

Abstract
Hepatocyte nuclear factor 4 alpha (HNF4α) is a transcription factor that plays a critical role in hepatocyte function, and HNF4α‐based reprogramming corrects terminal liver failure in rats with chronic liver disease. In the livers of patients with advanced cirrhosis, HNF4α RNA expression levels decrease as hepatic function deteriorates, and protein expression is found in the cytoplasm. These findings could explain impaired hepatic function in patients with degenerative liver disease. In this study, we analyzed HNF4α localization and the pathways involved in post‐translational modification of HNF4α in human hepatocytes from patients with decompensated liver function. RNA‐sequencing analysis revealed that AKT‐related pathways, specifically phospho‐AKT, is down‐regulated in cirrhotic hepatocytes from patients with terminal failure, in whom nuclear levels of HNF4α were significantly reduced, and cytoplasmic expression of HNF4α was increased. cMET was also significantly reduced in failing hepatocytes. Moreover, metabolic profiling showed a glycolytic phenotype in failing human hepatocytes. The contribution of cMET and phospho‐AKT to nuclear localization of HNF4α was confirmed using Spearman's rank correlation test and pathway analysis, and further correlated with hepatic dysfunction by principal component analysis. HNF4α acetylation, a posttranslational modification important for nuclear retention, was also significantly reduced in failing human hepatocytes when compared with normal controls. Conclusion: These results suggest that the alterations in the cMET‐AKT pathway directly correlate with HNF4α localization and level of hepatocyte dysfunction. This study suggests that manipulation of HNF4α and pathways involved in HNF4α posttranslational modification may restore hepatocyte function in patients with terminal liver failure.
Although drug induced liver injury (DILI) is a rare clinical event, it carries significant morbidity and mortality, leaving it as the leading cause of acute liver failure in the United States. It is one of the most challenging diagnoses encountered by gastroenterologists. DILI is also the most common single adverse event that has led to withdrawal of drugs from the marketplace, drug attrition and failure of implicated drugs to obtain FDA approval. The development of various drug injury networks have played a vital role in expanding our knowledge regarding drug, herbal and dietary supplement related liver injury. In this review, we discuss what defines liver injury, epidemiology of DILI, it's biochemical and pathologic patterns, and management.

Abbreviations
- AMPKα
adenosine monophosphate–activated protein kinase α
- CREB
cyclic adenosine monophosphate response element‐binding protein
- CYP
cytochrome P450
- EGFR
epidermal growth factor receptor
- GC
gas chromatography
- GC‐MS
gas chromatography–mass spectrometry
- HCC
hepatocellular carcinoma
- HNF4α
hepatocyte nuclear factor 4 alpha
- IPA
ingenuity pathway analysis
- NASH
nonalcoholic steatohepatitis
- PCA
principal component analysis
- PTM
posttranslational modification
- RNA‐Seq
RNA‐sequencing
- RXR
retinoid X receptor
- TCA
trichloroacetic acid
- Thr308
threonine 308
Terminal liver failure resulting from degenerative disease represented the twelfth leading cause of death in 2015.( 1 ) In the United States, the number of registered deaths coupled with chronic liver disease and cirrhosis in that year was 40,326.( 2 ) The most affected age range was 45‐64 year‐olds. and it was the fourth leading cause of death in that age group.( 2 ) The only definitive therapy for end‐stage liver failure is orthotopic liver transplantation, which—given the number of patients in need of liver transplants and the insufficient number of donor organs—makes it nearly untreatable for many patients.( 3 )
There are numerous causes of chronic liver disease, including chronic infection by hepatitis viruses, alcohol‐mediated cirrhosis, and nonalcoholic steatohepatitis (NASH),( 4 ) and each can produce hepatocellular failure.( 5 , 6 ) The mechanisms responsible for deterioration of hepatocyte function and ultimately hepatic failure in man are poorly understood. Chronic hepatic damage produces oxidative stress( 7 ) and endoplasmic stress,( 8 ) which can induce cell death( 8 , 9 , 10 ) and reduce the proliferative capacity of the hepatocytes.( 11 )
In previously published studies, we found that liver‐enriched transcription factors are stably down‐regulated in hepatocytes from rats with end‐stage cirrhosis,( 12 ) and that forced re‐expression of one of them, hepatocyte nuclear factor 4 alpha (HNF4α), reprograms dysfunctional hepatocytes to regain function, both in culture and in vivo.( 12 ) We have also shown in a large cohort of patients with advanced liver disease that the level of HNF4α messenger RNA (mRNA) expression in the diseased liver correlated with extent of hepatic dysfunction (Childs‐Pugh classification) and that HNF4α expression was not localized in the nucleus, as was the case in our rat studies.( 5 )
HNF4α is a transcription factor that plays a critical role in liver organogenesis and hepatocyte function in the mature liver,( 12 , 13 ) and its expression and function are regulated at multiple levels.( 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 ) It must be expressed in the nucleus to function properly; therefore, we analyzed the signaling pathways involved in nuclear localization of HNF4α in hepatocytes isolated from explanted human livers with decompensated function. In this analysis involving a significantly smaller cohort of patients, we were able to demonstrate that decompensated livers have significantly lower expression of nuclear HNF4α and higher expression of cytoplasmic HNF4α when compared with normal controls and correlated with low levels of cMET and phosphorylated AKT (protein kinase B). Moreover, we found that hepatic dysfunction correlated directly with a reduction in the nuclear acetylation of HNF4α. Thus, posttranslational modifications are important for HNF4α localization in the nucleus. Transcriptional activation may be responsible for deterioration of hepatocyte functional in human cirrhotic livers with terminal failure.
Materials and Methods
Human Samples and Hepatocyte Isolation
De‐identified normal human liver tissue and/or cells were obtained through the Liver Tissue Cell Distribution System (Pittsburgh, PA) after obtaining written, informed consent by a protocol approved by the Human Research Review Committee of the University of Pittsburgh, which was funded by National Institutes of Health Contract No. HSN276201200017C. Adult human liver tissue and/or cells were also obtained from the Fox Laboratory at the Children's Hospital of UPMC, after obtaining written informed consent by an expedited protocol approved by the Human Research Review Committee and the Institutional Review Board (IRB#: PRO12090466) of the University of Pittsburgh (Table 1). Hepatocytes were isolated using a three‐step collagenase digestion technique as previously described.( 24 ) Cell viability was assessed after isolation as previously described using trypan blue exclusion, and only cell preparations with viability > 80% were used for the analysis.
Table 1.
Clinical Parameters of the Patients in This Study
| Etiology/Code, n | Age (years) | Sex (M, F) | Total Bilirubin (mg/dL) | Albumin (9/dL) | MELD | Child‐Pugh |
|---|---|---|---|---|---|---|
| Healthy liver, 4 | 64 ± 13 | 2, 2 | — | — | — | — |
| NASH, 5 | 60 ± 10 | 2, 3 | 1.76 ± 0.6 | 2.9 ± 0.1 | 12.4 ± 6.1 | 9.4 ± 0.9 |
| NASH 1 | 71 | 0, 1 | 1.7 | 3.0 | 8 | 9 (B) |
| NASH 2 | 68 | 0, 1 | 1.7 | 2.9 | 8 | 9 (B) |
| NASH 3 | 53 | 1, 0 | 1.1 | 2.8 | 8 | 9 (B) |
| NASH 4 | 59 | 1, 0 | 1.6 | 2.8 | 21 | 11 (C) |
| NASH 5 | 48 | 0, 1 | 2.7 | 3.0 | 17 | 9 (B) |
| Alcohol, 2 | 55 ± 5 | 3, 0 | 2.03 ± 1.0 | 2.53 ± 0.6 | 14.0 ± 2.6 | 10.0 ± 1.0 |
| Alcohol 1 | 52 | 1, 0 | 1.5 | 1.9 | 16 | 11 (C) |
| Alcohol 2 | 61 | 1, 0 | 1.4 | 3.2 | 11 | 9 (B) |
Abbreviations: F, female; M, male; MELD, Model for End‐Stage Liver Disease.
In Silico HNF4α Posttranslational Modifications Analysis
To identify the posttranslational modifications (PTMs) that modulate HNF4α cellular localization, an in silico analysis was performed through computational searches in databases and publications (Supporting Fig. S1A). The process was divided into three phases: identification, screening, and selection. Initially, 51 PTMs were identified. Next, during the screening phase, 23 PTMs were selected by the application of two elimination criteria (Supporting Fig. S1B). Two phosphorylation and one acetylation modifications were identified in the selection phase as the most plausible PTMs related to HNF4α localization able to be evaluated.
Stable Isotope Analysis Using Gas Chromatography–Mass Spectrometry
One million human hepatocytes were cultured with Dulbecco's modified Eagle's medium F12 in the presence of 13‐C6‐labeled glucose and glutamine isotope tracers for 96 hours (Thermo Fisher Scientific, San Jose, CA). The medium was removed and cells were washed with ice‐cold phosphate‐buffered saline solution. Next, cells were quenched with 400 μL methanol and 400 μL water containing 1 μL norvaline, scraped, washed with 800 μL ice‐cold chloroform, vortexed at 4°C for 30 minutes, and centrifuged at 7,300 rpm for 10 minutes at 4°C. The upper aqueous phase was collected for metabolite analysis. Metabolite extracts were centrifuged at 14,000g for 10 minutes to separate the polar phase, protein interphase, and chloroform phase. The water/methanol phase–containing polar metabolites were transferred to fresh microcentrifuge tubes and dried in a SpeedVac and stored at −80°C until gas chromatography–mass spectrometry (GC‐MS) analysis. Then, 30 µL of methoxyamine hydrochloride (Thermo Scientific) was added to dried samples and incubated at 30°C for 2 hours with intermittent vortexing. A total of 45 µL of MBTSTFA + 1% tert‐butyldimethylchlorosilane was added to the samples and incubated at 55°C for 1 hour. Derivatized samples were transferred to gas chromatography (GC) vials with glass inserts and added to the GC‐MS autosampler. GC‐MS analysis was performed using an Agilent 7890 GC (Santa Clara, CA) equipped with a 30‐m HP‐5MSUI capillary column connected to an Agilent 5977B mass spectrometer. For polar metabolites, the following heating cycle was used for the GC oven: 100°C for 3 minutes, followed by a ramp of 5°C/minute to 300°C and held at 300°C for a total run time of 48 minutes. Data were acquired in scan mode. The relative abundance of metabolites was calculated from the integrated signal of all potentially labeled ions for each metabolite fragment. Mass isotopologue distributions were corrected for natural abundance using IsoCorrectoR prior to analysis with the model. Metabolite levels were normalized to internal standard Norvaline's signal. Fractional enrichment calculation represents the fractional contribution of 13C from a substrate to intermediate metabolite. It is calculated as follows:
where NC is the number of carbons that can be labeled as 13C, and xi is the fraction of (M + i)th isotopologue.
Immunohistochemistry and HNF4α Quantification
Paraffin‐embedded liver tissue was deparaffinized with xylenes and dehydrated with ethanol. Antigen unmasking was performed by boiling in citrate buffer at pH = 6.0. The slides were then incubated in 3% hydrogen peroxide, blocked with normal animal serum, and subsequently left incubating overnight at 4°C with primary antibodies. The primary antibodies used are listed in Supporting Table S1. Tissue sections were then incubated with biotinylated secondary antibody corresponding to the animal species of the primary antibody (BA‐1000; Vector Laboratories, Burlingame, CA) and exposed to 3,3′‐diaminobenzidine (SK‐4105; Vector Laboratories) to visualize the peroxidase activity. Counterstaining was performed with Richard‐Allan Scientific Signature Series Hematoxylin (Thermo Fisher Scientific). For quantification, immunoreactivities of nuclear and cytoplasmic HNF4α were independently graded by 2 liver pathologists, with 1,000 hepatocytes in three high‐power fields being counted per sample. Normal livers (n = 4), Child‐Pugh B (n = 5), and Child‐Pugh C (n = 2) were included for these analyses. The Child‐Pugh B and C livers were grouped as cirrhotic human liver, and the results are expressed as percentage over the total number of cells counted.
Protein Extraction and Western Blotting
To perform protein‐expression analysis, the isolated hepatocytes were divided in two fractions: One fraction was used for total protein extraction, according to standard procedures previously described,( 25 ) and the other fraction was used for nuclear protein isolation. For nuclear protein isolation, between 1 × 107 and 5 × 107 isolated hepatocytes per patient were washed and harvested in 40 mmol/L Tris (pH 7.6), 14 mmol/L NaCl, and 1 mmol/L ethylene diamine tetraacetic acid (EDTA), then centrifuged (5 minutes, 100g). Cell pellets were suspended in 2 mL of hypotonic buffer (10 mmol/L 4‐[2‐hydroxyethyl]‐1‐piperazine ethanesulfonic acid [HEPES; pH 7.9], 10 mmol/L NaH2PO4, 1.5 mmol/L MgCl2, 1 mmol/L dithiothreitol [DTT], 0.5 mmol/L spermidine, and 1 mol/L NaF with protease and phosphatase inhibitor cocktails [Sigma, St. Louis, MO]). Following 10 minutes of incubation on ice, samples were homogenized in a Dounce homogenizer and then centrifuged (5 minutes, 800g). Cell lysis was monitored with trypan blue stain. Supernatants were saved as cytoplasmic extracts. The nuclei pellets were washed two additional times in the same buffer.
Nuclear proteins were extracted in 50‐100 μL of hypertonic buffer (30 mmol/L HEPES [pH 7.9], 25% glycerol, 450 mmol/L NaCl, 12 mmol/L MgCl2, 1 mmol/L DTT, and 0.1 mmol/L EDTA with protease and phosphatase inhibitor cocktails [Sigma]) for 45 minutes at 4°C with continuous agitation. Extracts were centrifuged at 30,000g, and the supernatants were collected and dialyzed for 2 hours against the same solution but containing 150 mmol/L NaCl. Protein concentration was determined by the bicinchoninic acid assay (Sigma).
Western blot analysis was performed according to standard procedures.( 26 ) The intensity of each protein band was quantitated using National Institutes of Health Image J software.( 27 ) The primary antibodies and dilutions are listed in Supporting Table S1.
RNA‐Sequencing and Analysis
Whole‐genome strand‐specific RNA‐sequencing (RNA‐seq) was used to profile RNA expression levels from human isolated primary hepatocytes (Supporting Table S2). RNA‐Seq libraries were prepared as described previously( 28 ) and in the literature.( 29 ) RNA was extracted from human hepatocytes using TRIzol, followed by column purification (Zymo RNA clean and concentrator column) according to the manufacturers' instructions. Total RNA was depleted of ribosomal RNA using pooled antisense oligo hybridization and depletion through RNaseH digestion as previously described.( 30 , 31 ) Following purification over a Zymo RNA clean and concentrator column, first‐strand complementary DNA (cDNA) was synthesized. Subsequently, second ‐strand cDNA was synthesized, purified, and fragmented. RNA‐seq libraries were prepared using Illumina technology. Briefly, end repair, A‐tailing, and barcoded adapter ligation were followed by polymerase chain reaction amplification and size selection. The integrity of the libraries was confirmed by quBit quantification, fragment analyzer size distribution assessment, and Sanger sequencing of about 10 fragments from each library. Libraries were sequenced using paired‐end Illumina sequencing.
Paired‐end reads were aligned to hg38 using QIAGEN's CLC Genomics workbench and assessed as transcript per million. To sort the data, K‐means clustering was performed using Cluster 3.0,( 32 ) and heatmaps were generated using Java Treeview.( 33 ) The default settings for mismatch,( 2 ) insertion cost,( 3 ) were deletion cost( 3 ) were used. Ingenuity pathway analysis (IPA) was used to identify differentially expressed genes, predict downstream effects, and identify targets (QIAGEN Bioinformatics; http://www.qiagen.com/ingenuity). Regulatory effects analysis within IPA was used to identify the relationships between upstream regulators and biological functions. The default setting was used in the analysis (i.e., the upstream regulators were limited to genes, RNA, and proteins). The RNA‐seq data are available at Gene Expression Omnibus (accession number http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE134422).
AKT Inhibition in Normal Human Hepatocytes
Normal human hepatocytes (1 million cells/well) were cultured on collagen‐coated wells. Cells were culture for 6 hours in the absence of growth factors or serum. Cells then were treated with 5 µM of MK‐2206 (Cayman Chemical, Ann Arbor, Michigan), an AKT inhibitor, for 24 hours. Total, cytoplasmatic, and nuclear protein were extracted for western blotting as described previously.
Statistical Analysis
Data were expressed as mean ± SD. Results from western blot analysis for two statistical groups were evaluated by Mann‐Whitney nonparametric test and for three statistical groups by Kruskal–Wallis nonparametric test. The comparisons among groups were performed by Dunn multiple comparisons test. The association among the analyzed proteins was evaluated using the Spearman's rank correlation test. Linear regression was used to describe the relation between protein expression and clinical status, measured as Child‐Pugh score. Statistics were performed using Prism 4.0 (GraphPad Software Inc., San Diego, CA). Differences were considered significant when P < 0.05.
To identify the direct dependence between the proteins analyzed by western blot, the path analysis (structural equation model) was used. The objective of the path analysis model is to explain a possible causal association between the observed correlations among a dependent variable and multiple independent variables. The path model was tested and modified by adding and removing a path based on the research framework and the results of regression weights and model fit. The results are plotted as diagrams that show the direct and indirect effects of the variables on the study system. The degree of correlation and the linear relation between variables is determined by a P value less than 0.05 and an arbitrary coefficient that shows the level of importance (larger number represent a larger relation). The path analysis was performed using InfoStat version 2013 (Grupo InfoStat, FCA, Universidad Nacional de Córdoba, Córdoba, Argentina). Unsupervised multivariate principal component analysis (PCA) was applied to western blot data to reveal the group of proteins that distinguish among the sample's clinical status. Scatter plots of the principal components that explain most of the variance were drawn. The statistical software JMP version 14 (SAS Institute, Cary, NC) was used for PCA analysis.
Results
HNF4α Nuclear Localization Is Decreased in Human Livers With End‐Stage Liver Failure, While Cytoplasmic Localization Is Increased
HNF4α functions as a transcription factor, and nuclear localization is required for activity.( 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 22 , 23 , 25 , 34 , 35 ) Therefore, we performed immunohistochemistry and western blot on hepatocytes from diseased liver specimens to determine the location of HNF4α and to correlate expression with hepatic decompensation. About 78% of hepatocytes from livers with terminal liver failure showed only cytoplasm expression or cytoplasm and weak nuclear expression of HNF4α, whereas normal human livers displayed 75% of hepatocytes with strong nuclear localization of HNF4α (Fig. 1A,B). Total HNF4α protein expression from isolated hepatocytes, as assessed by western blot, did not show any statistical difference between end‐stage livers and normal controls (P = 0.166; Fig. 1C and Supporting Fig. S1A). This result was not surprising, as the ability to discern a difference in HNF4α expression in livers from patients with degenerative disease and controls based on degree of functional decompensation required the study of a large cohort of patients.( 5 ) However, in this study, a statistically significant difference was observed based on HNF4α location. HNF4α was detected at high levels in the cytoplasm (P = 0.023; Fig. 1D and Supporting Fig. S1B) and at low levels in the nucleus (P = 0.023; Fig. 1E and Supporting Fig. S1C) of hepatocytes isolated from functionally decompensated livers when compared with hepatocytes isolated from normal controls.
Fig. 1.

HNF4α location in hepatocytes from normal and cirrhotic livers. (A) Immunohistochemical staining micrographs of HNF4α show cytoplasmic and nuclear decrease of HNF4α expression in NASH and alcohol‐mediated Laennec's cirrhotic decompensated human livers (n = 6) compared with healthy human livers (n = 3). (B) Quantification of nuclear or/and cytoplasmic HNF4α expression in immunohistochemical staining micrographs. (C) Representative photographs of HNF4α immunofluorescence of isolated human hepatocytes from NASH decompensated liver and normal human hepatocytes. Western blot analysis and quantification of HNF4α normalized to B‐actin in hepatocytes isolated from functionally decompensated livers (NASH and alcohol‐mediated Laennec's cirrhosis) (n = 6) and hepatocytes isolated from healthy liver controls (n = 2) (P = 0.166). (D) Western blot analysis and quantification of cytoplasm HNF4α: healthy human hepatocytes versus decompensated human hepatocytes, normalized to B‐actin (P = 0.023). (E) Western blot analysis and quantification of nuclear HNF4α: healthy human hepatocytes versus decompensated human hepatocytes, normalized to nuclear marker histone deacetylase 1 (HDAC1; P = 0.023). Bar graphs from (C)‐(E) are plotted as the mean ± SD; P < 0.05. Abbreviation: DAPI, 4′,6‐diamidino‐2‐phenylindole.
Because the function and stability of HNF4α are regulated by a number of posttranslational modifiers,( 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 ) and because its nuclear localization is critical to its activity, we performed an in silico analysis to evaluate which modifiers regulate HNF4α localization. We found that adenosine monophosphate–activated protein kinase α (AMPKα) activation controls HNF4α transcription.( 16 ) In addition, acetylation of HNF4α, which may be mediated by the AKT pathway, stabilizes the molecule, favoring its retention in the nucleus (Supporting Fig. S2A,B).( 19 )
HNF4α Is the Major Regulator of Human Hepatocyte Function in Advanced Liver Disease
Next, we examined gene‐expression differences between human hepatocytes from normal controls (n = 4) and those recovered from patients with cirrhosis and terminal liver failure (n = 4) (Child‐Pugh C), limiting our study to patients with NASH and alcohol‐mediated Laennec's cirrhosis. Hierarchical clustering of RNA‐seq data revealed three major dynamic patterns associated with cirrhosis and liver dysfunction (Fig. 2A), as shown in the heatmap of a k‐means clustering (log2 fold change over controls; k = 3). Cluster 1 (3,478 genes) and cluster 3 (1,669 genes) represented genes that were moderately to highly up‐regulated in hepatocytes from patients with terminal liver failure relative to control human hepatocytes. Most genes in these clusters were related to autophagy and apoptotic signaling (Fig. 2A).
Fig. 2.
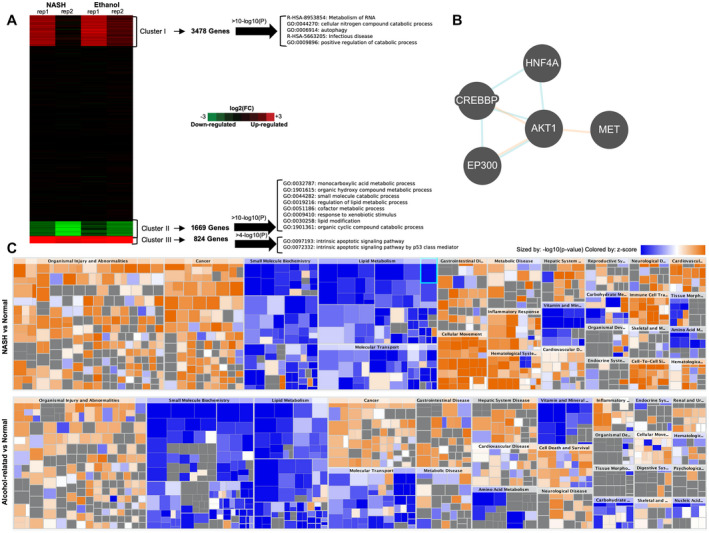
RNA‐seq analysis from human hepatocytes recovered from healthy liver specimens, NASH, and alcohol‐mediated Laennec's cirrhotic explanted livers. (A) K‐means clustering of log2 fold change differentially expressed genes from hepatocytes isolated from livers with NASH (n = 2) or alcohol‐mediated Laennec's cirrhosis (n = 2) and terminal liver failure relative to healthy hepatocytes (n = 3) demonstrated significant gene‐expression changes. (B) The RNA‐seq results suggest that the cirrhotic hepatocytes are expressing genes that simultaneously drive both proliferation and apoptosis, with an effected metabolism (clusters 1, 2, and 3), under the control of upstream regulators (cluster 2). This regulatory process involves the actions of HNF4α and AKT1. (C) Heatmaps for the top molecular and cellular functions altered in NASH and alcohol‐mediated Laennec's cirrhotic hepatocytes included down‐regulation of functions related to small molecules biochemistry, lipid metabolism and molecular transports, and up‐regulation of molecular functions related to cancer and organismal injury and abnormalities.
Cluster 2, however, consisted of 1,669 genes that were significantly down‐regulated in end‐stage hepatocytes, and included genes encoding the serine‐threonine protein kinase (AKT1), cytochrome P450s (cytochrome P450 [CYP]c8, CYP2c9, CYP2e1, and CYP3A4), and hepatocyte nuclear factors (HNF4α and forkhead box a1 [FOXa1]). The top pathways represented in this cluster included farnesoid X receptor/retinoid X receptor (RXR) and liver X receptor/RXR activation, mitochondrial dysfunction, oxidative phosphorylation, and inhibition of RXR function (Supporting Figures S3 and S4). A core pathway analysis using IPA on these down‐regulated genes showed that HNF4α was a central upstream regulator (Fig. 2B), and the heatmap confirmed many similarities in the gene‐expression profile of hepatocytes from patients with NASH and alcohol‐mediated Laennec's cirrhosis (Fig. 2C). These results were nearly identical to the gene‐expression profile that we previously described of rat hepatocytes that were recovered from cirrhotic livers with terminal failure.( 36 )
Human Hepatocytes From Livers With End‐Stage Liver Failure Rely on Pyruvate Dehydrogenase and Reductive Carboxylation of Glutamine to Maintain Trichloroacetic Acid Cycle Activity
Previously, we postulated that rat hepatocytes undergo an adaptive metabolic shift from generating energy predominantly from oxidative phosphorylation to glycolysis, allowing maintenance of energy homeostasis during stages of liver injury.( 36 , 37 ) Because HNF4α is a positive transcriptional regulator for controlling energy metabolism,( 38 , 39 , 40 ) we investigate functionally the metabolite sources of energy in the trichloroacetic acid (TCA) cycle in human hepatocytes from livers with end‐stage liver failure by performing [U‐13C6]glucose and glutamine tracing analysis. Central carbon metabolism of cirrhotic hepatocytes relies on distinct carbon sources compared with healthy hepatocytes. Glucose contribution to glycolytic intermediates and alanine are markedly increased in cirrhotic hepatocytes compared with normal hepatocytes (Fig. 3, P < 10−4, two‐tailed t test). Although glucose contribution to other amino acids and TCA cycle intermediates are similar across the two groups, cirrhotic failing hepatocytes rely on increased pyruvate dehydrogenase flux as an anaplerotic source for the TCA cycle, due to a commensurate decrease in pyruvate carboxylase flux (Supporting Fig. S5). On the other hand, glutamine contribution to glutamate and α‐ketoglutarate decreased in cirrhotic hepatocytes compared with normal human hepatocytes (Fig. 3, P < 0.01, two‐tailed t test), but the 13‐carbon enrichment of downstream TCA metabolites is similar across the two groups. This indicates an effective increase in the net glutamine contribution to downstream TCA metabolites. Importantly, the reductive carboxylation of glutamine is increased in cirrhotic hepatocytes compared with normal human hepatocytes, (Supporting Fig. S5B). Interestingly, the malic enzyme flux is decreased in cirrhotic hepatocytes compared with healthy controls (Supporting Fig. S5C). Together, this indicates the increased use of glutamine in cirrhotic hepatocytes in the TCA cycle, which indicates dysfunction of oxidative TCA. These results are in accordance to the metabolic profiling that we previously described of rat hepatocytes that were recovered from cirrhotic livers with terminal failure.( 36 , 37 )
Fig. 3.
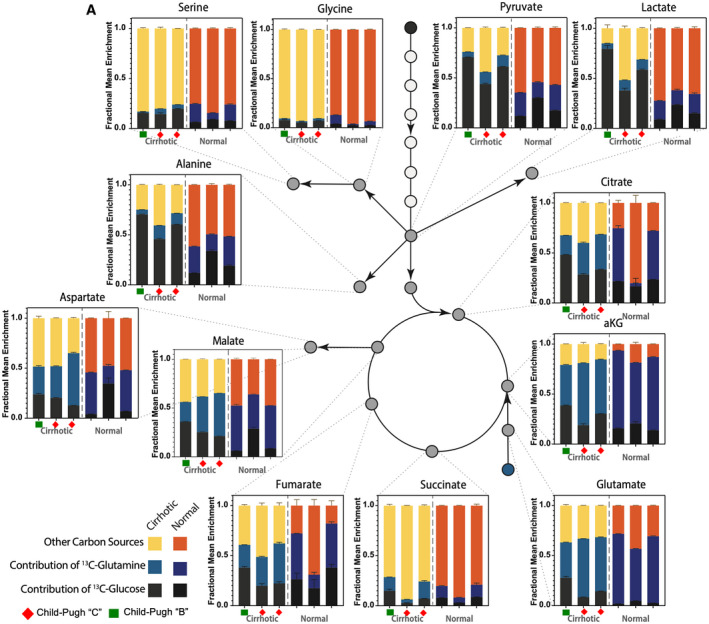
Mapping of carbon atom transitions using uniformly labeled 13‐C6‐glucose and glutamine. (A) Estimation of carbon contribution from various substrates in human cirrhotic (n = 3) and normal hepatocytes (n = 3). Fractional mean enrichment of central carbon metabolites and amino acids are estimated by measuring 13C enrichment of these metabolites in hepatocytes cultured with U[13C6]glucose or U[13C6]glutamine. Fraction of metabolites not labeled by either U[13C6]glucose or U[13C6]glutamine are assumed to be derived from other carbon sources.
cMET and AKT Phosphorylation Correlate With HNF4α Nuclear Localization in Human Hepatocytes From Patients With End‐Stage Liver Failure
Because epidermal growth factor receptor (EGFR) and cMET have been shown to regulate the AMPK and AKT pathways( 41 , 42 , 43 ) that were identified in our in silico and RNA‐seq analysis as important modulators of HNF4α, we performed antibody‐based assays for these molecules in our liver specimens. cMET expression in decompensated liver specimens (Fig. 4) was markedly reduced when measured by both immunohistochemistry and western blot (P = 0.023; Fig. 5A,B) when compared with isolated control human hepatocytes. Interestingly, there was no significant difference in the EGFR expression between normal and diseased liver specimens (Figs. 4 and 5A,B). However, the active form of EGFR, phospho‐EGFR (Y1086), was highly expressed in hepatocytes derived from diseased livers of patients with decompensated disease when compared with normal human hepatocytes (Supporting Fig. S6). In addition, as continuous cycles of cell death and hepatocyte replication are hallmarks of cirrhosis,( 1 ) we corroborated this observation in hepatocytes from patients with terminal liver failure, and demonstrated expression of replicative (phospho‐H3[Ser10]) and cell death (active caspase 3) markers in end‐stage hepatocytes using immunohistochemistry and western blot (Supporting Fig. S7A,B).
Fig. 4.
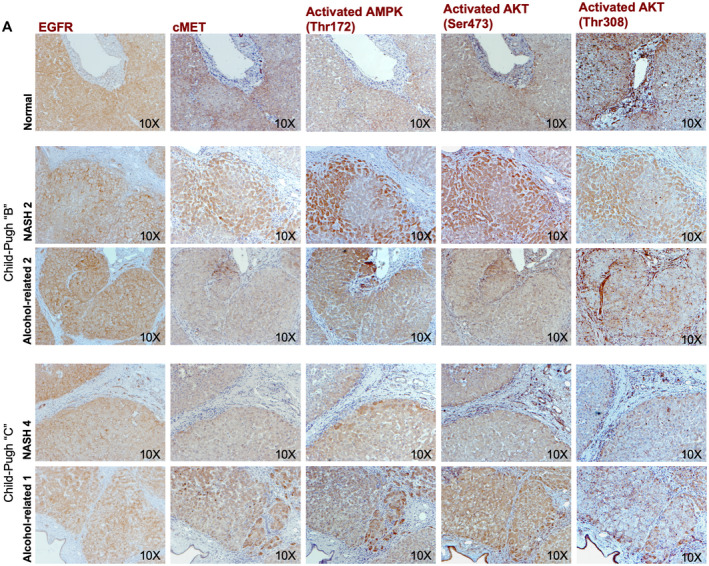
Histological expression of molecular candidates that regulates HNF4α PTMs. (A) Representative images of serial immunohistochemistry analysis of EGFR, cMET, activated AMPK(Thr172), activated AKT(Ser473), and activated AKT(Thr308) in decompensated NASH human livers (n = 2), and alcohol‐mediated Laennec's cirrhotic livers (n = 2) compared with healthy human livers (n = 2). EGFR expression did not demonstrate any difference in its expression pattern among the groups, whereas cMET and activated AKT(Thr308) expression levels were decreased in all decompensated human livers.
Fig. 5.
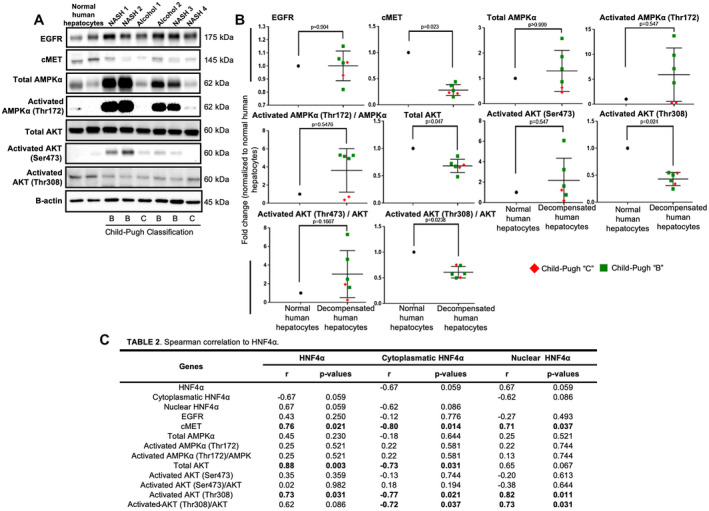
Protein expression and Spearman's rank correlation test of HNF4α PTMs. (A,B) Western blot analysis and quantification of EGFR (P = 0.904), cMET (P = 0.023), total AMPKα (P > 0.999), activated AMPKα(Thr172) (P = 0.547), total AKT (P = 0.047), activated AKT(Ser473) (P = 0.547), activated AKT(Thr308) (P = 0.024), activated AMPKα(Thr172)/AMPKα ratio (P = 5476), activated AKT(Ser473)/AKT ratio (P = 0.1667), and activated AKT(Thr308)/total AKT ratio (P = 0.0238) in decompensated NASH (n = 4) and alcohol‐mediated Laennec's cirrhotic freshly isolated hepatocytes (n = 2) and healthy control hepatocytes (n = 2). (C) Spearman's rank correlation test of the western blot analysis and quantification showing significant correlation of nuclear HNF4α with cMET (r = 0.71; P = 0.037), total AKT (r = 0.71; P = 0.037), activated AKT(Thr308) (r = 0.82; P = 0.011), and activated AKT(Thr308)/total AKT ratio (r = 0.73; P = 0.031). Moreover, cytoplasmic HNF4α showed a significant correlation with cMET (r = −0.80; P = 0.014), total AKT (r = −0.73; P = 0.031), activated AKT(Thr308) (r = −0.77; P = 0.021, and activated AKT(Thr308)/total AKT ratio (r = −0.72; P = 0.037). Dot plots are shown as the mean ± SD; P < 0.05.
Because cMET can control AMPKα and AKT, and we found that cMET significantly down‐regulated in hepatocytes from functionally decompensated livers, we analyzed the activation processes for AMPKα and AKT. Total AMPKα, activated AMPKα(Thr172), and its ratio did not differ statistically between hepatocytes with decompensated function and healthy human hepatocytes (Figs. 4 and 5A,B). However, total AKT, activated AKT(threonine 308 [Thr308]), and its ratio decreased significantly in liver specimens and isolated hepatocytes from patients with decompensated hepatic function (Figs. 4 and 5A,B). Another AKT phosphorylation site (Ser473) was unchanged in liver specimens or isolated hepatocytes from healthy controls or decompensated specimens (Fig. 4 and Fig. 5A,B).
To further analyze the relationship among HNF4α, its nuclear localization, and PTM, we performed a Spearman's rank correlation test. cMET expression showed a positive and statistically significant correlation with total HNF4α (r = 0.76; P = 0.021; Fig. 5C) and nuclear HNF4α (r = 0.71; P = 0.037; Fig. 5C). Activated AKT(Thr308) also showed a positive and statistically significant correlation with total HNF4α (r = 0.73; P = 0.031; Fig. 5C) and nuclear HNF4α (r = 0.82; P = 0.011; Fig. 5C), whereas cytoplasmic HNF4α correlated negatively with cMET (r = −0.80; P = 0.014; Fig. 5C) and activated‐AKT(Thr308) (r = −0.77; P = 0.021; Fig. 5C). In addition, the ratio of activated‐AKT(Thr308)/total AKT correlated positively with cMET (r = 0.80; P = 0.014; Supporting Fig. S6A) and total AKT (r = 0.71; P = 0.037; Supporting Fig. S6A). Thus, reduced cMET was associated with reduced activation of the AKT pathway, reduced HNF4α in the nucleus, and more expression of HNF4α in the cytoplasm.
Nuclear Localization of HNF4α Is Affected by the cMET/AKT Axis and Correlates With Extent of Liver Dysfunction
As seen in Fig. 6A, pathway analysis revealed a significant causal relationship between cMET expression and nuclear HNF4α expression (0.56; P = 0.004) and a direct relationship between levels of nuclear HNF4α and the activated AKT(Thr308)/total AKT ratio (0.05; P = 0.006). Modeling also demonstrated that total HNF4α expression levels contribute to nuclear localization (0.60; P = 0.042). However, cMET expression was negatively associated with total HNF4α expression (−0.37; P = 0.024) (Fig. 6A), indicating that cMET expression does not directly affect total HNF4α expression but only its nuclear localization. To assess whether the levels of nuclear expression correlate with extent of liver dysfunction (Child‐Pugh score), we performed a linear regression analysis and found that nuclear HNF4α expression levels have a significant inverse relationship with the Child‐Pugh score (R2 = 0.80; P = 0.007) (Fig. 6B). Together, these pathway and linear regression statistical analyses of protein expression suggest that HNF4α localization is associated with hepatic disease progression and that cMET expression and AKT phosphorylation may play a central role in maintaining hepatocyte HNF4α nuclear localization and function.
Fig. 6.
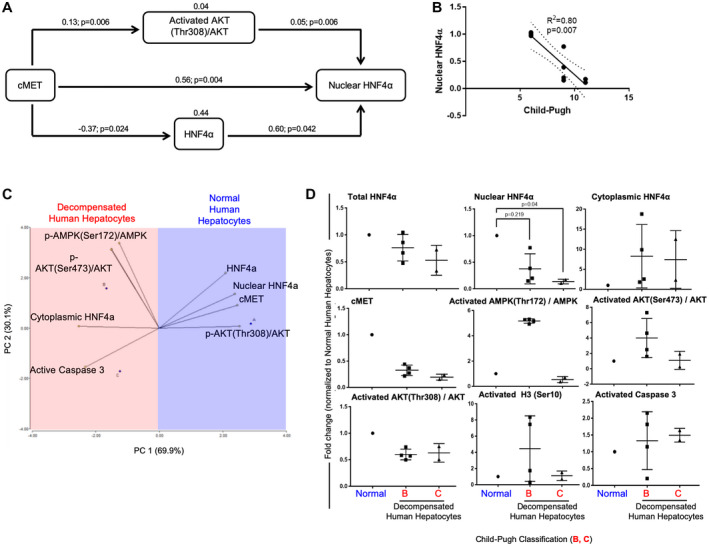
Relationship of PTMs and HNF4α cellular localization. (A) Schematic diagramming of the path analysis performed with protein expression revealed a significant direct relationship between nuclear HNF4α and cMET (0.56; P = 0.004), activated AKT(Thr308)/total AKT ratio (0.05; P = 0.006), and total HNF4α levels (0.60; P = 0.042). Path analysis also shows a negative significant relationship between cMET and total HNF4α (−0.37; P = 0.024). (B) Linear regression graph analysis shows the significant relationship of nuclear HNF4α expression and the degree of liver dysfunction (Child‐Pugh score) (R2 = 0.80, P = 0.007). (C,D) PCA and dot plots of the protein profile showing correlations with hepatocyte functional characteristics. cMET, activated AKT(Thr308)/total AKT ratio, and total and nuclear HNF4α expression positively explain the characteristics of healthy human hepatocytes (n = 2), whereas cytoplasmic HNF4α, active caspase 3, activated AKT(Ser473)/total AKT ratio, and p‐AMPK(Ser172)/AMPK explain the characteristics of decompensated human hepatocytes from NASH (n = 4) and alcohol‐mediated Laennec's cirrhotic livers (n = 2). (D) Dot plots are shown as the mean ± SD; P < 0.05.
Next, PCA was performed to validate our previous findings on HNF4α posttranslation modifier‐related molecules, and to delineate whether a pattern of molecules exist that correlates with hepatic function in end‐stage hepatocytes (Child‐Pugh score) (Fig. 6C, D). As shown in Fig. 6C, principal component 1 (PC1; 69.9%) and PC2 (30.1%) discriminate 100% of the variability in level of hepatic function (Child‐Pugh score) in the isolated hepatocytes studied. The vectors that characterized normal human hepatocytes were cMET and activated AKT(Thr308)/total AKT ratio, total HNF4α, and nuclear HNF4α, whereas the negative characteristics, which characterized failing human hepatocytes, were cytoplasmic HNF4α and active caspase 3 expression (Fig. 6C,D). Together, this statistical analysis corroborates our molecular profiling of human hepatocytes with terminal liver failure and establishes a causal connection among the expression of cMET, activated AKT(Thr308), and total and nuclear HNF4α.
Retention of HNF4α in the Nucleus Is Reduced in Patients With End‐Stage Liver Failure Through Decreased Acetylation
One of the targets of AKT is activation of the cyclic adenosine monophosphate response element‐binding protein (CREB).( 44 ) It is well known that the CREB‐binding protein has an intrinsic acetylation activity on nucleosomal histones, which increase the access of transcription factors to nucleosomal DNA and therefore activate transcription and retention of transcription factors in nuclei. Thus, this axis could be related to HNF4α nuclear retention.( 19 ) As genome‐wide transcriptome analysis and our in silico analysis suggested that the cMET/AKT kinase axis pathway could control HNF4α localization and stability through the activation of the CREB‐binding protein,( 39 ) we measured nuclear expression of acetylated HNF4α (Fig. 7A‐C) and found that HNF4α acetylation in the nucleus and nuclear acetylated HNF4α/total HNF4α ratio were significantly reduced in hepatocytes from patients with terminal liver failure when compared with healthy controls (P = 0.024 and P = 0.0001; Fig. 7B). In addition, to confirm that acetylation of HNF4α relates to the extent of liver dysfunction (Child‐Pugh score), we performed a linear regression analysis and found that a decrease in acetylated HNF4α correlates directly and significantly with liver dysfunction (R2 = 0.71; P = 0.004; Fig. 7C). As a proof of principle, we conducted preliminary experiments in freshly isolated normal human hepatocytes by inhibiting AKT signaling to corroborate the role of activated‐AKT(Thr308) in HNF4α nuclear localization. Freshly isolated normal human hepatocytes were treated with MK‐2206, a potent allosteric pan‐AKT inhibitor.( 45 ) After 24 hours of AKT‐inhibitory treatment, 80% of activated AKT(Thr308), 25% of nuclear HNF4α, and 14% of acetylated nuclear HNF4α expression were reduced when compared with the nontreated controls.
Fig. 7.
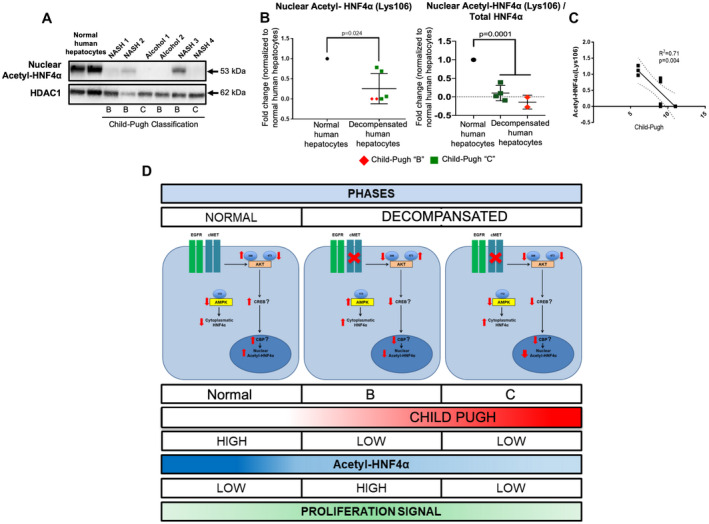
Acetylation of nuclear HNF4α is altered in human decompensated hepatocytes from NASH and alcohol‐mediated Laennec's cirrhotic explanted livers. (A,B) Western blots and quantification of the acetylated form of HNF4α(Lys106) in the nuclear fraction of human hepatocytes from decompensated NASH (n = 4) and alcohol‐mediated Laennec's cirrhotic (n = 4) hepatocytes, normalized to nuclear marker HDAC1 (P = 0.024) or total HNF4α expression (P = 0001). (C) Linear regression graph analysis shows a significant correlation of reduced acetylated form of HNF4α(Lys106) and liver dysfunction (R2 = 0.71, P = 0.004). (D) Diagram of the proposed model of regulation of HNF4α location and its association with terminal liver failure. cMET and activated AKT(Thr308) are down‐regulated in hepatocytes from patients with NASH and alcohol‐mediated Laennec's cirrhosis‐related terminal liver failure, which decreases HNF4α acetylation and retention in the nucleus. Dot plot (B) is shown as the mean ± SD; P < 0.05.
Discussion
HNF4α is the master regulator of liver function.( 13 , 14 , 15 , 17 , 18 , 19 , 20 , 21 , 23 , 25 ) Alterations in HNF4α expression have been described in hepatocellular carcinoma (HCC), hepatitis B, hepatitis C, alcohol‐mediated hepatitis and cirrhosis, as well as NASH.( 13 , 14 , 15 , 17 , 18 , 19 , 20 , 21 , 23 , 25 , 46 ) In an animal model of terminal chronic liver failure, HNF4α expression was found to be severely reduced, and restoration of HNF4 expression using gene therapy resulted in normalization of hepatocyte function.( 12 ) To assess whether this observation applies to humans, we studied liver‐enriched transcription factor expression in the livers of a large cohort of patients with decompensated liver function.( 5 ) We found that HNF4α mRNA levels were down‐regulated and correlated with the extent of liver dysfunction based on Child‐Pugh classification. Interestingly, in those studies, nuclear localization of HNF4α was not uniform.( 5 )
In the present study, we isolated human hepatocytes from the explanted livers of patients with cirrhosis and end‐stage (Child‐Pugh B and C) liver failure caused by NASH and alcohol‐mediated Laennec's cirrhosis. In these isolated hepatocytes, there was an increase in HNF4α located in the cytoplasm and a decrease in HNF4α in the nucleus compared with that seen in control healthy hepatocytes. In addition, localization of HNF4α to the cytoplasm or nucleus in human‐failing cirrhotic hepatocytes correlated with the degree of hepatocyte dysfunction. These findings suggest that pathways regulating HNF4α nuclear transport or retention could potentially be targets for the treatment of terminal liver failure.
The role of metabolic alterations is becoming evident in the development of terminal liver failure and HCC.( 37 , 47 ) We previously found that rat hepatocytes in early stages of cirrhosis switch to glycolysis to meet their energy requirements as a compensation for the decreased oxidative phosphorylation. We found similar metabolic profiling in human hepatocytes from livers with end‐stage liver failure. Human normal hepatocytes were able to use both pyruvate carboxylase and pyruvate dehydrogenase to fuel the TCA cycle, whereas human cirrhotic decompensated hepatocytes rely primarily on pyruvate dehydrogenase to fuel TCA from glucose. Moreover, we found that human cirrhotic decompensated hepatocytes showed a dysfunctional oxidative TCA, a phenomenon previously observed only in rat cirrhotic decompensated hepatocytes.( 37 ) It is important to note that HCC normally rises from liver cirrhosis and relies primarily on a glycolytic phenotype.( 47 ) Thus, it would be important to determine in future studies the metabolic mechanistic links among liver cirrhosis, terminal liver failure, and HCC.
AMPK and AKT kinases were identified by in silico and RNA‐seq analyses as molecular pathways that might regulate HNF4α localization.( 16 , 18 , 19 ) AMPK plays a central role in maintaining energy homeostasis, promoting adenosine triphosphate (ATP) production, and reducing ATP consumption,( 48 ) whereas AKT activation promotes cell proliferation, survival, and growth.( 49 , 50 ) AKT activation is mediated by phosphorylation at threonine 308 and/or serine 473.( 46 , 47 ) In our studies, we found a significant correlation between activated‐AKT(Thr308) and HNF4α localization, as activated‐AKT(Thr308) levels were significantly decreased in end‐stage human hepatocytes. These findings indicate that AKT phosphorylation at Thr308 may play an important role in hepatocyte failure in terminal stages of liver disease.
We then analyzed two central receptors that are upstream regulators of AKT and AMPK and related to liver function and regeneration: cMET and EGFR.( 26 , 41 , 42 , 43 ) We have reported in mice that combined disruption of cMET and EGFR alters liver homeostasis and leads to terminal liver failure.( 43 ) In human failing cirrhotic hepatocytes, there was reduced expression of cMET, and decreased expression correlated directly with HNF4α localization. In contrast, there was no difference in total EGFR expression in either human failing cirrhotic hepatocytes or control human hepatocytes. Thus, in human liver, cMET might play an important role in regulating AKT pathway activation and HNF4α localization and function.
Moreover, by using different statistical analyses (Spearman's rank correlation test, pathway analysis, linear regression analysis, and PCA), we were able to establish the association between the PTMs analyzed in human failing cirrhotic and healthy hepatocytes. Not surprisingly, these statistical analyses revealed that cMET and activated‐AKT(Thr308) were directly related to the expression levels of nuclear HNF4α. An intriguing finding is the negative association found between cMET protein expression and total HNF4α, indicating that cMET expression does not directly affect total HNF4α expression but only its nuclear localization. Based on the low expression levels of activated AKT(Thr308) in human failing cirrhotic hepatocytes and the direct effect of AKT on CREB‐binding protein, a molecule that has an intrinsic acetylation activity that increases transcription factor binding to nucleosomal DNA, we hypothesized that the acetylation of HNF4α might be affected in the cirrhotic failing hepatocytes. Indeed, nuclear HNF4α acetylation was strikingly reduced in human hepatocytes from cirrhotic livers, and its level was associated with the degree of hepatic dysfunction. Moreover, although preliminary experiments presented here using healthy human hepatocytes and allosteric pan‐AKT inhibitor corroborated the relationship of AKT and the nuclear expression of HNF4α. These observations suggest that AKT phosphorylation at Thr308 mediates the HNF4α nuclear retention through acetylation, possibly by controlling the CREB‐binding protein( 19 ) (Fig. 6D). Future experiments should explore the forced expression of these additional targets in conjunction with HNF4α in human hepatocytes in end‐stage cirrhotic livers, to determine what role they may play in causing terminal hepatocyte failure, and potentially treating it.
In summary, these results suggest that localization of HNF4α in the cytoplasm results from alterations of the molecular pathways, which maintain HNF4α in the nucleus during advanced stages of liver disease. cMET and activated AKT(Thr308) are down‐regulated and affect acetylation and nuclear retention of HNF4α (Fig. 6D). These findings may have therapeutic implications for the restoration of hepatocyte function in chronic liver diseases.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Fig S8
Table S1‐S2
Supplementary Material
Financial Disclosure: This work was supported by grants from NIH, DK099257, DK117881, DK119973, DK096990 and TR002383 to A.S.‐G. and CA227622, CA222251, & CA204969 to D.N.
Potential conflict of interest: A.S.‐G., A.B. and I.J.F., are inventors on a patent application that describes the use of transcription factors to treat chronic liver failure (US20140249209). N.A.F., K.T., A.O., J.G.‐L., E.N.T., G.K.M., A.B., S.J.H., I.J.F. and A.S.‐G., are inventors on a provisional patent application related to methods to enhance hepatic functions in human failing livers (Application No.:62/915,765). A.S.‐G., J.G.‐L., K.T., A.C.‐D, Y.W. and I.J.F., are co‐founders and have a financial interest in Von Baer Wolff, Inc. a company focused on biofabrication of autologous human hepatocytes from stem cells technology and programming liver failure and their interests are managed by the Conflict of Interest Office at the University of Pittsburgh in accordance with their policies.
References
Author names in bold designate shared co‐first authorship.
- 1. Tsochatzis EA, Bosch J, Burroughs AK. Liver cirrhosis. Lancet 2014;383:1749‐1761. [DOI] [PubMed] [Google Scholar]
- 2. Murphy SL, Xu J, Kochanek KD, Curtin SC, Arias E. Deaths: final data for 2015. Natl Vital Stat Rep 2017;66:1‐75. [PubMed] [Google Scholar]
- 3. Lopez PM, Martin P. Update on liver transplantation: indications, organ allocation, and long‐term care. Mt Sinai J Med 2006;73:1056‐1066. [PubMed] [Google Scholar]
- 4. Donato F, Gelatti U, Limina RM, Fattovich G. Southern Europe as an example of interaction between various environmental factors: a systematic review of the epidemiologic evidence. Oncogene 2006;25:3756‐3770. [DOI] [PubMed] [Google Scholar]
- 5. Guzman‐Lepe J, Cervantes‐Alvarez E, Collin de l'Hortet A, Wang Y, Mars WM, Oda Y, et al. Liver‐enriched transcription factor expression relates to chronic hepatic failure in humans. Hepatol Commun 2018;2:582‐594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut 2015;64:830‐841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Simoes ICM, Fontes A, Pinton P, Zischka H, Wieckowski MR. Mitochondria in non‐alcoholic fatty liver disease. Int J Biochem Cell Biol 2018;95:93‐99. [DOI] [PubMed] [Google Scholar]
- 8. Zhang XQ, Xu CF, Yu CH, Chen WX, Li YM. Role of endoplasmic reticulum stress in the pathogenesis of nonalcoholic fatty liver disease. World J Gastroenterol 2014;20:1768‐1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang K. Molecular mechanisms of hepatic apoptosis. Cell Death Dis 2014;5:e996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol 2011;54:795‐809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Michalopoulos GK, Khan Z. Liver stem cells: experimental findings and implications for human liver disease. Gastroenterology 2015;149:876‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nishikawa T, Bell A, Brooks JM, Setoyama K, Melis M, Han B, et al. Resetting the transcription factor network reverses terminal chronic hepatic failure. J Clin Invest 2015;125:1533‐1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Babeu JP, Boudreau F. Hepatocyte nuclear factor 4‐alpha involvement in liver and intestinal inflammatory networks. World J Gastroenterol 2014;20:22‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chellappa K, Jankova L, Schnabl JM, Pan S, Brelivet Y, Fung CL, et al. Src tyrosine kinase phosphorylation of nuclear receptor HNF4alpha correlates with isoform‐specific loss of HNF4alpha in human colon cancer. Proc Natl Acad Sci U S A 2012;109:2302‐2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guo H, Gao C, Mi Z, Wai PY, Kuo PC. Phosphorylation of Ser158 regulates inflammatory redox‐dependent hepatocyte nuclear factor‐4a transcriptional activity. Biochem J 2014;461:347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hong YH, Varanasi US, Yang W, Leff T. AMP‐activated protein kinase regulates HNF4alpha transcriptional activity by inhibiting dimer formation and decreasing protein stability. J Biol Chem 2003;278:27495‐27501. [DOI] [PubMed] [Google Scholar]
- 17. Lu H. Crosstalk of HNF4alpha with extracellular and intracellular signaling pathways in the regulation of hepatic metabolism of drugs and lipids. Acta Pharm Sin B 2016;6:393‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Song Y, Zheng D, Zhao M, Qin Y, Wang T, Xing W, et al. Thyroid‐stimulating hormone increases HNF‐4alpha phosphorylation via cAMP/PKA pathway in the liver. Sci Rep 2015;5:13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Soutoglou E, Katrakili N, Talianidis I. Acetylation regulates transcription factor activity at multiple levels. Mol Cell 2000;5:745‐751. [DOI] [PubMed] [Google Scholar]
- 20. Sun K, Montana V, Chellappa K, Brelivet Y, Moras D, Maeda Y, et al. Phosphorylation of a conserved serine in the deoxyribonucleic acid binding domain of nuclear receptors alters intracellular localization. Mol Endocrinol 2007;21:1297‐1311. [DOI] [PubMed] [Google Scholar]
- 21. Xu Z, Tavares‐Sanchez OL, Li Q, Fernando J, Rodriguez CM, Studer EJ, et al. Activation of bile acid biosynthesis by the p38 mitogen‐activated protein kinase (MAPK): hepatocyte nuclear factor‐4alpha phosphorylation by the p38 MAPK is required for cholesterol 7alpha‐hydroxylase expression. J Biol Chem 2007;282:24607‐24614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yokoyama A, Katsura S, Ito R, Hashiba W, Sekine H, Fujiki R, et al. Multiple post‐translational modifications in hepatocyte nuclear factor 4alpha. Biochem Biophys Res Commun 2011;410:749‐753. [DOI] [PubMed] [Google Scholar]
- 23. Zhou W, Hannoun Z, Jaffray E, Medine CN, Black JR, Greenhough S, et al. SUMOylation of HNF4alpha regulates protein stability and hepatocyte function. J Cell Sci 2012;125:3630‐3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gramignoli R, Green ML, Tahan V, Dorko K, Skvorak KJ, Marongiu F, et al. Development and application of purified tissue dissociation enzyme mixtures for human hepatocyte isolation. Cell Transplant 2012;21:1245‐1260. [DOI] [PubMed] [Google Scholar]
- 25. Bell AW, Michalopoulos GK. Phenobarbital regulates nuclear expression of HNF‐4alpha in mouse and rat hepatocytes independent of CAR and PXR. Hepatology 2006;44:186‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Natarajan A, Wagner B, Sibilia M. The EGF receptor is required for efficient liver regeneration. Proc Natl Acad Sci U S A 2007;104:17081‐17086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rasband WS. ImageJ. Bethesda, MD: U.S. National Institutes of Health. [Google Scholar]
- 28. Hainer SJ, Gu W, Carone BR, Landry BD, Rando OJ, Mello CC, et al. Suppression of pervasive noncoding transcription in embryonic stem cells by esBAF. Genes Dev 2015;29:362‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kumar R, Ichihashi Y, Kimura S, Chitwood DH, Headland LR, Peng J, et al. A high‐throughput method for Illumina RNA‐seq library preparation. Front Plant Sci 2012;3:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Morlan JD, Qu K, Sinicropi DV. Selective depletion of rRNA enables whole transcriptome profiling of archival fixed tissue. PLoS One 2012;7:e42882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Adiconis X, Borges‐Rivera D, Satija R, DeLuca DS, Busby MA, Berlin AM, et al. Comparative analysis of RNA sequencing methods for degraded or low‐input samples. Nat Methods 2013;10:623‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics 2004;20:1453‐1454. [DOI] [PubMed] [Google Scholar]
- 33. Saldanha AJ. Java Treeview—extensible visualization of microarray data. Bioinformatics 2004;20:3246‐3248. [DOI] [PubMed] [Google Scholar]
- 34. Tanaka T, Jiang S, Hotta H, Takano K, Iwanari H, Sumi K, et al. Dysregulated expression of P1 and P2 promoter‐driven hepatocyte nuclear factor‐4alpha in the pathogenesis of human cancer. J Pathol 2006;208:662‐672. [DOI] [PubMed] [Google Scholar]
- 35. Walesky C, Apte U. Role of hepatocyte nuclear factor 4alpha (HNF4alpha) in cell proliferation and cancer. Gene Expr 2015;16:101‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu L, Yannam GR, Nishikawa T, Yamamoto T, Basma H, Ito R, et al. The microenvironment in hepatocyte regeneration and function in rats with advanced cirrhosis. Hepatology 2012;55:1529‐1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nishikawa T, Bellance N, Damm A, Bing H, Zhu Z, Handa K, et al. A switch in the source of ATP production and a loss in capacity to perform glycolysis are hallmarks of hepatocyte failure in advance liver disease. J Hepatol 2014;60:1203‐1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol Cell Biol 2001;21:1393‐1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rui L. Energy metabolism in the liver. Compr Physiol 2014;4:177‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Watt AJ, Garrison WD, Duncan SA. HNF4: a central regulator of hepatocyte differentiation and function. Hepatology 2003;37:1249‐1253. [DOI] [PubMed] [Google Scholar]
- 41. Komposch K, Sibilia M. EGFR signaling in liver diseases. Int J Mol Sci 2015;17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Paranjpe S, Bowen WC, Mars WM, Orr A, Haynes MM, DeFrances MC, et al. Combined systemic elimination of MET and epidermal growth factor receptor signaling completely abolishes liver regeneration and leads to liver decompensation. Hepatology 2016;64:1711‐1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tsagianni A, Mars WM, Bhushan B, Bowen WC, Orr A, Stoops J, et al. Combined systemic disruption of MET and epidermal growth factor receptor signaling causes liver failure in normal mice. Am J Pathol 2018;188:2223‐2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dekker FJ, Haisma HJ. Histone acetyl transferases as emerging drug targets. Drug Discov Today 2009;14:942‐948. [DOI] [PubMed] [Google Scholar]
- 45. Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, et al. MK‐2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 2010;9:1956‐1967. [DOI] [PubMed] [Google Scholar]
- 46. Argemi J, Latasa MU, Atkinson SR, Blokhin IO, Massey V, Gue JP, et al. Defective HNF4alpha‐dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis. Nat Commun 2019;10:3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang X, Zhang A, Sun H. Power of metabolomics in diagnosis and biomarker discovery of hepatocellular carcinoma. Hepatology 2013;57:2072‐2077. [DOI] [PubMed] [Google Scholar]
- 48. Woods A, Williams JR, Muckett PJ, Mayer FV, Liljevald M, Bohlooly YM, et al. Liver‐specific activation of AMPK prevents steatosis on a high‐fructose diet. Cell Rep 2017;18:3043‐3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell 2017;169:381‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Morales‐Ruiz M, Santel A, Ribera J, Jimenez W. The role of Akt in chronic liver disease and liver regeneration. Semin Liver Dis 2017;37:11‐16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Fig S8
Table S1‐S2
Supplementary Material