

Abstract
Mutations in RNF216 have been found to be associated with autosomal recessive Huntington‐like disorder. Here, we describe a patient with Huntington‐like disorder caused by a novel de novo RNF216 mutation. The patient started to have choreatic movements of both hands, slowly progressing to head, face, and four extremities, with prominent cognitive deterioration. White matter lesions in cerebral hemispheres and brainstem, cerebellar atrophy, and low gonadotropin serum levels have been demonstrated. We have identified a homozygous deletion of exon 2 in the RNF216 gene by whole‐exome sequencing. Our findings increased genetic knowledge of autosomal recessive Huntington‐like disorder and extended the ethnic distribution of RNF216 mutations.
Introduction
Huntington‐like disorder (HDL) was described with characteristic clinical features of chorea, behavioral disturbances, dementia, and lack of the CAG repeat expansion in the HTT gene. 1 In the last few years, a number of different mutations in genes, such as C9orf72, ATXN1, ATXN2, ATXN3, PRNP, TBP, JPH3, DRPLA, PANK2, and FTL, have been shown to associate with HDL. 1 , 2 Recently, Santens et al. 3 have reported that RNF216 mutations were a rare cause of autosomal recessive HDL. Here, we report a Chinese patient with autosomal recessive HDL caused by a novel de novo mutation in the RNF216 gene.
Methods
Subjects
The HDL patient and her family members were referred to the neurological ward at Huashan Hospital affiliated to Fudan University in Shanghai for clinical evaluation and diagnostic investigations. All the subjects, including the proband and her parents, provided written informed consent.
Clinical evaluation
Medical histories and physical examinations were analyzed. Ancillary tests, such as hormonal evaluation, brain MRI, and neurologic examination, were performed in the proband and her parents. Brain DTI images were acquired and compared with those from three gender‐ and age‐ (<5 years apart) matched healthy controls. The parameters assessed in the patient were defined as abnormal when the mean values were more than 2 standard deviations lower or higher than those of the normal controls.
Genetic analyses
First, the proband had been previously screened by our targeted sequencing panel (HTT, C9orf72, ATXN1, ATXN2, ATXN3, FRDA, PRNP, TBP, JPH3, DRPLA, CHAC, XK, TITK1, PANK2, and FTL). Mutations in these genes were excluded. Subsequently, whole‐exome sequencing was performed using the Illumina Hiseq sequencing platform. Three exons (exons 2, 3, and 17) of the RNF216 gene were amplified by real‐time quantitative PCR and analyzed by electrophoresis in her parents.
Results
The proband was a 38‐year‐old female with normal physical and intellectual development. After graduating from a technical secondary school, she became a kindergarten teacher. She developed choreatic movements of both hands at the age of 29. In the next year, she progressed to choreatic movements of head, face, and four extremities, which incapacitated her for work. Her mother also noted mastatrophy and intermittent lactation, but could not remember more details. The patient was diagnosed with dysmyotonia in another hospital and began treatment with haloperidol, trihexyphenidyl, and tiapride. Symptoms were controlled in short time. Then her disease progressively worsened, and there was a need for support in daily activities to the degree that she became totally dependent. She also developed prominent cognitive deterioration 2 years ago. A comprehensive neuropsychological battery was administrated at the local hospital on 21 November 2018. The intelligence quotient and memory quotient were 57 and 15, respectively, based on Wechsler Adult Intelligence Scale‐Revised China (WAIS‐RC) 4 and Wechsler Memory Scale. 5 The Hamilton Anxiety and Depression Rating Scale (HAMA and HAMD) indicated that she displayed mild anxiety and depression (HAMA score = 12 and HAMD score = 14).
On examination, she looked skinny and body mass index was 18.49 (156 cm in height and 45 kg in weight). The Mini‐Mental State Examination (MMSE) and Montreal Cognitive Assessment (MoCA) scores were 10 and 6, respectively, suggesting that she had dementia. Facial and appendicular choreatic movements were present. Saccadic eye movements were characterized by variable onset latency and speed. Dysarthria was marked. The basal hormonal evaluation revealed inappropriately low luteinizing hormone (<0.1 mUI/mL), as well as low follicular‐stimulating hormone (1.19 mUI/mL) and estradiol (<18.4 pg/mL). Prolactin and cortisol levels were within normal range. Pelvic echography revealed a postmenopausal uterus (33 × 21 × 35 mm) and the ovaries were not seen. Brain MRI was also performed. The results showed moderate cerebellar atrophy on T1‐weighted image, and extensive bilateral white matter lesions in both cerebral hemispheres, as well as in the brainstem on T2‐weighted fluid‐attenuated inversion recovery image. Susceptibility‐weighted imaging showed no cerebral microbleeds in cortical and subcortical areas (Fig. 1A). DTI images displayed widespread white matter deterioration as reflected by the decreased fractional anisotropy values and increased radial diffusivity and axial diffusivity values in the voxels of both hemispheres compared with those of controls, while cerebellar white matter fibers relatively spared (Fig. 1B and Table S1).
Figure 1.
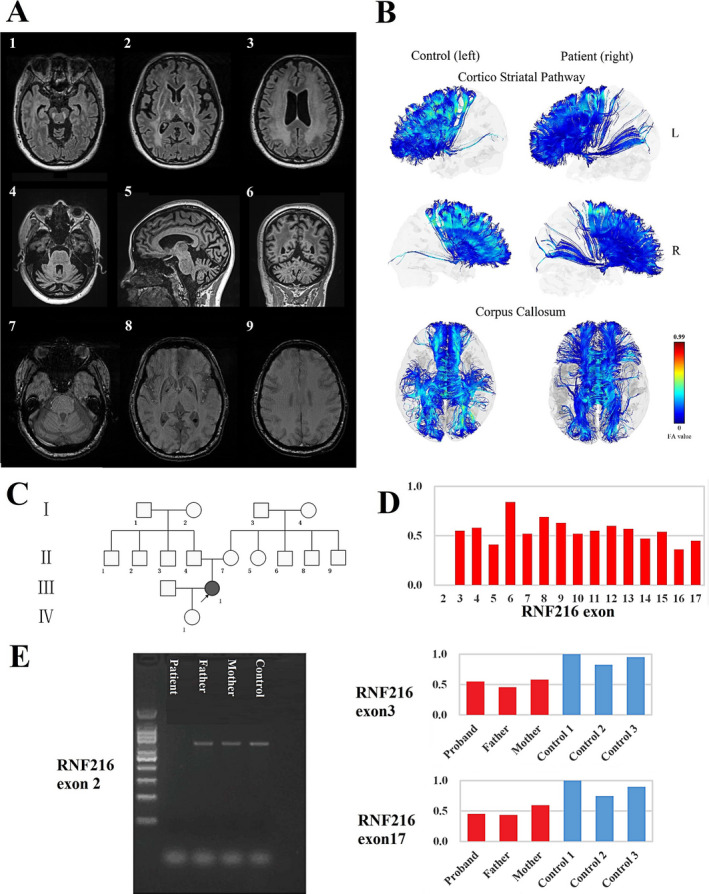
Clinical features of the proband and segregation analysis of the RNF216 mutations. (A) MRI scan showing extensive bilateral white matter lesions in both cerebral hemispheres and brainstem on T2‐weighted fluid‐attenuated inversion recovery image (1, 2, 3), cerebellar atrophy on T1‐weighted image (4, 5, 6), and no cerebral microbleeds on susceptibility‐weighted imaging (7, 8, 9). (B) Decreased fractional anisotropy (FA) was detected in cortico‐striatal pathway and corpus callosum, when compared with controls. (C) The pedigree of the Huntington‐like disorder family. (D) RNF216 mutations by whole‐exome sequencing. (E) Segregation analysis of deletions of exons 2, 3, and 17 in the RNF216 gene by quantitative polymerase chain reaction.
She was born through a normal full‐term delivery of nonconsanguinous parents (Fig. 1C). No cognitive impairment was noted in her parents based on their MMSE and MoCA scores (Father: MMSE = 27 and MoCA = 28, Mother: MMSE = 25 and MoCA = 25). Their hormone levels were within normal range. The brain MRI was normal for both of them, consistent with the normal results from a neurologic exam.
We found a homozygous deletion of exon 2 in the RNF216, which her parents did not show, determining a novel de novo RNF216 mutation. No pathogenetic mutations in other HDL‐associated genes were found in the proband. Heterozygous deletions of every exon from 3 to 17 were also found and the parental studies demonstrated that both parents carried heterozygous deletions of exons 3 and 17 (Fig. 1D and 1E).
Discussion
RNF216 encodes E3 ubiquitin ligase which recognizes and facilitates the ubiquitination of its target for degradation by the ubiquitin‐proteasome system. RNF216‐mediated neurodegeneration has been discovered very recently. Margolin et al. 6 were the first to identify RNF216 mutations or a combination of mutations in RNF216 and OTUD4 in five Gordon Holmes syndrome (GHS) families of Middle Eastern origin. Four subsequent studies with a total of nine patients reported RNF216 mutations in Belgian, Indian, Middle Eastern, and Argentinean populations. 3 , 7 , 8 , 9 Our patient was the first RNF216 mutation carrier identified in Chinese population, suggesting that RNF216 mutations are pan‐ethnic.
All previously reported cases with homozygous mutations in RNF216 were individuals with point mutations from consanguineous families or a common ancestor. 3 , 6 , 7 , 8 , 9 This patient showed a novel homozygous deletion of exon 2 in the RNF216 gene, which has not been previously reported. The parental studies found no corresponding deletion in both parents, indicating that this was a de novo mutation. As per the guidelines of the American College of Medical Genetics and Genomics for sequence variant interpretation, 10 this variation was interpreted as pathogenic. Several reasons account for this. (1) The variant led to exon deletion, which is possibly a deleterious effect (PVS1); (2) her parents were not affected, indicating it was a de novo mutation (PM6); and (3) it was absent in control databases (gnomAD, 1000 Genomes Project, ClinVar). The patient carried heterozygous deletions of exons 3–17, which were not pathogenic based on the parental studies. It is important to emphasize that the ethnic distribution of RNF216 mutations might have not been fully described yet due to the small number of cases.
The patient displayed cerebellar atrophy, which is consistent with previous research. 3 In bilateral cerebral hemispheres and brainstem, high T2 signal was found and no cerebral microhemorrhage foci were found, together suggesting no cerebrovascular pathology. White matter fiber tracts in corpus callosum and surrounding the basal ganglia, which have been reported related to Huntington's disease, 11 were injured on DTI images. While cerebellar white matter fibers, which were associated with ataxia, relatively spared. This suggests that white matter deterioration was related to her movement disorder.
Evidence of hypogonadotropic hypogonadism also links our patient to GHS, an autosomal recessive neurodegenerative disease with characteristic clinical features of ataxia and hypogonadism. 12 It should be noted that chorea was also described in GHS patients with RNF216 mutations, and ataxia was also found in HDL patients with RNF216 mutations. 3 , 6 The GHS diagnosis was also supported by other features, including dementia, cerebellar atrophy, and extensive white matter lesions.
In summary, we reported one patient with autosomal recessive HDL as part of the clinical spectrum of the RNF216‐mediated neurodegeneration and extended the ethnic distribution of RNF216 mutations.
Author Contributions
JTY, CM, and QD contributed to the conception and design, analysis and interpretation, and drafting and revision of manuscript. KLC and GXZ contributed to the genetic analysis, data interpretation, writing, drafting, and revising the manuscript. SDC, YYH, and BYL contributed to cognitive data acquisition and interpretation. HW and LW contributed to the image collection and interpretation.
Conflict of Interest
The authors have no financial conflict of interest.
Patient Consent
Parental/Guardian consent obtained.
Provenance and Peer Review
Not commissioned; externally peer reviewed.
Supporting information
Table S1. Fractional anisotropy (FA), radial diffusivity (RD), and axial diffusivity (AD) values of the patient and controls. Controls are mean (SD).
Acknowledgments
The authors thank the patient and her family for their cooperation in this study.
Funding Information
This study was supported by grants from the National Natural Science Foundation of China (91849126).
Funding Statement
This work was funded by National Natural Science Foundation of China grant 91849126.
References
- 1. Wild EJ, Tabrizi SJ. Huntington's disease phenocopy syndromes. Curr Opin Neurol 2007;20:681–687. [DOI] [PubMed] [Google Scholar]
- 2. Hensman Moss DJ, Poulter M, Beck J, et al. C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology 2014;82:292–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Santens P, Van Damme T, Steyaert W, et al. RNF216 mutations as a novel cause of autosomal recessive Huntington‐like disorder. Neurology 2015;84:1760–1766. [DOI] [PubMed] [Google Scholar]
- 4. Gong Y‐X. The Manual of Wechsler Adult Intelligence Scale Revised in China. China: Chang‐sha Hunan Medical University Press, 1992. [Google Scholar]
- 5. Gong Y‐X. The Manual of Wechsler Memory Scale Revised in China: Chang‐sha Hunan. China: Medical University Press, 1989. [Google Scholar]
- 6. Margolin DH, Kousi M, Chan YM, et al. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med 2013;368:1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alqwaifly M, Bohlega S. Ataxia and hypogonadotropic hypogonadism with intrafamilial variability caused by RNF216 mutation. Neurol Int 2016;8:6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ganos C, Hersheson J, Adams M, et al. The 4H syndrome due to RNF216 mutation. Parkinsonism Relat Disord 2015;21:1122–1123. [DOI] [PubMed] [Google Scholar]
- 9. Calandra CR, Mocarbel Y, Vishnopolska SA, et al. Gordon Holmes syndrome caused by RNF216 novel mutation in 2 Argentinean siblings. Mov Disord Clin Pract 2019;6:259–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang J, Gregory S, Scahill RI, et al. In vivo characterization of white matter pathology in premanifest huntington's disease. Ann Neurol 2018;84:497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Holmes G. A form of familial degeneration of the cerebellum. Brain 1908;30:466–489. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Fractional anisotropy (FA), radial diffusivity (RD), and axial diffusivity (AD) values of the patient and controls. Controls are mean (SD).