

SUMMARY
Fetal hematopoietic stem cells (HSCs) undergo a developmental switch to become adult HSCs with distinct functional properties. To better understand the molecular mechanisms underlying the developmental switch, we have conducted deep sequencing of the 3D genome, epigenome, and transcriptome of fetal and adult HSCs in mouse. We find that chromosomal compartments and topologically associating domains (TADs) are largely conserved between fetal and adult HSCs. However, there is a global trend of increased compartmentalization and TAD boundary strength in adult HSCs. In contrast, intra-TAD chromatin interactions are much more dynamic and wide-spread, involving over a thousand gene promoters and distal enhancers. These developmental-stage-specific enhancer-promoter interactions are mediated by different sets of transcription factors, such as TCF3 and MAFB in fetal HSCs, versus NR4A1 and GATA3 in adult HSCs. Loss-of-function studies of TCF3 confirm the role of TCF3 in mediating condition-specific enhancer-promoter interactions and gene regulation in fetal HSCs.
In Brief
A developmental transition occurs between fetal and adult hematopoietic stem cells. How the 3D genome folding contributes to this transition is poorly understood. Chen et al. show global genome organization is largely conserved, but a large fraction of enhancer-promoter interactions is reorganized and regulate genes contributing to the phenotypic differences.
Graphical Abstract
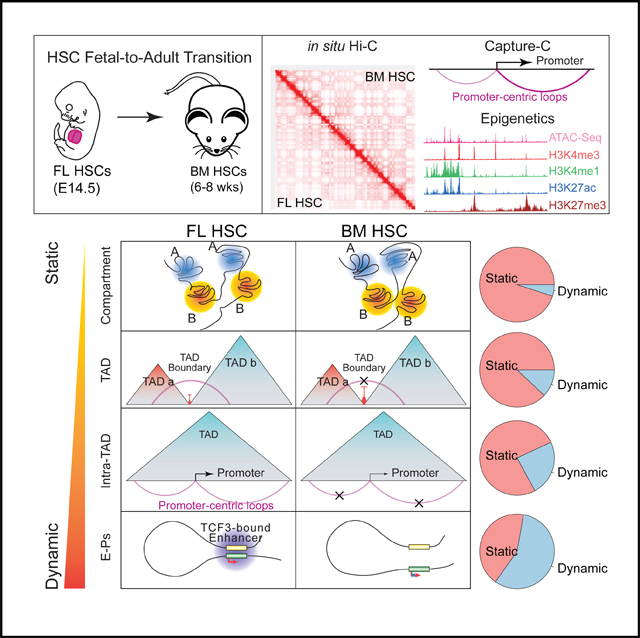
INTRODUCTION
During development, hematopoietic stem cells (HSCs) first appear in major arteries of the mouse embryo at embryonic day 11 (E11) and migrate to the fetal liver (FL) at E12 where they expand in number by 10- to 30-fold (Ema and Nakauchi, 2000). Right before birth, FL HSCs migrate to bone marrow (BM) to take up permanent residence. The physiological properties and functions of FL and adult BM HSCs are distinct. FL HSCs must support rapid blood development and hence rapidly expand, while BM HSCs support homeostatic blood production and respond to injury and external stress. Phenotypic differences between FL and BM HSCs enable them to fulfill these different physiological needs. Most (>70%) BM HSCs exist in a quiescent G0 state (Passegué et al., 2005; Wilson et al., 2008) to prevent HSC exhaustion, whereas the majority of FL HSCs are actively cycling (Bowie et al., 2006). Second, the relative lineage outputs of lymphoid and myeloid cells change between FL and BM HSCs and during the process of aging. FL HSCs tend to have balanced lymphoid and myeloid lineage outputs whereas BM HSCs tend to have a myeloid-biased lineage output that becomes more prevalent during the aging process in mouse (Benz et al., 2012; Busch et al., 2015). Finally, FL HSCs more robustly engraft mice when transplanted and display a greater self-renewal activity when stimulated to proliferate in vivo. FL HSCs regenerate daughter HSCs in irradiated recipients more quickly and outcompete the production of HSCs from adult BM HSCs (Bowie et al., 2007a; He et al., 2011). These phenotypic differences correlate with changes in HSC gene expression (Bowie et al., 2007b), indicating that FL and BM HSCs are sustained by distinct transcriptional programs.
The 3D genome organization plays an important role in transcription via multiple mechanisms, from long-range interactions between gene promoters and enhancers to higher-order chromosome compartments and domains that can act as expression domains (Bonev and Cavalli, 2016; Sexton and Cavalli, 2015). Global reorganization of 3D genome structures have been studied in different developmental systems, including human embryonic stem cell (ESC) differentiation(Dixon et al., 2015), mouse neural development (Bonev et al., 2017), B cell reprogramming process (Krijger et al., 2016), T cell linage commitment (Hu et al., 2018), and fetal versus adult erythroid cells (Huang et al., 2017) revealing new insights into the interplay between genome organization, gene expression, and cellular identity.
To date, little is known about how 3D genome organization contributes to the phenotypic difference between fetal and adult HSCs. Better understanding of the 3D genome organization may provide new ways to manipulate gene expression and HSC behavior for translational research. Here, we characterize the phenotypic differences between FL and BM HSCs. We next map the differences in 3D genome organization, epigenomic state, and gene expression between FL and BM HSCs. We reveal a general trend of increased dynamics in 3D genome organization as one moves down the organizational hierarchy. Moreover, our data suggest a high degree of intra-TAD promoter interactome dynamics during the fetal-to-adult HSC transition. We further identify a set of transcription factors potentially involved in mediating developmental-stage-specific promoterenhancer interactions.
RESULTS
Differences in Hematopoietic Lineage Potential and Cell-Cycle Status between Fetal and Adult HSCs
To examine the difference in lineage potentials of fetal and adult HSCs, we purified fetal and adult murine HSCs from embryonic (E) day 14.5 FL and adult (6–8 weeks) BM using established cell surface phenotype Lin− Sca-1+ c-Kit+ CD135− (LSKCD135−) (Figure S1A; Adolfsson et al., 2001; Christensen and Weissman, 2001; Må nsson et al., 2007; Wilson et al., 2015; Yang et al., 2005). We analyzed colonies derived from single HSCs for the generation of granulocyte-macrophage (GM), B, and T cells (Figures S1B and S1C). Among all plated cells, 33% and 39% cells produced colonies (Figure S1D). Of the single-cell-derived colonies with lineage readout (Figure S1D), ~53% produced mature cells of all three GM, B, and T cells (GM/B/T) for both FL and BM HSCs (Figure S1E). We found that FL HSCs gave rise to more lymphoid clones (B and/or T cells) whereas adult HSCs gave rise to more GM clones (p < 0.05, Figure S1E). To determine the long-term multilineage potential of the prospectively purified HSCs, we transplanted purified FL/BM HSCs competitively into lethally irradiated recipient mice. We analyzed GM, B and T cells in peripheral blood using flow cytometry16 weeks post-transplantation. The transplanted FL and BM HSCs engrafted 7/9 and 8/10 of the mice, respectively. Moreover, consistent with our single-cell analysis, peripheral blood reconstituted from BM HSCs has a higher GM/(B+T) ratio than that reconstituted from FL HSCs, again suggesting a myeloid lineage bias in BM HSCs (Figure S1F). Taken together, these results demonstrate the multilineage potential of the HSCs purified in this study and confirm previous reports that BM HSCs have a myeloid lineage bias.
To examine the cell cycle difference between FL and BM HSCs, we costained FL and BM samples with HSC surface markers (LSKCD135−), anti-Ki-67 antibody, and propidium iodide (Figure S1G), to simultaneously purify HSCs and profile their cell cycle status. Consistent with previous studies, the fractions of cells in each cell cycle phase are significantly different between FL and BM HSCs. In particular, the vast major of FL HSCs are cycling (only 0.19% in G0 phase) whereas the majority of BM HSCs (66%) are in G0 phase (Figure S1G).
Next, we performed RNA sequencing (RNA-seq) to profile the transcriptomes of fetal and adult HSCs. Using a false discovery rate (FDR) cutoff of 0.05 and fold change cutoff of 2, we identified 3,464 differentially expressed genes, including 1,630 and 1,834 genes expressed higher in either FL HSCs or BM HSCs, respectively (Table S1). Genes expressed higher in FL HSCs are enriched for Gene Ontology (GO) terms of “cell cycle process,” “ribosome biogenesis,” “G1/S transition of mitotic cell cycle,” “chromosome organization,” consistent with the larger fraction of FL HSCs in cell cycle (Figure S1H). In contrast, genes expressed higher in BM HSCs are enriched for GO terms of “regulation of immune response,” “cell differentiation,” “cell cycle arrest,” “hematopoietic or lymphoid organ development,” consistent with the phenotypic differences.
Chromosome Compartments Are Largely Unchanged, but Compartmentalization Is Strengthened during the Fetal-to-Adult Transition
To compare the global 3D genome organizations, we profiled genome-wide chromatin interactions using in situ Hi-C (Rao et al., 2014). We also conducted Capture-C (Hughes et al., 2014) to investigate the dynamics of promoter-centric chromatin interactions, focusing on 4,052 promoters that are highly expressed in HSCs compared to a compendium of 20 other mouse tissues (Figures 1A, S2A, and S2B; Table S2; STAR Methods). Using several metrics, we confirmed that our Hi-C and Capture-C data have sufficient sequencing depth and high reproducibility (Figures S2C–S2F; Table S3). To understand the relationship between the epigenome and 3D genome organization, we also generated chromatin immunoprecipitation sequencing (ChIP-seq) data for four histone marks, H3K4me1, H3K4me3, H3K27ac, and H3K27me, as well as assay for transposase-accessible chromatin using sequencing (ATAC-seq) data (Figure 1A). These data also have sufficient sequencing depth and high reproducibility (Figures S2G and S2H).
Figure 1. Limited Change in Global 3D Genome Organization during Fetal-to-Adult HSC Transition.

(A) Schematic diagram of experimental design.
(B) Fraction of genomic regions with compartment switching during fetal to adult transition. B → A, regions switching from compartment B to compartment A; static, regions without compartment switching.
(C) Gene expression change is correlated with compartment switching.
(D and E) Increased compartmentalization during fetal to adult transition.
(D) Shown are log ratios of observed versus expected contact frequencies between TADs from the same (A versus A, B versus B) or different compartments (A versus B).
(E) An example heatmap of contact frequencies along chromosome 2, showing increased contacts among regions of the same compartment. Compartment assignment is indicated along the top and left. Several examples of more frequent interactions between the same compartments are highlighted by rectangles. Color is proportional to the difference in contact frequency (BM HSC-FL HSC). Panel was generated by Juicebox.
(F) Scatterplot of TAD boundary strength in FL HSCs and BM HSCs. Boundaries with significantly increased and decreased strength (FDR <0.1) are highlighted in blue and red, respectively.
(G) 3D distance is larger between adjacent TADs with increased boundary strength during the fetal-to-adult transition. y axis, difference in 3D distance of adjacent TADs between BM HSCs and FL HSCs. Normalized distance was calculated based on the 3D structure model of each chromosome.
(H) An example TAD boundary with significantly increased strength during the transition. TAD heatmap color is proportional to SHAMAN score.
p values in (C), (D), and (G) were calculated using t test.
Previous studies have revealed that the 3D genome is organized in a hierarchical fashion. At the top level are so-called A and B compartments with an average size of 3 Mb. The A compartments correlate with early replicating, euchromatic regions whereas the B compartments correlate with heterochromatin (Dixon et al., 2015; Lieberman-Aiden et al., 2009). We found that in both FL and BM HSCs, the genome is equally divided into the A and B compartments (Figures S3A and S3B). Compartment A is associated with higher levels of active epigenetic marks including H3K4me3, H3K4me1, and H3K27ac, higher chromatin accessibility, and higher gene expression. In contrast, compartment B is associated with lower chromatin accessibility and lower gene expression (Figures S3C and S3D). Overall, 95% of the genome falls into the same compartment in both FL and BM HSCs, suggesting limited change in the global 3D genome organization during the fetal-to-adult HSC transition (Figure 1B). Of the 5% of the genome that switches compartment, genes are expressed significantly higher when the corresponding region switches from compartment B to compartment A and vice versa (Figure 1C). Despite the limited change in the locations of compartment boundaries, the overall compartmentalization is strengthened in BM HSCs, as indicated by the increased interactions among TADs from the same compartments and decreased interactions among TADs from different compartments in BM HSCs (Figures 1D and 1E).
Locations of TADs Boundaries Are Largely Conserved but Boundary Strength Increases during Fetal-to-Adult Transition
Topologically associating domains (TADs) (Dixon et al., 2015), ranging in size from 100 kb to 3 Mb and averaging 1 Mb, have been suggested to be important organization units of the 3D genome. Using the GMAP algorithm (Yu et al., 2017), we identified 2,393 and 2,391 TADs in FL and BM HSCs, respectively. Similar to compartment boundaries, 88% of TAD boundaries are shared between the two cell types (Figure S4A). Although the change in boundary location is small, we observed a global trend of increased TAD boundary strength, as measured by the difference in intra-TAD and inter-TAD interactions (see STAR Methods for details), during the fetal-to-adult transition (Figure S4B). This observation is further supported by our Capture-C data because the fraction of inter-TAD promoter-centric interactions is significantly reduced during the transition (Figure S4C). This global increase in boundary strength could be due to the difference in the fraction of cells in different cell cycle phases. A recent single-cell Hi-C study suggests that TAD boundary strength is highest in G1 phase and decreases as the cell enters S phase (Nagano et al., 2017). By comparing published Hi-C data of pure proliferating and quiescent human fibroblast cells, we also found that TAD boundary strength is higher in the quiescent cells than in proliferating cells (Figure S4D; Criscione et al., 2016). Taken together, these data suggest that the higher fraction of G0 cells (Figure S1G) in BM HSCs potentially account for the higher TAD boundary strength in these cells. Besides this general trend of increased boundary strength, we found 58 (3%) TADs whose boundaries exhibit significantly increased strength in BM HSCs compared to FL HSCs (FDR < 0.1, Figure 1F). Interestingly, 3D modeling of the genome shows that the two adjacent TADs of those boundaries are farther apart in BM HSCs than in FL HSCs (Figure 1G; STAR Methods), suggesting a positive correlation between boundary strength and physical distance between adjacent TADs. An example TAD boundary with significant increased strength is shown in Figure 1H. Additional examples are shown in Figure S4H. Using our Capture-C data, we also found inter-TAD enhancer-promoter interactions across those boundaries are significantly reduced (Figures S4F and S4G), suggesting the stronger boundaries may further impede the inter-TAD enhancer-promoter interactions in BM HSCs.
Intra-TAD Promoter Interactome Exhibits Substantial Dynamics during Fetal-to-Adult Transition
The analyses above suggest limited change of the 3D genome at the compartment and TAD levels. We therefore investigated the dynamics of chromatin interactions within TADs. We used our Hi-C data to identify TADs and Capture-C data to identify promoter-centric interactions. To identify statistically significant Capture-C interactions, we developed the LiMACC algorithm (local iterative modeling approach for Capture-C data) (STAR Methods). Performance benchmarking shows that LiMACC has better performance compared to the state-of-the-art method, CHiCAGO (Cairns et al., 2016; Figures S5A and S5B), in terms of identifying higher fractions of functional interactions, including enhancer-promoter interactions, promoter-promoter interactions, and promoter-ATAC-seq peak interactions. At an FDR cutoff of 0.01, 89,545 significant interactions were identified (Table S5). Among them, 32,814 (36.6%) are FL HSC-specific, 29,209 (32.6%) are BM HSC-specific, and 27,522 (30.8%) are shared (Figure S5C).
By comparing the interaction frequencies of the set of promoters in the same TAD, we identified 242 TADs exhibiting significant dynamics of the intra-TAD promoter interactome between FL and BM HSCs (Figure 2A; Table S4; STAR Methods). Genes in these TADs are enriched for GO terms such as cell cycle, metabolism, chromosome maintenance, and apoptosis (Figure 2B), consistent with the phenotypic difference between FL and BM HSCs. Interestingly, we found dynamic TADs are marked by significantly higher levels of active chromatin marks (H3K4me1, H3K4me3, and H3K27ac) and higher chromatin accessibility, compared to static TADs (Figures S6A and S6B). A dramatic example of such dynamic TADs is located around the Hmga2 locus. Hmga2 has multiple roles including chromatin architectural protein and transcription factor. It has been shown to play a critical role in the higher level of self-renewal activity in FL HSCs (Copley et al., 2013). We found that multiple interactions involving the Hmga2 promoter and two super enhancers in FL HSCs disappear in BM HSCs. This loss of enhancer interaction is associated with a significant decrease in Hmga2 expression (Figures 2C and 2E) in BM HSCs. Previous studies have shown that Hmga2 is post-transcriptionally regulated by the Lin28b-let7 axis (Copley et al., 2013). Our data suggest Hmga2 is also regulated at the chromatin interaction and transcriptional level. The other gene Llph in this TAD also loses three enhancer interactions but interestingly gains two promoter interactions in BM HSCs. Another example of dynamic intra-TAD interaction involves the Smarca2 gene, a member of the SWI/ SNF chromatin remodeling complex (Figure 2D). The Smarca2 promoters gain several interactions with active enhancers and Smarca2 expression is upregulated during the fetal-to-adult transition (Figure 2E). Additional examples are shown in Figure S6. Taken together, these data suggest that intra-TAD chromatin interaction dynamics plays a major role in driving the phenotypic differences between FL and BM HSCs.
Figure 2. Intra-TAD Promoter-Centric Interactions Exhibit Large Dynamics.

(A) Venn diagram of TADs with dynamic intra-TAD interactions during fetal-to-adult HSC transition.
(B) Enriched GO terms among genes in the TADs with dynamic intra-TAD interactions.
(C) An example TAD with more promoter-centric interactions in FL HSCs than BM HSCs. Gene promoters with Capture-C baits are highlighted in red. TAD is indicated with a navy green bar. The normalized signals of ATAC-Seq, H3K4me1, H3K27ac, and Capture-C are displayed for FL HSCs (upper tracks) and BM HSCs (lower tracks). Two super enhancers are indicated with an orange bar.
(D) An example TAD with more promoter-centric interactions in BM HSCs than FL HSCs (indicated by arrows).
(E) Expression levels of three genes, Hmga2, Llph, and Smarca2 in the TAD. p value for differential expression was computed using the edgeR software.
Dynamic Enhancer-Promoter Interactions Target Genes Underlying Phenotypic Differences
Using our histone mark ChIP-seq data and the CSI-ANN algorithm (Firpi et al., 2010), we identified active enhancers and promoters in both cell types (Figure S5F; STAR Methods). Approximately 20% and 30% of the 89,545 significant promoter-centric interactions are enhancer-promoter (EP) and promoter-promoter (PP) interactions (Figure S5D), respectively. Among the EP interactions, 57% are cell-type-specific (Figures 3A and S5E). Genes targeted by cell-type-specific EP interactions are expressed significantly higher in the same cell type (p = 7.8e–6, Figure 3B). They are also involved in multiple biological processes that underlying the phenotypic differences (Figure 3C).
Figure 3. Dynamic Enhancer-Promoter (EP) Interactions Account for Phenotypic Differences between FL and BM HSCs.

(A) Venn diagram of EP interactions detected by Capture-C. >60% EP interactions are cell-type-specific.
(B) Expression change of genes with cell-specific EP interactions. Expression change was calculated as fragments per kilobase of transcript per million mapped reads (FPKM) ratio of BM HSCs to FL HSCs. p value was calculated using t test.
(C) Enriched GO terms of genes with FL HSC-specific and BM HSC-specific EP interactions.
(D) An example of FL HSC-specific EP interactions involving the promoter of Ccna2. Difference in normalized Capture-C signal is shown in the middle track. Normalized ATAC-seq signal, H3K4me1, and H3K27ac ChIP-seq signals are displayed in the rest of the tracks. Gene whose promoter was used as Capture-C bait is marked as red.
(E) DNA FISH confirms the de novo FL HSC-specific EP interaction. Left: representative DNA FISH images of the Ccna2 promoter (red) and enhancer (green) in FL HSCs (left panel) and BM HSCs (right panel). Interaction is denoted by a white arrow. Right: frequency of the quantified distance distribution between Ccna2 promoter and the enhancer (μm) (# nuclei imaged: 90 and 59 for FL HSC and BM HSC, respectively). p value was calculated using t test. Scale bars, 2 μm.
(F) Gene expression level of Ccna2. p value of differential expression was calculated using edgeR.
(G) An example of BM HSC-specific EP interaction involving the promoter of Cdkn2c.
(H) DNA FISH confirmation of the EP interaction. Scale bars, 2 μm.
(I) Gene expression level of Cdkn2c.
(J) Enriched TF DNA binding motifs at enhancers of FL HSC-specific and BM HSC-specific EP interactions. Bottom plots, expression levels of the TFs with enriched motifs.
(K) Co-localization of enriched TF motifs at enhancers of cell-specific EP interactions. Color of heatmap is proportional to the p value of co-localization. Heatmap was clustered using hierarchical clustering.
An example FL HSC-specific EP interaction involving the gene Ccna2 is shown in Figures 3D–3F. Ccna2 gene is a positive regulator of G1/S and G2/M transitions and is expressed significantly higher in FL HSCs (Figure 3F). An active enhancer located 700 kb downstream of the Ccna2 promoter forms an interaction with the Ccna2 promoter in FL HSCs but not in BM HSCs. An example of BM HSC-specific EP interaction involving the gene Cdkn2c is shown in Figures 3G–3I. Cdkn2c is a negative regulator of cell cycle G1 phase progression. DNA fluorescence in situ hybridization (DNA-FISH) confirms both cell-specific EP interactions (Figures 3E and 3H). Additional examples are shown in Figures S5G–S5J.
To identify transcription factors that are involved in cell-type-specific EP interactions, we conducted TF motif analysis of enhancers involved in cell-specific EP interactions. We identified 22 and 6 TFs whose motifs are enriched at enhancers of FL HSC-specific and BM HSC-specific EP interactions (p < 0.01), respectively. Among those TFs, 10 and 4 are differentially expressed in FL and BM HSCs, respectively (Figure 3J). Moreover, many of the enriched TFs have co-localized binding sites within the same enhancers involved in the EP interactions (Figure 3K), suggesting combinatorial binding of these TFs may be required for the stage-specific EP interactions.
TCF3 Mediates Developmental-Stage-Specific Enhancer-Promoter Interactions
Transcription factor 3 (TCF3, also known as E2A) ranks as the top TF whose motif is enriched at enhancers of FL HSC-specific EP loops (Figure 3J). TCF3 is required for B and T cell development (Belle and Zhuang, 2014) and HSC maintenance (Semerad et al., 2009). It is also implicated in chromatin organization in B cells (Lin et al., 2010; Ribeiro de Almeida et al., 2012). We found Tcf3 is expressed 1.7-fold higher in FL compared to BM HSCs (p = 7e–3). FL HSC-specific TCF3 targets (genes targeted by FL HSC-specific EP loops that have TCF3 DNA binding sites in the enhancers) were also expressed higher in FL HSCs (Figure 4A). We confirmed TCF3 binding to a number of these enhancers using ChIP-qPCR (Figure 4B). The FL HSC-specific TCF3 targets are enriched for GO terms such as “cell-cycle phase,” “chromatin organization,” “regulation of cell proliferation,” and “lymphocyte activation” (Figure 4C). Taken together, these data suggest that TCF3 occupies FL HSC specific EP loops and regulates the expression of genes underlying the phenotypic differences.
Figure 4. TCF3 Occupies Developmental-Stage-Specific Enhancer-Promoter Loops and Affects Cell-Cycle Phase and Lineage Potential.

(A) FL HSC-specific TCF3 targets have significantly higher expression.
(B) ChIP-qPCR confirmation of TCF3 binding to enhancers involved in FL HSC-specific EP loops.
(C) Enriched GO terms among genes targeted by FL HSC-specific EP loops occupied by TCF3.
(D) Western blot showing knock down of TCF3 by CRISPR-Cas9.
(E and F) Cell cycle phase analysis by co-staining with propidium iodide and anti-Ki-67 antibody.
(E) Representative fluorescence-activated cell sorting (FACS) plots of wild-type and Tcf3 knockout HPC-7 cells.
(F) Quantification of cell cycle phases, mean ± SD of three biological replicates. p values were computed using t test.
(G) Limiting dilution assay showing significantly reduced lymphoid potential in Tcf3 knockout HPC-7 cells. y axis, frequencies of CD45+CD25+CD90+ T progenitors and CD45+B220+CD19+ B progenitors produced by wild-type and Tcf3 knockout HPC-7 cells after 10–12 days of co-culturing with OP9/OP9-DL1 cells. Values are mean of 2 biological replicate experiments. Error bar, SD.
To further confirm if TCF3 can mediate developmental-stage-specific enhancer-promoter interactions, we performed Capture-C experiment comparing wild-type HPC-7 cells and HPC-7 cells with Tcf3 knockout using CRISPR-Cas9 technology (Figure 4D). HPC-7 is a murine multi-potent hematopoietic precursor cell line that has been used as a model of HSCs (Pintodo O, et al., 1998; Schuütte et al., 2016; Wilson et al., 2010). Tcf3 knockout has a modest effect on the overall cell cycle duration of HPC-7 cells (Figures S7A–S7C). However, it significantly increases the fraction of cells in G0 phase (p = 1.1e–4) and reduces the fraction of cells in G2/S phase (p = 1.8e–3) (Figures 4E and 4F). Using limiting dilution assay, we further investigated the differentiation potential of HPC-7 cells with Tcf3 knockout. We found that the lymphoid potential of these cells is dramatically reduced (Figure 4G). The frequencies of CD45+CD19+B220+ B cells and CD45+CD25+CD90+ T cells are reduced from 1/4 to 1/12 (p = 0.011) and from 1/9 to 1/115 (p = 0.029), respectively. Taken together, these results suggest that TCF3 plays a role in cell cycle and lymphoid potential in HPC-7 cells.
We found that knocking out Tcf3 significantly reduces the contact frequency of enhancer-promoter interactions in which the enhancers are occupied by TCF3 (n = 93, Figure 5A) in FL HSCs. Of these FL HSC-specific EP loops that are bound by TCF3, several of them target key TFs in FL HSCs, such as Hmga2; cell cycle genes, such as Rcc2 (regulator of chromosome condensation 2), Cenpn (centromere protein N); and metabolic genes, such as Cryl1 (crystallin lambda 1), Psat1 (phosphoserine aminotransferase 1), Prkag1 (protein kinase AMP-activated non-catalytic subunit gamma 1), and Scd1 (stearoyl-CoA desaturase). Genome browser view of Capture-C signal and TCF3 ChIP-seq signal for the Hmga2, Rcc2, and Psat1 loci are shown in Figures 5C–5E. Additional examples are provided in Figures S7D–S7F. The relative expression levels of those genes are significantly decreased in Tcf3 knockout cells (Figure 5B, p < 0.05). In summary, these results confirm that TCF3 can mediate developmental-stage-specific enhancer-promoter interactions in fetal HSCs and the genes targeted by these interactions are responsible for fetal HSC-specific phenotypes.
Figure 5. Loss of TCF3 Results in Loss of Cell-Specific Enhancer-Promoter Loops and Deregulation of Target Gene Expression.

(A) Reduced interaction frequency among TCF3 bound enhancer-promoter loops after knocking down Tcf3.
(B) qRT-PCR of target genes of TCF3 bound enhancer-promoter loops.
(C–E) Capture-C data showing loss of enhancer-promoter interaction after Tcf3 knock down for Hmga2 (C), Rcc2 (D), and Psat1 (E).
DISCUSSION
Fetal and adult HSCs have dramatic phenotypic differences, especially in their cycling behavior, lineage output, and metabolic state. Our RNA-seq analysis revealed over 3,000 genes that are differentially expressed between these two types of HSCs. Here, we investigated how changes in the different hierarchical levels of 3D genome organization contribute to the differences in gene expression and phenotype. We found an increasing amount of changes going down the genome architectural hierarchy: 5% at the chromosome compartment level, 12% at TADs, 23% at subTADs, and 57% at enhancer-promoter interactions.
Although the location of compartment and TAD remain relatively unchanged during the fetal-to-adult transition of HSCs, we observed a general trend of increased compartmentalization and TAD boundary strength. To further corroborate our finding, we analyzed Hi-C data from four other developmental systems and observed a similar trend (Figures S4D and S4E), including human fibroblast senescence (Criscione et al., 2016), ESC differentiation to neurons (Bonev et al., 2017), differentiation of cardiovascular muscle cells from induced pluripotent stem cells (iPSCs) (Montefiori et al., 2018), and reprogramming of pre-B cells to pluripotent stem cells (Stadhouders et al., 2018). The mechanism for the increased strength of TAD boundary and compartmentalization during development is unclear. A potential factor may be the cohesin complex. Recent studies have suggested critical and distinct roles of the cohesin complex in compartmentalization versus formation of TADs (Rao et al., 2017; Schwarzer et al., 2017; Vian et al., 2018). Further investigation of different cohesin subunits and other architecture proteins such as condensin (Terakawa et al., 2017) and their interplay may uncover the mechanisms for the dynamics of compartmentalization and TAD boundaries.
The role of transcription factors in enhancer-promoter interactions is poorly understood during HSC development. Our analysis identified several TFs that are potential mediators of EP interactions in HSCs. We tested one of the predictions, TCF3, by a loss-of-function approach. We observed a significant decrease of FL HSC-specific EP contact frequency in TCF3 knockout HPC-7 cells compared to the wild-type HPC-7 cells, suggesting TCF3 is involved in mediating stage-specific EP interactions. Moreover, we identified TCF3 as a regulator of Hmga2, a key gene distinguishing fetal and adult HSCs. Knockout of Tcf3 significantly reduces the interactions between Hmga2 promoter and its enhancers, along with significant downregulation of Hmga2, which may contribute to the loss of lymphoid potential of HPC-7 cells.
The observed changes in EP interaction and gene expression after Tcf3 knockout are significant but modest, suggesting additional TFs might also contribute to EP interactions and phenotypic differences. This is corroborated by the enrichment of colocalized TFs at HSC-specific EP interactions. Multiple lines of evidence further support a potential role in EP interactions for these predicted TFs. For instance, MAFB is known to restrict myeloid lineage choice in HSCs (Sarrazin et al., 2009). We found Mafb is expressed 11-fold higher in fetal compared to adult HSCs, and it binds to an FL HSC-specific enhancer that targets to the Igf2 promoter (Figure S7G), an important gene that controls FL HSC cycling activity (Zhang and Lodish, 2004). Several TFs in the SP/KLF family are also enriched at FL HSC-specific EP interactions. These factors have potential of mediating cell-specific EP interactions, for example KLF1 in erythrocytes (Drissen et al., 2004; Stadhouders et al., 2012) and KLF4 in ESCs (Wei et al., 2013).
EGR1 negatively regulates HSC proliferation and mobilization (Min et al., 2008). Consistent with this role, Egr1 expression is >30-fold higher in BM HSCs than FL HSCs. Two nuclear receptors, NR3C1 and NR4A1, are enriched at adult HSC-specific EP interactions. NR4A1 was shown to regulate HSC quiescence in adult HSCs (Freire and Conneely, 2018; Land et al., 2015), which is consistent with our and previous findings. NR4A1 was shown to restrict the HSC proliferation through inflammatory response. GATA3 has been shown to promote cell cycle entry and proliferation in murine bone marrow HSCs (Ku et al., 2012).
In summary, our study suggests that the fetal-to-adult transition of HSCs is accompanied by a large-scale promoter interactome change within TADs, impacting many gene pathways relevant to phenotypic differences between the two types of HSCs. The newly identified transcription factors and their target genes via EP interactions may present novel targets for developing protocols for HSC mobilization for therapeutic purposes.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kai Tan (tank1@email.chop.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse strains
Female B6129SF1/J mice were mated with male C57BL/6J mice for FL HSCs. Fetal livers were dissected from embryonic (E) day 14.5 (E14.5) embryos. Pairs of female B6129SF1/J and male C57BL/6J (6–8 weeks old) were dissected for bone marrow HSCs. The Children’s Hospital of Philadelphia Office of the Institutional Animal Care and Use Committee review board approved these studies.
Cell Lines
Hematopoietic precursor cell-7 (HPC-7) cells were grown in IMDM medium (12440-053, Invitrogen) with 10% FBS, 10% stem cell factor conditional medium, 1% Pen/Strep, and 7.48×10−5 MTG (M6145, Sigma). Stem cell factor conditional medium was produced by BHK/NKL cell line. HPC-7 cells were maintained at the density of 5×105 ~2×106 cells per mL. OP9 and OP9-DL1 cells were grown in a-MEM (12571-063, Invitrogen) medium with 20% FBS, and 1% Pen/Strep.
METHOD DETAILS
Purification of HSCs from adult bone marrow and E14.5 fetal liver
Marrow of long bones (tibias and femurs) were flushed out with staining buffer (1 × PBS + 2% FBS) and stained with anti-mouse CD16/32 antibodies to block non-antigen-specific binding. Stained cells were washed twice with staining buffer and applied to autoMACS to enrich CD117+ cells. Enriched cells were stained with the antibody cocktail against CD117 (c-kit), Ly-6A/E (Sca-1), CD135, Ly6G/Ly-6C (Gr-1), CD11b, TER-119, CD4, CD8a, CD45R/B220, CD3ε and CD11c (Key Resources Table) at 4°C for 15 min in the dark. Cells were first subjected to yield sort for live HSC (Lin−Sca-1+c-Kit+CD135−) (Månsson et al., 2007; Woolthuis and Park, 2016; Yang et al., 2005) and collected into 500 μL 1 × IMDM + 20% FBS in a 12 × 75-mm polystyrene tube. Collected cells were sorted by purity sort using the same gating strategy and sorted into 0.8 mL 1 × IMDM+50% FBS in a 1.5 mL DNA LoBind tube. Purity of sorted cells is more than 95%.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Purified CD16/32 (Clone 93) | Biolegend | Cat# 101302; RRID:AB_312801 |
| APC anti-mouse CD117 (Clone 2B8) | eBioscience | Cat# 17-1171-81; RRID:AB_469429 |
| PE anti-mouse Sca-1 (Clone D7) | eBioscience | Cat# 12-5981-81; RRID:AB_466085 |
| BV421 anti-mouse CD135 (Clone A2F10) | Biolegend | Cat# 135313; RRID:AB_2562338 |
| AF488 anti-mouse CD3ε (Clone 145-2C11) | Biolegend | Cat# 100321; RRID:AB_389300 |
| AF488 anti-mouse Ter119 (Clone TER-119) | Biolegend | Cat# 116215; RRID:AB_493402 |
| AF488 anti-mouse CD8a (Clone 53-6.7) | Biolegend | Cat# 100723; RRID:AB_389304 |
| AF488 anti-mouse B220 (Clone RA3-6B2) | Biolegend | Cat# 103228; RRID:AB_492874 |
| AF488 anti-mouse CD4 (Clone RM4-5) | Biolegend | Cat# 100532; RRID:AB_493373 |
| AF488 anti-mouse Gr-1 (Clone RB6-8C5) | Biolegend | Cat# 108419; RRID:AB_493480 |
| AF488 anti-mouse CD19 (Clone 6D5) | Biolegend | Cat# 115521; RRID:AB_389307 |
| AF488 anti-mouse CD11b (Clone M1/70) | Biolegend | Cat# 101217; RRID:AB_389305 |
| AF488 anti-mouse CD11c (Clone N418) | Biolegend | Cat# 117311; RRID:AB_389306 |
| PE-Cy7 anti-mouse CD45 (Clone 30-F11) | Biolegend | Cat# 103113; RRID:AB_312978 |
| APC anti-mouse CD90.2 (Clone 30-H12) | Biolegend | Cat# 105311; RRID:AB_313182 |
| PE anti-mouse CD25 (Clone 3C7) | Biolegend | Cat# 101903; RRID:AB_312846 |
| APC anti-mouse CD19 (Clone 1D3) | Biolegend | Cat# 152409; RRID:AB_2629838 |
| PE anti-mouse B220 (Clone RA3-6B2) | Biolegend | Cat# 103208; RRID:AB_312993 |
| AF700 anti-mouse Ki67 (Clone SolA15) | eBioscience | Cat# 56-5698-80; RRID:AB_2637479 |
| AF700 anti-mouse Mac-1 (Clone M1/70) | Biolegend | Cat# 101222; RRID:AB_493705 |
| AF488 anti-mouse NK1.1 (Clone PK136) | Biolegend | Cat# 108717; RRID:AB_493184 |
| PE-Cy5 anti-mouse Gr-1 (Clone RB6-8C5) | Biolegend | Cat# 108409; RRID:AB_313374 |
| FITC anti-mouse CD45.2 (Clone 104) | eBioscience | Cat# 11-0454-82; RRID:AB_465061 |
| PE-Cy7 anti-mouse CD45.1 (Clone A20) | eBioscience | Cat# 25-0453-82; RRID:AB_469629 |
| Anti-mouse Histone H3 (mono-methyl K4) | Abcam | Cat# ab8895; RRID:AB_306847 |
| Anti-mouse Histone H3 (tri-methyl K4) | Millipore | Cat# 07-473; RRID:AB_1977252 |
| Anti-mouse Histone H3 (acetyl K27) | Abcam | Cat# ab4729; RRID:AB_2118291 |
| Anti-mouse Histone H3 (tri-methyl K27) | Millipore | Cat# 07-449; RRID:AB_310624 |
| Anti-mouse TCF3/E2A | R&D | Cat# AF7650 |
| Anti-mouse CAS9 (Clone 7A9-3A3) | Cell Signaling | Cat# 14697; RRID:AB_2750916 |
| Anti-mouse ACTB (Clone C4) | Millipore | Cat# MAB1501; RRID:AB_2223041 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human IL-7 | PeproTech | Cat# 200-07 |
| Human FLT3L | PeproTech | Cat# 300-19 |
| Mouse IL-7 | PeproTech | Cat# 217-17 |
| Mouse FLT3L | PeproTech | Cat# 250-31L |
| Mouse SCF | PeproTech | Cat# 250-03 |
| Critical Commercial Assays | ||
| Protease inhibitor | Sigma | Cat# P8340 |
| MboI | NEB | Cat# R0147L |
| DpnII | NEB | Cat# R0543M |
| T4 DNA ligase | Roche | Cat# 10799009001 |
| AMPure XP beads | BECKMAN COULTER | Cat# A63880 |
| Dynabeads MyOne Streptavidin C1 beads | Invitrogen | Cat# 65001 |
| NEBNext Ultra DNA Library Prep Kit | NEB | Cat# E7370S |
| SureSelectXT2 Reagent Kit | Agilent | Cat# G9621A |
| Bioanalyzer High Sensitivity DNA Analysis Kit | Agilent | Cat# 5067-4626 |
| Nextera DNA Library Prep Kit | Illumina | Cat# FC-121-1030 |
| MinElute PCR Purification Kit | QIAGEN | Cat# 28004 |
| ChIP-IT high sensitivity Kit | Active Motif | Cat# 53040 |
| ThruPLEX-FDPrep Kit | Rubicon Genomics | Cat# R40048 |
| KAPA Library Quantification Kit | Roche | Cat# KK4844 |
| DAPI | Invitrogen | Cat# P36935 |
| FITC BrdU flow kit | BD PharMingen | Cat# 559619 |
| Deposited Data | ||
| Raw and analyzed Hi-C data | This paper | GEO: GSE119347 |
| Raw and analyzed Capture-C data | This paper | GEO: GSE119339 |
| Raw and analyzed ATAC-Seq data | This paper | GEO: GSE119198 |
| Raw and analyzed ChIP-Seq data | This paper | GEO: GSE119200 |
| Raw and analyzed RNA-Seq data | This paper | GEO: GSE122908 |
| Human fibroblast senescence Hi-C data | Criscione et al., 2016 | DDBJ: SRP055421 |
| ESC differentiation to neurons Hi-C data | Bonev et al., 2017 | GEO: GSE96107 |
| Differentiation of cardiovascular muscle cells from induced pluripotent stem cells Hi-C data | Montefiori et al., 2018 | ArrayExpress: E-MTAB-601 |
| Reprogramming of pre-B cells to pluripotent stem cells HiC-data | Stadhouders et al., 2018 | GEO: GSE96553 |
| Mouse ENCODE RNA-Seq | Shen et al., 2012 | GEO: GSE29184 |
| Experimental Models: Cell Lines | ||
| HPC-7 | Pinto do O, et al., 1998 | RRID:CVCL_RB19 |
| Experimental Model: Organisms/Strains | ||
| B6129SF1/J | Jackson Laboratory | 101043; RRID:IMSR_JAX:101043 |
| C57BL6/J | Jackson Laboratory | 000664; RRID:IMSR_JAX:000664 |
| Software and Algoritms | ||
| HiC-Pro | Servant et al., 2015 | https://github.com/nservant/HiC-Pro |
| Shaman R package | Bonev et al., 2017 | https://bitbucket.org/tanaylab/shaman |
| GMAP | Yu et al., 2017 | https://github.com/wbaopaul/rGMAP |
| FIMO | Grant et al., 2011 | http://meme-suite.org/doc/fimo.html |
| DAVID | Huang et al., 2009 | https://david.ncifcrf.gov/ |
| CSI-ANN | Firpi et al., 2010 | https://github.com/tanlabcode/CSI-ANN |
| Bowtie2 | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| CCanalyser2.pl | Davies et al., 2016; Hughes et al., 2014 | https://github.com/telenius/captureC/releases |
| LorDG | Trieu and Cheng, 2017 | https://github.com/BDM-Lab/LorDG |
| Juicebox | Durand et al., 2016 | https://github.com/aidenlab/Juicebox |
| edgeR | Robinson et al., 2010 | http://bioconductor.org/packages/release/bioc/html/edgeR.html |
| LiMACC | This paper | https://github.com/wbaopaul/limacc |
Fetal livers were dissected from E14.5 embryos. Single cell suspension was prepared by dissociating mechanically followed by red blood cell lysis. Cells were stained with an antibody cocktail of CD117 (c-kit), Ly-6A/E (Sca-1), CD135, Ly6G/Ly-6C (Gr-1), TER-119, CD4, CD8a, CD45R/B220, CD3ε and CD19 (Key Resources Table). Yield sort was followed by purity sort for FL HSCs (Lin−Sca-1+c-Kit+CD135−) as described above for BM HSCs.
Lymphoid limiting dilution assays
Lymphoid limiting dilution assay was performed as previously described with a few modifications (Tober et al., 2018). OP9 or OP-DL1 cells were seeded 1 day before co-culture at 4,000 cells/well in a flat 96-well tissue culture plate. For T cell differentiation, cells were cultured with OP9-DL1 in 1ng/mL IL7 and 5ng/mL Flt3L with serial dilution of 50, 25, 12, 6, 3, 2, 1, 0.5 cells per well. Five replicates per dilution were performed. For B cell differentiation, cells were cultured with OP9 in 10ng/mL IL-7 and 5ng/mL Flt3L with the same serial dilution. At days10–12, cells were stained with CD45, CD19, B220 for B differentiation and CD45, CD25, CD90 for T differentiation. Antibody and Cytokine information is listed in Key Resources Table.
Combined lineage potential assay
Single cells (E14.5 fetal liver HSC and adult bone marrow HSC) were directly sorted onto OP9 stromal cells with 25ng/mL SCF, 25ng/ ml FLT3L, and 20ng/mL IL-7. Cultures were transferred from OP9 to OP9-DL1 stromal cells after 7 days of culture with 1ng/mL IL-7 and 5ng/mL FLT3L and analyzed by FACS after a total of 15 days. Clones were defined based on the following markers; B cells, NK1.1− CD19+; T cells, NK1.1− CD19− CD25+ Thy1.2hi; GM cells, NK1.1− CD19− CD25− Thy1.2− Gr1+/−Mac1+ and NK cells, NK1.1+ CD19− CD25− Thy1.2−. Antibody and Cytokine information is listed in Key Resources Table.
Transplantation experiment
50 FACS-sorted E14.5 fetal liver HSCs or adult bone marrow HSCs (6–8 wks) were transplanted together with 250,000 competitor CD45.1/CD45.2 spleen (from B6.SJL mice) cells into B6.SJL Ptprca Pep3b/BoyJ mice by retroorbital injection. Host mice were irradiated with two split doses administered 3–4 hours apart of 4.3–4.5 Gy from a Cs-137 source. Peripheral blood (PB) analyses were conducted at 15–17 weeks. Mice were considered reconstituted if R 0.1% donor contribution to total CD45+ cells were achieved. Antibody information is listed in Key Resources Tables.
Cell Cycle Assay
E14.5 fetal liver cells and bone marrow cells were enriched with anti-CD117 microbeads and labeled with antibody cocktail for HSCs. Cells were then fixed with 4% paraformaldehyde (43368, Alfa Aesar) in PBS, permeabilized with 1% saponin (47036-50G-F, Sigma). Cells were washed with staining buffer and then stained with anti-Ki-67 antibody conjugated with AlexaFluor700, followed by resuspension with 50ug/mL PI solution (421301, Biolegend). Stained cells were analyzed with BD LSRFortessa.
BrdU incorporation assay
BrdU incorporation assay was performed based on the instruction of FITC BrdU flow kit (559619, BD PharMingen). Briefly, cells were seeded into 6-well plates 1 day before BrdU incorporation at the density of 5×105/mL. BrdU (final concentration 10μM) was directly added into culture medium. Cells were fixed, permeabilized, DNase treated, and analyzed at 1 hr, 2 hr, 4 hr, 8 hr, 12 hr, and 24 hr.
In situ Hi-C
In situ Hi-C was performed based on previous publication with a few modifications (Rao et al., 2014). Half million sorted cells were cross-linked with 1% formaldehyde for 10 min at RT and quenched with glycine. Nuclei were then permeabilized with 250 μL of cold lysis buffer (10 mM Tris, 10 mM NaCl, 0.2% Igepal CA630) and 50 μL of protease inhibitors (P8340, Sigma). Chromatin was digested with 100 units of MboI overnight at 37°C with rotation. Restriction fragment ends were labeled with biotinylated nucleotides and proximity ligation in a small volume with 5 μL of 400 U/μL T4 DNA ligase. After reversal of cross-link, DNA was sheared to a length of 300~500 bp and size-selected using AMPure XP beads. The ligated junctions were pulled down with 150 μL of Dynabeads MyOne Streptavidin C1 magnetic beads. End repair, A-tailing, and addition of Illumina index adaptors were performed on beads. Libraries were size-selected and purified using AMPure XP beads. Libraries were sequenced with 75bp paired-end reads on an Illumina NextSeq and/or Hiseq 2000.
Selection of promoters and design of Capture-C probes
To identify genes that are developmentally regulated during the fetal-to-adult transition, we analyzed RNA-Seq data of E14.5 fetal liver HSCs and adult bone marrow HSCs. Using EBSeq (Leng et al., 2013), we identified 7174 differentially expressed transcripts (corresponding to 3464 genes) at a false discovery rate (FDR) cutoff of 0.05. To focus on genes that are specifically expressed in HSCs, we compared the HSC RNA-Seq data to an RNA-Seq compendium of 100 mouse tissues/cells generated by the mouse ENCODE project. Using a Z-score cutoff of 2, we identified 1921 transcripts that are highly expressed in both fetal liver and bone marrow HSCs. By overlapping the two sets of transcripts, we identified 715 transcripts that are expressed at high levels in HSCs and are differentially expressed between fetal liver and bone marrow HSCs. We also included additional 238 genes that are implicated in HSC biology based on literatures evidence (Ali et al., 2009; Deneault et al., 2009; Hope et al., 2010; Huang et al., 2013; Moignard et al., 2015; Rossi et al., 2012). We merged transcripts whose TSSs are located in the same DpnII restriction fragment. In total, 4052 transcripts were selected (Table S2). For each transcript, we defined the 1kb upstream and 1kb downstream of the TSS as the promoter region.
Capture probes for selected promoter regions were designed using the online tools (http://apps.molbiol.ox.ac.uk/CaptureC/cgi-bin/CapSequm.cgi) (Davies et al., 2016; Hughes et al., 2014). Briefly, the genomic coordinates of DpnII sites overlapping the target promoters were identified and 120-bp sequences from both Dpn II sites were generated for each fragment. Candidate probe sequences were filtered based on repeat density score % 10 and simple repeat content % 30. The remaining probe sequences were submitted to custom design of SureDesign capture oligos by Agilent.
Capture-C
Capture-C assay was performed based on previous publication with a few modifications (Davies et al., 2016; Hughes et al., 2014). Half million FACS-sorted cells were cross-linked with 2% formaldehyde for 10 min at RT, quenched with glycine. The cross-linked cells were washed with pre-chilled PBS and lysed with 1mL cold lysis buffer (10 mM Tris, 10 mM NaCl, 0.5% NP-40, and 1x Protease inhibitors) for 10 min on ice. Nuclei were centrifuged at 1600x g for 5 min at 4°C and washed with ddH2O. The nuclei pellet was digested with three aliquots of 500 U DpnII and incubated at 37°C for 16~24hr. DpnII was heat-inactivated by incubating samples at 65°C for 20 min. Chromatin fragments were ligated with 100 U T4 DNA ligase at 16°C for 8 hr with slow rotation. Samples were decross-linked with 3 μL Proteinase K at 65°C overnight, followed by RNase A treatment for 30 min at 37°C. DNA was purified using phenol-chloroform extraction and precipitated using 70% ethanol.
3C DNA was sonicated to 200~300bp using Covaris S220 ultra-sonicator (6 min; duty cycle, 10%; intensity, 5; cycle per burst, 200). Sequencing libraries were constructed using NEBNext Ultra Kit. The libraries were size-selected using AMPure XP beads.
Oligonucleotide capture was performed using the SureSelect XT2 protocol (G9621A, Agilent). The post-captured library was amplified with Herculase II Master Mix and purified using AMPure XP beads. The libraries were sequenced with 150 bp paired-end reads on Illumina NextSeq 500 or HiSeq 2000.
ATAC-Seq and histone modification ChIP-Seq
Assay for Transposase-Accessible Chromatin using Sequencing (ATAC-Seq) was performed based on previous study with minor modification (Buenrostro et al., 2013). Briefly, 50,000 FACS-sorted cells were centrifuged at 1600 g for 5 min at 4°C, followed by one wash using 50 μL of pre-chilled 1x PBS and centrifugation at 1600 g for 5 min at 4°C. Cells were lysed using pre-chilled lysis buffer (10 mM Tris-HCl, pH 7.4, 10mM NaCl, 3mM MgCl2 and 0.1% IGEPAL CA-630). Nuclei were centrifuged at 1600 g for 10 min at 4°C. Nuclei were re-suspended in transposase reaction mix (25 μL 2 × TD buffer, 2.5 μL transposase (FC-121-1030, Illumina) and 22.5 μL nuclease-free water) and incubated for 30 min at 37°C. The sample was purified using a QIAGEN MinElute kit (28004, QIAGEN). Following purification, libraries were amplified using 1x NEBNext PCR master mix and custom Nextera PCR primers. Libraries were size-selected at 100–700 bp by gel extraction (28604, QIAGEN). Libraries were quantified with KAPA qPCR and bioanalyzer prior to pair-end sequencing on Illumina HiSeq 2000.
Low-Cell-Number ChIP-Seq was performed as following. Briefly, 50,000 cells for each IP were cross-linked with 1% formaldehyde (28906, Thermo Scientific) for 5 min at RT. Cells were re-suspended in 1x shearing buffer and sonicated with Covaris E220 for 780 s. 5% sheared chromatin was used as the input and the remaining chromatin was used for IP. IP was performed using ChIP-IT high sensitivity kit (53040, Active Motif) with some modifications. IP and input samples were treated with RNase A followed by proteinase K treatment. Cross-linking was reversed by incubating overnight at 65°C. Reverse crosslinked DNA was purified using a MinElute PCR purification kit (QIAGEN, 28004) and re-suspended in 10 μL nuclease-free water. All IPed DNA and 1 ng input DNA were used for library preparation using the ThruPLEX-FDPrep kit (R40048, Rubicon Genomics) with 12 cycles of amplification for IP DNA and 9 cycles for input DNA. Libraries were quantified with KAPA qPCR and bioanalyzer prior to single-end sequencing on Illumina HiSeq 2000.
DNA fluorescence in situ hybridization (DNA-FISH)
Sorted cells were washed once with 1 mL PBS. Cells were fixed with 1 mL of MAA (methanol: acetic acid = 3:1) for 15 min on ice and spun down and re-suspended in 1 mL of MAA. This process is repeated for at least three times. Five million fixed cells were re-suspended in 1 mL of MAA. Cells were immobilized on the slide and denatured at 72°C for 3 min. Hybridization was performed in a dark humidity chamber for 3 days. Slides were washed with SSC buffer (S6639, Sigma), followed by staining with DAPI (P36935, Invitrogen). Slides were stored at −20°C or imaged immediately.
CRISPR-mediated knockout of Tcf3 in HPC-7 cells
Guide RNA sequences targeting Tcf3 were designed using Deskgen tools (Table S6). Annealed sgRNAs were cloned into lenti-CRISPR v2 (52961, Addgene). Lentivirus was produced by co-transfecting with pMD2.G (12259, Addgene) and psPAX2 (12260, Addgene) into HEK293FT cells. HPC-7 cells were transduced by lentivirus and positive cells were selected by culturing with 0.5 ug/mL puromycin for 21 days. Knockout of Tcf3 was confirmed using Western Blot.
QUANTIFICATION AND STATISTICAL ANALYSIS
Hi-C data processing
Hi-C read mapping, detection of valid interactions, correction of systematic noise, and calculation of normalized contact matrices were performed using HiC-Pro (Servant et al., 2015) with default parameters. Paired-end reads were mapped to the mm9 version of the mouse genome. Normalized contact matrices at 10kb resolution were computed using the ICE (Imakaev et al., 2012) algorithm with default parameters.
Analysis of chromatin compartments
Chromosome compartments were identified using principal component analysis (PCA). We first calculated the contact matrix for each chromosome using TAD as the unit. A cell of the contact matrix Oij represents the total number of contacts between the ith and jth TADs. We adjusted the contact matrix according to the TAD sizes and distance as Oij/sisjEij, where si, sj and Eij are the sizes of the ith and jth TADs, and the averaged contact frequency between genomic loci with distance dij, which is the distance between the middle points of the ith and jth TADs. Next, we converted the above contact matrix to Pearson’s correlation matrices and PCA was conducted on the correlation matrices. The sign of first principle component, denoted as PC1, was used to assign compartment label. Because the sign of PC1 was arbitrary, additional information was used for compartment assignment. As suggested by Dixon et al. (2015) and Schmitt et al. (2016), genomic regions with high gene density were assigned to positive PC1 values and correspond to compartment A. The rest of the genome were assigned to negative PC1 value and correspond to compartment B. Degree of compartmentalization was measured by the contact frequency between all possible pairs of TADs from the same (AA or BB) or from different types of compartments (AB). The normalized contact frequency of each pair of TADs was computed as the log2 ratio of the total number of observed inter-TAD contacts to the total number of expected inter-TAD interactions. The expected number of contacts between any pair of loci was calculated using the Shaman R package (https://bitbucket.org/tanaylab/shaman).
Analysis of topologically associating domains (TADs)
TADs were called using normalized Hi-C data and the GMAP algorithm (Yu et al., 2017). Two TAD boundaries were considered shared if they are within 50kb of each other.
Calculation of TAD boundary strength
To compute the score for TAD boundary strength, we first calculated the log2 ratio of observed to expected contact frequency between any two genome loci using the Shaman R package which we referred to as the Shaman ratio hereafter. The boundary strength score was then defined as the difference between the intra-TAD Shaman ratio and the inter-TAD Shaman ratio between the 600kb up- and down-stream regions flanking a TAD boundary. To identify boundaries with significantly altered strength between FL HSCs and BM HSCs, we first computed a null distribution of boundary strength difference using 40,000 randomly selected genomic loci that do not overlap with any observed TAD boundaries. The p value for altered boundary strength was then computed based on the null distribution. Multiple testing correction was conducted using the Benjamini-Hochberg procedure. Dscore, a statistic provided by the Shaman package, was used to visualize TADs and TAD boundaries.
Dynamics of promoter-centered intra-TAD interactions
We studied the dynamics of promoter-centered intra-TAD interactions by taking advantage of our high-resolution Capture-C data. Promoter-centered chromatin interactions were identified using the LiMACC algorithm with an FDR cutoff of 0.01. For each significant interaction identified in at least one cell type, the normalized interaction frequencies in both cell types were paired. TADs with fewer than 5 interaction pairs were excluded. P values for TADs with significant changes in promoter interactions was computed using paired t test. P values were adjusted for multiple testing using the Benjamini-Hochberg procedure.
3D genome structure modeling
ICE (Imakaev et al., 2012) normalized Hi-C contact matrix was further normalized by quantile normalization between FL HSCs and BM HSCs at 25kb resolution. Whole chromosome models were reconstructed on the quantile normalized Hi-C contact matrix using LorDG (Trieu and Cheng, 2017). The method is robust to noise and inconsistency in Hi-C data. It works by first translating contact frequencies into spatial distances and then solving an optimization problem to build 3D models consistent with the spatial distances. The 3D coordinates of each binned locus were used for calculating Euclidean distance between two adjacent TADs associated with a given TAD boundary (Figure 2F).
Capture-C data processing
Capture-C data were processed using the pipeline from Hughes et al. (2014). Briefly, Illumina TruSeq adaptor sequences were trimmed from raw reads using trim galore version 0.41 with default parameter setting. Paired-end reads were merged into one single fastq file to ensure each pair of reads interleaved in strict order. Reads were mapped to the mouse genome (mm9) using Bowtie2 (v2.2.2). Mapped reads were analyzed using the script CCanalyser2.pl (https://github.com/telenius/captureC/releases). Unique informative reads were extracted for each captured bait as the input for calling significant interactions.
Enhancer and promoter prediction using CSI-ANN
Enhancers and promoters were predicted using the CSI-ANN algorithm. The inputs to the algorithm are normalized ChIP-Seq signals of four histone marks (H3K4me1, H3K4me3, H3K27ac, and H3K27me3). The algorithm combines signals of all histone marks and uses an artificial neural network-based classifier to make predictions.
Interaction calling using Capture-C data
We developed a local iterative modeling approach for identifying chromatin interactions using Capture-C or Capture Hi-C data (LiMACC). The basic idea is to categorize all capture bait and other end interactions (BOEIs) into different groups based on the distance between the two ends, and then fit a negative binomial model in each group. To estimate the null distribution, an iterative model fitting approach is used.
Suppose there are m baits, and N BOEIs, and each BOEI is supported by at least one read.
For each BOEI, calculate the distance between the corresponding bait and other end, as d1;… dN.
- Classify each BOEI into one of the following G groups based on the distance:
- a. Let qk be the 100k% quantile of the N distances, k = 0, 1,…G, and
- b. The ith BOEI is pooled into group k if qk–1≤di < qk.
To generate the null distribution, in each group, we iteratively define high confidence random contacts (HCRC) and fit a negative binomial distribution using the HCRCs.
Calculate raw p values for each BOEI using the null distribution obtained from step 3 and pool the raw N p values and adjust them based on the Independent Hypothesis Weighting (IHW) procedure (Ignatiadis et al., 2016).
The detailed iterative procedure for model fitting is given as follows:
In each group, define the initial HCRCs as those with the number of reads in the bottom 95 percentile.
Fit the null distribution in each group using the corresponding HCRCs and calculate the raw p values.
Pool all raw p values and adjust them by either the BH procedure or IHW procedure.
For each group, define HCRCs as those whose adjusted p values are greater than a given FDR cutoff.
Repeat step 2–4 until there is no change in the set of HCRCs for each group and output the corresponding adjusted p values for each interaction.
Normalization of raw read counts
Some BOEIs may have larger number of reads than others due to experimental biases. We therefore normalized the average number of reads per bait to a fixed number. Suppose Nij is the raw read count between bait i and the other end j, let and M as the median number of , then the normalized reads are defined as:
Adjusting bait to bait bias
Given the nature of the Capture-C protocols, the interactions between two baits are more likely to be captured than interactions between a bait and a non-bait. We propose to adjust this bait to bait bias in the following way:
For a given bait i, normalize the median contacts of i to another bait to be the same as the median contacts of bait i to other non-bait ends. We adjust such effect separately for each bait. Let Oi be the median number of and Bi be the median number of then we adjust nij between bait i and j as:
Promoter interacting regions (PIRs)
We re-binned the other end fragments of significant interactions into 2kb windows. The score of each window was defined as the largest LiMACC score (negative log-transformation in base 10 of the adjusted p value) of all other ends that mapped to that window. We denoted those other end windows as Promoter Interacting Regions (PIRs). Downstream analyses such as transcription factor enrichment analysis and clustering analysis were based on PIRs.
ChIP-Seq data processing
Sequencing reads were mapped to the mouse genome (mm9) using Bowtie2 (v2.2.2) (Langmead et al., 2009) with default parameter setting. Uniquely mapped reads from both ChIPed and input DNA were used to compute a normalized signal for each 200 bp bin across the genome. Normalized signal is defined as following:
ATAC-Seq data processing
Ilumina Nextera transposase adaptor sequences were trimmed from raw reads using trim galore version 0.41 with default parameter setting. Trimmed reads were mapped to the mouse genome (mm9) using Bowtie2 (v2.2.2) (Langmead et al., 2009) and default parameter setting. ATAC-Seq peaks were called by MACS using default parameter setting.
TF motif analysis of enhancers involved in cell-specific enhancer-promoter interaction
The DNA binding motifs of 718 TFs were downloaded from the CIS-BP database. The FIMO software (Grant et al., 2011) was used to scan the enhancer regions that overlap with ATAC-seq peaks. Significant motif hits were called using a p value cutoff 0.01 with Bonferroni correction. Hypergeometric test was used to determine the enrichment of a given TF motif in the set of enhancers involved in stage-specific enhancer-promoter interactions. Raw hypergeometric p values are corrected for multiple testing using the Benjamini-Hochberg procedure.
Gene ontology analysis
GO term enrichment analyses were performed using the DAVID tool (Huang et al., 2009) (version 6.8). Raw p values were adjusted using the Benjamin-Hochberg procedure.
DATA AND CODE AVAILABILITY
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE119201. LiMACC algorithm is freely available on GitHub (https://github.com/tanlabcode/limacc).
Supplementary Material
Highlights.
Comparative analysis of 3D genome, epigenome, and transcriptome of fetal and adult HSCs
Compartments and TADs are conserved but compartmentalization and boundary strengthen
Dynamic enhancer-promoter interactions target genes involved in phenotypic differences
TCF3 mediates developmental-stage-specific enhancer-promoter interactions
ACKNOWLEDGMENTS
We thank the Research Information Services at the Children’s Hospital of Philadelphia for providing computing support. This work was supported by NIH USA (GM104369, GM108716, and HG006130 to K.T., HD089245 to K.T. and N.A.S., and HL129998 to G.A.B.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.11.065.
REFERENCES
- Adolfsson J, Borge OJ, Bryder D, Theilgaard-Mönch K, Astrand-Grund-ström I, Sitnicka E, Sasaki Y, and Jacobsen SE (2001). Upregulation of Flt3 expression within the bone marrow Lin(−)Sca1(+)c-kit(+) stem cell compartment is accompanied by loss of self-renewal capacity. Immunity 15, 659–669. [DOI] [PubMed] [Google Scholar]
- Ali N, Karlsson C, Aspling M, Hu G, Hacohen N, Scadden DT, and Larsson J. (2009). Forward RNAi screens in primary human hematopoietic stem/progenitor cells. Blood 113, 3690–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belle I, and Zhuang Y. (2014). E proteins in lymphocyte development and lymphoid diseases. Curr. Top. Dev. Biol 110, 153–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz C, Copley MR, Kent DG, Wohrer S, Cortes A, Aghaeepour N, Ma E, Mader H, Rowe K, Day C, et al. (2012). Hematopoietic stem cell subtypes expand differentially during development and display distinct lymphopoietic programs. Cell Stem Cell 10, 273–283. [DOI] [PubMed] [Google Scholar]
- Bonev B, and Cavalli G. (2016). Organization and function of the 3D genome. Nat. Rev. Genet 17, 661–678. [DOI] [PubMed] [Google Scholar]
- Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot JP, Tanay A, et al. (2017). Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 171, 557–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie MB, McKnight KD, Kent DG, McCaffrey L, Hoodless PA, and Eaves CJ (2006). Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. J. Clin. Invest 116, 2808–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie MB, Kent DG, Copley MR, and Eaves CJ (2007a). Steel factor responsiveness regulates the high self-renewal phenotype of fetal hematopoietic stem cells. Blood 109, 5043–5048. [DOI] [PubMed] [Google Scholar]
- Bowie MB, Kent DG, Dykstra B, McKnight KD, McCaffrey L, Hoodless PA, and Eaves CJ (2007b). Identification of a new intrinsically timed developmental checkpoint that reprograms key hematopoietic stem cell properties. Proc. Natl. Acad. Sci. USA 104, 5878–5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch K, Klapproth K, Barile M, Flossdorf M, Holland-Letz T, Schlenner SM, Reth M, Höfer T, and Rodewald HR (2015). Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature 518, 542–546. [DOI] [PubMed] [Google Scholar]
- Cairns J, Freire-Pritchett P, Wingett SW, Várnai C, Dimond A, Plagnol V, Zerbino D, Schoenfelder S, Javierre BM, Osborne C, et al. (2016). CHiCAGO: robust detection of DNA looping interactions in Capture Hi-C data. Genome Biol. 17, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen JL, and Weissman IL (2001). Flk-2 is a marker in hematopoietic stem cell differentiation: a simple method to isolate long-term stem cells. Proc. Natl. Acad. Sci. USA 98, 14541–14546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copley MR, Babovic S, Benz C, Knapp DJ, Beer PA, Kent DG, Wohrer S, Treloar DQ, Day C, Rowe K, et al. (2013). The Lin28b-let-7-Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells. Nat. Cell Biol 15, 916–925. [DOI] [PubMed] [Google Scholar]
- Criscione SW, De Cecco M, Siranosian B, Zhang Y, Kreiling JA, Sedivy JM, and Neretti N. (2016). Reorganization of chromosome architecture in replicative cellular senescence. Sci. Adv 2, e1500882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JO, Telenius JM, McGowan SJ, Roberts NA, Taylor S, Higgs DR, and Hughes JR (2016). Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nat. Methods 13, 74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deneault E, Cellot S, Faubert A, Laverdure JP, Fréchette M, Chagraoui J, Mayotte N, Sauvageau M, Ting SB, and Sauvageau G. (2009). A functional screen to identify novel effectors of hematopoietic stem cell activity. Cell 137, 369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. (2015). Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drissen R, Palstra RJ, Gillemans N, Splinter E, Grosveld F, Philipsen S, and de Laat W. (2004). The active spatial organization of the beta-globin locus requires the transcription factor EKLF. Genes Dev. 18, 2485–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, and Aiden EL (2016). Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 3, 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ema H, and Nakauchi H. (2000). Expansion of hematopoietic stem cells in the developing liver of a mouse embryo. Blood 95, 2284–2288. [PubMed] [Google Scholar]
- Firpi HA, Ucar D, and Tan K. (2010). Discover regulatory DNA elements using chromatin signatures and artificial neural network. Bioinformatics 26, 1579–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire PR, and Conneely OM (2018). NR4A1 and NR4A3 restrict HSC proliferation via reciprocal regulation of C/EBPa and inflammatory signaling. Blood 131, 1081–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Kim I, Lim MS, and Morrison SJ (2011). Sox17 expression confers self-renewal potential and fetal stem cell characteristics upon adult hematopoietic progenitors. Genes Dev. 25, 1613–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope KJ, Cellot S, Ting SB, MacRae T, Mayotte N, Iscove NN, and Sauvageau G. (2010). An RNAi screen identifies Msi2 and Prox1 as having opposite roles in the regulation of hematopoietic stem cell activity. Cell Stem Cell 7, 101–113. [DOI] [PubMed] [Google Scholar]
- Hu G, Cui K, Fang D, Hirose S, Wang X, Wangsa D, Jin W, Ried T, Liu P, Zhu J, et al. (2018). Transformation of Accessible Chromatin and 3D Nucleome Underlies Lineage Commitment of Early T Cells. Immunity 48, 227–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Huang HT, Kathrein KL, Barton A, Gitlin Z, Huang YH, Ward TP, Hofmann O, Dibiase A, Song A, Tyekucheva S, et al. (2013). A network of epigenetic regulators guides developmental haematopoiesis in vivo. Nat. Cell Biol 15, 1516–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Keller CA, Giardine B, Grevet JD, Davies JOJ, Hughes JR, Kurita R, Nakamura Y, Hardison RC, and Blobel GA (2017). Comparative analysis of three-dimensional chromosomal architecture identifies a novel fetal hemoglobin regulatory element. Genes Dev. 31, 1704–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JR, Roberts N, McGowan S, Hay D, Giannoulatou E, Lynch M, De Gobbi M, Taylor S, Gibbons R, and Higgs DR (2014). Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat. Genet 46, 205–212. [DOI] [PubMed] [Google Scholar]
- Ignatiadis N, Klaus B, Zaugg JB, and Huber W. (2016). Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat. Methods 13, 577–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, and Mirny LA (2012). Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods 9, 999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijger PH, Di Stefano B, de Wit E, Limone F, van Oevelen C, de Laat W, and Graf T. (2016). Cell-of-Origin-Specific 3D Genome Structure Acquired during Somatic Cell Reprogramming. Cell Stem Cell 18, 597–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku CJ, Hosoya T, Maillard I, and Engel JD (2012). GATA-3 regulates hematopoietic stem cell maintenance and cell-cycle entry. Blood 119, 2242–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land RH, Rayne AK, Vanderbeck AN, Barlowe TS, Manjunath S, Gross M, Eiger S, Klein PS, Cunningham NR, Huang J, et al. (2015). The orphan nuclear receptor NR4A1 specifies a distinct subpopulation of quiescent myeloid-biased long-term HSCs. Stem Cells 33, 278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, Smits BM, Haag JD, Gould MN, Stewart RM, and Kendziorski C. (2013). EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 29, 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Jhunjhunwala S, Benner C, Heinz S, Welinder E, Mansson R, Sigvardsson M, Hagman J, Espinoza CA, Dutkowski J, et al. (2010). A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat. Immunol 11, 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Månsson R, Hultquist A, Luc S, Yang L, Anderson K, Kharazi S, Al-Hashmi S, Liuba K, Thorén L, Adolfsson J, et al. (2007). Molecular evidence for hierarchical transcriptional lineage priming in fetal and adult stem cells and multipotent progenitors. Immunity 26, 407–419. [DOI] [PubMed] [Google Scholar]
- Min IM, Pietramaggiori G, Kim FS, Passegué E, Stevenson KE, and Wagers AJ (2008). The transcription factor EGR1 controls both the proliferation and localization of hematopoietic stem cells. Cell Stem Cell 2, 380–391. [DOI] [PubMed] [Google Scholar]
- Moignard V, Woodhouse S, Haghverdi L, Lilly AJ, Tanaka Y, Wilkinson AC, Buettner F, Macaulay IC, Jawaid W, Diamanti E, et al. (2015). Decoding the regulatory network of early blood development from single-cell gene expression measurements. Nat. Biotechnol 33, 269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montefiori LE, Sobreira DR, Sakabe NJ, Aneas I, Joslin AC, Hansen GT, Bozek G, Moskowitz IP, McNally EM, and Nóbrega MA (2018). A promoter interaction map for cardiovascular disease genetics. eLife 7, e35788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Lubling Y, Várnai C, Dudley C, Leung W, Baran Y, Mendelson Cohen N, Wingett S, Fraser P, and Tanay A. (2017). Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 547, 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passegué E, Wagers AJ, Giuriato SWC, and Weissman IL (2005). Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J. Exp. Med 202, 1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintodo O P, Kolterud A, and Carlsson L. (1998). Expression of the LIM-homeobox gene LH2 generates immortalized steel factor-dependent multipotent hematopoietic precursors. EMBO J. 17, 5744–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, and Aiden EL (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, et al. (2017). Cohesin Loss Eliminates All Loop Domains. Cell 171, 305–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro de Almeida C, Stadhouders R, Thongjuea S, Soler E, and Hendriks RW (2012). DNA-binding factor CTCF and long-range gene interactions in V(D)J recombination and oncogene activation. Blood 119, 6209–6218. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bio-conductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi L, Lin KK, Boles NC, Yang L, King KY, Jeong M, Mayle A, and Goodell MA (2012). Less is more: unveiling the functional core of hematopoietic stem cells through knockout mice. Cell Stem Cell 11, 302–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrazin S, Mossadegh-Keller N, Fukao T, Aziz A, Mourcin F, Vanhille L, Kelly Modis L, Kastner P, Chan S, Duprez E, et al. (2009). MafB restricts M-CSF-dependent myeloid commitment divisions of hematopoietic stem cells. Cell 138, 300–313. [DOI] [PubMed] [Google Scholar]
- Schmitt AD, Hu M, and Ren B. (2016). Genome-wide mapping and analysis of chromosome architecture. Nat. Rev. Mol. Cell Biol 17, 743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuütte J, Wang H, Antoniou S, Jarratt A, Wilson NK, Riepsaame J, Calero-Nieto FJ, Moignard V, Basilico S, Kinston SJ, et al. (2016). An experimentally validated network of nine haematopoietic transcription factors reveals mechanisms of cell state stability. eLife 5, e11469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe-Mie Y, Fonseca NA, Huber W, Haering CH, Mirny L, and Spitz F. (2017). Two independent modes of chromatin organization revealed by cohesin removal. Nature 551, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semerad CL, Mercer EM, Inlay MA, Weissman IL, and Murre C. (2009). E2A proteins maintain the hematopoietic stem cell pool and promote the maturation of myelolymphoid and myeloerythroid progenitors. Proc. Natl. Acad. Sci. USA 106, 1930–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, Heard E, Dekker J, and Barillot E. (2015). HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton T, and Cavalli G. (2015). The role of chromosome domains in shaping the functional genome. Cell 160, 1049–1059. [DOI] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, and Ren B. (2012). A map of the cis-regulatory sequences in the mouse genome. Nature 488, 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadhouders R, Thongjuea S, Andrieu-Soler C, Palstra RJ, Bryne JC, van den Heuvel A, Stevens M, de Boer E, Kockx C, van der Sloot A, et al. (2012). Dynamic long-range chromatin interactions control Myb protooncogene transcription during erythroid development. EMBO J. 31, 986–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadhouders R, Vidal E, Serra F, Di Stefano B, Le Dily F, Quilez J, Gomez A, Collombet S, Berenguer C, Cuartero Y, et al. (2018). Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat. Genet 50, 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terakawa T, Bisht S, Eeftens JM, Dekker C, Haering CH, and Greene EC (2017). The condensin complex is a mechanochemical motor that trans-locates along DNA. Science 358, 672–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tober J, Maijenburg MMW, Li Y, Gao L, Hadland BK, Gao P, Minoura K, Bernstein ID, Tan K, and Speck NA (2018). Maturation of hematopoietic stem cells from prehematopoietic stem cells is accompanied by up-regulation of PD-L1. J. Exp. Med 215, 645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trieu T, and Cheng J. (2017). 3D genome structure modeling by Lorentzian objective function. Nucleic Acids Res. 45, 1049–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vian L, Pekowska A, Rao SSP, Kieffer-Kwon KR, Jung S, Baranello L, Huang SC, El Khattabi L, Dose M, Pruett N, et al. (2018). The Energetics and Physiological Impact of Cohesin Extrusion. Cell 173, 1165–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z, Gao F, Kim S, Yang H, Lyu J, An W, Wang K, and Lu W. (2013). Klf4 organizes long-range chromosomal interactions with the oct4 locus in reprogramming and pluripotency. Cell Stem Cell 13, 36–47. [DOI] [PubMed] [Google Scholar]
- Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, et al. (2008). Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135, 1118–1129. [DOI] [PubMed] [Google Scholar]
- Wilson NK, Foster SD, Wang X, Knezevic K, Schütte J, Kaimakis P, Chilarska PM, Kinston S, Ouwehand WH, Dzierzak E, et al. (2010). Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell 7, 532–544. [DOI] [PubMed] [Google Scholar]
- Wilson NK, Kent DG, Buettner F, Shehata M, Macaulay IC, Calero-Nieto FJ, Sánchez Castillo M, Oedekoven CA, Diamanti E, Schulte R, et al. (2015). Combined Single-Cell Functional and Gene Expression Analysis Resolves Heterogeneity within Stem Cell Populations. Cell Stem Cell 16, 712–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolthuis CM, and Park CY (2016). Hematopoietic stem/progenitor cell commitment to the megakaryocyte lineage. Blood 127, 1242–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Bryder D, Adolfsson J, Nygren J, Månsson R, Sigvardsson M, and Jacobsen SE (2005). Identification of Lin(−)Sca1(+)kit(+)CD34(+)Flt3-short-term hematopoietic stem cells capable of rapidly reconstituting and rescuing myeloablated transplant recipients. Blood 105, 2717–2723. [DOI] [PubMed] [Google Scholar]
- Yu W, He B, and Tan K. (2017). Identifying topologically associating domains and subdomains by Gaussian Mixture model And Proportion test. Nat. Commun 8, 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CC, and Lodish HF (2004). Insulin-like growth factor 2 expressed in a novel fetal liver cell population is a growth factor for hematopoietic stem cells. Blood 103, 2513–2521. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE119201. LiMACC algorithm is freely available on GitHub (https://github.com/tanlabcode/limacc).