

Abstract
We have previously demonstrated that ischemia/reperfusion (I/R) impairs endoplasmic reticulum (ER)-based protein folding in the heart and thereby activates an unfolded protein response sensor and effector, activated transcription factor 6α (ATF6). ATF6 then induces mesencephalic astrocyte-derived neurotrophic factor (MANF), an ER-resident protein with no known structural homologs and unclear ER function. To determine MANF's function in the heart in vivo, here we developed a cardiomyocyte-specific MANF-knockdown mouse model. MANF knockdown increased cardiac damage after I/R, which was reversed by AAV9-mediated ectopic MANF expression. Mechanistically, MANF knockdown in cultured neonatal rat ventricular myocytes (NRVMs) impaired protein folding in the ER and cardiomyocyte viability during simulated I/R. However, this was not due to MANF-mediated protection from reactive oxygen species generated during reperfusion. Because I/R impairs oxygen-dependent ER protein disulfide formation and such impairment can be caused by reductive stress in the ER, we examined the effects of the reductive ER stressor DTT. MANF knockdown in NRVMs increased cell death from DTT-mediated reductive ER stress, but not from nonreductive ER stresses caused by thapsigargin-mediated ER Ca2+ depletion or tunicamycin-mediated inhibition of ER protein glycosylation. In vitro, recombinant MANF exhibited chaperone activity that depended on its conserved cysteine residues. Moreover, in cells, MANF bound to a model ER protein exhibiting improper disulfide bond formation during reductive ER stress but did not bind to this protein during nonreductive ER stress. We conclude that MANF is an ER chaperone that enhances protein folding and myocyte viability during reductive ER stress.
Keywords: endoplasmic reticulum stress (ER stress), protein folding, cardiomyocyte, ischemia, chaperone, cardiomyocyte, heart function, ischemia/reperfusion, mesencephalic astrocyte-derived neurotrophic factor (MANF), reductive stress, unfolded protein response
Introduction
The endoplasmic reticulum (ER) is a major site of the synthesis of proteins that are critical for proper function of the heart, including many calcium-handling proteins, receptors, and secreted proteins, such as hormones, stem cell homing factors, and growth factors (1, 2). Therefore, proper folding of proteins synthesized in the ER of cardiac myocytes is essential for maintaining optimal cardiac function. Furthermore, many post-translational modifications occur in the ER, including disulfide bond formation, which are critical for protein stability and function (3). Disulfide bond formation in the ER, also known as oxidative protein folding, is an oxygen-dependent process (3–7). Some time ago, this led us to the hypothesis that ER protein folding would be impaired in cardiac myocytes in response to a lack of oxygen during pathophysiological conditions of ischemia and ischemia/reperfusion (3, 4). Subsequently, it was demonstrated in cultured cardiac myocytes and in mouse hearts, in vivo, that I/R disrupts protein folding in the ER, leading to activation of the ER stress response, also called the unfolded protein response (UPR) (3–5).
The UPR is controlled in all mammalian cells by several ER-transmembrane sensors of ER protein misfolding, including the adaptive transcription factor, ATF6 (8, 9). When protein synthesis surpasses the capacity of the protein-folding machinery, increases in misfolded proteins cause the translocation of the ER-transmembrane, 90-kDa form of ATF6 to the Golgi, where it is clipped, liberating an N-terminal fragment that, after nuclear translocation, serves as a transcription factor. This 50-kDa active form of ATF6 regulates a gene program that is responsible for the expression of numerous proteins that enhance ER protein folding, which adaptively restores the balance between protein synthesis and folding (8, 9). We previously generated a transgenic mouse line in which ATF6 could be activated at will, selectively in cardiac myocytes; using this mouse line, we showed that ATF6 protected hearts from I/R damage (10). Transcript profiling of these Atf6 transgenic mouse hearts, as well as Atf6 knockout mouse hearts, defined the ATF6 gene program in the heart; we posited that these genes might contribute to the protective effects of ATF6 (11, 12). One of those genes encodes mesencephalic astrocyte-derived neurotrophic factor (MANF), which was originally isolated from astrocytes (13) but later found in all eukaryotic cells examined to date. One of the striking features of MANF is that it has a noncanonical but functional ER retention sequence at the C terminus but otherwise shares little structural homology with other proteins (14, 15). This finding spawned our hypothesis that MANF exerts a unique function within the ER to maintain ER protein folding and prevent myocyte death during I/R; however, such a concept has not been studied.
Accordingly, here we examined the function of MANF in the ER of cardiac myocytes. We found that, in cardiac myocytes, MANF is protective under specific forms of pharmacological and pathophysiological ER stress and that MANF exerts its protective effects by enhancing ER protein folding, thus maintaining ER proteostasis. Mechanistically, we showed that MANF exerts this effect, at least partly, by virtue of its ability to serve as a chaperone. This finding was unexpected, because MANF does not share significant structural features with other chaperones. Further studies demonstrated that the eight cysteine residues within the 158-aa MANF structure, whose positions are conserved among all species of MANF examined to date, are critical for its chaperone function, mainly under reductive ER stress, consistent with the importance of disulfide bond formation in ER protein folding. This study establishes a new protective role for MANF in the ER of cardiac myocytes in the heart and provides evidence that MANF mediates protection and enhances ER protein folding selectively during reductive ER stress.
Results
MANF loss of function in the heart increases cardiac damage during ischemia/reperfusion injury
To determine the effects of MANF loss of function in the heart, we generated a mouse model in which the α-MHC promoter drives expression of a Manf-specific microRNA in a cardiac myocyte–restricted manner. We elected to knock down endogenous MANF instead of completely deleting it because the deletion of many ER stress response genes has been shown to lead to embryonic lethality (16). Immunoblotting of mouse hearts showed that, compared with WT mice, MANF knockdown (KD) mice exhibited a 4-fold reduction in MANF (Fig. 1, A and B). As the effects of MANF knockdown on mouse heart function have not been previously examined, basal cardiac function was assessed by echocardiography. Compared with WT mouse hearts, MANF KD mice exhibited increased ejection fraction; MANF KD female mice had slightly decreased left ventricular systolic volume, whereas male MANF KD mice had slightly increased left ventricular diastolic volume (Table 1). To assess whether the increase in cardiac contractility elicited cardiac pathology, mRNA levels of cardiac pathology markers Nppa, Nppb, and Col1a1 and protein levels of ER stress markers (i.e. GRP94 and GRP78) as well as hearts and lung weights from WT and MANF KD mice were measured. Expression of ER stress and cardiac pathology markers (Fig. 1 (A and C–G)) and heart and lung weights (Fig. 1, H–J) were unaffected by MANF knockdown. Overall, these results show that knocking down MANF in cardiac myocytes of mouse hearts by ∼80% increases contractility but does not induce any overt cardiac pathology. Because no overt cardiac pathology was observed, we moved forward to study the effects of MANF knockdown in pathophysiological models of ER stress.
Figure 1.

Effect of MANF knockdown in the heart on expression of GRP94 and GRP78, expression of fetal genes, heart and lung weights, viability of isolated adult mouse ventricular myocytes, and cardiac contractility. A, MANF, GRP94, GRP78, and β-actin immunoblots of mouse heart extracts from 10-week-old female WT (n = 5) or MANF KD mice (n = 5). *, band of interest that was quantified in B–D. IgGH indicates position of immunoglobulin heavy chain. B–D, densitometry of immunoblots shown in A. Band intensities were normalized to those for β-actin and displayed as -fold WT control level. E–G, fetal gene mRNA levels were determined by RT-qPCR. H and I, heart weights were normalized to tibia length (H) or body weight (I). J, lung weights were normalized to body weight. Echocardiography data and statistical analysis can be found in Table 1. K, myocytes were isolated from 10-week-old adult WT and MANF KD mice and then subjected to sI/R followed by determination of cell viability. To determine percentage cell viability, the number of calcein AM–positive cells per field was divided by the total number of cells in the same field. L–P, ex vivo I/R of WT and MANF KD mouse hearts. Hearts from female WT (n = 3) or MANF KD (n = 4) were subjected to ex vivo ischemia for 20 min, followed by 60 min of reperfusion (I/R). L and M, LVDP upon reperfusion was normalized to the LVDP obtained during equilibration, the latter of which was set to 100%. L, plot of individual LVDP time courses from different mouse hearts. M, average of the plots shown in L. N and O, heart sections were stained with TTC to assess the extent of myocardial damage (N); shown is the average infarct size divided by area at risk (O). P, samples of ex vivo heart perfusates were obtained after 45 min of reperfusion and then assayed for LDH activity relative to LDH activity in the equilibrium perfusate. *, statistically significant difference by Student's unpaired t test, p ≤ 0.05. Note that GRP78 and GRP94 immunoblotting was performed using an anti-KDEL antibody. Error bars, S.E.
Table 1.
Echocardiographic parameters of WT and transgenic (MANF KD) mice
FS, fractional shortening; EF, ejection fraction; LVEDV, left ventricular end diastolic volume; LVESV, left ventricular end systolic volume; LVIDD, left ventricular inner diameter in diastole; LVIDS, left ventricular inner diameter in systole; PWTD, left ventricular posterior wall thickness in diastole; PWTS, left ventricular posterior wall thickness in systole; AWTD, left ventricular anterior wall thickness in diastole; AWTS, left ventricular anterior wall thickness in systole; LV mass, left ventricular mass; HR, heart rate in beats per minute (bpm). Statistical analyses used Student's unpaired t test. *, p ≤ 0.05, difference between WT and transgenic MANF KD mice of the same sex.
| Parameter | Female WT (n = 6) | Female MANF KD (n = 7) | Male WT (n = 6) | Male MANF KD (n = 7) |
|---|---|---|---|---|
| FS (%) | 39.80 ± 3.11 | 50.47 ± 0.75* | 36.89 ± 1.87 | 45.96 ± 1.72* |
| EF (%) | 70.61 ± 2.72 | 82.62 ± 0.74* | 67.91 ± 2.35 | 77.82 ± 1.83* |
| LVEDV (μl) | 53.17 ± 2.37 | 49.82 ± 3.25 | 41.83 ± 3.02 | 56.93 ± 3.86* |
| LVESV (μl) | 15.97 ± 2.18 | 8.74 ± 0.87* | 13.18 ± 0.72 | 12.99 ± 1.94 |
| LVIDD (mm) | 3.58 ± 0.09 | 3.40 ± 0.06 | 3.22 ± 0.10 | 3.66 ± 0.10 |
| LVIDS (mm) | 2.10 ± 0.14 | 1.65 ± 0.11* | 2.02 ± 0.04 | 1.98 ± 0.11 |
| PWTD (mm) | 0.89 ± 0.05 | 0.77 ± 0.06 | 0.92 ± 0.07 | 1.05 ± 0.14 |
| PWTS (mm) | 1.15 ± 0.06 | 1.32 ± 0.08 | 1.25 ± 0.12 | 1.45 ± 0.07 |
| AWTD (mm) | 0.85 ± 0.06 | 0.95 ± 0.11 | 1.03 ± 0.07 | 0.94 ± 0.04 |
| AWTS (mm) | 1.45 ± 0.07 | 1.58 ± 0.06 | 1.43 ± 0.07 | 1.56 ± 0.08 |
| LV mass (mg) | 99.16 ± 8.22 | 92.65 ± 7.72 | 106.75 ± 6.94 | 124.01 ± 14.04 |
| HR (bpm) | 472.96 ± 2.98 | 458.69 ± 5.51 | 473.88 ± 11.14 | 453.20 ± 4.58 |
To determine the consequences of MANF knockdown on myocyte viability during conditions known to induce ER stress, adult mouse ventricular myocytes (AMVMs) were isolated from WT and MANF KD mice and subjected to simulated ischemia/reperfusion (sI/R), a model of pathophysiological ER stress. Compared with WT myocytes, MANF KD myocytes exhibited more death in response to sI/R (Fig. 1K). To examine functional roles for MANF in the heart, WT and MANF KD mouse hearts were subjected to ex vivo I/R (17). Compared with WT mouse hearts, MANF KD mouse hearts exhibited significantly lower functional recovery, significantly increased tissue damage, and greater LDH release, the last of which is an indicator of necrotic tissue damage (Fig. 1, L–P) (10), although in this experiment this measure did not reach statistical significance. These results indicate that MANF knockdown in the heart decreases myocyte viability during I/R, resulting in increased tissue damage and decreased cardiac function.
Re-expression of MANF reverses the effects of MANF knockdown on cardiac damage following ischemia/reperfusion
To determine whether ectopic expression of FLAG-MANF could restore the functional and structural defects observed in the MANF KD mice, we engineered a recombinant adeno-associated virus serotype 9 (AAV9) (5, 12, 16) encoding FLAG-MANF using nucleotide sequences that are not targeted by the Manf-specific microRNA that is expressed in MANF KD mouse hearts. We found that the hearts of MANF KD mice that were treated with AAV9-FLAG-MANF had about the same level of MANF as WT hearts (Fig. 2A). When hearts were subjected to ex vivo I/R, compared with AAV9-Con–treated mice, AAV9-FLAG-MANF–treated mice exhibited smaller infarcts, greater contractile function, and less necrosis (Fig. 2, B–F). In fact, the AAV9-FLAG-MANF completely restored these cardiac parameters to those seen in WT mice. Thus, it is the depletion of endogenous MANF in the MANF KD mouse hearts that results in their greater I/R injury and decreased cardiac function.
Figure 2.

Effect of MANF re-expression in MANF KD mouse hearts on cardiac damage and contractility following I/R. AAV9-Con or AAV9-FLAG-MANF was administered to WT and MANF KD mice by tail vein injection. Seven days later, male hearts were extracted and subjected to MANF and β-actin immunoblotting (A), and female hearts were subjected to ex vivo I/R (B–F). For ex vivo I/R, hearts from WT mice injected with AAV-Con (n = 3) or MANF KD mice injected with AAV-Con (n = 4) or AAV-FLAG-MANF (n = 4) were subjected to 20 min of ex vivo global ischemia and then 60 min of reperfusion. B and C, LVDP upon reperfusion was normalized to the LVDP obtained during equilibration, the latter of which was set to 100%. B, plot of individual LVDP time courses from different mouse hearts. C, average of the plots shown in B. D and E, heart sections were stained with TTC to assess myocardial damage (D); shown is the average infarct size divided by area at risk (E). F, samples of perfusate were obtained after 45 min of reperfusion to assess LDH activity relative to LDH activity in the equilibrium perfusate. *, statistically significant difference from all other groups by two-way ANOVA followed by Tukey's post hoc analysis, p ≤ 0.05. These experiments were performed twice using separate cohorts of mice. Error bars, S.E.
MANF improves ER proteostasis during reperfusion injury
Myocyte death can occur during ischemia as well as reperfusion. To determine whether MANF could provide protection under one or both of these conditions, we examined MANF levels in cultured neonatal rat ventricular myocytes (NRVMs) subjected to sI or sI/R. MANF levels increased upon sI and further upon sI/R, suggesting that MANF could contribute to preserving myocyte viability under either or both conditions (Fig. 3A). To further examine functional roles for MANF, it was knocked down using siRNA (Fig. 3B); we found that MANF knockdown had no effect on cell death during sI (Fig. 3C); however, MANF knockdown increased cell death during reperfusion, as indicated by the increased number of propidium iodide (PI)-positive cells and release of HMGB1 into the culture medium during reperfusion (Fig. 3, D, E (top), and F). Because cell death in cardiac myocytes can be caused by reactive oxygen species (ROS) generated during reperfusion injury (5, 18), we examined whether MANF knockdown affected ROS during sI/R, finding that MANF knockdown significantly increased ROS under these conditions (Fig. 3G).
Figure 3.

Effect of sI and sI/R on MANF expression and the effect of MANF knockdown on myocyte death, ROS generation, and expression of ER stress markers during sI/R. A, immunoblots of MANF and GAPDH from NRVMs subjected to 8 h of sI or 8 h of sI followed by 24 h of simulated reperfusion (sI/R). Band intensities were normalized to those for GAPDH and displayed as -fold control level. *, statistically significant difference from control by Student's unpaired t test, p ≤ 0.05. #, statistically significant difference from all other groups by one-way ANOVA followed by Newman–Keuls post hoc analysis. B, immunoblots of MANF and GAPDH from NRVMs transfected with siCon or siManf demonstrating MANF knockdown. Band intensities were normalized to those for GAPDH and displayed as -fold control level. *, statistically significant difference by Student's unpaired t test, p ≤ 0.05. C and D, siCon- or siManf-transfected NRVMs were subjected to 12 h of sI (C) or 6 h of sI followed by 24 h of reperfusion (sI/R) (D), and percentage cell death was assessed by staining cell cultures with Hoechst 33342 and PI and dividing the number of PI-positive cells by the number of Hoechst-positive cells in a given field. E, HMGB1 immunoblots of the culture medium indicating cell death and immunoblots of GRP94, GRP78, PDIA6, and GAPDH from NRVMs transfected with siCon or siManf and subjected to sI/R. F, densitometry of HMGB1 immunoblot shown in E. G, siCon- or siManf-transfected NRVMs were subjected to sI/R followed by ROS measurement with CellROX. H, densitometry of GRP94, GRP78, PDIA6, and GAPDH immunoblots shown in E. Band intensities of GRP94, GRP78, and PDIA6 were normalized to those for GAPDH and displayed as -fold control level. * and #, statistically significant difference from all other groups by two-way ANOVA followed by Tukey's post hoc analysis, p ≤ 0.05. Note that GRP78, GRP94, and PDIA6 immunoblots were performed using an anti-KDEL antibody. Error bars, S.E.
Because ROS increased when MANF was knocked down in sI/R-treated NRVMs, and because accumulation of cellular ROS can result in ER stress (19), we investigated whether MANF knockdown increased indicators of ER protein misfolding during sI/R. One way to measure ER protein-folding status is to examine the levels of ER chaperones, which are known to increase when ER protein folding is impaired. Indeed, MANF knockdown impaired ER protein folding during sI/R, as evidenced by increased expression of GRP94, GRP78, and PDIA6 (Fig. 3, E (bottom) and H). Thus, endogenous MANF enhances myocyte viability, at least partly by contributing to maintaining ER protein folding during sI/R. Because the increase in ROS upon MANF knockdown might be responsible for increases in protein misfolding and decreases in myocyte viability during sI/R, we examined the effects of MANF knockdown on cell viability in NRVMs treated with the ROS generator, H2O2 (5, 18, 20). Whereas MANF knockdown and H2O2 each decreased myocyte viability, when examined together, their effects were approximately additive (Fig. 4A), suggesting that MANF does not directly protect against protein misfolding caused by ROS. The probable independence of the effects of MANF knockdown and H2O2 on NRVM viability was further supported by a two-way ANOVA, which statistically demonstrated that MANF knockdown and H2O2 independently affected viability. Because MANF-mediated protection of myocytes did not appear to involve mitigating the effects of ROS generated during reperfusion, we examined whether MANF could protect myocytes against other types of ER protein misfolding that occur during I/R injury.
Figure 4.

Effect of MANF loss of function and pharmacological ER stressors on myocyte viability, effect of TG or DTT on FLAG-MANF and α1AT co-immunoprecipitation, and effect of rMANF on protein aggregation and folding. A–D, NRVMs were transfected with siCon or siManf, and after 72 h, cells were treated with H2O2 (A), DTT (B), TG (C), TM (D), or vehicle. Viability was then assessed by an MTT assay. * and #, statistically significant difference from all other groups by two-way ANOVA followed by Tukey's post hoc analysis, p ≤ 0.05. E, NRVM cultures were co-infected with adenoviruses encoding FLAG-MANF and α1AT ΔCT, as shown, and treated with TG or DTT for 1 h. Cell extracts were subjected to SDS-PAGE followed by immunoblotting for FLAG or α1AT ΔCT. The cell extracts were also subjected to FLAG IP followed by SDS-PAGE and then IB for FLAG or α1AT ΔCT, as shown. F, HeLa cell cultures were co-transfected with plasmid constructs encoding FLAG-MANF and/or α1AT-HA ΔCT, as shown. The cell extracts were subjected to SDS-PAGE followed by immunoblotting for HA or FLAG. The cell extracts were also subjected to FLAG IP followed by SDS-PAGE and then IB for FLAG or HA, as shown. G, effect of rMANF at low (7.8 μm) or high (23.4 μm) concentrations on the aggregation of insulin (113 μm). H, effect of rMANF at low (14 μm) or high (28 μm) concentrations on the aggregation of α-lactalbumin (14 μm). I, effect of recombinant GRP78 (1 μm) or MANF (1 μm) on activity of heat-denatured citrate synthase (1 μm). Citrate synthase activity is displayed as fold heat-denatured control level. * and #, statistically significant difference from all other groups by one-way ANOVA, p ≤ 0.05, followed by Newman–Keuls post hoc analysis. Error bars, S.E.
MANF acts as a chaperone to reduce ER protein misfolding caused by reductive stress
Whereas ROS generated during I/R can result in protein misfolding, protein misfolding can also be caused by impaired disulfide bond formation. Disulfide bonds, formed in nascent ER proteins, are dependent on the presence of oxygen and the optimal redox environment of the ER, which is likely to shift toward a reductive environment during reperfusion, as the ER redox machinery needs time to recover during oxygen reintroduction, much like how mitochondria need time to recover (6, 21–29). Accordingly, we assessed whether MANF is protective during reductive stress by treating NRVMs with the reducing agent, DTT. Additionally, for the sake of comparison with nonreductive forms of ER stress that also occur during I/R (15, 30, 31), NRVMs were treated with thapsigargin, which inhibits SERCA, reducing ER Ca2+ and causing ER protein misfolding, or tunicamycin, which impairs ER protein glycosylation required for folding and trafficking of secreted and membrane-bound proteins through the classical secretory pathway (32–34). MANF knockdown and DTT treatment separately decreased myocyte viability and together appeared to synergistically decrease myocyte viability (Fig. 4B), suggesting that MANF is required for optimal myocyte viability during protein misfolding caused by reductive ER stress. In contrast to DTT, MANF knockdown and thapsigargin (TG) treatment separately decreased myocyte viability, and when combined, their effects on viability were approximately additive (Fig. 4C), and MANF knockdown had no effect on NRVM viability when under tunicamycin (TM) treatment (Fig. 4D) These results suggest that MANF is not required for optimal myocyte viability during protein misfolding caused by nonreductive ER stress. Furthermore, the synergistic effects of MANF knockdown and DTT treatment, and lack thereof with TG or TM treatment, were also supported by a two-way ANOVA.
We next considered whether MANF might bind to misfolded ER proteins and perhaps act as a chaperone during reductive stress. Accordingly, to examine whether MANF binds to misfolded ER proteins during reductive stress, NRVMs were infected with adenoviruses encoding FLAG-MANF and a known misfolded ER protein, α1-antitrypsin (α1AT ΔCT) (35–38), and then treated with TG or DTT. The decreased expression of FLAG-MANF and α1AT ΔCT during treatment with TG and DTT (Fig. 4E, FLAG IB and α1AT ΔCT IB) is probably due to the global repression of translation of mRNAs encoding non-ER stress response genes that takes place during ER stress (4, 39, 40). When cell lysates were subjected to FLAG immunoprecipitation (IP) followed by FLAG or α1AT ΔCT immunoblotting (IB), it was apparent that MANF co-immunoprecipitated with misfolded α1AT ΔCT, but only upon treatment with DTT and not TG (Fig. 4E). Thus, MANF appears to improve ER protein folding during DTT-mediated (reductive) ER stress, but not during ER Ca2+ depletion-mediated (nonreductive) ER stress. Additionally, these results were also observed in HeLa cells, indicating that MANF also exerts this function in noncardiac cells (Fig. 4F).
We next examined whether MANF acts as a chaperone. Chaperones are defined as any protein that interacts with, stabilizes, or helps another protein to acquire its functionally active conformation, without being present in its final structure (41). In this regard, the two well-established properties of chaperones are their abilities to inhibit aggregation of unfolded proteins and to fold proteins into their final functional conformations (41, 42). Accordingly, we tested the ability of recombinant MANF (rMANF) to inhibit the aggregation of insulin and α-lactalbumin and to refold citrate synthase into its active conformation, as these are commonly employed in vitro chaperone assays (42–44). rMANF decreased the aggregation of both insulin and α-lactalbumin in a concentration-dependent manner (Fig. 4, G and H) and was able to restore activity to unfolded, inactive citrate synthase, as did the positive control, recombinant GRP78 (Fig. 4I). Taken together, the results in Fig. 4 indicate that during reductive ER stress, MANF binds to misfolded ER proteins and acts as a chaperone.
The conserved cysteine residues of MANF are required for its chaperone function
To more deeply dissect the chaperone function of MANF, we wished to introduce mutations in regions of MANF predicted to contribute to its chaperone function as a loss-of-function approach. To determine where to make such mutations, we assessed whether MANF bears any structural similarity to regions of other, well-studied chaperones, with an aim to mutate such areas in MANF. However, after extensive informatics assessment using standard techniques (Table S1), we found that, compared with the known chaperones (35, 37, 42, 45–55), which align well and cluster together (Fig. 5 (A and B), black), MANF exhibited poor alignment with most of the chaperone families tested, falling outside the 95% confidence interval of sequence similarity to the chaperones tested (Fig. 5 (A and B), red). Because our comparative informatic analysis did not reveal any obvious domains that are shared between MANF and other chaperones, we aligned MANF across several different species in search of conserved potentially functional regions. In doing so, we observed that the cysteine residues of MANF are conserved across MANF from all species (Fig. 6A). Because MANF protects against DTT-induced reductive ER stress, and because cysteine residues are susceptible to reduction during DTT treatment, we focused on examining the functional roles of the cysteine residues in MANF. Accordingly, we generated a construct encoding FLAG-MANF in which the 8 cysteine residues are mutated to alanine (Fig. 6B, FLAG-MANFMut.). Immunocytofluorescence demonstrated that both FLAG-tagged forms of MANF co-localized with the ER-resident chaperone, GRP78 (Fig. 6, C and D), confirming that ectopically expressed MANF is properly located. We found that whereas FLAG-MANFWT can form a complex with misfolded α1AT ΔCT, in the ER, FLAG-MANFMut. was unable to form this complex (Fig. 6E). Moreover, we found that, in contrast to rMANFWT, rMANFMut. did not exhibit chaperone activity (Fig. 6, F and G). These results are consistent with the hypothesis that the cysteine residues of MANF contribute to its chaperone function, at least in part by forming complexes with misfolded proteins in the ER.
Figure 5.
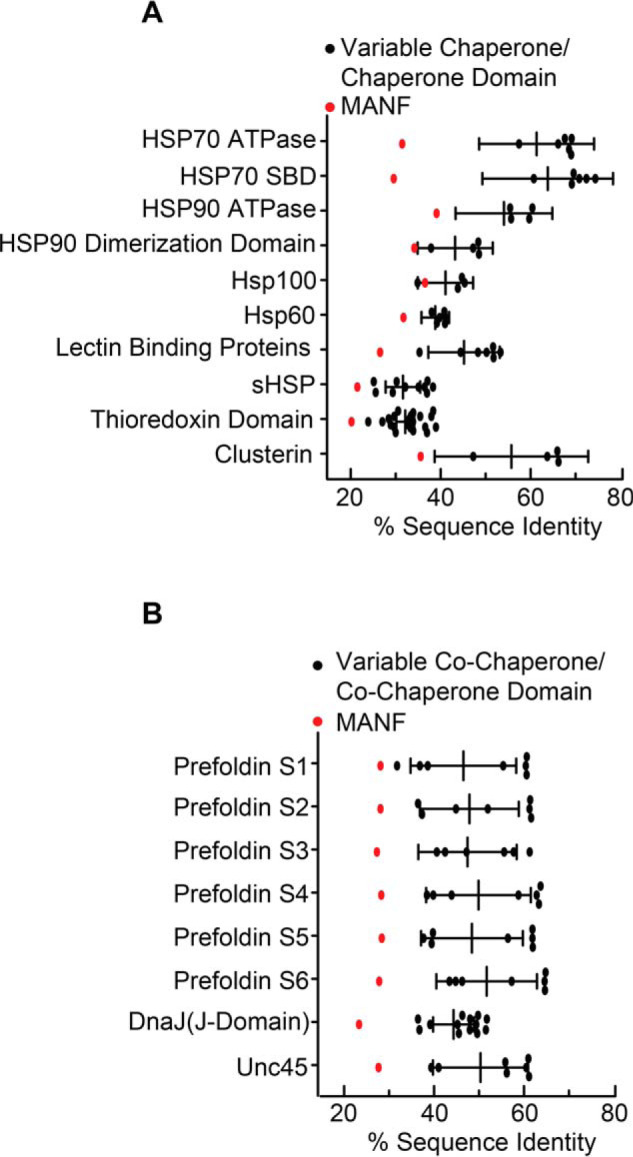
Sequence homology between MANF and known chaperone and co-chaperone orthologs or homologues. A and B, Clustal sequence identity analysis of MANF and various chaperones (A) and co-chaperones (B). Error bars, 95% confidence intervals.
Figure 6.

Effect of mutation of the conserved cysteine residues in MANF on MANF redox status and chaperone function. A, alignment of MANF sequences from different species. Highlighted in yellow are the positions of cysteine residues, the positions of which are conserved across the species shown. B, diagram of FLAG-MANFWT and FLAG-MANFMut. constructs indicating cysteine-to-alanine mutations. C and D, immunocytofluorescence of FLAG-MANF. NRVMs were transfected with siManf targeted to the 3′-UTR of the Manf transcript, followed by infection with AdV-FLAG-MANFWT (C) or AdV-FLAG-MANFMut. (D) and then treated with tunicamycin to induce GRP78 expression. NRVMs were then examined by immunocytofluorescence for GRP78 (green) and FLAG-MANF (red). Nuclei are indicated by TOPRO staining (blue). E, duplicate HeLa cell cultures were co-transfected with plasmid constructs encoding FLAG-MANFWT or FLAG-MANFMut. and α1AT-HA ΔCT and treated with DTT. The cell extracts were subjected to reducing SDS-PAGE followed by immunoblotting for α1AT-HA ΔCT (∼45 kDa) or FLAG-MANF (∼20 kDa). The cell extracts were also subjected to FLAG IP followed by nonreducing SDS-PAGE to maintain possible disulfide bonds between MANF and other proteins and then IB HA (bottom). (Note the FLAG-MANF/α1AT-HA ΔCT complex shown at ∼65 kDa). F, effect of recombinant MANFWT (23.4 μm) or MANFMut. (23.4 μm) on aggregation of insulin (113 μm). G, effect of recombinant MANFWT (21 μm) or MANFMut. (21 μm) on aggregation of α-lactalbumin (14 μm). * and #, statistically significant difference from all other groups by one-way ANOVA, p ≤ 0.05, followed by Newman–Keuls post hoc analysis. H, diagram depicting the function and mechanism of action of endogenous MANF in the heart resulting from this study. Error bars, S.E.
Whereas we observed expression of mutant MANFMut. in Escherichia coli, which allowed us to make enough of the recombinant protein to test in this study, and we were able to demonstrate its proper localization in NRVMs, we were unable to demonstrate significant expression of FLAG-MANFMut. in the hearts of AAV9-FLAG-MANFMut.–treated mice or in NRVMs infected with AAV9-FLAG-MANFMut. (data not shown). Accordingly, because of the very low expression levels of FLAG-MANFMut. in cultured cardiac myocytes and in the mouse heart, in vivo, we were unable conduct experiments assessing whether the cysteine residues of MANF are required for its function in vivo or in NRVMs.
Discussion
Although MANF was discovered more than 15 years ago, details about its function, especially in ER protein folding, remain unclear. We have previously demonstrated that, like several ATF6-inducible ER-resident proteins, MANF possesses a C-terminal retention signal that contributes in part to its ER localization (14, 15). Additionally, several ATF6-inducible ER-resident proteins interact with the chaperone GRP78 to maintain ER proteostasis (35, 46), and we previously demonstrated that MANF is also induced by ATF6 and directly interacts with GRP78 (15). Accordingly, here we focused our studies of MANF in cardiac myocytes, in vitro and in vivo, under reductive and nonreductive ER stress to determine what role MANF plays in ER protein folding.
MANF maintains proteostasis in cardiac myocytes during reperfusion injury
We have previously demonstrated that during ischemia, ATF6 up-regulates genes that encode ER-resident chaperones, such as GRP78 and GRP94, which are protective by contributing to ER protein folding, and we have shown that during I/R, ATF6 up-regulates genes encoding antioxidant proteins, such as catalase (4, 5), which neutralize ROS generated during reperfusion injury. Here, in vivo we observed that MANF loss of function decreases myocyte viability and cardiac function following I/R injury. Because the function of MANF to this point was unknown, it was important to dissect during which phase of I/R MANF exerts its function, because ATF6 up-regulates genes that are protective during both phases (4, 5, 56). Here we found that MANF was dispensable for cardiac myocyte viability during simulated ischemia but was critical for myocyte viability following I/R. Additionally, we observed that MANF knockdown did not sensitize NRVMs to oxidative stress mediated by H2O2, suggesting that the protective function of MANF was separate from oxidative stress and ROS generation associated with reperfusion injury. Furthermore, we observed that I/R in combination with MANF knockdown increased expression of GRP78, GRP94, and another ATF6-inducible chaperone and protein disulfide isomerase, PDIA6 (55), demonstrating for the first time that MANF exerts its protective function by maintaining ER protein folding during reperfusion.
MANF exerts its chaperone and protective functions in a stimulus-specific manner
We have recently demonstrated that the genes in cardiac myocytes that are induced by ATF6 are stimulus-specific (i.e. the ER stress response is tuned depending on what type of ER protein folding challenge the cell is facing) (12). Additionally, it has been demonstrated previously that various components of the ER protein-folding machinery have dedicated roles in maintaining ER protein folding and/or dedicated client proteins in the ER (45). To determine how MANF could maintain ER protein folding during reperfusion injury, we pharmacologically mimicked nonreductive and reductive ER stress using TG or TM and DTT, respectively (23, 25, 27, 30, 57). We observed that MANF was required for myocyte viability during reductive ER stress, but it was dispensable during nonreductive ER stress. Furthermore, we found that MANF forms a complex with the misfolded protein α1AT ΔCT during reductive, but not during nonreductive, ER stress. These results are consistent with findings from our laboratory and others that the ER stress response can be protective in a stimulus-specific manner (58, 59) and demonstrate that MANF exerts its protective function under specific proteotoxic stimuli. Because we also showed that MANF prevents the aggregation of disulfide-containing proteins (35, 43, 60), it seems likely that MANF plays a role in the folding of client proteins that form disulfide bonds as part of their final functional conformations.
MANF represents a novel class of chaperones
One of the most studied ER chaperones is GRP78, also known as BiP (37, 52). GRP78 plays a central role in ER protein folding in almost all cells in which it has been studied, including in cardiac myocytes (61, 62). Whereas it has been known for some time that ER stress, and in particular, ischemia, induces Grp78 in the heart (3–5), the importance of this chaperone in heart function has not been studied in depth until relatively recently, where Grp78 overexpression and deletion have demonstrated the adaptive and protective roles for Grp78 in mouse models of ischemic and hypertrophic heart disease (61, 62). We previously showed that Grp78 and MANF interact directly with each other in cardiac myocytes (14, 15). Moreover, another study has shown that MANF interacts with Grp78 in vitro in a way that affects the chaperone activity of Grp78 in a manner predicted to improve ER proteostasis, in vivo (63). In combination with that study, our results here demonstrate that MANF can improve ER protein folding by interacting with Grp78, and MANF can act as a chaperone itself, suggesting that there may be multiple ways that MANF improves ER protein folding and cardiac myocyte viability during reductive stresses, such as I/R and DTT. This is an unanticipated role for MANF, because our informatics analysis indicated that, from a structural viewpoint, MANF does not share domains with other well-known chaperones, indicating that MANF may be a new type of chaperone. However, because MANF resides in the ER of cardiac myocytes, the chaperone function of MANF complements the roles of other ER chaperones with which MANF likely collaborates to maintain ER proteostasis.
In conclusion, our study extends our understanding of MANF function in an in vivo model of cardiac pathology, demonstrating that MANF plays an important role in ER protein folding within cardiac myocytes, where it protects myocytes from reductive stress (Fig. 6H). This protective function likely depends on the cysteine residues in MANF, which are required for its chaperone activity in vitro and for its ability to bind to misfolded proteins in the ER in cells, consistent with a role for MANF as an adaptive responder to reductive stress in the ER of cardiac myocytes during cardiac pathology.
Experimental procedures
Laboratory animals
The research reported in this paper has been reviewed and approved by the San Diego State University Institutional Animal Care and Use Committee, and it conforms to the Guide for the Care and Use of Laboratory Animals published by the National Research Council.
Plasmid generation
Construct 1: pcDNA3.1(−) mouse MANF
The ORF of mouse Manf (NM_029103) was amplified by PCR and then ligated into the XhoI and HindIII sites of the multiple cloning site of pcDNA3.1(−).
Construct 2: pcDNA3.1(−) mouse MANF signal sequence
Using construct 1 as a template, PCR was carried out to introduce an NheI restriction site and Kozak sequence and an ApaI restriction site into the 5′ and 3′ ends, respectively, of the PCR-amplified product coding for the signal sequence MWATRGLAVALALSVLPDSRA. This PCR product and pcDNA3.1(−) were digested with NheI and ApaI and ligated together. The ApaI site is immediately followed by sequential XbaI and XhoI restriction sites.
Construct 3: pcDNA3.1(−) 3x-FLAG
A version of pcDNA3.1(−) containing an N-terminal 3x-FLAG sequence was constructed by annealing the following oligonucleotides: 5′-ctagcGCCATGGACTACAAAGACCACGACGGTGATTATAAAGATCACGATATCGATTACAAGGATGACGATGACAAGt-3′ and 5′-ctagaCTTGTCATCGTCATCCTTGTAA-TCGATATCGTGATCTTTATAATCACCGTCGTGGTCTTTGTAGTCCATGGCg-3′.
The NheI and XbaI overhangs of the annealed oligonucleotides were used to ligate the product into the XbaI site of pcDNA3.1(−), resulting in the creation of 3x-FLAG-pcDNA3.1, as described previously (64).
Construct 4: pcDNA3.1(−) mouse MANF signal sequence + 3x-FLAG
Using construct 3 as a template, PCR was performed to introduce XbaI and XhoI sites to the 5′ and 3′ ends, respectively, of the region coding for the 3x-FLAG tag, DYKDHDGDYKDHDIDYKDDDDK. This PCR product and construct 2 were digested with XbaI and XhoI and ligated together to create a construct encoding the MANF signal sequence followed by 3x-FLAG.
Construct 5: pcDNA3.1(−) mouse MANF signal sequence + 3x-FLAG + MANFWT (referred to hereafter as FLAG-MANFWT)
Using construct 1 as a template, PCR was performed with the following primers: forward, 5′-GGAACGCTCGAGCTGCGGCCAGGA-GAC-3′; reverse, 5′-GGAGCTGACACGGAAGAT-3′ (pcDNA3.1(−) reverse primer).
The resulting PCR product and construct 4 were digested with XhoI and HindIII and ligated together to generate a construct encoding the MANF signal sequence followed by 3x-FLAG and MANF, with the following amino acid sequence: MWATRGLAVALALSVLPDSRAGPSRDYKDHDGDYKDHDIDYKDDDDKLELRPGDCEVCISYLGRFYQDLKDRDVTSSPATIEEELIKFCREARGKENRLCYYIGATDDAATKIINEVSKPLAHHIPVEKICEKLKKKDSQICELKYDKQIDLSTVDLKKLRVKELKKILDDWGEMCKGCAEKSDYIRKINELMPKYAPKAASARTDL.
Construct 6: pcDNA3.1(−) mouse MANF signal sequence + 3x-FLAG + MANFMut. (referred to hereafter as FLAG-MANFMut.)
To mutate all eight cysteines in mouse MANF to alanine, serial site-directed mutagenesis was performed with the following primers: cysteines at positions 27 and 30, 5′-CGGCCAGGAGACGCTGAAGTTGCTA-TTTCTTATCTGG-3′ and 5′-CCAGATAAGAAATAGCAACTTCAGCG-TCTCCTGGCCG-3′; cysteines at positions 61 and 72, 5′-CTTATAAAGTTTGCCCGTGAAGCAAGAGGCAAAGAGAATCGGTTGGCCTACTACATTGGAG-3′ and 5′-CTCCAATGTAGTAGGCCAACCGATTCTCTTTGCCTCTTGCTTCACGGGCAAACTTTATAAG-3′; cysteines at positions 103 and 114, 5′-GTGGAAAAGATCGCTGAGAAGCTGAAGAAGAAAGACAGCCAGATCGCTGAACTAAAATC-3′ and 5′-GTATTTTAGTTCAGCGATCTGGCTGTCTTTCTTCTTCAGCTTCTCAGCGATCTTTTCCAC-3′; cysteines at positions 148 and 151, 5′-GACTGGGGGGAGATGGCCAAAGGCGCTGCAGAAAAGTCTG-3′ and 5′-CAGACTTTTCTGCAGCGCCTTTGGCCA-TCTCCCCCCAGTC-3′.
Construct 7: pcDNA3.1(−) 3x-HA
A version of pcDNA3.1(−) encoding a C-terminal 3x-HA sequence was constructed by sequentially annealing and ligating the following oligonucleotides into pcDNA3.1(−). The first set of oligonucleotides were annealed and digested with EcoRI and BamHI before ligation into pcDNA3.1(−) to generate a form of pcDNA3.1(−) encoding a C-terminal 1x-HA: 5′-GAATTCTACCCATACGATGTTCCAG-ATTACGCTTAATGAGGATCC-3′ and 5′-GGATCCTCATTAAGCGTAATCTGGA-ACATCGTATGGGTAGAATTC-3′.
The second set of oligonucleotides were annealed and directly ligated into the EcoRI site into the above vector pcDNA3.1(−) encoding a C-terminal 1x-HA: 5′-AATTCTATCCGTATGACGTACCTGACTATGCGGGCTATCCCTATGACGTGCCGGACTATGCAC-3′ and 5′-AATTGTGCATAGTCCGGCACGTCATAGGGATAGCCCGCATAGTCAGGTACGTCATACGGATAG-3′.
Constructs 8 and 9: α1AT
A construct encoding human α-1 antitrypsin (A1AT, NCBI RefSeq NM_000295) was generated by PCR using the appropriate primers to create an amplicon with XhoI on the 5′ end of the start site and with a termination codon and EcoRI on the 3′ end. This PCR product was cloned into the pCDNA3.1 vector (construct 8) or pcDNA3.1 vector modified to encode a C-terminal 3x-HA epitope (construct 9).
Constructs 10 and 11
To generate constructs encoding mutant α1 antitrypsin (36), α1AT ΔCT (construct 10) and α1AT-HA ΔCT (construct 11), site-directed mutagenesis was performed with the following primers: 5′-CAATGGGGCTGACCTCCGGGGTCACAG-3′ and 5′-CTGTGACCCCGGAGGTCAGCCCCATTG-3′.
Generation of MANF knockdown (MANF KD) transgenic mice
An oligonucleotide coding for an RNA hairpin targeting the mouse MANF transcript was cloned into the α-MHC vector, a gift from J. Robbins (University of Cincinnati). The αMHC-MANF KD construct was linearized and injected into the pronuclei of fertilized B6D2F1 (Harlan Sprague–Dawley) embryos. The resulting TG mice were back-crossed into the FVB background strain for at least 10 generations: MANF KD hairpin sequence, 5′-TGCTGTTATCTTCCGGATATAGTCAGGTTTTGGCCACTGACTGACCTGACTATCCGGAAGATAA-3′.
Animals were genotyped by isolating DNA from tail biopsies and performing PCR with the following primers: forward primer targeting the αMHC promoter, 5′-CGGCACTCTTAGCAAACCTC-3′; reverse primer, 5′-CAGATCTGGGCCATTTGTTC-3′.
AAV9 preparation and tail vein injection
To generate recombinant AAV9-control and AAV9-FLAG-MANFWT, shuttle vectors for these recombinants were constructed and co-transfected with AAV9 helper, pDG-9 (a gift from Dr. Roger Hajjar) into HEK293T cells to produce virus, as described previously (65). The shuttle vector pTRUF12-CMV was constructed by modifying pTRUF12 (a gift from Dr. Roger Hajjar) by first removing the region encoding GFP that was downstream of the internal ribosome entry site. New restriction sites were inserted into the multiple cloning site to include NheI, PmeI, XhoI, and MluI. The coding region of FLAG-MANFWT (construct 5, above) was excised and ligated into the NheI and HindIII restriction sites of the shuttle vector pTRUF12-CMV. To reduce recognition of the AAV9-mediated FLAG-MANF transcripts by the MANF KD hairpin, silent mutations were introduced into the pTRUF12-CMV plasmid by site-directed mutagenesis with the following primers: 5′-GCAGAAAAGTCTGATTACATTAGGAAAATCAATGAACTGATGC-3′ and 5′-GCATCAGTTCATTGATTTTCCTAATGTAATCAGACTTTTCTGC-3′.
To prepare the recombinant AAV9, HEK293T cells were plated at a density of 8 × 106 cells/T-175 flask and maintained in Dulbecco's modified Eagle's medium (DMEM)/F-12 containing 10% FBS, penicillin/streptomycin at 37 °C, and 5% CO2. For each virus preparation, 48 flasks were used. Twenty-four hours after plating, cultures were transfected using polyethyleneimine “Max” (molecular weight 40,000, Polysciences, catalog no. 24765) as follows. For each T-175 flask, 15 μg of helper plasmid and 5 μg of pTRUF12 plasmid were mixed with 1 ml of DMEM/F-12 (no antibiotics) and 160 μl of polyethyleneimine (0.517 mg/ml), vortexed for 30 s, and incubated for 15 min at room temperature. This was then mixed with 18 ml of DMEM/F-12 containing 2% FBS, penicillin/streptomycin and then used to replace the medium on the cultures. The cultures were then rocked intermittently for 15 min before being placed in a CO2 incubator. Three days later, the cells collected from six T-175 flasks were centrifuged at 500 × g for 10 min and then resuspended in 10 ml of lysis buffer (150 mm NaCl, 50 mm Tris-HCl). The resuspended cells were then subjected to three rounds of freeze-thaw, followed by treatment with Benzonase (1500 units of Benzonase; Novagen) and 1 mm MgCl2 at 37 °C for 30 min. The cell debris was collected by centrifugation at 3400 × g for 20 min. The supernatant obtained from six T-175 flasks containing the AAV9 was then purified on an iodixanol gradient comprised of the following four phases: 7.3 ml of 15%, 4.9 ml of 25%, 4 ml of 40%, and 4 ml of 60% iodixanol (Optiprep, Sigma-Aldrich) overlaid with 10 ml of cell supernatant. The gradients were centrifuged in a 70Ti rotor (Beckman Coulter) at 69,000 rpm for 1 h using OptiSeal Polyallomer Tubes (Beckman Coulter). Virus was collected by inserting a needle 2 mm below the 40–60% interface and collecting four or five fractions (∼4 ml) of this interface and most of the 40% layer. The fractions were analyzed for viral content and purity by examining 10 μl of each fraction on a 12% SDS-polyacrylamide gel (Bio-Rad), followed by staining with InstantBlue (Expedeon) to visualize the viral capsid proteins, VP1, VP2, and VP3. The virus was then collected from the fractions of several gradients, and the buffer was exchanged with lactated Ringer's using an ultrafiltration device, Vivaspin 20, 100 kDa molecular weight cutoff (GE Healthcare). The final viral preparation was then fractionated on a 12% SDS-polyacrylamide gel, stained with Coomassie Blue (Expedeon, catalog no. ISB1L), and then compared with a similarly stained gel of a virus of a known titer (an analogous control AAV9 with a CMVenhMLC800 composite promoter with no downstream ORF). To administer recombinant AAV, mice were injected via tail vein with 100 μl of 37 °C heated lactated Ringer's solution containing 1011 genome-containing units/mouse.
Adenovirus (AdV) generation
The AdEasy system was used for preparing recombinant adenoviral strains using methods described previously (2, 3). Recombinant AdV encoding FLAG-MANFWT was produced by excising the coding region of FLAG-MANFWT (constructs 5 and 6 above) and then ligating it into a version of the adenovirus shuttle vector, pAdTrack-CMV, that does not contain the GFP coding region. Recombinant AdV encoding α1AT ΔCT was produced by excising the coding region of α1AT ΔCT (construct 10 above) and then ligating it into a version of the adenovirus shuttle vector, pAdTrack-CMV, containing the GFP coding region. pAdTrack-CMV-FLAG-MANFWT and pAdTrack-CMV-α1AT ΔCT (36) were linearized and then co-transformed with the adenoviral vector, pAdEasy-1, into E. coli strain BJ5183. This strain of E. coli allows for homologous recombination of pAdEasy-1 and the pAdTrack-CMV shuttle vector containing the gene of interest. Recombinants were selected on kanamycin and screened by restriction digestion with PacI. Recombinant plasmids were then retransformed into E. coli DH5α for propagation purposes. To generate recombinant adenoviruses, these recombinant adenoviral plasmids were linearized with PacI and then transfected into 293 human embryonic kidney cells using Lipofectamine® (Life Technologies, Inc.). The recombinant viruses were then harvested 7–10 days post-infection. Viral titers were determined by qPCR as compared with a virus of a known titer.
Culturing of neonatal rat ventricular myocytes (5, 16)
NRVMs were prepared from 1–3-day-old Sprague–Dawley rat hearts using a neonatal cardiomyocyte isolation system (catalog no. LK003300, Worthington). Myocytes were then purified on a discontinuous Percoll density gradient. Briefly, isolated cells were counted and then collected by centrifugation at 250 × g for 5 min in an Eppendorf 5810R using the swinging bucket rotor. 40–60 million cells were then resuspended in 2 ml of 1× ADS buffer (116 mm NaCl, 18 mm HEPES, 845 μm NaHPO4, 5.55 mm glucose, 5.37 mm KCl, 831 μm MgSO4, 0.002% phenol red, pH 7.35 ± 0.5). Stock Percoll was prepared by combining 9 parts of Percoll (catalog no. 170891-02, GE Healthcare) with 1 part of clear (without phenol red) 10× ADS. The stock Percoll was used to make the Percoll for the top (density = 1.059 g/ml; 1 part Percoll stock added to 1.2 parts clear 1× ADS) and bottom (density = 1.082 g/ml; 1 part Percoll stock added to 0.54 parts red 1× ADS) layers. The gradient, consisting of 4 ml of top Percoll and 3 ml of bottom Percoll, was set in a 15-ml conical tube by pipetting the top Percoll first and layering the bottom Percoll gently underneath, and the cells (in 2 ml of red 1× ADS buffer) were layered on the top. Subsequently, the Percoll gradient was centrifuged at 1500 × g for 30 min with no deceleration brake at 4 °C. The isolated myocytes, which concentrated in the layer located between the lower red ADS layer and the middle clear ADS layer, were carefully collected and washed twice with 50 ml of 1× ADS and were then resuspended in plating medium and counted. This procedure is also effective for purifying myocytes that have been isolated by trypsin digestion, as described previously (1). Following Percoll purification, myocytes were plated at the desired density on plastic culture plates that had been pretreated with 5 μg/ml fibronectin in DMEM/F-12 (catalog no. 11330-32, Invitrogen) at 37 °C for 1 h in a 5% CO2 incubator. Cultures were then maintained in DMEM/F-12, supplemented with 10% FBS and antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin).
NRVM plating density
In experiments where cell viability was assessed by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, NRVMs were plated at 7.5 × 104 cells/well on 48-well dishes with 250 μl of culture medium/well (Corning, catalog no. 3548; 0.95 cm2 of growth area/well). For sI/R experiments, NRVMs were plated at 6 × 105 cells/well on 12-well dishes with 1 ml of culture medium/well (Corning, catalog no. 3513; 3.8 cm2 of growth area/well).
siRNA transfection of neonatal rat ventricular myocytes
Twenty-four hours after plating, NRVMs were transfected with control siRNA or siRNA targeting the Manf mRNA. In brief, siRNAs were diluted to a concentration of 120 nm in DMEM/F-12 supplemented with 0.5% (v/v) FBS 0.675% (v/v) HiPerfect reagent (Qiagen, catalog no./ID 301704) without antibiotics. The resulting solutions were incubated for 15 min at room temperature (21 °C). NRVMs were incubated with the relevant siRNA solutions for 16 h (125 μl/well on 48-well plates, 500 μl/well on 12-well plates), after which the medium was changed to DMEM/F-12 supplemented with 2% FBS and antibiotics (250 μl/well on 48-well plates, 1 ml/well on 12-well plates) for 72 h, after which cultures were subjected to ER stress treatments, simulated ischemia, or simulated ischemia/reperfusion.
siRNA sequences
siRNA sequences were as follows: Integrated DNA Technologies negative control DsiRNA, 5 nmol, catalog no. 51-01-14-04; Manf siRNA sequences, Integrated DNA Technologies siRNA, 20 μm stock, 3′-UTR custom synthesis, 5′-UGGGCUCCUGACAAUGAGAUGUGAA-3′. siRNAs were dissolved in molecular grade H2O to a 20 μm stock concentration.
NRVM cell viability assay following ER stress or H2O2 treatments (16)
Following siRNA transfection, medium from NRVMs plated on 48-wells was replaced with DMEM/F-12 supplemented with antibiotics and 10 μg/ml TM, 4 μm TG, 2 mm DTT (250 μl/well) for 12–24 h or 180 μm H2O2 for 5 h. Following treatment, culture medium was replaced with fresh 250 μl/well DMEM/F-12 supplemented with antibiotics and 180 μg/ml MTT labeling reagent (Millipore–Sigma/Roche catalog no. 11465007001) from a 5 mg/ml stock solution using PBS. Cultures were incubated for a further 4 h at 37 °C. The MTT-containing medium was manually pipetted away from the wells, and 50 μl of DMSO was added to each culture well. The absorbance at 560 nm of the resulting solution was measured on clear round-bottom 96-well plates using a VersaMax microplate reader.
Simulated ischemia/reperfusion (4)
To simulate ischemia, NRVM cultures (6 × 105 cells/well on 12-well culture dishes) were washed twice with 0.5 ml of prewarmed (37 °C) serum-free and glucose-free DMEM (Thermo Fisher Scientific, catalog no. A1443001) supplemented with antibiotics; after washing, cultures were incubated in 1 ml of serum-free and glucose-free DMEM with antibiotics at 0.1% O2 for 6–12 h in a hypoxia chamber with an oxygen controller. Parallel control cultures were maintained at ∼20–21% O2 in serum-free and glucose-free DMEM supplemented with antibiotics and 17.5 mm glucose. To simulate reperfusion, serum-free and glucose-free medium was replaced with DMEM/F-12, supplemented with 1 mg/ml BSA (Sigma–Aldrich, catalog no. A6003) and antibiotics for 24 h.
Intracellular reactive oxygen species measurement (5)
The levels of intracellular ROS were determined with the CellROX Orange fluorescent dye (Thermo Fisher Scientific, catalog no. C10443). After 8 h of sI and 1 h of reperfusion, NRVMs were incubated with 5 μm CellROX Orange for 20 min at 37 °C in DMEM/F-12, supplemented with 1 mg/ml BSA (Sigma–Aldrich, catalog no. A6003) and antibiotics and then washed three times with PBS (1 ml/well). PBS was replaced with DMEM/F-12 supplemented with 1 mg/ml BSA (Sigma–Aldrich, catalog no. A6003) and antibiotics. Cultures were then imaged using an IX70 fluorescence microscope (Olympus, Melville, NY). Fluorescence intensity in a field was measured with ImageJ software, which was normalized to the number of cells in that field to yield the mean cell fluorescence.
NRVM cell death assay (4)
Following 12 h of sI or 6–8 h sI and 24 h of simulated reperfusion (sI/R treatment), 2 μl of a 10 mg/ml PI stock solution and 1 μl of a 1 mg/ml stock solution of Hoechst 33342 were added to 22 μl of DPBS. The total 25 μl were then added directly to a well of a 12-well culture dish, which was then incubated for no more than 5 min at 37 °C. Cultures were then imaged using an IX70 fluorescence microscope (Olympus, Melville, NY). Numbers of PI- and Hoechst-positive cells were counted using ImageJ. For cultures subjected to simulated ischemia alone, we observed that it was important to keep the imaging time under 30 min to prevent increased cell death from the rapid onset of reperfusion injury (66).
NRVM immunocytofluorescence
NRVMs were plated at ∼1.25 × 105 cells on fibronectin-coated 4-chamber glass slides (Falcon). After siRNA transfection and adenovirus treatment as described above, slides were washed two times with 0.5 ml/well ice-cold DPBS. Slides were then fixed with 4% paraformaldehyde in DPBS on ice for 15 min. Slides were then washed three times with ice-cold DPBS (5 min/wash), followed by permeabilization with 0.5% Triton X-100, 3 mm EDTA for 10 min on ice, and then washed three times with DPBS (5 min/wash) and blocked for 1 h with Superblock. Slides were then incubated with primary antibodies diluted in Superblock (Thermo Fisher Scientific, catalog no. 37515) for 16 h at 4 °C. Primary antibodies used for staining NRVMs were anti-FLAG (1:200; Sigma catalog no. F1804) and anti-GRP78 (C-20, 1:30; catalog no. SC-1051, Santa Cruz Biotechnology). Slides were subsequently washed six times with ice-cold DPBS (5 min/wash) and then incubated at room temperature, in the dark for 90 min, with the appropriate fluorophore-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) diluted in Superblock, including Cy3-conjugated anti-mouse IgG (1:250) or FITC-conjugated anti-goat IgG (1:250). Slides were subsequently washed six times with ice-cold DPBS (5 min/wash). Nuclei were counterstained for 1 min with Topro-3 (1:1000; Thermo Fisher Scientific). Slides were subsequently washed six times with ice-cold DPBS. Images were obtained using laser-scanning confocal microscopy on an LSM 710 confocal laser-scanning microscope (Carl Zeiss, Oberkochen, Germany).
Immunoblotting
Medium was removed from 12-well culture dishes, and adherent cells were washed with ice-cold DPBS and then lysed with 60–100 μl of cell lysis buffer composed of 20 mm Tris (pH 7.5), 150 mm NaCl, 1% Triton X-100, 0.1% SDS, 1× protease/phosphatase inhibitor mixture (Roche Applied Science, catalog no. 05892791001). Lysates were scraped and transferred to microcentrifuge tubes and stored at −80 °C. Mouse hearts were excised, washed briefly in ice-cold DPBS, and snap-frozen in liquid nitrogen. Approximately 20 mg of frozen tissue was extracted in 250 μl of ice-cold tissue homogenization buffer composed of 20 mm Tris (pH 7.5), 150 mm NaCl, 1% Triton X-100, 1% SDS, and 0.5% sodium deoxycholate with 1× protease/phosphatase mixture. Following determination of protein concentration of clarified cell or tissue extracts using the BCA protein assay kit (Bio-Rad, catalog no. 5000111), between 5 and 40 μg of protein extracts or 25 μl of cell culture medium were subjected to reducing SDS-PAGE and then electroeluted onto polyvinylidene difluoride membranes. Membranes were blocked for 30 min at room temperature in 5% nonfat instant dry milk dissolved in TBS containing 1% Tween 20 (TBST) with gentle rocking. Membranes were probed with rabbit MANF antiserum at 1:1000 (anti-ARMET, catalog no. ab67203, Abcam; anti-MANF, catalog no. SAB3500384, Sigma-Aldrich), a mouse KDEL antiserum at 1:8000 (ENZO Life Sciences, catalog no. ADI-SPA-827), a mouse FLAG antibody at 1:8000 (Sigma–Aldrich, catalog no. F1804), an HMGB1 antiserum at 1:1000 (Abcam, catalog no. 18256), an HA antibody at 1:1000 (Santa Cruz Biotechnology, catalog no. sc-7392), an α1AT antiserum at 1:2000 (Dako, catalog no. A0012), and a mouse GAPDH antiserum at 1:6,000,000 (Fitzgerald, catalog no. 10R-G109a). Antibodies were diluted in 5% milk dissolved in TBST. Membranes were incubated with antibody solutions for 12–16 h at 4 °C. Membranes were then washed three times for 15 min in TBST and then incubated for 1 h at room temperature with the appropriate horseradish peroxidase–conjugated anti-IgG (Jackson ImmunoResearch Laboratories, Inc.) diluted at 1:2000 in 5% milk dissolved in TBST. Membranes were then washed three times for 15 min with gentle rocking in TBST, subjected to enhanced chemiluminescence, and exposed to autoradiography film or imaging using an ImageQuant 4000 from GE Healthcare Life Sciences. Immunoblots were quantified using ImageJ software densitometry.
FLAG immunoprecipitation
HeLa Cells were maintained in DMEM supplemented with 10% FBS and antibiotics not allowing confluency to surpass 80%. HeLa cells were resuspended at 6 × 106 cells/400 μl of ice-cold Dulbecco's PBS and electroporated with 1–20 μg of the relevant plasmids in a 0.4-cm gap electroporation cuvette at 250 V and 950 microfarads using a GenePulser II Electroporator (Bio-Rad). The cells were then plated at a density of 3 × 106 cells on a 10 cm dishes and incubated for 24 h in DMEM supplemented with 10% FBS and antibiotics. Transfected HeLa cell cultures were then treated for 1 h with vehicle or 2.5 mm DTT in serum-free DMEM/F-12 supplemented with antibiotics. NRVMs plated at 1 × 106 cells per well on 6-well culture dishes were incubated with with the relevant adenoviruses suspended in DMEM/F:12 supplemented with 2% FBS and antibiotics for 24 h. NRVMs were then treated for 1 h with vehicle, 2 μm TG, or 2.5 mm DTT in serum-free DMEM/F-12 supplemented with antibiotics. The culture medium containing TG or DTT was then removed, and cultures were briefly washed with ice-cold DPBS containing 20 mm N-ethylmaleimide (NEM; Sigma–Aldrich, catalog no. E376). Cultures were then lysed with cell lysis buffer composed of 20 mm Tris (pH 7.5), 150 mm NaCl, 1% Triton X-100, 20 mm NEM, and 1× protease/phosphatase inhibitors. The resulting cell lysates were then clarified by centrifugation at 20,000 × g. Between 80 and 200 μg of clarified protein cell extracts were diluted to 0.5–1 μg of protein/μl using cell lysis buffer composed of 20 mm Tris (pH 7.5), 150 mm NaCl, 1% Triton X-100, 20 mm NEM, and 1× protease/phosphatase inhibitors (maximum volume of 200 μl in 1.5-ml microcentrifuge tubes). 1 μg of FLAG antibody (Sigma–Aldrich; catalog no. F1804) was added to the resulting solutions, which were then gently rocked overnight (∼16 h) at 4 °C. To this mixture, 20 μl of protein A–agarose beads (50% slurry) that had been prewashed with ice-cold lysis buffer (20 mm Tris (pH 7.5), 150 mm NaCl, 1% Triton X-100) were added to each microcentrifuge tube. The resulting mixture was gently rocked for 2 h at 4 °C. The beads were sedimented to the bottom of the microcentrifuge tubes by centrifugation at 4000 × g for 1–2 min at 4 °C. The supernatant was removed with a 1-ml Pipetman pipette without disrupting the sedimented beads. The beads were then washed with 1 ml of ice-cold lysis buffer and sedimented by centrifugation at 4000 × g for 1–2 min at 4 °C. This process was repeated two more times. To elute the FLAG-immunoprecipitated complexes, the isolated beads were incubated with 30 μl of cell lysis buffer composed of 20 mm Tris (pH 7.5), 150 mm NaCl, 1% Triton X-100, 1× protease/phosphatase inhibitors, and 300 μg/ml FLAG peptide. The resulting mixture was gently rocked for 3 h at 4 °C, and the eluate was separated from the beads by centrifugation at 4000 × g for 1–2 min at room temperature (19–21 °C). The eluates were then subjected to reducing or nonreducing SDS-PAGE on a 4–12% gradient polyacrylamide gel and then electroeluted onto polyvinylidene difluoride membranes. Membranes were blocked for 30 min at room temperature in 5% nonfat instant dry milk dissolved in TBS containing 1% Tween 20 (TBST) with gentle rocking. Membranes were probed with a mouse FLAG antibody at 1:8000 (Sigma–Aldrich, catalog no. F1804), an HA antibody at 1:1000 (Santa Cruz Biotechnology, catalog no. sc-7392), or an α1AT antiserum at 1:2000 (Dako, catalog no. A0012).
Purification of recombinant WT and mutant forms of 6x-His–tagged MANF
BL21 E. coli were transformed with constructs (prSET-B, Invitrogen catalog no. V351-20) encoding 6x-His–tagged MANFWT or MANFMut and then maintained in 1 liter of Luria broth with 50 mg/liter ampicillin at 37 °C and 300 rpm until reaching an A600 of 0.6–0.7. Isopropyl β-d-1-thiogalactopyranoside was added to the culture flask to a final concentration of 1 mm, and cultures were incubated at 37 °C and 300 rpm for an additional 4 h. Cells were then collected by centrifugation at 4 °C and 4000 rpm for 30 min in an Eppendorf 5810R using the swinging bucket rotor. The cells were then frozen at −20 °C until the next step in the process. Cell pellets were thawed and then resuspended in 100 ml of Buffer Z (8 m urea, 100 mm NaCl, 20 mm HEPES, pH 8.0, and 20 mm imidazole), after which they were subjected to 10 rounds of sonication. Each round consisted of 20 s of sonication, followed by placement of the cells in an ice-water bath for at least 1 min. Lysates were clarified by centrifugation at 3220 relative centrifugal force for 15 min at 10 °C. The lysate was then applied to a pre-equilibrated 2.5-ml nickel-nitrilotriacetic acid nickel affinity column at room temperature (Qiagen, Valencia, CA). The column was then washed with 200 ml of Buffer Z and then eluted with 10 ml of Buffer Z to which 250 mm imidazole had been added. The eluate was then applied to PD-10 desalting columns (GE Healthcare Life Sciences, catalog no. 17085101) (2.5 ml/column) that had been equilibrated with PBS. The PD-10 eluates were then concentrated using Centricon Plus-70 centrifugal filter units (Millipore–Sigma, catalog no. UFC701008). The protein concentration was calculated using the BCA protein assay kit as described above and verified by SDS-PAGE and Coomassie Blue staining (Expedeon, catalog no. ISB1L).
Lactalbumin aggregation assay (44)
Reduced α-lactalbumin (rLA) was prepared by dissolving α-lactalbumin (Sigma–Aldrich, catalog no. L6010) to a concentration of 18.28 μm (2.55 mg/ml) in phosphate buffer (14 mm KH2PO4, 86 mm Na2HPO4, and 150 mm KCl, pH 7.5) containing 2.5 mm EDTA and 5 mm DTT for 30 min at 21 °C.
Denatured BSA was prepared by dissolving BSA (Sigma–Aldrich, catalog no. A6003) to a concentration of 210 μm (13.97 mg/ml) in 7.2 m guanidine hydrochloride dissolved in phosphate buffer and incubation at 21 °C overnight.
To initiate rLA aggregation, rLA was mixed with the denatured BSA at a final concentration of 14 μm rLA and 2.6 μm BSA in phosphate buffer. rMANF forms were added to the rLA and BSA mixture at final concentrations of 14–28 μm, after which samples were added to a clear 96-well round-bottom plate (100 μl/well), and absorbance at 360 nm was observed as a function of time using a VersaMax microplate reader set to 37 °C.
Insulin aggregation assay (43)
Insulin (Sigma–Aldrich, catalog no. I0516) was diluted to a final concentration of 113 μm (0.652 mg/ml) in phosphate buffer composed of 100 mm potassium phosphate (KH2PO4), 2.5 mm EDTA, pH 7.0, with or without 330 μm DTT, with or without 7.8–23.4 μm rMANF. Samples were transferred to a 96-well clear round-bottom plate (100 μl/well), and absorbance at 360 nm was observed as a function of time using a VersaMax microplate reader set to 21 °C.
Citrate synthase activity assay (42)
Citrate synthase (catalog no. C3260, Sigma–Aldrich) was suspended at 1 μm in TSC buffer composed of 10 mm Tris-HCl (pH 7.2), 150 mm NaCl, and 5 mm CaCl2 and kept on ice. An aliquot of the TSC/citrate synthase solution was incubated at 50 °C for 1 h to cause denaturation. Recombinant GRP78 (ProSpec, catalog no. HSP-037, HSPA5) or MANF was added at 1 μm to the TSC/citrate synthase solution after denaturation and incubated at 25 °C for 0.5–1 h. To measure the enzyme activity of citrate synthase, the TSC/citrate synthase solution was rapidly diluted 80-fold into a solution of TE (50 mm Tris, 2 mm EDTA, pH 8.0) containing 0.204 mm 5,5′-dithiobis-(2-nitrobenzoic acid) (Sigma-Aldrich, catalog no. D8130), 0.204 mm oxaloacetic acid (Sigma–Aldrich, catalog no. O9504), and 0.306 mm acetyl-CoA (Sigma–Aldrich, catalog no. A2056). Citrate synthase activity was assessed by measuring the absorbance of the solution at 412 nm over time, using a VersaMax microplate reader set to 29 °C.
RT-qPCR
Total RNA was isolated from mouse hearts using the RNeasy Mini kit (Qiagen). cDNA synthesis was performed using the SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific). RT-qPCR was performed using Maxima SYBR Green/ROX qPCR Master Mix in a StepOnePlus RT-PCR system (Thermo Fisher Scientific). The following primers were used: Manf, 5′-TGGGTGCGTTCTTCGACAT-3′ (forward) and 5′-GACGGTTGCTGGATCATTGAT-3′ (reverse); β-actin, 5′-GACGGCCAGGTCATCACTAT-3′ (forward) and 5′-GTACTTGCGCTCAGGAGGAG-3′ (reverse); Gapdh, 5′-ATGTTCCAGTATGACTCCACTCACG-3′ (forward) and 5′-GAAGACACCAGTAGACTCCACGACA-3′ (reverse); Nppa, 5′-TTGTGGTGTGTCACGCAGCT-3′ (forward) and 5′-TGTTCACCACGCCACAGTG-3′ (reverse); Nppb, 5′-AAGTCGGAGGAAATGGCCC-3′ (forward) and 5′-TTGTGAGGCCTTGGTCCTTC-3′ (reverse); Col1a1, 5′-AAGACGGGAGGGCGAGTGCT-3′ (forward) and 5′-TCTCACCGGGCAGACCTCGG-3′ (reverse).
Isolation and simulated ischemia treatment of AMVMs (5)
Briefly, hearts were rapidly cannulated via the ascending aorta, mounted on a perfusion apparatus, and retrograde-perfused at 3 ml/min for 4 min at 37 °C with heart medium (Joklik modified minimum essential medium, catalog no. M-0518 (Sigma–Aldrich), supplemented with 10 mm HEPES, 30 mm taurine, 2 mm dl-carnitine, 20 mm creatine, 5 mm inosine, 5 mm adenosine, and 10 mm butanedione monoxime, pH 7.36). Collagenase digestion of hearts was performed by perfusing for 13 min with heart medium supplemented with type 2 collagenase (50–60 mg; ∼320 units/ml, catalog no. LS004176, Worthington) and 12.5 μm CaCl2. Hearts were removed from the cannula and submerged in 2.5 ml of effluent collected off the heart during the collagenase digestion and dissociated using forceps. Collagenase was neutralized by adding 2.5 ml of heart medium supplemented with 10% FBS, and the final concentration of CaCl2 was adjusted to 12.5 μm. Cells were dissociated further by gently triturating for 4 min. The cell suspension was then filtered through a 100-μm mesh filter, and myocytes were allowed to sediment by gravity for 6 min at room temperature. The supernatant containing nonviable cells and nonmyocytes was discarded, and the remaining myocytes were resuspended in 5 ml of heart medium containing 5% FBS and 37.5 μm CaCl2. The concentration of CaCl2 in this suspension was slowly increased in a careful stepwise manner as follows: step 1, 50 μl of 10 mm CaCl2 added, mixed gently, and allowed to sit for 4 min; step 2, step 1 repeated; step 3, 100 μl of 10 mm CaCl2 added, followed by a 4-min wait; step 4, 80 μl of 100 mm CaCl2 added, followed by a 4-min wait. Cells were resuspended in plating medium (MEM (catalog no. 12350-039, Thermo Fisher Scientific), 1× insulin-transferrin-selenium (catalog no. 41400-045, Thermo Fisher Scientific), 10 mm HEPES, 100 units/ml penicillin, 100 μg/ml streptomycin, 10 mm butanedione monoxime, and 4% FBS). Cells were plated at 5 × 104 cells/well in 12-well culture plates coated with laminin (10 μg/ml). After at least 2 h, the medium was changed to maintaining medium (MEM, 1× insulin-transferrin-selenium, 10 mm HEPES, 1.2 mm CaCl2, 0.01% BSA, 25 μm blebbistatin). Cells were used for experiments 12–18 h later. To subject myocytes to simulated ischemia/reperfusion, the medium was changed to glucose-free DMEM (catalog no. A14430-01) with dialyzed FBS and antibiotics and incubated at 0.1% O2 for 3 h in a hypoxia chamber with an oxygen controller. Following 3 h of simulated ischemia, the medium was switched to reperfusion medium for 24 h (1× MEM containing 1× insulin, transferrin, and selenium in 10 mm HEPES, 1.2 mm CaCl2, and 0.1 mg/ml BSA with no blebbistatin).
AMVM cell viability assay (Calcein AM staining) (5)
Following TG or sI/R treatment, Calcein AM (Thermo Fisher Scientific, catalog no. C1430) dissolved at 1 mg/ml in DMSO was diluted 1:1000 directly into culture wells. Cultures were then incubated for 10 min at 37 °C. Viable NRVMs or AMVMs were identified as calcein AM–positive, and images were obtained using an IX70 fluorescence microscope (Olympus, Melville, NY). Numbers of viable, calcein AM-positive cells were counted using ImageJ. Parallel control cultures were maintained at ∼20–21% O2 in simulated ischemia medium supplemented with 17.5 mm glucose.
Ex vivo global ischemia/reperfusion (5, 10)
Briefly, age-matched (10–11 weeks of age) WT and MANF KD TG mice were injected intraperitoneally with heparin (500 units/kg), and after 10 min, mice were anesthetized with sodium pentobarbital (150 mg/kg). Hearts were isolated and rinsed with ice-cold modified Krebs–Henseleit buffer, the aortas were cannulated, and the hearts were mounted onto a Langendorff perfused heart apparatus. Hearts were perfused by gravity at a constant pressure of 80 mm Hg, and a pressure sensor balloon was inserted into the left ventricle through the left atrium. Left ventricular developed pressure (LVDP, mm Hg) was assessed using Powerlab software (10). Hearts were equilibrated for 30 min while submersed in buffer at 37 °C and paced at 400 Hz and 0.5 mA. Hearts were subjected to global no-flow ischemia without pacing for 20 min and then reperfused for 60 min.
LDH assay (10)
LDH release from isolated perfused hearts was assayed using a CytoTox 96® nonradioactive cytotoxicity assay from Promega. From the kit, 12 ml of room temperature assay buffer were used to dissolve one bottle of substrate mix. 100 μl of perfusate from cultures were transferred to a clear round-bottom 96-well plate. 50 μl of assay buffer mixed with assay substrate were added to each perfusate sample. The absorbance of the resulting solution was measured at 490 nm (5, 10), using a VersaMax microplate reader set at 29 °C.
Triphenyl tetrazolium chloride staining (10)
Hearts exposed to global ischemia/reperfusion were briefly frozen (5 min) at −80 °C. Hearts were partially thawed and sliced into 1-mm sections. Sections were incubated in 1% triphenyl tetrazolium chloride (TTC) in phosphate buffer (88 mm Na2HPO4, 1.8 mm NaH2PO4) at 37 °C for 10 min. Sections were then incubated in 10% formalin diluted in phosphate buffer overnight at 4 °C. Sections were placed on glass slides and scanned on a Canon scanner. Images were quantified using ImageJ to outline the infarct and area at risk.
Clustal analysis of MANF and chaperone/co-chaperone families
For a given family of chaperones, co-chaperones, or annotated chaperone functional domains, amino acid sequences (Table S1) were analyzed using the Clustal Omega Multiple Sequence Alignment tool with default settings (RRID:SCR_001591) and compared with the sequence of mouse mature MANF (Uniprot entry Q9CXI5, aa 22–179). The resulting percentage identity matrices (54) were used to generate the average sequence identities. The average sequence identities of the chaperone/co-chaperone/chaperone domain family members subjected to Clustal Omega analysis with MANF were plotted on a 95% confidence interval (Fig. 5).
Echocardiography
Echocardiography was carried out on anesthetized mice using a Visualsonics Vevo 770, or a Visualsonics Vevo 2100 high-resolution echocardiograph, as described previously (67).
Statistics
Unless otherwise stated, values shown are mean ± S.E., and statistical treatments are t tests, one-way ANOVA followed by Newman–Keuls post hoc analysis, or two-way ANOVA followed by Tukey post hoc analysis.
Data availability
All data are contained within the article.
Supplementary Material
This article contains supporting information.
Author contributions—A. A. and C. C. G. conceptualization; A. A., E. A. B., W. T. S., M. S. D., A. S. B., D. J. T., A. N. P., C. A., A. V. S., S. D., and C. C. G. data curation; A. A., E. A. B., W. T. S., A. S. B., D. J. T., A. N. P., A. V. S., S. D., and C. C. G. formal analysis; A. A., E. A. B., and C. C. G. funding acquisition; A. A., E. A. B., W. T. S., A. S. B., D. J. T., and C. C. G. validation; A. A., E. A. B., S. D., and C. C. G. investigation; A. A. and C. C. G. visualization; A. A., E. A. B., S. D., and C. C. G. methodology; A. A. and C. C. G. writing-original draft; A. A., E. A. B., W. T. S., D. J. T., S. D., and C. C. G. writing-review and editing; D. J. T., S. D., and C. C. G. supervision; C. C. G. project administration.
Funding and additional information—This work was supported by American Heart Association Grant 17PRE33670796; National Institutes of Health Grant 1F31HL140850 (to E. A. B.) and Grants R01 HL135893, R01 HL141463, and R01 HL149931 (to C. C. G.); the San Diego State University (SDSU) Heart Institute (to W. T. S., A. A., E. A. B., and C. C. G.); the Inamori Foundation (to E. A. B.); and the ARCS Foundation, Inc., San Diego Chapter (to W. T. S. and E. A. B). Additionally, W. T. S., A. A., and E. A. B are Rees-Stealy Research Foundation Phillips Gausewitz, M.D., Scholars of the SDSU Heart Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- ER
- endoplasmic reticulum
- AAV9
- adeno-associated virus serotype 9
- ANOVA
- analysis of variance
- ATF6
- activating transcription factor 6α
- GRP78 and -94
- 78- and 94-kDa glucose-regulated protein, respectively
- HMGB1
- high-mobility group box 1 protein
- I/R
- ischemia/reperfusion
- LVDP
- left ventricular developed pressure
- MANF
- mesencephalic astrocyte derived neurotrophic factor
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NEM
- N-ethylmaleimide
- sI
- simulated ischemia
- sI/R
- simulated ischemia/reperfusion
- TG
- thapsigargin
- TTC
- 2,3,5-triphenyl tetrazolium chloride
- UPR
- unfolded protein response
- aa
- amino acids
- KD
- knockdown
- AMVM
- adult mouse ventricular myocyte
- NRVM
- neonatal rat ventricular myocyte
- PI
- propidium iodide
- ROS
- reactive oxygen species
- α1AT
- α1-antitrypsin
- IP
- immunoprecipitation
- IB
- immunoblotting
- rMANF
- recombinant MANF
- TM
- tunicamycin
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- AdV
- adenovirus
- qPCR
- quantitative PCR
- DPBS
- Dulbecco's PBS
- HA
- hemagglutinin
- rLA
- reduced α-lactalbumin
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- LDH
- lactate dehydrogenase
- MEM
- minimal essential medium.
References
- 1. Glembotski C. C. (2012) Roles for the sarco-/endoplasmic reticulum in cardiac myocyte contraction, protein synthesis, and protein quality control. Physiology 27, 343–350 10.1152/physiol.00034.2012 [DOI] [PubMed] [Google Scholar]
- 2. Gidalevitz T., Stevens F., and Argon Y. (2013) Orchestration of secretory protein folding by ER chaperones. Biochim. Biophys. Acta 1833, 2410–2424 10.1016/j.bbamcr.2013.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thuerauf D. J., Marcinko M., Gude N., Rubio M., Sussman M. A., and Glembotski C. C. (2006) Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ. Res. 99, 275–282 10.1161/01.RES.0000233317.70421.03 [DOI] [PubMed] [Google Scholar]
- 4. Doroudgar S., Thuerauf D. J., Marcinko M. C., Belmont P. J., and Glembotski C. C. (2009) Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J. Biol. Chem. 284, 29735–29745 10.1074/jbc.M109.018036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jin J. K., Blackwood E. A., Azizi K., Thuerauf D. J., Fahem A. G., Hofmann C., Kaufman R. J., Doroudgar S., and Glembotski C. C. (2017) ATF6 decreases myocardial ischemia/reperfusion damage and links ER stress and oxidative stress signaling pathways in the heart. Circ. Res. 120, 862–875 10.1161/CIRCRESAHA.116.310266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Koritzinsky M., Levitin F., van den Beucken T., Rumantir R. A., Harding N. J., Chu K. C., Boutros P. C., Braakman I., and Wouters B. G. (2013) Two phases of disulfide bond formation have differing requirements for oxygen. J. Cell Biol. 203, 615–627 10.1083/jcb.201307185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lodish H. F., and Kong N. (1993) The secretory pathway is normal in dithiothreitol-treated cells, but disulfide-bonded proteins are reduced and reversibly retained in the endoplasmic reticulum. J. Biol. Chem. 268, 20598–20605 [PubMed] [Google Scholar]
- 8. Zhu C., Johansen F. E., and Prywes R. (1997) Interaction of ATF6 and serum response factor. Mol. Cell. Biol. 17, 4957–4966 10.1128/MCB.17.9.4957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haze K., Yoshida H., Yanagi H., Yura T., and Mori K. (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799 10.1091/mbc.10.11.3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Martindale J. J., Fernandez R., Thuerauf D., Whittaker R., Gude N., Sussman M. A., and Glembotski C. C. (2006) Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ. Res. 98, 1186–1193 10.1161/01.RES.0000220643.65941.8d [DOI] [PubMed] [Google Scholar]
- 11. Belmont P. J., Tadimalla A., Chen W. J., Martindale J. J., Thuerauf D. J., Marcinko M., Gude N., Sussman M. A., and Glembotski C. C. (2008) Coordination of growth and endoplasmic reticulum stress signaling by regulator of calcineurin 1 (RCAN1), a novel ATF6-inducible gene. J. Biol. Chem. 283, 14012–14021 10.1074/jbc.M709776200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Blackwood E. A., Hofmann C., Santo Domingo M., Bilal A. S., Sarakki A., Stauffer W., Arrieta A., Thuerauf D. J., Kolkhorst F. W., Müller O. J., Jakobi T., Dieterich C., Katus H. A., Doroudgar S., and Glembotski C. C. (2019) ATF6 regulates cardiac hypertrophy by transcriptional induction of the mTORC1 activator, Rheb. Circ. Res. 124, 79–93 10.1161/CIRCRESAHA.118.313854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Petrova P., Raibekas A., Pevsner J., Vigo N., Anafi M., Moore M. K., Peaire A. E., Shridhar V., Smith D. I., Kelly J., Durocher Y., and Commissiong J. W. (2003) MANF: a new mesencephalic, astrocyte-derived neurotrophic factor with selectivity for dopaminergic neurons. J. Mol. Neurosci. 20, 173–188 10.1385/JMN:20:2:173 [DOI] [PubMed] [Google Scholar]
- 14. Glembotski C. C. (2011) Functions for the cardiomyokine, MANF, in cardioprotection, hypertrophy and heart failure. J. Mol. Cell Cardiol. 51, 512–517 10.1016/j.yjmcc.2010.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Glembotski C. C., Thuerauf D. J., Huang C., Vekich J. A., Gottlieb R. A., and Doroudgar S. (2012) Mesencephalic astrocyte-derived neurotrophic factor protects the heart from ischemic damage and is selectively secreted upon sarco/endoplasmic reticulum calcium depletion. J. Biol. Chem. 287, 25893–25904 10.1074/jbc.M112.356345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Doroudgar S., Volkers M., Thuerauf D. J., Khan M., Mohsin S., Respress J. L., Wang W., Gude N., Müller O. J., Wehrens X. H., Sussman M. A., and Glembotski C. C. (2015) Hrd1 and ER-associated protein degradation, ERAD, are critical elements of the adaptive ER stress response in cardiac myocytes. Circ. Res. 117, 536–546 10.1161/CIRCRESAHA.115.306993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morrison L. E., Whittaker R. J., Klepper R. E., Wawrousek E. F., and Glembotski C. C. (2004) Roles for αB-crystallin and HSPB2 in protecting the myocardium from ischemia-reperfusion-induced damage in a KO mouse model. Am. J. Physiol. Heart Circ. Physiol. 286, H847–H855 10.1152/ajpheart.00715.2003 [DOI] [PubMed] [Google Scholar]
- 18. Marshall K. D., Edwards M. A., Krenz M., Davis J. W., and Baines C. P. (2014) Proteomic mapping of proteins released during necrosis and apoptosis from cultured neonatal cardiac myocytes. Am. J. Physiol. Cell Physiol. 306, C639–C647 10.1152/ajpcell.00167.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Malhotra J. D., and Kaufman R. J. (2007) Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid. Redox Signal. 9, 2277–2293 10.1089/ars.2007.1782 [DOI] [PubMed] [Google Scholar]
- 20. Siman R., McIntosh T. K., Soltesz K. M., Chen Z., Neumar R. W., and Roberts V. L. (2004) Proteins released from degenerating neurons are surrogate markers for acute brain damage. Neurobiol. Dis. 16, 311–320 10.1016/j.nbd.2004.03.016 [DOI] [PubMed] [Google Scholar]
- 21. Dimmeler S., and Zeiher A. M. (2007) A “reductionist” view of cardiomyopathy. Cell 130, 401–402 10.1016/j.cell.2007.07.028 [DOI] [PubMed] [Google Scholar]
- 22. Rajasekaran N. S., Connell P., Christians E. S., Yan L. J., Taylor R. P., Orosz A., Zhang X. Q., Stevenson T. J., Peshock R. M., Leopold J. A., Barry W. H., Loscalzo J., Odelberg S. J., and Benjamin I. J. (2007) Human αB-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell 130, 427–439 10.1016/j.cell.2007.06.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yu Q., Lee C. F., Wang W., Karamanlidis G., Kuroda J., Matsushima S., Sadoshima J., and Tian R. (2014) Elimination of NADPH oxidase activity promotes reductive stress and sensitizes the heart to ischemic injury. J. Am. Heart Assoc. 3, e000555 10.1161/JAHA.113.000555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chin K. T., Kang G., Qu J., Gardner L. B., Coetzee W. A., Zito E., Fishman G. I., and Ron D. (2011) The sarcoplasmic reticulum luminal thiol oxidase ERO1 regulates cardiomyocyte excitation-coupled calcium release and response to hemodynamic load. FASEB J. 25, 2583–2591 10.1096/fj.11-184622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Handy D. E., and Loscalzo J. (2017) Responses to reductive stress in the cardiovascular system. Free Radic. Biol. Med. 109, 114–124 10.1016/j.freeradbiomed.2016.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cuozzo J. W., and Kaiser C. A. (1999) Competition between glutathione and protein thiols for disulphide-bond formation. Nat. Cell Biol. 1, 130–135 10.1038/11047 [DOI] [PubMed] [Google Scholar]
- 27. Eaton P., Byers H. L., Leeds N., Ward M. A., and Shattock M. J. (2002) Detection, quantitation, purification, and identification of cardiac proteins S-thiolated during ischemia and reperfusion. J. Biol. Chem. 277, 9806–9811 10.1074/jbc.M111454200 [DOI] [PubMed] [Google Scholar]
- 28. Ellgaard L., Sevier C. S., and Bulleid N. J. (2018) How are proteins reduced in the endoplasmic reticulum? Trends Biochem. Sci. 43, 32–43 10.1016/j.tibs.2017.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sevier C. S., and Kaiser C. A. (2002) Formation and transfer of disulphide bonds in living cells. Nat. Rev. Mol. Cell Biol. 3, 836–847 10.1038/nrm954 [DOI] [PubMed] [Google Scholar]
- 30. Garcia-Dorado D., Ruiz-Meana M., Inserte J., Rodriguez-Sinovas A., and Piper H. M. (2012) Calcium-mediated cell death during myocardial reperfusion. Cardiovasc Res. 94, 168–180 10.1093/cvr/cvs116 [DOI] [PubMed] [Google Scholar]
- 31. Wang Z. V., Deng Y., Gao N., Pedrozo Z., Li D. L., Morales C. R., Criollo A., Luo X., Tan W., Jiang N., Lehrman M. A., Rothermel B. A., Lee A. H., Lavandero S., Mammen P. P. A., et al. (2014) Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell 156, 1179–1192 10.1016/j.cell.2014.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Poet G. J., Oka O. B., van Lith M., Cao Z., Robinson P. J., Pringle M. A., Arnér E. S., and Bulleid N. J. (2017) Cytosolic thioredoxin reductase 1 is required for correct disulfide formation in the ER. EMBO J. 36, 693–702 10.15252/embj.201695336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vincenz L., and Hartl F. U. (2014) Sugarcoating ER stress. Cell 156, 1125–1127 10.1016/j.cell.2014.02.035 [DOI] [PubMed] [Google Scholar]
- 34. Glembotski C. C. (2014) Finding the missing link between the unfolded protein response and O-GlcNAcylation in the heart. Circ. Res. 115, 546–548 10.1161/CIRCRESAHA.114.304855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ushioda R., Hoseki J., Araki K., Jansen G., Thomas D. Y., and Nagata K. (2008) ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 321, 569–572 10.1126/science.1159293 [DOI] [PubMed] [Google Scholar]
- 36. Belmont P. J., Chen W. J., San Pedro M. N., Thuerauf D. J., Gellings Lowe N., Gude N., Hilton B., Wolkowicz R., Sussman M. A., and Glembotski C. C. (2010) Roles for endoplasmic reticulum-associated degradation and the novel endoplasmic reticulum stress response gene Derlin-3 in the ischemic heart. Circ. Res. 106, 307–316 10.1161/CIRCRESAHA.109.203901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Preissler S., Chambers J. E., Crespillo-Casado A., Avezov E., Miranda E., Perez J., Hendershot L. M., Harding H. P., and Ron D. (2015) Physiological modulation of BiP activity by trans-protomer engagement of the interdomain linker. Elife 4, e08961 10.7554/eLife.08961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sifers R. N., Brashears-Macatee S., Kidd V. J., Muensch H., and Woo S. L. (1988) A frameshift mutation results in a truncated α1-antitrypsin that is retained within the rough endoplasmic reticulum. J. Biol. Chem. 263, 7330–7335 [PubMed] [Google Scholar]
- 39. Yang Q., and Sarnow P. (1997) Location of the internal ribosome entry site in the 5′ non-coding region of the immunoglobulin heavy-chain binding protein (BiP) mRNA: evidence for specific RNA-protein interactions. Nucleic Acids Res. 25, 2800–2807 10.1093/nar/25.14.2800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rainbolt T. K., Saunders J. M., and Wiseman R. L. (2014) Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol. Metab. 25, 528–537 10.1016/j.tem.2014.06.007 [DOI] [PubMed] [Google Scholar]
- 41. Balchin D., Hayer-Hartl M., and Hartl F. U. (2016) In vivo aspects of protein folding and quality control. Science 353, aac4354 10.1126/science.aac4354 [DOI] [PubMed] [Google Scholar]
- 42. Buchner J., Grallert H., and Jakob U. (1998) Analysis of chaperone function using citrate synthase as nonnative substrate protein. Methods Enzymol. 290, 323–338 10.1016/S0076-6879(98)90029-5 [DOI] [PubMed] [Google Scholar]
- 43. Holmgren A. (1979) Thioredoxin catalyzes the reduction of insulin disulfides by dithiothreitol and dihydrolipoamide. J. Biol. Chem. 254, 9627–9632 [PubMed] [Google Scholar]
- 44. Kubota K., Niinuma Y., Kaneko M., Okuma Y., Sugai M., Omura T., Uesugi M., Uehara T., Hosoi T., and Nomura Y. (2006) Suppressive effects of 4-phenylbutyrate on the aggregation of Pael receptors and endoplasmic reticulum stress. J. Neurochem. 97, 1259–1268 10.1111/j.1471-4159.2006.03782.x [DOI] [PubMed] [Google Scholar]
- 45. Behnke J., Mann M. J., Scruggs F. L., Feige M. J., and Hendershot L. M. (2016) Members of the Hsp70 family recognize distinct types of sequences to execute ER quality control. Mol. Cell 63, 739–752 10.1016/j.molcel.2016.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ellgaard L., and Ruddock L. W. (2005) The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 6, 28–32 10.1038/sj.embor.7400311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kozlov G., Pocanschi C. L., Rosenauer A., Bastos-Aristizabal S., Gorelik A., Williams D. B., and Gehring K. (2010) Structural basis of carbohydrate recognition by calreticulin. J. Biol. Chem. 285, 38612–38620 10.1074/jbc.M110.168294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee A. S. (2014) Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 14, 263–276 10.1038/nrc3701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lee C. F., Melkani G. C., and Bernstein S. I. (2014) The UNC-45 myosin chaperone: from worms to flies to vertebrates. Int. Rev. Cell Mol. Biol. 313, 103–144 10.1016/B978-0-12-800177-6.00004-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu Q., and Hendrickson W. A. (2007) Insights into Hsp70 chaperone activity from a crystal structure of the yeast Hsp110 Sse1. Cell 131, 106–120 10.1016/j.cell.2007.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Poon S., Easterbrook-Smith S. B., Rybchyn M. S., Carver J. A., and Wilson M. R. (2000) Clusterin is an ATP-independent chaperone with very broad substrate specificity that stabilizes stressed proteins in a folding-competent state. Biochemistry 39, 15953–15960 10.1021/bi002189x [DOI] [PubMed] [Google Scholar]
- 52. Preissler S., Rohland L., Yan Y., Chen R., Read R. J., and Ron D. (2017) AMPylation targets the rate-limiting step of BiP's ATPase cycle for its functional inactivation. Elife 6, e29428 10.7554/eLife.29428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Saibil H. (2013) Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 14, 630–642 10.1038/nrm3658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Taylor R. P., and Benjamin I. J. (2005) Small heat shock proteins: a new classification scheme in mammals. J. Mol. Cell Cardiol. 38, 433–444 10.1016/j.yjmcc.2004.12.014 [DOI] [PubMed] [Google Scholar]
- 55. Vekich J. A., Belmont P. J., Thuerauf D. J., and Glembotski C. C. (2012) Protein disulfide isomerase-associated 6 is an ATF6-inducible ER stress response protein that protects cardiac myocytes from ischemia/reperfusion-mediated cell death. J. Mol. Cell Cardiol 53, 259–267 10.1016/j.yjmcc.2012.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Blackwood E. A., Azizi K., Thuerauf D. J., Paxman R. J., Plate L., Kelly J. W., Wiseman R. L., and Glembotski C. C. (2019) Pharmacologic ATF6 activation confers global protection in widespread disease models by reprograming cellular proteostasis. Nat. Commun. 10, 187 10.1038/s41467-018-08129-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kranias E. G., and Hajjar R. J. (2012) Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 110, 1646–1660 10.1161/CIRCRESAHA.111.259754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tam A. B., Roberts L. S., Chandra V., Rivera I. G., Nomura D. K., Forbes D. J., and Niwa M. (2018) The UPR activator ATF6 responds to proteotoxic and lipotoxic stress by distinct mechanisms. Dev. Cell 46, 327–343.e7 10.1016/j.devcel.2018.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stauffer W. T., Arrieta A., Blackwood E. A., and Glembotski C. C. (2020) Sledgehammer to scalpel: broad challenges to the heart and other tissues yield specific cellular responses via transcriptional regulation of the ER-stress master regulator ATF6α. Int. J. Mol. Sci. 21, E1134 10.3390/ijms21031134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li J., Zhang S., and Wang C. (2001) Only the reduced conformer of α-lactalbumin is inducible to aggregation by protein aggregates. J. Biochem. 129, 821–826 10.1093/oxfordjournals.jbchem.a002925 [DOI] [PubMed] [Google Scholar]
- 61. Bi X., Zhang G., Wang X., Nguyen C., May H. I., Li X., Al-Hashimi A. A., Austin R. C., Gillette T. G., Fu G., Wang Z. V., and Hill J. A. (2018) Endoplasmic reticulum chaperone GRP78 protects heart from ischemia/reperfusion injury through Akt activation. Circ. Res. 122, 1545–1554 10.1161/CIRCRESAHA.117.312641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang G., Wang X., Bi X., Li C., Deng Y., Al-Hashimi A. A., Luo X., Gillette T. G., Austin R. C., Wang Y., and Wang Z. V. (2019) GRP78 (glucose-regulated protein of 78 kDa) promotes cardiomyocyte growth through activation of GATA4 (GATA-binding protein 4). Hypertension 73, 390–398 10.1161/HYPERTENSIONAHA.118.12084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yan Y., Rato C., Rohland L., Preissler S., and Ron D. (2019) MANF antagonizes nucleotide exchange by the endoplasmic reticulum chaperone BiP. Nat. Commun. 10, 541 10.1038/s41467-019-08450-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Thuerauf D. J., Hoover H., Meller J., Hernandez J., Su L., Andrews C., Dillmann W. H., McDonough P. M., and Glembotski C. C. (2001) Sarco/endoplasmic reticulum calcium ATPase-2 expression is regulated by ATF6 during the endoplasmic reticulum stress response: intracellular signaling of calcium stress in a cardiac myocyte model system. J. Biol. Chem. 276, 48309–48317 10.1074/jbc.M107146200 [DOI] [PubMed] [Google Scholar]
- 65. Hoover H. E., Thuerauf D. J., Martindale J. J., and Glembotski C. C. (2000) αB-crystallin gene induction and phosphorylation by MKK6-activated p38: a potential role for αB-crystallin as a target of the p38 branch of the cardiac stress response. J. Biol. Chem. 275, 23825–23833 10.1074/jbc.M003864200 [DOI] [PubMed] [Google Scholar]
- 66. Vanden Hoek T. L., Shao Z., Li C., Zak R., Schumacker P. T., and Becker L. B. (1996) Reperfusion injury on cardiac myocytes after simulated ischemia. Am. J. Physiol. 270, H1334–H1341 10.1152/ajpheart.1996.270.4.H1334 [DOI] [PubMed] [Google Scholar]
- 67. Tsujita Y., Kato T., and Sussman M. A. (2005) Evaluation of left ventricular function in cardiomyopathic mice by tissue Doppler and color M-mode Doppler echocardiography. Echocardiography 22, 245–253 10.1111/j.0742-2822.2005.04014.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the article.