

Abstract
Investigations of Alzheimer’s disease (AD), traumatic brain injury (TBI), and related brain disorders have provided extensive evidence for involvement of cathepsin B, a lysosomal cysteine protease, in mediating the behavioral deficits and neuropathology of these neurodegenerative diseases. This review integrates findings of cathepsin B regulation in clinical biomarker studies, animal model genetic and inhibitor evaluations, structural studies, and lysosomal cell biological mechanisms in AD, TBI, and related brain disorders. The results together indicate the role of cathepsin B in the behavioral deficits and neuropathology of these disorders. Lysosomal leakage occurs in AD and TBI, and related neurodegeneration, which leads to the hypothesis that cathepsin B is redistributed from the lysosome to the cytosol where it initiates cell death and inflammation processes associated with neurodegeneration. These results together implicate cathepsin B as a major contributor to these neuropathological changes and behavioral deficits. These findings support the investigation of cathepsin B as a potential drug target for therapeutic discovery and treatment of AD, TBI, and TBI-related brain disorders.
Keywords: cathepsin B, Alzheimer’s disease (AD), traumatic brain injury (TBI), neurodegeneration, brain, memory, cognition, behaviors, pathology, human brain, biomarker, gene knockout, inhibitors, active site binding, lysosomal leakage
1. Introduction
Neurodegeneration of Alzheimer’s disease (AD), traumatic brain injury (TBI), and related dementia (ADRD) disorders displays behavioral deficits in memory, cognition, problem solving, executive function, language, emotion, and related brain functions (1–15). Severe cognitive decline, dementia, occurs in ADRD disorders of Alzheimer’s disease (1–3), traumatic brain injury (TBI) (4–6), brain ischemia and stroke (7–9), and related neurodegenerative brain disorders (10–15). These disorders are widespread affecting more than 47 million people worldwide, with high prevalence in the aged population.
There have been tremendous efforts in the field of drug discovery for AD, TBI, and numerous related ADRD disorders, but decades of drug development research have not yet yielded effective therapeutics to attenuate or reverse these neurodegenerative diseases. Advancements in dementia therapeutics strategies will necessitate elucidation of novel molecular mechanisms in ADRD which can be modulated through drug intervention to ameliorate the behavioral dysfunctions and neuropathology of ADRD disorders. Investigation of novel targets of ADRD will advance opportunities for therapeutics development to extend prior efforts for targeting neuropathological amyloid-β (Aβ) (16–20), phospho-tau (21–23), and related mechanisms (24–27).
Cathepsin B, a lysosomal cysteine protease, represents a candidate drug target for AD, TBI, and related brain disorders. Cathepsin B has been shown by numerous studies to participate in AD and TBI cognitive and behavioral deficits, and in neuropathology. This review highlights the multi-disciplinary research of cathepsin B in human clinical studies, and in AD, TBI, and related animal models using cathepsin B gene knockout and chemical inhibition. Findings from these studies support the hypothesis that lysosomal leakage of cathepsin B to the cytosol leads to neurodegeneration and behavioral deficits in AD, TBI, and related brain disorders. These results provide the rationale for investigation of cathepsin B as a mechanistic drug target for therapeutics discovery and development for AD, TBI, and related dementia.
2. Cathepsin B elevation in human clinical AD, TBI, and related neurodegenerative disorders.
Several studies have shown increased levels of cathepsin B protein or activity in plasma, CSF, and amyloid plaques in human AD, TBI, and related neurodegenerative diseases (Table 1) (28–39). Importantly, increased plasma cathepsin B correlates with cognitive dysfunction in AD (29) and severe trauma outcomes (36).
Table 1.
Cathepsin B elevation in AD, TBI, and related dementia patients
| Clinical Condition | Biofluid or Tissue | Cathepsin B Regulation | Features | Reference |
|---|---|---|---|---|
| Alzheimer’s disease (AD) | plasma | ↑ | AD patients had 49% increase in CTSB protein above controls | (28) |
| AD | serum | ↑ | AD patients had ~45% increase in CTSB protein vs. controls ↑ CTSB correlated with MMSE* cognitive function scores indicating dementia |
(29) |
| AD | plasma | ↑ | elevated CTSB protein in mild and severe AD by 50–80% above controls | (30) |
| AD | CSF | ↑ | CTSB protein was elevated by 1.9-fold in AD compared to control | (31) |
| AD | brain amyloid plaque | abnormal localization at amyloid plaques of AD brains | high CTSB activity and protein were abnormally localized in senile plaques of AD brains | (32) |
| Aging | CSF | ↑ | increased CTSB protein correlated positively with age | (35) |
| Severe Trauma | plasma | ↑ | CTSB activity was elevated 5–6 fold in severe trauma leading to organ failure | (36) |
| Multiple Trauma | plasma | ↑ | elevated CTSB associated with trauma and sepsis | (37) |
| Traumatic Brain Injury (TBI) | CSF | ↑ | elevated CTSB protein by 2-fold increase compared to controls | (38) |
| Inflammatory Neurological Diseases | CSF | ↑ | elevated CTSB activity in Guilllain-Barrer syndrome (GBS), chronic inflammatory demyelinating polyneuroopathy (CIDP), multiple sclerosis (MS), and meningitis | (39) |
Mini-mental state examination scores are utilized to monitor cognitive dysfunction, dementia (29).
2.1. Human AD in aging and cathepsin B regulation.
In AD patients, cathepsin B levels are elevated in serum compared to age-matched healthy controls (28–30) (Figure 1). Notably, the elevated cathepsin B correlated with MMSE (Mini-Mental State Exam) scores of dementia, cognitive impairment, in AD patients (29) (Figure 1). Elevated plasma cathepsin B occurs in both mild and severe AD compared to healthy controls. In patients with mild cognitive impairment (MCI), plasma cathepsin B levels were similar to controls (30). These findings show that increases in plasma cathepsin B correlate with AD cognitive status, and occur in mild and severe stages of AD.
Figure 1. Elevation of cathepsin B in serum correlates with cognitive dysfunction in Alzheimer’s disease patients.
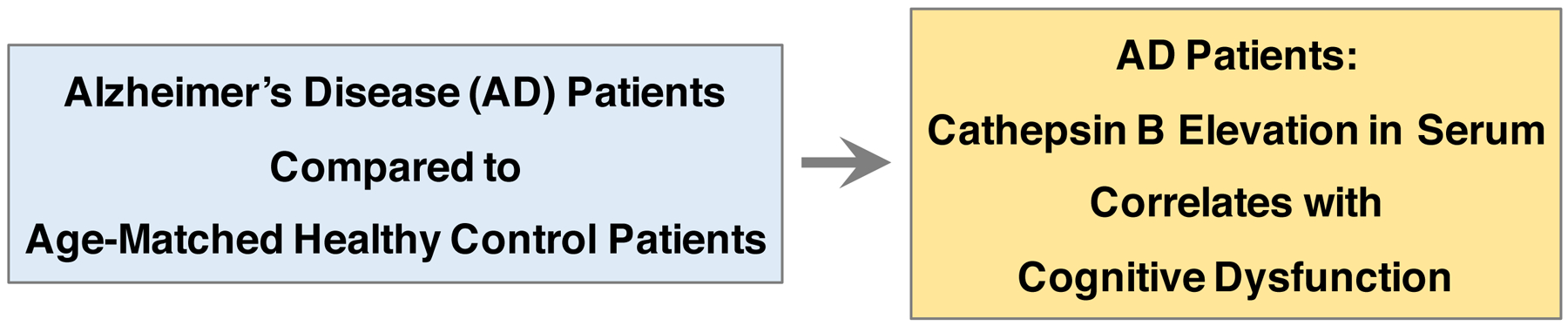
Cathepsin B levels were significantly elevated by ~50% in serum from AD patients compared to age-matched control patients, shown by studies of Sun et al., 2015 (29). Furthermore, the increased cathepsin B in serum was significantly correlated with cognitive dysfunction, measured the Mini-Mental State Exam which tests for cognitive functions of memory, attention, language, and orientation (29).
Notably, cathepsin B in human AD brain displays abnormal extracellular localization with amyloid plaque pathology (31). Extracellular cathepsin B contrasts with its intracellular location in lysosomes. These findings demonstrate re-distribution of lysosomal cathepsin B to abnormal extracellular locations in AD brain. The extracellular location suggests that cathepsin B levels in CSF may become elevated. Indeed, CSF analyses have demonstrated higher cathepsin B levels in AD patients compared to controls (32)
AD is an age-related neurodegenerative disease occurring at elderly ages. Aging is a main risk factor for AD and numerous neurodegenerative diseases (33, 34). Age-related increases in cathepsin B levels in CSF occur in elderly patients (35).
2.2. TBI and related neurodegenerative diseases exhibit cathepsin B regulation.
In numerous studies of clinical TBI and related conditions, cathepsin B is significantly elevated in plasma and CSF. Severe trauma patients show elevated cathepsin B in plasma associated with organ failure, compared to patients without complications (36, 37). In TBI patients, cathepsin B protein and activity in CSF are elevated compared to controls (38). Furthermore, cathepsin B activity in CSF is elevated in the inflammatory neurological diseases of Guillain-Barre syndrome (GBS), inflammatory demyelinating polyneuropathy (CIDP), and multiple sclerosis (MS) (39). These findings demonstrate elevated cathepsin B in TBI injury and related neurological diseases.
3. Human brain gene expression of cathepsin B at young to adult ages
Analyses of cathepsin B expression in human brain illustrates its abundant expression in hippocampal and cortical regions which are responsible for cognitive and behavioral functions (40). Cathepsin B mRNA expression is illustrated as RNAseq data overlaid on a human brain MRI image (from the Allen human brain atlas, https://human.brain-map.org/) (41, 42) in Figure 2a.
Figure 2. Expression of cathepsin B and lysosomal cathepsins in young and adult human brains.
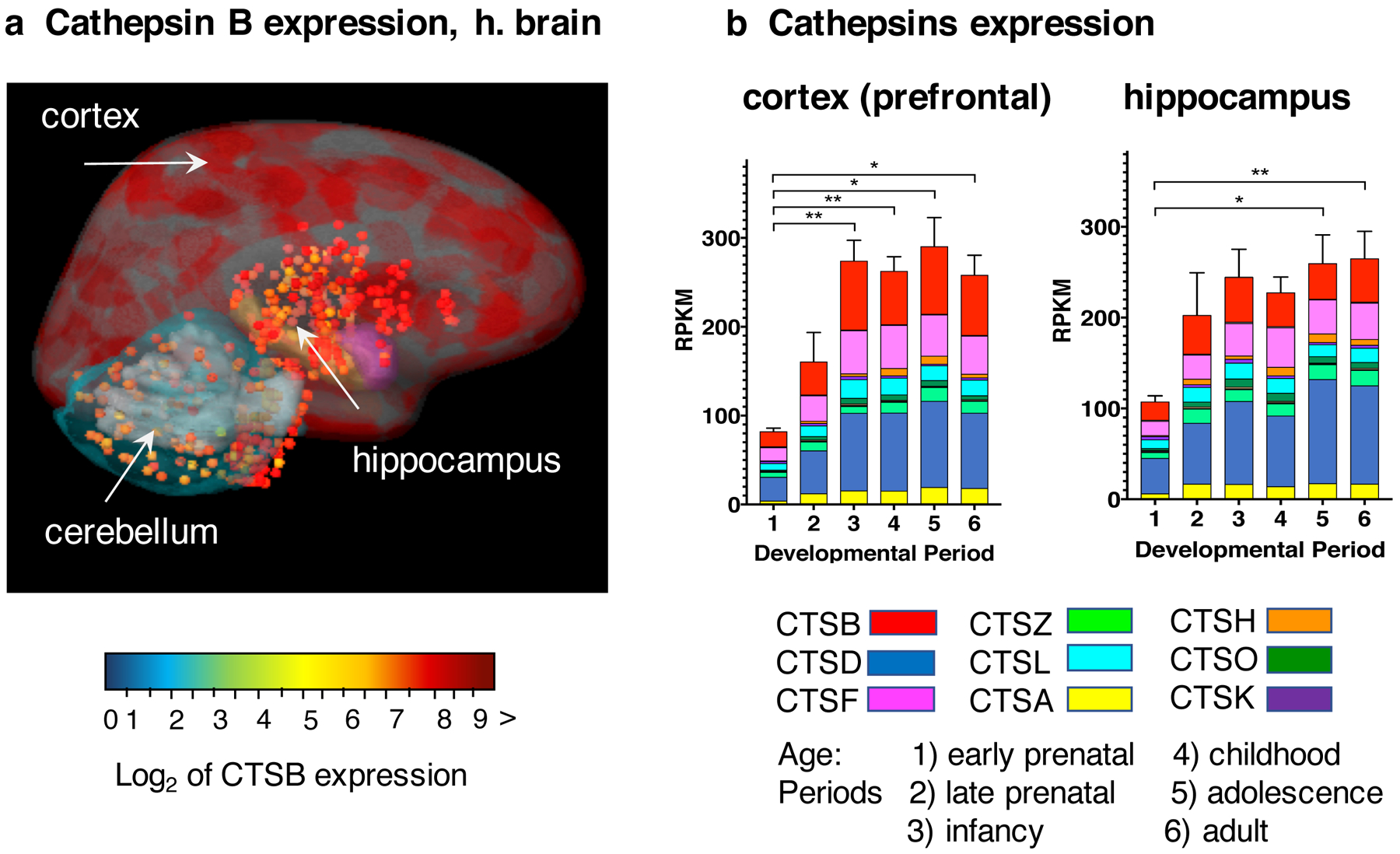
a. Cathepsin B expression in human adult brain. Cathepsin B mRNA expression levels were determined by RNAseq analyses by the Allen Human Brain Atlas (https://human.brain-map.org/). The log2 normalized expression values of cathepsin B mRNA levels are illustrated overlaid on an MRI image of adult human brain, in color-coded ranges of high (dark red), medium (yellow), and lower (green-blue) levels of expression. The cortex regions display very high levels of cathepsin B expression and the hippocampus shows high levels of expression.
b. Expression of cathepsin B and lysosomal cathepsin proteases in human brain regions at young through adult ages. Gene expression levels of cathepsin B and the majority of the lysosomal cathepsin proteases. Cathepsins D, F, Z, L, A, H, O, and K, representing the most abundantly expressed among 15 cathepsin proteases (40) are illustrated. Gene expression data are assessed as reads per kilobase million (RPKM) for six age periods of early prenatal, late prenatal, infancy, childhood, adolescence, and adult. Bar graphs show the total cathepsin gene expression as the average RPKM + s.e.m., with statistical significance (*p < 0.05, **p<0.01). Cathepsins B, F, and D are the most abundantly expressing cathepsins.
Cathepsin B is one of the most abundant lysosomal cathepsin proteases expressed in human brain compared to the 15 member lysosomal cysteine protease family, consisting of cathepsins B, C, F, H, K, L, O, S, V, W, Z, D, E, A, and G (43) (Figure 2b). Cathepsin B expression levels represents 12–32% of total cathepsin expression composed of 15 cathepsin proteases, in 16 human brain regions (40). The abundant expression of cathepsin B occurs throughout human brain development from infancy to childhood, adolescence, and adult life. These findings indicate the presence of cathepsin B in brain at all ages for normal lysosomal functions in health and in neurological diseases of brain injuries, neurodegeneration, and related dementia and behavioral disorders.
4. Cathepsin B participates in behavioral deficits and neuropathology in animal models of AD, TBI, and related brain disorders.
Numerous investigations of animal models of AD, TBI, and related dementia disorders demonstrate that cathepsin B is elevated during brain dysfunction and pathogenesis (29, 44–74). Furthermore, gene knockout of cathepsin B in these animal models of brain disorders results in amelioration of behavioral deficits and reduction in neuropathology (75–86, 88–91). Compelling evidence from numerous studies demonstrate that chemical inhibition of cathepsin B improves behavioral dysfunctions and neuropathology in animal models of AD, TBI, and related brain disorders (92–114). These findings are discussed in this section.
4.1. Cathepsin B elevation in animal models of AD, TBI, and related brain disorders.
AD models.
In AD animal models, cathepsin B gene expression is elevated in the 5X familial (FAD) AD mouse model which expresses three APP mutations (APP KM670/671NL, Swedish; APP (716V, Florida; APP V171I, London) and human presenilin 1 (PS1) containing two mutations (PSEN1 M146L; PSEN1 L286V (44) (Table 2). Cathepsin B protein levels are increased by 50% in the cortex and hippocampus brain regions of the APPSwe/PS1 mouse model, which expresses human APP with the Swedish (Swe) FAD mutation and PS1 with FAD mutations, relative to controls (29) (Table 2).
Table 2.
Regulation of cathepsin B in animal models of AD, TBI, and related brain disorders
| Animal Model | Species | Cathepsin B Regulation | mRNA, Protein, or Activity of Cathepsin B | Tissue | Refs. |
|---|---|---|---|---|---|
| AD, 5XFAD model | mouse | ↑ | elevated gene expression in brain | brain | (44) |
| AD, APPSwe/PS1 | mouse | ↑ | increased protein | brain | (29) |
| TBI Trauma | mouse rat |
↑ | elevated mRNA, protein, and proteolytic activity | brain | (45–50) |
| Trauma SCC | rat | ↑ | increased mRNA, protein, and activity | brain | (51, 52) |
| Trauma surgery | mouse | ↑ | elevated activity in extracellular matrix | intestine trauma model | (53) |
| Subarachnoid hemorrhage | rat | ↑ | increased protein | brain | (54, 55) |
| Brain aneurysm | rat | ↑ | elevated mRNA and activity | cerebral aneurysm walls | (56) |
| ALS (amyotrophic lateral sclerosis) | mouse | ↑ | increased mRNA | spinal cord motoneurons | (59, 60) |
| Excitatory epilepsy | rat | ↑ | increased protein | brain and spinal cord | (62) |
| Excitatory Huntington’s disease | rat | ↑ | elevated protein | brain | (63) |
| Ischemia, acute | rat | ↑ | elevated protein and activity | brain | (64) |
| Ischemia | monkey rat |
↑ | increased protein and activity | brain | (64–67) |
| Meningitis brain infection | mouse | ↑ | increased proteolytic activity | human THP-1 cells | (68) |
| Sepsis infection | rat | ↑ | increased proteolytic activity | skeletal muscle | (69, 70) |
| Inflammation, aging | mouse | ↑ | increased mRNA and protein | brain | (71–73) |
| Inflammatory pain | mouse | ↑ | elevated protein | spinal cord | (74) |
TBI models.
In TBI animal models, cathepsin B is significantly elevated in brains subjected to the controlled cortical impact (CCI) TBI mouse model (45), and in the penetrating ballistic-like brain injury (PBBI) TBI model (38). TBI related conditions of trauma (45–50), spinal cord contusion (SCC) (51, 52), surgery (53), hemorrhage (54, 55), and brain aneurysm (56) result in increased levels of brain cathepsin B.
The time-course of cathepsin B elevation occurs soon after the injury event and the increase is sustained during the course of the injury. In the CCI mouse model of TBI, cathepsin B is elevated at 2 hours and 24 hours post-injury (45). In the severe penetrating ballistic-like brain injury (PBBI) in rats, cathepsin B enzyme levels and activity are increased in the cortex and hippocampus during 3–7 days after PBBI (38). In the weight drop mouse model of brain injury, brain cathepsin B is elevated as early as 6 hours after injury, and peaks at 2 days post-trauma, with sustained elevation for up to one week (57, 58). In a model of TBI in rat, cathepsin B is elevated in the brain within one hour after injury and remains elevated for more than one month; the maximum 3-fold increase of cathepsin B occurs at 8 days post-injury (48). These models of moderate to severe TBI together demonstrate that increased cathepsin B occurs shortly after brain injury and is sustained for weeks to months post-TBI.
ALS, epilepsy, and related neurodegeneration.
In ALS (amyotrophic lateral sclerosis) neurodegeneration, cathepsin B expression is elevated by 3-fold in spinal cord in ALS mouse models compared to controls (59, 60). Cathepsin B is also up-regulated in ALS patients (61). In animal models of epilepsy, which is associated with TBI, cathepsin B protein levels are elevated (62). Furthermore, in excitatory Huntington’s disease, cathepsin B is elevated (63).
Ischemia and inflammatory injury models.
Ischemia and inflammatory brain injuries result in cognitive deficits and display elevated cathepsin B. Ischemia results in increased cathepsin B in the brain, demonstrated in acute and chronic ischemia in rat and monkey animal models (64–67). Meningitis (68), sepsis (69, 70), inflammation (71–73), and inflammatory pain (74) also result in increased cathepsin B.
4.2. Cathepsin B gene knockout improves behavioral deficits and ameliorates neuropathology in animal models of AD, TBI, and related disorders.
Studies of cathepsin B gene knockout in animal models of AD, TBI, infection, and aging in cognitive deficits show that the absence of cathepsin B preserves normal behaviors and ameliorates brain neuropathology and associated biomarkers (summarized in Figure 3).
Figure 3. Cathepsin B gene knockout in animal models of AD, TBI, PgLPS, and aging.
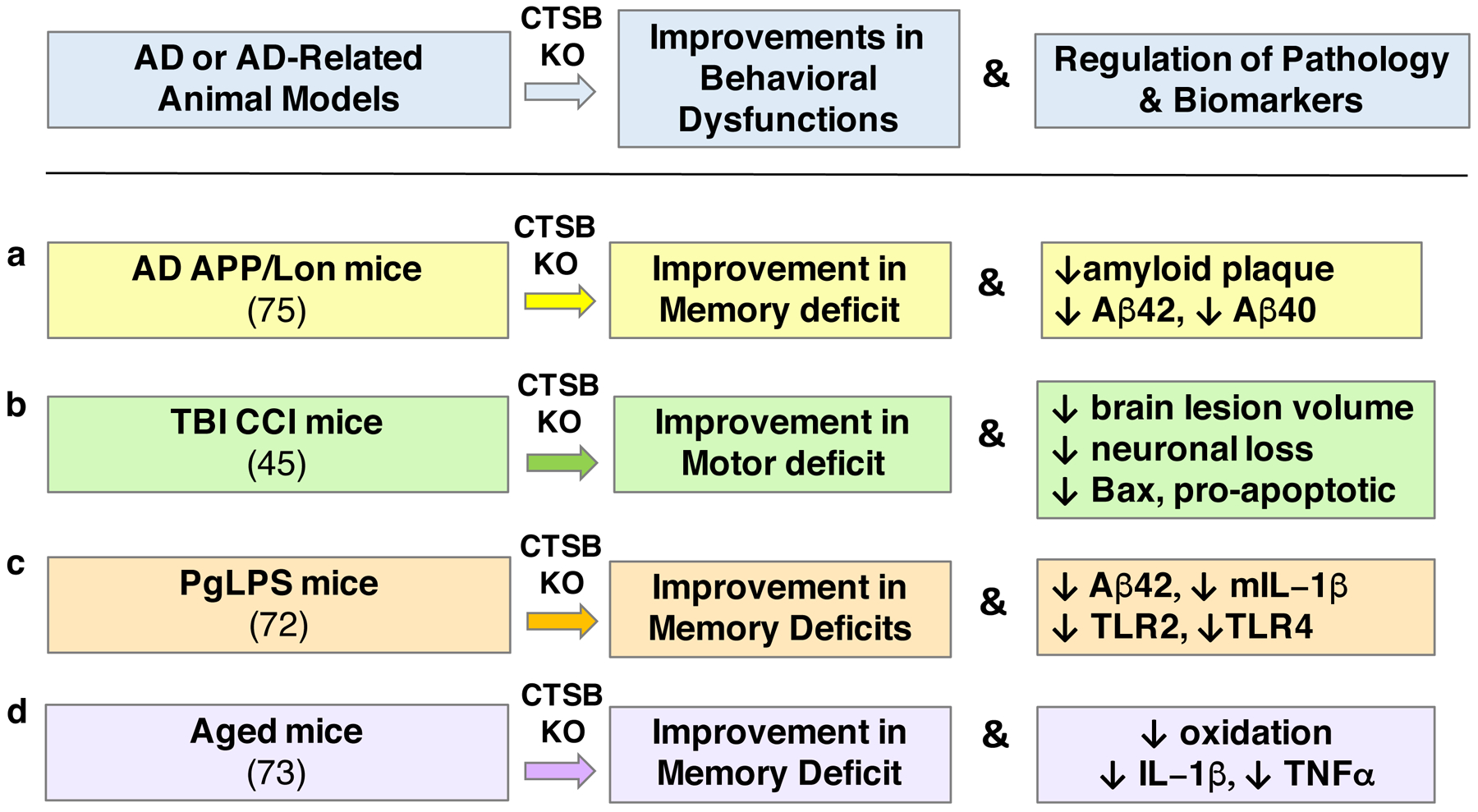
Knockout (KO) of the cathepsin B gene (CTSB) results in alleviation of memory deficits in animal models of AD (75), PgLPS (72), and aging (73), and improvement in motor dysfunction in the CCI-TBI animal model (45), shown in panels a, b, c, and d, respectively. These improvements are accompanied by reduced neuropathology and reductions in biomarkers associated with the behavioral deficits. (a) In the APP/Lon mouse model of AD, CTSB KO results in substantial improvements in memory deficit measured by the Morris water maze test, and reduces amyloid plaque pathology with reduced brain levels of Aβ(1–40) and Aβ(1–42) (75), as well as reductions in pGlu-Aβ(3–40) and pGlu-Aβ(3–42) (77). (b) In the controlled cortical impact (CCI) model of traumatic brain injury (TBI), CTSB KO results in amelioration of motor deficits and allevation of brain tissue lesion pathology, combined with reduced neuronal loss and decreased pro-apoptotic Bax levels (45). (c) In mice suffering from cognitive decline due to infection by lipopolysaccharide from Porphyromonas gingivalis (PgLPS), CTSB KO results in improvement in memory deficit and reductions in Aβ42 and pro-inflammatory IL-1β, TLR2, and TLR4 (72). (d) CTSB KO in aged mice improves cognitive dysfunction, and ameliorates age-dependent increases in oxidation, IL-1β, and TNFα (73).
AD APP/Lon model.
The APP/Lon mouse model of AD expresses the human APP gene with the V717I London mutation, resulting in memory deficits and amyloid plaque brain neuropathology (75). Knockout of the cathepsin B gene in APP/Lon mice results in preservation of memory function (measured by the Morris water maze behavioral assay) to nearly normal function (75) (Figure 3a). Deletion of the cathepsin B gene results in reduction of amyloid plaque load, concomitant with decreases in Aβ(1–40) and Aβ(1–42), and reduced pGlu-Aβ(3–40) and pGlu-Aβ(3–42), in brains of the APP/Lon mice (75–77). The N-truncated and pGlu-modified pGlu-Aβ(3–40) and pGlu-Aβ(3–42) peptides participate in seeding neurotoxic oligomers of Aβ peptides (78, 79). These findings support a role for cathepsin B in memory deficits and amyloid plaque brain pathology in the APP/Lon mouse model of AD.
CCI-TBI model.
TBI results in cognitive deficits (80, 81) and TBI increases the risk for development of AD (26, 82–84) (Figure 3b). Using the CCI (controlled cortical impact) mouse model of TBI, deletion of the cathepsin B gene results in reduced motor dysfunction after CCI compared to sham controls. Notably, the absence of the cathepsin B gene results in recovery to normal function by 7 days post-CCI, compared to the motor deficits displayed by shams (45). Brain tissue lesions resulting from CCI are ameliorated in the cathepsin B knockout mice (45). Cathepsin B knockout blocks the CCI-induced elevation in the pro-apoptotic cell death Bax protein (45). These data provide evidence for participation of cathepsin B in the behavioral dysfunction and brain lesions in the CCI mouse model of TBI.
Porphyromonas gingivalis (PgLPS) model.
Several studies reveal a strong association of peripheral infection by periodontitis with accelerated cognitive decline of AD (85, 86). In a mouse model of periodontitis-induced cognitive dysfunction, middle-aged mice subjected to chronic treatment with lipopolysaccharide from Porphyromonas gingivalis (PgLPS) display cathepsin B-dependent learning and memory deficits with Aβ accumulation (72). Cathepsin gene knockout results in improvement of the PgLPS-induced memory deficit, reduction of Aβ42 levels in brain, and reduction of elevated pro-inflammatory IL-1β, TLR2, and TLR4 factors (72) (Figure 3c). These findings demonstrate that cathepsin B participates in PgLPS-induced AD-like cognitive deficits involving elevation of Aβ42 and inflammation.
In addition, recent studies show that the Porphyromonas gingivalis (P. gingivalis) gingipain cysteine proteases are present in brains of AD patients, and P. gingivalis infection of mice result in neuronal loss in brain combined with increased Aβ42 and TNFα inflammatory factor (87). Thus, cathepsin B-mediated neurodegeneration in P. gingivalis-related AD may occur in the presence of bacterial gingipain cysteine proteases.
Aging model.
Advanced age is a major risk factor for dementia of AD and related neurodegenerative diseases (34, 88). Aged mice develop cognitive deficits associated with elevated cathepsin B, neuroinflammation, and ROS (reactive oxygen species) (73). Notably, cathepsin B gene knockout ameliorated cognitive dysfunction in the aged mice. Deletion of cathepsin B also resulted in amelioration of age-dependent elevations in oxidative stress and inflammation (IL-1β and TNFα) (73) (Figure 3d). These findings demonstrate a role for cathepsin B in age-dependent cognitive decline.
4.3. Cathepsin B knockout mice are healthy.
The Cathepsin B knockout mice have been found to be healthy and are indistinguishable from normal animals in behavior, histology, and fertility (89, 90). While there is some decrease in thyroglobulin solubilization, there is no apparent behavioral effect (91). The normal health of mice in the absence of the cathepsin B gene suggests that pharmacological inhibition of cathepsin B will generally be safe.
4.4. Chemical inhibition of cathepsin B alleviates behavioral deficits and reduces neuropathology in AD, TBI, and related animal models
The effectiveness of cathepsin B gene knockout to ameliorate behavioral deficits and neuropathology predicts that chemical inhibitors of cathepsin B can improve deficits of AD, TBI, and related conditions. Indeed, numerous studies show that cathepsin B inhibitors improve cognitive dysfunctions and reduce neuropathology of AD, TBI, and TBI-related animal models (summarized in Table 3).
Table 3.
Chemical inhibition of cathepsin B improves behavioral dysfunction and neuropathology in AD, TBI, and related animal models
| Disease Model | Inhibitor (route) | Behavior | Pathology | Biomarkers | Refs. |
|---|---|---|---|---|---|
| AD APPLon | E64d (oral) E64d (icv) CA-074Me (icv) |
↓ memory deficits | ↓ amyloid plaque | ↓ Aβ(1–40/42) ↓ pGluAβ(3–40/42) |
(92–94) |
| TBI Trauma | E64d (oral) | ↓ motor deficits | ↓ brain lesion | ↓ CTSB, ↓ Bax | (45) |
| Ischemia | E64c (ip, iv) E64d (ip) CA-074 (iv) CA-074Me (ip) |
nd | ↓ neuronal death, brain | ↓ CTSB ↓ MAP2 degrad. ↓ caspase 3 |
(104–109) |
| Trauma cerebral bleeding | CP-1 (iv) | ↓ motor deficits | ↓ neuronal death, brain | ↑ synaptophysin, brain | (110) |
| Excitatory Huntington’s disease | Z-FA-FMK (is) | nd | ↓ brain lesion | nd | (111) |
| Excitatory epilepsy | CA-074Me (ip) | ↑ learning ↑ neurolog. scores |
nd | ↓ CTSB ↓ LC3, ↓ beclin-1 ↓ bcl-2, ↓ PRG-1 |
(112) |
| Pain | CA-074Me (it) | ↓ inflammatory pain | nd | ↓ IL-1β, IL-18 | (113) |
| Inflammatory pain, edema | K11777 (ip) | ↓ inflammatory pain | ↓ edema ↓ necrosis |
↓ CTSB, CTSL, & CTSS ↓ c-fos |
(114) |
| Meningitis infection | CA-074Me (ip) | nd | ↑ clinical score | ↓ IL-1β | (68) |
Refs. indicate Reference citations
nd: not determined.
LTP, long-term potentiation; MS, multiple sclerosis; EAE, experimental atuoimmune encephalomyelitis; SCC, spinal cord contusion
AD model.
In an AD mouse model, APP/Lon mice, administration of E64d (oral) results in substantial improvement in memory deficits (92, 93), amelioration of amyloid plaque pathology, and reduction of brain levels of Aβ(1–40) and Aβ(1–42) as well as the truncated pGlu-Aβ(3–40) (pGlu, pyroglutamate) and pGlu-Aβ(3–42) peptides (92–94). E64d is a pro-drug which is converted in vivo by esterases to the active E64c inhibitor which potently inhibits cathepsin B (95, 96), as well as other cysteine cathepsin proteases (95, 97). It will be desirable to evaluate a selective inhibitor of cathepsin B to assess the role of this protease in the APP/Lon mice.
CA-074 is a potent and selective inhibitor of cathepsin B that can be used to evaluate cathepsin B inhibition in AD mice (APP/Lon) (98–100). This inhibitor is administered as the pro-drug form of CA-074Me, which is converted in vivo by esterases to the active CA-074 inhibitor (101). CA-074Me has greater membrane permeability than CA-074 and, therefore, enhanced penetration into cells. When administered (icv) to APP/Lon AD mice, memory deficits were improved, amyloid pathology was reduced, and Aβ(1–40) and Aβ(1–42) peptides in brains were reduced (93). The effectiveness of the CA-074 and E64c inhibitors to improve AD deficits in an animal model provides proof-of-concept that effective small molecule inhibitors targeting cathepsin B can be developed as candidate therapeutic agents for AD.
TBI models.
Investigation of the CCI model of TBI in mice examined the effectiveness of administering E64d (oral) immediately after CCI injury. E64d reduced the severity of motor dysfunction resulting from CCI and facilitated full recovery to normal motor function more rapidly than vehicle-treated controls (45). E64d reduced brain lesion volume resulting from CCI and reduced neuronal loss. E64d was effective when administered up to 8 hours after CCI, demonstrating a clinically plausible time period for acute therapeutic interventions. TBI trauma-induced deficits in cognitive and motor functions have also been improved by CA-074Me, as well as by the broad cysteine protease inhibitors z-DEVD-FMK and LHVS inhibitors (102, 103).
Related neurodegeneration and brain injury models.
Inhibitors of cathepsin B improve cognitive and behavioral deficits in animal models of various brain neurodegeneration and injuries which include ischemia, cerebral hemorrhage, excitatory Huntington’s disease, excitatory epilepsy, inflammatory pain, and meningitis (Table 3). In ischemia models, inhibitors of cathepsin B consisting of E64c, E64d, CA-074, and CA-074Me (administered intraperitoneally (ip) or intravenously (iv)) resulted in decreased neuronal death in the brain (104–109). In trauma of cerebral bleeding, motor deficits were improved with administration of the broad-acting cysteine protease inhibitor CP-1 (iv), accompanied with decreased neuronal cell death (110). In Huntington’s disease and excitatory epilepsy, memory and learning deficits are improved with Z-FA-FMK and CA-074Me inhibitors, respectively (111, 112). Pain is ameliorated by CA-074 (administered as CA-074Me), and inflammatory pain is improved with the inhibitor K11777, a cysteine protease inhibitor (74, 113). In addition, meningitis infection is improved by CA-074 assessed by clinical score (68).
Brain penetrance of cathepsin B inhibitors in animal model studies of AD and ischemia.
Information on the penetrance of cathepsin B inhibitors across the blood brain barrier has been demonstrated by inhibition of cathepsin B activity in the brain (92, 106). In AD mouse model studies, oral administration of E64d resulted in 80% inhibition of brain cathepsin B activity at 10 mg/kg E64d which was efficacious for improvement of memory deficits in the APPLon mice (92). E64d is a pro-drug which is converted in vivo by esterases to the active E64c inhibitor (92). In monkeys subjected to ischemic brain injury, administration of CA-074 or E64c inhibitors (4–5 mg/kg, iv) resulted in significant inhibition of cathepsin B activity in brain by 85–50% in sub-regions of the hippocampus and substantially reduced neuronal loss (106)”.
While inhibition of cathepsin B activity in brain by CA-074 or E64c was observed subsequent to peripheral administration (oral or iv) (92, 106), the brain concentrations of these these inhibitors have not been measured. It will be important in future studies to assess the pharmacokinetics of the concentrations of these inhibitors in the brain.
Overall, the findings of these studies indicate that inhibitors of cathepsin B, and inhibitors of cysteine cathepsins, improve behavioral deficits and ameliorate neuropathology in AD, TBI, and related brain disorders. Therefore, development of inhibitors of cathepsin B is a logical strategy for discovery of effective therapeutics for AD, TBI, and related brain dysfunctions.
5. Maturation of preprocathepsin B, member of the cysteine cathepsin protease family.
Cathepsin B is a lysosomal cysteine protease belonging to the clan CA and the papain-like family C1A. The other C1A papain-like cysteine proteases in humans are cathepsins C, F, H, K, L, O, S, V, W, and Z (also known as cathepsin X) (40, 43, 114). The cathepsin family also includes the aspartyl proteases cathepsins D and E, and the serine proteases cathepsins A and G. These cathepsins comprise the 15 protease members of lysosomal cathepsin proteases. Cathepsin B is among the most abundantly expressed cathepsins in the human brain (40).
Cathepsin B is synthesized as an inactive prepro-enzyme which undergoes sequential proteolytic processing for maturation of active cathepsin B (Figure 4a) (115, 116). The preprocathepsin is synthesized from its mRNA and undergoes removal of the NH2-terminal signal sequence (17 residues) in the rough endoplasmic reticulum (43, 114–116); the resultant procathepsin B is translocated to the Golgi apparatus and routed to lysosomes. Procathepsin B is processed by removal of the propeptide to form the mature, active enzyme (117–120). The cathepsin B enzyme can undergo additional processing to generate heavy and light chains with disulfide linkage.
Figure 4. Processing of preprocathepsin B zymogen to active cathepsin B.
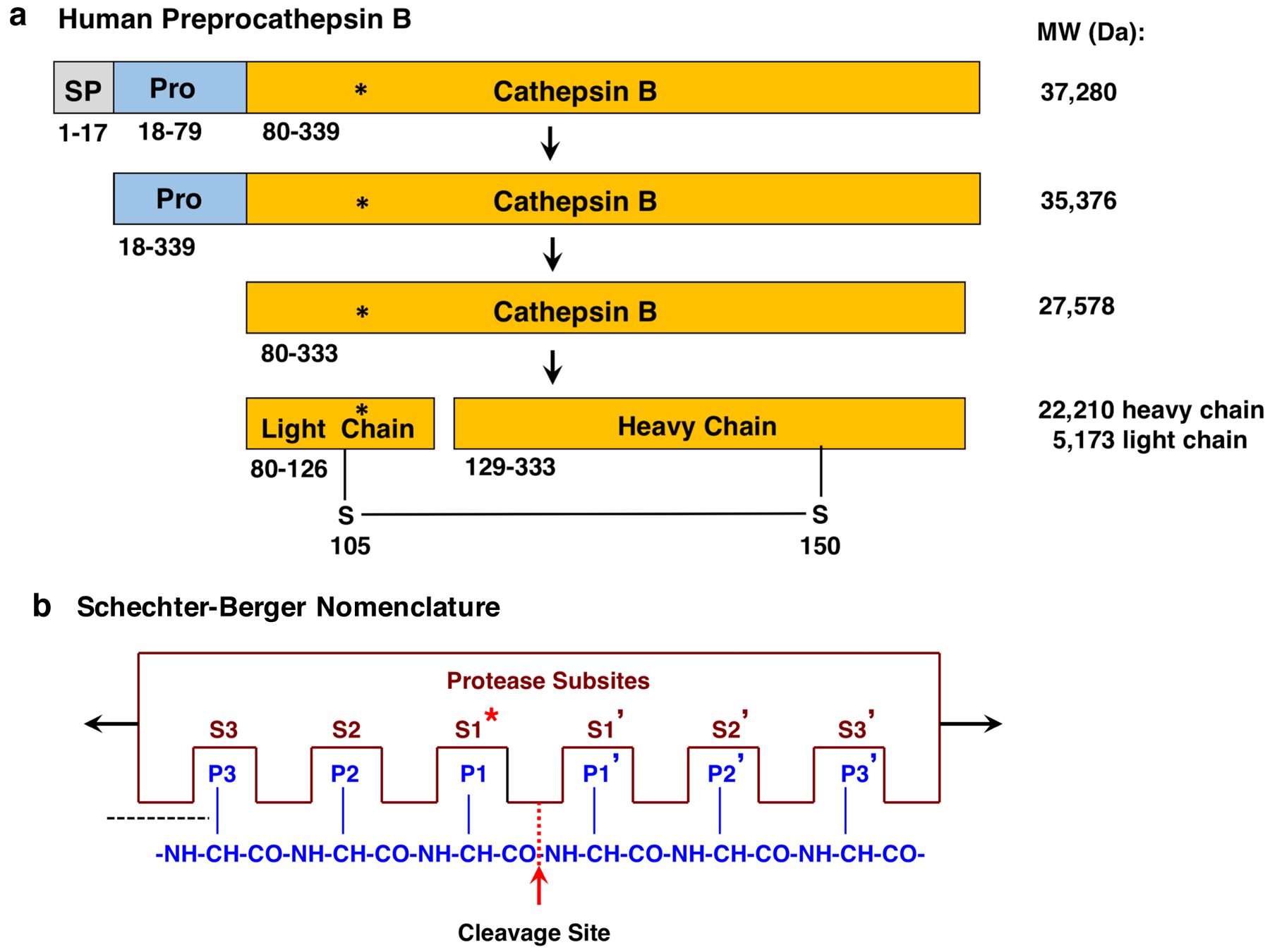
a. Preprocathepsin B processing to active protease. Preprocathepsin B (339 residues) (116) is translated from its mRNA at the rough endoplasmic reticulum (RER). The N-terminal signal sequence (SP, 17 residues) is removed at the RER by signal peptidase. The resultant procathepsin B (residues 18–333) is routed through the Golgi apparatus to lysosomes. The procathepsin B undergoes autoprocessing to remove the propeptide (Pro) to generate the active, mature cathepsin B (residues 80–333). Cathepsin B can undergo further processing into light and heavy chains linked by a disulfide bond. The active site Cys108 is indicated by the asterisk (*). Sequences of these forms of cathepsin B were obtained from the NCBI (196) and UniProt (197) protein sequence databases. It is noted that the NH2-terminal amino acid of mature cathepsin B (residue 80 of preprocathepsin B) may be referred as residue 1, with active site Cys29.
b. Schechter-Berger Nomenclature for protease and peptide interactions at the substrate cleavage site. According to the Schechter-Berger nomenclature (123), protease and substrate interactions are defined by the enzyme (i) non-prime subsites of S1, S2, etc. which are located at the N-terminal side of the cleaved scissile bond, and the (ii) prime subsites of S1’, S2’, etc. which are located at the C-terminal side of the cleaved peptide bond. Amino acid residues located at the cleaved scissile bond (P1-↓P1’) are represented by P1, P2, and Pn residues located at the C-terminal side of the cleavage site, and by P1’, P2’, and Pn’ residues located at the N-terminal side of the scissile bond. The active site Cys108, shown by the asterisk (*), resides in the S1 subsite of the enzyme.
Activation of procathepsin B occurs by removal of the propeptide to generate the mature active cathepsin B (43, 114–120). Procathepsin B can undergo autoprocessing since the proenzyme has low catalytic activity (117). Activation of procathepsin B has been shown to occur in unimolecular (117–119) and intermolecular mechanisms (120). Removal of the propeptide of procathepsin B occurs in one or several proteolytic steps which can involve dipeptidylpeptidase to generate the mature single-chain cathepsin B (117–121). Procathepsin B can also be activated by exogenous proteases consisting of the lysosomal cathepsin D and cathepsin L proteases (120), as well as by the serine proteases cathepsin G, elastase, and urokinase-type plasminogen activator (122). The mature cathepsin B can be further processed to the heavy chain and light chain (5 kDa) linked by disulfide bonds, which is enzymatically active (43, 114).
Cathepsin B possesses the active site Cys108 residue which mediates proteolysis (shown by asterisk * in Figure 4a). The Cys108 is located at the S1 subsite of the enzyme which interacts with the substrate at the P1-↓P1’ residues for cleavage of the peptide bond. The Schechter-Berger nomenclature (123) designates residues at the N-terminal side of the cleavage site as the non-prime (P1, P2, etc) residues, and prime (P1’, P2’, etc) residues are those located at the C-terminal side of the cleavage site (Figure 4b). Corresponding enzyme non-prime subsites (S1, S2, etc) interact with the non-prime amino acid side chains, and the prime subsites (S1’, S2’, etc) interact with the prime residues designated according to the Schechter-Berger nomenclature.
6. Selective CA-074 inhibitor of cathepsin B
Development of inhibitors of cathepsin B may lead to novel therapeutic agents for the treatment of the devastating AD, TBI, and related cognitive and behavioral deficits. Selectivity of inhibitors for cathepsin B compared to other members of the cysteine cathepsin family is important for targeted inhibition of cathepsin B without disturbing other related proteases and associated functions. Distinct structural features of cathepsin B have resulted in design and synthesis of the highly selective CA-074 inhibitor of cathepsin B, developed by the seminal research of Professor Nobuhiko Katunuma, renowned scientist in the field of proteolysis and their inhibitors (124).
Design and features of CA-074 selective inhibition of cathepsin B.
The CA-074 selective inhibitor of cathepsin B was developed by structural analyses of E64 and E-64c (98, 99, 125) which are irreversible inhibitors of cysteine cathepsins, as well as papain (98, 99, 125, 126,) and calpains (127). Crystal structure analysis of papain with E-64c and derivatives of E-64 (98, 99, 128) suggested that an L-trans-epoxy-succinic acid group would lead to a selective inhibitor of cathepsin B, and a series of epoxysuccinyl peptides were designed, synthesized, and evaluated (98). Novel L-trans-epoxysuccinyl peptides were identified as potent and selective inhibitors of cathepsin B, consisting of the inhibitors CA-074 (N-(L-3-trans-propyl-carbamoyloxirane-2-carbonyl)-L-isoleucyl-L-proline) and CA-030 (N-(L-3-trans-ethoxycarbonyloxirane-2-carbonyl)-L-isoleucyl-L-proline) (98). CA-074 and CA-030 display potent Ki values of 2–5 nM for inhibition of cathepsin B (98, 99). Both inhibitors were highly selective for cathepsin B, indicated by the high Ki values of these inhibitors for cathepsins L and H at 40 μM to 200 μM (98, 99). In vivo studies showed that CA-074 selectively inhibited cathepsin B in rat liver (99); however, CA-030 was not selective in the in vivo studies (99).
These results identified CA-074 as a highly selective and potent inhibitor of cathepsin B (98, 99, 129). CA-074 and its methylated form CA-074Me have been extensively utilized to examine the roles of cathepsin B in human disease models in cells and animals, including AD, TBI, and related animal models (Table 3). CA-074Me has improved cellular penetration and is converted by abundant cellular esterases to CA-074 (101). While CA-074Me has been found to inhibit both cathepsin B and cathepsin L, its conversion by CA-074 in situ supports the use of this inhibitor to investigate cathepsin B in biological functions.
CA-074 interactions with cathepsin B.
The binding interactions CA-074 for inhibition of cathepsin B has been analyzed by the x-ray crystal structure of the CA-074 and cathepsin B complex (100), illustrated by the Molecular Operating Environment (MOE) platform for visualization and modeling of molecules to protein structures (130, 131) (shown in Figure 5). Cathepsin B (mature) possesses the catalytic active site residue Cys29 which is part of an extended substrate binding groove that is mostly hydrophobic with some polar regions (Figure 5a). Binding of CA-074 to cathepsin B subsites utilizes the Schechter-Berger nomenclature (123) for interactions with the enzyme prime (S1’, S2’, etc.) and non-prime (S1, S2, etc.) subsites (Figure 4b).
Figure 5. Interaction of the CA-074 inhibitor to the active site pocket of cathepsin B.
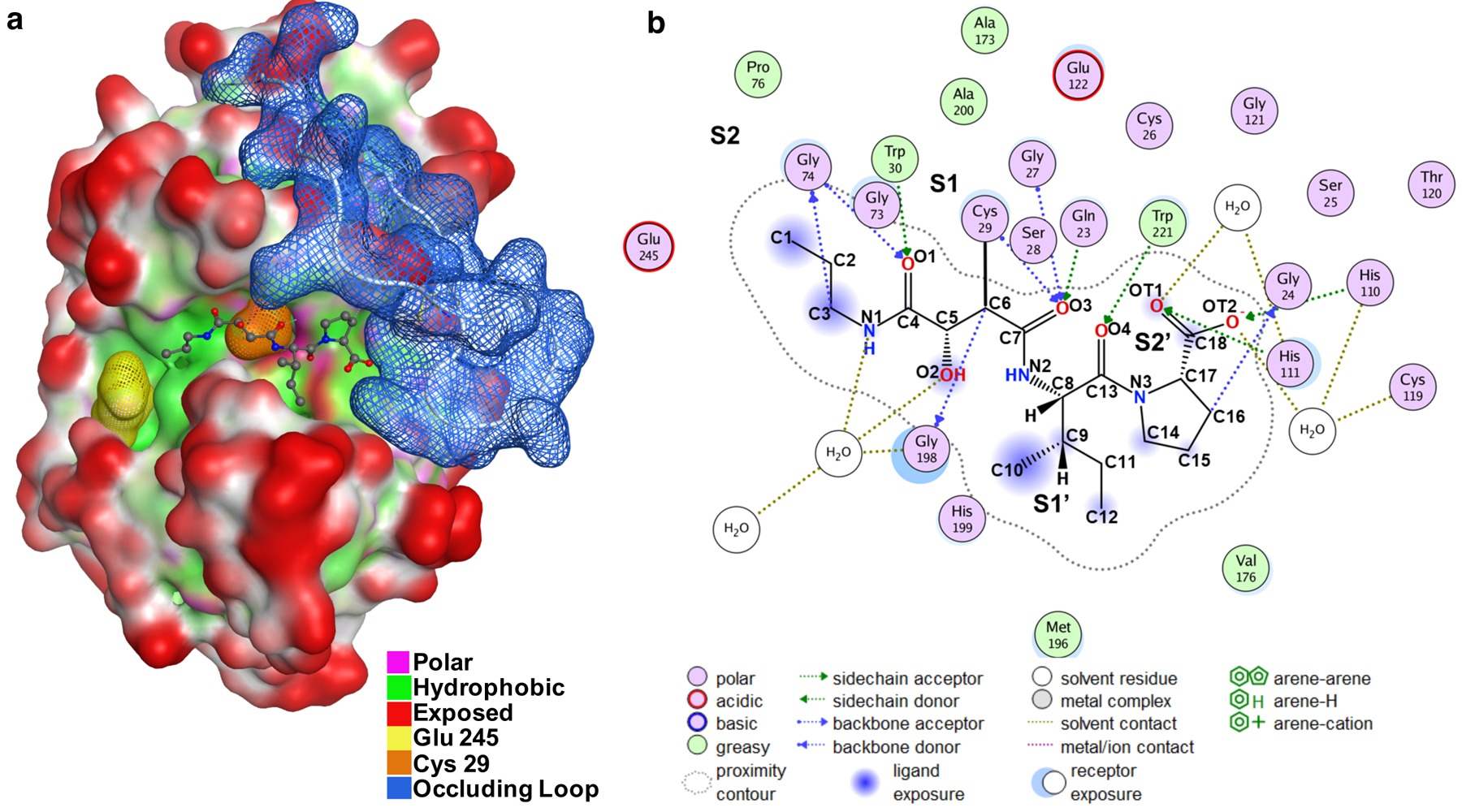
a. Structure of the cathepsin B/CA-074 complex. The structure of bovine cathepsin B complexed with CA-074 has been determined by x-ray crystallography by Yamamoto et al., 2000 (100); the PDB ID of this structural data of cathepsin B complexed with CA-074 is ID 1QdQ. This structural data was utilized by us with the Molecular Operating Environment (MOE) (130, 131) for visualization of the interacting features of CA-074 with the cathepsin B structure. The catalytic residue Cys29 (orange) is part of an extended substrate binding groove that is mostly hydrophobic (green) with some polar regions (pink). Using the Schechter and Berger nomenclature (123), Glu245 (yellow) sits in the S2 pocket and the occluding loop blue is orientated close to the S2ʹ pocket. The CA-074 inhibitor is orientated in the substrate binding groove and shown to extend from the S2 to S2ʹ pockets.
b. Illustration of two-dimensional (2D) CA-074 inhibitor and cathepsin B interactions. Interactions of CA-074 with cathepsin B (bovine) were determined from their crystal structure (100); CA-074 interacts with S2’ to S2 subsites of the enzyme. The S2’ subsite interacts with the C-terminal Pro of CA-074 with His110 and His111 of the occluding loop by hydrogen bonding; the S2’ subsite involves Gln23, Gly24, His110, His111 of the occluding loop domain of cathepsin B. The occluding loop is located in manner to block substrate accessibility to Sn’ subsites (n > 3). The S1’ subsite interacts with the Ile of the Ile-Pro C-terminal region of CA-074; the S1’ site is composed of a hydrophobic core of Val176, Leu181, Met196, His199, and Trp221 which holds the Ile side chain of CA-074 through hydrogen bonding with Trp221. The S1 subsite contains the active site Cys29 which forms a covalent bond with the C6 oxirane carbon of CA-074; the S1 pocket is composed of Cys29, Gly27, Gly74, and Gly198. The S2 subsite consists of Glu245, Pro76, Ala173, and Ala200 which form a hydrophobic pocket which accommodates the propyl chain of CA-074. These features of CA-074 covalent linkage to enzyme and interactions with the substrate binding pocket in the vicinity of the occluding loop provide the basis for selective CA-074 inhibition of cathepsin B.
The CA-074 inhibitor is oriented in the substrate active site binding groove of cathepsin B, extending from the S2 to S2’ pockets of the enzyme (Figure 5a, b). CA-074 is covalently bound by its C6 oxirane atom to the Cys-29 active site residue (S1 subsite) of cathepsin B for irreversible inhibition (100). The S2 site of the enzyme interacts with the propyl chain of CA-074 and involves Glu245 at the S2 subsite. The Ile-Pro-OH at the C-terminus of CA-074, important for selective inhibition, binds to the S1’ and S2’ subsites. The C-terminal proline of CA-074 interacts with His110 and Hs111 of the occluding loop of cathepsin B, which participates in specific inhibition of cathepsin B. Overall, CA-074 covalently binds to the active site Cys-29, with the inhibitor situated at S2 to S2’ sites near the occluding loop of the enzyme.
7. Regulation of cathepsin B proteolysis by its distinct occluding loop
Cathepsin B uniquely possesses the occluding loop compared to other cysteine cathepsins (132–137). The occluding loop is located near the enzyme’s substrate binding pocket containing the active site Cys-29 residue (100, 132) (Figure 5) which catalyzes endopeptidase and dipeptidyl carboxypeptidase activities. Thus, these activities are influenced by the occluding situated near the primed subsites which favor binding of peptide substrates with two residues carboxy-terminal to the cleaved scissile peptide bond. The His-110 and His-111 residues of the occluding loop provide positive charged anchors for the C-terminal carboxylate group of peptide substrates, which explains the dipeptidyl carboxypeptidase activity of cathepsin B. The occluding loop blocks substrate binding to the Sn’ (n ≥ 3) subsites of the protease.
The role of the occluding loop for endopeptidase compared to exopeptidase activities of cathepsin B has been assessed by deletion mutagenesis of a central 12-residue domain of the loop (133). The deletion mutant lacked exopeptidase activity (monitored with dansyl-Phe-Arg-Phe(NO2)-Leu substrate). The mutant proenzyme was autoprocessed to yield the mature enzyme with endopeptidase activity (monitored with Z-Phe-Arg-AMC substrate) comparable to wild-type cathepsin B. These findings demonstrate that the occluding loop restricts access to the active site and endows the dipeptidyl carboxypeptidase activity of cathepsin B.
Further evidence for regulation of endopeptidase activity by the occluding loop was illustrated by site-directed mutagenesis of selected residues within the loop. Such mutations resulted in increased endopeptidase activity (134). The triple mutant D22A/H110A/R116A resulted in the greatest increase in endopeptidase activity of 600-fold increase compared to WT cathepsin B (endopeptidase Abz-AFRSAAQ-Eddnp substrate was used). These findings show that the increase in endopeptidase activity of the mutant may reflect an increase in loop flexibility when an endopeptidase substrate binds to the enzyme.
The occluding loop regulates the pH dependence of propeptide inhibition of cathepsin B endopeptidase activity (135). At pH 6.0, propeptide inhibition had a Ki of 3.7 nM, and at pH 4.0 the propeptide was less effective with a Ki of 82 nM. The pH dependence of inhibition was eliminated by deletion mutagenesis of a segment of the occluding loop, and by the His110Ala mutation in the loop which increased the potency of propeptide inhibition to Ki of 2 nM at pH 4.0, similar to the nM Ki for propeptide inhibition at pH 6.0. These findings indicate that the occluding loop regulates the pH dependence of propeptide inhibition of cathepsin B (132, 134–136).
The occluding loop of cathepsin B can be displaced by the stefin protein inhibitors (137). The crystal structure of the complex of human cathepsin B and human stefin A showed that the occluding loop residues were displaced. These findings indicate that the occluding loop can exist in different positions and may become displaced by stefin or other endogenous protein inhibitors.
Clearly, the occluding loop distinguishes cathepsin B from other cysteine cathepsins. These structurally distinct features of the occluding loop provides the rationale for development of selective inhibitors of cathepsin B, as shown by CA-074 selective inhibition of cathepsin B.
8. Lysosomal leakage of cathepsin B to the cytosol in cell death and inflammation of AD, TBI, and related brain disorders.
Abnormal lysosomal leakage of cathepsin B into the cytosol of cells occurs as an integral mechanism for cell death and inflammation in AD, TBI, and related brain disorders. Cathepsin B and cathepsin proteases are normally sequestered within lysosome organelles for homeostasis by degradation cellular proteins into amino acids (138, 139). In AD, TBI, and related brain disorders, lysosomal membranes lose their integrity to result in lysosomal leakage and redistribution of cathepsin B to the cytosol (illustrated in Figure 6) (140–142). Notably, cytosolic cathepsin B activates cell death (143–147) and inflammatory (148–152) pathways which participate in AD, TBI, and related disorders (153–159). Cytosolic cathepsin B directs caspase-dependent cell death through cleavage of bid and anti-apoptotic Bcl-2 homologues, and removal of apoptosis-preventing Bcl-xl (143, 144). Furthermore, cytosolic cathepsin B increases inflammatory IL-1β cytokine production via activation of the neuroinflammasome (148–152). Lysosomal leakage results in pathogenic, cytosolic of cathepsin B in neurodegeneration of AD, TBI, and related brain disorders.
Figure 6. Lysosomal leakage results in cytosolic cathepsin B and activation of cell death neurodegeneration and inflammation in AD, TBI, and related.
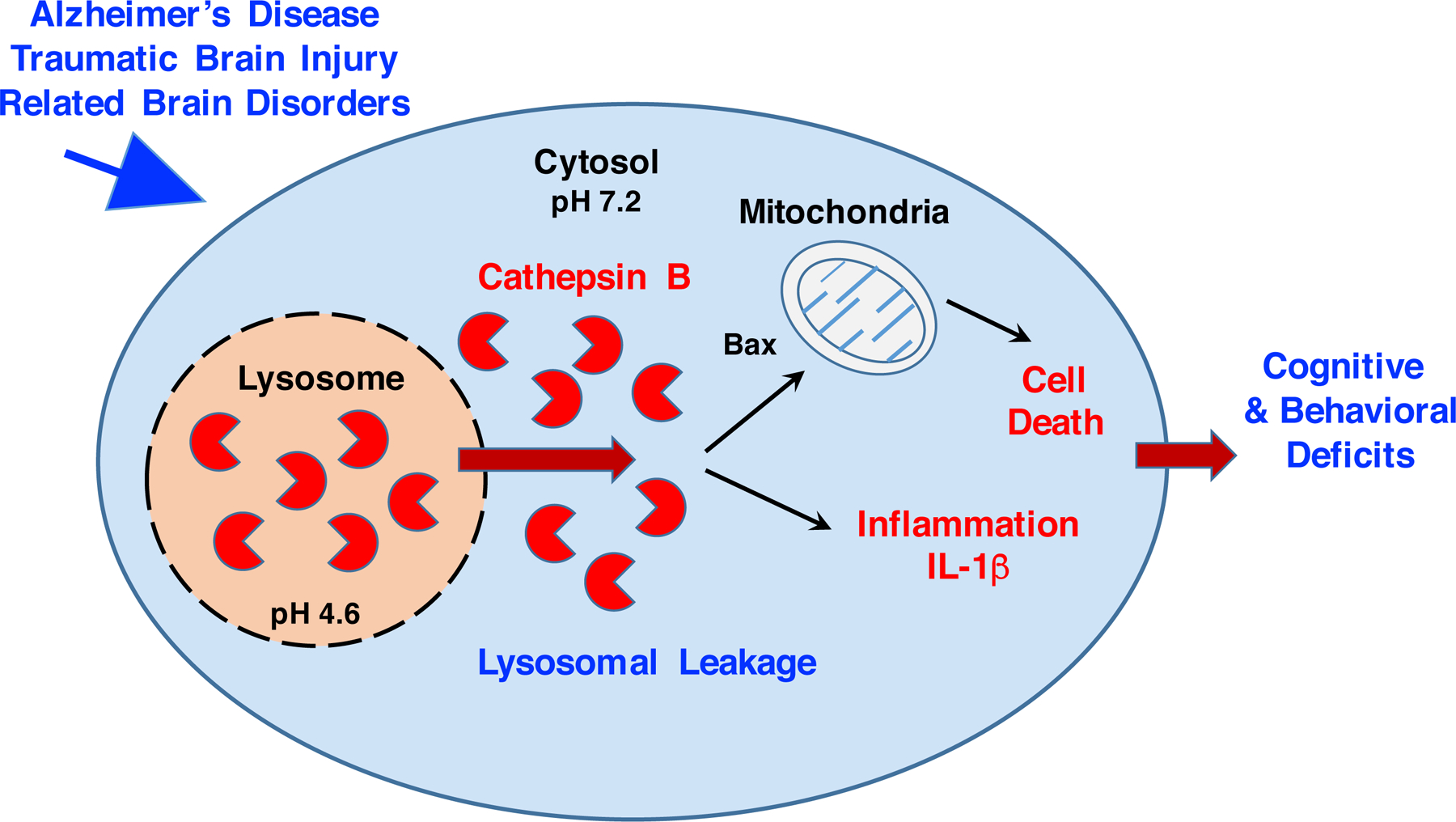
Lysosomal leakage occurs in AD (160–163), TBI (166, 167), and related brain disorders (168–176). Lysosomal leakage results from membrane permeabilization, resulting in relocation of cathepsin B from the lysosome to the cytosol. Cathepsin B is active at the neutral pH 7.2 of the cytosol (184–186), which differs from the acidic pH 4.6 within lysosomes (180–183). Cathepsin B proteolysis of cytosolic substrates initiates apoptotic cell death (143–146) and activation of IL-1β production in inflammation (148–155).
AD and lysosomal leakage.
In AD, oligomeric Aβ42 induces lysosomal leakage and relocation of lysosomal enzymes, including cathepsin B, to the cytosol to result in cell death of neuronal cells (160–163). Oligomers of pGlu-Aβ(3–42) also induce lysosomal leakage and apoptosis (162). The N-truncated and modified pGlu-Aβ(3–42) peptide increases in the aging AD brain and facilitates seeding of oligomeric Aβ peptide neurotoxicity (78, 79).
The Aβ42 induction of lysosomal leakage and cell death is exacerbated by ApoE4 (apolipoprotein E4), the major genetic risk factor for AD (164, 165). In ApoE4 transgenic mice, elevation of Aβ brain levels, achieved by inhibition of the Aβ-degrading enzyme neprilysin, results in cognitive deficits (165). These findings link oligomeric Aβ with lysosomal leakage and behavioral cognitive dysfunction.
TBI and TBI-related lysosomal leakage.
Numerous studies demonstrate that TBI and related neurodegenerative disorders result in lysosomal leakage of cathepsin B leading to cell death and neuronal damage (166–176). TBI modeled by the central fluid percussion injury (CFPI) model of TBI (in rats) results in elevation of intracranial pressure (ICP) (166). During increased ICP, brain neurons display a redistribution of cathepsin B from the lysosome to the cytosol, with mechanoporation of neurons, observed at 6 hours after CFPI. Mechanical injury to neurons in culture results in immediate cathepsin B release from the lysosome and increased cell death accompanied by increased tBid and release of cytochrome c from mitochondria (167); inhibition of cathepsin B improved cell viability.
TBI results in vascular disruption and brain ischemia. Lysosomal membrane permeabilization occurs in ischemia, indicated by cathepsin B release from lysosomes at 2 hours after ischemia in brain hippocampus (ischemia induced by oxygen-glucose deprivation) (168). Ischemia produced by middle cerebral artery occlusion results in lysosomal leakage of cathepsin B to the cytosol in 30–45% of neurons (coronal sections) at 1–4 hours after ischemia (169). The cytosolic cathepsin B was co-localized with caspase-3, tBid, and cytochrome-c of the apoptotic pathway.
Parkinson’s disease (PD) neurodegeneration can be modeled in mice and cells by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) treatment (170–172). The MPTP-treated mice display lysosomal membrane permeability and leakage of cathepsins to the cytoplasm, triggering caspase-dependent apoptotic cell death (172). PD brains accumulate neurotoxic α-synuclein aggregates in Lewy bodies (173, 174), and such a-synuclein aggregates induce lysosomal rupture combined with induction of cathepsin B-dependent increase in reactive oxygen species (ROS) and inflammation (175). It is hypothesized that the pathogenesis of PD involves α-synuclein and lysosomal dysfunction in neuronal cell death (176).
9. Hypothesis for pathogenic, cytosolic cathepsin B in AD and TBI-related neurodegeneration.
Translocation of lysosomal cathepsin B to the cytosol in neurodegeneration.
Lysosomal leakage results in the pathogenic redistribution of cathepsin B from the lysosome to the cytosol in neurodegeneration, in contrast to the normal intra-lysosomal location of cathepsin B for protein degradation and homeostasis, which leads to the hypothesis that abnormal cytosolic cathepsin B participates in AD and TBI-related dysfunctions (illustrated in Figure 6). The key question is what are the cytosolic substrates of cathepsin B that lead to neurodegeneration? The profiles of cytosolic substrates of cathepsin B in brain have not yet been determined. However, it is known that cytosolic cathepsin B cleaves Bid to generate tBid which leads to mitochondrial cytochrome c release and apoptosis. Also important is the role of cytosolic cathepsin B in regulating the neuroinflammasome for activation of IL-1β production in inflammation. Neurodegeneration resulting from cell death and inflammation leads to deficits in cognition, memory, thinking, executive functions, and related behaviors.
With respect to the mechanism of lysosomal leakage, studies have demonstrated involvement of the calpain cysteine protease which cleaves carbonylated Hsp70.1 to result in lysosomal permeabilization and release of lysosomal cathepsin proteases (141, 177–179). The HSP70.1 chaperone functions as a guardian of lysosomal integrity. The role of calpain in lysosomal rupture and the redistribution of cathepsin proteases suggests the dual participation of ‘calpain-cathepsin’ in neurodegeneration (141, 177–179). The consequence of lysosomal permeabilization is the translocation of lysosomal cathepsin B to the cytosol to initiate cell death and inflammation occurring in neurodegeneration of AD, TBI, and related brain injuries.
Cathepsin B is active at the neutral pH of the cytosol.
The redistribution of cathepsin B to the cytosol changes its normal acidic pH environment of lysosomes (pH 4.6) (180–183) to the neutral pH of the cytosol (pH 7.2) (184–186). Despite the large change in proton concentration at cytosolic pH 7.2 compared to lysosomal pH 4.6, cathepsin B retains proteolytic activity at neutral pH (187–189). Furthermore, the stability of cathepsin B at alkaline pH can be enhanced by binding to heparin of membranes to potentiate the endopeptidase activity at neutral pH (189).
The large 400-fold difference in proton concentration from lysosomal pH 4.6 to cytosolic pH 7.2 results in an altered charge state of cathepsin B (Table 4). Mature cathepsin B has estimated charge states of +9 at pH 4.6, and −10.3 at pH 7.2 (calculated using protein calculator 3.4, http://protcalc.sourceforge.net/). These distinct charge states can result in different protein-substrate interactions with cathepsin B at the cytosol compared to lysosomal pH.
Table 4.
Changes in cathepsin B charge states at cytoslic compared to lysosomal pH conditons
| Enzyme Domain | Charge State: | |
|---|---|---|
| pH 4.6 (lysosome) | pH 7.2 (cytosol) | |
| Cathepsin B, mature | + 9.5 | − 10.3 |
| Occluding loop | + 2.2 | − 0.9 |
Estimated charge states for human cathepsin B and its occluding loop domain were calculated using Protein Calculator 3.4 (http://protcalc.sourceforge.net/)
Furthermore, the occluding loop may possess very different charge states at lysosomal compared to cytosolic pH. The occluding loop has an estimated charge state of +2.2 at lysosomal pH 4.6, which contrasts with a predicted charge state of −0.9 at cytosolic pH 7.2. These different charge states of the occluding loop can influence substrate binding to the active site pocket at Sn’ sites located near the occluding loop and modulate proteolysis.
Cytosolic cathepsin B and cell death.
Lysosomal leakage and release of cathepsin B to the cytosol initiates apoptosis by proteolysis of Bcl-2 family members Bcl-xl, Bax, and Bid (143), resulting in apoptosis-inducing Bid and removes apoptosis-preventing Bcl-Xl. Furthermore, apoptosis-promoting Bax is elevated through cathepsin B proteolysis of a Bax-degrading protease.
The E-64d inhibitor of cathepsin B, as well as cysteine proteases, prevents apoptosis, Bid cleavage, and degradation of the anti-apoptotic Bcl-2, BCl-xL, and Mcl-1 (144). A pan caspase inhibitor (N-benzyloxycarbonyl-Val-Ala-Asp9OMe)fluoromethyl ketone) prevents apoptosis, but has no effects on cathepsin B cleavage of Bid to tBid, indicating that caspase-mediated apoptosis occurs downstream of cathepsin B. Production of tBid leads to its mitochondrial binding to BAK to release cytochrome c in the apoptotic pathway (146). These findings illustrate the role of cathepsin B in the regulation of anti-apoptotic Bcl-2 members and Bid to trigger the mitochondrial pathway to apoptosis.
Cytosolic cathepsin B and inflammation.
Cathepsin B in the cytosol activates the neuroinflammasome to elevate IL-1β production (68, 71, 73,148–152, 190, 191). The NLRP3 inflammasome in microglia participates in AD and related neurodegenerative diseases (148–152). Oligomeric Aβ (16–20) and oxidative stress (150, 192–195) of AD and neurodegeneration, induce cathepsin B-mediated activation of the NLRP3 inflammasome to initiate processing of pro-caspase-1 to caspase-1, and subsequent caspase-1 conversion of pro-IL-1β to active IL-1β inflammatory factor (71, 73, 150). The detailed mechanism of how cathepsin B mediates activation of the NLRP3 inflammasome complex to activate IL-1β production has not yet been defined.
10. Conclusions and future perspectives
The hypothesis that lysosomal leakage of pathogenic cathepsin B to the cytosol results in neurodegeneration and behavioral deficits of AD, TBI, and related brain disorders (Figure 6) is supported by ample evidence from clinical and animal model studies of AD, TBI, and related brain disorders. This leads to the question of what are the cytosolic substrates of cathepsin B that lead to neurodegeneration? Current data implicate cathepsin B substrates involved in cell death and inflammatory pathways. It will be advantageous to advance understanding of the distinct functions of cytosolic cathepsin B contributing to neurodegeneration and brain dysfunctions of AD, TBI, and related disorders. Such findings may lead to novel targets for drug discovery to address the unmet need for therapeutics to treat these neurodegenerative diseases.
Highlights.
Cathepsin B is elevated in human Alzheimer’s disease (AD) and traumatic brain injury (TBI) patients, and correlates with behavioral and injury outcomes
Cathepsin B gene expression in human AD brains is prevalent in numerous regions throughout young to adult ages
Elevated cathepsin B occurs in animal models of AD, TBI, and related brain disorders
Inhibition of cathepsin B in AD and TBI animal models results in alleviation of cognitive and behavioral deficits with improvement in neuropathology
Preprocathepsin B undergoes processing to generate the active, mature cathepsin B
Selective inhibition of cathepsin B by CA-074 via irreversible linkage to the active site of cathepsin B
The occluding loop of cathepsin B regulates dipeptidylcarboxypeptidase and endopeptidase activities of cathepsin B
Lysosomal leakage of cathepsin B to the cytosol in cell death and inflammation of AD, TBI, and related brain disorders
Acknowledgments
This work was supported by NIH grant R01NS109075 to VH, and NIH grant R41NS110147 to GH. The authors thank Michael Gilson and the Center for Drug Discovery Innovation (cDDI) at UC San Diego for advice and resources for the Molecular Operating Environment (MOE) analyses.
V. Hook organized manuscript for participation by coauthors and wrote the manuscript. MY, CM, and AJO conducted the MOE representation of the cathepsin B structure complexed with the CA-074 inhibitors, and conducted literature analyses for this manuscript. SP conducted the analyses of cathepsin B and lysosomal proteases in human brain using the Allen Brain Atlas resource. GI contributed to extensive literature searches for this manuscript. BH and RR contributed current knowledge of AD and TBI research in the field. GH contributed to the outline of topics for inclusion in this manuscript and their integration for this manuscript. V. Hook and G. Hook have equity positions at American Life Science Pharmaceuticals, Inc.
V. Hook’s conflict has been disclosed and is managed by her employer, the University of California, San Diego. The other authors have no conflicts of interest.
Footnotes
Declaration of interests
The majority of authors (MC, CM, GI, SP, BH, RR, AO) declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Montine TJ, Koroshetz WJ, Babcock D, Dickson DW, Galpern WR, Glymour MM, Greenberg SM, Hutton ML, Knopman DS, Kuzmichev AN, Manly JJ, Marder KS, Miller BL, Phelps CH, Seeley WW, Sieber BA, Silverberg NB, Sutherland M, Torborg CL, Waddy SP, Zlokovic BV, Corriveau RA; ADRD 2013 Conference Organizing Committee. Recommendations of the Alzheimer’s disease-related dementias conference. Neurology. 2014. August 26;83(9):851–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corriveau RA, Koroshetz WJ, Gladman JT, Jeon S, Babcock D, Bennett DA, Carmichael ST, Dickinson SL, Dickson DW, Emr M, Fillit H, Greenberg SM, Hutton ML, Knopman DS, Manly JJ, Marder KS, Moy CS, Phelps CH, Scott PA, Seeley WW, Sieber BA, Silverberg NB, Sutherland ML, Taylor A, Torborg CL, Waddy SP, Gubitz AK, Holtzman DM. Alzheimer’s Disease-Related Dementias Summit 2016: National research priorities. Neurology. 2017. December 5;89(23):2381–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aisen PS, Cummings J, Jack CR Jr, Morris JC, Sperling R, Frölich L, Jones RW, Dowsett SA, Matthews BR, Raskin J, Scheltens P, Dubois B. On the path to 2025: understanding the Alzheimer’s disease continuum. Alzheimers Res Ther. 2017. August 9;9(1):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramalho J, Castillo M. Dementia resulting from traumatic brain injury. Dement Neuropsychol. 2015. Oct-Dec;9(4):356–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brayne C. Traumatic brain injury and dementia. Lancet Psychiatry. 2018. May;5(5):383–384. [DOI] [PubMed] [Google Scholar]
- 6.LoBue C, Munro C, Schaffert J, Didehbani N, Hart J, Batjer H, Cullum CM. Traumatic Brain Injury and Risk of Long-Term Brain Changes, Accumulation of Pathological Markers, and Developing Dementia: A Review. J Alzheimers Dis. 2019;70(3):629–654. [DOI] [PubMed] [Google Scholar]
- 7.Mijajlović MD, Pavlović A, Brainin M, Heiss WD, Quinn TJ, Ihle-Hansen HB, Hermann DM, Assayag EB, Richard E, Thiel A, Kliper E, Shin YI, Kim YH, Choi S, Jung S, Lee YB, Sinanović O, Levine DA, Schlesinger I, Mead G, Milošević V, Leys D, Hagberg G, Ursin MH, Teuschl Y, Prokopenko S, Mozheyko E, Bezdenezhnykh A, Matz K, Aleksić V, Muresanu D, Korczyn AD, Bornstein NM. Post-stroke dementia - a comprehensive review. BMC Med. 2017. January 18;15(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corraini P, Henderson VW, Ording AG, Pedersen L, Horváth-Puhó E, Sørensen HT. Long-Term Risk of Dementia Among Survivors of Ischemic or Hemorrhagic Stroke. Stroke. 2017. January;48(1):180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Charidimou A, Shams S, Romero JR, Ding J, Veltkamp R, Horstmann S, Eiriksdottir G, van Buchem MA, Gudnason V, Himali JJ, Gurol ME, Viswanathan A, Imaizumi T, Vernooij MW, Seshadri S, Greenberg SM, Benavente OR, Launer LJ, Shoamanesh A; International META-MICROBLEEDS Initiative. Clinical significance of cerebral microbleeds on MRI: A comprehensive meta-analysis of risk of intracerebral hemorrhage, ischemic stroke, mortality, and dementia in cohort studies (v1). Int J Stroke. 2018. July;13(5):454–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gratwicke J, Jahanshahi M, Foltynie T. Parkinson’s disease dementia: a neural networks perspective. Brain. 2015. June;138(Pt 6):1454–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gomperts SN. Lewy Body Dementias: Dementia With Lewy Bodies and Parkinson Disease Dementia. Continuum (Minneap Minn). 2016. April;22(2 Dementia):435–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olney NT, Spina S, Miller BL. Frontotemporal Dementia. Neurol Clin. 2017. May;35(2):339–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Couratier P, Corcia P, Lautrette G, Nicol M, Marin B. ALS and frontotemporal dementia belong to a common disease spectrum. Rev Neurol (Paris). 2017. May;173(5):273–279. [DOI] [PubMed] [Google Scholar]
- 14.Ballard C, Mobley W, Hardy J, Williams G, Corbett A. Dementia in Down’s syndrome. Lancet Neurol. 2016. May;15(6):622–36. [DOI] [PubMed] [Google Scholar]
- 15.Whitlock EL, Diaz-Ramirez LG, Glymour MM, Boscardin WJ, Covinsky KE, Smith AK. Association Between Persistent Pain and Memory Decline and Dementia in a Longitudinal Cohort of Elders. JAMA Intern Med. 2017. August 1;177(8):1146–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002. July 19;297(5580):353–6. [DOI] [PubMed] [Google Scholar]
- 17.Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M. Alzheimer’s disease: clinical trials and drug development. Lancet Neurol. 2010. July;9(7):702–16. [DOI] [PubMed] [Google Scholar]
- 18.Sperling RA, Jack CR Jr, Aisen PS. Testing the right target and right drug at the right stage. Sci Transl Med. 2011. November 30;3(111):111cm33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schilling S, Rahfeld JU, Lues I, Lemere CA. Passive Aβ Immunotherapy: Current Achievements and Future Perspectives. Molecules. 2018. May 3;23(5). pii: E1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ricciarelli R, Fedele E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr Neuropharmacol. 2017;15(6):926–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khanna MR, Kovalevich J, Lee VM, Trojanowski JQ, Brunden KR. Therapeutic strategies for the treatment of tauopathies: Hopes and challenges. Alzheimers Dement. 2016. October;12(10):1051–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li C, Götz J. Tau-based therapies in neurodegeneration: opportunities and challenges. Nat Rev Drug Discov. 2017. December;16(12):863–883. [DOI] [PubMed] [Google Scholar]
- 23.Congdon EE, Sigurdsson EM. Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol. 2018. July;14(7):399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cummings J, Aisen PS, DuBois B, Frölich L, Jack CR Jr, Jones RW, Morris JC, Raskin J, Dowsett SA, Scheltens P. Drug development in Alzheimer’s disease: the path to 2025. Alzheimers Res Ther. 2016. September 20;8:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loera-Valencia R, Cedazo-Minguez A, Kenigsberg PA, Page G, Duarte AI, Giusti P, Zusso M, Robert P, Frisoni GB, Cattaneo A, Zille M, Boltze J, Cartier N, Buee L, Johansson G, Winblad B. Current and emerging avenues for Alzheimer’s disease drug targets. J Intern Med. 2019. October;286(4):398–437. [DOI] [PubMed] [Google Scholar]
- 26.Becker RE, Kapogiannis D, Greig NH. Does traumatic brain injury hold the key to the Alzheimer’s disease puzzle? Alzheimers Dement. 2018. April;14(4):431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kochanek PM, Jackson TC, Jha RM, Clark RSB, Okonkwo DO, Bayır H, Poloyac SM, Wagner AK, Empey PE, Conley YP, Bell MJ, Kline AE, Bondi CO, Simon DW, Carlson SW, Puccio AM, Horvat CM, Au AK, Elmer J, Treble-Barna A, Ikonomovic MD, Shutter LA, Taylor DL, Stern AM, Graham SH, Kagan VE, Jackson EK, Wisniewski SR, Dixon CE. Paths to Successful Translation of New Therapies for Severe Traumatic Brain Injury in the Golden Age of Traumatic Brain Injury Research: A Pittsburgh Vision. J Neurotrauma. 2019. February 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sundelöf J, Sundström J, Hansson O, Eriksdotter-Jönhagen M, Giedraitis V, Larsson A, Degerman-Gunnarsson M, Ingelsson M, Minthon L, Blennow K, Kilander L, Basun H, Lannfelt L. Higher cathepsin B levels in plasma in Alzheimer’s disease compared to healthy controls. J Alzheimers Dis. 2010;22(4):1223–30. [DOI] [PubMed] [Google Scholar]
- 29.Sun Y, Rong X, Lu W, Peng Y, Li J, Xu S, Wang L, Wang X. Translational study of Alzheimer’s disease (AD) biomarkers from brain tissues in AβPP/PS1 mice and serum of AD patients. J Alzheimers Dis. 2015;45(1):269–82. [DOI] [PubMed] [Google Scholar]
- 30.Morena F, Argentati C, Trotta R, Crispoltoni L, Stabile A, Pistilli A, di Baldassarre A, Calafiore R, Montanucci P, Basta G, Pedrinolla A, Smania N, Venturelli M, Schena F, Naro F, Emiliani C, Rende M, Martino S. A Comparison of Lysosomal Enzymes Expression Levels in Peripheral Blood of Mild-and Severe-Alzheimer’s Disease and MCI Patients: Implications for Regenerative Medicine Approaches. Int J Mol Sci. 2017. August 19;18(8). pii: E1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cataldo AM, Nixon RA. Enzymatically active lysosomal proteases are associated with amyloid deposits in Alzheimer brain. Proc Natl Acad Sci U S A. 1990. May;87(10):3861–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang J, Goodlett DR, Quinn JF, Peskind E, Kaye JA, Zhou Y, Pan C, Yi E, Eng J, Wang Q, Aebersold RH, Montine TJ. Quantitative proteomics of cerebrospinal fluid from patients with Alzheimer disease. J Alzheimers Dis. 2005. April;7(2):125–33; discussion 173–80. Erratum in: J Alzheimers Dis. 2006 Mar;9(1):81–8. [DOI] [PubMed] [Google Scholar]
- 33.Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, Bohr VA. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019. October;15(10):565–581. [DOI] [PubMed] [Google Scholar]
- 34.Hara Y, McKeehan N, Fillit HM. Translating the biology of aging into novel therapeutics for Alzheimer disease. Neurology. 2019. January 8;92(2):84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nilsson E, Bodolea C, Gordh T, Larsson A. Cerebrospinal fluid cathepsin B and S. Neurol Sci. 2013. April;34(4):445–8. [DOI] [PubMed] [Google Scholar]
- 36.Assfalg-Machleidt I, Jochum M, Nast-Kolb D, Siebeck M, Billing A, Joka T, Rothe G, Valet G, Zauner R, Scheuber HP, et al. Cathepsin B-indicator for the release of lysosomal cysteine proteinases in severe trauma and inflammation. Biol Chem Hoppe Seyler. 1990. May;371 Suppl:211–22. [PubMed] [Google Scholar]
- 37.Neugebauer E, Holaday JW. Handbook of mediators in septic shock CRC Press, 1993, pp. 336–356 [Google Scholar]
- 38.Boutte A, Hook V, Thangavelu B, Sarkis G, Brittany A, Hook G, Jacobsen JS, Robertson C, Gilsdorf, Yang Z, Wang K, Shear D. Penetrating traumatic brain injury triggers subacute dysregulation of cathepsin B protein levels independent of cysteine protease activity in brain and cerebral spinal fluid. J. Neurotrauma 2020. January 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagai A, Murakawa Y, Terashima M, Shimode K, Umegae N, Takeuchi H, Kobayashi S. Cystatin C and cathepsin B in CSF from patients with inflammatory neurologic diseases. Neurology. 2000. December 26;55(12):1828–32. [DOI] [PubMed] [Google Scholar]
- 40.Hsu A, Podvin S, Hook V. Lysosomal Cathepsin Protease Gene Expression Profiles in the Human Brain During Normal Development. J Mol Neurosci. 2018. August;65(4):420–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sunkin SM, Ng L, Lau C, Dolbeare T, Gilbert TL, Thompson CL, Hawrylycz M, Dang C. Allen Brain Atlas: an integrated spatio-temporal portal for exploring the central nervous system. Nucleic Acids Res. 2013. January;41(Database issue):D996–D1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shen EH, Overly CC, Jones AR. The Allen Human Brain Atlas: comprehensive gene expression mapping of the human brain. Trends Neurosci. 2012. December;35(12):711–4. [DOI] [PubMed] [Google Scholar]
- 43.Barrett AJ, Rawlings ND, Woessner JF (2004). Handbook of Proteolytic Enzymes, 2nd edition, Elsevier Academic Press, Amsterdam. [Google Scholar]
- 44.Bouter Y, Kacprowski T, Weissmann R, Dietrich K, Borgers H, Brauß A, Sperling C, Wirths O, Albrecht M, Jensen LR, Kuss AW, Bayer TA. Deciphering the molecular profile of plaques, memory decline and neuron loss in two mouse models forAlzheimer’s disease by deep sequencing. Front Aging Neurosci. 2014. April 16;6:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hook GR, Yu J, Sipes N, Pierschbacher MD, Hook V & Kindy MS 2014. The cysteine protease cathepsin B is a key drug target and cysteine protease inhibitors are potential therapeutics for traumatic brain injury. J Neurotrauma, 31, 515–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Natale JE, Ahmed F, Cernak I, Stoica B & Faden AI 2003. Gene expression profile changes are commonly modulated across models and species after traumatic brain injury. J Neurotrauma, 20, 907–27. [DOI] [PubMed] [Google Scholar]
- 47.Luo CL, Chen XP, Yang R, Sun YX, Li QQ, Bao HJ, Cao QQ, Ni H, Qin ZH, Tao LY. Cathepsin B contributes to traumatic brain injury-induced cell death through a mitochondria-mediated apoptotic pathway. J Neurosci Res. 2010. October;88(13):2847–58. [DOI] [PubMed] [Google Scholar]
- 48.Zhang YB, Chen XP, Tao LY, Qin ZH, Li SX, Yang L, Yang J, Zhang YG, Liu R. [Expression of cathepsin-B and -D in rat’s brain after traumatic brain injury]. Fa Yi Xue Za Zhi. 2006. December;22(6):404–6, 410. [PubMed] [Google Scholar]
- 49.Martinez-Vargas M, Soto-Nuñez M, Tabla-Ramon E, Solis B, Gonzalez-Rivera R, Perez-Arredondo A, Estrada-Rojo F, Castell A, Molina-Guarneros J, Navarro L. Cystatin C has a dual role in post-traumatic brain injury recovery. Int J Mol Sci. 2014. April 4;15(4):5807–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun YX, Dai DK, Liu R, Wang T, Luo CL, Bao HJ, Yang R, Feng XY, Qin ZH, Chen XP, Tao LY. Therapeutic effect of SN50, an inhibitor of nuclear factor-κB, in treatment of TBI in mice. Neurol Sci. 2013. March;34(3):345–55. [DOI] [PubMed] [Google Scholar]
- 51.Ellis RC, Earnhardt JN, Hayes RL, Wang KK, Anderson DK. Cathepsin B mRNA and protein expression following contusion spinal cord injury in rats. J Neurochem. 2004. February;88(3):689–97. [DOI] [PubMed] [Google Scholar]
- 52.Ellis RC, O’steen WA, Hayes RL, Nick HS, Wang KK, Anderson DK. Cellular localization and enzymatic activity of cathepsin B after spinal cord injury in the rat. Exp Neurol. 2005. May;193(1):19–28. [DOI] [PubMed] [Google Scholar]
- 53.Vreemann A, Qu H, Mayer K, Andersen LB, Stefana MI, Wehner S, Lysson M, Farcas AM, Peters C, Reinheckel T, Kalff J, Brix K. Cathepsin B release from rodent intestine mucosa due to mechanical injury results in extracellular matrix damage in early post-traumatic phases. Biol Chem. 2009. May-Jun;390(5–6):481–92. [DOI] [PubMed] [Google Scholar]
- 54.Yu ZQ, Jia Y, Chen G. Possible involvement of cathepsin B/D and caspase-3 in deferoxamine-related neuroprotection of early brain injury after subarachnoid haemorrhage in rats. Neuropathol Appl Neurobiol. 2014. April;40(3):270–83. [DOI] [PubMed] [Google Scholar]
- 55.Wang Y, Gao A, Xu X, Dang B, You W, Li H, Yu Z, Chen G. The Neuroprotection of Lysosomotropic Agents in Experimental Subarachnoid Hemorrhage Probably Involving the Apoptosis Pathway Triggering by Cathepsins via Chelating Intralysosomal Iron. Mol Neurobiol. 2015. August;52(1):64–77. [DOI] [PubMed] [Google Scholar]
- 56.Aoki T, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Cathepsin B, K, and S are expressed in cerebral aneurysms and promote the progression of cerebral aneurysms. Stroke. 2008. September;39(9):2603–10. [DOI] [PubMed] [Google Scholar]
- 57.Luo CL, Chen XP, Yang R, Sun YX, Li QQ, Bao HJ, Cao QQ, Ni H, Qin ZH, Tao LY. Cathepsin B contributes to traumatic brain injury-induced cell death through a mitochondria-mediated apoptotic pathway. J Neurosci Res. 2010. October;88(13):2847–58. [DOI] [PubMed] [Google Scholar]
- 58.Zhang M, Shan H, Chang P, Wang T, Dong W, Chen X, Tao L. Hydrogen sulfide offers neuroprotection on traumatic brain injury in parallel with reduced apoptosis and autophagy in mice. PLoS One. 2014. January 23;9(1):e87241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Offen D, Barhum Y, Melamed E, Embacher N, Schindler C, Ransmayr G. Spinal cord mRNA profile in patients with ALS: comparison with transgenic mice expressing the human SOD-1 mutant. J Mol Neurosci. 2009. June;38(2):85–93. [DOI] [PubMed] [Google Scholar]
- 60.Ferraiuolo L, Heath PR, Holden H, Kasher P, Kirby J, Shaw PJ. Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J Neurosci. 2007. August 22;27(34):9201–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saris CG, Groen EJ, Koekkoek JA, Veldink JH, van den Berg LH. Meta-analysis of gene expression profiling in amyotrophic lateral sclerosis: a comparison between transgenic mouse models and human patients. Amyotroph Lateral Scler Frontotemporal Degener. 2013. April;14(3):177–89. [DOI] [PubMed] [Google Scholar]
- 62.Ni H, Yan JZ, Zhang LL, Feng X, Wu XR. Long-term effects of recurrent neonatal seizures on neurobehavioral function and related gene expression and its intervention by inhibitor of cathepsin B. Neurochem Res. 2012. January;37(1):31–9. [DOI] [PubMed] [Google Scholar]
- 63.Wang Y, Gu ZL, Cao Y, Liang ZQ, Han R, Bennett MC, Qin ZH. Lysosomal enzyme cathepsin B is involved in kainic acid-induced excitotoxicity in rat striatum. Brain Res. 2006. February 3;1071(1):245–9. [DOI] [PubMed] [Google Scholar]
- 64.Tsubokawa T, Yamaguchi-Okada M, Calvert JW, Solaroglu I, Shimamura N, Yata K, Zhang JH. Neurovascular and neuronal protection by E64d after focal cerebral ischemia in rats. J Neurosci Res. 2006. September;84(4):832–40. [DOI] [PubMed] [Google Scholar]
- 65.Seyfried D, Han Y, Zheng Z, Day N, Moin K, Rempel S, Sloane B, Chopp M. Cathepsin B and middle cerebral artery occlusion in the rat. J Neurosurg. 1997. November;87(5):716–23. [DOI] [PubMed] [Google Scholar]
- 66.Yamashima T, Kohda Y, Tsuchiya K, Ueno T, Yamashita J, Yoshioka T, Kominami E. Inhibition of ischaemic hippocampal neuronal death in primates with cathepsin B inhibitor CA-074: a novel strategy for neuroprotection based on ‘calpain-cathepsin hypothesis’. Eur J Neurosci. 1998. May;10(5):1723–33. [DOI] [PubMed] [Google Scholar]
- 67.Tsuchiya K, Kohda Y, Yoshida M, Zhao L, Ueno T, Yamashita J, Yoshioka T, Kominami E, Yamashima T. Postictal blockade of ischemic hippocampal neuronal death in primates using selective cathepsin inhibitors. Exp Neurol. 1999. February;155(2):187–94. [DOI] [PubMed] [Google Scholar]
- 68.Hoegen T, Tremel N, Klein M, Angele B, Wagner H, Kirschning C, Pfister HW, Fontana A, Hammerschmidt S, Koedel U. The NLRP3 inflammasome contributes to brain injury in pneumococcal meningitis and is activated through ATP-dependent lysosomal cathepsin B release. J Immunol. 2011. November 15;187(10):5440–51. [DOI] [PubMed] [Google Scholar]
- 69.Ruff RL, Secrist D. Inhibitors of prostaglandin synthesis or cathepsin B prevent muscle wasting due to sepsis in the rat. J Clin Invest. 1984. May;73(5):1483–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hummel RP 3rd, James JH, Warner BW, Hasselgren PO, Fischer JE. Evidence that cathepsin B contributes to skeletal muscle protein breakdown during sepsis. Arch Surg. 1988. February;123(2):221–4. [DOI] [PubMed] [Google Scholar]
- 71.Terada K, Yamada J, Hayashi Y, Wu Z, Uchiyama Y, Peters C, Nakanishi H. Involvement of cathepsin B in the processing and secretion of interleukin-1beta in chromogranin A-stimulated microglia. Glia. 2010. January 1;58(1):114–24. [DOI] [PubMed] [Google Scholar]
- 72.Wu Z, Ni J, Liu Y, Teeling JL, Takayama F, Collcutt A, Ibbett P, Nakanishi H. Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav Immun. 2017. October;65:350–361 [DOI] [PubMed] [Google Scholar]
- 73.Ni J, Wu Z, Stoka V, Meng J, Hayashi Y, Peters C, Qing H, Turk V, Nakanishi H. Increased expression and altered subcellular distribution of cathepsin B in microglia induce cognitive impairment through oxidative stress and inflammatory response in mice. Aging Cell. 2019. February;18(1):e12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sun L, Wu Z, Hayashi Y, Peters C, Tsuda M, Inoue K, et al. 2012. Microglial cathepsin B contributes to the initiation of peripheral inflammation-induced chronic pain. J Neurosci, 32, 11330–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kindy MS, Yu J, Zhu H, El-Amouri SS, Hook V, Hook GR. Deletion of the cathepsin B gene improves memory deficits in a transgenic Alzheimer’s disease mouse model expressing AβPP containing the wild-type β-secretase site sequence. J Alzheimers Dis. 2012;29(4):827–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hook VY, Kindy M, Reinheckel T, Peters C, Hook G. Genetic cathepsin B deficiency reduces beta-amyloid in transgenic mice expressing human wild-type amyloid precursor protein. Biochem Biophys Res Commun. 2009. August 21;386(2):284–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hook G, Yu J, Toneff T, Kindy M, Hook V. Brain pyroglutamate amyloid-β is produced by cathepsin B and is reduced by the cysteine protease inhibitor E64d, representing a potential Alzheimer’s disease therapeutic. J Alzheimers Dis. 2014;41(1):129–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schilling S, Lauber T, Schaupp M, Manhart S, Scheel E, Böhm G, Demuth HU. On the seeding and oligomerization of pGlu-amyloid peptides (in vitro). Biochemistry. 2006. October 17;45(41):12393–9. [DOI] [PubMed] [Google Scholar]
- 79.Saido TC, Iwatsubo T, Mann DM, Shimada H, Ihara Y, Kawashima S. Dominant and differential deposition of distinct beta-amyloid peptide species, A beta N3(pE), in senile plaques. Neuron. 1995. February;14(2):457–66. [DOI] [PubMed] [Google Scholar]
- 80.Wang ML, Li WB. Cognitive impairment after traumatic brain injury: The role of MRI and possible pathological basis. J Neurol Sci. 2016. November 15;370:244–250. [DOI] [PubMed] [Google Scholar]
- 81.McInnes K, Friesen CL, MacKenzie DE, Westwood DA, Boe SG. Mild Traumatic Brain Injury (mTBI) and chronic cognitive impairment: A scoping review. PLoS One. 2017. April 11;12(4):e0174847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dams-O’Connor K, Guetta G, Hahn-Ketter AE, Fedor A. Traumatic brain injury as a risk factor for Alzheimer’s disease: current knowledge and future directions. Neurodegener Dis Manag. 2016. October;6(5):417–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Julien J, Joubert S, Ferland MC, Frenette LC, Boudreau-Duhaime MM, Malo-Véronneau L, de Guise E. Association of traumatic brain injury and Alzheimer disease onset: A systematic review. Ann Phys Rehabil Med. 2017. September;60(5):347–356. [DOI] [PubMed] [Google Scholar]
- 84.LoBue C, Wadsworth H, Wilmoth K, Clem M, Hart J Jr, Womack KB, Didehbani N, Lacritz LH, Rossetti HC, Cullum CM. Traumatic brain injury history is associated with earlier age of onset of Alzheimer disease. Clin Neuropsychol. 2017. January;31(1):85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ide M, Harris M, Stevens A, Sussams R, Hopkins V, Culliford D, Fuller J, Ibbett P, Raybould R, Thomas R, Puenter U, Teeling J, Perry VH, Holmes C. Periodontitis and Cognitive Decline in Alzheimer’s Disease. PLoS One. 2016. March 10;11(3):e0151081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW, Hong JT. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation. 2008. August 29;5:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, Nguyen M, Haditsch U, Raha D, Griffin C, Holsinger LJ, Arastu-Kapur S, Kaba S, Lee A, Ryder MI, Potempa B, Mydel P, Hellvard A, Adamowicz K, Hasturk H, Walker GD, Reynolds EC, Faull RLM, Curtis MA, Dragunow M, Potempa J. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. 2019. January 23;5(1):eaau3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Guerreiro R, Bras J. The age factor in Alzheimer’s disease. Genome Med. 2015. October 20;7:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reinheckel T, Deussing J, Roth W, Peters C. Towards specific functions of lysosomal cysteine peptidases: phenotypes of mice deficient for cathepsin B or cathepsin L. Biol Chem. 2001. May;382(5):735–41. [DOI] [PubMed] [Google Scholar]
- 90.Deussing J, Roth W, Saftig P, Peters C, Ploegh HL, Villadangos JA. Cathepsins B and D are dispensable for major histocompatibility complex class II-mediated antigen presentation. Proc Natl Acad Sci U S A. 1998. April 14;95(8):4516–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Friedrichs B, Tepel C, Reinheckel T, Deussing J, von Figura K, Herzog V, Peters C, Saftig P, Brix K. Thyroid functions of mouse cathepsins B, K, and L. J Clin Invest. 2003. June;111(11):1733–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hook G, Hook V, Kindy M. The cysteine protease inhibitor, E64d, reduces brain amyloid-β and improves memory deficits in Alzheimer’s disease animal models by inhibiting cathepsin B, but not BACE1, β-secretase activity. J Alzheimers Dis. 2011;26(2):387–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hook VY, Kindy M, Hook G. Inhibitors of cathepsin B improve memory and reduce beta-amyloid in transgenic Alzheimer disease mice expressing the wild-type, but not the Swedish mutant, beta-secretase site of the amyloid precursor protein. J Biol Chem. 2008. March 21;283(12):7745–53. [DOI] [PubMed] [Google Scholar]
- 94.Hook G, Yu J, Toneff T, Kindy M, Hook V. Brain pyroglutamate amyloid-β is produced by cathepsin B and is reduced by the cysteine protease inhibitor E64d, representing a potential Alzheimer’s disease therapeutic. J Alzheimers Dis. 2014;41(1):129–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hashida S, Towatari T, Kominami E, Katunuma N. Inhibitions by E-64 derivatives of rat liver cathepsin B and cathepsin L in vitro and in vivo. J Biochem. 1980. December;88(6):1805–11. [DOI] [PubMed] [Google Scholar]
- 96.Tamai M, Matsumoto K, Omura S, Koyama I, Ozawa Y, Hanada K. In vitro and in vivo inhibition of cysteine proteinases by EST, a new analog of E-64. J Pharmacobiodyn. 1986. August;9(8):672–7. [DOI] [PubMed] [Google Scholar]
- 97.Tamai M, Hanada K, Adachi T, Oguma K, Kashiwagi K, Omura S, Ohzeki M. Papain inhibitions by optically active E-64 analogs. J Biochem. 1981. July;90(1):255–7. [DOI] [PubMed] [Google Scholar]
- 98.Murata M, Miyashita S, Yokoo C, Tamai M, Hanada K, Hatayama K, Towatari T, Nikawa T, Katunuma N. Novel epoxysuccinyl peptides. Selective inhibitors of cathepsin B, in vitro. FEBS Lett. 1991. March 25;280(2):307–10. [DOI] [PubMed] [Google Scholar]
- 99.Towatari T, Nikawa T, Murata M, Yokoo C, Tamai M, Hanada K, Katunuma N. Novel epoxysuccinyl peptides. A selective inhibitor of cathepsin B, in vivo. FEBS Lett. 1991. March 25;280(2):311–5. [DOI] [PubMed] [Google Scholar]
- 100.Yamamoto A, Tomoo K, Hara T, Murata M, Kitamura K, Ishida T. Substrate specificity of bovine cathepsin B and its inhibition by CA074, based on crystal structure refinement of the complex. J Biochem. 2000. April;127(4):635–43. [DOI] [PubMed] [Google Scholar]
- 101.Buttle DJ, Murata M, Knight CG, Barrett AJ. CA074 methyl ester: a proinhibitor for intracellular cathepsin B. Arch Biochem Biophys. 1992. December;299(2):377–80. [DOI] [PubMed] [Google Scholar]
- 102.Knoblach SM, Alroy DA, Nikolaeva M, Cernak I, Stoica BA, Faden AI. Caspase inhibitor z-DEVD-fmk attenuates calpain and necrotic cell death in vitro and after traumatic brain injury. J Cereb Blood Flow Metab. 2004. October;24(10):1119–32. [DOI] [PubMed] [Google Scholar]
- 103.Xu J, Wang H, Ding K, Lu X, Li T, Wang J, Wang C, Wang J. Inhibition of cathepsin S produces neuroprotective effects after traumatic brain injury in mice. Mediators Inflamm. 2013;2013:187873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Inuzuka T, Tamura A, Sato S, Kirino T, Toyoshima I, Miyatake T. Suppressive effect of E-64c on ischemic degradation of cerebral proteins following occlusion of the middle cerebral artery in rats. Brain Res. 1990. August 27;526(1):177–9. [DOI] [PubMed] [Google Scholar]
- 105.Yamashima T, Kohda Y, Tsuchiya K, Ueno T, Yamashita J, Yoshioka T, Kominami E. Inhibition of ischaemic hippocampal neuronal death in primates with cathepsin B inhibitor CA-074: a novel strategy for neuroprotection based on ‘calpain-cathepsin hypothesis’. Eur J Neurosci. 1998. May;10(5):1723–33. [DOI] [PubMed] [Google Scholar]
- 106.Tsuchiya K, Kohda Y, Yoshida M, Zhao L, Ueno T, Yamashita J, Yoshioka T, Kominami E, Yamashima T. Postictal blockade of ischemic hippocampal neuronal death in primates using selective cathepsin inhibitors. Exp Neurol. 1999. February;155(2):187–94. [DOI] [PubMed] [Google Scholar]
- 107.Seyfried DM, Veyna R, Han Y, Li K, Tang N, Betts RL, Weinsheimer S, Chopp M, Anagli J. A selective cysteine protease inhibitor is non-toxic and cerebroprotective in rats undergoing transient middle cerebral artery ischemia. Brain Res. 2001. May 18;901(1–2):94–101. [DOI] [PubMed] [Google Scholar]
- 108.Yoshida M, Yamashima T, Zhao L, Tsuchiya K, Kohda Y, Tonchev AB, Matsuda M, Kominami E. Primate neurons show different vulnerability to transient ischemia and response to cathepsin inhibition. Acta Neuropathol. 2002. September;104(3):267–72. [DOI] [PubMed] [Google Scholar]
- 109.Tsubokawa T, Yamaguchi-Okada M, Calvert JW, Solaroglu I, Shimamura N, Yata K, Zhang JH. Neurovascular and neuronal protection by E64d after focal cerebral ischemia in rats. J Neurosci Res. 2006. September;84(4):832–40. [DOI] [PubMed] [Google Scholar]
- 110.Yang D, Han Y, Zhang J, Ding C, Anagli J, Seyfried DM. Improvement in recovery after experimental intracerebral hemorrhage using a selective cathepsin B and L inhibitor. J Neurosurg. 2011. April;114(4):1110–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang Y, Gu ZL, Cao Y, Liang ZQ, Han R, Bennett MC, Qin ZH. Lysosomal enzyme cathepsin B is involved in kainic acid-induced excitotoxicity in rat striatum. Brain Res. 2006. February 3;1071(1):245–9. [DOI] [PubMed] [Google Scholar]
- 112.Ni H, Yan JZ, Zhang LL, Feng X, Wu XR. Long-term effects of recurrent neonatal seizures on neurobehavioral function and related gene expression and its intervention by inhibitor of cathepsin B. Neurochem Res. 2012. January;37(1):31–9. [DOI] [PubMed] [Google Scholar]
- 113.Lyo V, Cattaruzza F, Kim TN, Walker AW, Paulick M, Cox D, Cloyd J, Buxbaum J, Ostroff J, Bogyo M, Grady EF, Bunnett NW, Kirkwood KS. Active cathepsins B, L, and S in murine and human pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2012. October 15;303(8):G894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114., Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B, Turk D. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta. 2012. January;1824(1):68–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115., Chan SJ, San Segundo B, McCormick MB, Steiner DF. Nucleotide and predicted amino acid sequences of cloned human and mouse preprocathepsin B cDNAs. Proc Natl Acad Sci U S A. 1986. October;83(20):7721–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mordier S, Béchet D, Roux MP, Obled A, Ferrara M. Nucleotide sequence of bovine preprocathepsin B. A study of polymorphism in the protein coding region. Biochim Biophys Acta. 1993. September 23;1174(3):305–11. [DOI] [PubMed] [Google Scholar]
- 117.Pungercar JR, Caglic D, Sajid M, Dolinar M, Vasiljeva O, Pozgan U, Turk D, Bogyo M, Turk V, Turk B. Autocatalytic processing of procathepsin B is triggered by proenzyme activity. FEBS J. 2009. February;276(3):660–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Quraishi O, Storer AC. Identification of internal autoproteolytic cleavage sites within the prosegments of recombinant procathepsin B and procathepsin S. Contribution of a plausible unimolecular autoproteolytic event for the processing of zymogens belonging to the papain family. J Biol Chem. 2001. March 16;276(11):8118–24. [DOI] [PubMed] [Google Scholar]
- 119.Mach L, Mort JS, Glössl J. Maturation of human procathepsin B. Proenzyme activation and proteolytic processing of the precursor to the mature proteinase, in vitro, are primarily unimolecular processes. J Biol Chem. 1994. April 29;269(17):13030–5. [PubMed] [Google Scholar]
- 120.Rowan AD, Mason P, Mach L, Mort JS. Rat procathepsin B. Proteolytic processing to the mature form in vitro. J Biol Chem. 1992. August 5;267(22):15993–9.. [PubMed] [Google Scholar]
- 121.Rowan AD, Feng R, Konishi Y, Mort JS. Demonstration by electrospray mass spectrometry that the peptidyldipeptidase activity of cathepsin B is capable of rat cathepsin B C-terminal processing. Biochem J. 1993. September 15;294 (Pt 3):923–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dalet-Fumeron V, Guinec N, Pagano M. In vitro activation of pro-cathepsin B by three serine proteinases: leucocyte elastase, cathepsin G, and the urokinase-type plasminogen activator. FEBS Lett. 1993. October 18;332(3):251–4. [DOI] [PubMed] [Google Scholar]
- 123.Schechter I. Mapping of the active site of proteases in the 1960s and rational design of inhibitors/drugs in the 1990s. Curr Protein Pept Sci. 2005. December;6(6):501–12. [DOI] [PubMed] [Google Scholar]
- 124.Kido H, Ishidoh K. Nobuhiko Katunuma: an outstanding scientist in the field of proteolysis and warm-hearted ‘Kendo Fighter’ biochemist. J Biochem. 2010. November;148(5):527–31. [DOI] [PubMed] [Google Scholar]
- 125.Barrett AJ, Kembhavi AA, Brown MA, Kirschke H, Knight CG, Tamai M, Hanada K. L-trans-Epoxysuccinyl-leucylamido(4-guanidino)butane (E-64) and its analogues as inhibitors of cysteine proteinases including cathepsins B, H and L. Biochem J. 1982. January 1;201(1):189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tamai M, Hanada K, Adachi T, Oguma K, Kashiwagi K, Omura S, Ohzeki M. Papain inhibitions by optically active E-64 analogs. J Biochem. 1981. July;90(1):255–7. [DOI] [PubMed] [Google Scholar]
- 127.Sugita H, Ishiura S, Suzuki K, Imahori K. Inhibition of epoxide derivatives on chicken calcium-activated neutral protease (CANP) in vitro and in vivo. J Biochem. 1980. January;87(1):339–41. [DOI] [PubMed] [Google Scholar]
- 128.Matsumoto K, Mizoue K, Kitamura K, Tse WC, Huber CP, Ishida T. Structural basis of inhibition of cysteine proteases by E-64 and its derivatives. Biopolymers. 1999;51(1):99–107. [DOI] [PubMed] [Google Scholar]
- 129.Katunuma N. Structure-based development of specific inhibitors for individual cathepsins and their medical applications. Proc Jpn Acad Ser B Phys Biol Sci. 2011;87(2):29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Vilar S, Cozza G, Moro S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr Top Med Chem. 2008;8(18):1555–72. [DOI] [PubMed] [Google Scholar]
- 131.Roy U, Luck LA. Molecular modeling of estrogen receptor using molecular operating environment. Biochem Mol Biol Educ. 2007. July;35(4):238–43. [DOI] [PubMed] [Google Scholar]
- 132., Musil D, Zucic D, Turk D, Engh RA, Mayr I, Huber R, Popovic T, Turk V, Towatari T, Katunuma N, et al. The refined 2.15 A X-ray crystal structure of human liver cathepsin B: the structural basis for its specificity. EMBO J. 1991. September;10(9):2321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Illy C, Quraishi O, Wang J, Purisima E, Vernet T, Mort JS. Role of the occluding loop in cathepsin B activity. J Biol Chem. 1997. January 10;272(2):1197–202. [DOI] [PubMed] [Google Scholar]
- 134.Nägler DK, Storer AC, Portaro FC, Carmona E, Juliano L, Ménard R. Major increase in endopeptidase activity of human cathepsin B upon removal of occluding loop contacts. Biochemistry. 1997. October 14;36(41):12608–15. [DOI] [PubMed] [Google Scholar]
- 135.Quraishi O, Nägler DK, Fox T, Sivaraman J, Cygler M, Mort JS, Storer AC. The occluding loop in cathepsin B defines the pH dependence of inhibition by its propeptide. Biochemistry. 1999. April 20;38(16):5017–23. [DOI] [PubMed] [Google Scholar]
- 136.Turk D, Podobnik M, Kuhelj R, Dolinar M, Turk V. Crystal structures of human procathepsin B at 3.2 and 3.3 Angstroms resolution reveal an interaction motif between a papain-like cysteine protease and its propeptide. FEBS Lett. 1996. April 22;384(3):211–4. [DOI] [PubMed] [Google Scholar]
- 137.Renko M, Požgan U, Majera D, Turk D. Stefin A displaces the occluding loop of cathepsin B only by as much as required to bind to the active site cleft. FEBS J. 2010. October;277(20):4338–45. [DOI] [PubMed] [Google Scholar]
- 138.De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–92. [DOI] [PubMed] [Google Scholar]
- 139.Lawrence RE, Zoncu R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol. 2019. February;21(2):133–142. [DOI] [PubMed] [Google Scholar]
- 140.Hook G, Jacobsen JS, Grabstein K, Kindy M, Hook V. Cathepsin B is a New Drug Target for Traumatic Brain Injury Therapeutics: Evidence for E64d as a Promising Lead Drug Candidate. Front Neurol. 2015. September 2;6:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yamashima T. Can ‘calpain-cathepsin hypothesis’ explain Alzheimer neuronal death? Ageing Res Rev. 2016. December;32:169–179. [DOI] [PubMed] [Google Scholar]
- 142.Nakanishi H. Microglial cathepsin B as a key driver of inflammatory brain diseases and brain aging. Neural Regen Res. 2020. January;15(1):25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.de Castro MA, Bunt G, Wouters FS. Cathepsin B launches an apoptotic exit effort upon cell death-associated disruption of lysosomes. Cell Death Discov. 2016. February 29;2:16012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Droga-Mazovec G, Bojic L, Petelin A, Ivanova S, Romih R, Repnik U, Salvesen GS, Stoka V, Turk V, Turk B. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J Biol Chem. 2008. July 4;283(27):19140–50. [DOI] [PubMed] [Google Scholar]
- 145., Kavčič N, Pegan K, Turk B. Lysosomes in programmed cell death pathways: from initiators to amplifiers. Biol Chem. 2017. March;398(3):289–301. [DOI] [PubMed] [Google Scholar]
- 146., Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB, Korsmeyer SJ. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000. August 15;14(16):2060–71. [PMC free article] [PubMed] [Google Scholar]
- 147., Aits S, Jäättelä M. Lysosomal cell death at a glance. J Cell Sci. 2013. May 1;126(Pt 9):1905–12. [DOI] [PubMed] [Google Scholar]
- 148.Campden RI, Zhang Y. The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Arch Biochem Biophys. 2019. July 30;670:32–42. [DOI] [PubMed] [Google Scholar]
- 149.Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019. February 12;10(2):128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.H, Yang B, Yu W, Xiao Y, Yu D, Zhang Q. Cathepsin B links oxidative stress to the activation of NLRP3 inflammasome. Exp Cell Res. 2018. January 1;362(1):180–187. [DOI] [PubMed] [Google Scholar]
- 151.Lian D, Lai J, Wu Y, Wang L, Chen Y, Zhang Y, Boini KM, Huang Y, Chen Y. Cathepsin B-mediated NLRP3 inflammasome formation and activation in angiotensin II -Induced hypertensive mice: role of macrophage digestion dysfunction. Cell Physiol Biochem. 2018;50(4):1585–1600. [DOI] [PubMed] [Google Scholar]
- 152., Amaral EP, Riteau N, Moayeri M, Maier N, Mayer-Barber KD, Pereira RM, Lage SL, Kubler A, Bishai WR, D’Império-Lima MR, Sher A, Andrade BB. Lysosomal cathepsin release is required for NLRP3-inflammasome activation by mycobacterium tuberculosis in infected macrophages. Front Immunol. 2018. June 21;9:1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ardura-Fabregat A, Boddeke EWGM, Boza-Serrano A, Brioschi S, Castro-Gomez S, Ceyzériat K, Dansokho C, Dierkes T, Gelders G, Heneka MT, Hoeijmakers L, Hoffmann A, Iaccarino L, Jahnert S, Kuhbandner K, Landreth G, Lonnemann N, Löschmann PA, McManus RM, Paulus A, Reemst K, Sanchez-Caro JM, Tiberi A, Van der Perren A, Vautheny A, Venegas C, Webers A, Weydt P, Wijasa TS, Xiang X, Yang Y. Targeting neuroinflammation to treat Alzheimer’s Disease. CNS Drugs. 2017. December;31(12):1057–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Minter MR, Taylor JM, Crack PJ. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J Neurochem. 2016. February;136(3):457–74. [DOI] [PubMed] [Google Scholar]
- 155.Skaper SD, Facci L, Zusso M, Giusti P. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front Cell Neurosci. 2018. March 21;12:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Simon DW, McGeachy MJ, Bayır H, Clark RS, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. 2017. March;13(3):171–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Faden AI, Wu J, Stoica BA, Loane DJ. Progressive inflammation-mediated neurodegeneration after traumatic brain or spinal cord injury. Br J Pharmacol. 2016. February;173(4):681–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chiu CC, Liao YE, Yang LY, Wang JY, Tweedie D, Karnati HK, Greig NH, Wang JY. Neuroinflammation in animal models of traumatic brain injury. J Neurosci Methods. 2016. October 15;272:38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Xiong Y, Mahmood A, Chopp M. Current understanding of neuroinflammation after traumatic brain injury and cell-based therapeutic opportunities. Chin J Traumatol. 2018. June;21(3):137–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yang AJ, Chandswangbhuvana D, Margol L, Glabe CG. Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Abeta1–42 pathogenesis. J Neurosci Res. 1998. June 15;52(6):691–8. [DOI] [PubMed] [Google Scholar]
- 161.Ditaranto K, Tekirian TL, Yang AJ. Lysosomal membrane damage in soluble Abeta-mediated cell death in Alzheimer’s disease. Neurobiol Dis. 2001. February;8(1):19–31. [DOI] [PubMed] [Google Scholar]
- 162.De Kimpe L, van Haastert ES, Kaminari A, Zwart R, Rutjes H, Hoozemans JJ, Scheper W. Intracellular accumulation of aggregated pyroglutamate amyloid beta: convergence of aging and Aβ pathology at the lysosome. Age (Dordr). 2013. June;35(3):673–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Umeda T, Tomiyama T, Sakama N, Tanaka S, Lambert MP, Klein WL, Mori H. Intraneuronal amyloid β oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J Neurosci Res. 2011. July;89(7):1031–42. [DOI] [PubMed] [Google Scholar]
- 164.Ji ZS, Miranda RD, Newhouse YM, Weisgraber KH, Huang Y, Mahley RW. Apolipoprotein E4 potentiates amyloid beta peptide-induced lysosomal leakage and apoptosis in neuronal cells. J Biol Chem. 2002. June 14;277(24):21821–8. [DOI] [PubMed] [Google Scholar]
- 165.Belinson H, Lev D, Masliah E, Michaelson DM. Activation of the amyloid cascade in apolipoprotein E4 transgenic mice induces lysosomal activation and neurodegeneration resulting in marked cognitive deficits. J Neurosci. 2008. April 30;28(18):4690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lafrenaye AD, McGinn MJ, Povlishock JT. Increased intracranial pressure after diffuse traumatic brain injury exacerbates neuronal somatic membrane poration but not axonal injury: evidence for primary intracranial pressure-induced neuronal perturbation. J Cereb Blood Flow Metab. 2012. October;32(10):1919–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Luo CL, Chen XP, Li LL, Li QQ, Li BX, Xue AM, Xu HF, Dai DK, Shen YW, Tao LY, Zhao ZQ. Poloxamer 188 attenuates in vitro traumatic brain injury-induced mitochondrial and lysosomal membrane permeabilization damage in cultured primary neurons. J Neurotrauma. 2013. April 1;30(7):597–607. [DOI] [PubMed] [Google Scholar]
- 168.Windelborn JA, Lipton P. Lysosomal release of cathepsins causes ischemic damage in the rat hippocampal slice and depends on NMDA-mediated calcium influx, arachidonic acid metabolism, and free radical production. J Neurochem. 2008. July;106(1):56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kilinc M, Gürsoy-Ozdemir Y, Gürer G, Erdener SE, Erdemli E, Can A, Dalkara T. Lysosomal rupture, necroapoptotic interactions and potential crosstalk between cysteine proteases in neurons shortly after focal ischemia. Neurobiol Dis. 2010. October;40(1):293–302. [DOI] [PubMed] [Google Scholar]
- 170.Smeyne RJ, Jackson-Lewis V. The MPTP model of Parkinson’s disease. Brain Res Mol Brain Res. 2005. March 24;134(1):57–66. [DOI] [PubMed] [Google Scholar]
- 171.Langston JW. The MPTP Story. J Parkinsons Dis. 2017;7(s1):S11–S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Dong H, Qin Y, Huang Y, Ji D, Wu F. Poloxamer 188 rescues MPTP-induced lysosomal membrane integrity impairment in cellular and mouse models of Parkinson’s disease. Neurochem Int. 2019. June;126:178–186. [DOI] [PubMed] [Google Scholar]
- 173.Rocha EM, De Miranda B, Sanders LH. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol Dis. 2018. January;109(Pt B):249–257. [DOI] [PubMed] [Google Scholar]
- 174.Burré J, Sharma M, Südhof TC. Cell Biology and Pathophysiology of α-Synuclein. Cold Spring Harb Perspect Med. 2018. March 1;8(3). pii: a024091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Freeman D, Cedillos R, Choyke S, Lukic Z, McGuire K, Marvin S, Burrage AM, Sudholt S, Rana A, O’Connor C, Wiethoff CM, Campbell EM. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One. 2013. April 25;8(4):e62143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bourdenx M, Bezard E, Dehay B. Lysosomes and α-synuclein form a dangerous duet leading to neuronal cell death. Front Neuroanat. 2014. August 14;8:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sahara S, Yamashima T. Calpain-mediated Hsp70.1 cleavage in hippocampal CA1 neuronal death. Biochem Biophys Res Commun. 2010. March 19;393(4):806–11. [DOI] [PubMed] [Google Scholar]
- 178.Zhu H, Yoshimoto T, Yamashima T. Heat shock protein 70.1 (Hsp70.1) affects neuronal cell fate by regulating lysosomal acid sphingomyelinase. J Biol Chem. 2014. October 3;289(40):27432–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yamashima T, Oikawa S. The role of lysosomal rupture in neuronal death. Prog Neurobiol. 2009. December;89(4):343–58. [DOI] [PubMed] [Google Scholar]
- 180.Mindell JA. Lysosomal acidification mechanisms. Annu Rev Physiol. 2012;74:69–86. doi: 10.1146/annurev-physiol-012110-142317. [DOI] [PubMed] [Google Scholar]
- 181.Ishida Y, Nayak S, Mindell JA, Grabe M. A model of lysosomal pH regulation. J Gen Physiol. 2013. June;141(6):705–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Myers BM, Tietz PS, Tarara JE, LaRusso NF. Dynamic measurements of the acute and chronic effects of lysosomotropic agents on hepatocyte lysosomal pH using flow cytometry. Hepatology. 1995. November;22(5):1519–26. [PubMed] [Google Scholar]
- 183.Ohkuma S, Poole B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci U S A. 1978. July;75(7):3327–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bright GR, Fisher GW, Rogowska J, Taylor DL. Fluorescence ratio imaging microscopy: temporal and spatial measurements of cytoplasmic pH. J Cell Biol. 1987. April;104(4):1019–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Madshus IH. Regulation of intracellular pH in eukaryotic cells. Biochem J. 1988. February 15;250(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Swietach P, Tiffert T, Mauritz JM, Seear R, Esposito A, Kaminski CF, Lew VL, Vaughan-Jones RD. Hydrogen ion dynamics in human red blood cells. J Physiol. 2010. December 15;588(Pt 24):4995–5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Linebaugh BE, Sameni M, Day NA, Sloane BF, Keppler D. Exocytosis of active cathepsin B enzyme activity at pH 7.0, inhibition and molecular mass. Eur J Biochem. 1999. August;264(1):100–9. [DOI] [PubMed] [Google Scholar]
- 188.Mort JS, Recklies AD, Poole AR. Extracellular presence of the lysosomal proteinase cathepsin B in rheumatoid synovium and its activity at neutral pH. Arthritis Rheum. 1984. May;27(5):509–15. [DOI] [PubMed] [Google Scholar]
- 189.Almeida PC, Nantes IL, Chagas JR, Rizzi CC, Faljoni-Alario A, Carmona E, Juliano L, Nader HB, Tersariol IL. Cathepsin B activity regulation. Heparin-like glycosaminogylcans protect human cathepsin B from alkaline pH-induced inactivation. J Biol Chem. 2001. January 12;276(2):944–51. [DOI] [PubMed] [Google Scholar]
- 190.Yu J, Zhu H, Taheri S, Mondy W, Bonilha L, Magwood GS, Lackland D, Adams RJ, Kindy MS. Serum Amyloid A-Mediated Inflammasome Activation of Microglial Cells in Cerebral Ischemia. J Neurosci. 2019. November 20;39(47):9465–9476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008. August;9(8):857–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Tönnies E, Trushina E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J Alzheimers Dis. 2017;57(4):1105–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Butterfield DA. Perspectives on Oxidative Stress in Alzheimer’s Disease and Predictions of Future Research Emphases. J Alzheimers Dis. 2018;64(s1):S469–S479. [DOI] [PubMed] [Google Scholar]
- 194.Khatri N, Thakur M, Pareek V, Kumar S, Sharma S, Datusalia AK. Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS Neurol Disord Drug Targets. 2018;17(9):689–695. [DOI] [PubMed] [Google Scholar]
- 195.Rodríguez-Rodríguez A, Egea-Guerrero JJ, Murillo-Cabezas F, Carrillo-Vico A. Oxidative stress in traumatic brain injury. Curr Med Chem. 2014. April;21(10):1201–11. [DOI] [PubMed] [Google Scholar]
- 196.NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016. January 4; 44:D7–Dd19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.The UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019. January8; 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]