefore paradoxically activate the protein by stabilizing it in the active form. Alternatively, compounds that stabilize the inactive confirmation of the pseudokinase (a type II kinase inhibitor) can inhibit downstream signaling.15–17 One previous report has found compounds that can either stabilize or destabilize a pseudokinase.18 ULK4 selective compounds could then be useful in understanding the functional role of the ULK4 pseudokinase, since no downstream activity has yet been discovered. ULK4 binders could even help correct defects from heterozygous mutants by increasing activity.
Here, we purify the kinase domain of ULK4 and solve the structure at high resolution. We discovered that ULK4 binds ATP with high affinity in the absence of magnesium, higher than known pseudokinases, and develop a high throughput assay and virtual screen to discover novel inhibitors of nucleotide binding, which could be the starting point for selective inhibitors of this pseudokinase.
We previously solved the structures of ULK1 and developed compounds that can inhibit ULK1 and ULK2, with varying degrees of selectivity.19–20 We now have developed a bacterial expression system for ULK4. We were able to express the pseudokinase domain of ULK4 as a sumo-tagged fusion. After cleaving the sumo tag, we were able to generate the pure pseudokinase domain. After modifying the construct to improve crystallization, we were able to crystallize the protein in the presence of a non-selective inhibitor we previously developed for ULK1, compound 1. Using ULK1 as a search model, we were able to solve the structure of ULK4 by molecular replacement (Figure 1 and Table S1). ULK4 exhibits a classic kinase fold with some unusual features in the active site. Most striking is that instead of the lysine:glutamate salt bridge, one of the most conserved features of all protein kinases, it instead has a tryptophan:leucine hydrophobic interaction (Figure 1 and Figure S1). This appears to be the only hydrophobic bridge in the kinome, as a few kinases have hydrophobic residues in this position in alignments but have a lysine or glutamate adjacent to those positions, which likely form the normal salt bridge in the protein structure.21 The hydrophobic residue at position 33 of ULK4 is conserved in different species, while the Trp 46 is mostly conserved except in fish, where it is a histidine residue (Figure S1).
Figure 1.
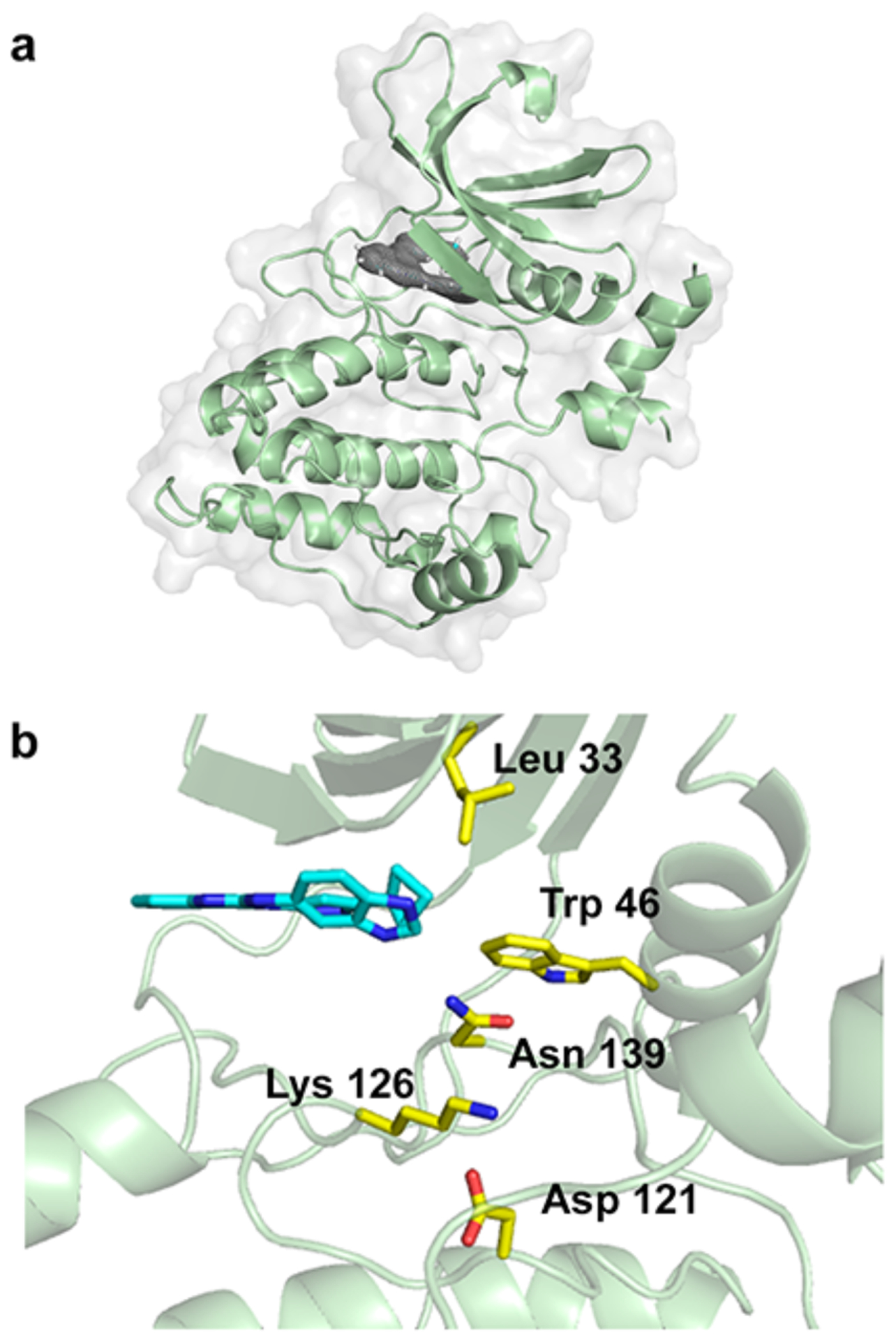
Structure of ULK4. (a) Overall structure of the kinase domain with density shown for the inhibitor, from a Fo-Fc omit map at 3 sigma. (b) Active site of the kinase, with Compound 1 shown in cyan and important residues shown in yellow.
While initially it was thought that pseudokinases have lost all activity, it is now known that they can sometimes transfer phosphate using alternative mechanisms22–24. Other pseudokinases can at least bind ATP tightly, using mechanisms other than those of conventional protein kinases. In these cases, nucleotide binding is critical for their function. We wondered, therefore, whether ULK4 had activity or could bind nucleotide despite missing several key motifs: the DFG motif, the HRD motif, and the lysine:glutamate pair in the active site (Figure S1). One report suggests that it can at least bind nucleotide.25 Initial tests with γ-32P-ATP revealed negligible activity for auto-phosphorylation or trans-phosphorylation for a generic substrate (data not shown). Therefore, we needed an assay to look at binding of nucleotide.
We developed two assays to measure binding of nucleotide to ULK4. The first assay uses the nucleotide analog ADP-Biotin, which can label kinases with biotin if they are able to bind ADP-Biotin. The biotinylated protein can then be detected by Western blot (Figure 2). Using this assay, we found that ULK4 can indeed bind nucleotide. We were also able to look at mutants of ULK4 and assess the role of those residues in nucleotide binding. Instead of the asparagine (N171 in PKA) that coordinates magnesium to bind the phosphates of ATP, ULK4 has two adjacent basic residues: Arg 125 and Lys 126, which could act as the cation for phosphate binding. This has been observed in other kinases, including TRIB2, which has a lysine in the Asn position and is capable of autophosphory lation.17 We performed mutagenesis studies on these residues and found that Lys 126 but not Arg 125 is critical for nucleotide binding, establishing that ULK4 can bind nucleotide using non-canonical residues. We examined other important residues that we thought could contribute to nucleotide binding, including Cys 141 and Asn 139, which is instead of the D in the DFG motif, and found both were important for nucleotide binding, although N139D was not completely inactive. Asp 195 occupies a similar position in the structure to Thr 180 in ULK1, which is the site of phoshorylation in the activation loop and enhances activity by locking the loop through ionic interactions with several arginines. For ULK4, Asp 195 acts as a mimic of the phosphothreonine in ULK1 and interacts with Lys 144. We hypothesized that a mutation at that position would decrease nucleotide binding, which we did indeed observe. Trp 46, which forms the hydrophobic bridge, also showed a large decrease in ADP-Biotin binding upon mutation to alanine. Another residue that had been thought to be important for activity in other pseudokinases, Lys 39, turned out to have no effect on nucleotide binding for ULK4. Thus, several mutants were able to provide insight on the mode of binding nucleotide.
Figure 2.

Nucleotide binding of ULK4 mutants. Using ADP-biotin, different mutants were monitored for their ability to bind ADP-Biotin by Western blot. Two color blot shows simultaneous His antibody signal as a loading control and streptavidin for measurement of biotinylation levels.
We also looked at other mutations that had been identified in cancer sequencing databases, with the thought that some of these mutations might have deleterious effects on the protein. One of the more frequently seen mutations in the kinase domain of ULK4 is P124S,26 which indeed abrogated nucleotide binding, most likely due to interfering with proper folding of the protein. Another mutation found in cancer, D201E, appears not to have a strong effect.
The second assay we developed for ULK4 relies on a fluorescent analog of ADP, Mant-ADP. In some cases, the fluorescent signal of Mant-nucleotide changes upon binding to the protein, allowing the measurement of nucleotide bound to protein compared to free nucleotide in solution. However, in our case, we noticed negligible change upon binding of the fluorescent signal at 445 nM. We also saw negligible change with another commonly used intrinsically fluorescent nucleotide analog, TNP-ATP (data not shown). Other studies have reported, however, that Mant-nucleotides can exhibit FRET between fluorescence of the protein excited at 280 nM and the Mant fluorophore27, which only occurs when the nucleotide is bound to protein. We found that ULK4 exhibits strong FRET between the protein and the Mant-ADP nucleotide, allowing us to monitor nucleotide binding to determine kinetic parameters. The fluorescent nucleotides can also enable high-throughput screening of inhibitors that can displace the Mant-ADP.28
We first determined the Kd of Mant-ADP by fixing the nucleotide concentration and varying the protein and measuring FRET at 445 nM emission after 280 nM excitation. Having the Kd value allows us to determine the inhibitor constants of other ligands, such as ADP, ATP, and inhibitors that compete with Mant-ADP for binding. As seen in Figure 3, we monitored Mant-ADP binding in the presence and absence of magnesium cation, to determine the role of metal ion in nucleotide binding, as many pseudokinases lack residues for cation coordination and instead use positively charged residues for binding nucleotide.29
Figure 3.

Binding of fluorescent nucleotide to ULK4. Using Mant-ADP, we measured FRET upon binding to protein, by fixing the nucleotide concentration and varying the protein concentration.
Our data shows that ULK4 is able to bind nucleotide in the absence of magnesium ions and less potently in the presence of magnesium. The FRET signal is also higher in the absence of nucleotide, which is consistent with the idea that Mg-nucleotide is binding in a different confirmation than nucleotide alone.
We then measured the inhibitory constants of ADP and ATP by varying their concentration in the presence of a fixed concentration of Mant-ADP (Table 1 and Figure S2). As with Mant-ADP, we found that ADP and ATP bind more tightly in the absence of magnesium. Interestingly, while about 40% of pseudokinases are thought to bind ATP, those that have had their affinity measured bind ATP in the single to double digit micromolar range.30 We note that some pseudokinases, including ULK4, were previously shown to have a large Tm shift in thermal-shift assays upon nucleotide binding,25 which has been shown to correlate well with binding affinity,31 indicating that other pseudokinases might have nanomolar affinity towards ATP, although these would need to be confirmed with binding assays. For now, ULK4 has one of the highest reported affinities for ATP for a pseudokinase
Table 1.
Kinetic parameters for ULK4
| With Mg | Without Mg | |
|---|---|---|
| Mant-ADP Kd | 28.6 μM | 4.69 μM |
| ADP Ki | 37.0 μM | 6.05 μM |
| ATP Ki | 4.67 μM | 0.306 μM |
We then performed two screens to identify inhibitors of ULK4. In the first approach, we performed virtual screens using our crystal structure of ULK4 and screened approximately 3.3 million purchasable lead-like compounds from ZINC15 using both OpenEye and Schrödinger suites32–33. We visually inspected the top 1000 compounds, focusing on those that can make similar contacts made by the key pharmacophores in compound 1 (Figure S3). We then selected approximately 40 compounds for purchase and tested them individually using the Mant-ADP assay at a single dose, and then selected those that showed inhibition for dose response curves. For comparison, we also evaluated compound 1 as well as compound 2, our previously optimized inhibitor for ULK1/2 that shows some selectivity towards these kinases (Figure 4a). As seen in Figure 4b, several of the compounds had inhibition of Mant-ADP binding in the double-digit micromolar range, suggestive of a starting point for further ULK4 compound development, including some conserved pharmacophores such as the purine and the spiro-lactam. We note that we were unable to determine the inhibitory constant for Compound 1 due to autofluorescence of this compound.
Figure 4.

Inhibitors of ULK4. Structures of compounds that were (a) identified for other ULK4 family members (b) discovered from a virtual screen, and (c) identified from a ka high-throughput screen of kinase inhibitors.
In order to find more potent inhibitors, we undertook a second approach, using a library of known commercial kinase inhibitors. The fluorescent assay of Mant-ADP enabled us to perform this as a small high-throughput screen, testing more than 300 commercial kinase inhibitors. We then selected the compounds that had the highest inhibition for dose response. As seen in Figure 4c, several kinase inhibitors showed potent displacement of mant-ADP, in the single-digit micromolar range.
To compare the inhibitors in an orthogonal assay, we then screened all of our confirmed hits at a single concentration (25 μM) using the ADP-biotin assay (Figure 5). This orthogonal assay allows us to confirm inhibition for autofluorescent compounds, such as compound 1, and established compound 1, R788, and PHA-848125 as the most potent ULK4 binders. To gain insight on how R788 binds potently, we docked the compound, which is a pro-drug, along with the active form of the compound, R406, which showed no inhibition. As seen in Figure S4, the phosphate likely makes contacts with the positively charged residues in the ATP-binding pocket, including R125. Although R788 would not make a suitable chemical probe for ULK4 because of the phosphate, it provides important information about optimizing the compounds further for potency, and provides a starting point as the most potent ULK4 binder. The phosphate interaction could also provide a guide for selectivity, because ULK4 uniquely has a lysine there instead of an asparagine, which is seen in almost all other kinases. Further improved compounds will allow us to study the role of ULK4 in cells, by selectively inhibiting or activating ULK4, and will help uncover its role in neurological development and schizophrenia. Future work will help elucidate the mechanism of ULK4’s biological role. Structural and biochemical information about the large unknown portions of the ULK4 protein will help shed light on its signaling mechanism.
Figure 5.

Inhibition of ULK4 from new inhibitors. Using ADP-biotin, different mutants were monitored for their ability to bind ADP-Biotin by Western blot. Two color blot shows simultaneous His antibody signal as a loading control and streptavidin for measurement of biotinylation levels. All inhibitors and nucleotides are at 25 μM concentration. The potency can be gauged by monitoring the ratio of the Streptavidin signal to the His signal, or looking at the redness in the overlay.
Supplementary Material
ACKNOWLEDGMENT
This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DEAC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). We appreciate OpenEye Scientific Software, Inc. for granting us access to its high-performance molecular modeling applications through its academic license program. This work was supported in part through the computational resources and staff expertise provided by the Department of Scientific Computing at the Icahn School of Medicine at Mount Sinai.
Funding Sources
No competing financial interests have been declared. This work was supported by K22CA201103 and GM124838 (M.B.L) and GM108911 (A.S.)
Footnotes
Supporting Information.
Supplemental figures and tables and experimental procedures(PDF) and structures factors and coordinates for the ULK4 crystal structure.
REFERENCES
- (1).Kamada Y; Funakoshi T; Shintani T; Nagano K; Ohsumi M; Ohsumi Y, Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 2000, 150 (6), 1507–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ogura K; Wicky C; Magnenat L; Tobler H; Mori I; Muller F; Ohshima Y, Caenorhabditis elegans unc-51 gene required for axonal elongation encodes a novel serine/threonine kinase. Genes Dev 1994, 8 (20), 2389–400. [DOI] [PubMed] [Google Scholar]
- (3).Wong PM; Puente C; Ganley IG; Jiang X, The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 2013, 9 (2), 124–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Noda NN; Fujioka Y, Atg1 family kinases in autophagy initiation. Cell Mol Life Sci 2015, 72 (16), 3083–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Joo JH; Wang B; Frankel E; Ge L; Xu L; Iyengar R; Li-Harms X; Wright C; Shaw TI; Lindsten T; Green DR; Peng J; Hendershot LM; Kilic F; Sze JY; Audhya A; Kundu M, The Noncanonical Role of ULK/ATG1 in ER-to-Golgi Trafficking Is Essential for Cellular Homeostasis. Mol Cell 2016, 62 (4), 491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wang B; Iyengar R; Li-Harms X; Joo JH; Wright C; Lavado A; Horner L; Yang M; Guan JL; Frase S; Green DR; Cao X; Kundu M, The autophagy-inducing kinases, ULK1 and ULK2, regulate axon guidance in the developing mouse forebrain via a noncanonical pathway. Autophagy 2018, 14 (5), 796–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Andrade MA; Petosa C; O’Donoghue SI; Muller CW; Bork P, Comparison of ARM and HEAT protein repeats. J Mol Biol 2001, 309 (1), 1–18. [DOI] [PubMed] [Google Scholar]
- (8).Perry J; Kleckner N, The ATRs, ATMs, and TORs are giant HEAT repeat proteins. Cell 2003, 112 (2), 151–5. [DOI] [PubMed] [Google Scholar]
- (9).Lang B; Pu J; Hunter I; Liu M; Martin-Granados C; Reilly TJ; Gao GD; Guan ZL; Li WD; Shi YY; He G; He L; Stefansson H; St Clair D; Blackwood DH; McCaig CD; Shen S, Recurrent deletions of ULK4 in schizophrenia: a gene crucial for neuritogenesis and neuronal motility. J Cell Sci 2014, 127 (Pt 3), 630–40. [DOI] [PubMed] [Google Scholar]
- (10).Lang B; Zhang L; Jiang G; Hu L; Lan W; Zhao L; Hunter I; Pruski M; Song NN; Huang Y; Zhang L; St Clair D; McCaig CD; Ding YQ, Control of cortex development by ULK4, a rare risk gene for mental disorders including schizophrenia. Sci Rep 2016, 6, 31126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Boudeau J; Miranda-Saavedra D; Barton GJ; Alessi DR, Emerging roles of pseudokinases. Trends Cell Biol 2006, 16 (9), 443–52. [DOI] [PubMed] [Google Scholar]
- (12).Byrne DP; Foulkes DM; Eyers PA , Pseudokinases: update on their functions and evaluation as new drug targets. Future Med Chem 2017, 9 (2), 245–265. [DOI] [PubMed] [Google Scholar]
- (13).Zeqiraj E; van Aalten DM, Pseudokinases-remnants of evolution or key allosteric regulators? Curr Opin Struct Biol 2010, 20 (6), 772–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kannan N; Taylor SS , Rethinking pseudokinases. Cell 2008, 133 (2), 204–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Dhawan NS; Scopton AP; Dar AC, Small molecule stabilization of the KSR inactive state antagonizes oncogenic Ras signalling. Nature 2016, 537 (7618), 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Tokarski JS; Zupa-Fernandez A; Tredup JA; Pike K; Chang C; Xie D; Cheng L; Pedicord D; Muckelbauer J; Johnson SR; Wu S; Edavettal SC; Hong Y; Witmer MR; Elkin LL; Blat Y; Pitts WJ; Weinstein DS; Burke JR, Tyrosine Kinase 2-mediated Signal Transduction in T Lymphocytes Is Blocked by Pharmacological Stabilization of Its Pseudokinase Domain. J Biol Chem 2015, 290 (17), 11061–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bailey FP; Byrne DP; Oruganty K; Eyers CE; Novotny CJ; Shokat KM; Kannan N; Eyers PA, The Tribbles 2 (TRB2) pseudokinase binds to ATP and autophosphorylates in a metal-independent manner. Biochem J 2015, 467 (1), 47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Foulkes DM; Byrne DP; Yeung W; Shrestha S; Bailey FP; Ferries S; Eyers CE; Keeshan K; Wells C; Drewry DH; Zuercher WJ; Kannan N; Eyers PA, Covalent inhibitors of EGFR family protein kinases induce degradation of human Tribbles 2 (TRIB2) pseudokinase in cancer cells. Sci Signal 2018, 11 (549). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lazarus MB; Novotny CJ; Shokat KM, Structure of the human autophagy initiating kinase ULK1 in complex with potent inhibitors. ACS Chem Biol 2015, 10 (1), 257–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lazarus MB; Shokat KM, Discovery and structure of a new inhibitor scaffold of the autophagy initiating kinase ULK1. Bioorg Med Chem 2015, 23 (17), 5483–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Manning G; Whyte DB; Martinez R; Hunter T; Sudarsanam S, The protein kinase complement of the human genome. Science 2002, 298 (5600), 1912–34. [DOI] [PubMed] [Google Scholar]
- (22).Mukherjee K; Sharma M; Urlaub H; Bourenkov GP; Jahn R; Sudhof TC; Wahl MC, CASK Functions as a Mg2+-independent neurexin kinase. Cell 2008, 133 (2), 328–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Min X; Lee BH; Cobb MH; Goldsmith EJ, Crystal structure of the kinase domain of WNK1, a kinase that causes a hereditary form of hypertension. Structure 2004, 12 (7), 1303–11. [DOI] [PubMed] [Google Scholar]
- (24).Eswaran J; Patnaik D; Filippakopoulos P; Wang F; Stein RL; Murray JW; Higgins JM; Knapp S, Structure and functional characterization of the atypical human kinase haspin. Proc Natl Acad Sci U S A 2009, 106 (48), 20198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Murphy JM; Zhang Q; Young SN; Reese ML; Bailey FP; Eyers PA; Ungureanu D; Hammaren H; Silvennoinen O; Varghese LN; Chen K; Tripaydonis A; Jura N; Fukuda K; Qin J; Nimchuk Z; Mudgett MB; Elowe S; Gee CL; Liu L; Daly RJ; Manning G; Babon JJ; Lucet IS, A robust methodology to subclassify pseudokinases based on their nucleotide-binding properties. Biochem J 2014, 457 (2), 323–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Barretina J; Caponigro G; Stransky N; Venkatesan K; Margolin AA; Kim S; Wilson CJ; Lehar J; Kryukov GV; Sonkin D; Reddy A; Liu M; Murray L; Berger MF; Monahan JE; Morais P; Meltzer J; Korejwa A; Jane-Valbuena J; Mapa FA; Thibault J; Bric-Furlong E; Raman P; Shipway A; Engels IH; Cheng J; Yu GK; Yu J; Aspesi P Jr.; de Silva M; Jagtap K; Jones MD; Wang L; Hatton C; Palescandolo E; Gupta S; Mahan S; Sougnez C; Onofrio RC; Liefeld T; MacConaill L; Winckler W; Reich M; Li N; Mesirov JP; Gabriel SB; Getz G; Ardlie K; Chan V; Myer VE; Weber BL; Porter J; Warmuth M; Finan P; Harris JL; Meyerson M; Golub TR; Morrissey MP; Sellers WR; Schlegel R; Garraway LA, The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483 (7391), 603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hammaren HM; Ungureanu D; Grisouard J; Skoda RC; Hubbard SR; Silvennoinen O, ATP binding to the pseudokinase domain of JAK2 is critical for pathogenic activation. Proc Natl Acad Sci U S A 2015, 112 (15), 4642–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Guarnieri MT; Blagg BS; Zhao R, A high-throughput TNP-ATP displacement assay for screening inhibitors of ATP-binding in bacterial histidine kinases. Assay Drug Dev Technol 2011, 9 (2), 174–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Knape MJ; Herberg FW, Metal coordination in kinases and pseudokinases. Biochem Soc Trans 2017, 45 (3), 653–663. [DOI] [PubMed] [Google Scholar]
- (30).Hammaren HM; Virtanen AT; Silvennoinen O, Nucleotide-binding mechanisms in pseudokinases. Biosci Rep 2015, 36 (1), e00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Rudolf AF; Skovgaard T; Knapp S; Jensen LJ; Berthelsen J, A comparison of protein kinases inhibitor screening methods using both enzymatic activity and binding affinity determination. PLoS One 2014, 9 (6), e98800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Spilman P; Podlutskaya N; Hart MJ; Debnath J; Gorostiza O; Bredesen D; Richardson A; Strong R; Galvan V, Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One 2010, 5 (4), e9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Schrödinger Release 2019–1: Induced Fit Docking protocol; Glide, Schrödinger, LLC, New York, NY, 2019; Prime, Schrödinger, LLC, New York, NY, 2019. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.