

Abstract
Oxalidaceae is one of the most important plant families in horticulture, and its key commercially relevant genus, Averrhoa, has diverse growth habits and fruit types. Here, we describe the assembly of a high-quality chromosome-scale genome sequence for Averrhoa carambola (star fruit). Ks distribution analysis showed that A. carambola underwent a whole-genome triplication event, i.e., the gamma event shared by most eudicots. Comparisons between A. carambola and other angiosperms also permitted the generation of Oxalidaceae gene annotations. We identified unique gene families and analyzed gene family expansion and contraction. This analysis revealed significant changes in MADS-box gene family content, which might be related to the cauliflory of A. carambola. In addition, we identified and analyzed a total of 204 nucleotide-binding site, leucine-rich repeat receptor (NLR) genes and 58 WRKY genes in the genome, which may be related to the defense response. Our results provide insights into the origin, evolution and diversification of star fruit.
Subject terms: Evolutionary biology, Evolution, Genome
Introduction
Wood sorrel (Oxalidaceae family) includes approximately 780 species and is distributed in both tropical and temperate areas. It contains species with various forms, including herbs, shrubs, and trees1. Wood sorrels are important economic crops and are utilized for both ornamental decoration and medicinal applications2,3. Based on morphological and molecular data, Oxalidaceae belongs to Oxalidales and is sister to the Connaraceae family. It can be divided into two main subfamilies: Oxalidoideae and Averrhooideae4. Averrhooideae differs from Oxalidoideae, an herbaceous subfamily, by having woody plants. It has four genera and is classified into two tribes: Biophyteae (Biophytum) and Averrhoeae (Dapania, Averrhoa and Sarcotheca). It is mostly distributed in tropical and subtropical regions5. Although these genera share synapomorphies with other wood sorrels, such as floral morphology, Averrhoa possesses several unique traits, including imparipinnate leaves, an herbaceous to papyraceous form, lateral petiolules that do not leave a stalk on the rachis after dropping, and the presence of 3–7 ovules per locule1. Therefore, Averrhoa is a key taxon in the evolutionary assessment of wood sorrel structure, and analysis of its genome should reveal new insights into the key adaptations that contribute to the diversification within Oxalidaceae6,7.
Averrhoa carambola, known as star fruit, originated in Asia and has been cultivated in Southeast Asia and Malaysia for many centuries8–10. Because the flesh is juicy and rich in vitamin C, star fruit is a commonly consumed tropical fruit11. In China, the total consumption of star fruit is approximately 2.6 million tons per year, while the annual production of star fruit in China is approximately two million tons11. In addition, star fruit is widely cultivated as a street tree in southern Chinese cities due to its dense foliage and star-like fruit12. Furthermore, the characteristics of bearing flowers and fruits on the main trunk and flowering year-round when temperatures exceed 27 °C in tropical regions13 make it an ideal species with which to explore economic and interesting traits (cauliflory, defined as flowering from the lower branch and trunk areas of woody plants, and high yield) at the whole-genome scale.
Cephalotus follicularis is a carnivorous plant native to southwest Australia that belongs to the monospecific family Cephalotaceae and is the only species of the order Oxalidales with a sequenced and annotated genome14. The relationship between star fruit and Cephalotus follicularis is not currently known and could be better understood through a comparison of their sequenced genomes.
Here, we present a complete genome sequence for A. carambola. Comparisons of genomic data with those from other flowering plants provide fundamental insights into the origin, evolution, adaptation, and diversification of star fruit.
Results and discussion
Genome sequencing and characterization
A. carambola has a karyotype of 2N = 2X = 22 chromosomes15. To sequence its genome, we utilized Illumina HiSeq short reads. We obtained a total of 131 Gb of raw reads with short inserts after library construction and sequencing (Supplementary Table 1). The A. carambola genome was estimated to be 357.79 Mb in size with a heterozygosity of 1.15% based on 17-K-mer analysis (Supplementary Fig. 1). Next, we utilized Oxford Nanopore Technology (ONT) and obtained a total of 52.33 Gb of long reads (Supplementary Table 1). The ONT reads were corrected and assembled to produce a 335.49 Mb genome with a contig N50 size of 4.22 Mb (Supplementary Table 2). Then, the draft assembly was polished using short reads, and BUSCO (Benchmarking Universal Single-Copy Orthologs, v3.1.0) assessment indicated that the completeness of the genome was 96.30%, suggesting that the A. carambola genome is nearly complete and of high quality (Supplementary Tables 2, 3).
We additionally used 42.76 Gb of Hi-C clean data to reconstruct physical maps by reordering and clustering the assembled scaffolds. We anchored 90.88% of the assembly (305.13 Mb) onto 11 pseudochromosomes using a hierarchical clustering strategy (Supplementary Table 4). Chromatin interaction data were used to assess the quality of the Hi-C assembly, which indicated that the assembly was of high quality (Supplementary Fig. 2). The length of the pseudochromosomes ranged from 17.89 Mb to 33.98 Mb with an N50 value of 31.25 Mb (Supplementary Tables 4, 5).
Approximately 61.3% of the A. carambola genome was found to be composed of repetitive elements (transposon elements, TEs) (Supplementary Figs. 3, 4 and Supplementary Table 6). We confidently annotated 25,419 protein-coding genes (Supplementary Table 7 and Supplementary Fig. 5), of which 21,316 (83.86%) were supported by transcriptome data (Supplementary Table 8). A total of 94.8% of annotated BUSCO gene models were identified, suggesting the near completeness of gene prediction (Supplementary Table 8). In addition, we identified 86 microRNAs, 581 transfer RNAs, 71 ribosomal RNAs and 212 small nuclear RNAs in the A. carambola genome (Supplementary Table 9).
Evolution of gene families
We then constructed a high-confidence phylogenetic tree and estimated the divergence times of 24 different plant species based on genes extracted from a total of 93 single-copy families (see Methods and Supplementary Table 10). The estimated divergence time of this set of Oxalidaceae species was 102 Mya (Supplementary Fig. 6). Next, we determined the expansion and contraction of orthologous gene families using CAFÉ 4.2 (Supplementary Fig. 7). Thirty-three gene families were expanded in the lineage leading to Oxalidales, whereas 904 families were contracted (Fig. 1). Four hundred ninety gene families were expanded in A. carambola, while 1021 gene families were contracted (Fig. 1).
Fig. 1. The expansion and contraction of gene families.

The green number indicates the number of expanded gene families, while the red number indicates the number of contracted gene families.
By comparing 24 different plant species, 504 gene families, including 8153 A. carambola genes, appeared to be unique to carambola (see Methods, Supplementary Figs. 7, 8 and Supplementary Table 10). We performed GO and KEGG enrichment analysis (Supplementary Table 11) and found that these gene families were enriched in several categories (Supplementary Tables 12–17).
Collinearity analysis
Genes are typically conserved both in function and order inside collinear fragments among closely related species. We utilized MCScanX16 to assess the collinearity among species related to Averrhoa carambola and found that all the predicted genes except TE-related genes were highly conserved in both function and order (Fig. 2 and Supplementary Figs. 9, 10).
Fig. 2. Collinear analysis.
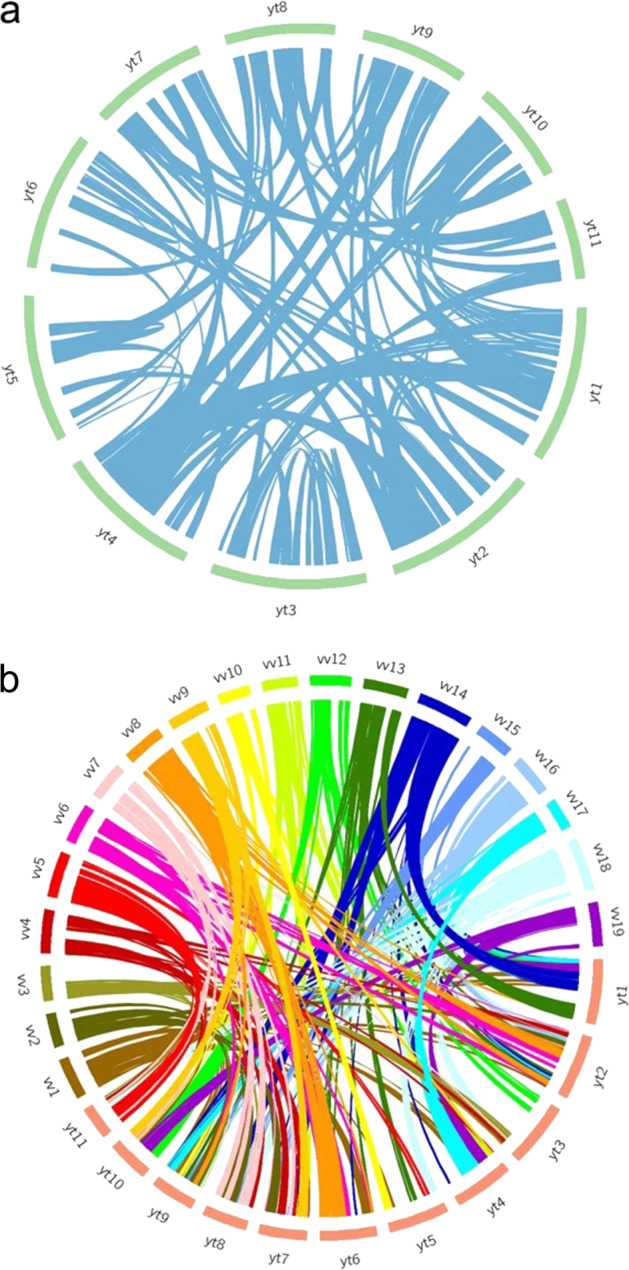
(a) Circos collinearity plot of A. carambola. (b) Circos collinearity plot of A. carambola with Vitis vinifera.
Whole-genome duplication
Whole-genome duplication (WGD) is a process of genome doubling that dramatically increases genome complexity. One of the striking features of plant genomes is that WGD has occurred many times17,18. WGD is a particularly important feature of angiosperm genomes. To determine whether the Averrhoa genome had undergone WGD during evolution, we used Ks (synonymous substitutions per site) distribution analysis. The paralogous gene pairs were extracted from the OrthoMCL results, and the Ks values were calculated using CodeML in the PAML package16,19.
There was a peak between Ks values of 1.6–1.8, indicating that the A. carambola genome had undergone a WGD event. Further analysis of A. carambola and C. follicularis revealed that the common ancestor prior to the differentiation of Averrhoa and C. follicularis experienced a WGD event. This event was likely the γ event shared by most core eudicots (Fig. 3). Comparative genomic analysis between the A. carambola and C. follicularis genomes revealed a one-to-one syntenic relationship, suggesting that no WGD events occurred after speciation (Supplementary Fig. 11).
Fig. 3. Ks distribution of Averrhoa carambola.
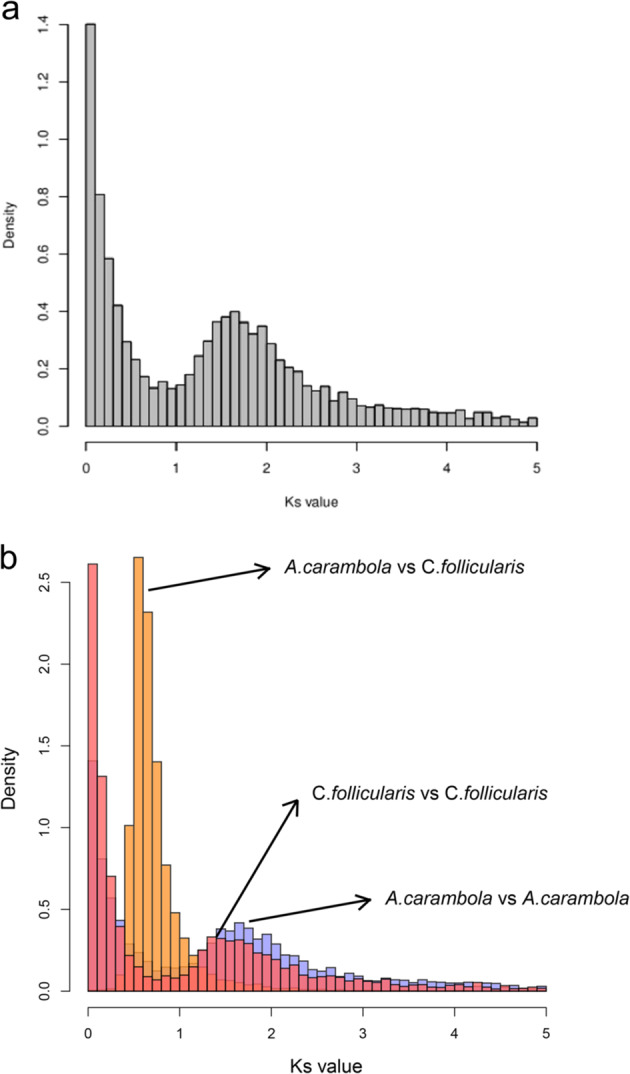
(a) Ks distribution of A. carambola paralogous genes. (b) Ks distribution of A. carambola and Cephalotus follicularis.
MADS-box genes of star fruit
MADS-box genes are known to be involved in many important processes during plant development and are especially known for their roles in flowering and flower development20. Because Averrhoa is well known for its cauliflory, defined as flowering from the lower branch and trunk areas of woody plants, we focused on identifying and characterizing the MADS-box genes in the Averrhoa genome in more detail.
In total, 74 putative functional MADS-box genes and two pseudogenes were identified in A. carambola (Table 1). This number is less than the number of MADS-box genes found in Arabidopsis thaliana21 and Theobroma cacao22,23 but greater than the number found in C. follicularis (Table 1). A. carambola has 48 type-II MADS-box genes, which is comparable to the number in A. thaliana (45) but less than the number found in T. cacao (67) (Table 1). Phylogenetic analysis (Supplementary Fig. 12) showed that many of the type-II MADS-box genes were duplicated, except those in the B-class, PI, FLC, AGL12, and Bs clades. The duplicated type-II clades included A-class (three members), B-class AP3 (two members), C/D-class (three members), E-class (four members), AGL6 (two members), SOC1 (three members), AGL15 (two members), ANR1 (five members), SVP (15 members), and MICK* (five members) clades (Table 1). Notably, we found that the SVP (15 members) clade in A. carambola contained more genes than that in A. thaliana, C. follicularis and cocoa tree (Table 1). The large number of SVP clade members might be due to tandem duplication. In Arabidopsis, the two SVP paralogs SVP and AGL24 are involved in floral transition and development. SVP suppresses flowering by acting with FLC to negatively regulate SOC1 and FT24. In contrast, AGL24 acts as a flowering activator to activate SOC1 expression during inflorescence development25. We detected transcripts of all SVP-like genes except Yangtao2024516 in vegetative leaves and shoots (Fig. 4). Interestingly, expression of an SVP-like gene (Yangtao2024516) was detected in the inflorescence and flower buds (Fig. 4). These results suggested that Yangtao2024516 functions in flowering activation, and the other 14 SVP-like genes might also be related to flowering suppression. The expanded SVP clade, including members with differential expression patterns in A. carambola, might contribute to flowering regulatory networks and relate to A. carambola cauliflory.
Table 1.
MADS-box genes in Averrhoa carambola, Arabidopsis thaliana, Cephalotus follicularis and Theobroma cacao.
| Category | A. carambola | A. thaliana21 | C. follicularis14 | T. cacao22,23 | ||||
|---|---|---|---|---|---|---|---|---|
| Functional | Pseudo | Functional | Pseudo | Functional | Pseudo | Functional | Pseudo | |
| Type II (Total) | 48a | 45 | 27 | 68 | ||||
| MIKCc | 43 | 39 | 24 | 58 | ||||
| A | 3 | 4 | 3 | 7 | ||||
| AGL12 | 1 | 1 | 1 | 1 | ||||
| C/D | 3 | 4 | 3 | 10 | ||||
| SOC1 | 3 | 6 | 3 | 7 | ||||
| SVP | 15 | 2 | 2 | 4 | ||||
| ANR1 | 5 | 4 | 2 | 3 | ||||
| Bs | 1 | 2 | 1 | 4 | ||||
| B-PI | 1 | 1 | 1 | 1 | ||||
| B-AP3 | 2 | 1 | 2 | 5 | ||||
| AGL6 | 2 | 2 | 1 | 5 | ||||
| E | 4 | 4 | 3 | 6 | ||||
| FLC | 1 | 6 | 0 | 3 | ||||
| AGL15 | 2 | 2 | 2 | 2 | ||||
| MIKCa | 5 | 6 | 3 | 10 | 2 | |||
| Type I (Total) | 28 | 2 | 61 | 24 | 28 | |||
| Mα | 20 | 25 | 15 | 8 | 3 | |||
| Mβ | 2 | 2 | 20 | 5 | 2 | 2 | 1 | |
| Mγ | 4 | 16 | 4 | 13 | 1 | |||
| Total | 74 | 2 | 106 | 51 | 2 | 91 | 7 | |
aThe number of MADS-box genes identified in this study. Genes with a stop codon in the MADS-box domain were categorized as pseudogenes.
Fig. 4. Expression patterns of genes involved in the flowering.
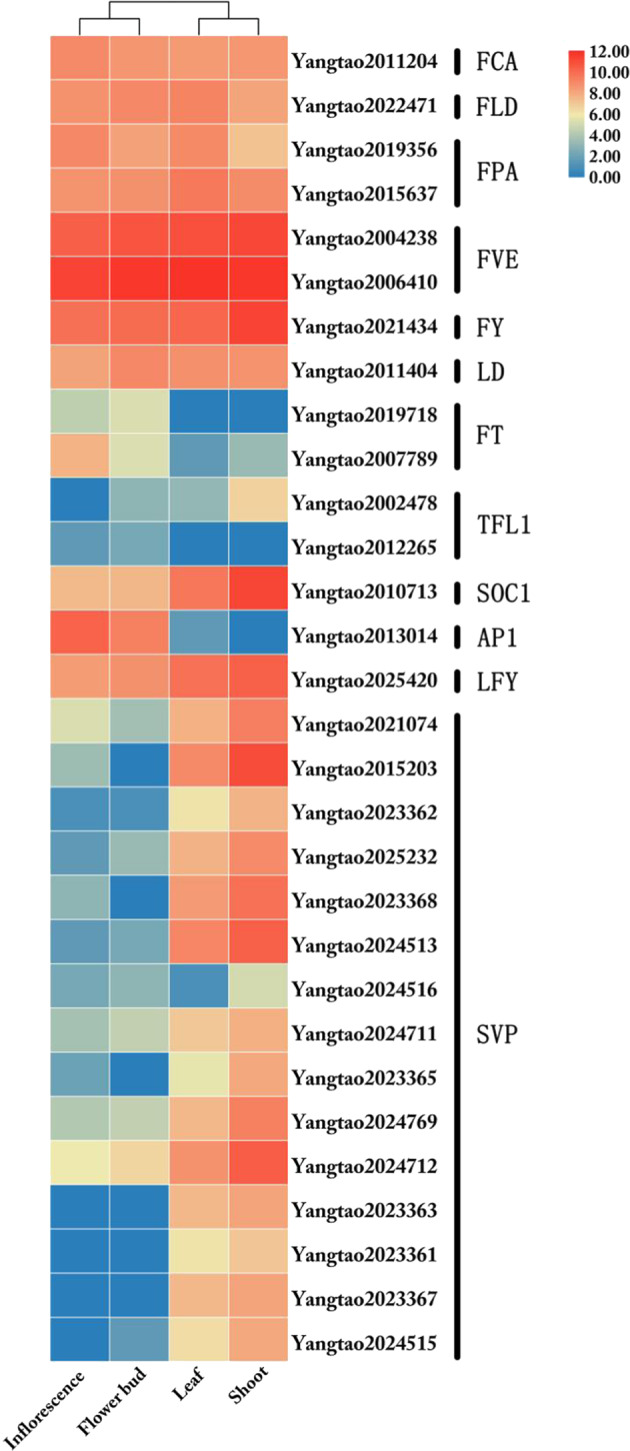
Pathway of Averrhoa carambola.
Cauliflorous flowering occurs due to the presence of adventitious buds that remain dormant and take years to develop after their formation, when the trunk develops and thickens due to secondary growth26. It has been reported that SVP2 functions in the suppression of meristem activity to prevent precocious budbreak in the perennial species kiwifruit27. The flowering repressor SVP is also controlled by the autonomous pathway and directly represses SOC1 transcription in Arabidopsis28. Interestingly, we found that genes involved in the autonomous flowering time pathway might repress the expression of SVP-like genes in reproductive tissues to promote flowering through activation of the floral integrator genes FLOWERING LOCUS T and SOC1 (Figs. 4, 5). Further study on the A. carambola flowering time pathways will improve our knowledge about causal genes, with applications in commercial variety selection.
Fig. 5. Putative autonomous flowering.
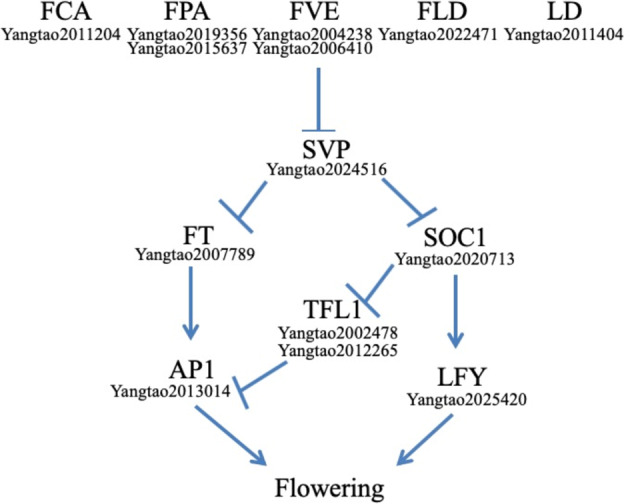
Time pathway in Averrhoa carambola.
Compared with type-II MADS-box genes, only 26 putative functional type-I genes and two pseudogenes were characterized (Table 1), which may have been due to the low duplication rate and high loss rate of type-I MADS-box genes. Tandem duplications seem to have contributed to more type-I genes, especially those in the Mα group. This suggests that type-I MADS-box genes have mainly been duplicated during smaller-scale and more-recent duplications21. Functional studies of type-I MADS-box genes in A. carambola will advance our understanding of their role in Oxalidaceae.
Analysis of NLR genes and the WRKY gene family
Resistance (R) genes produce R proteins to provide plant disease resistance against pathogens, most of which are nucleotide-binding site leucine-rich repeat (NLR) genes. NLRs contain three classes based on their N-terminal domain structures, namely, the toll/interleukin-1 receptor (TIR) NLR (TNL), coiled-coil (CC) NLR (CNL), and resistance to powdery mildew8 (RPW8) NLR (RLN)29. Here, a genome-wide identification of NLR genes in star fruit was carried out, resulting in the identification of 204 NLR genes, including homologues of known resistance genes: RNL (with 13 members), TNL (with 41 members) and CNL (with 150 members) (Table 2). There seem to be more NLR genes in A. carambola than in A. thaliana (174 genes)30 but fewer than in the basal angiosperm Nymphaea colorata (342 genes)31. Phylogenetic analysis of the NLR genes indicated that tandem duplication contributed to the increasing number of CNL genes. Remarkably, the NLR genes in the C. follicularis genome14 showed sharp contraction after the speciation of A. carambola and C. follicularis, with only one RNL and one TNL identified (Supplementary Fig. 13).
Table 2.
NLR genes in Averrhoa carambola, Arabidopsis thaliana, Cephalotus follicularis and Nymphaea colorata.
| A. carambola | A. thaliana30 | C. follicularis14 | N. colorata31 | |
|---|---|---|---|---|
| RNL | 13 | 9 | 1 | 22 |
| TNL | 41 | 101 | 1 | 57 |
| CNL | 150 | 64 | 26 | 263 |
| total | 204 | 174 | 28 | 342 |
RNL RPW8-NLR, TNL TIR-NLR, CNL CC-NLR, NLR nucleotide-binding site leucine-rich repeat, RPW8 resistance to powdery mildew 8, TIR toll/interleukin-1 receptor, CC coiled-coil.
Plant WRKY proteins are transcription factors (TFs) involved in regulating plant growth and development, as well as responses to many biotic and abiotic stresses32. An investigation of the star fruit genome revealed 58 WRKY genes. There appear to be fewer WRKY genes in star fruit than in the model plant A. thaliana (72 genes)33 and the basal angiosperm N. colorata (69 genes)31. The closely related species C. follicularis contains 42 WRKY genes14 (Table 3). Phylogenetic analysis showed that WRKY genes, especially type-IIc and type-I genes, underwent expansion in A. carambola, with the exception of those in the type-IIa clade. Among the 58 WRKY genes, type I (with 10 members), type II (33 members) and type III (eight members) contained more members than in C. follicularis (six members in type I, 29 members in type II and seven members in type III) (Supplementary Fig. 14). Interestingly, there was an unknown clade present in A. thaliana and C. follicularis, which contained three members in the star fruit genome and two members in the N. colorata genome.
Table 3.
WRKY genes in Averrhoa carambola, Arabidopsis thaliana, Cephalotus follicularis and Nymphaea colorata.
Conclusion
Although star fruit is well known as a delicacy in the tropics, research on it has been hampered by the absence of genetic resources. We identified new gene families and analyzed gene family expansion and contraction by comparisons with related species. Significant changes were found in the MADS-box gene family, which might be related to the cauliflory of A. carambola. Evolutionary analysis revealed one WGD event, which was the γ event experienced by most eudicots. The A. carambola assembly represents the first from the wood sorrel family and thus provides a valuable resource for evolutionary phylogenomic studies.
Material and methods
Plant sample preparation and sequencing
Diploid A. carambola was cultivated at the South China Botanical Garden, Guangzhou, Guangdong Province, China. Fresh young leaves were collected to extract genomic DNA for 400 bp paired-end library construction and Illumina sequencing. Genome size and heterozygosity were calculated using KmerFreq and GCE based on a 17-K-mer distribution. The high-molecular-weight DNA of fresh young leaves was extracted and randomly fragmented to construct a 20 kb library for Oxford Nanopore sequencing.
Genome assembly and chromosome anchoring
The raw fastq data from ONT sequencing were transformed and filtered using MinKNOW software. Canu (v1.7) was used to correct and trim the raw ONT reads with the default parameters. The corrected and trimmed ONT reads were assembled by SMARTdenovo using 21-mers34. The assembled contigs were then polished three times with Pilon (v1.22) using Illumina short reads. The quality of the genome assembly was estimated by searching for BUSCO v3.1.0 using the Embryophyta ODB 10 database.
Fresh leaves of A. carambola were used to construct a Hi-C sequencing library, including chromatin crosslinking, chromatin digestion with HindIII, biotin labeling and end repair, DNA purification and streptavidin pull-down of labeled Hi-C ligation products. The library was then sequenced using the Illumina platform, and the clean sequences were mapped to the draft genome, with valid Hi-C reads employed to correct the draft assembly. Finally, the draft genome of A. carambola was assembled into chromosomes using Lachesis (9).
Identification of repetitive sequences
Tandem repeats across the genome were predicted using Tandem Repeats Finder (v4.07b, http://tandem.bu.edu/trf/trf.html). Transposable elements (TEs) were first identified using RepeatMasker (http://www.repeatmasker.org, v3.3.0) and RepeatProteinMask based on the Repbase TE library35. Next, two de novo predication software programs, RepeatModeler (http://repeatmasker.org/RepeatModeler.html) and LTR_FINDER36, were used to identify TEs in the star fruit genome. Finally, repeat sequences with identities ≥ 50% were grouped into the same class.
Gene prediction and annotation
Three independent methods were used for gene prediction. Homologous sequence searching was performed by comparing protein sequences of five sequenced species against the star fruit genome using the TBLASTN algorithm with a cut-off E-value of ≤ 1e−5. Then, the corresponding homologous genome sequences were aligned against BLAST hits using GeneWise v2.4.1 to extract accurate exon–intron information37. Three ab initio prediction software programs, Augustus v3.0.238, FGENESH39 and GlimmerHMM40, were employed for de novo gene prediction. Then, the homology-based and ab initio gene structures were merged into a nonredundant gene model using GLEAN. The completeness of gene prediction was evaluated using BUSCO v3.1.0. Finally, the RNA sequencing (RNA-Seq) reads were mapped to the assembly using TopHat v2.0.1141, and Cufflinks v2.2.142 was applied to combine mapping results for transcript structural predictions.
The protein sequences of the consensus gene set were aligned to various protein databases, including GO (The Gene Ontology Consortium), KEGG43, InterPro44, Swiss-Prot and TrEMBL, for the annotation of predicted genes. The rRNAs were identified by aligning the rRNA template sequences from the Rfam45 database against the genome using the BLASTN algorithm at an E-value cut-off of 1e−5. The tRNAs were predicted using tRNAscan-SE46, and other ncRNAs were predicted by running Infernal 0.81 software against the Rfam database.
Genome evolution analysis
Gene families present in the 24 genomes were identified using OrthoMCL47. Peptide sequences from 93 single-copy gene families were used to construct phylogenetic relationships and estimate divergence times. Alignments from MUSCLE were then converted to coding sequences. Fourfold degenerate sites were concatenated and used to estimate the neutral substitution rate per year and the divergence time. PhyML48 was used to construct a phylogenetic tree. The Bayesian relaxed molecular clock approach was used to estimate species divergence times using the program MCMCTREE v4.0, which is part of the PAML package49. The ‘correlated molecular clock’ and ‘JC69’ models were used. Published Arabidopsis–Papaya (54–90 Mya), P. trichocarpa–Arabidopsis (100–129 Mya), monocot–dicot (140 Mya) and angiosperm (<200 Mya) divergence times were used for calibration50.
Transcriptome sequencing and expression analysis
The inflorescence, floral bud, leaf and shoot were collected independently from A. carambola. Total RNA was extracted according to the manufacturer’s protocol. Illumina RNA-Seq libraries were prepared and sequenced on a HiSeq 2500 system following the manufacturer’s instructions (Illumina, USA). Two biological replicates were analyzed for each sample. To estimate gene expression levels, clean reads of each sample were mapped onto the assembled genome to obtain read counts for each gene using HTSeq-count and normalized to FPKM counts42.
Phylogenetic reconstruction of the MADS-box gene family
For the identification of MADS-box family members in A. carambola, BLASTp was applied using known plant MADS-box proteins as reference sequences21,51, with an E-value cut-off of ≤ 1e−5. For phylogenetic analyses, a total of 333 protein sequences, including 76 A. carambola AcMADS-box, 106 A. thaliana AtMADS-box21, 53 C. follicularis CfMADS-box14 and 98 T. cacao TcMADS-box22,23, were aligned using MUSCLE52 (v3.8.31; http://www.drive5.com/muscle) with default parameters. A phylogenetic tree was drawn through RAxML53 with the GTRGAMMA substitution model and 1000 bootstraps on the CIPRES website (https://www.phylo.org/portal2/home.action)54. The phylogenetic tree was visualized using FigTree software (http://tree.bio.ed.ac.uk/software/figtree) (Supplementary Fig. 12).
NLR genes and WRKY TF identification
All the annotated protein sequences of Arabidopsis, C. follicularis, and N. colorata were downloaded. The NLR gene (PF00931) and WRKY TF (PF03106) models were obtained from the Pfam database (http://pfam.xfam.org/). The NLR genes and WRKY TFs were then identified using HMMER v3.2.1. The identified sequences were confirmed and filtered using the UniProt database (https://www.uniprot.org/) and the NCBI database (https://www.ncbi.nlm.nih.gov/). Then, MAFFT v7.407 was used to align multiple sequences of candidate proteins with default parameters. A maximum likelihood (ML) phylogenetic tree was constructed for the protein sequences of NLR genes and WRKY TFs from Arabidopsis30,33, C. follicularis14, N. colorata31 and star fruit by FastTree with default parameters.
Supplementary information
Acknowledgements
This work was supported by The National Key Research and Development Program of China (ref. 2019YFC1711103), the Fujian Agriculture and Forestry University Science and Technology Innovation Special Fund Project (ref. KFA17331A), the Natural Science Foundation of Fujian (ref. 2019J01410), and the Fujian Agriculture and Forestry University 2015 Outstanding Youth Fund Project (ref. xjq201620). The authors thank Junjie Wei and Mingtao Jiang for collecting needed samples.
Data availability
The whole-genome sequence data reported in this paper have been deposited in the Genome Warehouse of the National Genomics Data Center, Beijing Institute of Genomics (BIG), Chinese Academy of Sciences, under accession number GWHABKE00000000 and are publicly accessible at https://bigd.big.ac.cn/gwh.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
These authors contributed equally: Shasha Wu, Wei Sun, Zhichao Xu, Junwen Zhai
Contributor Information
Shi-lin Chen, Email: slchen@icmm.ac.cn.
Wen-Chieh Tsai, Email: tsaiwc@mail.ncku.edu.tw.
Siren Lan, Email: lkzx@fafu.edu.cn.
Zhong-Jian Liu, Email: zjliu@fafu.edu.cn.
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41438-020-0307-3).
References
- 1.Veldkamp, J. F. in Flora Malesiana (ed. van Steenis, C. G. C. J.) 155–178 (Noordhoff, Leyden, 1971).
- 2.Khare, C. P. Indian Medicinal Plants 74–75 (Springer, New York, 2007).
- 3.Gunawardena DC, Jayasinghe L, Fujimoto Y. Phytotoxic constituents of the fruits of Averrhoa carambola. Chem. Nat. Compd. 2015;51:532–533. doi: 10.1007/s10600-015-1332-6. [DOI] [Google Scholar]
- 4.The Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016;181:1–20. doi: 10.1111/boj.12385. [DOI] [Google Scholar]
- 5.Stevens, P. F. Angiosperm Phylogeny Website. Version 14, July 2017 [and more or less continuously updated since] (2001 onwards).
- 6.Heibl C, Renner SS. Distribution models and a dated phylogeny for Chilean Oxalis species reveal occupation of new habitats by different lineages, not rapid adaptive radiation. Syst. Biol. 2012;61:823–834. doi: 10.1093/sysbio/sys034. [DOI] [PubMed] [Google Scholar]
- 7.Sun M, Naeem R, Su JX, Cao ZY, Chen ZD. Phylogeny of the Rosidae: a dense taxon sampling analysis. J. Syst. Evol. 2016;54:363–391. doi: 10.1111/jse.12211. [DOI] [Google Scholar]
- 8.Fieischmann P, Watanabe N, Winterhalter P. Enzymatic carotenoid cleavage in star fruit (Averrhoa carambola) Phytochemistry. 2003;63:131–137. doi: 10.1016/S0031-9422(02)00657-X. [DOI] [PubMed] [Google Scholar]
- 9.Neto MM, et al. Intoxication by star fruit (Averrhoa carambola) in 32 uraemic patients: treatment and outcome. Nephrol. Dial. Transplant. 2003;18:120–125. doi: 10.1093/ndt/18.1.120. [DOI] [PubMed] [Google Scholar]
- 10.Carolino ROG, et al. Convulsant activity and neurochemical alterations induced by a fraction obtained from fruit Averrhoa carambola (Oxalidaceae: Geraniales) Neurochem. Int. 2005;46:523–531. doi: 10.1016/j.neuint.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 11.Shui GG, Leong LP. Residue from star fruit as valuable source for functional food ingredients and antioxidant nutraceuticals. Food Chem. 2005;97:277–284. doi: 10.1016/j.foodchem.2005.03.048. [DOI] [Google Scholar]
- 12.Bin-Ngah, A.W., Ahmad, I. & Hassan, A. Carambola Production, Processing and Marketing in Malaysia (Inter-American Society for Tropical Horticulture (IASTH), 1992).
- 13.Khoo H, et al. A review on underutilized tropical fruits in Malaysia. Guangxi Agr. Sci. 2010;41:698–702. [Google Scholar]
- 14.Fukushima K, et al. Genome of the pitcher plant Cephalotus reveals genetic changes associated with carnivory. Nat. Ecol. Evol. 2017;1:0059. doi: 10.1038/s41559-016-0059. [DOI] [PubMed] [Google Scholar]
- 15.Wu, Q. Study on Technology of Breeding polyploid of Averrhoa Carambola by Biotechnique. Master dissertation. Southwest Agricultural University, Chongqing (2002).
- 16.Wang C, et al. Genome-wide analysis of local chromatin packing in Arabidopsis thaliana. Genome Res. 2015;25:246. doi: 10.1101/gr.170332.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui L, et al. Widespread genome duplications throughout the history of flowering plants. Genome Res. 2006;16:738–749. doi: 10.1101/gr.4825606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van de Peer Y, Maere S, Meyer A. The evolutionary significance of ancient genome duplications. Nat. Rev. Genet. 2009;10:725–732. doi: 10.1038/nrg2600. [DOI] [PubMed] [Google Scholar]
- 19.Blanc G, Wolfe KH. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell. 2004;16:1667–1678. doi: 10.1105/tpc.021345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cai J, et al. The genome sequence of the orcid Phalaenopsis equestris. Nat. Genet. 2015;47:65–72. doi: 10.1038/ng.3149. [DOI] [PubMed] [Google Scholar]
- 21.Parenicova L, et al. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell. 2003;15:1538–1551. doi: 10.1105/tpc.011544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Argout X, et al. The cacao Criollo genome v2.0: an improved version of the genome for genetic and functional genomic studies. BMC Genomics. 2017;18:730. doi: 10.1186/s12864-017-4120-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Argout X, et al. The genome of Theobroma cacao. Nat. Genet. 2011;43:101–108. doi: 10.1038/ng.736. [DOI] [PubMed] [Google Scholar]
- 24.Willmann MR, Poethig RS. The effect of the floral repressor FLC on the timing and progression of vegetative phase change in Arabidopsis. Development. 2011;138:677–685. doi: 10.1242/dev.057448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gregis V, Sessa A, Dorca-Fornell C, Kater MM. The Arabidopsis floral meristem identity genes AP1, AGL24 and SVP directly repress class B and C floral homeotic genes. Plant J. 2009;60:626–637. doi: 10.1111/j.1365-313X.2009.03985.x. [DOI] [PubMed] [Google Scholar]
- 26.Oliveira GP, et al. Origin and development of reproductive buds in jabuticaba cv. Sabará (Plinia jaboticaba Vell) Sci. Horticulturae. 2019;249:432–438. doi: 10.1016/j.scienta.2019.02.020. [DOI] [Google Scholar]
- 27.Wu R. Kiwifruit SVP2, a dormancy regulator. Eur. J. Hort. Sci. 2018;83:231–235. doi: 10.17660/eJHS.2018/83.4.3. [DOI] [Google Scholar]
- 28.Li D, et al. A repressor complex governs the integration of flowering signals in Arabidopsis. Dev. Cell. 2008;15:110–120. doi: 10.1016/j.devcel.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 29.Shao Z, et al. Large-scale analyses of angiosperm nucleotide-binding site-leucine-rich repeat genes reveal three anciently diverged classes with distinct evolutionary patterns. Plant Physiol. 2016;170:2095–2109. doi: 10.1104/pp.15.01487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maleck K, et al. The transcriptome of Arabidopsis thaliana during systemic acquired resistance. Nat. Genet. 2000;26:403–410. doi: 10.1038/82521. [DOI] [PubMed] [Google Scholar]
- 31.Zhang LS, et al. The water lily genome and the early evolution of flowering plants. Nature. 2020;577:79–84. doi: 10.1038/s41586-019-1852-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Phukan U, et al. WRKY transcription factors: molecular regulation and stress responses in plants. Front Plant Sci. 2016;7:760. doi: 10.3389/fpls.2016.00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Q, Wang M, Zhang X, Hao B, Kaushik SK, Pan Y. WRKY gene family evolution in Arabidopsis thaliana. Genetica. 2011;139:973. doi: 10.1007/s10709-011-9599-4. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt MHW, et al. De novo assembly of a new Solanum pennellii accession using nanopore sequencing. Plant Cell. 2017;29:2336–2348. doi: 10.1105/tpc.17.00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jurka J, et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 36.Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35:W265–268. doi: 10.1093/nar/gkm286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Birney E, Clamp M, Durbin R. GeneWise and genomewise. Genome Res. 2004;14:988–995. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stanke M, Schoffmann O, Morgenstern B, Waack S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 2006;7:62. doi: 10.1186/1471-2105-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salamov AA, Solovyev VV. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 2000;10:516–522. doi: 10.1101/gr.10.4.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20:2878–2879. doi: 10.1093/bioinformatics/bth315. [DOI] [PubMed] [Google Scholar]
- 41.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trapnell C, et al. Differential gene and transcript expression analysis of RNA-Seq. experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ogata H, et al. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hunter S, et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Griffiths JS, et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33:D121–124. doi: 10.1093/nar/gki081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li L, Stoeckert CJ, Jr., Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 49.Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- 50.Zhang GQ, et al. The Apostasia genome and the evolution of orchid. Nature. 2017;549:379–383. doi: 10.1038/nature23897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zdobnov EM, Apweiler R. InterProScan-an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- 52.Letunic L, Doerks T, Bork P. SMART: recent updates, new develpoments and status in 2015. Nucleic Acids Res. 2014;43:D257–D260. doi: 10.1093/nar/gku949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miller, M. A., Pfeiffer, W. & Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. 2010 Gateway Computing Environments Workshop (GCE), (ed. Institute of Electrical and Electronic Engineers) 1–8 (New Orleans, LA, 2010).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The whole-genome sequence data reported in this paper have been deposited in the Genome Warehouse of the National Genomics Data Center, Beijing Institute of Genomics (BIG), Chinese Academy of Sciences, under accession number GWHABKE00000000 and are publicly accessible at https://bigd.big.ac.cn/gwh.