

Abstract
Objective:
Sepsis recognition is a clinical challenge in children. We aimed to determine if peripheral blood gene expression profiles were associated with pathogen type and sepsis severity in children with suspected sepsis.
Methods:
Prospective pilot observational study in a tertiary pediatric emergency department with a convenience sample of children enrolled. Participants were >56 days and <18 years of age, had suspected sepsis, and had not received broad spectrum antibiotics in the prior 4 hours. Primary outcome was source pathogen, defined as confirmed bacterial source from sterile body fluid or confirmed viral source. Secondary outcome was sepsis severity, defined as maximum therapy required for shock reversal in the first 3 hospital days. We drew peripheral blood for RNA isolation at the time of sepsis protocol activation, obtained gene expression measures with the GeneChip™ Human Gene 2.0 ST Array, and conducted differential expression analysis.
Results:
We collected RNA samples from a convenience sample of 122 children with suspected sepsis and 12 healthy controls. We compared the 66 (54%) children with confirmed bacterial or viral infection, and found 558 differentially expressed genes, many related to interferon signaling or viral immunity. We did not find statistically significant gene expression differences in patients according to sepsis severity.
Conclusions:
The study demonstrate feasibility of evaluating gene expression profiling data in children emergently evaluated for sepsis. Our results suggest that gene expression profiling may facilitate identification of source pathogen in children with suspected sepsis, which could ultimately lead to improved tailoring of sepsis treatment and antimicrobial stewardship.
Introduction
Each year, there are as many as 42,000 children with severe sepsis in the U.S., resulting in significant morbidity and mortality.1–3 The complex pathophysiology of sepsis, involving both innate and adaptive immunity, results in circulatory and bio-energetic dysfunction leading to organ failure.4–7 However, identifying which children with common infectious complaints such as fever will go on to develop severe sepsis is an ongoing challenge in children, and has been identified as a research priority in pediatric emergency medicine.8,9
In addition to appropriate sepsis recognition, timely fluid resuscitation, and vasoactive therapy when needed, antimicrobial therapy is a cornerstone of sepsis treatment, and decreased mortality is associated with timely antibiotic treatment appropriate for patients’ bacterial source of infection.10 Timely and accurate identification of pathogen type can ensure that antibiotics are utilized appropriately, and curtailed when unnecessary.11–13
RNA expression profiling from host peripheral blood has been proposed as a strategy to advance pathogen-specific diagnostics.14 In this technique, RNA is isolated from whole blood cells and sequenced, and patterns of gene expression can be discerned. Because peripheral blood RNA is almost exclusively from white blood cells, this technique allows a detailed evaluation of the host immune response to infection. Differences in RNA expression profiles have been observed in bacterial vs. viral infections in febrile young infants15 and children.16 Transcriptomic profiling has also been used to attempt to risk-stratify children with sepsis in the intensive care unit.17–19 Previous studies have not involved children with suspected sepsis prior to initial resuscitation. We present the first transcriptomic study that evaluates pathogen type and severity of disease in children with suspected sepsis in a pediatric ED.
Methods
Subjects and Sample Collection
We recruited study subjects prospectively at the Children’s Hospital of Philadelphia (CHOP) ED between June 1, 2014 and November 30, 2016 using a convenience sample of eligible subjects with suspected sepsis and control subjects. We included subjects if they were between 57 days and 18 years of age and had suspected sepsis, defined as treatment according to an existing institutional protocol, as previously described.20 Briefly, children are treated on the sepsis protocol if they have meet criteria for a-d described here: a. abnormal vital signs for age, b. concern for infection, c. abnormal perfusion, mental status, or underlying high risk condition, and d. clinician concern for possible septic shock. We have demonstrated previously that this method identifies >96% of children with septic shock.20 We excluded subjects if they received broad spectrum antibiotics within 4 hours prior to study eligibility. Control subjects were children with ages between 57 days and 18 years who had an intravenous line (IV) placed for orthopedic fracture reduction, had no fever or infection in the week prior to enrollment, and had not received any antibiotics in the 2 weeks prior to enrollment as determined by parental report. Controls were selected from this population because they are the most common cohort in the ED to require a IV placement for a non-infectious indication. We determined antibiotic timing cutoffs used for exclusion a priori based on biologic plausibility. We collected peripheral blood samples during the subjects’ ED visit (1ml if <1 year of age, 3ml otherwise) using Tempus blood RNA tubes (Thermo Fisher Scientific) and stored samples at −80°C within 24 hours of collection. Additional bacterial cultures and laboratory tests including viral testing were conducted at the discretion of the treating clinical team. We obtained demographic and clinical information from subjects’ electronic health records21. Clinical variables included age, race, ethnicity, sex, complex chronic conditions22, pediatric intensive care unit (PICU) admission, need for invasive mechanical ventilation, white blood cell count (WBC), platelet count, venous lactate level, c-reactive protein (CRP), and procalcitonin. The CHOP Institutional Review Board approved the study under a waiver of consent for RNA sample collection. We attempted to approach guardians after admission but within 72 hours to consent to continue in the study and allow clinical data collection (except when explicity requested by treating team not to approach due to ongoing critical medical managements). If patients/families did not consent, the blood sample associated with that patient was discarded and not included in further analysis. (diagram in Figure 1) We adhered to Strengthening of Reporting of Observational studies in Epidemiology (STROBE guidelines) in reporting these results.
Figure 1:
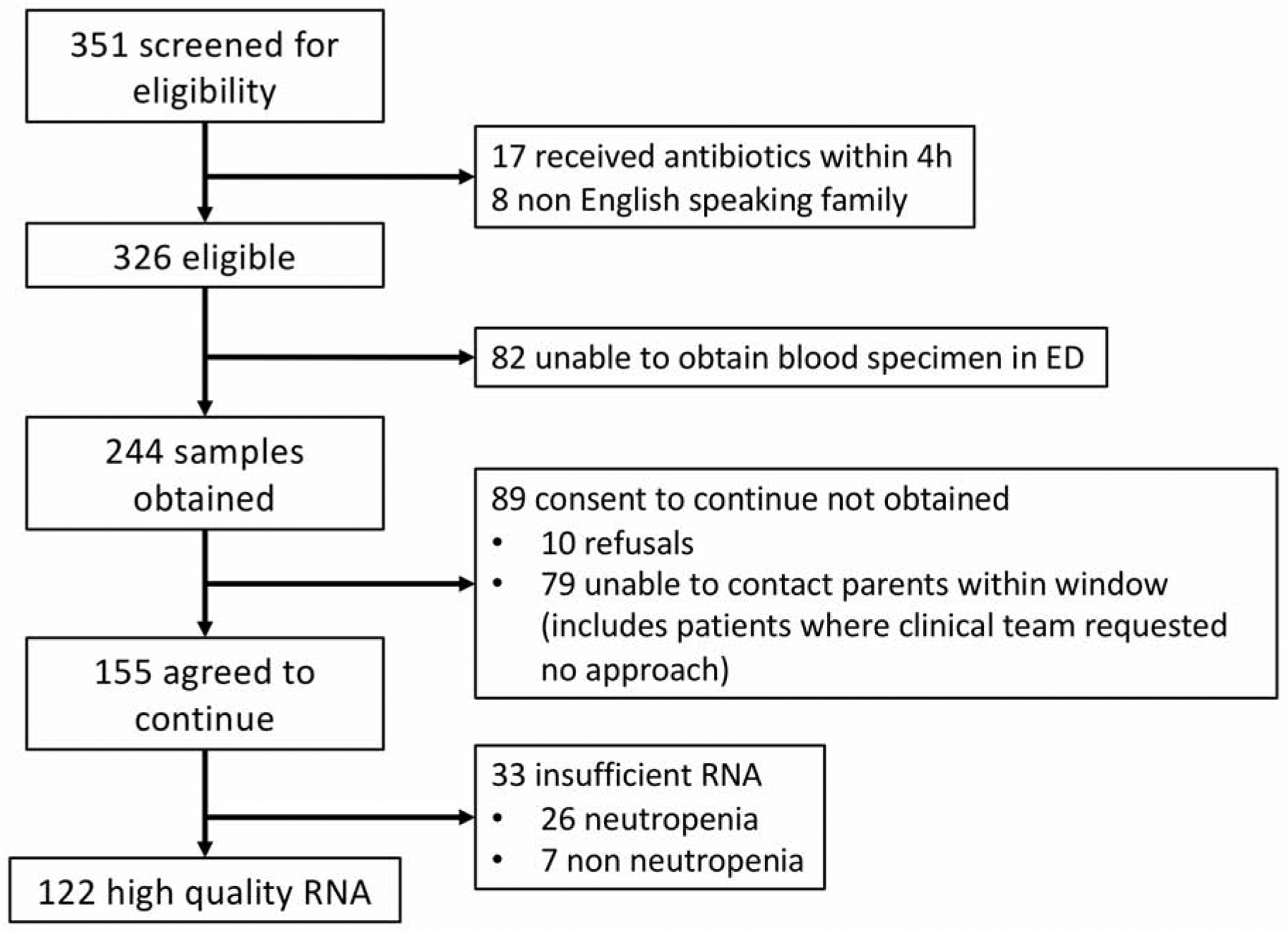
Enrollment diagram.
Sample Size Discussion
Microarray experiments are distinct from traditional biomedical studies where investigators typically test one or a few hypotheses. In microarray studies, each treatment or group comparison involves the testing of every gene on the chip, which in this case numbers more than 20,000. In these studies, novel strategies have been developed to facilitate statistical analysis. The term “Expected Discovery Rate” (EDR) is analogous to study power. Expected discovery rate is defined as the average power for all genes for which the null hypothesis is false in an experiment and can be interpreted as the expected proportion of genes that are truly differentially expressed that will appear to be differentially expressed in the assay.23 Microarray studies are also affected by multiple testing issues, so that one must consider both the alpha (α) level cut-off as well as the expected proportion of genes that are true positives. This is defined as the “False Discovery Rate” (FDR). Permutation formulas which include analysis of EDR, FDR, and α can be used to estimate sample size in microarray studies. These elements have been incorporated into a PowerAtlas”, which allows investigators to search a database of previously published microarray studies with similar characteristics to one’s proposed study in order to estimate sample size.23 Perusal of this database yielded several studies with characteristics similar to our proposed design which used a range of 7–15 patients per experimental group.24–26 We thus enrolled 12 control subjects, and estimated that the enrolled sample size would yield at least 12 patients with bacterial infections, 12 patients with viral infections, and 12 patients with the highest level of shock severity.
Determination of Infectious Pathogens
We determined type of infectious pathogen by medical record review and defined according to the following categories: bacterial: confirmed bacterial pathogen isolated from sterile body fluid; viral: positive viral testing from any body fluid; other: co-infections, suspected bacterial infection (e.g., treatment with a full course of antibiotics for pneumonia or skin/soft tissue infection in the absence of an identified organism) and/or fungal infections; no source: none of the above.
Determination of Shock Status and Sepsis Severity
We determined sepsis severity by medical record review by trained investigators as previously described, with established inter-rater reliability for each element of organ dysfunction as measured by Kendall’s co-efficient of concordance.27 Briefly, abstractors were trained in pre-study sessions using the established instrument described above. Each reviewed 3 pilot cases prior to full chart abstraction, and achieved 100% agreement with sepsis severity on these cases before abstraction began. As the same investigator reviewed records for both infectious pathogen and sepsis severity, these reviews were not blinded to additional outcomes. We defined septic shock using International Consensus Criteria modified by removing the requirement for treatment with 40 ml/kg of crystalloid fluid before shock determination,28 and compared samples from children with shock vs. no shock. These criteria remain the current definition of septic shock in children, despite the adoption of Sepsis-3 definitions for adults.29 We further classified samples by degree of sepsis as follows: vasopressor-responsive shock: septic shock resolved after treatment with vasoactive agents, including dopamine, epinephrine, or norepinephrine; fluid responsive shock: septic shock resolved after receipt of crystalloid fluid only; no shock: no septic shock. We determined shock status daily for the first 3 hospital days, and the worst value during this time period was used.
Descriptive Statistics
We used R software for statistical analyses.30 We assessed differences among demographic and clinical variables between groups with chi-squared tests for categorical variables and Kruskal-Wallis tests for continuous variables. We treated lab test results as categorical variables using the following a priori determined clinically relevant cut-off thresholds: white blood cell count >15,000/mm3, procalcitonin >0.5 ng/ml, c-reactive protein > 3.0 mg/dL, or venous lactate > 4 mmol/dL).
Gene Expression Microarray Sample Preparation and Analysis
We isolated and sequenced RNA from all study subjects. We extracted whole peripheral blood RNA using Tempus Spin RNA Isolation Kit (Thermo Fisher Scientific). We measured RNA quality with an Agilent 2100 Bioanalyzer (Agilent Technologies). Genome-wide gene expression measures were obtained using the Affymetrix GeneChip™ Human Gene 2.0 ST array, with standard quality assurance methods. Children with absolute neutrophil counts <1000 were excluded from the analysis given insufficient RNA. We analyzed raw intensity data using the RAVED pipeline (https://github.com/HimesGroup/raved) with steps that included obtaining quality metrics of raw intensity data using outlier scoring methods adapted from the arrayQualityMetrics R package,31 considering microarray scan date as a batch variable via principal component analysis, and processing raw intensities using robust multi-array average (RMA) with the oligo R package.32 We performed differential expression analysis with the limma R package,33 while using the Benjamini-Hochberg approach to correct for multiple comparisons made. We annotated microarray probes to official gene symbols using the hgu133plus2.db Bioconductor annotation package.31 Microarray data is available in the Gene Expression Omnibus (GEO) under accession GSE119217. We compared the following groups to identify expression patterns related to infectious pathogen type: 1) confirmed bacterial pathogen vs. control, 2) confirmed viral pathogens vs. control, and 3) confirmed bacterial vs. viral pathogens. Groups compared related to shock status were: 1) shock vs. no shock, 2) shock vs. control, and 3) no shock vs. control. Additionally, we compared vasopressor-responsive shock to fluid responsive shock to ensure shock status results were not driven by sepsis severity groups. We did not observe any appreciable microarray batch effects according to scan date [eFigure 1]. We included age (as a continuous variable) and sex as covariates in all differential expression analyses, as is typical in microarray experiments to account for their expected influence on gene expression. We used white blood cell count as an additional covariate in differential expression analyses to partially adjust for differences in cell composition of whole blood. We used a priori cut-offs of fold change >2 and adjusted p < 1 × 10 −4 to identify the top differentially expressed genes. We used the NIH Database for Annotation, Visualization and Integrated Discovery (DAVID) to perform gene ontological category enrichment analysis and functional annotation clustering. (options and annotation categories listed in the legend of eTable2)
Results
Subject Characteristics
We screened 351 patients with suspected sepsis in the appropriate age range for eligibility, and obtained gene expression data for 122 eligible subjects as well as 12 controls. Full enrollment diagram is depicted in Figure 1. We did not observe any statistically significant differences for sex or race/ethnicity between cases and controls, although controls were older (Table 1). Sixty-nine (57%) cases had at least one complex chronic condition, of which neuromuscular condition was the most common, while controls had none. Subjects with confirmed bacterial infection were more likely to have elevated CRP and to be admitted to the PICU than those with viral infection, while the distributions of age, sex, race/ethnicity, and presence of comorbid conditions were not significantly different between these two groups (Table 2). Sepsis patients had similar characteristics according to shock severity status, except for an expected difference in PICU admission rate, with all those requiring vasopressor therapy having a PICU admission, as well as 73% of those with shock responding to fluid therapy without vasopressor, vs. 37% of patients with no shock (Table e1).
Table 1.
Subject Characteristics. Overall characteristics of children enrolled in the study. For each category, N (%) are shown.
| Sepsis | Controls | |
|---|---|---|
| Age | ||
| 57 days - <1 year | 7 (5.7) | 0 (0) |
| 1 year - < 5 years | 39 (32) | 0 (0) |
| 5 years - <13 years | 39 (32) | 9 (75) |
| ≥ 13 years | 37 (30) | 3 (25) |
| Sex | ||
| Female | 67 (55) | 6 (50) |
| Race/Ethnicity | ||
| Black/African American | 24 (20) | 2 (17) |
| Non-Hispanic White | 53 (43) | 8 (67) |
| Hispanic | 24 (20) | 2 (17) |
| Asian | 13 (11) | 0 (0) |
| Missing | 8 (7) | 0 (0) |
| Payor | ||
| Medicaid | 50 (41) | - |
| Private | 67 (55) | - |
| Self-pay | 4 (3) | - |
| Any Complex Chronic Condition | 69 (57) | 0 (0) |
| Malignancy | 13 (11) | 0 (0) |
| Hematologic/Immune | 14 (11) | 0 (0) |
| Respiratory | 6 (5) | 0 (0) |
| Gastrointestinal | 7 (6) | 0 (0) |
| Metabolic | 11 (9) | 0 (0) |
| Neuromuscular | 34 (28) | 0 (0) |
| Cardiovascular | 13 (11) | 0 (0) |
| Renal | 8 (7) | 0 (0) |
| Other Congenital | 11 (9) | 0 (0) |
| Laboratory Test Results | ||
| WBC > 15 | 27 (22) | - |
| Platelet < 100 | 38 (31) | - |
| Lactate > 4 | 9 (7) | - |
| CRP > 3 | 62 (51) | - |
| Procal > 0.5 | 61 (50) | - |
| Invasive Ventilation | 11 (9) | - |
| PICU Admission | 63 (52) | - |
| Infectious Pathogen | ||
| Definite viral | 38 (31) | - |
| Definite bacterial | 28 (23) | - |
| Other* | 22 (18) | - |
| No source | 34 (28) | - |
| Shock Severity | ||
| No shock | 81 (66) | - |
| Fluid responsive | 30 (25) | - |
| Vasopressor responsive | 11 (9) | - |
Other includes suspected infection(s), fungal infection, definite co-infections
PICU: pediatric intensive care unit
Table 2.
Characteristics of Subjects According to Pathogen Status. Overall characteristics of children with a confirmed viral or bacterial infection. For each category, N (%) are shown. For variables with statistically significant differences between children with viral and bacterial infections, results are in italics with P-values reported.
| Bacterial | Viral | OR (95% CI) | |
|---|---|---|---|
| Age | |||
| 57 days - <1 year | 1 (4) | 1 (3) | reference |
| 1 year - < 5 years | 6 (21) | 17 (45) | 2.8 (−2.3, 4.4) |
| 5 years - <13 years | 12 (43) | 11 (29) | 0.9 (−3.4, 3.2) |
| ≥ 13 years | 9 (32) | 9 (24) | 1.0 (−3.3, 3.3) |
| Sex | |||
| Female | 14 (50) | 22 (58) | 1.4 (−0.7, 1.3) |
| Race/Ethnicity | |||
| Black/African American | 5 (18) | 8 (21) | reference |
| Non-Hispanic White | 14 (50) | 19 (50) | 0.9 (1.5, 1.1) |
| Hispanic | 4 (14) | 4 (11) | 0.6 (−2.3, 1.3) |
| Asian | 3 (11) | 4 (11) | 0.8 (−2.1, 1.8) |
| Missing | 2 (7) | 3 (8) | |
| Payor | |||
| Medicaid | 8 (29) | 20 (53) | reference |
| Private | 18 (64) | 18 (47) | 0.4 (−2.0,0.1) |
| Self-pay | 1 (4) | 0 (0) | NA |
| Any Complex Chronic Condition | 15 (54) | 19 (50) | 0.9 (−1.1, 0.8) |
| Malignancy | 2 (7) | 4 (11) | 1.5 (−1.3, 2.5) |
| Hematologic/Immune | 4 (14) | 5 (13) | 0.9 (−1.5, 1.4) |
| Respiratory | 2 (7) | 2 (5) | 0.7 (−2.5, 1.9) |
| Gastrointestinal | 2 (7) | 2 (5) | 0.7 (−2.5, 1.9) |
| Metabolic | 5 (18) | 2 (5) | 0.3 (−3.4, 0.3) |
| Neuromuscular | 8 (29) | 7 (18) | 0.6 (−1.8,0.6) |
| Cardiovascular | 2 (7) | 4 (11) | 1.5 (−1.3,2.5) |
| Renal | 4 (14) | 1 (3) | 0.2 (−4.8, 0.2) |
| Other Congenital | 1 (4) | 5 (13) | 4.1 (−0.5, 4.4) |
| Laboratory Test Results | |||
| WBC > 15 | 7 (25) | 4 (11) | 0.3 (−2.5, 0.4) |
| Platelet < 100 | 11 (39) | 11 (29) | 1.6 (−0.5, 1.7) |
| Lactate > 4 | 1 (4) | 3 (8) | 2.3 (−1.3, 3.9) |
| CRP > 3 | 21 (75) | 16 (42) | 0.2 (−3.2, −0.7) |
| Procalcitonin > 0.5 | 17 (61) | 21 (55) | 0.5 (−2.0, 0.5) |
| Invasive Ventilation | 4 (14) | 1 (3) | 0.2 (−4.8, 0.2) |
| PICU Admission | 18 (64) | 20 (53) | 0.4 (−1.5,0.5) |
| Shock Severity | |||
| No shock | 17 (61) | 28 (74) | reference |
| Fluid responsive | 9 (32) | 10 (26) | 0.7 (−1.5,0.7) |
| Vasopressor responsive | 2 (7) | 0 (0) | NA |
PICU: pediatric intensive care unit
Gene Expression Differences by Pathogen Type
Comparison of gene expression levels between children with confirmed viral vs. bacterial infection identified 558 differentially expressed probes with an adjusted p-value <0.05 (Figure 2A). Expression differences for children with confirmed viral infection vs. controls and confirmed bacterial infection vs. controls are shown in eFigure 2. Of the probes differentially expressed between children with a pathogen infection vs. controls, 5 probes were shared between children with confirmed viral or bacterial pathogen vs. controls (Figure 2B). Nearly half (263 of 558 genes) of the significantly differentially expressed genes in viral vs. bacterial infection were also significantly differentially expressed in viral infection vs. controls, while only 3 overlapped with the differentially expressed genes in bacterial infection vs. controls (Figure 2B).
Figure 2.
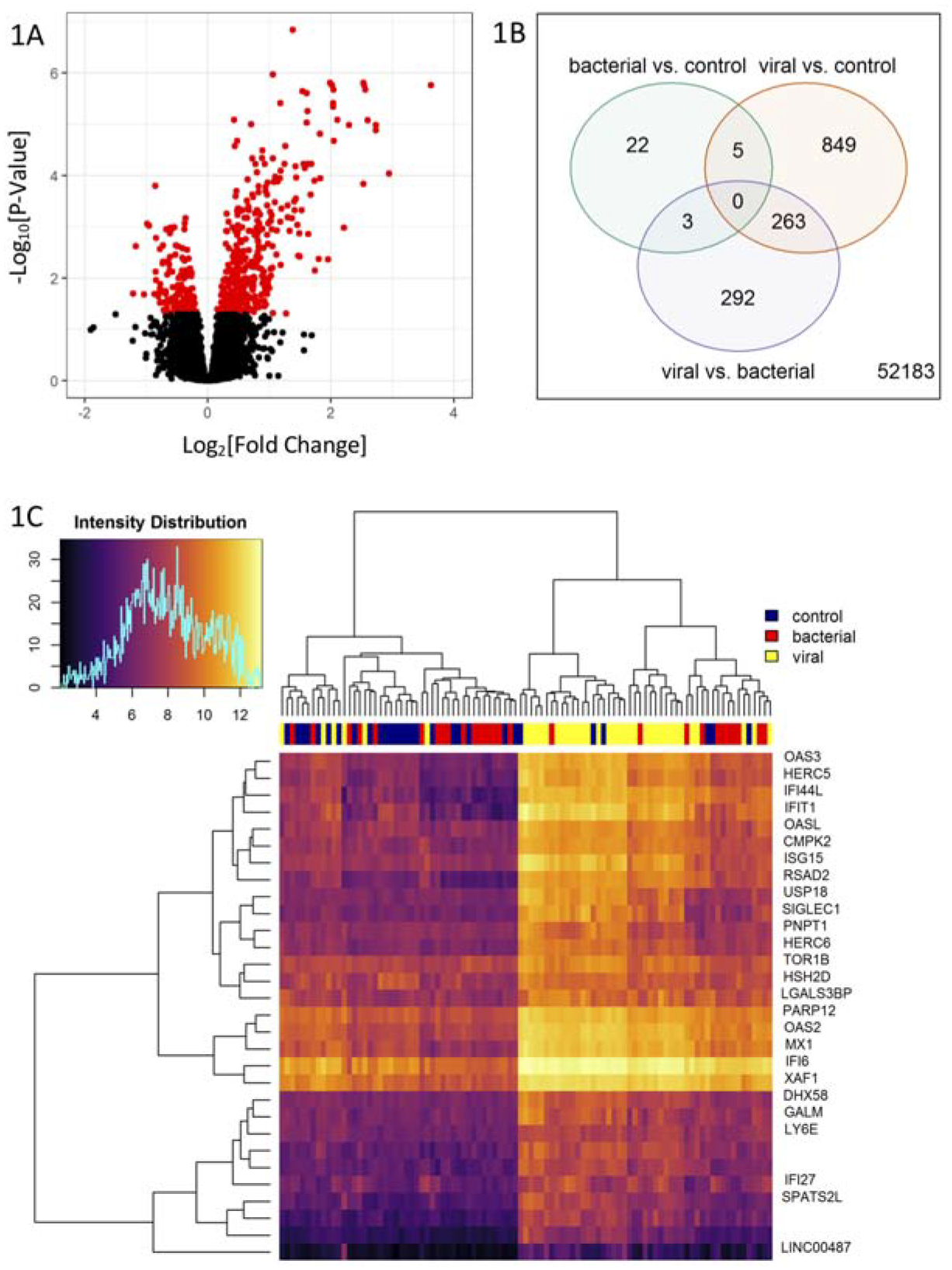
Gene Expression Differences According to Pathogen Type. A) Volcano plot of gene differential expression in children with confirmed viral vs. confirmed bacterial infections. The y-axis corresponds to the negative log (base 10) of adjusted P-values while the x-axis corresponds to the log (base 2) of the fold change of difference between categories. Differentially expressed genes according to an adjusted p-value <0.05 are colored in red. B) Venn diagram of the differential expressed genes from three two-group comparisons of children infected by bacterial pathogens vs. controls (green), children infected by viral pathogens vs. controls (orange), and children infected by viral pathogens vs. by bacterial pathogens (purple). C) Heatmap and dendrogram of 30 prioritized differential expressed genes in children infected by viral pathogens vs. by bacterial pathogens. Differentially expressed genes were selected according to an adjusted p-value <10−4 and an absolute log2 fold change >1.2. Hierarchical clustering is based on the Euclidean distance between the probe intensities.
To focus on genes that most distinguished viral from bacterial infection, we selected probes with an adjusted p-value <10−4 and an absolute log2 fold-expression change >1.2 for at least one of the three comparisons considered in Figure 1 (eTable 2 in the Supplement). Probes meeting these criteria were found for viral vs. bacterial (n=30) and viral vs. control (n=36) comparisons, but not for bacterial vs. control. Twenty-five probes met the criteria with similar effect size and significance in both the viral vs. bacterial and viral vs. control comparisons. Consistent with this overlap, hierarchical clustering of samples according to expression levels for the set of 30 probes in eTable 2 that differed between viral vs. bacterial infection showed that the expression pattern that distinguishes viral from bacterial infection tends to also distinguish viral infection from absence of sepsis (Figure 2C).
Ontological category enrichment analysis using the 263 genes that had an adjusted p-value <0.01 for differential expression in children with viral vs. bacterial infection identified six annotation clusters with enrichment scores >2.5. Gene Ontology categories related to viral infection, including response to virus, interferon-gamma-mediated signaling pathway, and double-stranded RNA binding (eTable 3 in the Supplement). A gene-based literature search for the function of the more stringently selected set of top viral vs. bacterial infection genes (adjusted p-value <10−4 and absolute log2 fold-expression change >1.2) found that most were interferon-pathway genes that respond to a broad range of viruses (eTable 4 in the Supplement).15
Repeating the differential expression analysis while adjusting for total peripheral WBC counts as a covariate found that although the number of significantly expressed genes (i.e., with adjusted p-value <0.05) was reduced from 558 to 490, the ranks of top probes were similar according to statistical significance, and the Pearson correlation coefficient of q-values between adjusted and unadjusted results was 0.92. Thus, the expression signatures identified in patients infected with viral vs. bacterial pathogens was not likely due to gross differences in white blood cell-type count.
Gene Expression Differences According to Shock Status and Sepsis Severity
Significant gene expression differences were observed between sepsis patients without shock vs. controls (Figure 4A) and between sepsis patients with shock vs. controls (Figure 4B). No significant differences in gene expression levels were observed between sepsis patients with shock vs. without shock (Figure 3D). A greater number of differentially expressed genes were found in patients without shock (2,681) than with shock (2,118) vs. controls, but an overlapping set of 1,550 probes significantly changed in both conditions (Figure 4D). Inspection of results for the probes with adjusted p-value <10−4 and absolute log2 fold-expression change >1.2 for at least one of the three comparisons considered in Figure 4, found that top gene expression changes in sepsis patients with and without shock vs. controls were consistent (i.e., similar effect size and significance), and thus, reflect sepsis status, rather than shock status (eTable 4 in the Supplement). Comparison of subgroups within shock: vasopressor-responsive to fluid-responsive shock subjects yielded no significant differences in gene expression.
Figure 3.
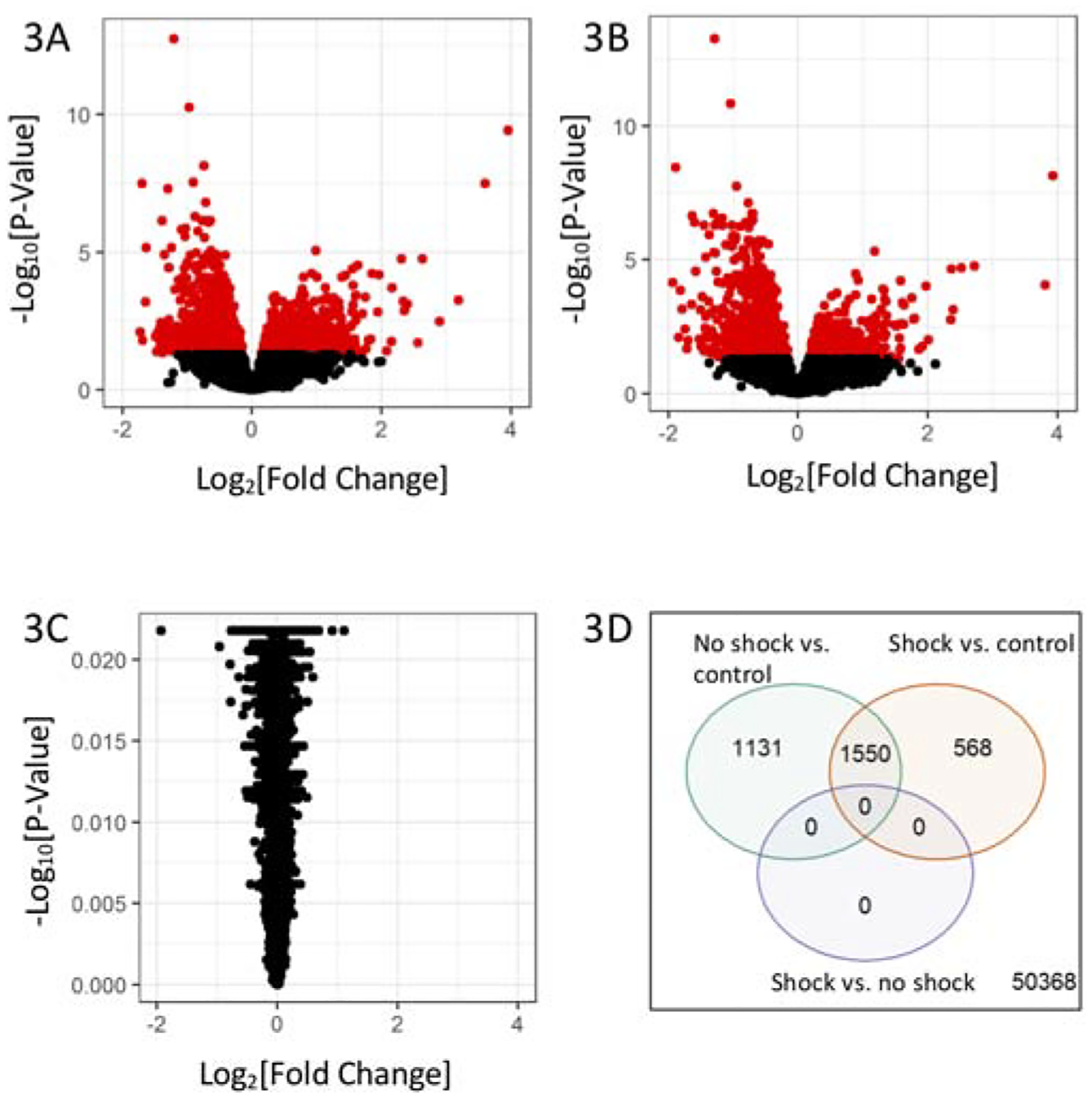
Gene Expression Differences According to Shock Status. A) Volcano plots of gene differential expression in children with no shock vs. controls, B) children with shock vs. control, and C) children with shock vs. with no shock. The y-axis corresponds to the negative log (base 10) of adjusted P-values while the x-axis corresponds to the log (base 2) of the fold change of difference between categories. Differentially expressed genes according to an adjusted p-value <0.05 are colored in red. D) Venn diagram of the differential expressed genes from three two-group comparisons of children with no shock vs. controls (green), children with shock vs. controls (orange), and children with shock vs. no shock (purple).
Limitations
There are several limitations to this study. First, it was a single center study with a relatively small sample size, and thus, generalizability outside a tertiary care specialty center with a complex patient population is limited. Second, we enrolled a convenience sample of subjects, and thus our findings may not be representative of all children with suspected sepsis. Of note, this convenience sample also excludes patients for whom samples were collected and later discarded due to caregiver refusal or failure to obtain consent for clinical data collection. However, the clinical data of our cohort compared to the entire suspected sepsis population at CHOP was similar in terms of age, gender, presence of comorbidity, and proportion requiring ICU care.20 Third, because we isolated RNA from whole blood, the expression signatures observed could represent differences in cell types, rather than gene expression. Although this concern is minimized because adjustment for WBC count did not change our results, we cannot entirely account for cell type-specific differences. Although this distinction is not critical when using whole blood as a diagnostic biomarker, it is critical to understand the biological mechanisms that are reflected in the gene expression changes observed. Fourth, this study represents RNA collected at one time point and does not account for duration of symptoms, thus we could not detect if RNA expression patterns change over time or if time of presentation relative to illness onset influenced expression patterns. However, as our goal was to evaluate gene expression profiles at the time of patient presentation, prior to therapeutic interventions, this information has its own import in emergency medicine evaluations. Finally, as these techniques and analytic platforms are relatively new, it is possible that some of the differences we found compared to previously published work were due either to differences in the gene chip or analytic platform utilized.
This study also had limitations related to pathogen and outcome determination. We were unable to perform comprehensive viral testing on all subjects, thus it is possible that there are some patients with undetected viral infections. Although this certainly could bias our results, this strategy has been utilized before in similar studies,15 and was not financially feasible in this study. Additionally, investigators reviewed medical records comprehensively and, were thus, not blinded to sepsis severity and pathogen determination or vice versa. Because sepsis severity is clinically distinct from pathogen (ie a patient can have bacteremia with or without septic shock), this is unlikely to have a large effect on our results. Second, and related, is that the large gene expression differences identified based on pathogen type could obscure differences based on sepsis severity because patients in each sepsis severity category had a mixture of pathogen types. One sensitivity analysis that could help to address this issue of multiple group comparisons is to further adjust q values for the 6 potential patient groups. Because the most significant genes identified had q values on the order of 10−6, the observed differences would still hold. Finally, although we have enrolled a heterogeneous group of patients with different ages, comorbidities, and timing of presentation which may limit our abilities to identify molecular mechanisms due to gene expression differences, this heterogeneity precisely describes the group of patients in which advanced diagnostic decisions must be made in the emergency department, and thus is an appropriate group to derive and validate gene expression based diagnostic tools.
Discussion
We present the first effort to characterize gene expression profiles of children with suspected sepsis in an emergency setting, demonstrating the feasibility of collecting high quality RNA samples from potentially critically ill pediatric patients prior to any emergent care resuscitation efforts. Although this study involved a relatively small sample size from a convenience sample of patients, we were able to identify gene expression patterns that distinguished samples according to viral vs. bacterial pathogen type. We did not observe significant gene expression changes according to shock status or sepsis severity.
The potential for an early and accurate mechanism to identify bacterial sepsis in pediatric ED patients carries important implications for appropriately targeting therapy as well as antibiotic stewardship. To that end, our current, and prior studies by others, have sought to identify peripheral blood biomarkers as a strategy to advance pathogen diagnostics and therapy.15,16,36–39 Among the previous studies that have proposed gene expression signatures for pathogen diagnostics is one by Herberg and colleagues, who described a two-transcript host RNA signature based on IFI44L and FAM89A that identified bacterial infection diagnosis in febrile children,16 and was later validated in febrile young infants.41 Consistent with their findings, IFI44L was among our 30 top differentially expressed transcripts; we were unable to validate FAM89A because this gene was not represented on our gene expression microarray. While the Herberg study differed from ours, as they investigated children with febrile illness regardless of clinician concern for sepsis and included a large number of children with meningococcal disease, replication of IFI44L suggests that its ability to distinguish viral illness is generalizable and supports our own findings.
Another proposed classifier by Sweeney et al. was based on a meta-analysis of several publicly available datasets.42 After identifying a seven-gene signature composed of IFI27, JUP, LAX1, HK3, TNIP1, GPAA1 and CTSB that robustly discriminated bacterial from viral infections, they used a metascore of the genes’ expression levels to create an antibiotic decision model that could be used for diagnostic purposes. Of their seven genes, only two (IFI27 and JUP) were significantly differentially expressed between children with viral vs. bacterial infections in our study. Because the Sweeney meta-analysis included several heterogeneous individual studies consisting of children and adults with diverse clinical conditions, the lack of full generalizability to our study may reflect differences in study designs.
In addition to searching for potential diagnostic biomarkers, gene expression studies offer hypotheses for understanding biological mechanisms underlying disease. Our study, as well as efforts such as those by Herberg et al. and Sweeney et al., have consistently found that most significant pathogen-type differences in gene expression are in viral immunity genes, including interferon signaling (e.g., IFI27, IFIT1)43 and (anti-)viral immune response (e.g., IFI44L, OAS1, OAS2, OAS3).16,37,38,43 This shared ontological enrichment, along with the observation that the gene expression signature separating viral-infection samples from controls is stronger than that separating bacterial-infection samples from controls suggests that the robust gene expression signatures are driven by viral infection.
We did not identify significant gene expression differences based on sepsis severity. In previous studies, Wong and colleagues were able to identify significant changes in RNA expression among critically ill children with sepsis that are associated with clinical outcomes.44,45 Specifically, they identified patterns of RNA expression associated with increased mortality and differential response to steroid therapy.46 Importantly, Wong’s work has focused on using RNA expression patterns as predictors of complicated clinical course, not as a predictor of infectious pathogen. An additional difference in recruitment between the studies by Wong and ours is that we enrolled children with suspected sepsis upon presentation to the ED, while Wong enrolled children on arrival to the PICU after initial resuscitation was complete. Future studies from large multicenter cohorts with numbers sufficient to evaluate classes of sepsis severity across individual pathogen types, may elucidate these patterns more clearly. In addition, collection of samples from well appearing children with fever, may enable the identification of expression changes across a broader range of sepsis severity levels. It is important to note that our main comparison was a nested analysis between sepsis patients with and without shock. We checked the difference between shock vs. control and no shock vs. control to confirm that the two sepsis groups had significant changes compared to healthy individuals. We did not compare all sepsis patients (N=122) versus the control group precisely because the demographic/clinical differences between groups would make results difficult to interpret. We controlled for age and sex in the differential expression analysis, and this yielded similar results as the unadjusted ones. We did not adjust for race because 8 cases had missing values and there were no Asian subjects among controls. While this is certainly a limitation, the proportions across remaining categories were comparable, and thus, expression results were not dependent on this confounder. Therefore, it is unlikely that the significant differences of shock vs. control and no shock vs. control were due to demographic differences
Although in the current state, RNA expression-based testing cannot be performed in a sufficiently timely manner to influence ED decision making, these techniques are on the horizon and have the potential to impact emergency care in coming years. Once these techniques are in place, it may be possible to use these assay types to appropriately tailor antimicrobial therapy and/or ensure appropriate disposition for patients based on risk of organ dysfunction. Although our current system of ED based sepsis recognition works well, with 96% sensitivity, assays such as RNA based tools could bring objectivity to some of the currently clinician based subjective elements and enhance generalizability. In addition, these novel tools, particularly those that incorporate both clinical and biomarker variable types, may improve differentiation of patients who require early treatment to avert poor outcome from those who would have improved without aggressive treatment. Alternately, pathogen based testing could be employed in the inpatient setting in antibiotic stewardship efforts to appropriately curtail unnecessary antibiotic use. Perhaps even more importantly, this type of detailed gene expression information could help to guide the use specific targeted therapies as they are developed, and thus could form the foundations of molecular precision medicine in the ED.
In conclusion, we identified gene expression changes that distinguished children with suspected sepsis infected with confirmed bacterial vs. viral pathogen types. Before it can be used as a diagnostic biomarker, larger independent studies are needed to validate this expression signature and to increase our ability to understand classes of sepsis severity across individual pathogen types.
Supplementary Material
Funding Source:
This study was funded in part by NIH grants K23 HD082368, R01 HL133433, R01 HL141992. Funders were not involved in design and conduct of the study; collection, management, analysis, interpretation of the data; preparation, review, or approval of the manuscript.
Financial Disclosure: The authors have no financial relationships relevant to this article to disclose.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.34,35,40
Meetings:
This work was presented in part at the Pediatric Academic Societies Meeting Toronto, CA May 2018.
Conflict of Interest:
The authors have no conflicts of interest to disclose.
References
- 1.Balamuth F, Weiss SL, Neuman MI, et al. Pediatric severe sepsis in U.S. children’s hospitals. Pediatr Crit Care Med. 2014;15(9). doi: 10.1097/PCC.0000000000000225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartman ME, Linde-Zwirble WT, Angus DC, Watson RS. Trends in the epidemiology of pediatric severe sepsis*. Pediatr Crit Care Med. 2013. doi: 10.1097/PCC.0b013e3182917fad [DOI] [PubMed] [Google Scholar]
- 3.Ruth A, McCracken CE, Fortenberry JD, Hall M, Simon HK, Hebbar KB. Pediatric Severe Sepsis. Pediatr Crit Care Med. 2014. doi: 10.1097/PCC.0000000000000254 [DOI] [PubMed] [Google Scholar]
- 4.Arulkumaran N, Deutschman CS, Pinsky MR, et al. Mitochondrial function in sepsis. Shock. 2016. doi: 10.1097/SHK.0000000000000463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernard AM, Bernard GR. The immune response: Targets for the treatment of severe sepsis. Int J Inflam. 2012. doi: 10.1155/2012/697592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wynn J, Cornell TT, Wong HR, Shanley TP, Wheeler DS. The Host Response to Sepsis and Developmental Impact. Pediatrics. 2010. doi: 10.1542/peds.2009-3301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wynn J, Cvijanovich N, Allen GL, et al. The influence of developmental age on the early transcriptomic response of children with septic shock. Mol Med. 2011. doi: 10.2119/molmed.2011.00169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thompson GC, Macias CG. Recognition and Management of Sepsis in Children: Practice Patterns in the Emergency Department. J Emerg Med. 2015;49(4):391–399. doi: 10.1016/j.jemermed.2015.03.012 [DOI] [PubMed] [Google Scholar]
- 9.Stoner MJ1, Mahajan P2, Bressan S3, Lam SHF4, Chumpitazi CE5, Kornblith AE6, Linakis SW1, Roland D7, Freedman SB8, Nigrovic LE9, Denninghoff K10, Ishimine P11KN. Pediatric Emergency Care Research Networks: A Research Agenda. Acad Emerg Med. 2018;25(12):1336–1344. [DOI] [PubMed] [Google Scholar]
- 10.Gaieski DF, Mikkelsen ME, Band RA, et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit Care Med. 2010. doi: 10.1097/CCM.0b013e3181cc4824 [DOI] [PubMed] [Google Scholar]
- 11.Pulia MS, Redwood R, Sharp B. Antimicrobial Stewardship in the Management of Sepsis. Emerg Med Clin North Am. 2017. doi: 10.1016/j.emc.2016.09.007 [DOI] [PubMed] [Google Scholar]
- 12.Masterton RG. Antibiotic De-Escalation. Crit Care Clin. 2011. doi: 10.1016/j.ccc.2010.09.009 [DOI] [PubMed] [Google Scholar]
- 13.Afshari A, Harbarth S. Procalcitonin as diagnostic biomarker of sepsis. Lancet Infect Dis. 2013. doi: 10.1016/S1473-3099(13)70026-4 [DOI] [PubMed] [Google Scholar]
- 14.Ramilo O, Mejias A, Mahajan P, Kuppermann N. RNA signature test to distinguish bacterial from viral infection. J Pediatr. 2017. doi: 10.1016/j.jpeds.2016.12.066 [DOI] [PubMed] [Google Scholar]
- 15.Mahajan P, Kuppermann N, Mejias A, et al. Association of RNA biosignatures with bacterial infections in febrile infants aged 60 days or younger. JAMA - J Am Med Assoc. 2016. doi: 10.1001/jama.2016.9207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herberg JA, Kaforou M, Wright VJ, et al. Diagnostic test accuracy of a 2-transcript host RNA signature for discriminating bacterial vs viral infection in febrile children. JAMA - J Am Med Assoc. 2016. doi: 10.1001/jama.2016.11236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong HR, Cvijanovich N, Allen GL, et al. Genomic expression profiling across the pediatric systemic inflammatory response syndrome, sepsis, and septic shock spectrum. Crit Care Med. 2009. doi: 10.1097/CCM.0b013e31819fcc08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cornell TT, Wynn J, Shanley TP, Wheeler DS, Wong HR. Mechanisms and Regulation of the Gene-Expression Response to Sepsis. Pediatrics. 2010. doi: 10.1542/peds.2009-3274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong HR, Weiss SL, Giuliano JS, et al. Testing the prognostic accuracy of the updated pediatric sepsis biomarker risk model. PLoS One. 2014. doi: 10.1371/journal.pone.0086242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balamuth F, Alpern ER, Abbadessa MK, et al. Improving Recognition of Pediatric Severe Sepsis in the Emergency Department: Contributions of a Vital Sign–Based Electronic Alert and Bedside Clinician Identification. Ann Emerg Med. 2017;70(6). doi: 10.1016/j.annemergmed.2017.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balamuth F, Alpern ER, Grundmeier RW, et al. Comparison of Two Sepsis Recognition Methods in a Pediatric Emergency Department. Acad Emerg Med. 2015;22(11). doi: 10.1111/acem.12814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feudtner C, Hays RM, Haynes G, Geyer JR, Neff JM, Koepsell TD. Deaths Attributed to Pediatric Complex Chronic Conditions: National Trends and Implications for Supportive Care Services. Pediatrics. 2001. doi: 10.1542/peds.107.6.e99 [DOI] [PubMed] [Google Scholar]
- 23.Page GP, Edwards JW, Gadbury GL, et al. The PowerAtlas: A power and sample size atlas for microarray experimental design and research. BMC Bioinformatics. 2006. doi: 10.1186/1471-2105-7-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bull TM, Coldren CD, Moore M, et al. Gene microarray analysis of peripheral blood cells in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2004. doi: 10.1164/rccm.200312-1686OC [DOI] [PubMed] [Google Scholar]
- 25.Reghunathan R, Jayapal M, Hsu LY, et al. Expression profile of immune response genes in patients with severe acute respiratory syndrome. BMC Immunol. 2005. doi: 10.1186/1471-2172-6-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Segman RH, Shefi N, Goltser-Dubner T, Friedman N, Kaminski N, Shalev AY. Peripheral blood mononuclear cell gene expression profiles identify emergent post-traumatic stress disorder among trauma survivors. Mol Psychiatry. 2005. doi: 10.1038/sj.mp.4001636 [DOI] [PubMed] [Google Scholar]
- 27.Balamuth F, Weiss SL, Hall M, et al. Identifying Pediatric Severe Sepsis and Septic Shock: Accuracy of Diagnosis Codes. J Pediatr. 2015;167(6). doi: 10.1016/j.jpeds.2015.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldstein B, Giroir B, Randolph A. International pediatric sepsis consensus conference: Definitions for sepsis and organ dysfunction in pediatrics. In: Pediatric Critical Care Medicine.; 2005. doi: 10.1097/01.PCC.0000149131.72248.E6 [DOI] [PubMed] [Google Scholar]
- 29.Singer M, Deutschman CS, Seymour C, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA - J Am Med Assoc. 2016. doi: 10.1001/jama.2016.0287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.TEam RC. R: A language and environment for statistical computing R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 31.Kauffmann A, Gentleman R, Huber W. arrayQualityMetrics - A bioconductor package for quality assessment of microarray data. Bioinformatics. 2009. doi: 10.1093/bioinformatics/btn647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carvalho BS, Irizarry RA. A framework for oligonucleotide microarray preprocessing. Bioinformatics. 2010. doi: 10.1093/bioinformatics/btq431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015. doi: 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liaw A, Wiener M. Classification and Regression by Random Forest. R news. 2002. doi: 10.1177/154405910408300516 [DOI] [Google Scholar]
- 35.Díaz-Uriarte R, Alvarez de Andrés S. Gene selection and classification of microarray data using random forest. BMC Bioinformatics. 2006. doi: 10.1186/1471-2105-7-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramilo O, Allman W, Chung W, et al. Gene expression patterns in blood leukocytes discriminate patients with acute infections. Blood. 2007. doi: 10.1182/blood-2006-02-002477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Herberg JA, Kaforou M, Gormley S, et al. Transcriptomic profiling in childhood H1N1/09 influenza reveals reduced expression of protein synthesis genes. J Infect Dis. 2013. doi: 10.1093/infdis/jit348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu X, Yu J, Crosby SD, Storch G a. Gene expression profiles in febrile children with defined viral and bacterial infection. Proc Natl Acad Sci U S A. 2013. doi: 10.1073/pnas.1302968110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sweeney TE, Wong HR, Khatri P. Robust classification of bacterial and viral infections via integrated host gene expression diagnostics. Sci Transl Med. 2016. doi: 10.1126/scitranslmed.aaf7165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mar KB, Rinkenberger NR, Boys IN, et al. LY6E mediates an evolutionarily conserved enhancement of virus infection by targeting a late entry step. Nat Commun. 2018. doi: 10.1038/s41467-018-06000-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaforou M, Herberg JA, Wright VJ, Coin LJM, Levin M. Diagnosis of bacterial infection using a 2-transcript host RNA signature in febrile infants 60 days or younger. JAMA - J Am Med Assoc. 2017. doi: 10.1001/jama.2017.1365 [DOI] [PubMed] [Google Scholar]
- 42.Sweeney TE, Azad TD, Donato M, et al. Unsupervised Analysis of Transcriptomics in Bacterial Sepsis Across Multiple Datasets Reveals Three Robust Clusters. Crit Care Med. 2018. doi: 10.1097/CCM.0000000000003084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schoggins JW, Wilson SJ, Panis M, et al. A diverse range of gene products are effectors of the type i interferon antiviral response. Nature. 2011. doi: 10.1038/nature09907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wong H, Freishtat R, Monaco M, Odoms K, Shanley TP. Leukocyte subset-derived genomewide expression profiles in pediatric septic shock. Pediatr Crit Care Med. 2010. doi: 10.1097/PCC.0b013e3181c519b4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wong HR, Shanley TP, Sakthivel B, et al. Genome-level expression profiles in pediatric septic shock indicate a role for altered zinc homeostasis in poor outcome. Physiol Genomics. 2007. doi: 10.1152/physiolgenomics.00024.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Menon K, Wong HR. Corticosteroids in pediatric shock: A call to arms. Pediatr Crit Care Med. 2015. doi: 10.1097/PCC.0000000000000513 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.