

Abstract
Enoyl-acyl carrier protein reductase (FabI) catalyzes a rate-controlling step in bacterial fatty-acid synthesis and is a target for antibacterial drug development. A phylogenetic analysis shows that FabIs fall into four divergent clades. Members of clades 1–3 have been structurally and biochemically characterized, but the fourth clade, found in members of phylum Bacteroidetes, is uncharacterized. Here, we identified the unique structure and conformational changes that distinguish clade 4 FabIs. Alistipes finegoldii is a prototypical Bacteroidetes inhabitant of the gut microbiome. We found that A. finegoldii FabI (AfFabI) displays cooperative kinetics and uses NADH as a cofactor, and its crystal structure at 1.72 Å resolution showed that it adopts a Rossmann fold as do other characterized FabIs. It also disclosed a carboxyl-terminal extension that forms a helix–helix interaction that links the protomers as a unique feature of AfFabI. An AfFabI·NADH crystal structure at 1.86 Å resolution revealed that this feature undergoes a large conformational change to participate in covering the NADH-binding pocket and establishing the water channels that connect the active site to the central water well. Progressive deletion of these interactions led to catalytically compromised proteins that fail to bind NADH. This unique conformational change imparted a distinct shape to the AfFabI active site that renders it refractory to a FabI drug that targets clade 1 and 3 pathogens. We conclude that the clade 4 FabI, found in the Bacteroidetes inhabitants of the gut, have several structural features and conformational transitions that distinguish them from other bacterial FabIs.
Keywords: bacteria, fatty acid synthase (FAS), microbiome, acyl carrier protein (ACP), crystal structure, Bacteroidetes, FabI, enoyl-acyl carrier protein reductase, fatty acid biosynthesis, Alistipes finegoldii, structural biology, gut microbiome, water channel, NADH binding
Introduction
Bacterial fatty acid biosynthesis (FASII3) consists of a conserved collection of individual proteins that produce the fatty acids required for membrane phospholipid biogenesis (1). The FASII substrates are linked to acyl carrier protein (ACP)3 and undergo successive rounds of condensation, reduction, dehydration, and reduction to extend the acyl chain by two carbons with each elongation cycle (Fig. 1A). The final reduction step of each elongation cycle is catalyzed by enoyl-ACP reductase (ENR) (Fig. 1A). There are two widely distributed, distinct ENR protein families, FabI and FabK (2–5). FabI proteins use NAD(P)H as the reductant and belong to the short-chain dehydrogenase superfamily of enzymes that use a Rossmann fold to bind the nucleotide cofactor (6, 7). Most short-chain dehydrogenases have an active site dyad consisting of Tyr-Xaa3-Lys, but the bacterial FabI subfamily is distinguished by having a differently spaced catalytic dyad consisting of Tyr-Xaa6-Lys (8, 9). FabK is a flavoprotein (FMN) that belongs to the NAD(P)H-dependent flavin oxidoreductase family (4). FabK adopts an overall TIM barrel fold (4, 5) and requires NAD(P)H as a reductant (2), but how the NADP(P)H and FMN cofactors collaborate in catalysis is not clear (3, 5). There are also two FabI-like ENRs (FabL and FabV) with a limited distribution in bacteria (10–14). These two reductases also belong to the short-chain dehydrogenase superfamily but are distantly related to FabI. The function of ENR in the FASII pathway is to pull each cycle of elongation to completion and is the rate-determining step of fatty acid elongation (15).
Figure 1.

Enoyl-ACP reductases in the Bacteroidetes. A, a diagram of the four-step elongation cycle of bacterial fatty acid synthesis (FASII). The acyl chains are covalently bound to ACP and are extended by two carbons with each turn of the cycle. The final step is catalyzed by an enoyl-ACP reductase that catalyzes the reduction of trans-2-acyl–ACP to acyl-ACP. FabI and FabK are two widely distributed enoyl reductase protein families. B, the percentage of individual species encoding FabI, FabK, or both, in five abundant human-associated Bacteroidetes genera (Alistipes, n = 10; Bacteroides, n = 41; Parabacteroides, n = 7; Porphyromonas, n = 15; Prevotella, n = 38). C, evolutionary relationship between the FabI family sequences. The distantly related FabL sequences were used to root the tree. FabI proteins fall into four clades: clade 1, prototypical Proteobacteria/Firmicute proteins; clade 2, Mycobacteria proteins; clade 3, plastidial enzymes; and clade 4, Bacteroidetes/Chlorobi enzymes.
The role of ENR as a pacemaker of the essential FASII pathway has made it the subject of intense investigation as a target for antibiotic drug discovery (16, 17). AFN-1252 (afabicin (Debio 1452)) is the most clinically advanced of these compounds and is designed to specifically target staphylococcal FabI (18–21). Most commonly used, broad-spectrum antibiotics not only attack the pathogen but also devastate the commensal gut microbiome, leading to multiple complications (22, 23). As a pathogen-specific antibiotic (16), AFN-1252 has the desirable property of having no effect on the size or composition of the gut microbiome during therapy (24). Firmicutes and Bacteroidetes are the two most abundant bacterial phyla in the gut microbiome. Most Firmicutes (85%) are in Class Clostridia that encode fabK and not fabI (25), therefore these organisms are expected to be refractory to growth inhibition by FabI-targeted drugs (2, 3). The Bacteroidetes occupants of the gut microbiome are more diverse. Some contain only a fabI homolog, some contain only a fabK homolog, and some contain both a fabI and fabK (Fig. 1B). This bioinformatic analysis suggests that some commensal Bacteroidetes may be susceptible to FabI-targeted therapeutics. We constructed a phylogenetic tree to understand the evolutionary relationships between the FabIs expressed in bacteria (Fig. 1C). This analysis shows that there are four distinct FabI clades. Representative FabIs from clades 1–3 have been biochemically and structurally characterized. Bacteroidetes/Chlorobi phyla are in the fourth FabI clade, suggesting that Bacteroidetes FabI may have unique features not found in structures from the other clades.
The goal of this study was to structurally and functionally characterize the FabI from Alistipes finegoldii (AfFabI), a human Bacteroidetes commensal anaerobe containing a clade 4 FabI. Like other FabIs, AfFabI is a tetramer and adopts a Rossmann fold to bind the nucleotide cofactor. The unique feature of AfFabI is the carboxyl-terminal α9 helix that forms a coiled-coil structure with the α9 helix of an adjacent protomer to form a protomer-protomer contact that is absent in the other three FabI clades. Upon NADH binding, the intertwined α9–α9 helices unravel to form a structured loop that is involved in sealing the lid over the active site of the opposite protomer. Deletion of the unique carboxyl-terminal domain results in a folded, but inactive enzyme because of severely compromised NADH affinity. The AfFabI active site uses the same Tyr-Xaa6-Lys catalytic dyad as other FabIs, but the surrounding residues create a unique active site environment that renders AfFabI refractory to a FabI therapeutic (AFN-1252) that effectively targets the clade 1 and 3 FabIs.
Results
Bioinformatics
The distribution of species in Bacteroidetes taxa encoding FabI, FabK, or both, was determined by counting the high homology FabI and FabK TBLASTN hits for each unique species entry in the RefSeq Representative Genomes database (12/22/2019) using the Alistipes FabI and FabK sequences and an e-value cut-off of 1e-100 for each hit. Environmental Bacteroidetes species encode for FabI only, FabK only, or both (not shown). Likewise, the individual species in the five major human-associated Bacteroidetes genera (Alistipes, Bacteroides, Parabacteroides, Porphyromonas, and Prevotella) encode for one of the three possible combinations of FabI and FabK (Fig. 1B). Prevotella, a prominent genus of the gut microbiome, had the highest number of species that encode only a FabI, but all three possible combinations were found in the individual species from all the commensal Bacteroidetes genera. A phylogenetic analysis was conducted to understand the evolutionary relationships between the FabIs (Fig. 1C). Clade 1 FabIs are encoded in the Proteobacteria and Firmicute phyla and are represented by the prototypical Escherichia coli (26) and Staphylococcus aureus (27) crystal structures. Clade 2 are the mycobacterial enzymes typified by the structure of InhA of Mycobacterium tuberculosis (28). Clade 3 FabIs consist of the plastid FabIs and include the structurally characterized FabIs of Chlamydia trachomatis (29) and Plasmodium falciparum (30). The FabI sequences from the closely related Bacteroidetes and Chlorobi phyla form a distinct fourth clade (Fig. 1C). The phylogenetic relationship between the various Bacteroidetes/Chlorobi FabI sequences is in overall agreement with the phylogenetic relationships between the organisms based on their 16S RNA sequences (31). These data show that Bacteroidetes/Chlorobi clade 4 FabIs have a divergent evolutionary history from the other clades, suggesting that there may be structural and/or functional differences between clade 4 FabIs and the other three characterized clades.
A. finegoldii FabI and FabK
We selected A. finegoldii as a prototypical gut commensal bacterium to examine the properties of the clade 4 FabIs. A. finegoldii is predicted to have both a FabI and FabK. A. finegoldii FabI (AfFabI) (UniProt ID: A0A174E195) has 32% identity with Neisseria gonorrheae FabI. The A. finegoldii FabK (AfFabK) (UniProt ID: I3YI65) has 42% identity with Streptococcus pneumoniae FabK and contains the signature FMN-binding motif. The ENR functions of AfFabI and AfFabK were verified by determining whether plasmids directing their synthesis would complement E. coli strain JP1111 (fabI(Ts)) (Fig. 2, A and B). The positive controls for the experiment were strain JP1111 carrying plasmids expressing E. coli FabI (EcFabI), C. trachomatis FabI (CtFabI), or Clostridium acetobutylicum FabK (CaFabK), and the negative control was the expression vector lacking a gene insert. All strains grew at the permissive temperature (30 °C) (Fig. 2A). Strain JP1111 containing the empty vector failed to grow at the nonpermissive temperature (42 °C), whereas the plasmids expressing AfFabI, AfFabK, and all the positive controls grew at 42 °C (Fig. 2B). These data show that A. finegoldii encodes both a FabI and FabK ENR as predicted from the bioinformatic analyses.
Figure 2.

A. finegoldii encodes two functional enoyl-acyl carrier protein reductases. A, growth of temperature-sensitive E. coli strain JP1111 (fabI(Ts)) mutant transformed with empty plasmid vector control (VC), or plasmids expressing either E. coli FabI (EcFabI), C. trachomatis FabI (CtFabI), A. finegoldii FabI (AfFabI; UniProt ID: A0A174E195), C. acetobutylicum FabK (CaFabK), or A. finegoldii FabK (AfFabK; UniProt ID: I3YI65) under the growth-permissive temperature (30 °C). B, growth of the same strain set at the nonpermissive temperature (42 °C). Five biological replicates were tested for each strain. C, microbroth dilution assay to rank the sensitivities of the FabIs and FabKs to AFN-1252 by determining the minimal inhibitory concentrations for AFN-1252 against E. coli strain ANS1 (ΔtolC) (33) transformed with the same series of plasmids described in panel A. Mean ± S.E. (n = 3).
AFN-1252 is a FabI inhibitor that is known to inhibit clade 1 and clade 3 FabIs (29, 32, 33). The sensitivity of AfFabI (clade 4) to AFN-1252 was compared with other FabIs using E. coli strain ANS1 (ΔtolC) expressing a series of FabIs as described previously (33). Strain ANS1 was transformed with a series of plasmids expressing different FabI and FabK enzymes and the AFN-1252 minimal inhibitory concentrations were determined using a microbroth dilution assay (Fig. 2C). Growth of strains ANS1/pEcFabI (clade 1) and ANS1/pCtFabI (clade 3) were both inhibited by AFN-1252 whereas all strains expressing a FabK were refractory to AFN-1252 growth inhibition. A key result was that strain ANS1/pAfFabI was refractory to AFN-1252 inhibition indicating that clade 4 FabIs may have an active site that is distinct from the clade 1 and clade 3 proteins.
Biochemical properties of AfFabI
An amino-terminal His-tagged version of AfFabI was expressed in E. coli and purified by affinity and gel filtration chromatography to obtain a homogeneous 34-kDa protein based on SDS gel electrophoresis (Fig. 3A, inset). AfFabI eluted as a single species on the calibrated XBridge BEH SEC column (Fig. 3A). Its Stokes radius was consistent with a molecular weight of 145 kDa (Fig. 3A, inset), indicating that like other characterized FabI proteins AfFabI exists as a tetramer (theoretical molecular weight 136 kDa) (7). Sedimentation velocity analysis confirmed AfFabI exists as a homotetramer in solution with an s20 value of 6.59 S corresponding to 141 kDa protein (Fig. 3B). AfFabI exhibited high-affinity, cooperative binding to NADH. The KD was estimated by surface plasmon resonance to be 225 nm with a Hill coefficient of 1.6 (Fig. 3C). The AfFabI affinity for its reaction product NAD+ was estimated in similar surface plasmon resonance experiments to be orders of magnitude lower (KD = 1.623 ± 0.008 mm) than for NADH (not shown). NADH increased the stability of AfFabI to thermal denaturation by 7 °C (Fig. 3D). These data indicated that NADH binding results in a more stable protein structure.
Figure 3.

AfFabI oligomerization and NADH binding. AfFabI was purified by affinity and gel filtration chromatography. A, AfFabI migrated as a single 145 kDa species (Ve = 5.058 min) on a calibrated XBridge BEH SEC 200 Å 3.5-μm column (136 kDa theoretical mass). Insets, gel electrophoresis shows the purity of AfFabI (34 kDa) along with the indicated molecular weight standards (Std). The standard curve for the XBridge BEH SEC column used thyroglobulin (669 kDa), immunoglobin (150 kDa), BSA (66.4 kDa), and myoglobulin (17 kDa). B, the sedimentation velocity profiles (fringe displacement) were fitted to a continuous sedimentation coefficient distribution model c(s). AfFabI sedimented as a 141 kDa tetramer with a sedimentation coefficient of 6.59 S. C, surface plasmon resonance sensorgrams depicting one experiment to determine NADH binding to AfFabI (top panel). The data from three independent titrations were fit to the Hill equation using GraphPad Prism software to calculate the KD and Hill coefficient (h). Mean ± S.E. (n = 3). D, protein thermal denaturation analysis was used to determine the stabilization of AfFabI by NADH. Assays contained 10 μm AfFabI (black) or 10 μm AfFabI + 100 μm NADH (red).
AfFabI enzymatic activity of NADH oxidation to NAD+ was monitored by spectrophotometry at 340 nm. AfFabI catalyzed the oxidation of NADH to NAD+ in the presence of crotonyl-CoA (Fig. 4A). The NADH apparent K0.5 was 22.3 ± 4.6 μm at 1.25 mm crotonyl-CoA and NADH exhibited positive cooperative behavior with a Hill number of 2.4 ± 0.2 (Fig. 4B). NADPH did not support the reaction (Fig. 4A, inset). Cooperative binding of the reduced nicotinamide substrate is characteristic of FabIs and has been studied in detail with S. aureus FabI (34). The crotonyl-CoA K0.5 was 457 ± 45 μm and exhibited some cooperative behavior with a Hill number of 1.5 (Fig. 4C). Mass spectrometry analysis confirmed that butyryl-CoA was the product in the enzymatic reaction (Fig. 4C, inset). These data demonstrate AfFabI catalyzes the FASII ENR reaction and that AfFabI exhibits kinetic properties that are like other characterized FabI enzymes. Although triclosan inhibited AfFabI with an IC50 of 1.76 ± 0.57 μm, which was similar to triclosan inhibition of EcFabI (35), AfFabI was refractory to AFN-1252 inhibition (Fig. 4D), suggesting its active site was significantly different from the active sites in the clade 1 and clade 3 FabIs that are sensitive to this drug.
Figure 4.

AfFabI displays prototypical FabI kinetics. A, the rate of 200 μm NADH oxidation in 1.25 mm crotonyl-CoA, 20 mm Tris-HCl, pH 8.0, as measured by the change in absorbance at 340 nm with (blue) or without (red) 100 nm AfFabI. The slope of the linear phase of the progress curve was converted to specific activity using the NADH extinction coefficient of 6220 m−1 cm−1. Inset, NADPH did not support the reaction. B, specific activities were calculated as a function of NADH concentration. The behavior of AfFabI with NADH is best described by the Hill equation (fitted line shown on graph). C, specific activities were calculated as a function of crotonyl-CoA concentration. The behavior of AfFabI with crotonyl-CoA is best described by the Hill equation (fitted line shown on graph). Inset, mass spectrum analysis of the reaction mixture shows reduction of crotonyl-CoA (m/z = 836.2) to butyryl-CoA (m/z = 838.3) either with (blue) or without (red) AfFabI. D, fractional activity of AfFabI versus increasing concentrations of AFN-1252 or triclosan. Data points were fit to the inhibitor versus response. Variable slope nonlinear regression equation using GraphPad Prism 8.2.1 software and the fitted lines are shown on the graph. AfFabI spectrophotometric assays were performed in triplicate as described under “Experimental procedures.” Mean ± S.E. (n = 3).
The AfFabI crystal structure
The 1.72 Å AfFabI structure was refined to R/Rfree 20.6/24.5 (Table 1). Two protomers were in the asymmetric unit and are similar to each other with a Cα RMSD of 0.34 Å across 260 residues. The protomers interact with two symmetry-related protomers around a crystallographic 2-fold axis to create the tetramer. The presence of tetramers in the crystal is consistent with the gel filtration and sedimentation experiments. The unique AfFabI protomer feature is α9, a carboxyl-terminal helix that protrudes from the α/β core structure that is absent in the clade 1–3 enzymes (Fig. 5A). Structural alignment of AfFabI protomer (lacking α9) with EcFabI (PDB ID: 5CFZ) (36, 81) showed the two structures are near identical with a Cα RMSD of 1.48 Å across 448 residues. Like the clade 1–3 FabIs (7), the AfFabI protomer consists of a seven-stranded parallel β-sheet flanked by three α helices on either side that adopts a Rossmann fold for dinucleotide substrate binding (Fig. 5A). The signature Tyr-Xaa6-Lys FabI catalytic dyad places the active site in the same location as other FabIs (8, 9). FabI is a dimer of dimers with three molecular 2-fold noncrystallographic symmetry axes (P, Q, and R) along the subunit interfaces (27, 38, 39) (Fig. 5B). Each protomer has a total surface area of ∼13,400 Å2, and ∼4800 Å2 (∼36%) is buried upon tetramerization. These values are similar to other FabIs (28, 36, 40–42, 81). There are two disordered regions in the structure. The residues constituting the putative α6 active site lid (Thr-197 to Gly-209) were expected to be absent from the model because the flexible lid is only resolved in FabI·NAD(P)H binary complexes in other FabI clades (27, 43, 44). The second unstructured region is the final 13 amino acids of the carboxyl terminus.
Table 1.
Data collection and model refinement statistics
| AfFabI | AfFabI·NADH | |
|---|---|---|
| PDB ID | 6VLX | 6VLY |
| Data collection | ||
| Space group | P21221 | P212121 |
| Cell dimensions | ||
| a, b, c (Å) | 60.6, 77.8, 127.6 | 75.3, 111.5, 150.5 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 35–1.72 (1.76–1.72)a | 29–1.86 (1.91–1.86) |
| Rmerge | 0.056 (0.483) | 0.077 (0.901) |
| CC1/2 | 99.7 (90.9) | 99.7 (77.0) |
| I/σ | 13.4 (1.8) | 12.6 (1.7) |
| Completeness (%) | 98.3 (87.7) | 94.4 (88.8) |
| Redundancy | 6.4 (5.4) | 4.9 (4.7) |
| Wilson B factor (Å2) | 25.8 | 24.7 |
| Refinement | ||
| Resolution (Å) | 30–1.72 (1.76–1.72) | 29–1.86 (1.91–1.86) |
| No. reflections | 59,692 (3142) | 100,112 (5169) |
| Rwork (%) | 20.6 (33.5) | 15.8 (42.8) |
| Rfree (%)b | 24.5 (34.3) | 19.7 (44.3) |
| Number of non-hydrogen atoms | ||
| Protein | 4110 | 8862 |
| Ligand/ion | 6 | 225 |
| Water | 402 | 1046 |
| RMSD | ||
| Bond lengths (Å) | 0.006 | 0.010 |
| Bond angles (°) | 1.380 | 1.463 |
| Average B factors (Å2) | 34.0 | 25.0 |
| Ramachandran plot | ||
| Favored (%) | 95.4 | 96.8 |
| Allowed (%) | 4.4 | 3.0 |
| Outliers (%) | 0.2 | 0.2 |
a Statistics for the highest-resolution shell are shown in parentheses.
bRfree test set uses ∼5% of the data.
Figure 5.
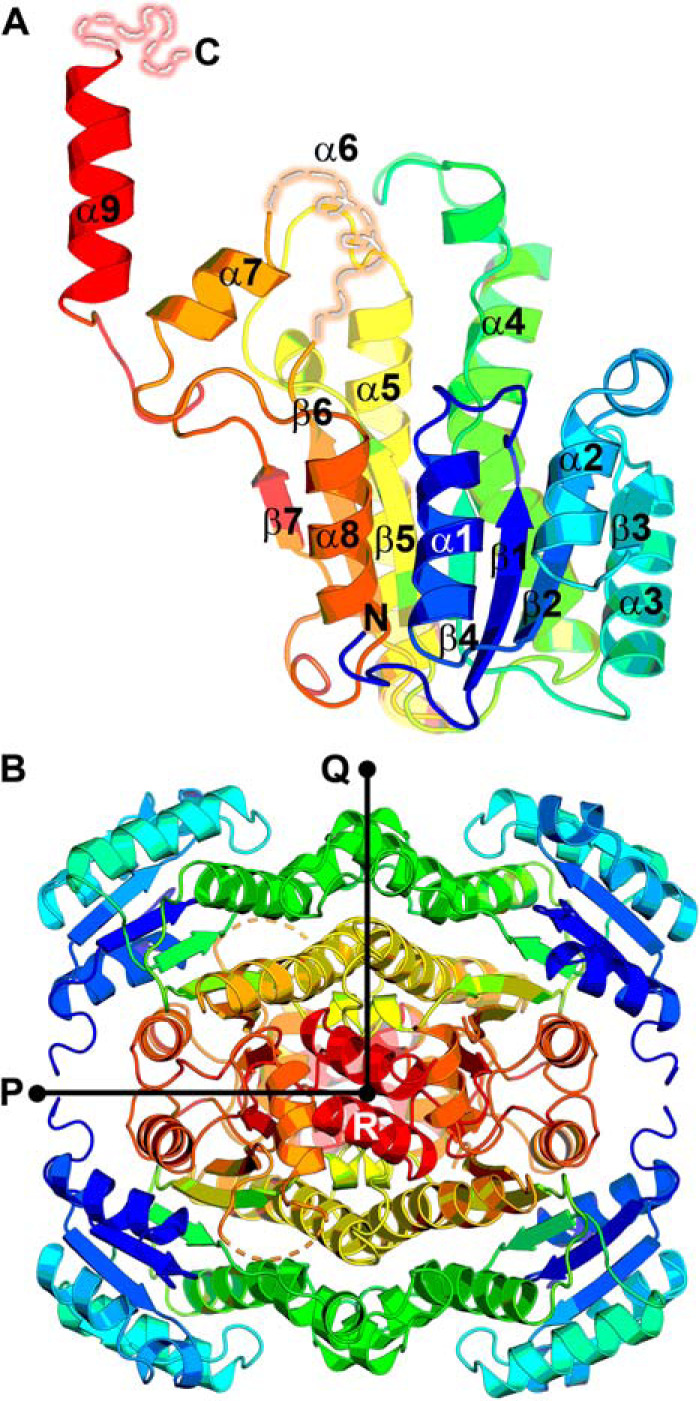
Overall crystal structure of AfFabI. A, the AfFabI monomer contains a Rossmann fold. The polypeptide chain is colored from blue (N terminus) to red (carboxyl terminus). The active site lid (α6) and terminal 13 residues are depicted as disordered (broken dashes) because these residues were not resolved in the electron density map. B, AfFabI is a tetramer with three perpendicular 2-fold noncrystallographic symmetry axes P, Q, and R.
FabI tetramers are stabilized by extensive intermolecular interactions along the protomer interfaces with conserved dimerization and tetramerization interfaces (27, 38, 45). Like clade 1–3 FabIs, AfFabI dimerization occurs along the P axis interface and consists of interacting helices α8 and strands β7 linking the A/B or C/D protomers (Fig. 6A). The AfFabI tetramerization domain links protomers along the Q axis interface and consists of an antiparallel four-helical bundle composed of interacting helices α4 (kinked) and α5 linking the A/C or B/D protomers (Fig. 6B). AfFabI α9–α9 interaction domain is a unique feature of clade 4 FabIs and links the A/D or B/C protomers along the R axis (Fig. 6C). Helix α9 extends from Ser-261 to Glu-275 and is amphipathic with the hydrophobic side chains forming the α9–α9 contact interface and the polar side chains extending into solvent.
Figure 6.

Subunit interface domains of AfFabI. Specific structural domains link the protomers along the P, Q, and R axes interfaces. A, the dimerization domain consists of interacting α8 helices and β7 strands that link protomers A/B and C/D along the P axis. B, the tetramerization domain is an antiparallel four-helix bundle composed of α4 and α5 that links protomers A/C and B/D along the Q axis. C, the carboxyl-terminal α9 helix–helix interaction domain is a unique feature of AfFabI. This domain links protomers A/D and B/C along the R axis. Rotation of the tetramer by 90° around the Q axis enables clear visualization of the protomer–protomer interaction mediated by α9.
The AfFabI·NADH binary complex
The 1.86 Å AfFabI·NADH complex structure was refined to R/Rfree 15.8/19.7 (Table 1). The asymmetric unit contained one homotetramer. The protomers are nearly identical with a Cα RMSD of 0.13 Å across 282 residues and the NADH molecules were well-resolved in all four active sites. The planarity of the cofactor carboxamide relative to its attached pyridine ring is indicative of the NADH redox state (46). The C2N-C3N-C7N-O7N torsion angles in the NADH molecules were between 150.8° and 162.5° and are consistent with reduced NADH being present in the crystals. NADH binding increases the overall protomer surface area to ∼15,000 Å2, and the buried surface area of the subunit interfaces to ∼5400 Å2 (∼36%). The active site lid containing α6 was not resolved in the AfFabI structure but was clearly seen in the AfFabI·NADH electron density map. The lid forms several hydrogen bond interactions with the pyrophosphate and nicotinamide of NADH (Fig. 7A). Nucleotide binding proteins often stabilize pyrophosphate binding using a positive helix dipole (47). Helix α1 creates this helix dipole in AfFabI and hydrogen bonds between NADH and lid residues Thr-199 side chain and Ala-201 backbone amide fix the cofactor in space. Lys-167 on α5 is part of the Tyr-Xaa6-Lys catalytic dyad and donates two hydrogen bonds to the nicotinamide ribose to position and stabilize the nicotinamide (Fig. 7A). In addition, the nicotinamide moiety is also stabilized by hydrogen bonds that form between lid residue Thr-197 and the nicotinamide ring. The adenine moiety sits in a solvent-exposed hydrophobic pocket in the active site cavity. Hydrogen bonds between the adenine nitrogen N-1 and Ala-67 backbone carbonyl, and the adenine ribose and Asn-41 and Ser-45 side chains orient the adenine moiety in this cavity. The locations and interactions between AfFabI and NADH are very similar to other FabI proteins (48–51).
Figure 7.

Active site and water channels in the AfFabI·NADH complex upon NADH binding. A, schematic diagram shows hydrogen bond interactions between the active site and NADH designated by purple lines. Lid residues that interact with NADH are shown by yellow highlight. B, molecular surface rendering of the central water well that connects to the four active sites was visualized in the AfFabI·NADH complex crystal structure along with the water channels that connect each active site with the water well using PyMOL. The molecular surface of the water channels and central water well are colored according to the contribution from each protomer. Segments of each water channel have more than one color because they are formed by the protomer interfaces along the R axis. The active sites in each protomer are designated with the letter A and the protomer superscript, and the letter W indicates the location of the central water well. C, each water channel contains ordered water molecules (red spheres) that form hydrogen bond interactions with each other and the peptide backbone. The line traces the chain of structured waters that begin at the carboxyamide of NADH in the active site (AC), exit into a narrow tunnel formed by the A/B/C protomer interfaces along the R axis, and empty into the central water well (W).
The NADH-induced conformational change creates four water-filled channels that connect the active sites to a central water well located in the core of the tetramer (Fig. 7B). These water channels enable efficient proton transfer to the active sites for catalysis (52). The resolution of the AfFabI·NADH crystal structure enabled the visualization of ordered water molecules within the individual channels making hydrogen bond interactions with the peptide backbone (Fig. 7C). The individual water channels exit the active sites, narrow into tunnels that are formed by residues along the R axis of the promoter interfaces and empty into the water well at the center of the protein (Fig. 7C). The active site cavity is hydrated in the AfFabI crystal structure, and few ordered water molecules are detected because the water channels are only partially formed and the active site lid is open to solvent. As with other FabIs (52), the lid isolates the active site from bulk solvent to create an environment that promotes hydride transfer from NADH.
Helix α9 and the carboxyl terminus
A unique feature of AfFabI is how the clade 4–specific α9 helix and disordered carboxyl-terminal tail participate in the conformational change required for NADH binding (Fig. 8). In the example shown, helix α9C and α9B intertwine to connect the C/B partner protomers in AfFabI and the 13 residues of the protein attached to carboxyl terminus of α9 are disordered (Fig. 8A). In the AfFabI·NADH structure, the interacting α9 helices unwind, and along with 10 of the 13 disordered carboxyl-terminal amino acids (Val-277 to Glu-286), form an ordered loop feature that extends to the active site lid of the partner protomer (Fig. 8B). The N terminus of α7 lies at the base of the α9–α9 interaction domain and forms the corner of the active site cavity. The conformational change relocates the Cα chain of α9 up to 20 Å toward the opposite active site and drives the formation of a new protomer–protomer interaction interface that closes the active site and creates the water channels.
Figure 8.
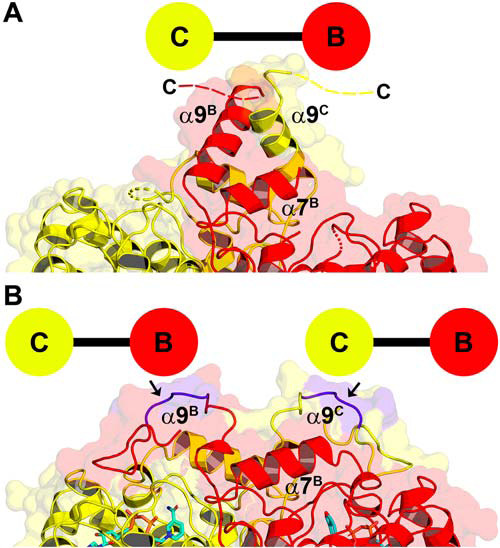
Conformational change in helix α9 and the carboxyl terminus. A, in the AfFabI crystal structure, the α9C and α9B helices are wound together, connecting the C and B protomers, and the last 13 residues of the protein are disordered. Broken dashes indicate the relative location of missing carboxyl terminus residues. B, in the AfFabI·NADH crystal structure, the α9C–α9B interaction domain unwinds, and 10 of the 13 carboxyl-terminal residues form a structured loop feature that connects to the active site lid of the partner protomer. The arrows denote the location of residues (purple) in the α9 turn that are removed in the AfFabI(1–269) truncation mutant.
The active site is a large solvent-exposed crevice in the AfFabI structure (Fig. 9A). In this conformation, Asn-213C and Asn-217C from the last two turns of α7C form hydrogen bonds with Tyr-268B from α9B. Upon NADH binding, the lid consisting of α6 and adjacent residues closes over NADH to convert the solvent-exposed crevice into an enclosed active site (Fig. 9B). Asp-210C from α7C forms a hydrogen bond with Thr-267B from α9B (Fig. 9B) as the unwound α9B residues slide along α7C. Lid closure leaves the adenine moiety surface-exposed and buries the pyrophosphate and nicotinamide regions of NADH. The solvent-accessible surface area of the active site crevice decreases from ∼500 Å2 to ∼300 Å2. Every residue in the first turn of α9 in AfFabI forms a new hydrogen bond interaction in the AfFabI·NADH complex (Fig. 9C). The Arg-262B backbone amide nitrogen forms a hydrogen bond interaction with the Tyr-179A side chain from α5A in the tetramerization domain. The Arg-263B side chain forms hydrogen bond interactions with backbone carbonyls from Ser-219B, Glu-216B, and Asn-217B from α7B in the corner of the protomer B active site. The Ala-264B backbone amide forms a hydrogen bond interaction with the Ser-261B side chain at the base of α9B, and the backbone carbonyls from Arg-263B and Met-265B form hydrogen bond interactions with the Lys-266C side chain from α9C. Two residues in the second turn of α9 form new hydrogen bond interactions in AfFabI·NADH. In addition to the Thr-267B–Asp-210C interaction shown in Fig. 9B, the side chain of Lys-266B forms hydrogen bonds with Arg-263C and Met-265C from α9C (Fig. 9D). There are no protein–protein interactions formed between the loop residues derived from the third turn of α9 in AfFabI and the protomers in the AfFabI·NADH structure. This loop extends away from the surface as seen in Fig. 8B.
Figure 9.

Creation of the NADH-binding site. The active site lid is disordered in the absence of NADH and helix α9B (red) is coiled around α9C (yellow) along the R axis. NADH (cyan) binding to the active-site crevice orders the lid. A, Tyr-268B in the second turn of helix α9 in the AfFabI structure forms hydrogen bond interactions with Asn-217C and Asn-213C in the second two turns of α7C. B, following NADH binding, the unwound α9B slips along α7C. The Thr-267B side chain on α9B forms a hydrogen bond interaction with Asp-210C in the first turn of α7C. C, residues in the first turn of α9 in the AfFabI structure become an ordered loop in AfFabI·NADH with multiple hydrogen bond interactions with the other protomer. D, Lys-266B from the second turn of α9 in AfFabI form hydrogen bond interactions with Arg-263C and Met-265C from α9C in AfFabI·NADH. E, hydrogen bond interactions between the disordered carboxyl terminus of AfFabI with the other protomers in AfFabI·NADH. The backbone amine of Glu-275B forms a hydrogen bond with the Tyr-158C side chain, the side chain of Asp-276B forms a hydrogen bonds to the side chain of Arg-183A (on α4A), the backbone amine of His-278B forms a hydrogen bond interaction with the Thr-106C and Asp-108C side chains and the backbone carbonyl and side chain from Gln-279B forms hydrogen bonds with the side chain and backbone carbonyl of Arg-134A from α5A.
Ten carboxyl-terminal–disordered residues after α9B in the AfFabI structure become ordered to create a channel connecting the active site to the water well in the AfFabI·NADH structure (Fig. 9E). These interactions form “latches” along the R axis that create the surface enclosing a water channel. The first latch is formed by Glu-275B backbone amide, and Asp-276B side chain hydrogen bonds to Arg-183A and Tyr-158C side chains (Fig. 9E). Asp-276B is oriented to coordinate a water molecule in the channel. Arg-262B in the first turn of α9B forms a planar stacking interaction (53) between the guanidinium group in the side chain and the aromatic ring of Tyr-158C. A van der Waals contact distance is formed between lid residue Leu-206C side chain and Glu-275B backbone carbonyl. The second latch is formed by His-278B backbone amide hydrogen bonding to the Thr-106C and Asp-108C side chains, and Gln-279B backbone carbonyl and side chain hydrogen bonds to the Arg-134A backbone carbonyl and side chain (Fig. 9E). A van der Waals contact is formed between the Val-277C and Ile-180A side chains (not shown in Fig. 9). The molecular latches convert a region of the tetramer that freely exchanges with bulk solvent in AfFabI into an enclosed channel that connects the active site with the central water well in AfFabI·NADH. These data indicate that the conformational changes associated with α9 and the carboxyl terminus are important for catalysis because they participate in the formation of the catalytic center and the water channel that connects the active site to the water well.
Function of the carboxyl terminus
We constructed a series of truncation mutants to test the role of α9 and the disordered carboxyl terminus in catalysis. All the truncated mutant enzymes had similar turnover numbers (Vmax/[ET]) but had reduced catalytic efficiencies (Vmax/[ET]/K0.5) that were driven by defects in the apparent K0.5 for NADH (Table 2). AfFabI(1–273) lacked the disordered carboxyl-terminal residues in the AfFabI structure and eliminated the interactions contributing to sealing the water channels in the AfFabI·NADH structure illustrated in Fig. 9E. AfFabI(1–273) was a tetramer that was stabilized by NADH, but the catalytic efficiency of the enzyme was reduced by 3-fold driven by an increase in the apparent K0.5 for NADH (Table 2). Although AfFabI(1–273) was catalytically compromised, it was able to complement the growth of the fabI(Ts) mutant when overexpressed. AfFabI(1–269) removed four resides (1 turn) from α9 in the AfFabI structure. This region did not form any specific protein contacts in the AfFabI·NADH structure because it loops away from the protein and the side chains point toward solvent (Fig. 8B, arrows). Thus, the biochemical properties of AfFabI(1–269) were similar to the properties of AfFabI(1–273). The AfFabI(1–265) truncation removed the second turn of the α9 helix and the important interactions depicted in Fig. 9, B and D. These two interactions are key to closing one side of the lid that lays over the active site in the AfFabI·NADH structure and would be predicted to compromise NADH binding. AfFabI(1–265) was a thermally stable tetramer but was not stabilized by the addition of NADH (Table 2). The apparent K0.5 for NADH increased 6-fold and the catalytic efficiency dropped by 5.5-fold leading to a protein that could not complement the fabI(Ts) mutant even when overexpressed (Table 2). AfFabI(1–261) removed the remaining turn of the α9 helix and the multiple interactions depicted in Fig. 9C. These interactions connect protomers A and C with protomer B and form a junction with the NADH lid. AfFabI(1–261) reduces AfFabI to the clade 1 structure. Like clade 1 enzymes it is stable and cooperative but had a 23-fold increase in the NADH apparent K0.5 and was 22.5-fold less catalytically efficient (Table 2). These data show the importance of α9 and the carboxyl terminus in creating the substrate binding lid and the water channels required for efficient catalysis.
Table 2.
Properties of AfFabI carboxyl-terminal truncation mutants
| Property | AfFabIa(1–289) | AfFabI(1–273) | AfFabI(1–269) | AfFabI(1–265) | AfFabI(1–261) |
|---|---|---|---|---|---|
| Complementation | |||||
| fabI(Ts) | Yes | Yes | Yes | No | No |
| Biophysical properties | |||||
| s20 | 6.59 | 6.40 | 6.37 | 6.18 | 6.11 |
| Thermal stability | |||||
| Ligand-free (°C) | 47.9 ± 0.1 | 46.0 ± 0.2 | 45.0 ± 0.1 | 48.8 ± 0.2 | 52.1 ± 0.4 |
| + NADH (°C) | 54.8 ± 0.2 | 52.1 ± 0.2 | 50.5 ± 0.3 | 49.3 ± 0.2 | 51.4 ± 0.2 |
| ΔT (°C) | 6.9 ± 0.2 | 6.1 ± 0.3 | 5.5 ± 0.4 | 0.5 ± 0.4 | −0.7 ± 0.6 |
| Biochemical properties | |||||
| NADH K0.5 (μm) | 52.8 ± 3.0 | 160.3 ± 22.2 | 127.1 ± 10.9 | 291.9 ± 31.8 | 1225.0 ± 47.5 |
| hb | 2.1 ± 0.2 | 1.4 ± 0.2 | 1.3 ± 0.1 | 1.0 ± 0.1 | 2.6 ± 0.2 |
| Vmax/[ET]c (min−1) | 6.45 ± 0.19 | 6.57 ± 0.24 | 7.26 ± 0.19 | 6.46 ± 0.21 | 6.63 ± 0.18 |
| Vmax/[ET]/K0.5d (10−3 μmol−1 min−1) | 122.12 ± 3.64 | 40.97 ± 1.51 | 57.13 ± 1.52 | 22.14 ± 0.71 | 5.41 ± 0.15 |
aAfFabI(1–289) is the full-length, wildtype protein.
bHill number.
cTurnover number. [ET] = total enzyme concentration.
dA measure of catalytic efficiency.
Discussion
This work defines the function of the structural features that are unique to clade 4 FabIs. The four clades of bacterial FabIs diverged from their common ancestor in the distant past and each have a long and unique evolutionary history. All FabIs have common features like a Rossmann fold, cofactor-induced conformational change, Tyr-Xaa6-Lys catalytic dyad, and dimerization and tetramerization interfaces. These basic structural features are embodied in the prototypical clade 1 EcFabI structure depicted in Fig. 10, upper panel. Using the clade 1 enzymes as the prototypical comparator, each of the other three clades possesses a unique structural feature. Clade 2 FabIs are the Mycobacterium InhA enzymes that have a deeper active site crevice and longer α6 lid than the other FabI clades. These features are thought to enable InhA to bind very long–chain enoyl-ACP substrates for the synthesis of cell wall mycolic acids (28, 54). Clade 3 FabIs have an insertion between β3 and α3 that creates peripheral loops that are related by the Q axis interface. A functional or structural role is yet to be described for this feature in the plastid FabIs. Clade 4 FabIs have an extension of the carboxyl terminus that provides a new protomer–protomer interaction surface that links protomers along the R axis. This study shows that the carboxyl-terminal extension is critical for catalysis in clade 4 FabIs by participating in the conformational change that creates the NADH-binding pockets and water channels that feed the active site. This unique feature is present in all the Bacteroidetes FabIs and has evolved into a domain that is essential for catalysis in clade 4 enzymes.
Figure 10.

Unique structural features of the FabI clades. Representative models for the four FabI clades are compared using clade 1 FabI as the standard for comparison. Top panel, clade 1 EcFabI has the basic Rossmann fold and tetrameric organization common to all FabIs. The unique features in clades 2–4 are highlighted in blue. Clade 2 enzymes are represented by the Mycobacterial enoyl-ACP reductase (MtInhA). Clade 2 enzymes contain an extended active site lid to accommodate the long acyl-ACP substrates (28 carbons) that arise during the synthesis of mycolic acids. Clade 3 plastid enzymes are represented by the Chlamydial CtFabI and contain peripheral loops of unknown function. Clade 4 enzymes are presented by AfFabI and contain carboxyl-terminal α9–α9 coiled coils that are required for high-affinity NADH binding. Bottom panel, active site volumes for each clade are depicted by a surface mesh. The catalytic lysine and tyrosine residues are shown in orange. Ternary complex structures of clade 1 (PDB ID: 4JQC) (77) and 3 (PDB ID: 4Q9N) (29, 83) enzymes with NAD(H) and AFN-1252 (shown in yellow) have been determined and contain active site volumes that bind AFN-1252. Binary complex structure of clades 2 (PDB ID: 4DRE) (78, 82) and 4 (PDB ID: 6VLY) enzymes with NAD(H) have been determined, and AFN-1252 was modeled in their active site volumes with respect to the tyrosine. In both cases, the 3-methylbenzofuran moiety of AFN-1252 extends outside the active site and the oxotetrahydronaphthyridine moiety clashes with amino acid residues (shown in black).
Further, our data explain why a potent antimicrobial drug that targets the FabI of clade 1 and three pathogens does not impact a major constituent of the microbiome. AFN-1252 is a S. aureus–selective (Fig. 10, lower panel) FabI inhibitor that was developed as a pathogen-specific drug (16, 17). Treatment of mice with AFN-1252 does not perturb the Bacteroidetes phylum in the gut microbiome, whereas broad-spectrum antibiotics devastate the microbiome (24). Bioinformatic analysis of the microbiome enoyl-ACP reductase distribution indicates the presence of FabK in the Clostridia and commensal Bacteroidetes, and the expression of FabK would account for the resistance of organisms to AFN-1252. However, many Bacteroidetes genera only express a FabI (Fig. 1B). Biochemical analysis shows that clade 4 FabIs are resistant to AFN-1252, explaining why Bacteroidetes genera are not impacted by AFN-1252 therapy. AFN-1252 is a rigid small molecule that contains an oxotetrahydronaphthyridine and 3-methylbenzofuran moiety connected by a cis-amide. All three components of the drug form interactions with the protein in AFN-1252–sensitive FabIs in clades 1 and 3 (19, 29). The AfFabI·NADH crystal structure shows that clade 4 FabIs have a differently shaped active site cavity than clade 1 and 3 enzymes (Fig 10, lower panel). In AfFabI, the lid residue Val-205 side chain occupies the space where the 3-methylbenzofuran moiety should reside and the Arg-102 side chain sterically clashes with the oxotetrahydronaphthyridine moiety. Similarly, modeling AFN-1252 in the clade 2 active site reveals a steric clash with Ile-202. These considerations suggest that FabI-targeted drugs against clade 1 or 3 pathogens would have the benefit of having little to no impact on the gut microbiome, sparing patients the complications that arise from current therapies that destroy the microbiome (55, 56).
Experimental procedures
Materials
All chemicals and reagents were obtained from Sigma-Aldrich or Fisher unless otherwise indicated. Strains and plasmids used in this work are listed in Table 3.
Table 3.
Bacterial strains and plasmids
| Description | Reference | |
|---|---|---|
| Strain | ||
| E. coli JP1111 | Hfr(PO1), galE45(GalS), λ−, fabI392(Ts), relA1, spoT1, thiE1 | 79 |
| E. coli ANS1 | metB1 relA1 spoT1 gyrA216 tolC::Tn10 λ− λr F− | 63 |
| Plasmid | ||
| pPJ131 | E. coli expression vector | 80 |
| pEcFabI | E. coli fabI in pPJ131 | 29 |
| pCtFabI | Chlamydia trachomatis fabI in pPJ131 | 29 |
| pAfFabI | A. finegoldii fabI (UniProt ID: A0A174E195) in pPJ131 | This study |
| pCaFabK | Clostridium acetobutylicum fabK in pPJ131 | 37 |
| pAfFabK | A. finegoldii fabK (UniProt ID: I3YI65) in pPJ131 | This study |
| pET_AfFabI | A. finegoldii fabI 1–289 (UniProt ID: A0A174E195) in pET28b | This study |
| pET_1–273 | A. finegoldii fabI 1–273 (UniProt ID: A0A174E195) in pET28b | This study |
| pET_1–269 | A. finegoldii fabI 1–269 (UniProt ID: A0A174E195) in pET28b | This study |
| pET_1–265 | A. finegoldii fabI 1–265 (UniProt ID: A0A174E195) in pET28b | This study |
| pET_1–261 | A. finegoldii fabI 1–261 (UniProt ID: A0A174E195) in pET28b | This study |
Phylogeny
Predicted FabI sequences were collected from the NCBI Reference Sequence database to include characterized FabI sequences and their homologs. Maximum likelihood phylogenetic trees were constructed using the DECIPHER and phangorn packages in R (57, 58). Briefly, sequences were aligned using the AlignSeqs and StaggerAlignment functions in DECIPHER. The LG + Γ (4) + I model (59) was best fitting by the Bayesian information criteria from the model. Test function in phangorn evaluating the WAG, JTT, LG, and Dayhoff amino acid replacement matrix models with and without gamma distributed rate variation among sites (Γ) and invariant sites (I). The initial neighbor-joining tree was constructed using the distance matrix, with the maximum likelihood tree generated from the neighbor-joining tree using the LG + Γ (4) + I model with stochastic branch rearrangement. Bootstrap method of 1000 replicate trees was used to determine the confidence of the tree topology. The maximum likelihood tree was visualized using ggtree (60).
Molecular biology
The predicted A. finegoldii FabI and FabK have been previously identified (61). Representative predicted A. finegoldii genes AffabI (UniProt ID: A0A174E195) and AffabK (UniProt ID: I3YI65) were optimized for gene expression in E. coli using GeneArt Gene Synthesis Technology (Life Technologies). An NdeI restriction site was engineered at the 5′-end of the gene with the start codon in the NdeI site, whereas a stop codon and an EcoRI restriction site were sequentially engineered at the 3′-end of the gene. The genes were cloned into the pPJ131 plasmid (a modified version of the pBluescript plasmid with the multiple cloning site from pET28a) via the NdeI and EcoRI (New England Biolabs) restriction sites (29, 80). To create the AffabI carboxyl-terminal truncation mutant constructs, premature stop codons were engineered into the gene using the QuikChange II Site-Directed Mutagenesis Kit (Agilent) and primers 5′-CAAGCATGGGTATGTCATGATGACGTCGTGCAATGAAAAC, 5′-TGTCACGTCGTGCAATGTGATGAAAAACCTATGAAAAAGG, 5′-CAATGAAAACCTATGAATGATGAAAAGGTATGCGCTTTGA, and 5′-ATGAAAAAGGTATGCGCTGATGATTTGAAGATGTGCACCA and their reverse complements (The bold letters emphasize the stop codons that were engineered into the gene sequence). The amino acid numbering convention used assumes the initiating methionine of the native protein as residue 1. For protein purification, the AffabI constructs were cloned into the pET28b plasmid via the NcoI and EcoRI (New England Biolabs) restriction sites.
Complementation assay
The AfFabK amino acid sequence contains an FMN-binding motif (5); the AfFabI does not. The trans-2-enoyl–ACP reductase function of AfFabI and AfFabK was confirmed using the previously established complementation method (3, 29). Expression plasmids were constructed to overexpress nothing (negative control), E. coli FabI (EcFabI), C. trachomatis FabI (CtFabI), AfFabI and its truncation mutants, C. acetobutylicum FabK (CaFabK) (37), or A. finegoldii FabK (AfFabK) and then transformed into E. coli strain JP1111 (fabI(Ts)) (79) and plated at 30 °C on LB plates containing 100 μg/ml carbenicillin. Individual colonies were streaked on plates containing 100 μg/ml carbenicillin and incubated at 30 °C and 42 °C. Carbenicillin stress slows growth at 42 °C. Strain JP1111 is nonviable at 42 °C unless complemented with a functional trans-2-enoyl–ACP reductase, such as EcFabI or CaFabK that serve as positive controls.
Minimum inhibitory concentration analysis
The minimum inhibitory concentrations for AFN-1252 and triclosan against E. coli strain ANS1 (ΔtolC) were determined using a broth microdilution method as described previously (62). ANS1 is a tolC knockout mutant that is used to eliminate the contribution of type 1 secretion systems to drug resistance (63). Briefly, pPJ131 expression plasmids containing nothing, EcFabI, CtFabI, AfFabI, CaFabK, or AfFabK were transformed into ANS1, and these strains were grown to A600 = 0.6 in LB before being backdiluted 1:30,000 in 1% DMSO in 1% tryptone media. Diluted cells (100 μl) were added to each well of a U-bottom 96-well plate except the first column of wells. Diluted cells (200 μl) were mixed with 50 μm AFN-1252 or 50 μm triclosan was added to the first column of wells, and 100 μl was serially diluted through the plate excluding the last column of wells, leaving 100 μl of cells in each well with the appropriate concentration of compound. The plate was incubated at 37 °C for 24 h and then read at 600 nm using a SPECTRAmax 340PC Microplate Reader. Cells from each strain grown with 0 μm compound were used as a reference (i.e. 100% fractional growth).
AfFabI protein expression and purification
BL21 (DE3) cells harboring the pET-AfFabI plasmid were grown in LB medium with 100 μg/ml carbenicillin at 37 °C and 200 rpm shaking to A600 = 0.6. The culture was cooled to 16 °C then induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside overnight. Cells were centrifuged and pellets were resuspended in 20 mm Tris-HCl, pH 8.0, 200 mm NaCl, 10 mm imidazole (30 ml/liter culture). Cells were lysed via a cell disruptor and the amino-terminal His6-tagged AfFabI was purified by standard nickel chelation chromatography (29). The protein was then gel filtered into 20 mm Tris-HCl, pH 8.0, 200 mm NaCl, 10 mm EDTA using a preparative Superdex 200 column with dimensions 16 mm × 60 cm. The AfFabI molecular weight was estimated using an analytical XBridge BEH SEC 200 Å 3.5-μm column with dimensions 7.8 mm × 150 mm. Approximately 26 mg of purified AfFabI was obtained per liter of culture. The same methods were used to overexpress and purify AfFabI mutant proteins.
Analytical ultracentrifugation
Sedimentation velocity experiments were conducted in a ProteomeLab XL-I analytical ultracentrifuge (Beckman Coulter) following standard protocols unless mentioned otherwise (64, 65). Samples in buffer containing 20 mm Tris-HCl, pH 8, 200 mm NaCl, 10 mm EDTA were loaded into cell assemblies comprised of double sector charcoal-filled centerpieces with a 12 mm path length and sapphire windows. The cell assemblies, containing identical sample and reference buffer volumes of 390 μl, were placed in a rotor and the samples and cells were incubated at 20 °C for 2 h before being accelerated from 0 to 50,000 rpm. Rayleigh interference optical data as well as absorbance data at 280 nm were collected at 1-min intervals for 12 h. The velocity data were modeled with diffusion-deconvoluted sedimentation coefficient distributions c(s) in SEDFIT (RRID: SCR_018365) using algebraic noise decomposition and with signal-average frictional ratio and meniscus position refined with nonlinear regression (66). The s-value was corrected for time and finite acceleration of the rotor was accounted for in the evaluation of Lamm equation solutions (67). Maximum entropy regularization was applied at a confidence level of p = 0.68. The partial specific volumes of the proteins, based on its amino acid composition, was calculated in SEDFIT.
Enzymology
The AfFabI enzymatic activity was determined by measuring the conversion of NADH to NAD+ at 340 nm. The enzyme reactions were 100 μl in volume and monitored in Costar UV half-area 96-well plates with a SPECTRAmax 340PC instrument taking 340-nm readings at 10-s intervals at 30 °C. In these experiments, enzyme concentrations are reported relative to monomer concentration. The velocity of the AfFabI enzyme (100 nm) was measured by adding 1.25 mm crotonyl-CoA and 100 μm NADH in 20 mm Tris-HCl, pH 8.0. The apparent K0.5 of crotonyl-CoA was determined by adding 100 nm AfFabI to 200 μm NADH and 0, 0.05, 0.1, 0.25, 0.5, 1, 1.5, 2, or 2.5 mm crotonyl-CoA. The apparent K0.5 of NADH was determined by adding 100 nm AfFabI to 1.25 mm crotonyl-CoA and 0, 5, 10, 15, 20, 30, 40, 50, or 70 μm NADH. The reactions were mixed for 10 s by the mix function on the plate reader, and data were acquired at 10-s intervals for 30 min. Initial velocity was calculated from the linear phase of the progress curve and fit using the Hill equation to determine the apparent K0.5 (68). The IC50 of AFN-1252 and triclosan were measured above saturating substrate concentrations (100 nm AfFabI, 1.25 mm crotonyl-CoA, and 200 μm NADH in 20 mm Tris-HCl, pH 8.0) against 0, 0.15, 0.31, 0.62, 1.25, 2.5, 5, and 10 μm compound. Initial velocities comparing AfFabI full-length and truncated proteins were determined using a Waters e2695 Separations Module HPLC system. Protein (100 nm) was added to 1.25 mm crotonyl-CoA and 0, 5, 10, 15, 20, 30, 40, 50, 60, 125, 250, 500, 1000, 2000, 4000 μm NADH in 20 mm Tris-HCl, pH 8.0. The enzyme reactions were 40 μl in volume and incubated for 10 min at 30 °C. Reactions were stopped by adding equal volume methanol and applied to a Gemini C18, 3 μm 100 Å, 4.6 mm × 150 mm column at a 0.5 ml/min flow rate. Butyryl-CoA product was separated from crotonyl-CoA substrate by gradient elution and monitored at A260. The 30-min gradient was from 90% 50 mm KH2PO4, pH 4.6, acetonitrile (90/10, v/v) to 40% 50 mm KH2PO4, pH 4.6, acetonitrile (40/60, v/v). All kinetic experiments were run in triplicate.
Surface plasmon resonance experiments
Surface plasmon resonance experiments were conducted at 20 °C using a ForteBio Pioneer optical biosensor (ForteBio). Poly–His-tagged AfFabI constructs were immobilized on polycarboxylate hydrogel-coated gold chips preimmobilized with nitrilotriacetic acid (HisCap chips, ForteBio). The chip was primed in chelating buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 50 μm EDTA, 0.005% Tween 20) and was preconditioned at 10 μl/min with three 60-s injections of wash buffer (10 mm HEPES, pH 8.3, 150 mm NaCl, 350 mm EDTA, 0.05% Tween 20) and one 60-s injection of chelating buffer before being charged with a 60-s injection of 500 μm NiCl2 in chelating buffer. After priming into binding buffer (20 mm Tris-HCl, pH 8.0, 200 mm NaCl, 0.01% Brij-35, 5% DMSO), FabIs were injected until ∼1500–1700 resonance units of protein were captured. One flow cell on the chip was charged with Ni2+ without adding protein to be used as a reference cell.
NADH was prepared in binding buffer as a 2-fold dilution series with maximum concentration of 4 μm for binding to AfFabI and was injected in triplicate for each concentration at a flow rate of 30 μl/min. A series of buffer-only (blank) injections was included throughout the experiment to account for instrumental noise. NADH fully dissociated from the protein surfaces, eliminating the need for a regeneration step. The data were processed, double-referenced, microcalibrated, and analyzed using the software package Qdat (version 4.3.1.2, ForteBio). Equilibrium binding levels were determined and exported to GraphPad Prism for fitting to the Hill equation.
Protein thermal shift assays
Protein thermal shift analysis was conducted to investigate if NADH enhanced the thermal stability of the AfFabI constructs. Solutions (30 μl) of AfFabI (10 μm) and AfFabI (10 μm) + NADH (100 μm) in 20 μm HEPES, pH 7.5, 150 mm NaCl, 20 mm MgCl2, and 2.5× Sypro Orange Dye were added to wells of ThermoGrid optically clear PCR plates (Denville Scientific). The plates were centrifuged at 1000 × g for 5 min and then analyzed by the ABI 7300 real-time PCR system as described previously (29). The temperature was ramped from 25 °C to 95 °C at 1 °C/min with the fluorescence read six times at each temperature ramp. The resulting data were fit to a Boltzmann sigmoidal equation to determine the melting point of each AfFabI construct with and without substrate. Each enzyme or enzyme and substrate condition was replicated six times, and the thermal melting point of each replicate was determined independently. The melting points from each replicate were averaged to determine the reported thermal melting point. A representative thermal shift experiment is shown in Figure 3D and average thermal melting points from triplicate experiments are shown in Table 2.
Mass spectrometry analysis of the AfFabI enzyme reaction
Samples were diluted with an equal volume of 80% acetonitrile + 15 mm ammonium hydroxide and were analyzed by direct injection to a QTrap 4500 equipped with a Turbo V ion source (Sciex). The QTrap 4500 was operated in the positive mode using neutral loss scanning, and the ion source parameters were ion spray voltage, 5000 V; curtain gas, 15 psi; temperature, 275 °C; collision gas, medium; ion source gas 1, 15 psi; ion source gas 2, 20 psi; neutral loss, 507.0 m/z; declustering potential, 50 V; and collision energy, 40 V. The system was controlled and analyzed by the Analyst® software (Sciex).
Crystallization and structure determination
The AfFabI protein was concentrated to 12 mg/ml for crystallization. Initial screening was performed at 20 °C against the Protein Complex Suite (Qiagen) by hanging drop vapor diffusion method combining 300 nl protein and 200 nl precipitant. Diffraction quality crystals were obtained by combining 1.5 μl protein and 1 μl 1.2 m sodium potassium tartrate and 100 mm Tris-HCl, pH 8.0. Crystals were cryo-protected with 1.3 m sodium potassium tartrate, 100 mm Tris-HCl, pH 8.0, and 20% v/v glycerol, and then flash-frozen in liquid nitrogen for X-ray diffraction experiments. The diffraction datasets were collected at the SER-CAT beam line 22-ID at the Advanced Photon Source and processed using HKL2000 (69). The AfFabI structure was solved by the molecular replacement method using the program Phaser (70) and the coordinates of FabI from Aquifex aeolicus (PDB ID: 2P91) (71) as the search model. The AfFabI·NADH complex was achieved by incubating 5 mg/ml (150 μm) AfFabI with 500 μm NADH for 2 h at room temperature prior to crystallization. Crystals were grown by hanging drop vapor diffusion by combining 1.5 μl of the protein·substrate mixture and 1.5 μl 12.5% PEG 1000, 200 mm NaCl, and 100 mm MES, pH 6.0. Crystals were cryo-protected with 12.5% PEG 1000, 200 mm NaCl, 100 mm MES, pH 6.0, and 25% glycerol, and then flash-frozen in liquid nitrogen for X-ray analysis. Diffraction datasets were collected at the NSLS-II NYX beamline (19-ID) and AMX beamline (17-ID-1) at the Brookhaven National Laboratory and processed using XDS (72). The structure of the AfFabI·NADH complex was solved by molecular replacement using the AfFabI structure lacking α9 as a search model. The structures were completed by iterative rounds of refinement using REFMAC (73) and manual rebuilding using Coot (74). The refinement was monitored by following the Rfree value calculated from a random subset (5%) of omitted reflections. A tight turn resolved in the electron density map led to 0.2% of the atoms to be in the Ramachandran outliers (Asn-161A in the AfFabI structure, and Asn-161A, Asn-161C in the AfFabI·NADH complex structure). A summary of the data processing and structure refinement statistics is provided in Table 1. The coordinates have been deposited in the Protein Data Bank (accession code 6VLX AfFabI; 6VLY AfFabI·NADH complex). The figures related to the protein structure were generated with PyMOL (75).
In silico analyses
Cα RMSD calculations were performed using SSM Superpose in Coot (74). Surface areas were calculated in PyMOL as described previously (76). The buried surface area by dimerization was calculated by subtracting the surface area of two protomers linked along the P axis from the combined surface area of the two individual protomers. The buried surface by tetramerization was calculated by subtracting the surface area of two protomers linked along the Q axis from the combined surface area of the two individual protomers. The buried surface area from the helix–helix interaction domain was calculated by subtracting the surface area of two protomers linked along the R axis from the combined surface area of the two individual protomers. The water channels and water well were visualized using the PyMOL surface view in Cavities and Pockets (Culled) mode. AFN-1252 binding visualization across FabI clades was done by structural alignment of the catalytic dyads from clade I EcFabI·NAD+·AFN-1252 (PDB ID: 4JQC) (77), clade II MtInhA·NAD+ (PDB ID: 4DRE) (78, 82), clade III CtFabI·NADH·AFN-1252 (PDB ID: 4Q9N) (29, 83), and clade IV AfFabI·NAD+ (PDB ID: 6VLY).
Statistical analysis
Statistical analysis (i.e. standard error, K0.5) and mathematical modeling (i.e. Hill equation, variable slope nonlinear regression) were performed using GraphPad Prism software version 8.2.1.
Data availability
Coordinates and structure factors for AfFabI and AfFabI·NADH crystal structures have been deposited in the Protein Data Bank (PDB) under accession codes 6VLX and 6VLY, respectively. All remaining data are contained within the article.
Author contributions
C. D. R., J. Y., and C. O. R. conceptualization; C. D. R., M. W. F., J. Y., J. S., and D. J. M. data curation; C. D. R., M. W. F., J. Y., J. S., and D. J. M. formal analysis; C. D. R. and C. O. R. investigation; C. D. R., M. W. F., J. S., and D. J. M. methodology; C. D. R. writing-original draft; C. D. R., M. W. F., J. Y., J. S., D. J. M., and C. O. R. writing-review and editing; D. J. M. resources; D. J. M. software; C. O. R. supervision; C. O. R. funding acquisition; C. O. R. project administration.
Acknowledgments
We thank Dr. Amanda Nourse (Molecular Interaction Analysis Shared Resource, St. Jude Children's Research Hospital) for sample analysis by analytical ultracentrifugation and Brett Waddell (Molecular Interaction Analysis Shared Resource, St. Jude Children's Research Hospital) for NADH-binding experiments by surface plasmon resonance. We thank the St. Jude Structural Biology X-ray Center for crystallography support. The Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory is supported by its member institutions, and equipment grants (S10_RR25528 and S10_RR028976) from the National Institutes of Health. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31–109-Eng-38. The NYX (19-ID) and AMX (17-ID-1) beamlines of the National Synchrotron Light Source II is a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract DE-SC0012704. NYX is supported by the New York Structural Biology Center (NYSBC). AMX is part of the Center for BioMolecular Structure that is supported by the National Institute of General Medical Sciences (P30 GM133893), and by the DOE Office of Biological and Environmental Research (KP 1605010).
This work was supported by NIGMS, National Institutes of Health Grant GM034496 (to C. O. R.), Cancer Center Support Grant CA21765, and the American Lebanese Syrian Associated Charities. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ACP
- acyl carrier protein
- ENR
- enoyl-ACP reductase
- RMSD
- root mean square deviation
- FabI
- enoyl-(acyl-carrier-protein) reductase
- FabK
- enoyl-ACP reductase II
- FASII
- bacterial type II fatty acid synthesis
- InhA
- M. tuberculosis enoyl-(acyl-carrier-protein) reductase.
References
- 1. Parsons J. B., and Rock C. O. (2013) Bacterial lipids: Metabolism and membrane homeostasis. Prog. Lipid Res. 52, 249–276 10.1016/j.plipres.2013.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Heath R. J., and Rock C. O. (2000) A triclosan-resistant bacterial enzyme. Nature 406, 145–146 10.1038/35018162 [DOI] [PubMed] [Google Scholar]
- 3. Marrakchi H., DeWolf W. E. Jr., Quinn C., West J., Polizzi B. J., So C. Y., Holmes D. J., Reed S. L., Heath R. J., Payne D. J., Rock C. O., and Wallis N. G. (2003) Characterization of Streptococcus pneumoniae enoyl-[acyl carrier protein] reductase (FabK). Biochem. J. 370, 1055–1062 10.1042/bj20021699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Saito J., Yamada M., Watanabe T., Iida M., Kitagawa H., Takahata S., Ozawa T., Takeuchi Y., and Ohsawa F. (2008) Crystal structure of enoyl-acyl carrier protein reductase (FabK) from Streptococcus pneumoniae reveals the binding mode of an inhibitor. Protein Sci. 17, 691–699 10.1110/ps.073288808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hevener K. E., Santarsiero B. D., Lee H., Jones J. A., Boci T., Johnson M. E., and Mehboob S. (2018) Structural characterization of Porphyromonas gingivalis enoyl-ACP reductase II (FabK). Acta Crystallogr. F Struct. Biol. Commun. 74, 105–112 10.1107/S2053230X18000262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oppermann U., Filling C., Hult M., Shafqat N., Wu X., Lindh M., Shafqat J., Nordling E., Kallberg Y., Persson B., and Jornvall H. (2003) Short-chain dehydrogenases/reductases (SDR): The 2002 update. Chem. Biol. Interact. 143–144, 247–253 10.1016/s0009-2797(02)00164-3 [DOI] [PubMed] [Google Scholar]
- 7. White S. W., Zheng J., Zhang Y.-M., and Rock C. O. (2005) The structural biology of type II fatty acid biosynthesis. Annu. Rev. Biochem. 74, 791–831 10.1146/annurev.biochem.74.082803.133524 [DOI] [PubMed] [Google Scholar]
- 8. Baker M. E. (1995) Enoyl-acyl-carrier-protein reductase and Mycobacterium tuberculosis InhA do not conserve the Tyr-Xaa-Xaa-Xaa-Lys motif in mammalian 11β- and 17β-hydroxysteroid dehydrogenases and Drosophila alcohol dehydrogenase. Biochem. J. 309, 1029–1030 10.1042/bj3091029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Parikh S., Moynihan D. P., Xiao G., and Tonge P. J. (1999) Roles of tyrosine 158 and lysine 165 in the catalytic mechanism of InhA, the enoyl-ACP reductase from Mycobacterium tuberculosis. Biochemistry 38, 13623–13634 10.1021/bi990529c [DOI] [PubMed] [Google Scholar]
- 10. Heath R. J., Su N., Murphy C. K., and Rock C. O. (2000) The enoyl-[acyl-carrier-protein] reductases FabI and FabL from Bacillus subtilis. J. Biol. Chem. 275, 40128–40133 10.1074/jbc.M005611200 [DOI] [PubMed] [Google Scholar]
- 11. Kim K. H., Ha B. H., Kim S. J., Hong S. K., Hwang K. Y., and Kim E. E. (2011) Crystal structures of enoyl-ACP reductases I (FabI) and III (FabL) from B. subtilis. J. Mol. Biol. 406, 403–415 10.1016/j.jmb.2010.12.003 [DOI] [PubMed] [Google Scholar]
- 12. Massengo-Tiassé R. P., and Cronan J. E. (2008) Vibrio cholerae fabV defines a new class of enoyl acyl-carrier-protein reductase. J. Biol. Chem. 283, 1308–1316 10.1074/jbc.M708171200 [DOI] [PubMed] [Google Scholar]
- 13. Zhu L., Lin J., Ma J., Cronan J. E., and Wang H. (2010) Triclosan resistance of Pseudomonas aeruginosa PAO1 is due to FabV, a triclosan-resistant enoyl-acyl carrier protein reductase. Antimicrob. Agents Chemother. 54, 689–698 10.1128/AAC.01152-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hirschbeck M. W., Kuper J., Lu H., Liu N., Neckles C., Shah S., Wagner S., Sotriffer C. A., Tonge P. J., and Kisker C. (2012) Structure of the Yersinia pestis FabV enoyl-ACP reductase and its interaction with two 2-pyridone inhibitors. Structure 20, 89–100 10.1016/j.str.2011.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heath R. J., and Rock C. O. (1995) Enoyl-acyl carrier protein reductase (fabI) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli. J. Biol. Chem. 270, 26538–26542 10.1074/jbc.270.44.26538 [DOI] [PubMed] [Google Scholar]
- 16. Yao J., and Rock C. O. (2016) Resistance mechanisms and the future of bacterial enoyl-acyl carrier protein reductase (FabI) antibiotics. Cold Spring Harbor Perspect. Med. 6, a027045 10.1101/cshperspect.a027045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yao J., and Rock C. O. (2017) Bacterial fatty acid metabolism in modern antibiotic discovery. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 1300–1309 10.1016/j.bbalip.2016.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kaplan N., Awrey D., Bardouniotis E., Berman J., Yethon J., Pauls H. W., and Hafkin B. (2013) In vitro activity (MICs and rate of kill) of AFN-1252, a novel FabI inhibitor, in the presence of serum and in combination with other antibiotics. J. Chemother. 25, 18–25 10.1179/1973947812Y.0000000063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kaplan N., Albert M., Awrey D., Bardouniotis E., Berman J., Clarke T., Dorsey M., Hafkin B., Ramnauth J., Romanov V., Schmid M. B., Thalakada R., Yethon J., and Pauls H. W. (2012) Mode of action, in vitro activity, and in vivo efficacy of AFN-1252, a selective antistaphylococcal FabI inhibitor. Antimicrob. Agents Chemother. 56, 5865–5874 10.1128/AAC.01411-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hafkin B., Kaplan N., and Murphy B. (2015) Efficacy and safety of AFN-1252, the first Staphylococcus-specific antibacterial agent, in the treatment of acute bacterial skin and skin structure infections, including those in patients with significant comorbidities. Antimicrob. Agents Chemother. 60, 1695–1701 10.1128/AAC.01741-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Menetrey A., Janin A., Pullman J., Overcash J. S., Haouala A., Leylavergne F., Turbe L., Wittke F., and Nicolas-Métral V. (2019) Bone and joint tissue penetration of the Staphylococcus-selective antibiotic afabicin in patients undergoing elective hip replacement surgery. Antimicrob. Agents Chemother. 63, e01669–18 10.1128/AAC.01669-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Keeney K. M., Yurist-Doutsch S., Arrieta M. C., and Finlay B. B. (2014) Effects of antibiotics on human microbiota and subsequent disease. Annu. Rev. Microbiol. 68, 217–235 10.1146/annurev-micro-091313-103456 [DOI] [PubMed] [Google Scholar]
- 23. Croswell A., Amir E., Teggatz P., Barman M., and Salzman N. H. (2009) Prolonged impact of antibiotics on intestinal microbial ecology and susceptibility to enteric Salmonella infection. Infect. Immun. 77, 2741–2753 10.1128/IAI.00006-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yao J., Carter R. A., Vuagniaux G., Barbier M., Rosch J. W., and Rock C. O. (2016) A pathogen-selective antibiotic minimizes disturbance to the microbiome. Antimicrob. Agents Chemother. 60, 4264–4273 10.1128/AAC.00535-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rinninella E., Raoul P., Cintoni M., Franceschi F., Miggiano G. A. D., Gasbarrini A., and Mele M. C. (2019) What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms 7, E14 10.3390/microorganisms7010014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ward W. H., Holdgate G. A., Rowsell S., McLean E. G., Pauptit R. A., Clayton E., Nichols W. W., Colls J. G., Minshull C. A., Jude D. A., Mistry A., Timms D., Camble R., Hales N. J., Britton C. J., and Taylor I. W. (1999) Kinetic and structural characteristics of the inhibition of enoyl (acyl carrier protein) reductase by triclosan. Biochemistry 38, 12514–12525 10.1021/bi9907779 [DOI] [PubMed] [Google Scholar]
- 27. Schiebel J., Chang A., Lu H., Baxter M. V., Tonge P. J., and Kisker C. (2012) Staphylococcus aureus FabI: Inhibition, substrate recognition, and potential implications for in vivo essentiality. Structure 20, 802–813 10.1016/j.str.2012.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rozwarski D. A., Vilchèze C., Sugantino M., Bittman R., and Sacchettini J. C. (1999) Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate. J. Biol. Chem. 274, 15582–15589 10.1074/jbc.274.22.15582 [DOI] [PubMed] [Google Scholar]
- 29. Yao J., Abdelrahman Y. M., Robertson R. M., Cox J. V., Belland R. J., White S. W., and Rock C. O. (2014) Type II fatty acid synthesis is essential for the replication of Chlamydia trachomatis. J. Biol. Chem. 289, 22365–22376 10.1074/jbc.M114.584185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Belluti F., Perozzo R., Lauciello L., Colizzi F., Kostrewa D., Bisi A., Gobbi S., Rampa A., Bolognesi M. L., Recanatini M., Brun R., Scapozza L., and Cavalli A. (2013) Design, synthesis, and biological and crystallographic evaluation of novel inhibitors of Plasmodium falciparum enoyl-ACP-reductase (PfFabI). J. Med. Chem. 56, 7516–7526 10.1021/jm400637m [DOI] [PubMed] [Google Scholar]
- 31. Johnson E. L., Heaver S. L., Walters W. A., and Ley R. E. (2017) Microbiome and metabolic disease: Revisiting the bacterial phylum Bacteroidetes. J. Mol. Med. (Berl.) 95, 1–8 10.1007/s00109-016-1492-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Karlowsky J. A., Kaplan N., Hafkin B., Hoban D. J., and Zhanel G. G. (2009) AFN-1252, a FabI inhibitor, demonstrates a Staphylococcus-specific spectrum of activity. Antimicrob. Agents Chemother. 53, 3544–3548 10.1128/AAC.00400-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yao J., Maxwell J. B., and Rock C. O. (2013) Resistance to AFN-1252 arises from missense mutations in Staphylococcus aureus enoyl-acyl carrier protein reductase (FabI). J. Biol. Chem. 288, 36261–36271 10.1074/jbc.M113.512905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heath R. J., Li J., Roland G. E., and Rock C. O. (2000) Inhibition of the Staphylococcus aureus NADPH-dependent enoyl-acyl carrier protein reductase by triclosan and hexachlorophene. J. Biol. Chem. 275, 4654–4659 10.1074/jbc.275.7.4654 [DOI] [PubMed] [Google Scholar]
- 35. Heath R. J., Rubin J. R., Holland D. R., Zhang E., Snow M. E., and Rock C. O. (1999) Mechanism of triclosan inhibition of bacterial fatty acid synthesis. J. Biol. Chem. 274, 11110–11114 10.1074/jbc.274.16.11110 [DOI] [PubMed] [Google Scholar]
- 36. Jordan C. A., Sandoval B. A., Serobyan M. V., Gilling D. H., Groziak M. P., Xu H. H., and Vey J. L. (2015) Crystallographic insights into the structure-activity relationships of diazaborine enoyl-ACP reductase inhibitors. Acta Crystallogr. F Struct. Biol. Cryst. Commun. 71, 1521–1530 10.1107/S2053230X15022098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marrakchi H., Choi K.-H., and Rock C. O. (2002) A new mechanism for anaerobic unsaturated fatty acid formation in Streptococcus pneumoniae. J. Biol. Chem. 277, 44809–44816 10.1074/jbc.M208920200 [DOI] [PubMed] [Google Scholar]
- 38. Kim H. T., Kim S., Na B. K., Chung J., Hwang E., and Hwang K. Y. (2017) Structural insights into the dimer-tetramer transition of FabI from Bacillus anthracis. Biochem. Biophys. Res. Commun. 493, 28–33 10.1016/j.bbrc.2017.09.084 [DOI] [PubMed] [Google Scholar]
- 39. Kim S. J., Ha B. H., Kim K. H., Hong S. K., Shin K. J., Suh S. W., and Kim E. E. (2010) Dimeric and tetrameric forms of enoyl-acyl carrier protein reductase from Bacillus cereus. Biochem. Biophys. Res. Commun. 400, 517–522 10.1016/j.bbrc.2010.08.083 [DOI] [PubMed] [Google Scholar]
- 40. Priyadarshi A., Kim E. E., and Hwang K. Y. (2010) Structural insights into Staphylococcus aureus enoyl-ACP reductase (FabI), in complex with NADP and triclosan. Proteins 78, 480–486 10.1002/prot.22581 [DOI] [PubMed] [Google Scholar]
- 41. Rafferty J. B., Simon J. W., Baldock C., Artymiuk P. J., Baker P. J., Stuitje A. R., Slabas A. R., and Rice D. W. (1995) Common themes in redox chemistry emerge from the X-ray structure of oilseed rape (Brassica napus) enoyl acyl carrier protein reductase. Structure 3, 927–938 10.1016/S0969-2126(01)00227-1 [DOI] [PubMed] [Google Scholar]
- 42. Perozzo R., Kuo M., Sidhu A. S., Valiyaveettil J. T., Bittman R., Jacobs W. R. Jr., Fidock D. A., and Sacchettini J. C. (2002) Structural elucidation of the specificity of the antibacterial agent triclosan for malarial enoyl acyl carrier protein reductase. J. Biol. Chem. 277, 13106–13114 10.1074/jbc.M112000200 [DOI] [PubMed] [Google Scholar]
- 43. Luckner S. R., Liu N., am Ende C. W., Tonge P. J., and Kisker C. (2010) A slow, tight binding inhibitor of InhA, the enoyl-acyl carrier protein reductase from Mycobacterium tuberculosis. J. Biol. Chem. 285, 14330–14337 10.1074/jbc.M109.090373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pidugu L. S., Kapoor M., Surolia N., Surolia A., and Suguna K. (2004) Structural basis for the variation in triclosan affinity to enoyl reductases. J. Mol. Biol. 343, 147–155 10.1016/j.jmb.2004.08.033 [DOI] [PubMed] [Google Scholar]
- 45. Ghosh D., Wawrzak Z., Weeks C. M., Duax W. L., and Erman M. (1994) The refined three-dimensional structure of 3α,20β-hydroxysteroid dehydrogenase and possible roles of the residues conserved in short-chain dehydrogenases. Structure 2, 629–640 10.1016/S0969-2126(00)00064-2 [DOI] [PubMed] [Google Scholar]
- 46. Tanaka N., Nonaka T., Tanabe T., Yoshimoto T., Tsuru D., and Mitsui Y. (1996) Crystal structures of the binary and ternary complexes of 7α-hydroxysteroid dehydrogenase from Escherichia coli. Biochemistry 35, 7715–7730 10.1021/bi951904d [DOI] [PubMed] [Google Scholar]
- 47. Hol W. G., van Duijnen P. T., and Berendsen H. J. (1978) The α-helix dipole and the properties of proteins. Nature 273, 443–446 10.1038/273443a0 [DOI] [PubMed] [Google Scholar]
- 48. Muench S. P., Prigge S. T., McLeod R., Rafferty J. B., Kirisits M. J., Roberts C. W., Mui E. J., and Rice D. W. (2007) Studies of Toxoplasma gondii and Plasmodium falciparum enoyl acyl carrier protein reductase and implications for the development of antiparasitic agents. Acta Crystallogr. D Biol. Crystallogr. 63, 328–338 10.1107/S0907444906053625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Qiu X., Janson C. A., Court R. I., Smyth M. G., Payne D. J., and Abdel-Meguid S. S. (1999) Molecular basis for triclosan activity involves a flipping loop in the active site. Protein Sci. 8, 2529–2532 10.1110/ps.8.11.2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dessen A., Quémard A., Blanchard J. S., Jacobs W. R. Jr., and Sacchettini J. C. (1995) Crystal structure and function of the isoniazid target of Mycobacterium tuberculosis. Science 267, 1638–1641 10.1126/science.7886450 [DOI] [PubMed] [Google Scholar]
- 51. Mehboob S., Hevener K. E., Truong K., Boci T., Santarsiero B. D., and Johnson M. E. (2012) Structural and enzymatic analyses reveal the binding mode of a novel series of Francisella tularensis enoyl reductase (FabI) inhibitors. J. Med. Chem. 55, 5933–5941 10.1021/jm300489v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schiebel J., Chang A., Merget B., Bommineni G. R., Yu W., Spagnuolo L. A., Baxter M. V., Tareilus M., Tonge P. J., Kisker C., and Sotriffer C. A. (2015) An ordered water channel in Staphylococcus aureus FabI: Unraveling the mechanism of substrate recognition and reduction. Biochemistry 54, 1943–1955 10.1021/bi5014358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Flocco M. M., and Mowbray S. L. (1994) Planar stacking interactions of arginine and aromatic side-chains in proteins. J. Mol. Biol. 235, 709–717 10.1006/jmbi.1994.1022 [DOI] [PubMed] [Google Scholar]
- 54. Rozwarski D. A., Grant G. A., Barton D. H., Jacobs W. R. Jr., and Sacchettini J. C. (1998) Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science 279, 98–102 10.1126/science.279.5347.98 [DOI] [PubMed] [Google Scholar]
- 55. Langdon A., Crook N., and Dantas G. (2016) The effects of antibiotics on the microbiome throughout development and alternative approaches for therapeutic modulation. Genome Med. 8, 39 10.1186/s13073-016-0294-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Francino M. P. (2015) Antibiotics and the human gut microbiome: Dysbioses and accumulation of resistances. Front. Microbiol. 6, 1543 10.3389/fmicb.2015.01543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schliep K. P. (2011) Phangorn: Phylogenetic analysis in R. Bioinformatics 27, 592–593 10.1093/bioinformatics/btq706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wright E. S. (2015) DECIPHER: Harnessing local sequence context to improve protein multiple sequence alignment. BMC Bioinformatics 16, 322 10.1186/s12859-015-0749-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Le S. Q., and Gascuel O. (2008) An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320 10.1093/molbev/msn067 [DOI] [PubMed] [Google Scholar]
- 60. Yu G. C., Smith D. K., Zhu H. C., Guan Y., and Lam T. T. Y. (2017) GGTREE: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36 10.1111/2041-210X.12628 [DOI] [Google Scholar]
- 61. Radka C. D., Frank M. W., Rock C. O., and Yao J. (2020) Fatty acid activation and utilization by Alistipes finegoldii, a representative Bacteroidetes resident of the human gut microbiome. Mol. Microbiol. 113, 807–825 10.1111/mmi.14445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Parsons J. B., Frank M. W., Subramanian C., Saenkham P., and Rock C. O. (2011) Metabolic basis for the differential susceptibility of Gram-positive pathogens to fatty acid synthesis inhibitors. Proc. Natl. Acad. Sci. U.S.A. 108, 15378–15383 10.1073/pnas.1109208108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jackowski S., Zhang Y.-M., Price A. C., White S. W., and Rock C. O. (2002) A missense mutation in the fabB (β-ketoacyl-acyl carrier protein synthase I) gene confers thiolactomycin resistance to Escherichia coli. Antimicrob. Agents Chemother. 46, 1246–1252 10.1128/AAC.46.5.1246-1252.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhao H., Brautigam C. A., Ghirlando R., and Schuck P. (2013) Overview of current methods in sedimentation velocity and sedimentation equilibrium analytical ultracentrifugation. Curr. Protoc. Protein Sci. 71, 20.12.1–20.12.49 10.1002/0471140864.ps2012s71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schuck P., Brautigam Z. H. C. A., and Ghirlando R. (2015) Basic Principles of Analytical Ultracentrifugation, CRC Press, Inc., Boca Raton, FL. [Google Scholar]
- 66. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 10.1016/S0006-3495(00)76713-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhao H., Ghirlando R., Alfonso C., Arisaka F., Attali I., Bain D. L., Bakhtina M. M., Becker D. F., Bedwell G. J., Bekdemir A., Besong T. M., Birck C., Brautigam C. A., Brennerman W., Byron O., et al. (2015) A multilaboratory comparison of calibration accuracy and the performance of external references in analytical ultracentrifugation. PLoS One 10, e0126420 10.1371/journal.pone.0126420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yao J., Bruhn D. F., Frank M. W., Lee R. E., and Rock C. O. (2016) Activation of exogenous fatty acids to acyl-acyl carrier protein cannot bypass FabI inhibition in Neisseria. J. Biol. Chem. 291, 171–181 10.1074/jbc.M115.699462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Otwinowski Z., and Minor W. (1997) [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 70. McCoy A. J. (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63, 32–41 10.1107/S0907444906045975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chen L., Li Y., Ebihara A., Shinkai A., Kuramitsu S., Yokoyama S., Zhao M., Rose J. P., Wang B. C., Southeast Collaboratory for Structural Genomics (SECSG), and RIKEN Structural Genomics/Proteomics Initiative (RSGI). (2007) Crystal structure of enoyl-[acyl-carrier-protein] reductase (NADH) from Aquifex aeolicus VF5. Protein Data Bank. 10.2210/pdb2P91/pdb [DOI]
- 72. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Skubák P., Murshudov G. N., and Pannu N. S. (2004) Direct incorporation of experimental phase information in model refinement. Acta Crystallogr. D Biol. Crystallogr. 60, 2196–2201 10.1107/S0907444904019079 [DOI] [PubMed] [Google Scholar]
- 74. Emsley P., and Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 75. DeLano W. L. (2002) The PyMOL Molecular Graphics System. DeLano Scientific, Palo Alto, CA [Google Scholar]
- 76. Radka C. D., Chen D., DeLucas L. J., and Aller S. G. (2017) The crystal structure of the Yersinia pestis iron chaperone YiuA reveals a basic triad binding motif for the chelated metal. Acta Crystallogr. D Struct. Biol. 73, 921–939 10.1107/S2059798317015236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Subramanya H., Rao K. N., and Anirudha L. (2013) Crystal structure of E. coli enoyl reductase in complex with NAD and AFN-1252. Protein Data Bank. 10.2210/pdb4JQC/pdb [DOI]
- 78. Hartkoorn R. C., Sala C., Neres J., Pojer F., Magnet S., Mukherjee R., Uplekar S., Boy-Röttger S., Altmann K. H., and Cole S. T. (2012) Towards a new tuberculosis drug: Pyridomycin—nature's isoniazid. EMBO Mol. Med. 4, 1032–1042 10.1002/emmm.201201689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bergler H., Hogenauer G., and Turnowsky F. (1992) Sequences of the envM gene and of two mutated alleles in Escherichia coli. J. Gen. Microbiol. 138, 2093–2100 10.1099/00221287-138-10-2093 [DOI] [PubMed] [Google Scholar]
- 80. Lu Y.-J., Zhang Y.-M., Grimes K. D., Qi J., Lee R. E., and Rock C. O. (2006) Acyl-phosphates initiate membrane phospholipid synthesis in gram-positive pathogens. Molec. Cell 23, 765–772 10.1016/j.molcel.2006.06.030 [DOI] [PubMed] [Google Scholar]
- 81. Jordan C. A., and Vey J. L. (2015) Crystal structure of E. coli FabI in apo form. Protein Data Bank. 10.2210/pdb5CFZ/pdb [DOI]
- 82. Pojer F., Hartkoorn R.C., Boy. S., Cole S.T. (2012) Mycobacterium tuberculosis InhA in complex with NADH. Protein Data Bank. 10.2210/pdb4DRE/pdb [DOI]
- 83. Yao J., Abdelrahman Y., Robertson R.M., Cox J.V., Belland R.J., White S.W., Rock C.O. (2014) Crystal structure of Chlamydia trachomatis enoyl-ACP reductase (FabI) in complex with NADH and AFN-1252. Protein Data Bank. 10.2210/pdb4Q9N/pdb [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Coordinates and structure factors for AfFabI and AfFabI·NADH crystal structures have been deposited in the Protein Data Bank (PDB) under accession codes 6VLX and 6VLY, respectively. All remaining data are contained within the article.