

Summary
As three-dimensional cell culture formats gain in popularity, there emerges a need for tools that produce vast amounts of data on individual cells within the spheroids or organoids. Here, we present a microfluidic platform that provides access to such data by parallelizing the manipulation of individual spheroids within anchored droplets. Different conditions can be applied in a single device by triggering the merging of new droplets with the spheroid-containing drops. This allows cell-cell interactions to be initiated for building microtissues, studying stem cells’ self-organization, or observing antagonistic interactions. It also allows the spheroids’ physical or chemical environment to be modulated, as we show by applying a drug over a large range of concentrations in a single parallelized experiment. This convergence of microfluidics and image acquisition leads to a data-driven approach that allows the heterogeneity of 3D culture behavior to be addressed across the scales, bridging single-cell measurements with population measurements.
Keywords: droplet microfluidics, spheroids, organoids, tissue engineering, liver toxicity
Graphical Abstract
Highlights
-
•
Microfluidic droplet pairs sequentially trapped in capillary anchors before merging
-
•
1 spheroid/droplet, with microenvironment modulations driven by droplet merging
-
•
A wide range of drug concentrations tested on hepatic-like spheroids in a single chip
-
•
Data-driven approach unravels 3D tissue-level dynamic drug response
Tomasi et al. describe a microfluidic droplet approach to sequentially modulate culture conditions on 3D spheroids, enabling complex protocols such as co-culture, hydrogel encapsulation, or drug testing. When applied to a drug toxicity experiment on a chip, the platform yields time-resolved, single-cell data that uncover a dynamic response regulated at the spheroid level.
Introduction
Recent years have seen the emergence of many new cell culture approaches to improve the relevance of in vitro experiments to the behavior of the cells residing within living tissues. One of the main objectives of these methods is to recapitulate the native cells’ microenvironment, including biochemical signaling delivered from the blood stream or from neighboring cells, formation of intercellular junctions, interactions with the endogenous extra-cellular matrix (ECM), mechano-transduction, and effects such as diffusion gradients (Pampaloni et al., 2007). The three-dimensional (3D) culture formats that have emerged range from culturing individual cells in hydrogel matrices (Ranga et al., 2014) or de-cellularized scaffolds (Sart et al., 2016), to making functional aggregates such as spheroids (Bartosh et al., 2010) or organoids (Lancaster et al., 2017), to building more complex engineered structures that involve multiple cell types on a microfluidic device (Bhatia and Ingber, 2014). The combination of microfluidics and 3D cell culture has allowed the emergence of a range of organ-on-a-chip approaches that include many of these strategies (Zhang and Radisic, 2017).
These formats are not meant to replace two-dimensional (2D) culture. Instead, they will allow specific questions to be asked on more physiologically relevant culture models. Some of these questions can only be asked in specific 3D formats, such as questions related to embryogenesis (van den Brink et al., 2014), tumor-stromal interactions (Glentis et al., 2017), or the effect of vascularization on tumor growth (Chiew et al., 2017). In contrast, other applications depend on cellular phenotypes that are modified when the cells are cultured in 2D versus 3D, such as the function of hepatocytes (Fey and Wrzesinski, 2012), chondrocytes (Shi et al., 2015), pancreatic cells (Lee et al., 2018), neural cells (Cullen et al., 2011), or lung cells (Kim et al., 2014) and the impact of this function on their response to toxic compounds (Imamura et al., 2015). Therefore, the most suitable technological format for a particular question will balance the level of biological complexity that is required with the desired throughput and the necessary ease of use and reproducibility of the experiment.
In this context, spheroids present an appealing format for 3D culture, because they combine a moderately high level of biological complexity with simple production protocols (Fennema et al., 2013). The biological function is enhanced in spheroids compared with 2D cultures (Bartosh et al., 2010, Proctor et al., 2017, Bell et al., 2018, Vorrink et al., 2018), while cells have been shown to produce their own ECM and interact with it (Wang et al., 2009). However, despite the long history of spheroid cultures (Sutherland et al., 1971) and the ability to produce them in large quantities in bulk formats (Ungrin et al., 2008), the manipulation and observation of individual spheroids remains largely manual and labor intensive.
Two main approaches are used to form, culture, modulate, and image spheroids: multiwell plate-based systems and microfluidic devices. Methods based on modifications of multiwell plates (Tung et al., 2011, Vinci et al., 2012, Hou et al., 2018) enable reliable formation of a single spheroid per well but suffer from high volumes when using expensive reagents, like Matrigel or antibodies, and do not comply with perfusion protocols. Low-adhesion microwells in microfluidic chambers (Kwapiszewska et al., 2014, Mulholland et al., 2018) overcome these disadvantages but lose the compartmentalization of each spheroid, which prevents multiplexing and hydrogel encapsulation. Microfluidic encapsulation in liquid droplets (Alessandri et al., 2013, Chen et al., 2016, Siltanen et al., 2016) simplifies the use of hydrogels, but the lack of droplet control limits the implementation of sequential protocols, as well as the actual throughput of quantification because of the difficulty of imaging free droplets over time.
To address these limitations, we have demonstrated a data-driven approach to spheroid culture. This approach is based on a microfluidic platform that integrates many necessary operations for the regulation of spheroid behavior in vitro while providing several hundred independent cultures per experiment on a single microscope-slide format (Sart et al., 2017). The approach is uses so-called anchored droplets (Abbyad et al., 2011) in which spheroids are formed, manipulated, and observed over several days in culture. The ability to perform precise image analysis on the single-cell level, while combining results on thousands of spheroids, enables the mapping of cellular function depending on position within the spheroids, thus providing a link between the spheroid structure and the biological function of the cells within it. However, the platform could not address spheroids individually with a specific condition, nor did it allow a succession of operations to be performed on them.
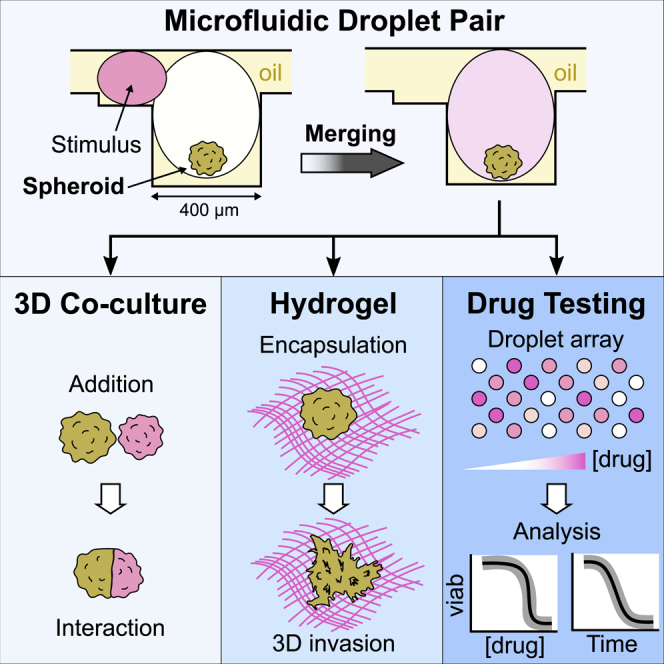
In the present paper, we build on our previous results to allow random and time-dependent operations on each spheroid. This is achieved by introducing a new asymmetric design for the anchors, which leads to a qualitative transformation in the functionality of the microfluidic approach for a range of applications. The physical principles of the devices and the protocols that allow combinatorial operations are first explained, followed by the description of two key classes of applications. First, we describe the ability to bring cells into contact for building complex tissues, studying the self-organization of stem cells, investigating antagonistic interactions between cell types, or incorporating a hydrogel like Matrigel into the droplets. These results have immediate applications in several areas of biological research, such as tissue engineering, models of immuno-therapies, or understanding host-pathogen interactions. Then, we describe how the platform can be used to investigate different conditions and to obtain a detailed drug dose-response on hepatocyte-like spheroids. The behavior is tracked by combining measurements of the time evolution of hundreds of spheroids with a single-cell resolution. The dynamic that emerges is fundamentally linked with the 3D structure of the spheroids and shows a strong effect of cell-cell interactions on their response to the drug.
Results
Physical Principles of Differential Anchoring
To understand the principles underlying the device operation and robustness, we recall that confined droplets (e.g., water-in-oil droplets) are subjected to a trapping force in regions where they reduce their surface area and thus their surface energy (Abbyad et al., 2011, Dangla et al., 2011). Therefore, by designing microfluidic devices in which the droplets are confined everywhere except in localized regions, as sketched in Figure 1A, one can define positions at which the droplets can be anchored. The efficiency of this immobilization depends on the relative strength of the anchoring force, which is given by the gradient of surface energy, and by the drag force resulting from the flow of the outer fluid: as long as the anchoring force is stronger than the drag force, the droplet will remain immobile even if the outer fluid is flowing (Dangla et al., 2011, Yamada et al., 2014).
Figure 1.

Physical Principles of Differential Anchor Strengths
(A) Side view and top view of a confined aqueous droplet near a capillary anchor. The brown arrows represent the direction of the external oil flow.
(B) Two anchoring strengths can be distinguished: for narrow anchors (blue regions), the droplet only partially enters the anchor, whereas for wide anchors (red regions), the droplet entirely enters the anchors. This leads to an anchoring efficiency that depends on droplet size for the narrow anchors and to nearly irreversible trapping in the wide anchors.
(C) Side view and top view of an asymmetric anchor. The wide (d1 > 2h) and narrow (d2 < 2h) regions can be combined into a single capillary anchor by designing asymmetric shapes. Thick dashed lines denote the planes of the corresponding side views.
When the droplet is above the anchor, the curvature of the interface will tend to homogenize to equilibrate the Laplace pressure jump between inside and outside. Therefore, geometric considerations define two limits that lead to two regimes (Figure 1B). First, in the case of wide anchors, i.e., when the anchor diameter d is larger than around 2h, the equilibrium position for the droplet is penetration of the anchor as long as the hole is sufficiently deep (Dangla et al., 2011). This leads to high trapping efficiency as a large reduction of the surface area is combined with a weak drag force, because the droplet exposes only a small region in the channel where the fluid is flowing. Conversely when , the equilibrium position corresponds to the droplet only partially entering the anchor. In this situation, the droplet is trapped by the anchor only if the outer fluid is flowing below a critical velocity that depends on the physical parameters (viscosity and surface tension) and on the droplet size. A detailed analysis shows that this critical velocity rapidly decreases as the droplet size increases (Dangla et al., 2011) (see STAR Methods for detailed discussion).
These principles can guide the design of anchors that have regions with different trapping efficiencies, as shown, for example, in Figure 1C. The red-shaded region of this anchor displays a large diameter d1 and thus can accommodate a large droplet and trap it with a high efficiency while keeping the blue region free to receive a second droplet. In contrast, the characteristic dimension d2 of this second region is smaller than 2h, so a second droplet will only be able to partially enter the blue region and therefore will be trapped with a weaker force. Depending on the design details of this shape, the contrast between the two trapping efficiencies can be very large, leading to nearly irreversible trapping in the red regions and much weaker trapping in the blue regions.
Protocol for Droplet Pairing and Fusion
This differential trapping system can be exploited to generate pairs of droplet with different contents. This is achieved in a microfluidic chip whose main feature is a large trapping chamber with an array of asymmetric anchors (Figure 2A). The protocol generally begins by bringing into the oil-filled chamber a population of large aqueous droplets and allowing them to randomly occupy the strong regions of the anchors, as shown in Figure 2B (left). Once all anchors are filled, a second population of smaller droplets is transported into the trapping region, where they are trapped in the triangular parts of the anchors (right). Beyond simply trapping the smaller droplets, the triangular shape of the anchors produces a local gradient of confinement that pushes the two droplets in each anchor into intimate contact (Fradet et al., 2013). As such, flushing an emulsion destabilization agent in the outer phase results in the quick merging of the two types of droplets (Figure 2C; Video S1). Because the excess droplets that are not trapped can be flushed away before triggering coalescence, one merging event reliably takes place in each anchor.
Figure 2.

Microfluidic Protocols for Pairing and Merging Different Droplet Populations (Aqueous Droplets in Oil)
(A) Design of the microfluidic chips.
(B–E) Images show portions of the trapping area. (B) First, a population of large droplets is injected and allowed to fill the large regions of each anchor, followed by a second population of smaller droplets. The small droplets (dark dye) then occupy the triangular regions of each anchor. Scale bar is 200 μm. (C) Flushing the device with an emulsion destabilization agent (PFO, perfluoro-octanol; Tullis et al., 2014, Akartuna et al., 2015) results in the merging of the touching droplets, which allows their contents to mix in a few seconds. Scale bar is 100 μm. (D and E) Droplet libraries can be produced in a different microfluidic device and re-injected into the trapping region. In the current example, the large droplet population contains variable concentrations of dye ranging from blue, to green, to yellow. The small droplets contain a gradient of red dye. Image of 80 anchors filled with 2 sets of colored droplets before (D) and after (E) merging. Scale bar is 1 mm.
(F and G) Quantification of the droplet colors in red, green, and blue (RGB) space before (F) and after (G) the coalescence (dot color corresponds to RGB coordinates; nchips = 1, ndroplets = 351).
All corresponding flows can be found in Table S1.
After coalescence, the droplet color fading indicates the reaction of the dark dye (2,6-DCPIP) with the oxygen carried by the oil. Time is shown in min:sec.
Alternatively, one or both populations can be produced in a different device (Funfak et al., 2009, Kaminski et al., 2012), stored off-chip, before being re-injected into the trapping region. In this way, droplet libraries can be generated independently and later brought into contact with a sample of interest that is immobilized in the capillary anchors. Such a protocol is demonstrated in Figures 2D and 2E, where two droplet populations, containing food dye as a proxy for chemical content, are merged. In this example, each of the libraries was produced on a separate chip through a confinement gradient (Dangla et al., 2013), as described in Figure S1. The large droplets were formed by mixing yellow and blue solutions, while the small droplets contained a gradient of red dye. The large and small droplets were then sequentially loaded into the anchors to yield more than 350 merged droplets each containing a unique color (Figures 2F and 2G).
The demonstrations of Figure 2 show the ability to bring together two droplets in each microfluidic anchor. Next, we show how this technology can be applied to cell manipulation, thus enabling unique operations on 3D cultures toward tissue engineering or applications requiring different conditions on a single device.
Cellular Interactions and Hydrogels in Anchored Droplets
When a suspension of cells is encapsulated in the droplets, the cells aggregate to form a single functional sphere of adherent cells in each anchored droplet (Sart et al., 2017). This protocol can be implemented in the current asymmetric anchors (Figure 3A) to produce a spheroid in the large droplet of each anchor. The second droplet can then be used to bring some fresh medium or a different cell population that can interact with the original spheroid in various ways, as illustrated in Figure 3. A few examples of synergistic or antagonistic interactions are described later.
Figure 3.

Regulation of the Biological Environment in Droplets
(A) Design of the asymmetric anchors adapted for the droplet spheroid formation and culture (side view corresponds to the thick dashed line in the top view).
(B and C) Protocol for spheroid merging scheme (B) and corresponding micrographs showing 2 consecutive merging events with H4-II-EC3 spheroids (C, scale bar is 50 μm).
(D) Micrographs of 2 heterospheroids with different configurations after the protocol shown in (C). Scale bar is 20 μm.
(E–G) Selected micrographs showing co-culture experiments with: hMSCs CD146bright (magenta) and hMSCs CD146dim (green), which were sorted by flow cytometry (E, scale bar is 20 μm); Jurkat cells and A673 spheroids (F, scale bar is 20 μm); H4-II-EC3 spheroids and E. coli (G, scale bar is 50 μm).
(H) Matrigel addition through the second droplet after spheroid formation.
(I) B16-F0 cells (mouse melanoma) were imaged just after Matrigel addition (D+2) and the next day (D+3). Black arrows highlight some cell protrusions through the hydrogel matrix. Scale bar is 20 μm.
Additional quantification is provided in Figure S2.
First, complex tissues can be constructed by successively, bringing into contact different cell populations. In the first example (Figures 3B and 3C), a single H4-II-EC3 cell spheroid (a rat hepatoma cell line) was formed in each droplet trapped in the strong region of an anchor. The operation was repeated with a smaller droplet in the triangular region to form a single magenta-stained spheroid. After droplet pair merging, the two spheroids of each anchor came into contact in the resulting merged droplet and initiated fusion to form a composite spheroid (D+3). Because the volume of the small droplet was much smaller than that of the large droplet, the triangular region of the anchor was left empty after this first droplet merging. In this way, another trapping/merging cycle was possible, for instance, with a new set of droplets containing cells stained green. This sequential three-step process resulted in the formation of a single composite microtissue in each anchor of the microfluidic chamber. The resulting microtissues had various shapes (Figure 3D), though a statistical analysis showed that the green spheroids were more likely to merge with the first two along their elongated side, resulting in a dominance of triangular structures (Figures S2A–S2C).
The ability to merge two spheroids can be used to address the self-organization of stem/progenitor cells in 3D by working with a primary population of human mesenchymal stem cells. As with most progenitor cell populations, these cells presented a varying degree of commitment toward differentiated states (Hough et al., 2014, Singer et al., 2014, Kim et al., 2016). It is not known how this heterogeneity is reflected in the structural organization of the spheroid.
To address this question, the cells were sorted, using flow cytometry, based on their level of expression of a characteristic marker for the differentiation (CD146) (Sacchetti et al., 2007). This sorting resulted in the isolation of low (CD146dim) and high (CD146bright) fluorescent cells, which were then used to form spheroids in the large and the small droplets, respectively. After droplet merging, the two spheroids fused and the cells self-reorganized into a core-shell structure, with the CD146bright cells in the core and the CD146dim cells forming the shell (Figure 3E). The transition from side-by-side spheroids to the core-shell structure was found to reproducibly take place within a 24 h period (Figures S2D and S2E).
The structural organization that emerged from these spheroid fusions was identical to the self-arrangement obtained when the different cell populations were seeded together in the same droplet (Sart et al., 2020). However, those experiments could not distinguish between kinetic effects, related to the speed of aggregation of the different cell types, and the biophysical effects, related to the affinity of different cells. By avoiding the kinetic effects, the experiments shown here confirmed that human mesenchymal stromal cells (hMSCs) self-organized in a hierarchical manner in 3D because of cell-cell affinities.
In addition to these synergistic interactions, the droplet merging can be used to explore host-pathogen or other antagonistic interactions between cell types. For instance, the microfluidic approach is well suited for studying the interaction of immune-like cells with a cancer spheroid in vitro. A simple example of such an interaction is obtained by bringing into contact a spheroid of A-673 cells (Ewing’s sarcoma) with a suspension of individualized Jurkat cells (Figure 3F). These immune cells form robust junctions with the cancer spheroid and begin to interact with the cancerous cells in each of the anchors.
Another example is shown in Figure 3G, where a well-formed spheroid (H4-II-EC3 cells) is brought into contact with a droplet containing a dilute suspension of E. coli (MG-1655-GFP, green; Figure 3G). After one day in culture. E. coli colonized the whole volume of the droplets concomitantly with a significant decline of H4-II-EC3 viability (Figures S2F–S2I).
Beyond cell-cell interactions, the second droplet can modify the physical properties of the spheroid microenvironment, for example, by introducing a hydrogel into the droplet. The hydrogel can then be solidified to initiate interactions with the cells, as shown in Figure 3H. Here, B16-F0 cells (mouse melanoma) showed clear protrusions through the matrix (Figure 3I), oriented in different directions (Figures S2J–S2L), one day after hydrogel incorporation. Finally, combinations of an inert gel (e.g., agarose) with cells in the second droplet can also be used as a porous barrier that allows paracrine signaling without allowing the cells to migrate through them, which functions as a Transwell in a droplet (Figure S3).
The preceding examples are meant to illustrate the range of interactions that are possible in the device. Learning new biology from these protocols will require more detailed experiments and analysis, including the choice of the most relevant cell types. Nevertheless, the examples of Figure 3 confirm that the microfluidic droplet format is compatible with a range of cell types in various situations and that it can be used to observe their interactions.
Dynamic Measurement of Drug Toxicity on Spheroids
Beyond looking at cellular interactions and hydrogels, the microfluidic platform lends itself to investigating the influence of different chemical conditions in each droplet. As a demonstration of the type of approaches that can be applied, we show how the asymmetric anchors can be used to test several culture environments on H4-II-EC3 cell spheroids (Figure 4A). Spheroids are first formed in the large droplets and allowed to incubate overnight. In parallel, a library of secondary droplets is produced in a different device, following protocols similar to those described in Figure 2. The secondary droplets are then injected into the microfluidic chamber and paired with the spheroid-containing drops (Figure 4B). To recognize the individual conditions in each drop, different fluorescent barcodes (Duncombe and Dittrich, 2019) are prepared by combining 3 concentrations of 3 fluorescent dyes (CF™350, CF™488A, and CF™647; see STAR Methods).
Figure 4.

Multiplexed Conditions on Liver Spheroids
(A and B) Timeline (A) and schematic top view of an anchor (B) showing the experimental protocol.
(C–E) Chemical droplet library. Droplets with 27 color barcodes were randomly trapped in capillary anchors and merged with H4-II-EC3 spheroid droplets (C, montage of 224 droplets, nchips = 2, scale bar is 1 mm). (D) Distribution of the 27 barcodes in all droplets. Each bar corresponds to one barcode, with matching colors. APAP was only added in the droplets with barcode 9. (E) Micrograph of 2 droplets 24 h after library merging. Barcode 9 corresponds to the APAP droplet. PI fluorescent intensity is shown in red. Scale bar is 100 μm.
(F) Polydispersity histogram of the spheroids produced in this study (nchips = 4, nspheroids = 685). CV = standard deviation/mean.
(G) Calibration of the fluorescent signal of the CF™488A dye (green, ndroplets = 429) and CF™647 dye (magenta, ndroplets = 455) for the drug concentration determination. The detailed protocol is explained in STAR Methods. The black circles and error bars represent the mean over all droplets of each concentration and the standard deviation, respectively. The colored lines represent a linear fit of the data.
(H and I) Montages of 6 micrographs showing anchors with single liver spheroids (H4-II-EC3 cells) before (H, scale bar is 200 μm) and after (I) drug droplet trapping. The green and magenta fluorescent dyes correspond to the APAP stock solutions at low and high concentrations, respectively, used for creating the droplet library.
(J) Micrograph of the entire chip array after droplet coalescence (nspheroids = 252, montage of the cropped anchors). The white rectangle represents the location of the droplets displayed in (C) and (D). Scale bar is 1 mm.
After droplet trapping and merging, the 27 conditions were randomly distributed in the array (Figure 4C) and each condition could be identified from the color of the fluorescent signal (Figure S4). Given the number of conditions and anchors, each barcode was represented between 1 and 15 times (Figure 4D; see STAR Methods for statistical design of the library). At the production stage of the droplets’ fluorescent barcodes, acetaminophen (APAP, a drug known for its hepatotoxicity; Vale, 2012) was added in a single barcode solution, resulting in the specific killing of the corresponding spheroids 1 day after exposition (Figure 4E).
In this proof-of-concept experiment, APAP was identified as hepatotoxic; therefore, we investigated the concentration-dependent APAP toxicity on H4-II-EC3 cell spheroids. We first verified that the spheroid size was well controlled by measuring the coefficient of variation (CV) of the diameter, which was around 10% for this cell type (Figure 4F). Then, we subjected the spheroids to a large gradient of APAP concentrations, which was prepared as described in STAR Methods. The APAP stock solution was marked with a magenta fluorescent probe, while the dilutant solution contained a green fluorescent probe. In this way, the two fluorescent signals could be used to determine the APAP concentration in each of the droplets (Figure 4G). These drug droplets were injected into the chip, trapped, and merged with the spheroid droplets (Figures 4H and 4I). Consequently, the spheroids on a single device were exposed to a range of APAP concentrations covering three decades (Figure 4J), and the evolution of cell death was dynamically monitored for 36 h in each spheroid by live viability staining. Diffusion of the mortality dye (propidium iodide [PI]) was verified to occur within a few tens of minutes (Figure S4).
The results at the low and high extremes of the concentration range were as expected: spheroids that were exposed to control droplets or low APAP concentrations remained viable for the duration of the experiment (Figure 5A), whereas the viability of the spheroids exposed to high APAP concentrations was altered (Figure 5B). The mean viability at 24 h displayed the typical sigmoidal shape on a logarithmic scale (Figure 5C), with a half maximal inhibitory concentration (IC50) of 18.0 mM (a similar value is found in 2D; Figure S5). The variability among microfluidic chips was very low, because the IC50 CV was only 3% (nchips = 4). The IC50 value of 18.0 mM was close to the value reported in the literature (Fey and Wrzesinski, 2012, Ramaiahgari et al., 2014), for similar cells like HepG2 (Schoonen et al., 2012). Because cytochrome (CYP) enzymes play a major role in APAP toxicity (Jaeschke and Bajt, 2006), liver cells that express higher levels of CYP enzymes, like HepaRG cells (Anthérieu et al., 2010), are more sensitive to APAP, with an IC50 close to 5 mM (Gunness et al., 2013, Wang et al., 2015).
Figure 5.

Measurment of APAP Toxicity on H4-II-EC3 Spheroids
(A and B) Time-lapse images showing a spheroid without drug (A, after merging with a control droplet) and a spheroid exposed to a 48.7 mM APAP concentration (B), in bright field (bottom) and with fluorescent viability staining (top).
(C) Viability values at the spheroid level after a 24 h exposure (nspheroids = 685). Each black dot represents one spheroid, and the red and blue curves represent the mean behavior and a sigmoidal fit of the data, respectively, with the blue dashed lines highlighting the IC50 value of 18.0 mM.
(D) Time-lapse images showing a spheroid exposed to a 22.1 mM APAP concentration with PI (red). White dots are the locations of the detected dead nuclei, and the cross represents the spheroid center. R is the equivalent radius of the spheroid, and r is the distance to the spheroid center.
(E and F) Time evolution of the number of dead cells detected on one spheroid image (E) and of the mean normalized distance of the dead cells to the spheroid center (F) depending on the drug concentration. Scale bars are 50 μm.
More interestingly, the experiments yielded a deeper understanding of the drug response when the time evolution of each spheroid was tracked at the level of individual cells (Figure 5D). For this purpose, we detected the apparition of dead cells as a function of time (Figure 5E) and measured their distance to the spheroid center for each of the 685 spheroids (Figure 5F; see STAR Methods for the image analysis details, with individual curves shown in Figure S6). At low APAP concentrations, the number of dead cells remained below five cells per image and their mean location in the spheroid was constant and close to the spheroid edge (, light and dark blue lines). For higher APAP concentrations, the number of dead cells increased significantly after 10 h to reach up to 30 dead cells at the end of the experiment, corresponding to all cells on an epifluorescence image. In addition, the position of these dead cells shifted toward the spheroid center with time and concentration, with dropping from 0.68 to 0.55 for an APAP concentration higher than 40 mM (red line). This indicated that the drug concentration influenced the number and the location of dead cells within the spheroids in a time-dependent manner.
The single-cell level of detail allowed us to address the wide spread that was observed for intermediate concentrations of APAP. For APAP concentrations between 15 and 23 mM, spheroids could have a very low, very high, or intermediate viability (Figure 6A). One major parameter for explaining this spread was the presence of dead cells in the spheroids at the outset of the experiment. Spheroids with at least one dead cell at t = 0 h displayed, at t = 24 h, a significantly lower viability (24%) than the spheroids without dead cells initially (60%, Figure 6B). Moreover, the location of these first detected dead cells was significantly correlated to the viability after 24 h (Figure 6C): the spheroid was more likely to have a low viability at t = 24 h when the first dead cell was close to the spheroid center ( < 0.7). The signature of the initial state was again visible when observing the time evolution of the viability of each spheroid (Figure 6D; Figure S6), in which the curves displayed different trends depending on the presence or absence of a dead cell initially. This observation motivated us to question what part of the viability dynamics was determined by the drug concentration and what part depended on the eigenstate (the structure, interactions, and initial spheroid state) of each spheroid in time. To evaluate the effects, we defined two timescales to describe the evolution of each spheroid, as sketched in Figure 6E: the time to reach 75% viability and the time required to drop from 75% to 25% viability . The first timescale could be considered the time required for the cells to begin responding to the drug, while the second one described the time required to kill the spheroid once the process began. These two parameters were found to be independent of each other (Figure 6F). They also had different mean values and variances (Figure 6G), such that could be considered nearly constant compared with . Indeed, showed a significant dependence on APAP concentration, dropping from 18 to 8 h for APAP concentration from 10 to 70 mM (Figure 6H), while remained constant close to 7 h for all concentrations (Figure 6I). These measurements suggested that was the more relevant parameter to understand the effect of the drug, while was characteristic of the response of these spheroids to a change in their microenvironment.
Figure 6.

Analysis of the Viability
(A) Toxicity values after a 24 h exposure. Data points corresponding to spheroids exposed to a 14–23 mM range of APAP concentrations are highlighted in purple (same dataset as Figure 5C).
(B) Influence of the number of initial dead cells on the viability after a 24 h exposure for an APAP concentration between 15 and 23 mM (red, at least one initial dead cell; blue, no initial dead cell; nspheroids = 98).
(C) Correlation between the viability at t = 24 h and the mean normalized distance of the first detected dead cells (whatever its time of appearance) to the spheroid center (nspheroids = 308), for an APAP concentration between 10 and 40 mM.
(D) Dynamic evolution of the spheroid viability for an APAP concentration between 15 and 23 mM. Each thin line represents one spheroid (nspheroids = 98); the red and blue curves correspond to the spheroids that had at least one detected dead cell and no detected dead cell at t = 0 h, respectively; and the thick black line represents the overall mean.
(E) Definition of the time needed to reach a 75% viability and the time to go from a 75% to a 25% viability ( = − ) on a viability followup corresponding to a spheroid exposed to a high APAP concentration (above 40 mM).
(F) Low correlation (Pearson’s correlation coefficient = −0.06) between and (nspheroids = 215) shows that the two parameters are independent.
(G) Evaluation of (nspheroids = 262) and (nspheroids = 215).
(H and I) Evolution of (H) and (I) with the APAP concentration.
N.S., nonsignificant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Statistical test details are provided in Table S2.
Discussion
As the demand for highly relevant culture models becomes stronger (Horvath et al., 2016), many approaches have been explored for structuring 3D cultures that capture essential aspects of the in vivo microenvironment (Picollet-D’hahan et al., 2016). Among these approaches, spheroids constitute an interesting format that balances biological relevance while remaining simple to produce in large quantities. Nevertheless, spheroid manipulation remains laborious and technical, which severely limits the ability to generate large datasets on various culture conditions (Fennema et al., 2013). In this context, we have recently demonstrated the ability to obtain multiscale cytometry by performing phenotypic measurements (in situ immuno-cyto-chemistry, qRT-PCR on recovered spheroids, and ELISA on the chip supernatant) on 105–106 individual cells in situ within thousands of spheroids (Sart et al., 2017). We showed elsewhere that similar microfluidic techniques can be applied to the study of single CHO-S (Chinese ovary) cells (Vitor et al., 2018). Here we complete the toolbox by developing a protocol to manipulate independently the hundreds of spheroids present on a microfluidic device in a time-dependent manner.
The ability to generate the cellular interactions and data on large numbers of spheroids requires protocols that combine many individual operations, as shown earlier. Although most of these operations are available individually on alternative platforms or with other 3D culture formats (Yu et al., 2018), integrating them into a complete protocol requires a high level of robustness and tolerance to errors, because each new step can introduce new sources of error. The current platform is expected to display particularly strong performance for a range of applications, such as for generating large amounts of single-cell data on a few well-controlled conditions. This improvement in performance stems from the use of microfluidics to handle the fluids, the encapsulation offered by the droplet methods, the tolerance of stationary droplet methods to experimental error, and the integration of all operations on a single device.
The protocols shown here are based on the ability to reliably pair two droplets at any moment over a period of several days. For this, the current microfluidic design uses anchors composed of regions with two widely varying trapping strengths, such that the primary droplet can be trapped nearly irreversibly while other droplets are brought into contact at later times. This contrast in trapping efficiencies makes the device operation possible for a range of physical and experimental parameters (viscosities, flow rates, droplet sizes, etc.), which translates into stronger robustness and stability compared with existing designs (e.g., Bai et al., 2010, Huebner et al., 2011, Fradet et al., 2013, Tullis et al., 2014). These qualities bring droplet microfluidics closer to the functionalities of multiwell plates in which any multistep chemical or biological assay is possible (Kaminski and Garstecki, 2017).
If the first droplet contains a spheroid, the second droplet can be used to bring fresh culture medium to increase the culturing time in confined droplets. More interestingly, the user can add different cell types, for example, to build microtissues within the droplets (Chen et al., 2016), to study cell-cell interactions (Pawlizak et al., 2015), or to reproduce crucial steps in development (Bagley et al., 2017). Applying our microfluidic approach to these problems leads to the emergence of non-trivial cellular organizations within the fused spheroids: when the cell types constituting each spheroid are homogeneous, the fusion of two spheroids display a side-by-side juxtaposition (e.g., Figure 3D). In contrast, when the cell types have different phenotypes, a core-shell structure is observed (e.g., Figure 3E). The microfluidic format is unique in its ability to easily scale up these fusions to hundreds of parallel microtissues per experiment.
Alternatively, the format allows the study of antagonistic interactions as in immuno-therapy models (Gravelle et al., 2014) or host-pathogen interactions (Voznica et al., 2017), with potential applications to drug development or basic biology. Furthermore, the second droplet can contain a hydrogel that is gelled after the droplet fusion, which can also be used for a range of applications, such as for metastasis invasion assays (Glentis et al., 2017) or as functionalized scaffolds to capture secreted molecules (Ranga et al., 2014).
Beyond these illustrative examples, the ability to manipulate individual spheroids allows us to test the toxicity of a drug on 3D cultures. For this, we adapt previous work that uses fluorescent droplet barcoding (Gielen et al., 2015, Eduati et al., 2018, Kulesa et al., 2018) to identify APAP droplets inside a pool of 26 other colors and confirm its toxicity on liver spheroids. Although such multiplexing is not yet sufficient for high-throughput screening, the amount of detail that can be obtained by the image analysis allows us to address fundamental questions on 3D cell culture.
To this end, we perform a complete analysis of the toxicity of APAP over a range of three decades in concentration. This analysis includes generating the droplet library, merging the library with the spheroids, following the response of individual cells within hundreds of spheroids, and analyzing the results at the single-cell level and as a function of time. The results obtained in this section are noteworthy. First, such detailed data would have been more difficult to obtain using the current state of the art (Gunness et al., 2013, Messner et al., 2013, Sirenko et al., 2016). Here, the integrated microfluidic format makes the experiments simple to perform and the measurements straightforward. Second, the large number of spheroids involved in the study highlights the presence of a strong heterogeneity of responses near the IC50 value, in which some spheroids are viable and others are dead after 24 h. By using the single-cell longitudinal measurements, this variety of responses is found to correlate with the presence of dead cells within the spheroids at the initial moment, indicating that the structural integrity of the spheroid plays a role in its ability to resist a drug treatment.
The viability measurements show complex dynamics, with the emergence of two timescales from the time-resolved single-cell measurements: that depends on the drug concentration and that does not. These observations suggest that once a spheroid has reached a 75% viability, it will proceed to die at a rate that is proper to the spheroid and independent of the external stimulus. If we consider a particular cell whose neighbors die because of the drug, leading it to lose its focal adhesions and to be subjected to toxic hydrolases, the change in its microenvironment can induce (or at least precipitate) its death for purely structural reasons. Such collective structural effects, which are not present in 2D monolayers, could only be detected by the combination of live imaging and detailed quantitative analysis (intra-spheroid heterogeneity) provided by our technology.
Looking ahead, the technological tools presented here can be transposed to other 3D culture formats, including organoids (Takebe et al., 2017), blastoids (Rivron et al., 2018), or embryoid bodies (Vrij et al., 2016), following some adaptations of the protocols. In addition, it is straightforward to combine the different operations, such as building 3D microenvironments in the microfluidic anchors and then testing the effects of different molecules on these complex models or testing combination treatments involving small molecules, cellular therapy, and other approaches in a single device.
Finally, as stated in the introduction, the most suitable 3D model depends on the question being addressed. So future work will involve a careful choice of the cell types, hydrogels and combinations, the 3D culture format, and validation steps to ensure that the results obtained from the model system are relevant to the in vivo conditions. The microfluidic platform described here is designed to make such studies and validation more efficient and reproducible.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa Fluor® 647 Mouse Anti-Human CD146 (clone P1H12) | BD | Cat#563619, RRID: AB_2738323 |
| Biological Samples | ||
| Umbilical Cord-Derived Mesenchymal Stem Cells | ATCC | PCS-500-010 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Acetaminophen | Sigma-Aldrich | Cat#A7085 |
| Experimental Models: Cell Lines | ||
| H4-II-EC3 cells | ATCC | CRL-1600 |
Resource Availability
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Charles N. Baroud (charles.baroud@pasteur.fr).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The code supporting the current study has not been deposited in a public repository but is available from the corresponding author on request.
Experimental Model and Subject Details
Cell Culture
A rat H4-II-EC3 hepatoma cell line (American Type Culture Collection, CRL-1600™, LGC), A-673, an muscle Ewing’s Sarcoma cell line (ATCC, CRL-1598™, LGC) and B16-F0, a mouse melanoma cell line (ATCC, CRL-6322™, LGC) were maintained on T-25 cm2 flasks (Corning) in a standard CO2 incubator (5% (v/v) CO2, C150 incubator, Binder), following the instructions provided by the manufacturer. For the H4-II-EC3 and A-673, the culture medium was composed of Dulbecco’s Modified Eagle’s Medium (DMEM, ThermoFischer) containing high glucose supplemented with 10% (v/v) fetal bovine serum (FBS, ThermoFischer) and 1% (v/v) penicilin-streptamicine (ThermoFischer). For the B16-F0, the culture medium was composed of RPMI 1640 (ThermoFischer), with a similar amount of FBS and antibiotics. The cells were seeded at 5.104 cells/cm2 and sub-cultivated every 3 days.
Human mesenchymal stromal cells derived from the Wharton’s jelly of umbilical cord (UC-hMSCs) (ATCC, PCS-500-010, lot #63516504, LGC) were obtained at passage 2. UC-hMSCs were maintained in T-175 cm2 flasks (Corning) and cultivated in a CO2 incubator. The culture medium was composed of Alpha Modified Eagle’s medium (α-MEM) (GIBCO, ThermoFischer) supplemented with 10% (v/v) fetal bovine serum (GIBCO) and 1% (v/v) penicilin-streptamicine (GIBCO). The cells were seeded at 5.103 cells/cm2, sub-cultivated every week, and the medium was refreshed every 2 days. UC-hMSCs at passage 2 were first expanded until passage 4 (for about 5-6 populations doublings, PDs), then cryopreserved in 90% (v/v) FBS / 10% (v/v) DMSO and stored in a liquid nitrogen tank. The experiments were carried out with UC-hMSCs at passage 4 to 8 (about 24-35 PDs, after passage 2).
Bacteria culture
Esherichia coli (K12 - MG 1655-GFP strain) expressing constitutively GFP were cultivated as colonies on LB-agar plates. The day of the fusion experiments, E.coli were cultivated in suspension using LB medium up to reaching an O.D. of 0.5.
Method Details
Estimation of the trapping and drag forces
Let us consider the anchors described in the main text in Figures 1 and 2. The microfluidic channel has a height h and the circular parts of these anchors have a diameter d and a depth . In order to estimate the trapping force of the first droplets in the circular parts of the anchors, the variation of surface area before and after trapping must be estimated. The surface area of a confined droplet is estimated by considering pancake shape, meaning a cylinder of radius , height h surrounded by the outer half of a torus of small radius . As we consider the case where the droplet size matches the circular part volume of the asymmetric anchor, the surface area of the trapped droplet is estimated by calculating the area of a cylinder whose section is the circular part of the anchor and whose height is . Thus, the trapping force of the first droplets in the circular parts of the anchors is:
where the characteristic length l over which the surface energy changes is estimated by .
For estimating the trapping force of the first droplets in the triangular parts, the triangular parts are assimilated to small circular anchors, whose radius is smaller than the channel height. According to Dangla et al. (2011), the resulting trapping force is:
With the experimental parameters used for Figure 2 ( = 125 μm, = 250 μm, = 95 μm, = 50 μm) and assuming = 150 μm (equal to the length and width of the anchor triangle), we have:
So, for the first droplets, the trapping in the circular parts of the anchors is much more efficient than the trapping in the triangular parts.
Also, the drag force exerted by the fluid on a confined droplet scales as , where and are respectively the mean velocity and viscosity of the outer fluid. Therefore, the ratio between the drag force exerted on the large first ( = 170 μm) and small second ( = 80 μm) confined droplets before trapping is:
Consequently, if the small droplets experience a similar trapping force in the triangular parts of the anchors than the large droplets, they are exposed to a smaller drag at constant flow rate, meaning that their trapping in the triangular parts of the anchors is more robust.
Droplet library statistics
Since secondary droplets get injected and trapped randomly in the anchor array, the number of different barcodes in a chemical library and the number of traps used in the experiment need to be careful chosen. The probability to have one particular barcode in one anchor after droplet merging is , n being the number of different barcodes. Assuming a completely random trapping of the library droplets in the array, if X is the number of times that a particular barcode is found in an anchor array, the probability P of having exactly i times this particular barcode follows a binomial distribution:
As a consequence, the probability of finding at least m times one particular barcode in the array is:
As there are n different barcodes, the probability to have at least m times each of the n possible barcodes in the array is:
In Figures 4C–4E, the probability to get at least 1 time each of the 27 barcodes in 224 traps is 0.99.
Microfabrication
Molds were mainly fabricated using standard dry film soft lithography. Up to five layers of dry film photoresist, consisting of 50 and 33 μm Eternal Laminar (respectively E8020 and E8013, Eternal Materials, Taiwan) and 15 μm Alpho NIT215 (Nichigo-Morton) negative films, were successively laminated using an office laminator (PEAK pro PS320) at a temperature of 100°C until the desired channel height, from 50 to 200 μm depending on the different cases, was reached. After each laminating step, the photoresist film was exposed to UV (LightningCure, Hamamatsu) through a photomask of the junction, channels, trapping chamber boundaries or anchors. The masters were revealed after washing in a 1% (w/w) K2CO3 solution (Sigma-Aldrich). For the 3D anchors fabrication (Figures 3, 4, and 5), a specific method was developed. In these cases, the top of the chip consisted of the flow-focusing device and chambers and the anchors were located at the bottom of these chips. The anchors mold was designed with RhinoCAM software (MecSoft Corporation) and was fabricated by micro-milling a brass plate (CNCMini-Mill/GX, Minitech Machinery). That was also the case for the droplet library producing chips with an aqueous injector and a slope (see Figure S1). The topography of the molds and masters were measured using an optical profilometer (VeecoWyco NT1100, Veeco). For the fabrication of the top of the chip, poly(dimethylsiloxane) (PDMS, SYLGARD 184, Dow Corning, 1 g of curing agent for 10 g of bulk material) was poured over the master and cured for 2 h at 70°C. For the 3D anchors, the metallic mold was first covered with PDMS. Then, a glass slide was immersed into uncured PDMS, above the anchors. The mold was finally heated on a hot plate at 180°C for 15 minutes before extraction of the glass slides covered by a thin layer of PDMS with the anchor pattern. In all cases, the top and the bottom of chip were sealed after plasma treatment (Harrick).
Chip Design
Two main different chip designs were used in this study, depending on the presence of cells. For the experiences involving cells (Figures 3, 4, and 5), the chip design is shown on Figure 2A. In this case, there were 252 anchors disposed along an hexagonal pattern in the 2 cm2 trapping chamber that has a 165 μm height. For the non-biological experiments (Figure 2), the design was similar but with different dimensions. Notably, the chamber height is 95 μm. In this case, contrary to the cellular chip, the 50 μm deep anchors were patterned on the top of the chamber.
Experimental Microfluidic Protocol
The chips were filled 3 times with Novec Surface Modifer (3M), a fluoropolymer coating agent, for 30 min at 110°C on a hot plate. All experiments were conducted using the FC40 fluorinated oil (3M) implemented with a biocompatible FluoroSurfactant (Ran Biotechnologies) at different concentrations. The solutions were loaded in glass (SGE) or plastic (Terumo) syringes, that were actuated with programmable and computer controlled syringe pumps (neMESYS, Cetoni). The syringes were directly connected to the PDMS chips with PTFE tubing (Adtech). For the merging of droplet pairs, the trapping chambers were perfused with a 20% (v/v) 1H,1H,2H,2H-perfluoro-1-octanol (Sigma-Aldrich) solution dissolved in Novec™-7500 Engineered Fluid (3M) at the flowrate indicated in Table S1. The uncolored and dark droplets seen in Figure 2 are respectively made of pure water and of a 6 mM 2,6-dichlorophenolindophenol (2,6-DCPIP, Sigma-Aldrich) aqueous solution.
Spheroid Formation on Chip
The chips were first filled with a 3% (w/w) FluoroSurfactant solution. All air bubbles were discarded. H4-II-EC3 cells were detached from the culture flasks with a 2-3 minutes incubation in TrypLE™ Express enzyme (ThermoFischer), that was then inactivated by addition of warm medium. The resulting cell solution was centrifuged (centrifuge 5702 R, Eppendorf) at 2,400 rpm for 6 minutes while the cell concentration was determined using a haemocytometer (Marienfeld). The supernatant was discarded and the cell pellet was resuspended at a 6.106 cells/mL for direct use, or 8.6.106 cells/mL before addition of agarose, in culture medium supplemented with gentamicin (Sigma-Aldrich) to a final concentration of 50 mg/L. When needed, the agarose stock solution was prepared in parallel. Ultra-low gelling agarose (Type IX-A, Sigma-Aldrich) was dissolved at a 3% (w/w) concentration in warm sterile PBS implemented with gentamicin to a final concentration of 50 mg/L and kept at 37°C. 30 μL of the agarose stock solution and 70 μL of the cell solution were mixed to obtain a final cell concentration of 6.106 cells/mL in a 0.9% (w/w) agarose solution. One glass syringe was loaded with this solution and droplets were produced according to the flowrates displayed in Table S1. Spheroids of hMSCs, A673, PC-3 and H4-II-EC3 were formed in droplets containing DMEM medium, while Jurkat and B16-F0 were loaded on chip in RMPI medium. The cell loading was performed at 37°C in a microscope incubator (Okolab) in which all chips, syringes, connectics and solutions were pre-heated. After the loading, all flowrates were stopped, the tubings were cut and the chips were kept immersed in PBS in the CO2 incubator. Cells started sedimenting at the bottom of each droplet when the flowrates were stopped. They reorganized overnight in the liquid agarose droplets into spheroids. For the toxicity experiments (Figures 4 and 5), the gelation allowed to immobilize the spheroids at the bottom of their droplets, facilitating live imaging.
Spheroid Staining for Co-culture Experiments
A 10 mM solution of CellTracker™ Red and Green (ThermoFischer) was prepared in sterile DMSO (PAN Biotech). H4-II-EC3 cells were incubated for 30 min in culture medium with 10 μM of CellTracker™ in the culture flask, before PBS washing and exposition to the TrypLE™ express enzyme. Alternatively, Jurkat and hMSCs were stained for Vybrant™ Dil (red) and PC-3 were labeled with Vybrant™ Dio (green), following the manufacturer instructions (ThermoFisher).
Cell sorting
hMSCs were harvested by scrapping or trypsinization from T-175 cm2 flasks. Then, the cells were incubated in staining buffer (2% (v/v) FBS in PBS), stained with a mouse anti-human CD146-Alexa Fluor®647 (Clone P1-H12, BD Bioscience) for 30 min. hMSCs were then sorted based on their level of expression of CD146 using a FACSAria III (BD Bioscience, San Jose, CA): CD146bright constitured 20% of cell population expressiong the highest levels of CD146, while CD146dim constitute 20% of cell population expressiong the lowest levels of CD146.
Matrigel Droplets
Matrigel (Matrigel Matrix Basement Membrane, Corning) was aliquoted and kept at −20°C. One aliquot was thawed on ice for about 30 min before the experiment. Droplets were produced, trapped close to the spheroid droplets and merged at 4°C in a cold room. The chip was then kept in the cell incubator at 37°C allowing the gelation to take place.
Protocol For Droplet Library Production
Droplet libraries were produced following two successive steps. First, the two solutions of interest were mixed at a PEEK cross junction (Upchurch) in known ratios controlled by programmable syringe pumps (neMESYS, Cetoni). Between each different ratio, a plug of oil (FC40 with a 0.25% (w/w) concentration of FluoroSurfactant) was injected at the cross junction for physically separating the different droplets in the exit tubing. This technique is called micro-segmented flow (Funfak et al., 2009) and results in a train of microliter droplets separated by oil, each of them with a different ratio of the two aqueous stock solutions (see Figures S2A–S2C). In the present study, the ratios followed either a linear (Figure 2; Figure S1) or logarithmic (Figures 4 and 5) progression.
Second, these segments were partitioned into nanoliter droplets (Kaminski et al., 2012) using a specific droplet producing chip (Figure S1). The segments were injected in a chamber filled with oil through a slope (see Figure S1A). The slope allows to continuously deconfine the aqueous phase that spontaneously breaks into monodisperse droplets, without the need of an external oil flow (Figure S1B). The droplet size is governed by the geometrical parameters of the injector, namely its height and width, as well as the angle of the slope. The nanoliter droplets resulting from this production were brought in the storage chamber of the chip thanks to a small continuous oil flow (FC40 with 6% (w/w) FluoroSurfactant) at the corner of the slope (Figures S1C and S1D). The droplets ascended to the top of this very deep storage chamber resulting in the trapping of the produced droplet library.
Droplet Library Injection in the Anchors Array
During the segment partitioning, the droplets were kept in the storage chamber of the microfluidic droplet producing chip. Before injection into a trapping chamber with capillary anchors, the droplet producing chip was manually flipped over several times to mix the different droplet types inside the storage chamber (Figure S2G). Then, the chip was connected to the aqueous inlet of anchor array chip, and it was maintained upside down to allow the droplets to escape the storage chamber (Figure S1M). Using the aqueous inlet for the droplet injection presented several advantages. The separation between the droplets in the trapping chip was controlled by the oil flowrate at the junction inlet and it also broke down possible large droplets coming from a coalescence event during the droplet transfer. In addition, the flowrate in the chamber was controlled independently with the chamber inlet.
Food Dye Droplet Library Production
The color droplets shown in Figure 2 and Figure S1 were produced by mixing commercially available yellow, blue, red food dyes (Vahiné) and pure water in known ratios. The syringe pumps were programmed to create 11 segments (5 with a linear increase of the first solution, 1 purely made of the first solution, and 5 with a linear decrease of first solution), each of 2 μL, separated by 1 μL of oil at a global constant flowrate of 20 μL/min. To partition these segments at the desired volumes, two droplet producing chips were used. For the first and second droplets, the geometrical parameters of the chip were respectively a slope of 8 and 11%, an injector width of 100 and 90 μm and an injector height of 40 μm for both. The flowrates are indicated in Table S1.
Droplet library production with fluo barcodes
3 fluorescent dyes at 3 concentrations were mixed in a combinatorial manner to obtain 27 barcodes. CF™647 hydrazide was used at 70.0 μM, 18.7 μM and 5.00 μM. CF™488A hydrazide was used at 100 μM, 9.49 μM and 900 nM. CF™405 hydrazide was used at 350 μM, 102 μM and 30.0 μM. These signal of these 3 dyes is respectively represented in Figures 4C–4E in red, green and blue. Acetaminophen (APAP, Sigma-Aldrich) was added at a 225 mM concentration in the solution corresponding to barcode #09. All solutions were implemented with DMSO (8% (v/v)), gentamicin (50 mg/L) and propidium iodide (3.0 μM, Sigma-Aldrich).
1 μL of each barcode solution was aspirated sequentially, with 1 μL of fluorinated oil in between each plug. These segments were injected in a droplet production chip with the following geometrical parameters: an injector width and height respectively of 100 and 40 μm and a slope of 8%. The volume of the first (with the cells) and second (with the drug) droplets was respectively estimated to 12 nL and 60 nL, so the dilution factor from the library to the post-merging droplets was approximately 6.
Acetaminophen Droplet Library Production
Two stock solutions were prepared for the acetaminophen (APAP, Sigma-Aldrich) droplet library production, with a 3 mM and 300 mM drug concentration. Both of them were implemented with DMSO (8% (v/v)) and gentamicin (50 mg/L). For the live viability staining, the 2 solutions of the manufacturer (ReadyProbes™ Cell Viability Imaging Kit (Blue/Red), ThermoFischer) were diluted to a 17.5% (v/v) concentration. The low and high APAP drug concentration solutions were marked respectively with a green and red fluorescent dye (CF™488A hydrazide and CF™647 hydrazide, Sigma-Aldrich) at a 7 μM concentration. The dilutant was culture medium. These fluorescent dyes did not have any observable effect on the spheroids viability at the highest concentration used in this study. A solution was also prepared for the control droplets with the same composition as the drug solution (including DMSO), without the APAP and the CF™ fluorescent dyes.
12 segments of 3 μL each were produced to cover regularly a logarithmic scale between the low and high concentration solutions. An additional 3 μL segment of the control solution was added before partitioning. These segments were injected in a droplet production chip with the following geometrical parameters: an injector width and height respectively of 100 and 40 μm and a slope of 8%. The volume of the first (with the cells) and second (with the drug) droplets was respectively estimated to 12 nL and 60 nL, so the dilution factor from the library to the post-merging droplets was approximately 6. The concentration range tested here corresponded to a large excess of APAP molecules (about 108 APAP molecules per cell in a 1 mM APAP droplet).
Determination of the APAP Concentration in Droplets
The drug concentration in the droplets was determined right after the merging of the spheroid and drug droplets by measuring the signals from the CF™ dyes. For each anchor, the fluorescent signal was defined as the local background (average of the signal just outside this anchor) subtracted from the raw fluorescent intensity (average of the raw intensity in the middle of the deep part of the anchor). This signal was correlated to a drug concentration using the calibration curves of the CF™ dyes shown in Figure 4C. In order to replicate the conditions of the experiments, the calibration was made by trapping large droplets of culture medium in the deep parts of the anchors with a similar concentration of viability dyes, DMSO and a known concentration of each CF™ dye. 4 concentrations were loaded in each chip at the same time (Sart et al., 2017), giving about 60 fluorescent droplets per concentration. 2 chips were used for getting the 8 measurements of each calibration curve. The droplets were imaged with the same experimental parameters than for the toxicity experiments.
Diffusion of PI in the Spheroids
Spheroids were formed with H4-II-EC3 at 6.106 cells/mL in droplets containing 0.9% (w/w) agarose, as described in the section “Spheroid Formation on Chip.” After spheroid formation, the agarose was gelled by placing the chip at 4°C for 20 min. The oil phase was then exchanged to culture medium containing 50 mM APAP. The chip was then incubated for 24h. The day of the experiment, a solution of 1 mL of culture medium containing two drops of ReadyProbes Red (PI, ThermoFischer) was perfused in the culture culture chamber and image acquisition was started at the same time.
Image Acquisition
Images without cells were acquired either on a binocular (MZ16 FA, Leica) using a CCD camera (Insight camera, 4MP Firewire, SPOT), or, for the colored droplets, with a digital single-lens reflex camera (D7000, Nikon). The droplet production images of Figure S1B were acquired with a high speed camera (FASTCAM 1024 PCI, Photron). The cellular fluorescent images were taken with an inverted microscope (Eclispe Ti, Nikon), equipped of a motorized stage, an illumination system (Spectra-X, Lumencor) and a temperature controlled incubator (Okolab), with a CMOS camera (ORCA Flash 4.0, Hamamatsu).
Quantification and Statistical Analysis
Image Analysis
For quantifying the droplet colors in Figure 2, the entire array was imaged using the binocular and the reflex camera. Then, a custom MATLAB code (R2016a, Mathworks) allowed to detect each anchor and to compute the RGB values in the center of each droplet, before and after merging. For the droplet fluorescent barcode assignment in Figure 4 and Figure S5, a signal range was manually set for each dye concentration, leaving the few droplets having one signal out of range unassigned.
For the toxicity experiment, single images of the anchors were acquired automatically with the motorized stage of the microscope. The analysis was conducted on a montage of the detected anchors using a protocol previously described (Sart et al., 2017). Briefly, cells were detected using bright field and fluorescent intensities, and spheroids were selected based on morphological parameters. For each spheroid, the local background was used to determine a specific threshold for the fluorescent dead cells. The viability at the spheroid level was then defined as:
and being respectively the number of dead pixels and area of the spheroid. A similar analysis was conducted for determining the viability of the 2D cultures.
At the cellular level, dead cell centers were detected as the local maxima of the fluorescent mortality marker, above the local threshold. Then, the radial distance of each dead cell center was computed and compared to the equivalent radius R of the spheroid to define the mean normalized distance of dead cell centers to the spheroid center . It was close to 0 if all detected dead cells were close to the spheroid center and close to 1 if they were close to the spheroid edge.
Statistical analysis
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; N.S.: non-significant. p value ranges are only indicated for the highlighted comparisons. For each test, the normality and equality of variances (homoscedasticity) were tested using respectively Anderson-Darling and Bartlett’s tests. Then, the proper statistical test was chosen accordingly each case. Details of the hypothesis and p values can be found in Table S2. In the Tukey box-and-whiskers figures (Figure 6), the boxes represent the first (q1) and third (q3) quartiles with the median shown by the line bissecting the box, and the mean is shown with black circles. The whiskers represent 1.5 times the inter-quartile range (q3-q1) of the sample. Finally, the box width is proportional to . Exact values of n are reported in each figure legend.
Acknowledgments
Caroline Frot is acknowledged for her help with the microfabrication, and Gabriel Amselem is acknowledged for useful discussions. The A673 and B16-F0 cells were provided, respectively, by Karine Laud Duval from the U830 INSERM/Institut Curie unit and Philippe Bousseau from the U1223 INSERM/Institut Pasteur unit. The research leading to these results received funding from the European Research Council (ERC) grant agreement 278248 (Multicell) and ERC-POC grant agreement 768790 (Multiscreen). The Biomaterials and Microfluidics (BMCF) group of the Center for Innovation and Technological Research, as well as the Center for Translational Science (CRT)-Cytometry and Biomarkers Unit of Technology and Service (CB UTechS), are also acknowledged for access to the microfabrication and flow cytometry platforms at Institut Pasteur.
Author Contributions
Conceptualization, R.F.-X.T. and C.N.B.; Methodology, R.F.-X.T. and C.N.B.; Software, R.F.-X.T.; Formal Analysis, R.F.-X.T.; Investigation, R.F.-X.T., S.S., and T.C.; Writing – Original Draft, R.F.-X.T. and C.N.B.; Writing – Review & Editing, R.F.-X.T., S.S., and C.N.B.; Visualization, R.F.-X.T.; Supervision, C.N.B.; Funding Acquisition, R.F.-X.T. and C.N.B.
Declaration of Interests
R.F.-X.T., S.S., and C.N.B. are co-inventors of two patents related to this work, owned by CNRS and Ecole Polytechnique.
Published: May 26, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107670.
Supplemental Information
References
- Abbyad P., Dangla R., Alexandrou A., Baroud C.N. Rails and anchors: guiding and trapping droplet microreactors in two dimensions. Lab Chip. 2011;11:813–821. doi: 10.1039/c0lc00104j. [DOI] [PubMed] [Google Scholar]
- Akartuna I., Aubrecht D.M., Kodger T.E., Weitz D.A. Chemically induced coalescence in droplet-based microfluidics. Lab Chip. 2015;15:1140–1144. doi: 10.1039/c4lc01285b. [DOI] [PubMed] [Google Scholar]
- Alessandri K., Sarangi B.R., Gurchenkov V.V., Sinha B., Kießling T.R., Fetler L., Rico F., Scheuring S., Lamaze C., Simon A. Cellular capsules as a tool for multicellular spheroid production and for investigating the mechanics of tumor progression in vitro. Proc. Natl. Acad. Sci. USA. 2013;110:14843–14848. doi: 10.1073/pnas.1309482110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthérieu S., Chesné C., Li R., Camus S., Lahoz A., Picazo L., Turpeinen M., Tolonen A., Uusitalo J., Guguen-Guillouzo C., Guillouzo A. Stable expression, activity, and inducibility of cytochromes P450 in differentiated HepaRG cells. Drug Metab. Dispos. 2010;38:516–525. doi: 10.1124/dmd.109.030197. [DOI] [PubMed] [Google Scholar]
- Bagley J.A., Reumann D., Bian S., Lévi-Strauss J., Knoblich J.A. Fused cerebral organoids model interactions between brain regions. Nat. Methods. 2017;14:743–751. doi: 10.1038/nmeth.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y., He X., Liu D., Patil S.N., Bratton D., Huebner A., Hollfelder F., Abell C., Huck W.T. A double droplet trap system for studying mass transport across a droplet-droplet interface. Lab Chip. 2010;10:1281–1285. doi: 10.1039/b925133b. [DOI] [PubMed] [Google Scholar]
- Bartosh T.J., Ylöstalo J.H., Mohammadipoor A., Bazhanov N., Coble K., Claypool K., Lee R.H., Choi H., Prockop D.J. Aggregation of human mesenchymal stromal cells (MSCs) into 3D spheroids enhances their antiinflammatory properties. Proc. Natl. Acad. Sci. USA. 2010;107:13724–13729. doi: 10.1073/pnas.1008117107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell C.C., Dankers A.C.A., Lauschke V.M., Sison-Young R., Jenkins R., Rowe C., Goldring C.E., Park K., Regan S.L., Walker T. Comparison of hepatic 2D sandwich cultures and 3D spheroids for long-term toxicity applications: A multicenter study. Toxicol. Sci. 2018;162:655–666. doi: 10.1093/toxsci/kfx289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia S.N., Ingber D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014;32:760–772. doi: 10.1038/nbt.2989. [DOI] [PubMed] [Google Scholar]
- Chen Q., Utech S., Chen D., Prodanovic R., Lin J.M., Weitz D.A. Controlled assembly of heterotypic cells in a core-shell scaffold: organ in a droplet. Lab Chip. 2016;16:1346–1349. doi: 10.1039/c6lc00231e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiew G.G.Y., Wei N., Sultania S., Lim S., Luo K.Q. Bioengineered three-dimensional co-culture of cancer cells and endothelial cells: A model system for dual analysis of tumor growth and angiogenesis. Biotechnol. Bioeng. 2017;114:1865–1877. doi: 10.1002/bit.26297. [DOI] [PubMed] [Google Scholar]
- Cullen D.K., Wolf J.A., Vernekar V.N., Vukasinovic J., LaPlaca M.C. Neural tissue engineering and biohybridized microsystems for neurobiological investigation in vitro (Part 1) Crit. Rev. Biomed. Eng. 2011;39:201–240. doi: 10.1615/critrevbiomedeng.v39.i3.30. [DOI] [PubMed] [Google Scholar]
- Dangla R., Lee S., Baroud C.N. Trapping microfluidic drops in wells of surface energy. Phys. Rev. Lett. 2011;107:124501. doi: 10.1103/PhysRevLett.107.124501. [DOI] [PubMed] [Google Scholar]
- Dangla R., Kayi S.C., Baroud C.N. Droplet microfluidics driven by gradients of confinement. Proc. Natl. Acad. Sci. USA. 2013;110:853–858. doi: 10.1073/pnas.1209186110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncombe T.A., Dittrich P.S. Droplet barcoding: tracking mobile micro-reactors for high-throughput biology. Curr. Opin. Biotechnol. 2019;60:205–212. doi: 10.1016/j.copbio.2019.05.004. [DOI] [PubMed] [Google Scholar]
- Eduati F., Utharala R., Madhavan D., Neumann U.P., Longerich T., Cramer T., Saez-Rodriguez J., Merten C.A. A microfluidics platform for combinatorial drug screening on cancer biopsies. Nature Communications. 2018;9 doi: 10.1038/s41467-018-04919-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fennema E., Rivron N., Rouwkema J., van Blitterswijk C., de Boer J. Spheroid culture as a tool for creating 3D complex tissues. Trends Biotechnol. 2013;31:108–115. doi: 10.1016/j.tibtech.2012.12.003. [DOI] [PubMed] [Google Scholar]
- Fey S.J., Wrzesinski K. Determination of drug toxicity using 3D spheroids constructed from an immortal human hepatocyte cell line. Toxicol. Sci. 2012;127:403–411. doi: 10.1093/toxsci/kfs122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradet E., Abbyad P., Vos M.H., Baroud C.N. Parallel measurements of reaction kinetics using ultralow-volumes. Lab Chip. 2013;13:4326–4330. doi: 10.1039/c3lc50768h. [DOI] [PubMed] [Google Scholar]
- Funfak A., Hartunga R., Caoa J., Martin K., Wiesmüller K.-H., Wolfbeis O.S., Köhlera J.M. Highly resolved dose-response functions for drug-modulated bacteria cultivation obtained by fluorometric and photometric flow-through sensing in microsegmented flow. Sens. Actuators B Chem. 2009;142:66–72. [Google Scholar]
- Gielen F., Buryska T., Van Vliet L., Butz M., Damborsky J., Prokop Z., Hollfelder F. Interfacing microwells with nanoliter compartments: A sampler generating high-resolution concentration gradients for quantitative biochemical analyses in droplets. Analytical Chemistry. 2015;87:624–632. doi: 10.1021/ac503336g. [DOI] [PubMed] [Google Scholar]
- Glentis A., Oertle P., Mariani P., Chikina A., El Marjou F., Attieh Y., Zaccarini F., Lae M., Loew D., Dingli F. Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane. Nat. Commun. 2017;8:924. doi: 10.1038/s41467-017-00985-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravelle P., Jean C., Familiades J., Decaup E., Blanc A., Bezombes-Cagnac C., Laurent C., Savina A., Fournié J.J., Laurent G. Cell growth in aggregates determines gene expression, proliferation, survival, chemoresistance, and sensitivity to immune effectors in follicular lymphoma. Am. J. Pathol. 2014;184:282–295. doi: 10.1016/j.ajpath.2013.09.018. [DOI] [PubMed] [Google Scholar]
- Gunness P., Mueller D., Shevchenko V., Heinzle E., Ingelman-Sundberg M., Noor F. 3D organotypic cultures of human HepaRG cells: a tool for in vitro toxicity studies. Toxicol. Sci. 2013;133:67–78. doi: 10.1093/toxsci/kft021. [DOI] [PubMed] [Google Scholar]
- Horvath P., Aulner N., Bickle M., Davies A.M., Nery E.D., Ebner D., Montoya M.C., Östling P., Pietiäinen V., Price L.S. Screening out irrelevant cell-based models of disease. Nat. Rev. Drug Discov. 2016;15:751–769. doi: 10.1038/nrd.2016.175. [DOI] [PubMed] [Google Scholar]
- Hou S., Tiriac H., Sridharan B.P., Scampavia L., Madoux F., Seldin J., Souza G.R., Watson D., Tuveson D., Spicer T.P. Advanced Development of Primary Pancreatic Organoid Tumor Models for High-Throughput Phenotypic Drug Screening. SLAS Discov. 2018;23:574–584. doi: 10.1177/2472555218766842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hough S.R., Thornton M., Mason E., Mar J.C., Wells C.A., Pera M.F. Single-cell gene expression profiles define self-renewing, pluripotent, and lineage primed states of human pluripotent stem cells. Stem Cell Reports. 2014;2:881–895. doi: 10.1016/j.stemcr.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebner A.M., Abell C., Huck W.T., Baroud C.N., Hollfelder F. Monitoring a reaction at submillisecond resolution in picoliter volumes. Anal. Chem. 2011;83:1462–1468. doi: 10.1021/ac103234a. [DOI] [PubMed] [Google Scholar]
- Imamura Y., Mukohara T., Shimono Y., Funakoshi Y., Chayahara N., Toyoda M., Kiyota N., Takao S., Kono S., Nakatsura T., Minami H. Comparison of 2D- and 3D-culture models as drug-testing platforms in breast cancer. Oncol. Rep. 2015;33:1837–1843. doi: 10.3892/or.2015.3767. [DOI] [PubMed] [Google Scholar]
- Jaeschke H., Bajt M.L. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol. Sci. 2006;89:31–41. doi: 10.1093/toxsci/kfi336. [DOI] [PubMed] [Google Scholar]
- Kaminski T.S., Garstecki P. Controlled droplet microfluidic systems for multistep chemical and biological assays. Chem. Soc. Rev. 2017;46:6210–6226. doi: 10.1039/c5cs00717h. [DOI] [PubMed] [Google Scholar]
- Kaminski T.S., Jakiela S., Czekalska M.A., Postek W., Garstecki P. Automated generation of libraries of nL droplets. Lab Chip. 2012;12:3995–4002. doi: 10.1039/c2lc40540g. [DOI] [PubMed] [Google Scholar]
- Kim E., Jeon W.B., Kim S., Lee S.K. Decrease of reactive oxygen species-related biomarkers in the tissue-mimic 3D spheroid culture of human lung cells exposed to zinc oxide nanoparticles. J. Nanosci. Nanotechnol. 2014;14:3356–3365. doi: 10.1166/jnn.2014.8257. [DOI] [PubMed] [Google Scholar]
- Kim T.H., Saadatpour A., Guo G., Saxena M., Cavazza A., Desai N., Jadhav U., Jiang L., Rivera M.N., Orkin S.H. Single-Cell Transcript Profiles Reveal Multilineage Priming in Early Progenitors Derived from Lgr5(+) Intestinal Stem Cells. Cell Rep. 2016;16:2053–2060. doi: 10.1016/j.celrep.2016.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulesa A., Kehe J., Hurtado J.E., Tawde P., Blainey P.C. Combinatorial drug discovery in nanoliter droplets. Proceedings of the National Academy of Sciences. 2018;115:6685–6690. doi: 10.1073/pnas.1802233115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwapiszewska K., Michalczuk A., Rybka M., Kwapiszewski R., Brzózka Z. A microfluidic-based platform for tumour spheroid culture, monitoring and drug screening. Lab Chip. 2014;14:2096–2104. doi: 10.1039/c4lc00291a. [DOI] [PubMed] [Google Scholar]
- Lancaster M.A., Corsini N.S., Wolfinger S., Gustafson E.H., Phillips A.W., Burkard T.R., Otani T., Livesey F.J., Knoblich J.A. Guided self-organization and cortical plate formation in human brain organoids. Nat. Biotechnol. 2017;35:659–666. doi: 10.1038/nbt.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.H., Hong S., Song J., Cho B., Han E.J., Kondapavulur S., Kim D., Lee L.P. Microphysiological Analysis Platform of Pancreatic Islet β-Cell Spheroids. Adv. Healthc. Mater. 2018;7:1–11. doi: 10.1002/adhm.201701111. [DOI] [PubMed] [Google Scholar]
- Messner S., Agarkova I., Moritz W., Kelm J.M. Multi-cell type human liver microtissues for hepatotoxicity testing. Arch. Toxicol. 2013;87:209–213. doi: 10.1007/s00204-012-0968-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland T., McAllister M., Patek S., Flint D., Underwood M., Sim A., Edwards J., Zagnoni M. Drug screening of biopsy-derived spheroids using a self-generated microfluidic concentration gradient. Sci. Rep. 2018;8:14672. doi: 10.1038/s41598-018-33055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampaloni F., Reynaud E.G., Stelzer E.H.K. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007;8:839–845. doi: 10.1038/nrm2236. [DOI] [PubMed] [Google Scholar]
- Pawlizak S., Fritsch A.W., Grosser S., Ahrens D., Thalheim T., Riedel S., Kießling T.R., Oswald L., Zink M., Manning M.L. Testing the differential adhesion hypothesis across the epithelial-mesenchymal transition. New. J. Phys. 2015;17:83049. [Google Scholar]
- Picollet-D’hahan N., Dolega M.E., Liguori L., Marquette C., Le Gac S., Gidrol X., Martin D.K. A 3D Toolbox to Enhance Physiological Relevance of Human Tissue Models. Trends Biotechnol. 2016;34:757–769. doi: 10.1016/j.tibtech.2016.06.012. [DOI] [PubMed] [Google Scholar]
- Proctor W.R., Foster A.J., Vogt J., Summers C., Middleton B., Pilling M.A., Shienson D., Kijanska M., Ströbel S., Kelm J.M. Utility of spherical human liver microtissues for prediction of clinical drug-induced liver injury. Arch. Toxicol. 2017;91:2849–2863. doi: 10.1007/s00204-017-2002-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaiahgari S.C., den Braver M.W., Herpers B., Terpstra V., Commandeur J.N., van de Water B., Price L.S. A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver-like properties for repeated dose high-throughput toxicity studies. Arch. Toxicol. 2014;88:1083–1095. doi: 10.1007/s00204-014-1215-9. [DOI] [PubMed] [Google Scholar]
- Ranga A., Gobaa S., Okawa Y., Mosiewicz K., Negro A., Lutolf M.P. 3D niche microarrays for systems-level analyses of cell fate. Nat. Commun. 2014;5:4324. doi: 10.1038/ncomms5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivron N.C., Frias-Aldeguer J., Vrij E.J., Boisset J.C., Korving J., Vivié J., Truckenmüller R.K., van Oudenaarden A., van Blitterswijk C.A., Geijsen N. Blastocyst-like structures generated solely from stem cells. Nature. 2018;557:106–111. doi: 10.1038/s41586-018-0051-0. [DOI] [PubMed] [Google Scholar]
- Sacchetti B., Funari A., Michienzi S., Di Cesare S., Piersanti S., Saggio I., Tagliafico E., Ferrari S., Robey P.G., Riminucci M., Bianco P. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131:324–336. doi: 10.1016/j.cell.2007.08.025. [DOI] [PubMed] [Google Scholar]
- Sart S., Yan Y., Li Y., Lochner E., Zeng C., Ma T., Li Y. Crosslinking of extracellular matrix scaffolds derived from pluripotent stem cell aggregates modulates neural differentiation. Acta Biomater. 2016;30:222–232. doi: 10.1016/j.actbio.2015.11.016. [DOI] [PubMed] [Google Scholar]
- Sart S., Tomasi R.F., Amselem G., Baroud C.N. Multiscale cytometry and regulation of 3D cell cultures on a chip. Nat. Commun. 2017;8:469. doi: 10.1038/s41467-017-00475-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sart S., Tomasi R.F., Barizien A., Amselem G., Cumano A., Baroud C.N. Mapping the structure and biological functions within mesenchymal bodies using microfluidics. Sci. Adv. 2020;6:w7853. doi: 10.1126/sciadv.aaw7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoonen W.G., Stevenson J.C., Westerink W.M., Horbach G.J. Cytotoxic effects of 109 reference compounds on rat H4IIE and human HepG2 hepatocytes. III: Mechanistic assays on oxygen consumption with MitoXpress and NAD(P)H production with Alamar Blue™. Toxicol. in Vitro. 2012;26:511–525. doi: 10.1016/j.tiv.2012.01.004. [DOI] [PubMed] [Google Scholar]
- Shi Y., Ma J., Zhang X., Li H., Jiang L., Qin J. Hypoxia combined with spheroid culture improves cartilage specific function in chondrocytes. Integr. Biol. 2015;7:289–297. doi: 10.1039/c4ib00273c. [DOI] [PubMed] [Google Scholar]
- Siltanen C., Yaghoobi M., Haque A., You J., Lowen J., Soleimani M., Revzin A. Microfluidic fabrication of bioactive microgels for rapid formation and enhanced differentiation of stem cell spheroids. Acta Biomater. 2016;34:125–132. doi: 10.1016/j.actbio.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer Z.S., Yong J., Tischler J., Hackett J.A., Altinok A., Surani M.A., Cai L., Elowitz M.B. Dynamic heterogeneity and DNA methylation in embryonic stem cells. Mol. Cell. 2014;55:319–331. doi: 10.1016/j.molcel.2014.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirenko O., Hancock M.K., Hesley J., Hong D., Cohen A., Gentry J., Carlson C.B., Mann D.A. Phenotypic Characterization of Toxic Compound Effects on Liver Spheroids Derived from iPSC Using Confocal Imaging and Three-Dimensional Image Analysis. Assay Drug Dev. Technol. 2016;14:381–394. doi: 10.1089/adt.2016.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland R.M., McCredie J.A., Inch W.R. Growth of multicell spheroids in tissue culture as a model of nodular carcinomas. J. Natl. Cancer Inst. 1971;46:113–120. [PubMed] [Google Scholar]
- Takebe T., Sekine K., Kimura M., Yoshizawa E., Ayano S., Koido M., Funayama S., Nakanishi N., Hisai T., Kobayashi T. Massive and Reproducible Production of Liver Buds Entirely from Human Pluripotent Stem Cells. Cell Rep. 2017;21:2661–2670. doi: 10.1016/j.celrep.2017.11.005. [DOI] [PubMed] [Google Scholar]
- Tullis J., Park C.L., Abbyad P. Selective fusion of anchored droplets via changes in surfactant concentration. Lab Chip. 2014;14:3285–3289. doi: 10.1039/c4lc00558a. [DOI] [PubMed] [Google Scholar]
- Tung Y.-C., Hsiao A.Y., Allen S.G., Torisawa Y.S., Ho M., Takayama S. High-throughput 3D spheroid culture and drug testing using a 384 hanging drop array. Analyst (Lond.) 2011;136:473–478. doi: 10.1039/c0an00609b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungrin M.D., Joshi C., Nica A., Bauwens C., Zandstra P.W. Reproducible, ultra high-throughput formation of multicellular organization from single cell suspension-derived human embryonic stem cell aggregates. PLoS ONE. 2008;3:e1565. doi: 10.1371/journal.pone.0001565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale A. Paracetamol (acetaminophen) Medicine. 2012;40:144–146. [Google Scholar]
- van den Brink S.C., Baillie-Johnson P., Balayo T., Hadjantonakis A.K., Nowotschin S., Turner D.A., Martinez Arias A. Symmetry breaking, germ layer specification and axial organisation in aggregates of mouse embryonic stem cells. Development. 2014;141:4231–4242. doi: 10.1242/dev.113001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinci M., Gowan S., Boxall F., Patterson L., Zimmermann M., Court W., Lomas C., Mendiola M., Hardisson D., Eccles S.A. Advances in establishment and analysis of three-dimensional tumor spheroid-based functional assays for target validation and drug evaluation. BMC Biol. 2012;10:29. doi: 10.1186/1741-7007-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitor M.T., Sart S., Barizien A., Torre L.G., Baroud C.N. Tracking the Evolution of Transiently Transfected Individual Cells in a Microfluidic Platform. Sci. Rep. 2018;8:1225. doi: 10.1038/s41598-018-19483-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorrink S.U., Zhou Y., Ingelman-Sundberg M., Lauschke V.M. Prediction of drug-induced hepatotoxicity using long-term stable primary hepatic 3D spheroid cultures in chemically defined conditions. Toxicol. Sci. 2018;163:655–665. doi: 10.1093/toxsci/kfy058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voznica J., Gardella C., Belotserkovsky I., Dufour A., Enninga J., Stévenin V. Identification of Parameters of Host Cell Vulnerability during Salmonella Infection by Quantitative Image Analysis and Modeling. Infect. Immun. 2017;86 doi: 10.1128/IAI.00644-17. e00644-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrij E.J., Espinoza S., Heilig M., Kolew A., Schneider M., van Blitterswijk C.A., Truckenmüller R.K., Rivron N.C. 3D high throughput screening and profiling of embryoid bodies in thermoformed microwell plates. Lab Chip. 2016;16:734–742. doi: 10.1039/c5lc01499a. [DOI] [PubMed] [Google Scholar]
- Wang C.-C., Chen C.H., Hwang S.M., Lin W.W., Huang C.H., Lee W.Y., Chang Y., Sung H.W. Spherically symmetric mesenchymal stromal cell bodies inherent with endogenous extracellular matrices for cellular cardiomyoplasty. Stem Cells. 2009;27:724–732. doi: 10.1634/stemcells.2008-0944. [DOI] [PubMed] [Google Scholar]
- Wang Z., Luo X., Anene-Nzelu C., Yu Y., Hong X., Singh N.H., Xia L., Liu S., Yu H. HepaRG culture in tethered spheroids as an in vitro three-dimensional model for drug safety screening. J. Appl. Toxicol. 2015;35:909–917. doi: 10.1002/jat.3090. [DOI] [PubMed] [Google Scholar]
- Yamada A., Lee S., Bassereau P., Baroud C.N. Trapping and release of giant unilamellar vesicles in microfluidic wells. Soft Matter. 2014;10:5878–5885. doi: 10.1039/c4sm00065j. [DOI] [PubMed] [Google Scholar]
- Yu K.N., Nadanaciva S., Rana P., Lee D.W., Ku B., Roth A.D., Dordick J.S., Will Y., Lee M.Y. Prediction of metabolism-induced hepatotoxicity on three-dimensional hepatic cell culture and enzyme microarrays. Arch. Toxicol. 2018;92:1295–1310. doi: 10.1007/s00204-017-2126-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Radisic M. Organ-on-a-chip devices advance to market. Lab Chip. 2017;17:2395–2420. doi: 10.1039/c6lc01554a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
After coalescence, the droplet color fading indicates the reaction of the dark dye (2,6-DCPIP) with the oxygen carried by the oil. Time is shown in min:sec.
Data Availability Statement
The code supporting the current study has not been deposited in a public repository but is available from the corresponding author on request.