

Abstract
Microbes, commensals and pathogens, control numerous functions in host cells. They can alter host signaling and modulate immune surveillance by interacting with host proteins. To shed light on the contribution of microbes to health and disease, it is vital to discern how microbial proteins rewire host signaling and through which host proteins. Host-Microbe Interaction PREDictor (HMI-PRED) is a user-friendly web server for structural prediction of protein-protein interactions (PPIs) between host and a microbial species, including bacteria, viruses, fungi, and protozoa. HMI-PRED relies on “interface mimicry” through which the microbial proteins hijack host binding surfaces. Given the structure of a microbial protein of interest, HMI-PRED will return structural models of potential host-microbe interaction (HMI) complexes, the list of host endogenous and exogenous PPIs that can be disrupted, and tissue expression of the microbe-targeted host proteins. The server also allows users to upload homology models of microbial proteins. Broadly, it aims at large-scale efficient identification of HMIs. The prediction results are stored in a repository for community access. HMI-PRED is free and available at https://interactome.ku.edu.tr/hmi.
Keywords: host-microbe interaction, HMI, host-pathogen interaction, HPI, interface mimicry, computational prediction, modeling, protein-protein interaction, structure
Graphical Abstract
Introduction
Microbes modulate host physiological processes in health and can induce diverse diseases, including cancer. Exactly how they initiate and maintain the disease phenotype is often a mystery. Microbes can crosstalk with the host through proteins, metabolites, small molecules, and nucleic acids. They evolved efficient molecular mimicry strategies to usurp host immune surveillance by repurposing host signaling through protein-protein interactions (PPIs) with the host [1]. They hijack host proteins at four different levels: sequence, structure, motif, and interface [2–5]. By exploiting local structural similarity, interface mimicry allows proteins with distinct global structures to interact in similar ways [6, 7]. Interface mimicry appears the most common mimicry type and is observed frequently in all interactions including endogenous (intra-species) [8, 9] and exogenous (inter-species) ones [1]. The similarity in interface architectures enables microbial proteins to compete with host proteins to bind to a common target, and thereby alter host physiological signaling. A microbial protein that hijacks a host interface grapples with all host endogenous PPIs sharing this architecture. Elucidation of the complete set of host-microbe interactions (HMIs), along with their complex (bound) structures, will provide insights into microbes’ virulence strategies and action on host signaling. Revealing the map of structural host-microbe crosstalk can also foster a more potent therapeutic development.
Experimental HMI data, including structural details, are extremely limited and large-scale experimental detection is challenging. Yeast-2-hybrid (Y2H), protein arrays, and mass-spectrometry followed by affinity purification (AP-MS) or crosslinking are examples of high throughput experimental methods to determine HMIs [10]. Each method has downsides, such as Y2H and AP-MS have high false-negative [11] and false-positive rates [12], respectively. Structural determination is even more time consuming, expensive and difficult to apply on a large scale. There is an escalating demand for robust and efficient computational approaches to complete the HMI space and resolve the complex structures. Although predicting endogenous PPIs is common, modeling exogenous interactions – i.e. HMIs – is fairly new. Still, several computational approaches have been put forward to identify HMIs [13, 14], most of which rely on sequence homology [15–21]. Sequence-based methods are often successful only if the homology is high. However, not all microbial proteins have human homologs and they may still hijack host protein structure fully or partially [22] and impede host signal transduction. There are also methods that consider sequence motifs [23–25], global structural similarity combined with sequence similarity [26–30], and structural motif-domain interactions [31, 32]. To date, the only method that relies solely on interface mimicry is ours [3, 4]. Interface-based approaches have also been successful in predicting endogenous host PPIs [33–38]. One of the widely used pioneers of these approaches is PRISM [33, 34]. HMI-PRED’s protocol was inspired by PRISM’s protocol; however, several changes were made to accommodate the HMI prediction requirements, as well as using different structural alignment, refinement and scoring tools. Since interface mimicry appears the most prevalent type, it holds promise in identifying more HMIs compared to other methods and may absolve the need for sequence homology. In addition, most of the available HMI predictors give the list of potential HMIs, but not the structures of the complexes. The availability of the complex structures of both modeled HMIs and endogenous host PPIs is key to deduce the potential consequences of the HMIs – whether they are likely to inhibit or activate the host pathway.
Although several methods have been published on predicting HMIs, only one web server is available [39]. This server requires the user to provide the sequences of both the microbial protein and its potential interactor in the host. However, the potential host partners are often unknown, and the main purpose of the predictors is to identify them. To facilitate this task for researchers with no programming experience, we developed a Host-Microbe Interaction PREDictor (HMI-PRED), a user-friendly web server of the only available interface-based HMI prediction method, of predicting not only the interaction but also the structural model of the complex [3, 4]. Users can predict HMIs, model and visualize the structures of their complexes, retrieve interface residues, get the list of affected/hijacked endogenous human PPIs and tissue expression of the host proteins. We tested all structurally known HMIs in PDB with our webserver and found that our accuracy of recapitulating known HMIs through self-interfaces is 81% and through alternative interfaces is 78% (see Supplementary Materials for details). Below, we introduce the HMI-PRED and present case studies to illustrate its utility. Case studies include proteins from pathogenic and commensal microbes with and without experimentally resolved structures.
HMI-PRED Overview
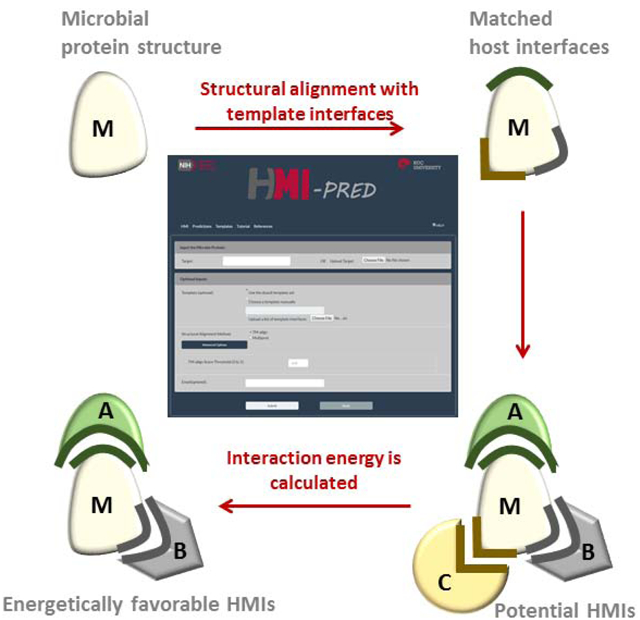
The rationale behind HMI-PRED is that if the microbial protein has a patch of surface that is structurally similar to one face of the template interface and similar evolutionarily conserved “hot spots”, it can interact with the host protein on the complementary face (Figure 1). Structural alignment of the microbial protein with all known human interfaces allows determining the potential HMI pairs, which are later scored with Rosetta docking [40, 41] to pick the energetically favorable ones. HMI-PRED performs online calculations and stores the results for community access. Our applications revealed that HMI-PRED generates promising HMI models that can explain the molecular mechanisms of how microbes evade host immunity, alter cytoskeletal dynamics, and modulate cell cycle regulation [3, 4].
Figure 1: Workflow of HMI-PRED.
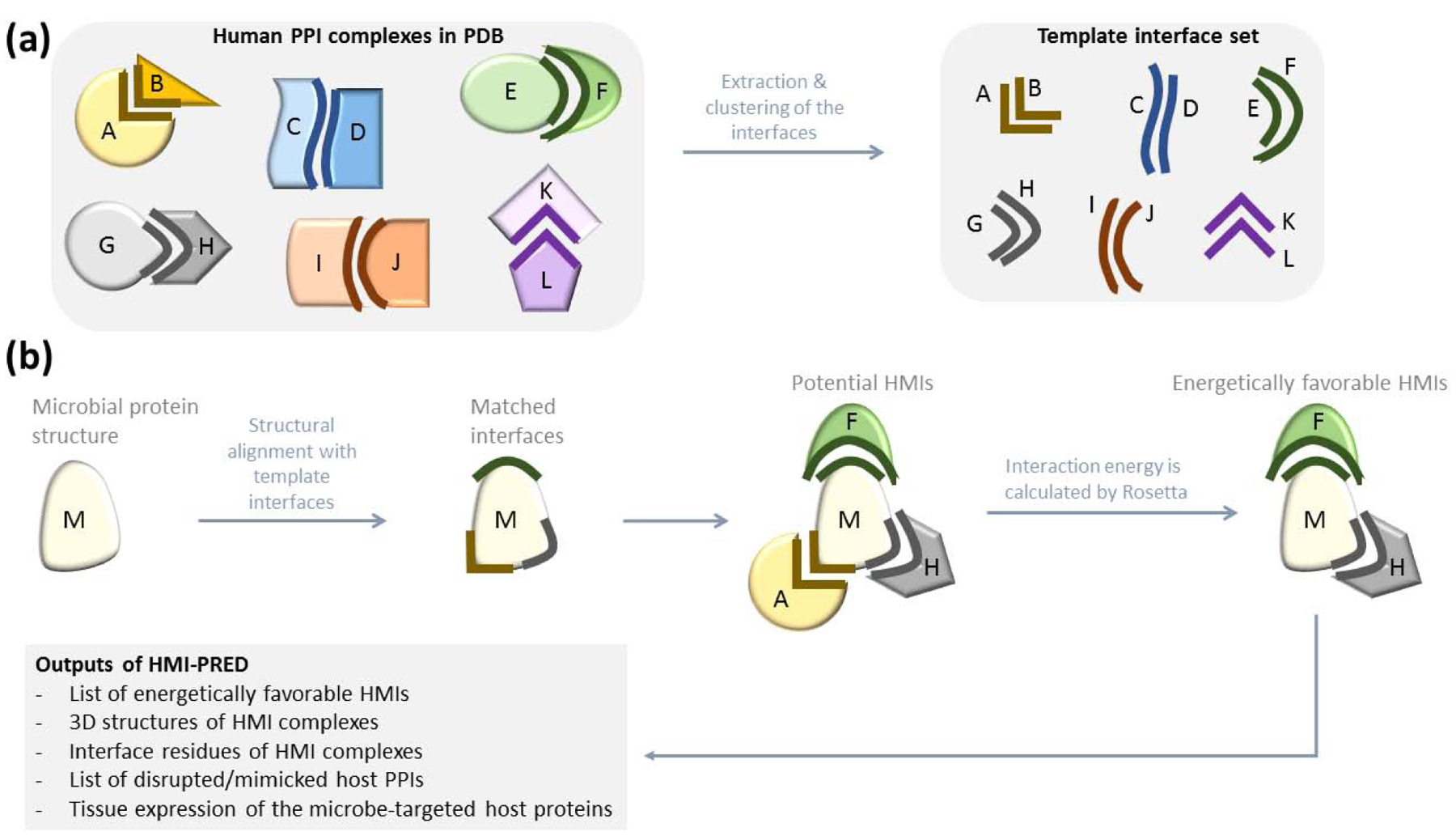
(a) Preparation of template interface set. We extract all human interfaces from PDB. Each interface has two faces and at least one face is from human. If both faces are from human, it is an endogenous interface. If one face is from human and the other face from another organism, it is an exogenous interface. Our non-redundant template interface set has 15.762 endogenous and 1.589 exogenous interfaces. (b) HMI-PRED workflow. Microbial protein structure is the only user input to HMI-PRED. If it has not been resolved yet, homology model can also be used. We first extract the surface of the microbial protein, then structurally align each template interface with the microbial protein surface and find some matches. If the microbial protein is aligned with the B-face of the interface, we assume that it can interact with the complementary face, A. We generate the potential HMI pairs based on this assumption. However, structural complementarity may not always guarantee electrochemical complementarity. Therefore, we calculate the interaction energy of the potential HMIs to pick the energetically favorable ones.
Workflow
Figure 1 illustrates the HMI-PRED workflow. First, users provide the PDB and chain IDs of the microbial (target) protein or upload their 3D structure (model) of the microbial protein in a PDB format. The surface of the target protein is extracted by NACCESS [42], which is then structurally aligned with the template interfaces by either TM-align [43] (default) or Multiprot [44]. For a structural match, at least 15 residues and 1 hotspot residue of the template interface should be aligned with the microbial protein. The aligned hotspot residue should be of the same type. The default threshold for TM-score is 0.25, but the user has an option to change it, in the “Advanced Options”. TM-score ranges between 0 and 1: higher thresholds allow more stringent and lower thresholds are for more relaxed alignment. The alignment threshold for Multiprot is 2 Å. Once a structural match is found, the microbial protein is transformed onto the aligned interface. The aligned protein of the template PPI is replaced with the microbial protein, creating the putative HMI pair between the microbial protein and the un-aligned face of the template. Potential HMI complexes have structural (geometrical) complementarity, but they may not have electrochemical complementarity, i.e. favorable interaction energy. To identify the energetically favorable HMI complexes, we exploit Rosetta (local refinement, Rosetta 2018.09.60072) [40]. HMIs with Rosetta interface scores (I_sc) smaller than −5 and total energy scores smaller than zero are assigned as energetically favorable. I_sc of template PPIs are also calculated to assess whether the microbial proteins will outcompete the bona fide partners of the microbe-targeted host proteins.
To help users to evaluate the HMI models further, we also provide details, such as (i) percent match (ratio of the number of aligned residues to the total number of residues in the aligned face of the interface), (ii) the assigned weight of the interface which depends on the aligned face size, (iii) the probability of template interfaces being real biological interfaces or crystal artifacts, and (iv) tissue expression of the host proteins. The larger the size of the interface, the larger its weight, under the premise that hijacking large human interfaces by microbes should be rewarded more than mimicry of small interfaces which may occur by chance: 0.5 for interfaces with less than 30 residues (n<30), 1 for 30<n<50, 1.5 for 50<n<80, and 2 for n>80 (users can assign different weights). We do not filter the results by weights or any other score, except I_sc. We exploited EPPIC (Evolutionary Protein-Protein Interface Classifier) [45] to deduce the probability of interfaces being biological. Tissue expression data are obtained from the Human Protein Atlas [46, 47]. For instance, if a microbe resides only in the gastrointestinal tract, the user can see which human proteins are expressed there. Please also see “Accuracy of structural alignment and scoring function” in Supplementary Materials, Tables S1–S4, and Figures S1–S2.
Template Interface Set
We extracted the interfaces from all cocrystal endogenous and exogenous human PPI structures in the PDB that are deposited as of January 3rd, 2019 (Figure 1a). Some PDB entries have huge protein assemblies and are unavailable in PDB format. We excluded them and chimeric proteins at the template generation step. Each interface has 2 faces (partners/chains/sides), one from each interacting partner in the host PPI. Interfaces are composed of interacting and nearby residues. If the distance between any atoms of two residues from two proteins is less than the sum of their van der Waals radii plus 0.5 Å, these residues are defined as interacting. Residues with Cα’s within a 6 Å radius of an interacting residue on the same protein are defined as ‘nearby residues’. Evolutionarily conserved interface residues are predicted as computational hotspots [48, 49]. All interfaces in our template set have at least 1 hotspot residue and more than 15 residues in at least one side for endogenous interfaces and in the non-human side of the exogenous interfaces.
Our initial template set included 53,339 redundant human interfaces. We clustered interfaces of the same human PPI and selected representatives of each cluster (see Supplementary Materials and Figure S3 for details). We ended-up with 17,351 structurally non-redundant interfaces in the template set that encompasses 15,762 endogenous and 1,589 exogenous host PPIs. 983 of 1,589 exogenous interactions include proteins from mouse, rat and other higher organisms. The remaining (606) exogenous interfaces correspond to HMIs with 199 microbial species, including bacteria, viruses, fungi, and protozoa. On average, only 3 HMIs per microbial species are known structurally (606/199). However, most microbes have more than 3 interactions with host proteins, and there are still hundreds of HMIs to be identified. This also points to the urgent need for computational approaches to complete the structural HMI space.
Since HMI-PRED finds favorable interactions with both faces of symmetric homodimer interfaces, the results become redundant. To avoid redundancy, we align the target protein with only one side of symmetric interfaces and with both sides of the non-symmetric interfaces. We determined the symmetric homodimer interfaces. 14,241 of the redundant 53,339 template interfaces and 3,617 of the 17,351 representative interfaces are symmetric homodimers (TM-threshold for symmetry is 0.80).
If the user wants to test only a subset of interfaces or an interface that is missing in our template set, such as an interface deposited to PDB after January 3rd, 2019, a list of up to 100 interfaces (PDB IDs with two chains, e.g. 1a0nAB) can be provided. HMI-PRED automatically downloads the corresponding PDBs and extracts the interfaces and hotspots.
Target Protein
The target is a microbial protein structure and it is the only mandatory user input to HMI-PRED. For available structures, the user provides the PDB ID of the target protein, including a chain identifier. Chain ID is case sensitive: “a” and “A” chains are not the same. HMI-PRED automatically downloads the structures.
If the microbial protein of interest has no resolved structure, the user can upload a homology model in a PDB format. If the microbial protein has a resolved structure and the user is interested in exploring the effects of a mutation on the microbial protein, the results from an uploaded in silico mutated structure in PDB format can be compared with those of the wild-type.
The HMI-PRED Web Server Usage
HMI-PRED has five pages:
the main “HMI” page, where users submit jobs;
the “Predictions” page where users can access the results of previous analyses;
the “Templates” page where the details of each template are found, such as the organism name, protein name and tissue expression data for each constituent face of the interface (Supplementary Materials and Figure S4).
the “Tutorial” page, where the detailed instructions can be found.
The “References” page, where “how to cite” information can be found.
HMI-PRED Main Page
This is the query submission page (Figure 2). PDB and chain ID of the target (microbial protein), or a homology model can be uploaded by the “Upload Target” button. In the default settings, our template interface set and TM-align will be used. The query will be placed in the HMI-PRED server queue. Users will be provided with a URL to track their job status. If users provide their emails, they will be notified upon the start and completion of the job.
Figure 2: HMI-PRED main page.

If the microbial protein has a resolved structure available in PDB, user enters its PDB and chain IDs. If it doesn’t have an available structure, the user can upload the homology model in PDB format. If the user wants to use the default template set and settings, s/he doesn’t need to change anything in “Template” and “Structural Alignment Method” parts; just entering the email address and clicking the “Submit” button is enough. When the run is completed, the user will receive a notification email with a URL which will take him/her to the results page. If the user wants to try an interface that is missing in our template set or try only a few interfaces instead of aligning 17.351 interfaces in the entire template set, s/he can give up to 100 interfaces. The default structural alignment tool is TM-align and the default alignment threshold is 0.25. The user can change the threshold: Higher thresholds will result in more stringent alignment. Users can also switch from TM-align to Multiprot.
Depending on the size of the target protein and number of matched interfaces, computation time can take several hours, but on average the results are returned to the user within 12–24 hours. If the target protein was analyzed before, the results from the previous query will be already in the database (i.e. “Predictions” page) and HMI-PRED will instantly direct the user to these, instead of re-modeling the HMIs.
Predictions Page
The results of all queries with the default settings and PDB identifiers (non-homology model targets) can be accessed on the Predictions page (Figure 3). However, the results of the homology models as targets or of non-default settings will not be stored. Results obtained through TM-align and Multiprot are stored separately. The default page shows TM-align results, but the user can switch to Multiprot by clicking on the “Multiprot” box.
Figure 3: “Predictions” tab, a repository for previous results.

Results of all jobs that use the default settings and a PDB ID as an input are stored for future access. TM-align and Multiprot results are stored separately. (a) This page shows a typical results table. The 10th and 11th columns have the interface scores (I_sc) for the HMI model and the template PPI, respectively. If the I_sc of the HMI is lower than the template PPI, it suggests that microbial protein has higher affinity to the human protein than the human protein’s bona fide partner in the template PPI. User can sort the columns that have PDB IDs, I_sc values, and microbial organism. (b) Clicking on the “View” button on the last column opens a pop-up page for visualizing the HMI model interactively. The user can rotate, change colors, download the structure in PDB format (“Download Structure” button), (c) retrieve the interface residues of the HMI, (d) visualize the HMI structure with highlighted interface residues, and (e) visualize the superimposed structures of the HMI and the template PPI. The superimposed structure will show how well the microbial protein hijacks the interface on the human protein (although they have different global structures).
The Predictions page displays a typical results table. Users can sort these and search by the microbial organism, PDB_IDs of the microbial/target protein and template interface. For each prediction, the PDB and chain IDs of the target, the microbial organism to which this target protein belongs, microbial protein name, its predicted interactor in the host, mimicked/disrupted human PPI (template PPI), I_sc for the HMI and I_sc of the template PPI, and tissue expression of the host protein are listed. For a given target, the Results table is also downloadable. There are additional details in the downloaded file, such as percent match, the weight of the interface, and the probability of template interfaces being biological interfaces (can be seen in Table S5). Functional annotation of each microbial protein results can also be accessed (Supplementary Materials and Figure S5).
The View button allows users to interactively visualize the HMI complex structures with a pop-up window. The structure and interface residues of the HMI model can also be downloaded through “Download Structure” and “Contacts of Interface Residues” links in the visualization window. “View More Detailed Structure” button opens another page, where the user can visualize the structures of the HMI model with highlighted interface residues and superimposed HMI and the corresponding template PPI structures. Both the HMIs and the superimposed structures are downloadable.
Case Studies
1- A pathogenic bacteria protein with a resolved 3D structure as an input
Subversion of host immunity is the most significant aim of pathogens. Epstein-Barr virus (EBV), an oncovirus, secretes lytic-cycle protein BARF1 to modulate host immunity [50]. BARF1 is present almost in all EBV-related malignancies. BARF1 is known to bind to human cytokine CSF1 (colony stimulating factor-1) with picomolar affinity (Figure S6a) [50]. The sequestration and inactivation of CSF1 by BARF1 also impairs the cooperative assembly of CSF1-CSF1R complex, which is a key to CSF1 function in innate and adaptive host immunity. HMI-PRED successfully identified the BARF1-CSF1 complex and the known interaction surface.
In our recent study where we investigated the HMIs of all oncoviruses, we presented 155 novel potential HMIs for EBV BARF1 protein, in addition to recovering the interaction between BARF1 and human CSF1 [51]. The results can be accessed in the “Predictions” tab, by filtering according to “Target” with 3uezA (PDB_ID of the target protein). Among the 155 potential targets, there are some host immunity proteins, such as T-cell receptor beta 1 chain C region (TRBC1) (Figure S6b–c), immunoglobulin constant heavy chains (IGHE), and tumor necrosis factor (TNFA). These HMIs potentially disrupt heterodimerization of TCR α and β chains, homodimerization of immunoglobulins, and homotrimerization of TNFA, impairing antigen recognition by TCR and immunoglobulins, and activation of TNFA signaling, respectively. These results suggest that BARF1 protein is capable of moonlighting: besides impacting the canonical pathway (CSF1R), it can also modulate alternative pathways of host immunity. These potential HMIs and their complex structures may provide a mechanistic understanding of how EBV circumvents recognition by the host and persists for decades. With only the list of pathogenic protein targets, without the structures of the HMI complexes and potentially affected endogenous human PPIs, it would be difficult to fully understand the underlying pathogenesis mechanisms of infections, host immune subversion, and malignant transformation of host cells by oncoviruses and oncobacteria.
2- A Commensal bacteria protein homology model as an input
Microbial anti-inflammatory molecule (MAM) protein from Faecalibacterium prausnitzii – one of the most abundant commensal bacteria in healthy individuals and deficient in Crohn’s disease patients – is able to inhibit the NF-κB pathway [52]. MAM protein does not have a resolved structure. We homology modeled it with I-TASSER [53] and modeled its HMIs. HMI-PRED predicted 35 HMIs for this protein (Table S5). MAM-targeted host proteins include polyubiquitin-C (UBC), Ubiquitin-like modifier-activating enzyme 1 (UBA1) and Ubiquitin conjugating enzyme E2N (UBE2N, Ubc13).
Ubiquitination is a reversible posttranslational modification and is integral to the activation of the NF-κB pathway. Distinct ubiquitination modes may lead to opposing results: while K48-linked ubiquitination targets a protein for degradation, the K63-linked ubiquitin chain serves as a docking platform for assembly of large protein complexes. For instance, the K63-linked ubiquitin chain attached to TRAF6 recruits the downstream players, TAB2/3 and NEMO (IKK- γ), which is vital for NF-κB activation. UBE2N-UB2V2 ubiquitin conjugating enzyme complex catalyzes the addition of K63-linked to TRAF6.
Since this job used a homology model as the input, the results are not stored in the “Predictions” tab. We found that MAM can hijack the interface on UB2V2 to bind to UBE2N and impede the UBE2N-UB2V2 enzyme complex (Figure S6d–f). Consequently, this could result in blocking NF-κB activation, and the production of pro-inflammatory cytokines. Our HMI complex may provide the conceptual foundation of the anti-inflammatory functions of MAM protein at the atomistic level and why its absence contributes to chronic inflammation in Crohn’s disease patients. These two case studies demonstrate the ability of HMI-PRED to identify disease-related proteins or implicate new proteins in disease pathogenesis.
Conclusions
There is a paramount need for identification of HMIs and resolving their complex structures to grasp the pathogenesis mechanisms of infections at the molecular level. HMI-PRED is a pioneering web server that allows fast, proteome-wide, and robust prediction of structural HMIs through interface mimicry. Its accuracy in recovering structurally known HMIs is 81% (through self-interfaces) and 78% (through alternative interfaces). It is applicable to any microbial protein, including proteins from pathogens and commensals. As the repository grows, we anticipate that it will fill the gap of to-date unknown virulence strategies of microbes. With the comprehensive map of structural HMIs, researchers can take further steps to develop efficient therapies against microbial infections.
Supplementary Material
Research highlights.
HMI-PRED is a user-friendly web server for host-microbe interaction prediction.
HMI-PRED relies on interface mimicry – hijacking of host protein binding surfaces.
Any microbial proteins, both pathogenic and commensal proteins, can be analyzed.
Acknowledgement
This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Franzosa EA, Xia Y. Structural principles within the human-virus protein-protein interaction network. Proc Natl Acad Sci U S A. 2011;108:10538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Guven-Maiorov E, Tsai CJ, Nussinov R. Pathogen mimicry of host protein-protein interfaces modulates immunity. Semin Cell Dev Biol. 2016;58:136–45. [DOI] [PubMed] [Google Scholar]
- [3].Guven-Maiorov E, Tsai CJ, Ma B, Nussinov R. Prediction of Host-Pathogen Interactions for Helicobacter pylori by Interface Mimicry and Implications to Gastric Cancer. J Mol Biol. 2017;429:3925–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Guven-Maiorov E, Tsai CJ, Ma B, Nussinov R. Interface-Based Structural Prediction of Novel Host-Pathogen Interactions. Methods Mol Biol. 2019;1851:317–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Guven-Maiorov E, Tsai CJ, Nussinov R. Structural host-microbiota interaction networks. PLoS Comput Biol. 2017;13:e1005579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tsai CJ, Lin SL, Wolfson HJ, Nussinov R. Protein-protein interfaces: architectures and interactions in protein-protein interfaces and in protein cores. Their similarities and differences. Crit Rev Biochem Mol Biol. 1996;31:127–52. [DOI] [PubMed] [Google Scholar]
- [7].Keskin O, Nussinov R. Favorable scaffolds: proteins with different sequence, structure and function may associate in similar ways. Protein Eng Des Sel. 2005;18:11–24. [DOI] [PubMed] [Google Scholar]
- [8].Keskin O, Nussinov R. Similar binding sites and different partners: implications to shared proteins in cellular pathways. Structure. 2007;15:341–54. [DOI] [PubMed] [Google Scholar]
- [9].Muratcioglu S, Guven-Maiorov E, Keskin O, Gursoy A. Advances in template-based protein docking by utilizing interfaces towards completing structural interactome. Curr Opin Struct Biol. 2015;35:87–92. [DOI] [PubMed] [Google Scholar]
- [10].Nicod C, Banaei-Esfahani A, Collins BC. Elucidation of host-pathogen protein-protein interactions to uncover mechanisms of host cell rewiring. Curr Opin Microbiol. 2017;39:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Brito AF, Pinney JW. Protein-Protein Interactions in Virus-Host Systems. Front Microbiol. 2017;8:1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods. 2013;10:730–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nourani E, Khunjush F, Durmus S. Computational approaches for prediction of pathogen-host protein-protein interactions. Front Microbiol. 2015;6:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mariano R, Wuchty S. Structure-based prediction of host-pathogen protein interactions. Curr Opin Struct Biol. 2017;44:119–24. [DOI] [PubMed] [Google Scholar]
- [15].Dyer MD, Murali TM, Sobral BW. Computational prediction of host-pathogen protein-protein interactions. Bioinformatics. 2007;23:i159–66. [DOI] [PubMed] [Google Scholar]
- [16].Krishnadev O, Srinivasan N. Prediction of protein-protein interactions between human host and a pathogen and its application to three pathogenic bacteria. Int J Biol Macromol. 2011;48:613–9. [DOI] [PubMed] [Google Scholar]
- [17].Doxey AC, McConkey BJ. Prediction of molecular mimicry candidates in human pathogenic bacteria. Virulence. 2013;4:453–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Huo T, Liu W, Guo Y, Yang C, Lin J, Rao Z. Prediction of host - pathogen protein interactions between Mycobacterium tuberculosis and Homo sapiens using sequence motifs. BMC Bioinformatics. 2015;16:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Eid FE, ElHefnawi M, Heath LS. DeNovo: virus-host sequence-based protein-protein interaction prediction. Bioinformatics. 2016;32:1144–50. [DOI] [PubMed] [Google Scholar]
- [20].Lee SA, Chan CH, Tsai CH, Lai JM, Wang FS, Kao CY, et al. Ortholog-based protein-protein interaction prediction and its application to inter-species interactions. BMC Bioinformatics. 2008;9 Suppl 12:S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Beltran JF, Brito IL. Host-microbiome protein-protein interactions capture mechanisms in human disease. BiRxiv. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Standfuss J. Structural biology. Viral chemokine mimicry. Science. 2015;347:1071–2. [DOI] [PubMed] [Google Scholar]
- [23].Evans P, Dampier W, Ungar L, Tozeren A. Prediction of HIV-1 virus-host protein interactions using virus and host sequence motifs. BMC Med Genomics. 2009;2:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Becerra A, Bucheli VA, Moreno PA. Prediction of virus-host protein-protein interactions mediated by short linear motifs. BMC Bioinformatics. 2017;18:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Porter KA, Xia B, Beglov D, Bohnuud T, Alam N, Schueler-Furman O, et al. ClusPro PeptiDock: efficient global docking of peptide recognition motifs using FFT. Bioinformatics. 2017;33:3299–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Davis FP, Barkan DT, Eswar N, McKerrow JH, Sali A. Host pathogen protein interactions predicted by comparative modeling. Protein Sci. 2007;16:2585–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Doolittle JM, Gomez SM. Structural similarity-based predictions of protein interactions between HIV-1 and Homo sapiens. Virol J. 2010;7:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].de Chassey B, Meyniel-Schicklin L, Aublin-Gex A, Navratil V, Chantier T, Andre P, et al. Structure homology and interaction redundancy for discovering virus-host protein interactions. EMBO Rep. 2013;14:938–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Drayman N, Glick Y, Ben-nun-shaul O, Zer H, Zlotnick A, Gerber D, et al. Pathogens use structural mimicry of native host ligands as a mechanism for host receptor engagement. Cell Host Microbe. 2013;14:63–73. [DOI] [PubMed] [Google Scholar]
- [30].Mahajan G, Mande SC. Using structural knowledge in the protein data bank to inform the search for potential host-microbe protein interactions in sequence space: application to Mycobacterium tuberculosis. BMC Bioinformatics. 2017;18:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Segura-Cabrera A, Garcia-Perez CA, Guo X, Rodriguez-Perez MA. A viral-human interactome based on structural motif-domain interactions captures the human infectome. PLoS One. 2013;8:e71526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhang A, He L, Wang Y. Prediction of GCRV virus-host protein interactome based on structural motif-domain interactions. BMC Bioinformatics. 2017;18:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tuncbag N, Gursoy A, Nussinov R, Keskin O. Predicting protein-protein interactions on a proteome scale by matching evolutionary and structural similarities at interfaces using PRISM. Nat Protoc. 2011;6:1341–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Baspinar A, Cukuroglu E, Nussinov R, Keskin O, Gursoy A. PRISM: a web server and repository for prediction of protein-protein interactions and modeling their 3D complexes. Nucleic Acids Res. 2014;42:W285–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kundrotas PJ, Anishchenko I, Badal VD, Das M, Dauzhenka T, Vakser IA. Modeling CAPRI targets 110–120 by template-based and free docking using contact potential and combined scoring function. Proteins. 2018;86 Suppl 1:302–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hwang H, Dey F, Petrey D, Honig B. Structure-based prediction of ligand-protein interactions on a genome-wide scale. Proc Natl Acad Sci U S A. 2017;114:13685–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Petrey D, Chen TS, Deng L, Garzon JI, Hwang H, Lasso G, et al. Template-based prediction of protein function. Curr Opin Struct Biol. 2015;32C:33–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Viswanathan R, Fajardo E, Steinberg G, Haller M, Fiser A. Protein-protein binding supersites. PLoS Comput Biol. 2019;15:e1006704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Basit AH, Abbasi WA, Asif A, Gull S, Minhas F. Training host-pathogen protein-protein interaction predictors. J Bioinform Comput Biol. 2018:1850014. [DOI] [PubMed] [Google Scholar]
- [40].Wang C, Bradley P, Baker D. Protein-protein docking with backbone flexibility. J Mol Biol. 2007;373:503–19. [DOI] [PubMed] [Google Scholar]
- [41].Moretti R, Lyskov S, Das R, Meiler J, Gray JJ. Web-accessible molecular modeling with Rosetta: The Rosetta Online Server that Includes Everyone (ROSIE). Protein Sci. 2018;27:259–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hubbard SJ, Thornton JM. ‘NACCESS’, Computer Program Department of Biochemistry and Molecular Biology, University College London; 1993. [Google Scholar]
- [43].Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Shatsky M, Nussinov R, Wolfson HJ. A method for simultaneous alignment of multiple protein structures. Proteins. 2004;56:143–56. [DOI] [PubMed] [Google Scholar]
- [45].Duarte JM, Srebniak A, Scharer MA, Capitani G. Protein interface classification by evolutionary analysis. BMC Bioinformatics. 2012;13:334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Uhlen M, Bjorling E, Agaton C, Szigyarto CA, Amini B, Andersen E, et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol Cell Proteomics. 2005;4:1920–32. [DOI] [PubMed] [Google Scholar]
- [47].Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. [DOI] [PubMed] [Google Scholar]
- [48].Cukuroglu E, Gursoy A, Keskin O. HotRegion: a database of predicted hot spot clusters. Nucleic Acids Res. 2012;40:D829–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tuncbag N, Gursoy A, Keskin O. Identification of computational hot spots in protein interfaces: combining solvent accessibility and inter-residue potentials improves the accuracy. Bioinformatics. 2009;25:1513–20. [DOI] [PubMed] [Google Scholar]
- [50].Elegheert J, Bracke N, Pouliot P, Gutsche I, Shkumatov AV, Tarbouriech N, et al. Allosteric competitive inactivation of hematopoietic CSF-1 signaling by the viral decoy receptor BARF1. Nat Struct Mol Biol. 2012;19:938–47. [DOI] [PubMed] [Google Scholar]
- [51].Guven-Maiorov E, Tsai CJ, Nussinov R. Oncoviruses Can Drive Cancer by Rewiring Signaling Pathways Through Interface Mimicry. Front Oncol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Quevrain E, Maubert MA, Michon C, Chain F, Marquant R, Tailhades J, et al. Identification of an anti- inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn’s disease. Gut. 2016;65:415–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.