

HLA-B*27:05 and ERAP2 are associated with ankylosing spondylitis, a chronic inflammatory spondyloarthropathy. ERAP2 trims N-terminally extended residues of peptide precursors to their final length. The HLA-B*27:05 ligandomes from unedited and edited-ERAP2 isogenic cell clones were compared, which demonstrated alterations at P1, P2, P3, P7, and P9 peptide positions with enrichment of N-terminal basic residues and minority canonical P2 residues in the natural ligandome from edited-ERAP2 cells. Several ERAP2-dependent cellular peptides were highly homologous to multiple arthritogenic bacteria sequences. We propose that these findings highlight the pathogenic role of this aminopeptidase in the triggering of the autoimmune disease.
Keywords: HPLC, Immunology, Label-Free Quantification, Mass Spectrometry, Peptides, Ankylosing Spondylitis, Antigen Processing, HLA
Graphical Abstract

Highlights
HLA-B*27:05 and ERAP2 are risk factors for ankylosing spondylitis.
The effects of ERAP2 on the B*27:05 ligandome are defined.
P1, P2, P3, P7, and PΩ peptide positions are influenced by ERAP2.
These effects provide a basis for the association of ERAP2 with the autoimmune disease.
Abstract
The HLA-B*27:05 allele and the endoplasmic reticulum-resident aminopeptidases are strongly associated with AS, a chronic inflammatory spondyloarthropathy. This study examined the effect of ERAP2 in the generation of the natural HLA-B*27:05 ligandome in live cells. Complexes of HLA-B*27:05-bound peptide pools were isolated from human ERAP2-edited cell clones, and the peptides were identified using high-throughput mass spectrometry analyses. The relative abundance of a thousand ligands was established by quantitative tandem mass spectrometry and bioinformatics analysis. The residue frequencies at different peptide position, identified in the presence or absence of ERAP2, determined structural features of ligands and their interactions with specific pockets of the antigen-binding site of the HLA-B*27:05 molecule. Sequence alignment of ligands identified with species of bacteria associated with HLA-B*27-dependent reactive arthritis was performed. In the absence of ERAP2, peptides with N-terminal basic residues and minority canonical P2 residues are enriched in the natural ligandome. Further, alterations of residue frequencies and hydrophobicity profile at P3, P7, and PΩ positions were detected. In addition, several ERAP2-dependent cellular peptides were highly similar to protein sequences of arthritogenic bacteria, including one human HLA-B*27:05 ligand fully conserved in a protein from Campylobacter jejuni. These findings highlight the pathogenic role of this aminopeptidase in the triggering of AS autoimmune disease.
Self and pathogenic proteins are proteolytically degraded by proteasomes and other peptidases in the cytosol to generate an extremely diverse pool of peptides, both in sequence and in length. The transporter associated with antigen-processing molecules specifically translocates some of these degradation products to the endoplasmic reticulum lumen (1). The peptides with the correct length (8 to 11 residues) and interactions with specific pockets of the antigen recognition site of the HLA class I molecule (2), usually at position 2 (P2) as anchor residue and auxiliary residues at the C terminus and/or other positions of the peptide (3, 4) can stabilize their direct binding to HLA class I molecules. The fraction of amino-terminally extended precursors can be also utilized for antigen presentation after precursor editing and customization by endoplasmic-reticulum-resident aminopeptidase activities. In humans, two related aminopeptidases, ERAP1 (5) and ERAP2 (6, 7), with nonredundant specificities and different substrate preferences trims N-terminally extended residues of these peptide precursors to their final length. Of both, ERAP1 has a wider specificity since, with the exception of Pro, it cleaves virtually all N-terminal residues (8). In contrast, basic residues are the preferential target of ERAP2 activity (6, 9–11). The binding of peptide to HLA class I molecule in the ER stabilizes the nascent trimolecular peptide-HLA-β2-microglobulin complexes and allows for their subsequent transport to the cell membrane, where they are exposed to CD8+ cytotoxic T lymphocyte activity (12). The recognition of foreign or self-peptide ligands can lead to the beneficial killing of pathogen-infected cells or, instead, to initiate an autoimmune damage, respectively.
HLA-B*27 is an interesting HLA class I allele, whose prevalence varies significantly among different subpopulations, and it is more common in Caucasoids (http://www.allelefrequencies.net/). HLA-B*27 is strongly associated with ankylosing spondylitis (AS)1 (13), a chronic arthritis that cause inflammation, primarily of the spinal joints; although hip or shoulder joints can also be involved (14). Even though most of the HLA-B*27 subtypes are strongly associated with this chronic inflammatory spondyloarthropathy, HLA-B*27:06 and -B*27:09 subtypes are either not or perhaps only very weakly associated with this autoimmune disease, respectively (reviewed in (15)). This fact suggests that the polymorphism of HLA-B*27 subtypes modulates disease susceptibility. Previous, unambiguous functional distinction between the closely related AS-associated B*2704 and non-AS-associated B*2706 subtypes was found (16). However, after 40 years of scientific effort, the bases for this association remain largely unknown. Several hypotheses, based on different features of HLA-B*27, have been previously proposed to explain this intriguing association. However, to this day, none of them has reasonably explained the mechanism and the differential association between HLA-B*27 subtypes and the AS disease.
The classical arthritogenic peptide hypothesis (17) is focused in the antigen-presenting properties of HLA-B*27. In a bacterial infection, microbial peptide epitopes bound to HLA-B*27 elicit a normal CD8+ cytotoxic T lymphocyte response against the pathogen. This hypothesis assumes that some of these antimicrobial effector T cells would cross-react with autologous self-ligands, which are also presented by this HLA class I molecule, peptides showing a molecular mimicry with the primary bacterial epitope. Thus, this undesired cross-reaction triggers both autoimmune tissue injury and inflammation. Under this assumption, variations in HLA binding or immune recognition between mimetic self-peptides and bacterial epitopes with the HLA-B*27 subtypes would explain their differential association with AS. In the recent years, the two endoplasmic reticulum aminopeptidases involved in the trimming of peptides for HLA class I antigen presentation, ERAP1 and ERAP2, have been described as the main non-HLA susceptibility genes for AS (18–21), and thus, the interest on the arthritogenic peptide hypothesis has been highly renewed. ERAP1 is expressed in all individuals and shows a significant degree of polymorphism, which affects the activity and/or level of enzyme expression (22). Although the polymorphism of ERAP2 seems to be very limited, a differential splicing promotes nonsense-mediated RNA decay, and thus, about 25% of individuals fail to express this aminopeptidase (23). The highly active ERAP1 variants and the ERAP2 expression favor AS, while the less active variants of ERAP1 and the absence of ERAP2 are protective (24). Similarly to AS, the expression of functional ERAP2 is related with other different HLA class I molecule: HLA-A*29 is also associated to the birdshot chorioretinopathy, an eye-specific autoinflammatory disorder (25). Moreover, the role of ERAP2 in association with HLA-C*06 is more controversial in the triggering of the psoriasis, another immune-mediated disease (22).
Previously, only two studies using unrelated ERAP2-positive and -negative LCLs tried to determine the effect of ERAP2 protease on the generation of HLA-B*27 peptide ligands (26, 27). Moreover, although a rigorous pairwise comparison between both types of cell lines were carried (26, 27), these LCLs may vary in the expression of many proteins, including those related with antigen processing, and thus, the possibility of background differences cannot be formally ruled out. Thus, to elucidate conclusively the role of this aminopeptidase in the generation of HLA-B*27 ligands, high-throughput MS analyses isolated from ERAP2-edited cell clones were carried out in this study. In the unedited and edited ERAP2 isogenic cell clones, all the antigen presentation machinery and the rest of cellular proteins are common with the exception of the deleted ERAP2 protein. Our results determine that ERAP2 select HLA-B*27:05 ligands with a higher contribution of P2 anchor motif and lower influence of auxiliary residues.
EXPERIMENTAL PROCEDURES
Cells and antibodies
B27-C1R is an HLA-B*27:05 transfectant (28) of the human lymphoid cell line HMy2.C1R. HMy2.C1R cells are ERAP-2 positive and express the ERAP1 variant Hap8 not associated with AS (29). Cells were cultured in RPMI 1640 with 10% fetal bovine serum. The monoclonal antibodies used in this study were W6/32 (specific for a monomorphic HLA class I) (30), MAB3830 (R&D Systems, Minneapolis, MN) specific for ERAP2 protein, and GTU88 (Sigma-Aldrich, St. Louis, MO) specific for γ-tubulin.
CRISPR/Cas9-edited ERAP2−/− C1R-B*27:05 cell line
Using Benchling software (https://benchling.com) two pairs of guide RNAs targeting both the region 5′ of exon 5 and the region 3′ of exon 6 (coding the active site of ERAP2) were designed. 105 C1R-B*27:05 cells were transfected with the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 ribonucleoprotein complex (IDT Technologies, Coralville, IA) comprising the Cas9 protein and the pair of guide RNAs (exon 5: 5′…AATCAGTGCCCATTATCAAG…3′; exon 6: 5′…TCCTGTCAACACCTCTGCAA…3′) using the Neon electroporation system (Thermo Fisher Scientific, Waltham, MA). 48 h after transfection, DNA was extracted, and the ERAP2 locus was analyzed by PCR using primers that amplified the inside and outside of the deleted region: Ext_Fw (TGGCATACAGTTCCAGGCTT9, Ext_Rv (AACCTCTGCCCCTGACTTAC), Int_Fw (GCAGTAGGCACCCAAAATATT), and Int_Rv (ACTTGTTACGTGCCTAGACCT). Transfected cells were cloned, and the ERAP2 expression was analyzed by PCR and Western blotting as previously described (31). Two unedited (UN17 and UN20 with the complete ERAP2 sequence) and two homozygously edited (KO9 and KO19 without the ERAP2 exons 5 and 6) cell clones were selected.
Major histocompatibility complex class I-bound peptide isolation
HLA-bound peptides were isolated from three independent biological replicates of 1 × 109 cells from UN17, UN20, KO9, and KO19 cell clones as previously described (32). Cells were lysed in 1% Igepal CA-630 (Merck KGaA, Darmstadt, Germany), 20 mm Tris/HCl buffer, and 150 mm NaCl, pH 7.5, in the presence of the Protease Inhibitor Mixture (Merck KGaA). After centrifugation, the supernatants were passed first through a control precolumn containing CNBr-activated Sepharose 4B (GE Healthcare, Buckinghamshire, UK) to remove nonspecific peptides. The HLA-B*27:05/peptide complexes were isolated via affinity chromatography from the soluble cell extract fraction with the W6/32 monclonal antibody. The HLA-bound peptides were eluted with 1% aqueous trifluoroacetic acid, separated from the HLA molecules, and concentrated by ultrafiltration with a Vivaspin 2 filter, 5,000 MWCO HY (Sartorius Stedim Biotech, Goettingen, Germany), as previously described (33).
Electrospray-ion trap MS analysis
Peptide mixtures were desalted using OMIX Tips (C18, Agilent Technologies, Santa Clara, CA) (33, 34), and were analyzed by nanoLC-MS/MS using a Q-Exactive-Plus mass spectrometer fitted with an Ultimate 3000 RSLC nanocapillary UHPLC (Thermo Fisher Scientific), using the same parameters previously described (35). The peptides were resolved on homemade Reprosil C18-Aqua capillary columns (Dr. Maisch GmbH, Ammerbuch-Entringen, Germany) with a 5–28% acetonitrile linear gradient for 2 h in the presence of 0.1% formic acid at a flow rate of 0.15 μl/min. The dynamic exclusion was set to 20 s, and the automatic gain control value for the full MS was set to 3 × 106. The selected masses were fragmented from the survey scan of m/z 300–1,800 AMU at resolution 70,000. The 10 most intense masses from each full mass spectrum were fragmented by higher-energy collisional dissociation. MS/MS spectra were acquired with a resolution of 17,500 at m/z 200. The target value of the MS/MS was set to 1 × 105 and the isolation window to 1.8 m/z. The maximum injection time was set to 100 ms and normalized collision energy to 25 eV.
Database searches
Peptides were identified and quantified using the MaxQuant software (36) version 1.6.0.16 with the Andromeda search engine (37) using the human section of the UniProt/Swiss-Prot database (release 27.2.19, containing 73,101 entries). Methionine oxidation and N-acetylation were accepted as variable modifications. The peptide precursors and fragment mass tolerances were set at 6 and 20 ppm, respectively, and the false discovery rate was 0.01. Peptides with the previously defined HLA-B*27 motifs of Arg/Gln/Lys at P2 were selected if MaxQuant score was greater than 45. No peptides were found in a search of the reversed database. The MS data have been deposited to the MassIVE repository (http://massive.ucsd.edu) with the dataset identifier MSV000084718.
In silico binding prediction of HLA-B*27:05 ligands
The predicted binding of each peptide to HLA-B*27:05 class I molecules was calculated using NetMHCcons 1.1 Server (available at http://www.cbs.dtu.dk/services/NetMHCcons/).
Sequence alignments
Human peptide sequences were searched in the pan-proteome of the 12 arthritogenic bacterial species represented in the RefSeqdatabase (https://www.ncbi.nlm.nih.gov/refseq/) by an in-house Perl script. Only full-sequence peptide hits without any gap, with a maximum of two mismatches respect to the human peptide and Arg/Gln/Lys in the second peptide position were considered.
Experimental design and statistical rationale
The effect of ERAP2 depletion on the HLA-B*27:05 peptidome was analyzed by quantitative label-free MS from two unedited (with the complete ERAP2 sequence) and two homozygously edited (without the ERAP2 exons 5 and 6) cell clones. Three independent preparations of the HLA-B*27:05-bound peptides were obtained from each of the four cell lines in this study, and used as biological replicates, whose reproducibility was assessed by Pearson correlation analyses (Fig. S1). A precolumn was utilized to remove nonspecific binding proteins and peptides. Additionally, a 0.01 false discovery rate was utilized.
Since a majority of the identified peptides was found in both the UN and KO cells (Fig. S2), quantitative differences in peptide amounts between cell lines were assigned in pairwise comparisons as follows (38): In each experiment, the intensity of any given ion peak was normalized to the total intensity of all the identified B*27:05 ligands. The mean normalized intensity of each ion peak from the three individual experiments for each cell line was taken as the amount of that peptide relative to the total amount of ligands identified in that cell clone. The HLA-B*27:05 ligands in each pairwise comparison were classified based on the normalized IR of the corresponding ion peak in the cell clones. Peptides predominant in one cell clone, relative to the other (IR>1.0) were subdivided in two subsets as previously described (38). Peptides with IR>1.0 to 1.5 in either cell line were considered to be expressed in similar amounts in the two cell clones and therefore with little or no effect by ERAP2. Peptides with IR>1.5 in one cell clone relative to the other, including peptides found only in that cell clone, were considered to be up-regulated in that cell clone and consequently influenced by the ERAP2 context.
To analyze the statistical significance of the differences in residue frequencies the Bonferroni-corrected chi-square test was utilized. To analyze the statistical significance of the relative intensity differences among peptide sets a linear regression was carried out. Differences in the hydrophobicity at different peptide position among the HLA-B*27:05 ligands were assessed by multiple t tests. p values < 0.05 were considered to be statistically significant.
RESULTS
Homozygous ERAP2-deficient cell clones were generated by deleting a 4741-bp fragment of the ERAP2 gene, including the sequence encoding the active site of ERAP2 from B27-C1R cell line (Fig. 1A). The presence or absence of ERAP2 exons 5 and 6 and protein expression in unedited control (UN) or KO cells was confirmed by PCR analysis (Fig. 1B) and immunoblotting (Fig. 1C), respectively. The effect of ERAP2 on the surface expression of B*27:05 heterodimers was assessed by flow cytometry with ME1 Ab. No statistically significant differences in the surface expression of HLA-B*27:05 heterodimers from KO cell clones relative to the UN cell clones was observed.
Fig. 1.

Targeted deletion of exons 5–6 of the gene encoding ERAP2 protein in cell clones with homozygous deletion. (A) Schematic representation of the deletion of exons 5–6 of the gene encoding ERAP2 protein in human chromosome 5. gRNA cleavage sites are indicated by blue arrowheads. (B) Analysis of deletion of exons 5–6 detected by PCR from indicated ERAP2-positive (UN) and ERAP2-negative (KO) cell clones using specific flanking external (A and B) and internal (C) probes. (C) Representative Western blotting and expression levels of ERAP2 (closed bars) and γ-tubulin (open bars) of the indicated UN and KO cell clones relative to the cell line showing maximal values.
HLA-B*27:05 peptidome in presence or absence of ERAP2
The peptides pools bound to HLA-B*27:05 class I molecules isolated from two UN (UN17 and UN20) and two KO (KO9 and KO19) cell clones were subjected to LC-MS/MS analysis in biologic triplicate experiments. A total of 6,212 peptides, and 6,052 peptides were identified in UN17 and UN20 cell clones, respectively (supplemental Table S1). Similarly, 6,077 peptides, and 4170 peptides were identified bound to HLA-B*27:05 in KO9 and KO19 cell clones, respectively (supplemental Table S1). The anchor motifs for HLA-B*27:05 binding: Arg, Gln, or Lys at position 2 (P2) were detected for peptides from both UN and KO cell clones identified by MS (supplemental Table S1). In silico binding prediction differences between the HLA-B*27:05 ligands from UN and KO were not found (Fig. S3).
Global effect of ERAP2 on the HLA-B*27:05 ligandome
To analyze the influence of ERAP2 on the relative amounts of B*27:05 ligands expressed in the presence or absence of this aminopeptidase, comparison between the B*27:05 peptidomes from UN and KO cell clones was carried out. The HLA-B*27:05-bound peptides were classified based on the relative intensity of the UN versus KO in each two types of cell lines compared (supplemental Table S1). The enriched peptides in presence (UN cell clones) or absence (KO cell clones) of ERAP2, that is with a normalized IR > 1.0 relative to the other condition, were compared and split in two subgroups: 1.0 < IR ≤ 1.5 (mild enrichment) and IR > 1.5 (strong enrichment), because the statistical significance of IR values was previously established (38). In addition, the specific peptides only identified in UN or KO clones were included in their respective IR > 1.5 subgroup. No differences in MW were found when 1.0 < IR ≤ 1.5 subgroups from UN and KO cell clones were compared (Fig. 2, left panel). In contrast, the subset of HLA-B*27:05-bound peptides strongly enriched in absence of ERAP2 (IR > 1.5) showed a statistically significant MW increase of 56 Daltons relative to the corresponding ligands strongly enriched in presence of ERAP2 (Fig. 2, right panel).
Fig. 2.

Molecular weight (MW) of HLA-B*27:05 ligands from ERAP2-positive (UN) and ERAP2-negative (KO) cell clones. The peptides from UN or KO cell clones were subdivided based on their ion peak IR to the following conditions: 1.0 < IR ≤ 1.5 (A) and IR > 1.5 (B), and the equivalent subgroups were compared as previously described (38). The intensity assigned to each peptide in a cell line was normalized by dividing its mean intensity by the total intensity of all peptides from this cell line in the corresponding pairwise comparison. The differences in the mean MW (ΔMW) of the peptides in each subgroup from both UN and KO cell clones are indicated. Significant p values: *, p < 0.05; **, p < 0.01; and ***, p < 0.001 are indicated in the corresponding MW range, respectively. (C) volcano plots showing thee differences in peptide amounts between mean of UN and KO cell clones.
In absence of ERAP2 the HLA-B*27:05 ligandome was enriched in N-terminal basic residues
The residue frequencies at different peptide position of HLA-B*27:05 ligands identified in presence or absence of ERAP2 aminopeptidase were analyzed. First, no differences were found in the pairwise comparison of 1.0 < IR ≤ 1.5 subgroups from UN and KO cell clones at N-terminal (P1) position. In contrast, in the comparison of IR > 1.5 subgroup from both types of cell clones, a statistically significant increase in the frequencies of Arg and Lys was detected in absence of ERAP2 (Fig. 3). In addition, a simultaneous and statistically significant decrease in the frequencies of Ile, Ser, and Val was also detected in absence of ERAP2 (Fig. 3).
Fig. 3.

Effect of ERAP2 depletion on the individual residue usage from HLA-B*27:05 ligands. Percentage of residue frequencies at the indicated position in each individual panel among the peptides with IR > 1.5 from UN (open bars) and KO (closed bars) cell clones. These subgroups include also the peptides specifically found only in UN or KO cell clones. Significant p values: *, p < 0.05; **, p < 0.01; and ***, p < 0.001 between UN and KO cell clones are indicated, respectively. In addition, amino acids with statistically significant increase in UN or KO cell clones are indicated as open or closed box, respectively.
Qualitative and quantitative enrichment of HLA-B*27:05 ligands with Gln and Lys at anchor motif (P2) peptide position in absence of ERAP2
Next, the residue frequencies at anchor motif P2 peptide position of HLA-B*27:05 ligands from UN and KO cell clones were analyzed. As previously described with the N-terminal (P1) position, no differences were found in the pairwise comparison of 1.0 < IR ≤ 1.5 subgroups from ERAP2-positive and ERAP2-negative cell clones. However, in the comparison of IR > 1.5 subgroups statistically significant increases in the frequencies of Gln and Lys with concomitant decrease in the frequency of Arg at P2 peptide position were found in absence of ERAP2 (Fig. 3B).
In addition, LC-MS signal intensities of shared ligands with Gln and Lys at anchor motif P2 peptide position were statistically stronger in absence of ERAP2 relative to the same peptides identified in presence of this aminopeptidase when compared with shared ligands with Arg at P2 position (Fig. 4). No differences were found between shared ligands with Gln and Lys at the anchor motif position when peptide LC-MS signal intensities from UN and KO cell clones were compared (Fig. 4).
Fig. 4.
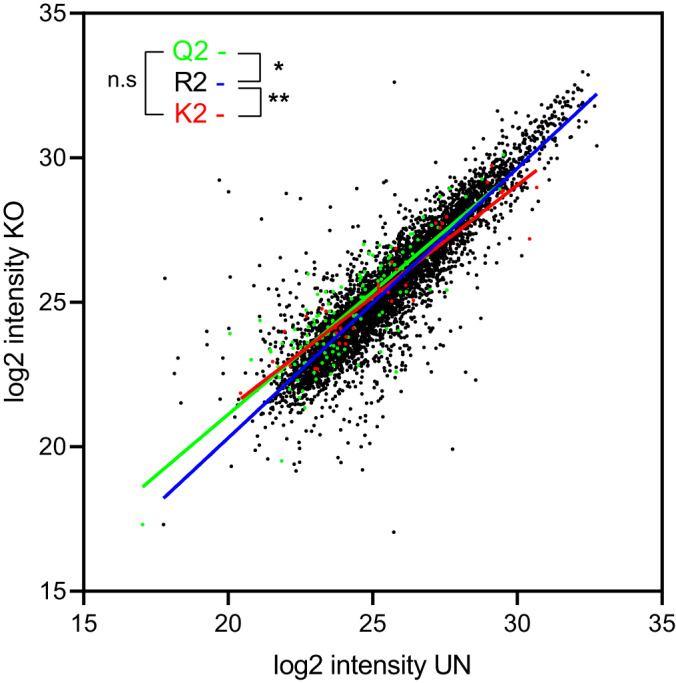
Quantitation of the shared HLA-B*27:05 ligandome from ERAP2-positive (UN) and ERAP2-negative (KO) cell clones. Scatter plots of the mean of LC-MS signal intensities of the HLA-B*27:05 ligandome from UN and KO cell clones. Peptides with Arg, Gln, and Lys at P2 position are colored in black, green, and red, respectively. Linear regression of peptides with Arg, Gln, and Lys at P2 position are colored in blue, green, and red, respectively. Significant p values: *, p < 0.05; and **, p < 0.01 between linear regression from UN and KO cell clones are indicated, respectively.
Qualitative differences in residue frequencies at P3, P7, and PΩ but not at P-1 peptide position of HLA-B*27:05 ligands from ERAP2-positive and ERAP2-negative cell clones
Similarly to P1 and P2 positions, the residue frequencies at P3 to PΩ peptide positions and P-1 position of HLA-B*27:05 ligands identified in the presence or absence of ERAP2 aminopeptidase were also analyzed. As previously described with the P1 and P2 positions, no differences were found in the pairwise comparison of 1.0 < IR ≤ 1.5 subgroups from ERAP2-positive and ERAP2-negative cell clones at all positions analyzed. When IR > 1.5 subgroups were compared, only the frequencies of some residues at P3, P7, and PΩ peptide positions were statistically significant. Leu and Phe were increased, and conversely, Ala, Asn, and Asp were decreased in absence of ERAP2 when P3 peptide position was assessed (Fig. 3C). In the comparison of residue frequencies at P7, Phe was increased whereas Asp, Gly, and Glu were decreased in the ERAP2-deficient cell clones (Fig. 3D). Finally, Leu was increased and concomitant Arg was decreased at PΩ position when ERAP2 is absent (Fig. 3E).
Influence of ERAP2 in the hydrophobicity of HLA-B*27:05 ligands
The hydrophobicity at the five relevant peptide positions (P1, P2, P3, P7, and PΩ) of HLA-B*27:05 ligands modified by ERAP2 expression was measured on the grand average of hydropathy scale. Statistically significant increases in the hydrophobicity at N-terminal position peptide with concomitant decrease in the hydrophobicity at P3, P7, and PΩ peptide position were found in absence of ERAP2 versus ERAP2-positive cell clones (Fig. S4).
Identity between a human HLA-B*27:05 ligand with a bacteria sequence from C. jejuni
The peptides specifically generated or enriched, IR > 1.5 subgroup, in presence of ERAP2 are candidates to be hypothetical arthritogenic peptides. Thus, a comparison of the sequences of these HLA-B*27:05 ligands identified in the current study with seven representative species of bacterial genera associated with HLA-B*27-dependent reactive arthritis (39) was carried out. A striking and complete identity was found between human HLA-B*27:05 ligands and a bacteria sequence (Table I). The IRPAINVGL ligand from human ATP synthase subunit alpha and a 9-mer from C. jejuni proteome, which spans amino acid residues 353–361 of the F0F1 ATP synthase subunit alpha, were identical (Table I). Although these two proteins share a moderate 64% identity using Basic Local Alignment Search Tool bioinformatics tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi), another seven additional 9-mers with P2 anchor motif for HLA-B*2705 were also identical between human synthase and the following F0F1 ATP synthase subunit alpha sequences: GRGQRELII, GQRELIIGD, QRELIIGDR, LRRPPGREA, RRPPGREAY, GREAYPGDV, and TQAGDVSAY.
Table I. Alignment of natural HLA-B*27:05 cellular peptides with sequences of bacteria associated with B*27-dependent reactive arthritis.
| Peptide | Protein | RefSeq | Species |
|---|---|---|---|
| IRPAINVGL | ATP synthase subunit alpha | NP_001001935 | Human |
| . . . . . . . . . | F0F1 ATP synthase subunit alpha | WP_002851836.1 | C. jejuni |
| KRALPFSLV | OCIA domain-containing protein 2 | NP_001273702 | Human |
| . . . . . . V . . | FkbM family methyltransferase | WP_038815436.1 | C. jejuni |
| SRHGLEQYL | Calcium signal-modulating cyclophilin ligand | NP_001736 | Human |
| . . . V . . . . . | Replication initiation and membrane attachment family protein | WP_021420237.1 | Clostridium difficile |
| SRTDLIELL | Protein furry homolog-like | XP_011511982 | Human |
| . . . . . D . . . | AAA family ATPase | WP_117695277.1 | Prevotella copri |
| RRALLLLLL | Lysosomal Pro-X carboxypeptidase | XP_005274150 | Human |
| . . N . . . . . . | DUF4468 domain-containing protein | WP_089544435.1 | P. copri |
| SRAAEKLYL | Signal recognition particle 9 kDa protein | NP_001123912 | Human |
| . . . . . . . R . | Y-family DNA polymerase | WP_004197540.1 | Salmonella typhimurium |
| ERLQEELNK | Origin recognition complex subunit 3 | NP_862820 | Human |
| . . . . . . N . . | DUF2326 domain-containing protein | WP_050162494.1 | Yersinia enterocolitica |
Similarity of ERAP2-dependent HLA-B*27:05 ligands with pathogenic bacterial sequences
In addition, in the search of homology between the HLA-B*27:05 ligands identified in the current study with the bacteria associated with HLA-B*27-dependent reactive arthritis, other multiple bacteria peptides were highly homologs (with only one or two amino acid changes) with several ERAP2-dependent cellular peptides (Table II). The higher and lower conservation rates between HLA human ligands and bacteria sequences were found with Yersinia enterocolitica and Chlamydia trachomatis, respectively (Table II). Six human HLA-B*27:05 ligands presented only one change versus sequences from five different bacteria (Tables I and II). These modifications affected to different peptide positions, which can interact with several pockets of HLA-B*27:05 molecule or be exposed to direct contact with the T cell receptor of effector cells. Also, multiple ERAP2-dependent cellular peptides bound to HLA-B*27:05 showed only two amino acid changes with different bacteria sequences (Table II). Forty-two of these human HLA ligands presented homology with sequences from representative species of at least two different bacterial genera, as for example the ERLQEELNK and SRTDLIELL ligands shown in supplemental Table S2. These changes affected practically all peptide positions, including the substitution of Arg for Gln or Lys at P2 anchor motif (supplemental Table S1).
Table II. Conservation of natural HLA-B*27 cellular peptides with sequences of bacteria associated with B*27-dependent reactive arthritis.
| Bacteria | Number of bacteria peptides with the indicated changes versus human liganda |
|
|---|---|---|
| 1 change | 2 changes | |
| C. jejuni | 1 | 18 |
| Chlamydia trachomatis | 0 | 4 |
| C. difficile | 1 | 34 |
| P. copri | 2 | 35 |
| S. typhimurium | 1 | 18 |
| Shigella flexneri | 0 | 24 |
| Y. enterocolitica | 1 | 47 |
a Only the peptides with P2 anchor motif for HLA-B*27:05 binding were considered.
DISCUSSION
In the current report, several key issues concerning the role of ERAP2 on the HLA-B*27:05 ligandome have been investigated. Previously, the only two studies using unrelated ERAP2-positive and -negative LCLs showed a shift toward higher MW from ligands and lower amounts of peptides with N-terminal basic residues in the absence of ERAP2 (26, 27). Here, we have confirmed the MW difference of HLA-B*27:05 ligands associated to ERAP2 with a high average: 56 Daltons using CRISPR/Cas9-edited cell clones versus a mean of 28 Daltons previously reported with LCLs (26, 27). Also, an enrichment of peptides with N-terminal basic residues in the absence of ERAP2 was detected in the MS analyses using ERAP2-edited cell clones as was previously described using ERAP2-positive and -negative LCLs (26, 27).
Moreover, several new concerns on the influence of ERAP2 on the HLA-B*27:05 ligandome have been identified in the current report. Concomitant with the decrease in the frequencies of Arg and Lys in P1 for ERAP2+ cells, the frequencies in the specific/enriched peptides of Ile, Ser, and Val at this peptide position were increased in unedited cell clones. These alterations on P1 residue usage were associated with an increase in the hydrophobicity at the N-terminal position of HLA-B*27:05 ligands. For HLA-B*27:05 ligands, the P1 residue is an auxiliary anchor motif, whose lateral chain interacts with different residues of the A pocket (40). A previous study using poly-A analogs containing the Arg2 anchor motif and different changes at P1 position concluded that Arg was the amino acid with the highest contribution to HLA binding; Ile and Lys presented a neutral contribution, whereas Ser and Val amino acids were detrimental residues at this P1 peptide position (41). Thus, the results obtained with ERAP2-edited cell clones suggest that this aminopeptidase favors ligands with low interactions with the A pocket of HLA-B*27:05 class I molecules.
The interaction between the B pocket of HLA-B*27:05 and the P2 peptide position is the major contribution to B*27:05 binding and thus, this position was defined as anchor motif (SYFPEITHI database: http://www.syfpeithi.de (4)). Arg is overwhelmingly the amino acid most utilized at this P2 peptide position, with residual presence of Gln (42–44) and Lys (45, 46) at this anchor position. Previously, computational simulations suggested a similar binding conformation between P2-Arg and P2-Lys peptides to the B pocket of the HLA-B*27:05 molecules but slightly different from the P2-Gln peptides (46). In this modeling, the hydrogen bond profiles were energetically and geometrically enhanced for Arg at P2 position relative to Gln or Lys amino acids (46). In addition, binding analysis with the same peptide sequence and different changes at P2 position concluded that the HLA affinity was substantially lower for peptides with Lys or Gln as anchor motif than the respective P2-Arg ligands (46). In the present study, we found that the absence of ERAP2 increases, in both quantity and quality, the frequencies of Gln and Lys amino acids at P2 anchor motif position. Thus, in ERAP2-deficient cell clones, the HLA-B*27:05 molecule appears to be more tolerant to the loss of interactions in its B pocket.
In the absence of ERAP2, the frequencies at P3 peptide position of Ala, Asn, and Asp decreased, and conversely, Leu and Phe were increased with a subsequent reduction in the hydrophobicity at this position of HLA-B*27:05 ligands. Previous studies using poly-A analogs containing the Arg2 anchor motif and different changes at P3 position concluded that Leu and Phe were favored residues to the interaction with the D pocket of HLA-B*27:05 class I molecules and, thus, increase the HLA binding. Moreover, in these analyses, Ala and Asn presented a neutral contribution, whereas Asp amino acid was a detrimental residue at this P3 peptide position (41, 47). Thus, in ERAP2-deficient cell clones, the interactions in the D pocket seems to be more relevant to HLA-B*27:05 binding than in ERAP2-positive cell clones.
The lateral chain of P7 residue interacts with the E pocket of HLA-B*27:05 class I molecules. Unfortunately, only one of the four amino acids with altered frequencies between ERAP2-UN and -KO cell clones identified in our screening was previously analyzed using poly-A analogs(47), and thus, no information about ERAP2 influence in this position could be obtained.
Finally, the lateral chain of PΩ residue interacts with the F pocket of HLA-B*27:05 class I molecules. At this position, the frequency of Leu was increased and concomitant Arg was decreased when ERAP2 is absent. In the two previous studies using poly-A analogs at PΩ residue, the relative binding affinity of Leu residue was fourfold (41) or eightfold (47) higher than the one of Arg residue. This scenario supports that interactions at pocket F are more important when ERAP2 is not available.
The global picture emerging from the current report is consistent with a remodeling of interactions between the diverse residues of ligands with the different pockets of HLA-B*27:05 class I molecule influenced by ERAP2. The absence of this aminopeptidase increases the presence of ligands with minority canonical P2 residues, which present low interactions in the B pocket but that are compensated by the contribution of contribution of otherwise weak interactions between the different auxiliary anchor motif P1, P3, or PΩ to A, D, or F pockets, respectively.
The widely spread ERAP2 deficiency (with an incidence of about a quarter of the human population) has a protective role for HLA-B*27 in AS (21, 23). The qualitative and quantitative changes in the HLA-B*27:05 ligandome, detected in the current report, may be relevant in the study of association between this aminopeptidase with this autoimmune type of arthritis in the context of an arthritogenic peptide hypothesis. The cellular peptides specifically generated by ERAP2, identified herein, may be tested as candidates to be the arthritogenic peptides cross-recognized by antibacterial T cells. Another possibility will be that quantitative differences by the ERAP2 influence detected by us in multiple HLA-B*27 ligands may alter the equilibrium between immunogenicity and tolerance. Such misbalance would trigger the autoimmune T cell response, expanding the universe of peptides to be tested as possible arthritogenic peptides.
In this context, since no arthritogenic peptides have been previously described, the human HLA-B*27:05 ligands akin to bacterial protein segments reported in the current study are interesting, and especially those fully conserved with arthritogenic bacteria as the IRPAINVGL ligand from the ATP synthase subunit alpha since the leakage of T cell tolerance after infection was previously described (48–50). In addition, it is also striking that the bacteria protein including this conserved sequence has other seven conserved sequences with P2 anchor motif for HLA-B*27:05 binding too. As currently, MS analyses are not able to identify all the ligands bound to HLA class I on the cell surface, the possible generation of these seven hypothetical natural HLA-B*27:05 ligands with identical sequence for the alpha subunit of the F0F1 ATP synthase peptides remains open and make this protein from C. jejuni a very attractive candidate as source for hypothetical arthritogenic peptides. Also, many of the 42 human HLA ligands that presented only two amino acid changes with sequences from representative species of at least two different bacterial genera shows amino acid changes in the P1, P2, P3, or PΩ peptide positions influenced by ERAP2 and thus, are also attractive candidates to be hypothetical arthritogenic peptides.
In addition to the arthritogenic peptide hypothesis, two additional features of HLA-B*27 have been proposed to explain the association of this class I molecule with AS: misfolded forms of the HLA-B*27 heavy chain which triggers first ER stress signaling pathways activation, and later the inflammatory response (51), and the presence of HLA-B*27 heavy chain homodimers at the cell surface (52), which can be recognized by NK cells or a subset of Th17 cells (53). In these last two hypothesis no contribution of ERAP1 and/or ERAP2 aminopeptidases is necessary to trigger the AS disease. In addition, recently similar levels of HLA-B*27 heavy chain homodimers for AS-associated and non-AS-associated subtypes were described (54). In contrast, the role of ERAP2 in AS would be more compatible with the hypothesis of the cross-reactive peptide epitopes, involving this enzyme with either the generation of pathogenic peptide(s) or the destruction of putative protective epitope one(s). Then, on the basis of our observations, the significant destruction of peptides with basic residues in P1 peptide position could have a direct contribution to the risk of AS, activating T cells or breaking tolerance. This mechanism, unlike the other two proposed, would also explain that other HLA-B alleles have been linked to AS (55).
Finally, the complexity of ERAP1 haplotypes, with influence of individual mutations (56) and the combined effects of co-occurring polymorphisms (57) is also relevant in the risk of AS. Thereby, Hap1 to Hap3 (but not Hap8 or Hap10) are AS-associated haplotypes (24). Therefore, future studies analyzing the combined effect of ERAP2 and the ERAP1 haplotypes associated with AS must be carried out to target more specifically the possible set of arthritogenic peptides.
DATA AVAILABILITY
The mass spectrometry data have been deposited to the MassIVE repository (http://massive.ucsd.edu) with the data set identifier MSV000084718.
Supplementary Material
Footnotes
* The Spanish Ministry of Economy grants SAF2014-58052 and “Acción Estratégica en Salud” MPY 388/18 to D. L., “Acción Estratégica en Salud” MPY 1366/13 and MPY 483 1346/16 to P. L., and by Israel Science Foundation, grant No. 1435/16 to A. A. The funding agencies had no role in the study design, data collection, analysis decision to publish, or preparation of the manuscript. The authors declare that they have no conflicts of interest with the contents of this article.
 This article contains supplemental material Tables S1 and S2 and Figs. S1–S4.
This article contains supplemental material Tables S1 and S2 and Figs. S1–S4.
1 The abbreviations used are:
- AS
- ankylosing spondylitis
- ERAP3
- endoplasmic reticulum aminopeptidase 2
- HLA
- human leukocyte antigen
- IR
- intensity ratio
- LCL
- lymphoblastoid cell line
- KO
- edited-ERAP2 isogenic cell clone
- MS
- mass spectrometry
- UN
- unedited-ERAP2 isogenic cell clone.
REFERENCES
- 1. Shastri N., Schwab S., and Serwold T. (2002) Producing nature's gene-chips: The generation of peptides for display by MHC class I molecules. Annu. Rev. Immunol. 20, 463–493 [DOI] [PubMed] [Google Scholar]
- 2. Bjorkman P. J., Saper M. A., Samraoui B., Bennett W. S., Strominger J. L., and Wiley D. C. (1987) Structure of the human class I histocompatibility antigen, HLA-A2. Nature 329, 506–512 [DOI] [PubMed] [Google Scholar]
- 3. Parker K. C., Bednarek M. A., and Coligan J. E. (1994) Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J. Immunol. 152, 163–175 [PubMed] [Google Scholar]
- 4. Rammensee H. G., Bachmann J., Emmerich N. P. N., Bachor O. A., and Stevanovic S. (1999) SYFPEITHI: Database for MHC ligands and peptide motifs. Immunogenetics 50, 213–219 [DOI] [PubMed] [Google Scholar]
- 5. Saric T., Chang S. C., Hattori A., York I. A., Markant S., Rock K. L., Tsujimoto M., and Goldberg A. L. (2002) An IFN-g-induced aminopeptidase in the ER, ERAP1, trims precursors to MHC class I-presented peptides. Nat. Immunol. 3, 1169–1176 [DOI] [PubMed] [Google Scholar]
- 6. Tanioka T., Hattori A., Masuda S., Nomura Y., Nakayama H., Mizutani S., and Tsujimoto M. (2003) Human leukocyte-derived arginine aminopeptidase—The third member of the oxytocinase subfamily of aminopeptidases. J. Biol. Chem. 278, 32275–32283 [DOI] [PubMed] [Google Scholar]
- 7. Hattori A., Matsumoto H., Mizutani S., and Tsujimoto M. (1999) Molecular cloning of adipocyte-derived leucine aminopeptidase highly related to placental leucine aminopeptidase/oxytocinase. J. Biochem. (Tokyo) 125, 931–938 [DOI] [PubMed] [Google Scholar]
- 8. Hearn A., York I. A., and Rock K. L. (2009) The specificity of trimming of MHC class I-presented peptides in the endoplasmic reticulum. J. Immunol. 183, 5526–5536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saveanu L., Carroll O., Lindo V., Del Val M., López D., Lepelletier Y., Greer F., Schomburg L., Fruci D., Niedermann G., and van Endert P. M. (2005) Concerted peptide trimming by human ERAP1 and ERAP2 aminopeptidase complexes in the endoplasmic reticulum. Nat. Immunol. 6, 689–697 [DOI] [PubMed] [Google Scholar]
- 10. Zervoudi E., Papakyriakou A., Georgiadou D., Evnouchidou I., Gajda A., Poreba M., Salvesen G. S., Drag M., Hattori A., Swevers L., Vourloumis D., and Stratikos E. (2011) Probing the S1 specificity pocket of the aminopeptidases that generate antigenic peptides. Biochem. J. 435, 411–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Birtley J. R., Saridakis E., Stratikos E., and Mavridis I. M. (2012) The crystal structure of human endoplasmic reticulum aminopeptidase 2 reveals the atomic basis for distinct roles in antigen processing. Biochemistry 51, 286–295 [DOI] [PubMed] [Google Scholar]
- 12. York I. A., Goldberg A. L., Mo X. Y., and Rock K. L. (1999) Proteolysis and class I major histocompatibility complex antigen presentation. Immunol. Rev. 172, 49–66 [DOI] [PubMed] [Google Scholar]
- 13. Brewerton D. A., Hart F. D., Nicholls A., Caffrey M., James D. C., and Sturrock R. D. (1973) Ankylosing spondylitis and HL-A 27. Lancet 1, 904–907 [DOI] [PubMed] [Google Scholar]
- 14. Braun J., and Sieper J. (2007) Ankylosing spondylitis. Lancet 369, 1379–1390 [DOI] [PubMed] [Google Scholar]
- 15. Marcilla M., and Lopez de Castro J. A. (2008) Peptides: The cornerstone of HLA-B27 biology and pathogenetic role in spondyloarthritis. Tissue Antigens 71, 495–506 [DOI] [PubMed] [Google Scholar]
- 16. López D., Rojo S., Calvo V., and Lopez de Castro J. A. (1992) Peptide-presenting similarities among functionally distant HLA-B27 subtypes revealed by alloreactive T lymphocytes of unusual specificity. J. Immunol. 148, 996–1002 [PubMed] [Google Scholar]
- 17. Benjamin R., and Parham P. (1990) Guilt by association: HLA-B27 and ankylosing spondylitis. Immunol. Today 11, 137–142 [DOI] [PubMed] [Google Scholar]
- 18. Wellcome Trust Case Control Consortium; Australo-Anglo-American Spondylitis Consortium (TASC), Burton P. R., Clayton D. G., Cardon L. R., Craddock N., Deloukas P., Duncanson A., Kwiatkowski D. P., McCarthy M. I., Ouwehand W. H., Samani N. J., Todd J. A., Donnelly P., Barrett J. C., Davison D., Easton D., Evans D. M., Leung H. T., Marchini J. L., Morris A. P., Spencer C. C., Tobin M. D., Attwood A. P., Boorman J. P., Cant B., Everson U., Hussey J. M., Jolley J. D., Knight A. S., Koch K., Meech E., Nutland S., Prowse C. V., Stevens H. E., Taylor N. C., Walters G. R., Walker N. M., Watkins N. A., Winzer T., Jones R. W., McArdle W. L., Ring S. M., Strachan D. P., Pembrey M., Breen G., St. Clair D., Caesar S., Gordon-Smith K., Jones L., Fraser C., Green E. K., Grozeva D., Hamshere M. L., Holmans P. A., Jones I. R., Kirov G., Moskivina V., Nikolov I., O'Donovan M. C., Owen M. J., Collier D. A., Elkin A., Farmer A., Williamson R., McGuffin P., Young A. H., Ferrier I. N., Ball S. G., Balmforth A. J., Barrett J. H., Bishop T. D., Iles M. M., Maqbool A., Yuldasheva N., Hall A. S., Braund P. S., Dixon R. J., Mangino M., Stevens S., Thompson J. R., Bredin F., Tremelling M., Parkes M., Drummond H., Lees C. W., Nimmo E. R., Satsangi J., Fisher S. A., Forbes A., Lewis C. M., Onnie C. M., Prescott N. J., Sanderson J., Matthew C. G., Barbour J., Mohiuddin M. K., Todhunter C. E., Mansfield J. C., Ahmad T., Cummings F. R., Jewell D. P., Webster J., Brown M. J., Lathrop M. G., Connell J., Dominiczak A., Marcano C. A., Burke B., Dobson R., Gungadoo J., Lee K. L., Munroe P. B., Newhouse S. J., Onipinla A., Wallace C., Xue M., Caulfield M., Farrall M., Barton A.; Biologics in R. A. Genetics and Genomics Study Syndicate (BRAGGS) Steering Committee, Bruce I. N., Donovan H., Eyre S., Gilbert P D., Hilder S. L., Hinks A. M., John S. L., Potter C., Silman A. J., Symmons D. P., Thomson W., Worthington J., Dunger D. B., Widmer B., Frayling T. M., Freathy R. M., Lango H., Perry J. R., Shields B. M., Weedon M. N., Hattersley A. T., Hitman G. A., Walker M., Elliott K. S., Groves C. J., Lindgren C. M., Rayner N. W., Timpson N. J., Zeggini E., Newport M., Sirugo G., Lyons E., Vannberg F., Hill A. V., Bradbury L. A., Farrar C., Pointon J. J., Wordsworth P., Brown M. A., Franklyn J. A., Heward J. M., Simmonds M. J., Gough S. C., Seal S.; Breast Cancer Susceptibility Collaboration (UK), Stratton M. R., Rahman N., Ban M., Goris A., Sawcer S. J., Compston A., Conway D., Jallow M., Newport M., Sirugo G., Rockett K. A., Bumpstead S. J., Chaney A., Downes K., Ghori M. J., Gwilliam R., Hunt S. E., Inouye M., Keniry A., King E., McGinnis R., Potter S., Ravindrarajah R., Whittaker P., Widden C., Withers D., Cardin N. J., Davison D., Ferreira T., Pereira-Gale J., Hallgrimsdo'ttir I. B., Howie B. N., Su Z., Teo Y. Y., Vukcevic D., Bentley D., Brown M. A., Compston A., Farrall M., Hall A. S., Hattersley A. T., Hill A. V., Parkes M., Pembrey M., Stratton M. R., Mitchell S. L., Newby P. R., Brand O. J., Carr-Smith J., Pearce S. H., McGinnis R., Keniry A., Deloukas P., Reveille J. D., Zhou X., Sims A. M., Dowling A., Taylor J., Doan T., Davis J. C., Savage L., Ward M. M., Learch T. L., Weisman M. H., and Brown M. (2007) Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 39, 1329–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Evans D. M., Spencer C. C., Pointon J. J., Su Z., Harvey D., Kochan G., Oppermann U., Dilthey A., Pirinen M., Stone M. A., Appleton L., Moutsianas L., Leslie S., Wordsworth T., Kenna T. J., Karaderi T., Thomas G. P., Ward M. M., Weisman M. H., Farrar C., Bradbury L. A., Danoy P., Inman R. D., Maksymowych W., Gladman D., Rahman P., Spondyloarthritis Research Consortium of Canada (SPARCC), Morgan A., Marzo-Ortega H., Bowness P., Gaffney K., Gaston J. S., Smith M., Bruges-Armas J., Couto A. R., Sorrentino R., Paladini F., Ferreira M. A., Xu H., Liu Y., Jiang L., Lopez-Larrea C., Díaz-Peña R., López-Vázquez A., Zayats T., Band G., Bellenguez C., Blackburn H., Blackwell J. M., Bramon E., Bumpstead S. J., Casas J. P., Corvin A., Craddock N., Deloukas P., Dronov S., Duncanson A., Edkins S., Freeman C., Gillman M., Gray E., Gwilliam R., Hammond N., Hunt S. E., Jankowski J., Jayakumar A., Langford C., Liddle J., Markus H. S., Mathew C. G., McCann O. T., McCarthy M. I., Palmer C. N., Peltonen L., Plomin R., Potter S. C., Rautanen A., Ravindrarajah R., Ricketts M., Samani N., Sawcer S. J., Strange A., Trembath R. C., Viswanathan A. C., Waller M., Weston P., Whittaker P., Widaa S., Wood N. W., McVean G., Reveille J. D., Wordsworth B. P., Brown M. A., Donnelly P., Australo-Anglo-American Spondyloarthritis Consortium (TASC), Wellcome Trust Case Control Consortium 2 (WTCCC2). (2011) Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat. Genet. 43, 761–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tsui F. W., Haroon N., Reveille J. D., Rahman P., Chiu B., Tsui H. W., and Inman R. D. (2010) Association of an ERAP1 ERAP2 haplotype with familial ankylosing spondylitis. Ann. Rheum. Dis. 69, 733–736 [DOI] [PubMed] [Google Scholar]
- 21. Robinson P. C., Costello M. E., Leo P., Bradbury L. A., Hollis K., Cortes A., Lee S., Joo K. B., Shim S. C., Weisman M., Ward M., Zhou X., Garchon H. J., Chiocchia G., Nossent J., Lie B. A., Førre Ø., Tuomilehto J., Laiho K., Jiang L., Liu Y., Wu X., Elewaut D., Burgos-Vargas R., Gensler L. S., Stebbings S., Haroon N., Mulero J., Fernandez-Sueiro J. L., Gonzalez-Gay M. A., Lopez-Larrea C., Bowness P., Gafney K., Gaston J. S., Gladman D. D., Rahman P., Maksymowych W. P., Xu H., van der Horst-Bruinsma I. E., Chou C. T., Valle-Oñate R., Romero-Sánchez M. C., Hansen I. M., Pimentel-Santos F. M., Inman R. D., Martin J., Breban M., Evans D., Reveille J. D., Kim T. H., Wordsworth B. P., and Brown M. A. (2015) ERAP2 is associated with ankylosing spondylitis in HLA-B27-positive and HLA-B27-negative patients. Ann. Rheum. Dis. 74, 1627–1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lopez de Castro J. A. (2018) How ERAP1 and ERAP2 Shape the peptidomes of disease-associated MHC-I proteins. Front Immunol. 9, 2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Andres A. M., Dennis M. Y., Kretzschmar W. W., Cannons J. L., Lee-Lin S. Q., Hurle B., Schwartzberg P. L., Williamson S. H., Bustamante C. D., Nielsen R., Clark A. G., and Green E. D. (2010) Balancing selection maintains a form of ERAP2 that undergoes nonsense-mediated decay and affects antigen presentation. PLoS. Genet. 6, e1001157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ombrello M. J., Kastner D. L., and Remmers E. F. (2015) Endoplasmic reticulum-associated amino-peptidase 1 and rheumatic disease: genetics. Curr. Opin. Rheumatol. 27, 349–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kuiper J. J. W., Setten J. V., Devall M., Cretu-Stancu M., Hiddingh S., Ophoff R. A., Missotten T. O. A. R., Velthoven M. V., Den Hollander A. I., Hoyng C. B., James E., Reeves E., Cordero-Coma M., Fonollosa A., Adan A., Martin J., Koeleman B. P. C., Boer J. H., Pulit S. L., Marquez A., and Radstake T. R. D. J. (2018) Functionally distinct ERAP1 and ERAP2 are a hallmark of HLA-A29-(Birdshot) uveitis. Hum. Mol. Genet. 27, 4333–4343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Martin-Esteban A., Guasp P., Barnea E., Admon A., and Lopez de Castro J. A. (2016) Functional iInteraction of the ankylosing spondylitis-associated endoplasmic reticulum aminopeptidase 2 with the HLA-B*27 peptidome in human cells. Arthritis Rheumatol. 68, 2466–2475 [DOI] [PubMed] [Google Scholar]
- 27. Martin-Esteban A., Sanz-Bravo A., Guasp P., Barnea E., Admon A., and Lopez de Castro J. A. (2017) Separate effects of the ankylosing spondylitis associated ERAP1 and ERAP2 aminopeptidases determine the influence of their combined phenotype on the HLA-B*27 peptidome. J. Autoimmun. 79, 28–38 [DOI] [PubMed] [Google Scholar]
- 28. Calvo V., Rojo S., López D., Galocha B., and Lopez de Castro J. A. (1990) Structure and diversity of HLA-B27-specific T cell epitopes. Analysis with site-directed mutants mimicking HLA-B27 subtype polymorphism. J. Immunol. 144, 4038–4045 [PubMed] [Google Scholar]
- 29. Garcia-Medel N., Sanz-Bravo A., Van N. D., Galocha B., Gomez-Molina P., Martin-Esteban A., Alvarez-Navarro C., and de Castro J. A. (2012) Functional interaction of the ankylosing spondylitis-associated endoplasmic reticulum aminopeptidase 1 polymorphism and HLA-B27 in vivo. Mol. Cell. Proteomics 11, 1416–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barnstable C. J., Bodmer W. F., Brown G., Galfre G., Milstein C., Williams A. F., and Ziegler A. (1978) Production of monoclonal antibodies to group A erythrocytes, HLA and other human cell surface antigens-new tools for genetic analysis. Cell 14, 9–20 [DOI] [PubMed] [Google Scholar]
- 31. Guasp P., Lorente E., Martin-Esteban A., Barnea E., Romania P., Fruci D., Kuiper J. J. W., Admon A., and Lopez de Castro J. A. (2019) Redundancy and complementarity between ERAP1 and ERAP2 revealed by their effects on the Behcet's disease-associated HLA-B*51 peptidome. Mol. Cell. Proteomics 18, 1491–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Infantes S., Lorente E., Barnea E., Beer I., Cragnolini J. J., García R., Lasala F., Jiménez M., Admon A., and López D. (2010) Multiple, non-conserved, internal viral ligands naturally presented by HLA-B27 in human respiratory syncytial virus-infected cells. Mol. Cell. Proteomics 9, 1533–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Johnstone C., Lorente E., Barriga A., Barnea E., Infantes S., Lemonnier F. A., David C. S., Admon A., and Lopez D. (2015) The viral transcription group determines the HLA class I cellular immune response against human respiratory syncytial virus. Mol. Cell. Proteomics 14, 893–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lorente E., Infantes S., Barnea E., Beer I., Garcia R., Lasala F., Jimenez M., Vilches C., Lemonnier F. A., Admon A., and López D. (2012) Multiple viral ligands naturally presented by different class I molecules in transporter antigen processing-deficient vaccinia virus-infected cells. J. Virol. 86, 527–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barnea E., Melamed K. D., Haimovich Y., Satumtira N., Dorris M. L., Nguyen M. T., Hammer R. E., Tran T. M., Colbert R. A., Taurog J. D., and Admon A. (2017) The human leukocyte antigen (HLA)-B27 peptidome in vivo, in spondyloarthritis-susceptible HLA-B27 transgenic rats and the effect of Erap1 deletion. Mol. Cell. Proteomics 16, 642–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p. p. b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 37. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., and Mann M. (2011) Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome. Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 38. Alvarez-Navarro C., Martin-Esteban A., Barnea E., Admon A., and Lopez de Castro J. A. (2015) Endoplasmic reticulum aminopeptidase 1 (ERAP1) polymorphism relevant to inflammatory disease shapes the peptidome of the birdshot chorioretinopathy-associated HLA-A*29:02 antigen. Mol. Cell. Proteomics 14, 1770–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Colmegna I., Cuchacovich R., and Espinoza L. R. (2004) HLA-B27-associated reactive arthritis: Pathogenetic and clinical considerations. Clin. Microbiol. Rev. 17, 348–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Madden D. R., Gorga J. C., Strominger J. L., and Wiley D. C. (1992) The three-dimensional structure of HLA-B27 at 2.1 A resolution suggests a general mechanism for tight peptide binding to MHC. Cell 70, 1035–1048 [DOI] [PubMed] [Google Scholar]
- 41. Lamas J. R., Paradela A., Roncal F., and Lopez de Castro J. A. (1999) Modulation at multiple anchor positions of the peptide specificity of HLA-B27 subtypes differentially associated with ankylosing spondylitis. Arthritis Rheum. 42, 1975–1985 [DOI] [PubMed] [Google Scholar]
- 42. Ben Dror L., Barnea E., Beer I., Mann M., and Admon A. (2010) The HLA-B*2705 peptidome. Arthritis Rheum. 62, 420–429 [DOI] [PubMed] [Google Scholar]
- 43. Alvarez I., Sesma L., Marcilla M., Ramos M., Marti M., Camafeita E., and de Castro J. A. (2001) Identification of novel HLA-B27 ligands derived from polymorphic regions of its own or other class I molecules based on direct generation by 20 S proteasome. J. Biol. Chem. 276, 32729–32737 [DOI] [PubMed] [Google Scholar]
- 44. Infantes S., Lorente E., Barnea E., Beer I., Barriga A., Lasala F., Jimenez M., Admon A., and Lopez D. (2013) Natural HLA-B*2705 protein ligands with glutamine as anchor motif: Implications for HLA-B27 association with spondyloarthropathy. J. Biol. Chem. 288, 10882–10889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fiorillo M. T., Meadows L., Damato M., Shabanowitz J., Appella E., and Sorrentino R. (1997) Susceptibility to ankylosing spondylitis correlates with the C-terminal residue of peptides presented by various HLA-B27 subtypes. Eur. J. Immunol. 27, 368–373 [DOI] [PubMed] [Google Scholar]
- 46. Yair-Sabag S., Tedeschi V., Vitulano C., Barnea E., Glaser F., Melamed K. D., Taurog J. D., Fiorillo M. T., Sorrentino R., and Admon A. (2018) The peptide repertoire of HLA-B27 may include ligands with lysine at P2 anchor position. Proteomics. 18, e1700249. [DOI] [PubMed] [Google Scholar]
- 47. Fruci D., Greco G., Vigneti E., Tanigaki N., Butler R. H., and Tosi R. (1994) The peptide-binding specificity of HLA-B27 subtype (B*2705) analyzed by the use of polyalanine model peptides. Hum. Immunol. 41, 34–38 [DOI] [PubMed] [Google Scholar]
- 48. Rocken M., Urban J. F., and Shevach E. M. (1992) Infection breaks T-cell tolerance. Nature 359, 79–82 [DOI] [PubMed] [Google Scholar]
- 49. Soulas P., Woods A., Jaulhac B., Knapp A. M., Pasquali J. L., Martin T., and Korganow A. S. (2005) Autoantigen, innate immunity, and T cells cooperate to break B cell tolerance during bacterial infection. J. Clin. Invest. 115, 2257–2267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Voehringer D., Blaser C., Grawitz A. B., Chisari F. V., Buerki K., and Pircher H. (2000) Break of T cell ignorance to a viral antigen in the liver induces hepatitis. J. Immunol. 165, 2415–2422 [DOI] [PubMed] [Google Scholar]
- 51. Colbert R. A., Tran T. M., and Layh-Schmitt G. (2014) HLA-B27 misfolding and ankylosing spondylitis. Mol. Immunol. 57, 44–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Allen R. L., O'Callaghan C. A., McMichael A. J., and Bowness P. (1999) HLA-B27 can form a novel b2-microglobulin-free heavy chain homodimer structure. J. Immunol. 162, 5045–5048 [PubMed] [Google Scholar]
- 53. Bowness P., Ridley A., Shaw J., Chan A. T., Wong-Baeza I., Fleming M., Cummings F., McMichael A., and Kollnberger S. (2011) Th17 cells expressing KIR3DL2+ and responsive to HLA-B27 homodimers are increased in ankylosing spondylitis. J. Immunol. 186, 2672–2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lim Kam Sian T. C. C., Indumathy S., Halim H., Greule A., Cryle M. J., Bowness P., Rossjohn J., Gras S., Purcell A. W., and Schittenhelm R. B. (2019) Allelic association with ankylosing spondylitis fails to correlate with human leukocyte antigen B27 homodimer formation. J. Biol. Chem. 294, 20185–20195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cortes A., et al. (2015) Major histocompatibility complex associations of ankylosing spondylitis are complex and involve further epistasis with ERAP1. Nat.Commun. 6, 7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stamogiannos A., Koumantou D., Papakyriakou A., and Stratikos E. (2015) Effects of polymorphic variation on the mechanism of endoplasmic reticulum aminopeptidase 1. Mol. Immunol. 67, 426–435 [DOI] [PubMed] [Google Scholar]
- 57. Reeves E., Edwards C. J., Elliott T., and James E. (2013) Naturally occurring ERAP1 haplotypes encode functionally distinct alleles with fine substrate specificity. J. Immunol. 191, 35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry data have been deposited to the MassIVE repository (http://massive.ucsd.edu) with the data set identifier MSV000084718.