

In December 2019, a pneumonia outbreak associated with a novel form of human coronavirus was reported in Wuhan, Hubei province, China.1, 2
As the causative agent of coronavirus disease 2019 (COVID‐19), the severe acute respiratory distress syndrome coronavirus 2 (SARS‐CoV‐2) is now responsible for the third coronavirus‐associated pandemic in recent human history.3, 4 The World Health Organization declared a public health emergency of international concern5 because of a growing number of deaths around the globe, as well as unprecedented economic and sociodemographic consequences.
SARS‐CoV‐2 is the seventh coronavirus known to infect humans.6 It belongs to the family of Coronaviridae, a group of large, enveloped, nonsegmented positive‐sense RNA viruses. The family includes some of the less pathogenic viruses, like HKU and 229E,7 but also highly pathogenic ones, such as SARS‐CoV and the Middle East respiratory (MERS)‐CoV, which emerged in 2002 and 2012, respectively, causing substantial human morbidity and mortality.8 Even though shortly after its emergence it was broadly compared with the H1N1 influenza virus (IAV), SARS‐CoV‐2 seems not to share molecular similarities with IAV. However, H1N1 and SARS‐CoV show some resemblance regarding immune system activation. Both viruses induce alterations of epigenetic control mechanisms, allowing interferon‐stimulated genes (ISGs) effector response, which provides the first defense against viral infection.9
The body of knowledge that the scientific community has gathered on SARS‐CoV‐2 is extremely recent, but is growing daily. Still, there are no antiviral treatments against this disease, nor are there vaccines for its prevention. The long term consequences of the infection on human health remain uncertain at this point. Nevertheless, some extrapolations can be made about the potential effects of the virus on cellular life span as well as on organismal health span. Here, we argue that SARS‐CoV‐2 infection may, in the long term, lead to accelerated aging phenotypes in survivors not only in affected tissues, but also in other organs, including the brain. Given that some of the effects could manifest months or years after infection, it will be necessary to follow carefully people affected by COVID‐19. Keeping accurate registries may enable us to, in the future, establish connections with aging‐associated disorders, such as Parkinson's disease (PD) and other neurodegenerative disorders.
Effects of SARS‐CoV‐2 on Aging Hallmarks
Although studies elucidating molecular details of SARS‐CoV‐2 infection are still missing, a recent study mapping the interactions between each viral protein and human proteome lends support to reasoning presented above.10 The study shows the interactions of SARS‐CoV‐2 proteins with human proteins from several aging‐related pathways, like vesicle trafficking (Nsp6, Nsp7, Nsp10, Nsp13, Nsp15, Orf3a, E, and Orf8), lipid modifications (Spike), RNA processing and regulation (Nsp8, N), ubiquitin ligases (Orf10), and mitochondrial activity (Nsp4, Nsp8, and Orf9c).
Nucleocapsid protein (N) interacts with stress granule marker protein G3BP1, a protein whose antiviral activity is based on the induction of innate immune response.11, 12, 13 Such interaction likely inhibits SG formation, thus leading also to manipulation of the host cell RNA biology and protein synthesis.14 The SARS‐CoV‐2 nucleocapsid protein also interacts with the mTOR translational repressor, LARP1.10 Importantly, all target proteins are expressed in both lungs as well as nonlung tissue. The available literature on cellular outcomes of SARS‐CoV and IAV infections reveals frequent modulations of the pathways involved in cellular aging (reviewed in an earlier work15), thus supporting the potential involvement of SARS‐CoV‐2 in similar pathways (Fig. 1).
Figure 1.
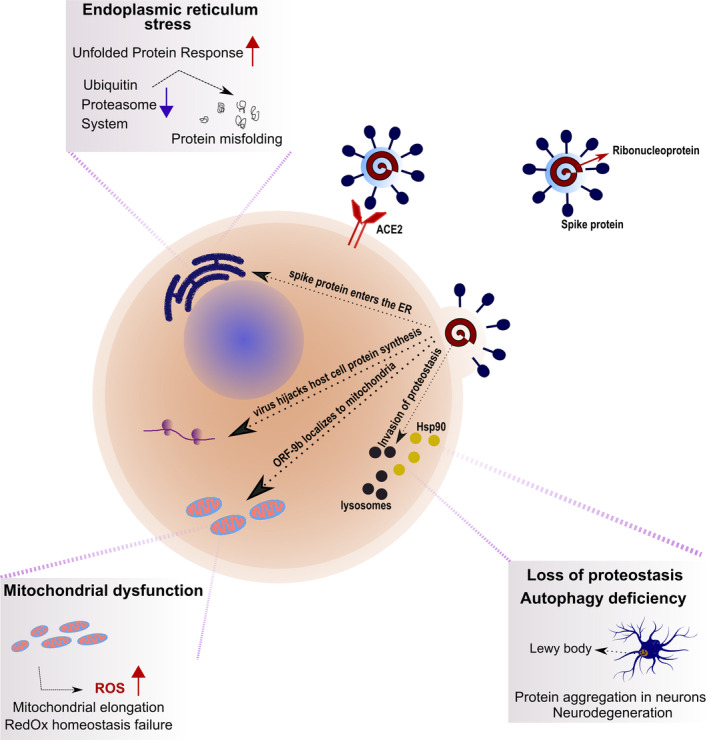
Putative modus operandi of SARS‐CoV‐2. Upon binding to the angiotensin‐converting enzyme 2 (ACE2) receptor, the virus enters the host cells and hijacks the cellular machineries for its own replication, affecting pathways relevant in the maintenance of cellular longevity. The virus seizes control over the host's Hsp90 (yellow circles) to enhance the function of its RNA polymerase. Lysosomal activity (black circles) is inhibited, leading to protein aggregation in target cells, including neurons, thus increasing the long‐term likelihood of neurodegenerative diseases, such as PD. Viral ORF‐9b localizes to mitochondria, propelling mitochondrial dysfunction, as well as an overall turmoil of redox homeostasis. The viral spike protein enters the ER and activates the UPR, which, together with the inhibition of the ubiquitin‐proteasome system, leads to cell‐wide protein misfolding. This model is based on literature currently available on SARS‐CoV, SARS‐Cov‐2, and influenza A virus. [Color figure can be viewed at http://wileyonlinelibrary.com]
Protein homeostasis (proteostasis) is the result of coordinated networks that act to maintain a dynamic equilibrium among protein translation, folding, and clearance. It includes molecular chaperones, predominantly the heat shock proteins (HSPs), which enable the correct protein folding, native conformation maintenance, and cooperation with the protein degradation machinery. However, preservation of proteome stability is challenging given that cells are frequently exposed to stresses. A viral infection is one such example given that viruses hijack the host's cellular machinery to replicate efficiently. In particular, viruses apply several strategies to manipulate proteostasis pathways at different stages and take advantage for their cycle progression. During the early stages of H1N1 infection, Hsp40 associates with two subunits of viral RNA polymerase, thus enhancing its activity.16 Hsp40 mediates the translocation of the viral genome into the nucleus attributed to the interaction with the viral nucleoprotein (NP), which encapsulates the viral genome (Fig. 1).
Moreover, Hsp40‐NP interaction plays a role at the late stages of infection by inhibiting protein kinase R (PKR) activation, essential in the antiviral response of the host.17, 18 Although previous studies have shown an adverse effect of Hsp70 in preventing the nuclear export of ribonucleoprotein in the H3N2 influenza virus,19 recent studies described that Hsp70 acts as a chaperon for viral polymerase.20 Hsp90 is another target of the IAV infection strategy. After viral infection, Hsp90 relocalizes in the nucleus and positively regulates the activity and structure of viral RNA polymerase.21, 22 HSPs are also negative regulators of cell death. Normally, Hsp70 directly binds Apaf‐1, preventing the recruitment of procaspase‐9 to the apoptosome. Using a similar mechanism, Hsp90 inhibits Apaf‐1 oligomerization and recruitment of procaspase. Both pathways block the initiation of apoptosis.23, 24 During an infection, viruses can prevent the formation of these complexes, facilitating caspase cascade activation to induce apoptosis to spread infection and evade host immune response.
Moreover, the SARS‐CoV virus uses the endoplasmic reticulum (ER) as a site for the synthesis and processing of viral proteins.25 The infection with SARS‐CoV induces the unfolded protein response (UPR) in the host cell. The SARS‐CoV spike (S) protein activates the transcription of several UPR effectors, including glucose‐regulated protein 78 (GRP78), GRP94, and C/EBP homologous protein. The spike protein accumulates in the ER, suggesting that it modulates the UPR explicitly to facilitate viral replication25 (Fig. 1).
The potential for degrading and recycling their components provides cells with a powerful means of killing intracellular pathogens.26 For this reason, autophagy represents an innate immune defense against viruses by delivering viruses and viral proteins to lysosomes for degradation. Therefore, viruses can interfere with protein degradation pathways to maintain the correct concentration and function of viral proteins. H1N1 blocks autophagic flux at early stages and leads to a decreased number of autophagosomes, whereas at the late stages, it inhibits autophagosome fusion with lysosomes.27 However, for (+) strand RNA viruses, autophagosomes can facilitate assembly of replicase proteins. In this context, it has been shown that nonstructural protein (NSP) 6 of the avian coronavirus, infectious bronchitis virus, generates autophagosomes from the ER of the host cell28 (Fig. 1). NSP6 protein limits autophagosome expansion, thus favoring coronavirus infection by impeding the delivery of viral components to lysosomes for degradation. SARS‐CoV open reading frame 9b (ORF‐9b) strongly induces the autophagy of the host cells.29
H1N1 can hijack the host ubiquitin‐proteasome system. Cells can ubiquitinate viral proteins to target them for degradation, but viruses present strategies to evade such response by inactivating host cell antagonists of viral replication.30, 31 Such a scenario triggers alterations in proteostasis that may lead to the accumulation of toxic insoluble proteins.32 As a response to this stress, cells shut down the translation of housekeeping genes to conserve energy for the synthesis of stress response proteins.
SARS‐CoV has been recognized to manipulate host cell mitochondria and mitochondrial function to avoid innate host immunity.29 ORF‐9b of SARS‐CoV localizes to mitochondria and causes mitochondrial elongation by enhancing proteasomal degradation of dynamin‐like protein 1, a human protein acting in mitochondrial fission (Fig. 1). Moreover, ORF‐9b targets the mitochondrial‐associated adaptor molecule MAVS signalosome to suppress antiviral cellular signaling. Furthermore, SARS‐CoV proteins—ORF‐3a, ORF‐3b, ORF‐6, and ORF‐7a—induce apoptosis of the host cell.33 Another adaptive mechanism cells turn to during stress is the sequestration of misfolded proteins into stress granules. H1N1 displays the potential to inhibit the translation, as well as the stress granule formation, by phosphorylation of the host's eukaryotic translation initiation factor 2α (eIF2α). Given that viral replication depends on functional host translation machinery, many viruses bind PKR to prevent eIF2α phosphorylation.34 A myriad of cellular malfunctions triggers redox imbalance, increased reactive oxygen species (ROS) production, as well as mitochondrial and lysosomal dysfunction. Finally, such a sequence of events creates a vicious circle by rendering the cells even less resistant to infection, which, in the long term, may lead to an increase in the biological age among COVID‐19 survivors by accelerated aging of the immune system and affected tissues.
A Possible Connection With PD
Age‐related loss of proteostasis has been strongly correlated with more severe consequences of IAV and SARS‐CoV‐2 in older adults. The loss of ability of properly activating stress response mechanisms in the elderly can lead to severe phenotypes, including a decrease of protein solubility and accumulation of aggregates, such as those characteristic of various age‐associated neurodegenerative disorders, including PD. Indeed, infection of dopaminergic cells expressing alpha‐synuclein (aSyn), the major protein component of Lewy bodies and Lewy neurites, with the H1N1 influenza virus, resulted in the formation of aSyn aggregates, but not of tau or TDP‐43, suggesting selectivity.27 In this study, the molecular mechanisms pointed to H1N1‐mediated blocking of autophagic flux, which has long been associated with aSyn accumulation in models of PD.
Interestingly, amantadine, an antiviral agent, is used in early and advanced PD to treat tremor.35, 36, 37, 38, 39 In addition, oseltamivir, an antiviral widely used to treat influenza, was reported to significantly improve parkinsonism, but, at the same time, to increase dyskinesia.40 Although the risk for idiopathic PD does not seem to be increased because of previous influenza infections, parkinsonism may be linked with more recent infections.41
aSyn may play a role in inducing innate and adaptive immunity in PD.42, 43 Therefore, investigating the molecular mechanisms connecting viral infections with alterations in cellular proteostasis pathways that may, in turn, potentiate aSyn aggregation could prove extremely valuable for the design of therapeutic strategies for PD and for adjusting therapies for PD patients who were infected by SARS‐CoV‐2.
Previous reports suggested a possible interaction of human CoV with the central nervous system, and with PD in particular.44 Interestingly, intracerebral injection of IAV in mice results in the presence of the virus in the SN and hippocampus.45
Importantly, aSyn was reported to act as an antiviral factor in neurons of patients with West Nile virus (WNV) encephalitis. In aSyn‐knockout mice, the WNV infectious titer in the brain is increased by 5 orders of magnitude, and the rate of WNV‐induced mortality is strongly aggravated. The cortical neurons of aSyn‐knockout mice also exhibit an earlier increase in the amount of virus‐induced caspase‐3 after the onset of infection, thus triggering neuronal death by apoptosis at an earlier time point.46
Mutations in the leucine‐rich repeat kinase (LRRK2) are, so far, the most common genetic determinant of PD. Interestingly, mice expressing G2019S mutant LRRK2 exhibit increased mortality triggered by reovirus‐induced encephalitis. Strikingly, brains from these animals contain higher levels of aSyn.47
LRRK2 is present in many cell types in the immune system, and its expression is increased in pathogen‐stimulated macrophages.48 Previous research brought sufficient evidence to hypothesize that the role of LRRK2 in the immune system provides a “glue” connecting the immune system function with the development and propagation of PD, as well as the biological age of the host cells.48
Concluding Remarks
At this point, it is indisputable that SARS‐CoV‐2 is causing a global medical emergency that is taking a substantial number of lives every day. Yet, enormous efforts are being made by the scientific community to develop treatments and vaccines that would help treat and eradicate this virus. Several interventions have already been proposed, targeting viral progression. Extensive biochemical studies will be essential for targeted drug design, resonating with valuable work on Coronaviridae interactions with the host cell pathways.
However, when the pandemic is over, what will the consequences be to the health of survivors? The findings described so far on SARS‐CoV‐2 echo those with SARS‐CoV and with H1N1 virus: Mitochondrial function, proteostasis, lipid metabolism, as well as stress responses are only some of the crucial cellular pathways affected by the infection. Strikingly, these processes also reverberate with multiple pathways relevant in cellular and organismal aging, and in neurodegenerative diseases such as PD, suggesting that accelerated aging in certain tissues might be a potential long‐term complication of the SARS‐CoV‐2 infections.
Author Roles
Manuscript: A. Writing of the First Draft, B. Review and Critique.
Alice Lippi: A
Renato Domingues: A
Cristian Setz: A
Tiago F. Outeiro: A
Anita Krisko: A
Financial Disclosures
A.K. and T.F.O. have received funding from DFG, in Germany.
Relevant conflicts of interest/financial disclosures: Nothing to report.
Full financial disclosures and author roles may be found in the online version of this article.
Contributor Information
Tiago F. Outeiro, Email: touteir@gwdg.de.
Anita Krisko, Email: anita.krisko@med.uni-goettingen.de.
References
- 1. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature 2020;579:265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen X, Yu B. First two months of the 2019 Coronavirus Disease (COVID‐19) epidemic in China: real‐time surveillance and evaluation with a second derivative model. Glob Health Res Policy 2020;5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Drosten C, Günther S, Preiser W, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 2003;348:1967–1976. [DOI] [PubMed] [Google Scholar]
- 4. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus ADME, Fouchier RAM. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 2012;367:1814–1820. [DOI] [PubMed] [Google Scholar]
- 5. Li Q, Guan X, Wu P, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia. N Engl J Med 2020;382:1199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Corman VM, Muth D, Niemeyer D, Drosten C. Hosts and sources of endemic human coronaviruses. Adv Virus Res 2018;100:163–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Su S, Wong G, Shi W, et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol 2016;24:490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. de Wit E, van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol 2016;14:523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Menachery VD, Eisfeld AJ, Schäfer A, et al. Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon‐stimulated gene responses. mBio 2014;5:e01174‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gordon DE, Jang GM, Bouhaddou M, et al. A SARS‐CoV‐2‐human protein‐protein interaction map reveals drug targets and potential drug‐repurposing. biorxiv 2020.03.22.002386v1. [Preprint]. [Google Scholar]
- 11. Reineke LC, Lloyd RE. The stress granule protein G3BP1 recruits protein kinase R to promote multiple innate immune antiviral responses. J Virol 2015;89:2575–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang W, Ru Y, Ren J, et al. G3BP1 inhibits RNA virus replication by positively regulating RIG‐I‐mediated cellular antiviral response. Cell Death Dis 2019;10:946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim SSY, Sze L, Lam KP. The stress granule protein G3BP1 binds viral dsRNA and RIG‐I to enhance interferon‐β response. J Biol Chem 2019;294:6430–6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Raaben M, Groot Koerkamp MJA, Rottier PJM, de Haan CAM. Mouse hepatitis coronavirus replication induces host translational shutoff and mRNA decay, with concomitant formation of stress granules and processing bodies. Cell Microbiol 2007;9:2218–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. López‐Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013;153:1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cao M, Wei C, Zhao L, et al. DnaJA1/Hsp40 is co‐opted by influenza a virus to enhance its viral RNA polymerase activity. J Virol 2014;88:14078–14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sharma K, Tripathi S, Ranjan P, et al. Influenza A virus nucleoprotein exploits Hsp40 to inhibit PKR activation. PLoS One 2011;6:e20215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Batra J, Tripathi S, Kumar A, et al. Human heat shock protein 40 (Hsp40/DnaJB1) promotes influenza A virus replication by assisting nuclear import of viral ribonucleoproteins. Sci Rep 2016;6:19063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hirayama E, Atagi H, Hiraki A, Kim J. Heat shock protein 70 is related to thermal inhibition of nuclear export of the influenza virus ribonucleoprotein complex. J Virol. 2004;78:1263–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Manzoor R, Kuroda K, Yoshida R, Tsuda Y, Fujikura D, Miyamoto H, et al. Heat shock protein 70 modulates influenza A virus polymerase activity. J Biol Chem. 2014;289:7599–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Momose F, Naito T, Yano K, Sugimoto S, Morikawa Y, Nagata K. Identification of Hsp90 as a stimulatory host factor involved in influenza virus RNA synthesis. J Biol Chem 2002;277:45306–45314. [DOI] [PubMed] [Google Scholar]
- 22. Naito T, Momose F, Kawaguchi A, Nagata K. Involvement of Hsp90 in assembly and nuclear import of influenza virus RNA polymerase subunits. J Virol 2007;81:1339–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, et al. Heat‐shock protein 70 inhibits apoptosis by preventing recruitment of procaspase‐9 to the Apaf‐1 apoptosome. Nat Cell Biol 2000;2:469–475. [DOI] [PubMed] [Google Scholar]
- 24. Pandey P. Negative regulation of cytochrome c‐mediated oligomerization of Apaf‐1 and activation of procaspase‐9 by heat shock protein 90. EMBO J 2000;19:4310–4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chan CP, Siu KL, Chin KT, Yuen KY, Zheng B, Jin DY. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J Virol 2006;80:9279–9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Levine B. Eating oneself and uninvited guests. Cell 2005;120:159–162. [DOI] [PubMed] [Google Scholar]
- 27. Marreiros R, Müller‐Schiffmann A, Trossbach SV, et al. Disruption of cellular proteostasis by H1N1 influenza A virus causes α‐synuclein aggregation. Proc Natl Acad Sci U S A 2020;117:6741–6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cottam EM, Whelband MC, Wileman T. Coronavirus NSP6 restricts autophagosome expansion. Autophagy 2014;10:1426–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shi CS, Qi HY, Boularan C, et al. SARS‐coronavirus open reading frame‐9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J Immunol 2014;193:3080–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gack MU, Albrecht RA, Urano T, et al. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG‐I. Cell Host Microbe 2009;5:439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Widjaja I, de Vries E, Tscherne DM, Garcia‐Sastre A, Rottier PJM, de Haan CAM. Inhibition of the ubiquitin‐proteasome system affects influenza A virus infection at a postfusion step. J Virol 2010;84:9625–9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu XD, Ko S, Xu Y, et al. Transient aggregation of ubiquitinated proteins is a cytosolic unfolded protein response to inflammation and endoplasmic reticulum stress. J Biol Chem 2012;287:19687–19698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ye Z, Wong CK, Li P, Xie Y. A SARS‐CoV protein, ORF‐6, induces caspase‐3 mediated, ER stress and JNK‐dependent apoptosis. Biochim Biophys Acta 2008;1780:1383–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li S, Min JY, Krug RM, Sen GC. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double‐stranded RNA. Virology 2006;349:13–21. [DOI] [PubMed] [Google Scholar]
- 35. Grelak RP, Clark R, Stump JM, Vernier VG. Amantadine‐dopamine interaction: possible mode of action in Parkinsonism. Science 1970;169:203–204. [DOI] [PubMed] [Google Scholar]
- 36. Bauer RB, McHenry JT. Comparison of amantadine, placebo, and levodopa in Parkinson's disease. Neurology 1974;24:715–720. [DOI] [PubMed] [Google Scholar]
- 37. Danielczyk W. Twenty‐five years of amantadine therapy in Parkinson's disease. J Neural Transm Suppl 1995;46:399–405. [PubMed] [Google Scholar]
- 38. Dolin R, Reichman RC, Madore HP, Maynard R, Linton PN, Webber‐Jones J. A controlled trial of amantadine and rimantadine in the prophylaxis of influenza A infection. N Engl J Med 1982;307:580–584. [DOI] [PubMed] [Google Scholar]
- 39. Blake GJ. Amantadine for influenza A. Nursing 1990;20:21. [DOI] [PubMed] [Google Scholar]
- 40. Kadowaki T, Komagamine T, Suzuki K, Hirata K. Oseltamivir‐induced dyskinesia in Parkinson's disease. Parkinsonism Relat Disord 2011;17:133–134. [DOI] [PubMed] [Google Scholar]
- 41. Toovey S, Jick SS, Meier CR. Parkinson's disease or Parkinson symptoms following seasonal influenza: influenza and Parkinson's disease. Influenza Other Respir Viruses 2011;5:328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Allen Reish HE, Standaert DG. Role of α‐synuclein in inducing innate and adaptive immunity in Parkinson disease. J Parkinsons Dis 2015;5:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Roodveldt C, Labrador‐Garrido A, Izquierdo G, Pozo D. Alpha‐synuclein and the immune response in Parkinson's disease. In: Finkelstein D, ed. Towards New Therapies for Parkinson's Disease [Internet]. InTech; 2011 [cited 2020. Apr 3]. Available from: http://www.intechopen.com/books/towards-new-therapies-for-parkinson-s-disease/alpha-synuclein-and-the-immune-response-in-parkinson-s-disease. .
- 44. Fazzini E, Fleming J, Fahn S. Cerebrospinal fluid antibodies to coronavirus in patients with Parkinson's disease. Mov Disord 1992;7:153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yamada T. Viral etiology of Parkinson's disease: focus on influenza A virus. Parkinsonism Relat Disord 1996;2:113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Beatman EL, Massey A, Shives KD, et al. Alpha‐synuclein expression restricts RNA viral infections in the brain. J Virol 2016;90:2767–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shutinoski B, Hakimi M, Harmsen IE, et al. Lrrk2 alleles modulate inflammation during microbial infection of mice in a sex‐dependent manner. Sci Transl Med 2019;11:eaas9292. [DOI] [PubMed] [Google Scholar]
- 48. Hakimi M, Selvanantham T, Swinton E, et al. Parkinson's disease‐linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J Neural Transm 2011;118:795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]