

Abstract
To identify novel D3 dopamine receptor (D3R) agonists, we conducted a high-throughput screen using a β-arrestin recruitment assay. Counter-screening of the hit compounds provided an assessment of their selectivity, efficacy, and potency. The most promising scaffold was optimized through medicinal chemistry resulting in enhanced potency and selectivity. The optimized compound, ML417 (20), potently promotes D3R-mediated β-arrestin translocation, G protein activation, and pERK phosphorylation while lacking activity at other dopamine receptors. Screening of ML417 against multiple GPCRs revealed exceptional global selectivity. Molecular modeling suggests that ML417 interacts with the D3R in a unique manner, possibly explaining its remarkable selectivity. ML417 was also found to protect against neurodegeneration of dopaminergic neurons derived from iPSCs. Together with promising pharmacokinetics and toxicology profiles, these results suggest that ML417 is a novel and uniquely selective D3R agonist that may serve as both a research tool and a therapeutic lead for the treatment of neuropsychiatric disorders.
Keywords: dopamine, D3 receptor, agonist, molecular modeling, neuroprotection
Graphical Abstract
INTRODUCTION
Dysregulation of dopamine receptors is linked to the etiology and/or therapy of many neuropsychiatric disorders including Parkinson’s disease (PD), schizophrenia, and substance use disorders.1–5 These receptors are categorized into two subfamilies: D1-like (D1R and D5R) and D2-like (D2R, D3R, and D4R) based on protein structure, pharmacology, and physiological signaling.1, 3, 5 Dopamine receptor subfamilies exhibit high sequence homology, particularly the D2R and D3R, which share 74% identity between their transmembrane domains (TMs) and 94% identity between their putative orthosteric binding sites, where the endogenous agonist dopamine (DA) binds.6 Not surprisingly, most currently available drugs that target these receptors, including antipsychotics, anti-PD medications, and research tool compounds, are not highly subtype selective but instead modulate both D2R and D3R subtypes to varying degrees.7, 8 Further, most known compounds that target the D2R and/or D3R also cross-react with other related G protein-coupled receptors (GPCRs), especially those for biogenic amines, creating the potential for profound off-target side effects. Thus, more selective agents are critically needed not only for the delineation of dopamine receptor action in vivo, but also for the development of more selective therapeutics with fewer side effects.
D3R activation is known to have important therapeutic effects. Agonists that are D3R-preferring (e.g., ~10-fold D3R>D2R selective), including pramipexole and ropinirole, are effective in treating both PD and restless legs syndrome (RLS). These compounds are clinically active in relieving motor deficits and slowing the loss of dopaminergic terminals upon long-term administration to PD patients.9, 10 Further, in animal models, D3R-preferring agonists are the most potent neuroprotective agents identified to date against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced neurodegeneration in mice11–14 and primates,15 and against 6-hydroxydopamine (6-OHDA)-induced lesions in rats.16 Importantly, studies with D3R-knockout mice support that the neuroprotective effects are mediated directly by the D3R.14, 17, 18 Unfortunately, clinical treatment with drugs such as pramipexole and ropinirole is frequently associated with impulse control disorders including pathological gambling, shopping, eating, and hypersexuality. These side effects typically occur at higher doses that would fully activate the D2R, which may underlie the loss of impulse control.19, 20 Thus, a highly selective D3R agonist which lacks D2R activity may hold significant therapeutic potential, as well as filling an important role as a probe compound for dissecting signaling pathways underlying D3R signaling from those of D2R.
Not surprisingly, extensive efforts have been directed toward the development of selective agonists and antagonists that target the D3R.21, 22 Significant progress has been made in the development of antagonists that are selective for the D3R vs. other dopamine receptors, which has been aided by the availability of a crystal structure of the D3R in an inactive state.6, 23 The most selective D3R antagonists are extended-length compounds that are structurally bivalent in that they possess a primary pharmacophore that binds to the orthosteric site and, connected via a linker, a secondary pharmacophore that binds to a secondary site, which may confer allosteric effects.23–30 Bivalent compounds that possess allosteric properties are considered to be bitopic in nature.31–35 No D3R-selective antagonists have yet reached the clinic,36, 37 however, newer antagonists have shown promise, especially for the treatment of substance use disorders.28, 29, 38–41
Less progress has been made in the development of D3R-selective agonists for clinical use. In part, this is due to the unavailability of a crystal structure of the D3R in an active state. As might be expected, virtual screens based on the inactive state D3R structure have yielded mostly compounds with antagonist-like effects.42, 43 In contrast, synthetic medicinal chemistry approaches to D3R-selective agonists have been more successful with some agents achieving good pharmacological separation between the D3R and D2R.22, 44–50 Various examples of D3R-preferring agonists and their structures have recently been described in Moritz et al.8 However, nearly all such compounds have been based on a single precursor scaffold – pramipexole, and their global GPCR selectivity and suitability for clinical advancement is unclear. Clearly, novel chemical scaffolds for designing highly selective D3R agonists are greatly needed. Very recently, the approach of designing extended-length bivalent compounds with D3R-selective agonist properties has been reported.34, 51
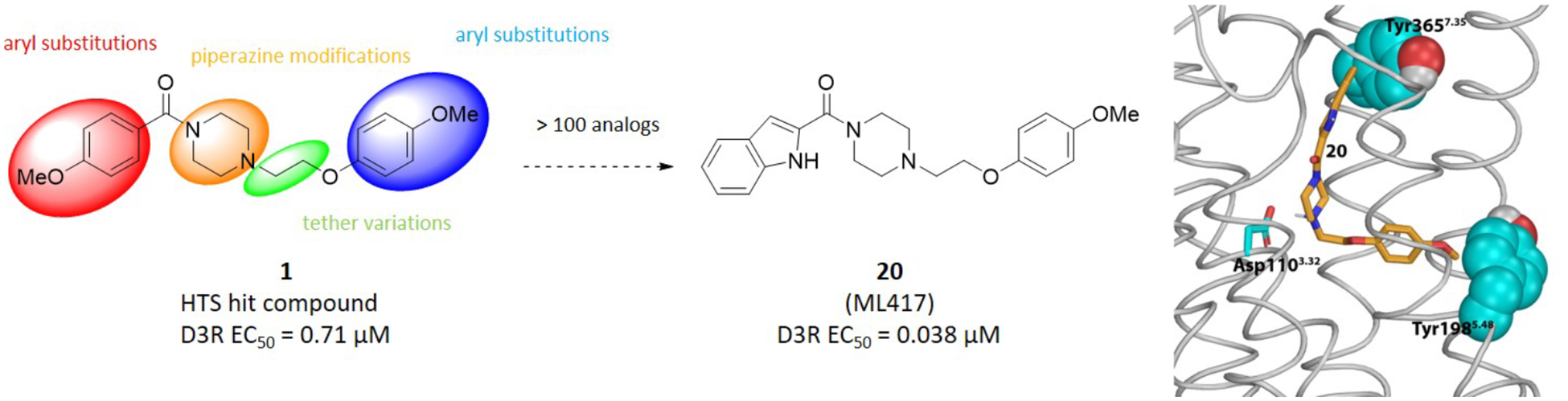
Here we report the identification and development of a novel agonist scaffold that exhibits high selectivity for the D3R. Starting from an unbiased high-throughput screening approach, we identified a hit compound with promising pharmacological and structural characteristics. From this initial hit compound, over 100 structural analogs were synthesized and characterized to develop a comprehensive structure–activity relationship (SAR) investigation and to establish the structural determinants for potency, efficacy, and selectivity at the D3R. An optimized lead compound, 20 (ML417), was identified that promotes potent D3R-mediated β-arrestin translocation, G protein-mediated signaling, and pERK phosphorylation with minimal effects on other GPCR-mediated signaling. In addition, 20 was found to exhibit neuroprotection against toxin-induced neurodegeneration of dopaminergic neurons. In summary, 20 is a novel and uniquely selective D3R agonist that will prove useful as a research tool and may show utility as a therapeutic lead for the treatment of neuropsychiatric disorders.
RESULTS AND DISCUSSION
High-throughput screening and hit compound identification.
To identify novel agonist scaffolds for the D3R, we conducted a high-throughput screen (HTS) of the NIH Molecular Libraries Probe Production Centers Network (MLPCN) small molecule library consisting of ~400,000 diverse compounds.52 The HTS utilized two concentrations (10 μM and 50 μM) of each library compound and measured their ability to stimulate β-arrestin recruitment to the D3R. The primary screen identified 4,165 compounds that stimulated β-arrestin recruitment greater than >30% (>3 standard deviations) over basal (vehicle control) at either the lower (10 μM) or higher (50 μM) concentration of library compound. Primary screening data were deposited in PubChem (AID 652050 and 652048). Hit compounds were first chemoinformatically triaged based on results from a previously executed D2R HTS of the same library53, 54 to eliminate compounds that also possess D2R agonist activity. In addition, compounds were further triaged if they contained known chemically reactive moieties and/or promiscuous scaffolds resulting in ~2,500 unique compounds that were then tested in secondary assays. These triaged hit compounds were characterized using 7-point concentration-response assays of β-arrestin recruitment to the D3R and D2R, as well as β-arrestin recruitment to the unrelated prostaglandin E receptor 2 (PTGER2) to assess selectivity and eliminate compounds that modulate β-arrestin recruitment in a nonspecific manner. 152 unique agonist compounds replicated in a concentration-dependent manner with demonstrated D3R>D2R selectivity (data not shown) and were selected for further characterization.
All 152 vetted hit compounds were evaluated using D3R radioligand binding competition assays that employed the orthosteric antagonist [3H]-methylspiperone. 90 of these compounds inhibited radioligand binding by greater than 50% at a dose of 40 μM and were thus initially classified as orthosteric. In contrast, 62 compounds failed to inhibit binding by greater than 50% (but were functionally active) and thus initially classified as potentially allosteric in nature. As allosteric compounds have the potential for global GPCR selectivity,55 we focused on those 62 compounds that were relatively ineffective at inhibiting [3H]-methylspiperone binding. These compounds were ranked via potency, D3R>D2R selectivity, and chemical tractability resulting in lead compound 1 (Figure 1).
Figure 1. Chemical structure of the HTS lead, compound 1.

.
The HTS lead 1 is a full agonist (compared to dopamine) in a D3R-mediated β-arrestin recruitment assay, displaying an EC50 of 710 nM (Figure 2A). No measurable agonist activity was detected at the D2R using the same β-arrestin recruitment assay at concentrations up to 100 μM (Figure 2B). Interestingly, when 1 was tested for its ability to inhibit DA-stimulated β-arrestin recruitment to the D2R, we found that it was able to antagonize the receptor with an IC50 of 16 μM (Figure 2C). We next sought to determine the affinity of 1 at the orthosteric binding site by conducting competition binding studies with [3H]-methylspiperone at both the D2R and D3R. As shown in Figure 2D, the compound weakly inhibited radioligand binding to the D3R with an IC50 >10 μM and no measurable ability to displace binding to the D2R. This observation suggests an apparent disconnect between the potency of 1 as an agonist in the D3R β-arrestin functional assay and its limited, but demonstrable, ability to inhibit the binding of [3H]-methylspiperone to the D3R. One possible explanation is that 1 is a weak partial (orthosteric) agonist of the D3R that appears as a potent full agonist in the functional assay due to spare receptors and/or signal amplification. This is not likely, however, as the β-arrestin recruitment assay only produces a signal when the receptor and β-arrestin are complemented 1:1–there are no spare receptors or signal amplification. Regardless of mechanism, the overall activity profile of 1 appeared to warrant further investigation and its optimization.
Figure 2. Pharmacological activity of lead compound 1 on the D3R and D2R.

A and B, Agonist concentration-response curves for stimulating β-arrestin recruitment in response to either dopamine (DA) or 1 using cells expressing either the D3R (A) or D2R (B). Cells were stimulated as indicated and β-arrestin recruitment was assessed using the DiscoverX assay as described in the Experimental Section. Data are expressed as a percentage of the maximum dopamine (DA) signal and are shown as means ± SEM of at least 3 experiments performed in triplicate. In A, the EC50 value for DA is 6.4 ± 0.6 nM (mean ± SEM, n = 12) and of 1 is 710 ± 150 nM (mean ± SEM, n = 13). In B, the EC50 value for DA is 140 ± 23 nM (mean ± SEM, n = 13). C, Antagonist concentration-response curves of either sulpiride or 1 for inhibiting β-arrestin recruitment to the D2R. Cells were incubated with the indicated concentrations of compound in the presence of an EC80 concentration of DA (1 μM). Data are expressed as a percentage of signal seen in the absence of test compounds and are shown as means ± SEM of at least 3 experiments performed in triplicate. The IC50 value of sulpiride is 42 ± 4.9 nM (mean ± SEM, n = 8) and of 1 is 16 ± 3.0 μM (mean ± SEM, n = 10). D, Radioligand binding assays using [3H]-methylspiperone were performed as described in the Experimental Section. Data are representative of six independent experiments and expressed as a percentage of the specific binding. The IC50 values of 1 were determined to be >100 μM for the D3R and D2R.
Chemistry.
We employed several complementary synthetic routes to construct the target analog compounds all based on a central strategy of iterative derivatization of the piperazine core. Our initial strategy to the HTS hit compound resynthesis and analog exploration coupled the N-acylated piperazines 111 with the alkyl bromides 112 (Scheme 1) utilizing General Procedures A or B (see Experimental Section for general procedure details and the synthesis of components 111 and 112). For specific target analogs, we utilized slightly modified conditions as shown in Scheme 1. Exploration of the N-acyl moiety was most efficiently achieved through a reversal in the order of piperazine functionalization. Thus, alkylation of 1-Boc-piperazine and subsequent Boc deprotection afforded the 1-(2-(aryloxy)ethyl)piperazines 114, which were then acylated utilizing General Procedures D or E (see Experimental Section) to provide the final analogs (Scheme 2). Again, specific target analogs required slightly modified conditions as shown in Scheme 2. We utilized this broadly defined acylation strategy to access to a total of 59 analogs—the most common approach that we employed in these SAR studies. The sulfonamide analog 44 was synthesized using an analogous protocol to acylation by replacing the acid chloride component with 4-methoxybenzene-1-sulfonyl chloride (Scheme 3). The racemic methyl derivative 46 was synthesized via reductive amination of carboxamide fragment 111a and 1-(4-methoxyphenoxy)propan-2-one (Scheme 4). To efficiently explore the aryl ether composition of (2-indoyl)-substituted piperazine analogs, we employed a nucleophilic displacement approach on the alkyl chloride 116 (Scheme 5) utilizing General Procedures F or G (see Experimental Section).This displacement protocol also furnished the piperidine analog 50 and 1,2-dimethylethylenediamine analog 51 using the corresponding amine starting materials 117 and 119 (Scheme 5). Select individual analog syntheses utilized a complementary Mitsunobu reaction protocol on the 1-(2-hydroxyethyl)piperazine 121 (Scheme 6). Minor modifications to these strategies were utilized to construct other target analogs. Thus, starting with 1-(2-hydroxyethyl)piperidine 117 and using a nucleophilic displacement approach allowed ready access to compound 108 (Scheme 7). In an analogous approach to the late-stage acylation used in Scheme 2, the alkylated piperazine 114a was subjected to a second alkylation to afford compound 109 (Scheme 8). Overall, the routes summarized here enabled the efficient and modular construction of the numerous analogs synthesized and facilitated the survey of structural modifications on all regions of compound 1. The activity of these analogs and our optimization strategy will be discussed in detail in the following section.
Scheme 1.

Summary of alkylation routes to SAR analogs.a
aReagents and conditions: (a) potassium iodide (0.1 or 1.0 equiv), K2CO3 (3.0 equiv), MeCN or DMF, 60 °C, 14–20 h, 27–81% yield; (b) 112a (1.1 equiv), Et3N (1.4 equiv), MeCN, 60 °C, 18 h, 68% yield; (c) 112a (1.1 equiv), K2CO3 (2.0 equiv), DMF, 100 °C, 5 h, 37% yield; (d) 1-(3-bromopropoxy)-4-methoxybenzene or 1-(4-bromobutoxy)-4-methoxybenzene, potassium iodide (1.0 equiv), K2CO3 (3.0 equiv), MeCN, 60 °C, 19 h, 76–83% yield.
Scheme 2.

Summary of late-stage acylation routes to SAR analogs.a
aReagents and conditions: (a) K2CO3 (2.0 equiv), potassium iodide (0.1 equiv), MeCN or DMF, 70 or 90 °C, 16–21 h, 70–79% yield; (b) Et3SiH (1.5–2.0 equiv), trifluoroacetic acid (15–20 equiv), CH2Cl2, rt, 4–26 h, 80–91% yield; (c) Ar1C(O)Cl (1.0–1.3 equiv), Et3N (1.5 equiv), CH2Cl2, rt, 16–20 h, 61–69% yield; (d) Ar1CO2H (1.0–1.3 equiv), PyBOP (1.2 equiv), i-Pr2EtN (3.0 equiv), DMF, rt, 16–20 h, 15–93% yield; (e) 1H-indole-2-carboxylic acid (1.2 equiv), diisopropylcarbodiimide (3.0 equiv), DMAP (0.1 equiv), THF, rt, 15 h, 73% yield; (f) 2-chloroproponyl chloride (1.1 equiv), K2CO3 (2.5 equiv), 4-methoxyphenol or 4-chlorophenol (1.2 equiv), DMF, 80 °C, 4 h, 24–28% yield; (g) BH3•THF (3.1–3.2 equiv), THF, 65 °C, 0.5–4 h, 86–88% yield.
Scheme 3.

Synthesis of the sulfonamide analog 44.a
aReagents and conditions: (a) 4-methoxybenzene-1-sulfonyl chloride (1.0 equiv), Et3N (2.0 equiv), toluene, rt, 19 h, 55% yield.
Scheme 4.

Reductive amination protocol for the synthesis of the methyl-substituted analog 46.a
aReagents and conditions: (a) 1-(4-methoxyphenoxy)propan-2-one (1.0 equiv), NaBH(OAc)3 (1.5 equiv), Cl2CH2CH2Cl2, AcOH (0.3 equiv), 50 °C, 7 d, 57% yield.
Scheme 5.

Summary of alkyl chloride displacement routes to SAR analogs.a
aReagents and conditions: (a) 1H-indole-2-carbonyl chloride (1.1 equiv), Et3N (1.2 equiv), CH2Cl2, rt, 20 h, 75% yield; (b) triphosgene (0.5 equiv), CH2Cl2, 0 °C to rt, 3 h, 53% yield; (c) Ar2OH (1.1–1.9 equiv), Ar2SH (1.6 equiv) or Ar2NHMe (1.6 equiv), K2CO3 (1.8–3.3 equiv), DMF, 50–80 °C, 7–19 h, 5–73% yield; (d) 4-methoxybenzoyl chloride (1.1 equiv), Et3N (1.5 equiv), CH2Cl2, rt, 16 h, 80% yield; (e) thionyl chloride (1.0–1.5 equiv), CHCl3, 50–55 °C, 0.5 or 46 h, 54–86% yield; (f) 2-(methylamino)ethanol (5.1 equiv), MeCN, 80 °C, 25 h, 67% yield.
Scheme 6.

Mitsunobu reaction route to SAR analogs 6, 8 and 10.a
aReagents and conditions: (a) 4-methoxybenzoyl chloride (1.1 equiv), Et3N (1.3 equiv), CH2Cl2, 0 °C to rt, 24 h, 68% yield; (b) Ar2OH (1.0 equiv), Ph3P (1.0 equiv), DIAD (1.3 equiv), CH2Cl2 or THF, 60 °C, 16–40 h, 15–46% yield.
Scheme 7.

Synthesis of the piperidine analog 108a
aReagents and conditions: (a) indole-2-carboxylic acid (1.0 equiv), HOBt (1.0 equiv), EDC•HCl (1.0 equiv), CH2Cl2, rt, 18 h, 49% yield; (b) TsCl (2.0 equiv), Et3N (4.0 equiv), CH2Cl2, rt, 2 h, 46% yield; (c) 4-methoxyphenol (3.0 equiv), K2CO3 (3.0 equiv), MeCN, 60 °C, 17 h, 70% yield.
Scheme 8.

Synthesis of the dibasic piperazine analog 109a
aReagents and conditions: (a) 2-chloro-1-(1H-indol-2-yl)ethan-1-one (1.0 equiv), KI (1.0 equiv), K2CO3 (4.0 equiv), MeCN, 65 °C, 18 h, 51% yield.
Structure–activity relationships and HTS hit compound 1 optimization.
In an effort to increase the functional potency of 1 at the D3R, while at the same time eliminate its D2R antagonist activity, we explored SAR studies of this scaffold by synthesizing over 100 unique analogs of 1. These analogs were designed to investigate four regions of the scaffold including substitutions of the aryl ether and aryl carboxamide groups, modifications of the piperazine core, and changes to the central tether of the molecule as illustrated in Figure 3. All analogs were analyzed in the D2R and D3R β-arrestin recruitment assays to generate a comprehensive SAR survey around the scaffold.
Figure 3.

Illustration of structural modifications to compound 1 to investigate structure–activity relationships.
We began our SAR studies by examining modifications to the 4-methoxyphenyl groups on the left and right termini of compound 1 (i.e., aryl carboxamide and aryl ether moieties, respectively). In the latter case, replacing the 4-methoxyphenyl ether with other aryl ethers afforded analogs possessing a wide range of D3R agonist potencies (Table 1). The 2-methoxyphenyl ether 2 was found to be more than twice as potent, while the 3-methoxyphenyl ether 3 was more than seven-fold as potent, compared to the compound 1. Other monosubstituted phenyl ether analogs were less potent (entries 4–6). In fact, the 4-(trifluoromethoxy)phenyl (6) and 3,5-dimethoxyphenyl (7) ethers were found to be inactive even at the highest tested concentrations. All compounds in this subset possessed no D2R agonism and only negligible, if any, D2R antagonism. The unsubstituted phenyl ether 8 was almost as potent as the 3-methoxyphenyl ether (EC50 = 115 nM), however this analog was found to also possess D2R agonism (EC50 = 2,300 nM). The aryl ether 9, which contained bridged 3,4-dialkoxy substitution possessed two-fold improved D3R agonist potency and excellent selectivity, albeit with reduced agonist efficacy at the D3R. The 3-pyridyl ether analog 10 was 35-fold more potent than the hit compound 1 in the D3R agonist assay (EC50 = 17 nM), however, it also exhibited modestly potent D2R agonist activity (EC50 = 2,900 nM).
Table 1.
Analogs exploring 2-(4-methoxyphenylether)ethyl replacement.
| Compound ID |  |
D3R agonist activity1 | D2R agonist activity1 | D2R antagonist activity1 | |||
|---|---|---|---|---|---|---|---|
| Ar1 = | EC50 (nM) | Emax (% control) | EC50 (nM) | Emax (% control) | IC50 (nM) | Imax (% control) | |
| 1 | 4-methoxyphen-1-yl | 710 ± 150 | 102 ± 4.2 | Inactive | Inactive | 15,700 ± 3,000 | 88 ± 7 |
| 2 | 2-methoxyphen-1-yl | 278 ± 62 | 36 ± 3.1 | Inactive | Inactive | 9,000 ± 3,700 | 99 ± 1 |
| 3 | 3-methoxyphen-1-yl | 98 ± 21 | 95 ± 6 | >100,000 | ND | 6,800 ± 1,400 | 63 ± 6 |
| 4 | 4-ethylphen-1-yl | 2,600 ± 550 | 44 ± 9.3 | Inactive | Inactive | >50,000 | ND |
| 5 | 4-chlorophen-1 -yl | 1,000 ± 275 | 103 ± 27 | Inactive | Inactive | >10,000 | 101 ± 2 |
| 6 | 4-(trifluoromethoxy) phen-1-yl | Inactive | Inactive | Inactive | Inactive | >50,000 | ND |
| 7 | 3,5-dimethoxyphen-1-yl | Inactive | Inactive | Inactive | Inactive | >50,000 | ND |
| 8 | phenyl | 115 ± 12 | 64 ± 4 | 2300 ± 900 | 38 ± 5 | >100,000 | ND |
| 9 | benzo[d][1,3]dioxol-5-yl | 310 ± 150 | 78 ± 9.4 | Inactive | Inactive | >10,000 | 87 ± 6.8 |
| 10 | 3-pyridyl | 17 ± 2.3 | 110 ± 15 | 2,900 ± 1,300 | 96 ± 7.4 | >100,000 | ND |
β-arrestin recruitment activity was assessed as described in Figure 2. Emax values are expressed as a percentage of the maximum dopamine response observed in the same assay. Imax values are expressed as a percentage of the maximum inhibition of a dopamine (EC80 concentration) response observed with the antagonist sulpiride in the same assay.
ND Curve did not plateau.
In a more extensive effort than above, we investigated replacements of the 4-methoxybenzamide on the left-hand portion of the compound 1 (Table 2). The unsubstituted benzamide analog 11 was approximately an equipotent D3R agonist compared to the hit compound 1, along with ablation of the D2R antagonism. The 2-methoxybenzamide (12) possessed only modest potency, while the 3-methoxybenzamide (13) was marginally more potent. Other mono substituted benzamides were less promising, with the 4-chloro analog 14 inactive as a D3R agonist and the 4-ethyl analog 15 on par in potency compared to the hit. However, both 14 and 15 also possessed limited D2R antagonist activity. We next examined a number of heterocyclic carboxamides at this position, beginning with the two pyridyl analogs 16 and 17, neither of which possessed significant D3R agonist activity. We next investigated a series of indole carboxamide analogs. While all three of the 5-, 3- and 2-indole carboxamide analogs (18, 19 and 20, respectively) tested possessed potent D3R agonist activity, only 20 was completely D3R-selective and also possessed the greatest potency (EC50 = 38 nM)–almost a 20-fold improvement compared to the hit. Thus, we delved more systematically into the substitution of 2-indole carboxamide analogs, examining methoxy, methyl and chloro substitution around the benzene of the indole moiety (i.e., 4-, 5-, 6- and 7-position substitution). The methoxy-substituted analogs 21 to 24 were all D3R agonists of reasonably high potency (EC50s = 155–980 nM), however, all these analogs also possessed measurable D2R agonist activity (EC50s = 5,200–7,800 nM). The methyl-substituted analogs 25 to 28 were also all fairly potent D3R agonists (EC50s = 130–611 nM), along with reduced D2R agonism (EC50s = 1,100 to >50,000 nM). The 5-methyl analog 27 was the most selective and possessed D3R agonist potency (EC50 = 611 nM) on par with the hit compound 1. Similarly, the chloro-substituted analogs 29 to 32 were all D3R agonists of modest potency (EC50s = 160–2,900 nM), with D2R agonist activity similar to the methyl-substituted series (EC50s = 3,500 to >50,000 nM). In this series, only the 5-chloro analog was fully D3R selective, however the D3R agonism was not very potent (EC50 = 2,900 nM). None of these substituted analogs approached the potency of the unsubstituted indole 20 and we did not further investigate substitution of this region of the indole.
Table 2.
Analogs exploring 4-methoxybenzamide replacement.
| Compound ID | D3R agonist activity1 | D2R agonist activity1 | D2R antagonist activity1 | ||||
|---|---|---|---|---|---|---|---|
| full structure or R = | EC50 (nM) | Emax (% control) | EC50 (nM) | Emax (% control) | IC50 (nM) | Hmax (% control) | |
| 11 | phenyl | 548 ± 165 | 70 ± 2.6 | Inactive | Inactive | >50,000 | ND |
| 12 | 2-methoxyphen-1-yl | 2,500 ± 1,000 | 113 ± 16 | Inactive | Inactive | >100,000 | ND |
| 13 | 3 -methoxyphen-1 -yl | 550 ± 57 | 112 ± 12 | 24,000 ± 3000 | 24 ± 7 | Inactive | Inactive |
| 14 | 4-chlorophen-1 -yl | Inactive | Inactive | Inactive | Inactive | 9,000 ± 3,000 | 107 ± 8.8 |
| 15 | 4-ethylphen-1-yl | 530 ± 130 | 95 ± 13 | Inactive | Inactive | 6,000 ± 1,200 | 106 ± 15 |
| 16 | 4-pyridyl | 22,000 ± 5,600 | 50 ± 7.9 | Inactive | Inactive | Inactive | Inactive |
| 17 | 5-methoxy-2-pyridyl | 2,100 ± 375 | 97 ± 5.7 | >50,000 | ND | >100,000 | ND |
| 18 | 5-indolyl | 208 ± 62 | 112 ± 11 | 2,500 ± 383 | 18.5 ± 3.2 | 4,600 ± 1,900 | 64 ± 11.2 |
| 19 | 3-indolyl | 210 ± 29 | 89 ± 9 | >100,000 | ND | 2,800 ± 500 | 86 ± 2 |
| 20 | 2-indolyl | 38 ± 4.0 | 103 ± 3 | >10,000 | ND | >10,000 | ND |
| 21 | 7-methoxy-2-indolyl | 980 ± 135 | 115 ± 6.5 | 6,000 ± 1,300 | 56 ± 9.7 | >10,000 | ND |
| 22 | 6-methoxy-2-indolyl | 155 ± 62 | 91 ± 18 | 5,200 ± 3,500 | 62 ± 11.5 | Inactive | Inactive |
| 23 | 5-methoxy-2-indolyl | 520 ± 25 | 109 ± 2.6 | 7,800 ± 1,900 | 46 ± 14.2 | >100,000 | ND |
| 24 | 4-methoxy-2-indolyl | 411 ± 144 | 116 ± 5.7 | 5,200 ± 255 | 33 ± 4.7 | >100,000 | ND |
| 25 | 7-methyl-2-indolyl | 473 ± 44 | 103 ± 15.5 | 10,100 ± 780 | 49 ± 18.6 | >100,000 | ND |
| 26 | 6-methyl-2-indolyl | 130 ± 40 | 117 ± 3 | 1,100 ± 400 | 72 ± 7 | Inactive | Inactive |
| 27 | 5-methyl-2-indolyl | 611 ± 79 | 119 ± 7.9 | >50,000 | ND | >100,000 | ND |
| 28 | 4-methyl-2-indolyl | 266 ± 82 | 106 ± 15 | 4,300 ± 1,700 | 37 ± 5.5 | 744 ± 282 | 30 ± 8.2 |
| 29 | 7-chloro-2-indolyl | 563 ± 138 | 116 ± 13 | 13,000 ± 5,200 | 70 ± 26 | >100,000 | ND |
| 30 | 6-chloro-2-indolyl | 225 ± 73 | 110 ± 30 | 3,500 ± 1,400 | 58 ± 12.4 | >100,000 | ND |
| 31 | 5-chloro-2-indolyl | 2,900 ± 1,100 | 117 ± 14.7 | Inactive | Inactive | Inactive | Inactive |
| 32 | 4-chloro-2-indolyl | 160 ± 56 | 105 ± 4 | 7,100 ± 3,300 | 57 ± 17 | >100,000 | ND |
| 33 | 1 -methyl-2-indolyl | 310 ± 52 | 88 ± 6 | Inactive | Inactive | 9,600 ± 2,400 | 105 ± 9 |
| 34 | 3-methyl-2-indolyl | 1,900 ± 600 | 92 ± 4 | 4,400 ± 1,200 | 31 ± 6 | >100,000 | ND |
| 35 | pyrrolo[2,3-b]pyridin-2-yl | 167 ± 21 | 109 ± 14 | 413 ± 105 | 94 ± 18 | Inactive | Inactive |
| 36 | pyrrolo[2,3-c]pyridin-2-yl | 810 ± 130 | 105 ± 9.8 | 7,800 ± 3,300 | 21 ± 3.1 | >100,000 | ND |
| 37 | pyrrolo[3,2-c]pyridin-2-yl | 576 ± 102 | 107 ± 901 | 5,500 ± 2,200 | 28 ± 5.6 | >100,000 | ND |
| 38 | pyrrolo[3,2-b]pyridin-2-yl | 2,800 ± 541 | 103 ± 9.4 | >100,000 | ND | Inactive | Inactive |
| 39 | benzo[d]imidazol-2-yl | 192 ± 76 | 95 ± 12.3 | Inactive | Inactive | >50,000 | ND |
| 40 | benzofuran-2-yl | 430 ± 117 | 80 ± 14.4 | Inactive | Inactive | 7,700 ± 2,500 | 111 ± 7 |
| 41 | benzothiophen-2-yl | 3,300 ± 1,000 | 51 ± 5.8 | Inactive | Inactive | 5,100 ± 1,000 | 96 ± 5.6 |
| 42 | cyclohexyl | >50,000 | ND | Inactive | Inactive | >50,000 | ND |
| 43 | methyl | 9,700 ± 2,500 | 35 ± 9.2 | Inactive | Inactive | Inactive | Inactive |
| 44 |  |
Inactive | Inactive | >100,000 | ND | Inactive | Inactive |
| 45 | Inactive | Inactive | Inactive | Inactive | >50,000 | ND | |
β-arrestin recruitment activity was assessed as described in Figure 2. Emax values are expressed as a percentage of the maximum dopamine response observed in the same assay. Imax values are expressed as a percentage of the maximum inhibition of a dopamine (EC80 concentration) response observed with the antagonist sulpiride in the same assay.
ND Curve did not plateau.
Next, we probed substitution on the nitrogen-containing ring of the indole by testing the N- and 3-methyl analogs 33 and 34, respectively. While the N-methyl analog retained D3R agonist activity on par with compound 1, it also exhibited D2R antagonism. The 3-methyl analog was nonselective, showing only weak activity in all three assays (EC50s = 1,900–4,400 nM). We next examined other bicyclic heterocycles attached to the carboxamide, beginning with additional nitrogen incorporation into the indole ring. Moving the additional nitrogen atom around the ring afforded analogs (35–39) of modest D3R agonist potency (EC50s = 167–2,800 nM), though often with modest (35, EC50 = 413 nM) to weak (36 and 37, EC50s = 7,800 and 5,500 nM, respectively) D2R agonism. Only the benzimidozyl analog 39 was both reasonably potent (EC50 = 192 nM) and D3R selective. Replacement of the indole with a benzofuran moiety afforded analog 40, a D3R agonist of modest potency (EC50 = 430 nM), though with weak D2R antagonism (IC50 = 7,700 nM). The benzothiophene analog 41 was both less potent and selective than the benzofuran. The cyclohexyl and methyl carboxamides (42 and 43, respectively) did not show any appreciable activity, with only 43 possessing any measurable D3R agonist potency (EC50 = 9,700 nM). The 4-methoxyphenyl sulfonamide 44 possessed no measurable activity across the three SAR assays, indicating that the sulfonamide isostere is not a suitable replacement for the carboxamide functional group in this instance. The directly attached 4-methoxyphenyl piperizine 45 was similarly inactive, again indicating the importance of the carboxamide for potency.
We explored a number of modifications to the piperazine or alkyl chain of the tethered ether, most often through the addition of a single methyl group to the HTS lead compound 1 structure (Table 3). Thus, analogs 46 and 47 contained an additional methyl group on the two-carbon tether bridging the piperazine and aryl ether groups. While both were D3R-selective agonists, the position of the methyl group was critical to the effect on potency. The analog 46 (where the methyl group was adjacent to the piperazine) possessed weak D3R agonist potency (EC50 = 9,300 nM) while the constitutional isomer 47 (with a methyl group adjacent to the ether moiety) was almost three-fold more potent than hit compound 1 (EC50 = 160 nM). The effect of methyl substitution on the piperazine ring exhibited an analogous dependence on position. Analog 48 (with the methyl group adjacent to the basic piperazine nitrogen) displayed greatly diminished potency and efficacy, while analog 49 (with a methyl group adjacent to the amide nitrogen) was slightly more potent than the hit compound 1 (EC50 = 510 nM), although 49 was not fully selective, possessing weak D2R agonism (EC50 = 4,200 nM). The profound detrimental effect observed from methyl group introduction adjacent to the basic piperazine nitrogen indicates the importance of this moiety for activity, although whether this arises from steric interactions or through the induction of an unfavorable conformation is not immediately clear. Other structural modifications were explored and found to cause an almost complete loss of any activity. The piperidine analog 50 was inactive in all assays and the ring opened analog 51 possessed only very weak D2R antagonist activity (IC50 = 18,000 nM).
Table 3.
Analogs exploring modification of the tether or piperazine core.
| Compound ID | Structure | D3R agonist activity1 | D2R agonist activity1 | D2R antagonist activity1 | |||
|---|---|---|---|---|---|---|---|
| EC50 (nM) | Emax (% control) | EC50 (nM) | Emax (% control) | IC50 (nM) | Imax (% control) | ||
| 46 |  |
9,300 ± 2,500 | 106 ± 11 | Inactive | Inactive | >100,000 | ND |
| 47 |  |
160 ± 33 | 86 ± 7 | Inactive | Inactive | 18,000 ± 2,900 | 89 ± 7 |
| 48 |  |
4,400 ± 2,300 | 55 ± 7 | Inactive | Inactive | >100,000 | ND |
| 49 |  |
510 ± 110 | 101 ± 7 | 4,200 ± 900 | 29 ± 1 | >100,000 | ND |
| 50 |  |
Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| 51 |  |
Inactive | Inactive | Inactive | Inactive | 18,000 ± 4,800 | 72 ± 4.6 |
β-arrestin recruitment activity was assessed as described in Figure 2. Emax values are expressed as a percentage of the maximum dopamine response observed in the same assay. Imax values are expressed as a percentage of the maximum inhibition of a dopamine (EC80 concentration) response observed with the antagonist sulpiride in the same assay.
ND Curve did not plateau.
Having identified potency-enhancing replacements for either the 4-methoxyphenyl ether or 4-methyoxybenzamide group of compound 1, we were interested in exploring simultaneous changes to both ends of the molecule (Table 4). The 3-methoxyphenyl (3) and 3-pyridyl ethers (10) were two of the most potent aryl ether analogs identified (EC50s = 98 and 17 nM, respectively (Table 1), though both also possessed weak D2R activity. We thus explored a range of carboxamide analogs to better identify SAR trends that could lead to potent and selective analogs. The 3-methoxyphenyl ether analogs 52–56 contained some of the most promising carboxamide groups from earlier SAR studies and, gratifyingly, all afforded potent D3R analogs (EC50s = 34–285 nM). The most potent was the 2-indolyl carboxamide analog 54 (EC50 = 34 nM), although it also possessed slight D2R agonist activity (EC50 = 2,300 nM). The other 3-methoxyphenyl ether analogs were both less potent and even less selective than 54. The 3-pyridyl ether analog 10 was the most potent ether replacement identified (EC50 = 17 nM) and we explored a more comprehensive survey of carboxamide replacements paired with this ether, primarily focused on substituted 2-indole carboxamides. The 4-methoxy-2-methylphenyl carboxamide 57 was almost 7-fold less potent than the corresponding 4-methoxyphenyl analog 10. In an effort that mirrored the original exploration of carboxamide groups, we tested a series of methoxy-, methyl- and chloro-substituted 2-indoles. These analogs (58–70) possessed good potency (EC50s = 3–116 nM), with six analogs (58, 61, 63, 66, 68 and 70) in the single-digit nanomolar range. Unfortunately, none of these analogs were highly selective D3R agonists and the more potent compounds were generally the least selective. Upon comparing the 3-pyridyl ether series to the 4-methoxyphenyl ether series, we also noted poor correlation between indole substitution position and rank order of potency, suggesting that the SAR trends might not be independent of the substitution at other positions of the molecule. Additional nitrogen incorporation on the indole moiety afforded the set of 3-pyridyl analogs (71–76) with excellent to good D3R agonist potency (EC50s = 13–200 nM) and all but analog 74 possessing EC50 values < 100 nM. However, as observed with the substituted indole set, none of these analogs were fully D3R>D2R selective. Three other heterocyclic carboxamide analogs were tested, the benzofuran 77, the benzothiophene 78 and the pyrrole 79, and all three were found to be potent D3R agonists. Contrary to the 4-methoxyphenyl ether series, in this set 78 was found to be the more potent (EC50 =13 nM) than 77 (EC50 = 42 nM), further supporting that SAR trends might be dependent upon all the groups in the molecule and a cautionary note against the extrapolation of SAR trends across all analogs. The pyrrole 79 was over 15-fold less potent (EC50 = 118 nM) than the corresponding indole 99 (EC50 = 7 nM, (Table 5). In line with all other 3-pyridyl ether analogs, these three compounds were not highly D3R>D2R selective.
Table 4.
Analogs exploring concurrent 4-methoxyphenyl ether and 4-methoxybenzamide replacement.
| Compound ID |  |
D3R agonist activity1 | D2R agonist activity1 | D2R antagonist activity1 | ||||
|---|---|---|---|---|---|---|---|---|
| Ar1 = | Ar2 = | EC50 (nM) | Emax (% control) | EC50 (nM) | Emax (% control) | IC50 (nM) | Imax (% control) | |
| 52 | 3-methoxyPhen-1-yl | pyrrolo[2,3-b]pyridin-2-yl | 50.0 ± 12 | 113 ± 2.8 | 128 ± 33 | 76 ± 8.3 | Inactive | Inactive |
| 53 | pyrrolo[3,2-c]pyridin-2-yl | 280 ± 19 | 111 ± 8 | 830 ± 170 | 71 ± 7 | 7,400 ± 3,700 | 30 ± 3 | |
| 54 | 2-indolyl | 33.8 ± 8.7 | 105 ± 9.3 | 2,300 nM ± 540 | 48 ± 5.1 | >100,000 | ND | |
| 55 | 5-methoxy-2-indolyl | 284 ± 100 | 110 ± 6.5 | 1,700 ± 324 | 67 ± 15 | Inactive | Inactive | |
| 56 | benzo[d]imidazol-2-yl | 285 ± 58 | 101 ± 12 | 660 ± 46 | 29 ± 7 | 2,000 ± 1,500 | 53 ± 10 | |
| 57 | 3-pyridyl | 4-methoxy-2-methylphen-1-yl | 116 ± 29 | 102 ± 7.7 | 4,600 ± 1,100 | 52 ± 8.3 | >100,000 | ND |
| 58 | 6-methoxy-2-indolyl | 6.4 ± 1.5 | 107 ± 7 | 364 ± 58 | 81 ± 3.0 | Inactive | Inactive | |
| 59 | 5-methoxy-2-indolyl | 59 ± 23 | 97 ± 5 | 2,600 ± 1,200 | 97 ± 2.8 | Inactive | Inactive | |
| 60 | 7-methyl-2-indolyl | 25.7 ± 8.1 | 108 ± 6 | 786 ± 210 | 81 ± 8.5 | Inactive | Inactive | |
| 61 | 6-methyl-2-indolyl | 4.3 ± 2.0 | 106 ± 13 | 138 ± 42 | 74 ± 4.3 | Inactive | Inactive | |
| 62 | 5-methyl-2-indolyl | 13 ± 3.2 | 113 ± 8 | 1,300 ± 270 | 101 ± 5.1 | Inactive | Inactive | |
| 63 | 4-methyl-2-indolyl | 6.9 ± 1.7 | 97 ± 3 | 180 ± 23 | 70 ± 7.1 | Inactive | Inactive | |
| 64 | 3-methyl-2-indolyl | 80 ± 32 | 92 ± 13 | 800 ± 80 | 102 ± 7 | Inactive | Inactive | |
| 65 | 1-methyl-2-indolyl | 37.6 ± 7.2 | 112 ± 8 | 6,200 ± 1,900 | 53 ± 8.7 | Inactive | Inactive | |
| 66 | 1-methyl-3-indolyl | 9.5 ± 1.1 | 94 ± 9 | 4,400 ± 1,500 | 91 ± 17.2 | Inactive | Inactive | |
| 67 | 7-chloro-2-indolyl | 33 ± 4.3 | 113 ± 3.4 | 617 ± 144 | 74 ± 7.8 | Inactive | Inactive | |
| 68 | 6-chloro-2-indolyl | 3.0 ± 0.3 | 96 ± 5.5 | 183 ± 57 | 89 ± 0.4 | Inactive | Inactive | |
| 69 | 5-chloro-2-indolyl | 98 ± 45 | 106 ± 8 | 1,400 ± 282 | 90 ± 6.3 | Inactive | Inactive | |
| 70 | 4-chloro-2-indolyl | 4.4 ± 0.8 | 107 ± 8.4 | 217 ± 67 | 82 ± 6.5 | Inactive | Inactive | |
| 71 | pyrrolo[2,3-b]pyridin-2-yl | 13.5 ± 6.5 | 98.6 ± 6.1 | 210 ± 83 | 99± 13.7 | Inactive | Inactive | |
| 72 | pyrrolo[2,3-c]pyridin-2-yl | 81 ± 26.8 | 116 ± 9.7 | 780 ± 240 | 66 ± 7.0 | Inactive | Inactive | |
| 73 | pyrrolo[3,2-c]pyridin-2-yl | 63 ± 20 | 108 ± 11 | 1,800 ± 460 | 61 ± 5.1 | Inactive | Inactive | |
| 74 | pyrrolo[3,2-b]pyridin-2-yl | 200 ± 53 | 105 ± 8 | 3,000 ± 1,100 | 64 ± 2 | Inactive | Inactive | |
| 75 | benzo[d]imidazol-2-yl | 33 ± 14.6 | 112 ± 9.8 | 2,400 ± 1,400 | 60.6 ± 4.3 | Inactive | Inactive | |
| 76 | 1H-indazol-3-yl | 45 ± 6.1 | 97 ± 8.5 | 4,100 ± 1,500 | 71 ± 2.8 | Inactive | Inactive | |
| 77 | benzofuran-2-yl | 42 ± 8.5 | 104 ± 9.5 | 1,700 ± 63 | 57 ± 7.4 | Inactive | Inactive | |
| 78 | benzothiophen-2-yl | 13.3 ± 2.6 | 102 ± 7.7 | 617 ± 40 | 34 ± 3 | 2,100 ± 1,200 | 60 ± 4.9 | |
| 79 | pyrrol-2-yl | 118 ± 47 | 110 ± 17.6 | 6,100 ± 1,300 | 34 ± 8.4 | >100,000 | ND | |
β-arrestin recruitment activity was assessed as described in Figure 2. Emax values are expressed as a percentage of the maximum dopamine response observed in the same assay. Imax values are expressed as a percentage of the maximum inhibition of a dopamine (EC80 concentration) response observed with the antagonist sulpiride in the same assay.
ND Curve did not plateau.
Table 5.
Analogs based on the 2-indolylcarboxamide scaffold.
| Compound ID |  |
D3R agonist activity1 | D2R agonist activity1 | D2R antagonist activity1 | |||
|---|---|---|---|---|---|---|---|
| full structure or Ar1 = | EC50 (nM) | Emax (% control) | EC50 (nM) | Emax (% control) | IC50 (nM) | Imax (% control) | |
| 80 | phenyl | 9.2 ± 0.6 | 83 ± 3.4 | 1,800 ± 1,100 | 49 ± 6.3 | 535 ± 220 | 37 ± 8 |
| 81 | 3,4-(methylenedioxy)phenyl | 290 ± 130 | 82 ± 5.2 | 7,000 ± 1,700 | 36 ± 8.8 | >50,000 | ND |
| 82 |  |
Inactive | Inactive | Inactive | Inactive | >100,000 | ND |
| 83 | 4-thiomethoxyphen-1-yl | 630 ± 149 | 89 ± 9.3 | Inactive | Inactive | >100,000 | ND |
| 84 | 4-ethoxyphen-1-yl | >100,000 | ND | Inactive | Inactive | >100,000 | ND |
| 85 | 4-(trifluoromethoxy)phen-1-yl | 3,800 ± 770 | 100 ± 16.7 | >100,000 | ND | >100,000 | ND |
| 86 | 4-methylphen-1-yl | 151 ± 30 | 91 ± 16.6 | >50,000 | ND | >100,000 | ND |
| 87 | 4-ethylphen-1-yl | 1,600 ± 640 | 98 ± 16 | Inactive | Inactive | >50,000 | ND |
| 88 | 4-tert-butylphen-1-yl | Inactive | Inactive | Inactive | Inactive | >100,000 | ND |
| 89 | 4-fluorophen-1-yl | 126 ± 35 | 90 ± 13.3 | >50,000 | ND | 9,700 ± 5,800 | 72 ± 4.6 |
| 90 | 4-chlorophen-1-yl | 114 ± 27 | 118 ± 10 | Inactive | Inactive | >50,000 | ND |
| 91 | 4-bromophen-1-yl | 105 ± 29 | 106 ± 21 | Inactive | Inactive | >100,000 | ND |
| 92 | 4-nitrophen-1-yl | 1,300 ± 280 | 92 ± 9.7 | Inactive | Inactive | Inactive | Inactive |
| 93 | 3,4-dimethoxyphen-1-yl | 710 ± 121 | 111 ± 18 | Inactive | Inactive | >50,000 | ND |
| 94 | 3,4-dimethylphenyl | 610 ± 37 | 70 ± 8.1 | Inactive | Inactive | 13,000 ± 1,600 | 88 ± 9.4 |
| 95 | 3-chloro-4-methylphen-1-yl | 997 ± 300 | 103 ± 12 | Inactive | Inactive | Inactive | Inactive |
| 96 | 4-chloro-3 -methylphen-1-yl | 114 ± 20 | 110 ± 1.3 | Inactive | Inactive | 3,800 ± 1,100 | 75 ± 3 |
| 97 | 2-chloro-4-methoxyphen-1-yl | 41 ± 12 | 113 ± 3.5 | Inactive | Inactive | >10,000 | ND |
| 98 | 2-pyridyl | 3,500 ± 900 | 94 ± 17 | Inactive | Inactive | >100,000 | ND |
| 99 | 3-pyridyl | 7.0 ± 1.8 | 98 ± 7 | 570 ± 220 | 70 ± 6 | Inactive | Inactive |
| 100 | 4-pyridyl | 472 ± 39 | 71 ± 6.3 | Inactive | Inactive | >100,000 | ND |
| 101 | 5-chloropyridin-3-yl | 5.0 ± 1.2 | 79 ± 9.3 | 790 ± 220 | 56 ± 2.5 | Inactive | Inactive |
| 102 |  |
82 ± 7.8 | 108 ± 6.4 | 3,300 ± 1,300 | 46 ± 7.6 | Inactive | Inactive |
| 103 |  |
32 ± 8.1 | 101 ± 4.1 | 1,900 ± 570 | 43 ± 4.9 | Inactive | Inactive |
| 104 |  |
1,900 ± 47 | 45 ± 3 | Inactive | Inactive | >100,000 | ND |
| 105 | 5-indolyl | 60.9 ± 20.6 | 84 ± 5.8 | Inactive | Inactive | 440 ± 138 | 100 ± 0 |
| 106 |  |
Inactive | Inactive | Inactive | Inactive | 691± 75 | 97 ± 3 |
| 107 |  |
Inactive | Inactive | Inactive | Inactive | 1200 ± 357 | 97 ± 6 |
| 108 |  |
Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| 109 |  |
Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
β-arrestin recruitment activity was assessed as described in Figure 2. Emax values are expressed as a percentage of the maximum dopamine response observed in the same assay. Imax values are expressed as a percentage of the maximum inhibition of a dopamine (EC80 concentration) response observed with the antagonist sulpiride in the same assay.
ND Curve did not plateau.
We hoped that the potency might be improved through further optimization of the 4-methoxyphenyl ether group. Of the 35 carboxamide analogs in Table 2, the 2-indolyl analog 20 was found to be by far the most potent and selective, and remained our lead compound. Keeping the 2-indole carboxamide in place, we systematically explored replacements for the 4-methoxyphenyl ether (Table 5). The unsubstituted phenyl ether 80 was indeed more potent (EC50 = 9 nM), however it possessed modest D2R agonist and antagonist activity. The 3,4-methylenedioxyphenyl analog 81 was a competent D3R agonist, though with slight D2R antagonism. The 4-methoxyphenyl thioether 82 was completely inactive across all three SAR assays, while the 4-thiomethoxyphenyl ether 83 was a modestly potent (EC50 = 630 nM) and a fully selective D3R agonist. The limited data on oxygen-to-sulfur replacement potentially suggest a steric requirement of the ether oxygen for effective interaction with the D3R. This contrasts with the data for the α-methyl ether analog 47, which possessed increased potency over the desmethyl hit compound 1. Additional focused analogs and computational experiments would be necessary to further elucidate the contributions of steric bulk and hydrogen-bond acceptor ability to overall analog potency. We prepared a set of other 4-substituted phenyl ether analogs 84–92. The 4-(trifluoromethoxy)phenyl ether 85 was a weakly potent (EC50 = 3,800 nM) and a fully selective D3R agonist, whereas the corresponding 4-(trifluoromethoxy)phenyl ether on the 4-methoxyphenyl carboxamide scaffold (i.e. 6) possessed no measurable activity. The 4-ethylphenyl ether analog 87 possessed comparable D3R potency (EC50 = 1,600 nM) to its counterpart on the 4-methoxyphenyl carboxamide scaffold, 4. However, the 4-chlorophenyl ether analog 90 did display a significant improvement to potency (EC50 = 114 nM) compared to its 4-methoxyphenyl carboxamide counterpart 5 (EC50 = 1,000 nM). Other 4-substituted phenyl ethers also afforded potent and selective D3R agonists, such as the methyl-, fluoro- and bromo-phenyl ether analogs (86, 89 and 91, respectively). The 4-ethoxyphenyl ether 84 and the 4-tert-butylphenyl ether 88 were completely inactive, possibly revealing a limit to the tolerated substituent size. A limited set of disubstituted phenyl ether analogs were explored (93–97), with the 2-chloro-4-methoxyphenyl ether 97 being the most potent (EC50 = 41 nM) and selective analog among the group. Analog 97 was only slightly less potent than the original 2-indolyl carboxamide 20, however the chlorine atom in 97 did not confer any additional benefit and we continued to focus on 20 for further evaluation. Other examples were notably less potent including the 3-chloro-4-methylphenyl ether 95. This is interesting in light of the potent D3R agonism of 4-methylphenyl analog 86, further suggesting size limitations of the aryl ether. A set of four pyridyl ether analogs were evaluated, ranging from the very potent 5-chloro-3-pyridyl ether 101 (EC50 = 5 nM) to the weakly potent 2-pyridyl ether 98 (EC50 = 3,500 nM). While not an exhaustive set, the potent analogs identified (i.e., 99 and 100) lacked full D3R>D2R selectivity and only the 4-pyridyl ether 100 was found to be a selective D3R agonist of modest potency (EC50 = 472 nM).
Having earlier noted a slight potency improvement from methyl incorporation adjacent to the aryl ether (i.e. 47), we tested the effect on two such 2-indole carboxamide exemplars (Table 5). We observed a contradictory effect between the two cases: while the effect was slightly detrimental in the 4-methoxyphenyl ether analog 102, it provided a three-fold potency enhancement for the 4-chlorophenyl ether analog 103. In both examples however, the analogs also exhibited D2R agonist activity. It may be worth noting that methyl incorporation onto the tether afforded analogs that were racemic mixtures. Separation and testing of the enantiopure analogs could markedly affect the potency and/or selectivity and may be pursued in future SAR investigations. Replacement of the ether with a N-methyl tertiary amine afforded the modestly potent (EC50 = 1,900 nM) though D3R-selective analog 104. The 5-indole ether 105 was foundto be a potent D3R agonist (EC50 = 61 nM) as well as a reasonably potent D2R antagonist (IC50 = 440 nM). Although this particular compound does not meet the criteria of a selective D3R agonist sought in the current study, 105 may be of particular interest as a lead in complementary studies examining D3R stimulation and, simultaneously, D2R antagonism.
Finally, within the context of the 2-indoylcarboxaminde analogs, we made additional modifications to the linker between the 2-indoylcarboxaminde and 4-methoxylphenyl moieties. Compounds 106 and 107 have one or two additional methylene groups, respectively, inserted between the piperazine and 4-methoxyl phenyl ether (Table 5). These modifications lead to a complete loss of D3R agonist activity, although both compounds retained low potency D2R antagonist activity. In 108, the piperazine ring was converted to a piperidine ring (Table 5), which, similar to that observed with compound 50 (Table 3), led to a complete loss in D3R and D2R activities, further highlighting the importance of this nitrogen for receptor activity. Lastly, we made analog 109 in which a methylene group was inserted between the 2-indoylcarboxaminde and piperazine moieties which lead to a complete loss of both D3R agonist and D2R antagonist activities (Table 5). Taken together, these results highlight the importance of linker length, especially for D3R agonist potency and efficacy.
In summary, we explored SAR trends for both termini of compound 1 and, to a lesser extent, modifications on the core piperazine and tether. We have identified a number of potent and selective D3R agonists with the greater challenge being to maintain D3R receptor selectivity without gaining D2R activity. The most potent and selective D3R agonist identified was the 2-indole carboxamide analog 20, containing the 4-methoxyphenyl ether found in hit compound 1. The closely related, but slightly less potent 2-chloro-4-methoxyphenyl ether 97 supports the chemotype as a valid D3R agonist scaffold. Therefore, we chose to further investigate compound 20 as a D3R-selective agonist and have designated it NIH Molecular Libraries Initiative probe molecule ML417 (Figure 4A).
Figure 4. Pharmacological activity of compound 20 on all dopamine receptor subtypes.

A, Chemical structure of 20. B, Agonist concentration-response curves for stimulating β-arrestin recruitment in response to 20 for the indicated DA receptor subtypes. β-arrestin recruitment was assessed using the DiscoverX assay as described in the Experimental Section and the data are expressed as a percentage of the maximum dopamine signal for each receptor (not shown) and represent means ± SEM of at least 3 experiments performed in triplicate. The EC50 for 20 at the D3R is reported in Table 2. C, Antagonist concentration response curves of 20 on cells expressing individual dopamine receptor subtypes as indicated. β-arrestin recruitment was stimulated with an ~EC80 concentration of dopamine for each receptor subtype and incubated with the indicated concentrations of 20. Data are expressed as a percentage of signal seen with the EC80 concentration of dopamine and represent means ± SEM of at least 3 experiments performed in triplicate. The IC50 for 20 at the D2R was estimated to be >10 μM.
Pharmacological characterization of lead compound 20.
Our lead compound 20 was subjected to further characterization using a variety of pharmacological assays. We initially wanted to assess the selectivity of compound 20 among all dopamine receptor subtypes using the β-arrestin recruitment functional assay. As shown in Figure 4B, 20 is a full agonist at the D3R with an EC50 of 38 nM while displaying minimal agonist efficacy at all other receptor subtypes. When tested as an antagonist, 20 displayed very limited activity (IC50 > 50 μM) at all subtypes except for the D2R where it exhibited some weak partial antagonism (IC50 >10 μM; Figure 4C). This is in contrast to the parent hit compound 1 that showed full antagonism at the D2R indicating that our medicinal chemistry efforts increased the potency of the compound for the D3R and decreased its activity at the D2R resulting in a potent and highly selective agonist of the D3R.
The activity of 20 on β-arrestin recruitment to the D3R was confirmed using an orthogonal assay that measures bioluminescence resonance energy transfer (BRET) when the D3R and β-arrestin, fused to complimentary biosensors (see Experimental Section), are in close proximity. As shown in Figure 5A using the BRET-based β-arrestin recruitment assay, 20 displayed full agonist activity at the D3R with an EC50 of 1.2 nM. These results confirm those obtained using the DiscoverX β-arrestin recruitment assay (Figure 4B), although 20 exhibited ~30-fold greater potency using the BRET-based assay. The reason for this discrepancy is not clear but may be related to differing assay time-courses, different cell types, and/or varying levels and types of G protein-coupled receptor kinases (GRKs) that can affect β-arrestin recruitment to GPCRs.56 Regardless, these findings confirm that 20 is a full and potent agonist for β-arrestin recruitment. We next sought to characterize the effects of 20 on G protein-mediated signaling. Initially, we used another BRET-based assay to examine D3R coupling to Go, an inhibitory G protein that the D3R has previously been shown to activate.57, 58 As shown in Figure 5B, 20 functioned as a full agonist in the D3R-mediated Go-BRET assay with an EC50 of 0.18 nM. The higher potency of 20 in this assay is likely due to spare receptors resulting in signal amplification in contrast to the β-arrestin recruitment assays which lack amplification.59 We next wished to examine G protein-mediated second messenger modulation and turned to a BRET-based cAMP assay using the Epac-based biosensor, CAMYEL.60–62 Figure 5C shows that 20 potently inhibits cAMP accumulation with an EC50 of 86 nM and an efficacy identical to that of dopamine. Lastly, we examined the activity of 20 in an ERK1/2 phosphorylation (pERK) assay (Figure 5D). GPCR-mediated phosphorylation of ERK1/2 has previously been shown to occur through G protein-dependent and independent signaling pathways,63, 64 which may be related to the GPCR and/or cell type. However, we have found that pretreatment of our cells with pertussis toxin, which inactivates Gαi/o proteins65, 66 completely ablates the D3R-mediated pERK response indicating that it is G protein-mediated (data not shown). Similar to the other signaling assays, we found that 20 functioned as a full agonist at the D3R with an EC50 of 21 nM (Figure 5D). Taken together, these data demonstrate that 20 is a full and potent agonist for multiple signaling pathways associated with D3R activation. Notably, in these signaling assays there are small variances in the potencies of 20 relative to those for dopamine suggesting that 20 may exhibit biased signaling properties; however, this will need to be evaluated in detail in future experiments.
Figure 5. Pharmacology of compound 20 on D3R-mediated signaling outputs.

Agonist mediated concentration-response curves of dopamine or 20 for stimulating a variety of D3R-mediated signaling pathways. A, Cells were stimulated as indicated and analyzed using the BRET-based β-arrestin recruitment assay (see the Experimental Section) resulting in EC50 values of 2.3 ± 0.9 nM and 1.2 ± 0.5 nM (mean ± SEM, n = 4) for dopamine and 20, respectively. B, Cells were stimulated as indicated and analyzed using the BRET-based Go activation assay as described in the Experimental Section resulting in EC50 values of 1.1 ± 0.04 nM and 0.18 ± 0.1 nM (mean ± SEM, n = 6) for dopamine and 20, respectively. C, Cells were incubated with the indicated concentrations of dopamine (DA) or 20 and inhibition of forskolin-stimulated cAMP accumulation was determined using the CAMYEL biosensor as described in the Experimental Section. Data are presented as the percentage of maximum inhibition by DA. 20 displays full agonist activity of D3R-mediated inhibition of cAMP production exhibiting an EC50 of 86 ± 26 nM (mean ± SEM, n = 4). Dopamine demonstrated an EC50 value of 3.5 ± 0.7 nM (mean ± SEM, n = 4). D, Cells were stimulated as indicated and pERK was assessed using the Alphascreen SureFire assay as described in the Experimental Section. EC50 values for dopamine and 20 are 2.9 ± 0.6 nM and 21 ± 6.6 nM (mean ± SEM, n = 4), respectively. All data were analyzed using nonlinear regression curve fits and expressed as a percentage of maximum dopamine signal.
To further characterize the pharmacological selectivity of 20, we evaluated its activity in large arrays of GPCRs and several transporters or ion channels. For comparison, we also evaluated the recently described D3R-selective agonist CJ-163945 and its parent scaffold the D3R-preferring agonist pramipexole. Initially, we screened the compounds using the NIMH Psychoactive Drug Screening Program,67 which uses radioligand binding assays to assess affinity values for ligands at 45 unique GPCRs, transporters and ion channels. The results of this screen are shown in Table 6. For the primary screen, a single high concentration (10 μM) of test compound was used to inhibit radioligand binding to the targets and those compounds that exhibited >50% inhibition were rescreened in full concentration-response format in order to estimate their affinity (Ki) values. Notably, 20 exhibited submicromolar affinity for only three of the targets, the β1-adrenergic, 5-HT2B serotoninergic, and σ-1 receptors. Pramipexole and CJ-1639 exhibited submicromolar affinity for 6 and 13 of the targets, respectively. Among the D2-like receptors, pramipexole was 826-fold D3R>D2R-selective and 32-fold D3R>D4R-selective. In parallel studies in our laboratory, we found that pramipexole inhibits [3H]-methylspiperone binding to the D3R with a Ki of 8.2 ± 2.0 nM (mean ± SEM, n = 3) and to the D2R with a Ki of 4.7 ± 1.3 μM (mean ± SEM, n = 3), exhibiting 573-fold D3R>D2R selectivity in good agreement with the PDSP data. In the PDSP screen, CJ-1639 exhibited 130-fold D3R>D2R selectivity and 1.5-fold D3R>D4R selectivity. In our laboratory, we found that CJ-1639 inhibits [3H]-methylspiperone binding to the D3R with a Ki of 5.6 ± 1.0 nM (mean ± SEM, n = 3) and to the D2R with a Ki of 21 ± 5.6 μM (mean ± SEM, n = 3) exhibiting 3,750-fold D3R>D2R selectivity. Thus, in our hands, CJ-1639 is significantly more D3R>D2R-selective than was observed in the PDSP screen and closer to the D3R>D2R selectivity initially described by Chen et al.45
Table 6.
Binding affinities1 of 20, CJ-1639 and pramipexole for inhibiting radioligand binding to the indicated drug targets as determined in the PDSP screen.
| Target | Compound | Target | Compound | ||||
|---|---|---|---|---|---|---|---|
| 20 (Ki, nM) | CJ-1639 (Ki, nM) | Pramipexole (Ki, nM) | 20 (Ki, nM) | CJ-1639 (Ki, nM) | Pramipexole (Ki, nM) | ||
| 5HT1A | 2,108 | 708 | 6514 | D3 | 1240 | 30 | 0.9 |
| 5HT1B | NA | NA | 3508 | D4 | NA | 45 | 29 |
| 5HT1D | NA | NA | >10,000 | D5 | NA | NA | >10,000 |
| 5HT1E | NA | NA | >10,000 | DAT | NA | 205 | NA |
| 5HT2A | NA | 2,841 | NA | DOR | NA | NA | >10,000 |
| 5HT2B | 674 | 1,178 | NA | GABAA | NA | NA | ND |
| 5HT2C | 5,997 | 762 | NA | H1 | NA | 110 | NA |
| 5HT3 | NA | NA | >10,000 | H2 | NA | 224 | 2,683 |
| 5HT5A | NA | NA | >10,000 | H3 | NA | 893 | NA |
| 5HT6 | NA | NA | >10,000 | H4 | NA | NA | >10,000 |
| 5HT7 | NA | 770 | 1,188 | KOR | NA | >10,000 | NA |
| Alpha1A | NA | NA | >10,000 | M1 | NA | NA | >10,000 |
| Alpha1B | NA | 666 | NA | M2 | NA | NA | >10,000 |
| Alpha1D | NA | 1,184 | NA | M3 | NA | NA | >10,000 |
| Alpha2A | >10,000 | NA | 75.7 | M4 | NA | 2,279 | NA |
| Alpha2B | N/A | NA | 67.7 | M5 | NA | 2,297 | NA |
| Alpha2C | 2,841 | NA | 52.2 | MOR | NA | 745 | NA |
| Beta 1 | 77 | NA | NA | NET | NA | 436 | NA |
| Beta 2 | NA | ND | >10,000 | PBR | NA | NA | ND |
| Beta 3 | NA | NA | >10,000 | SERT | NA | 32 | NA |
| BZP site | NA | NA | ND | Sigma 1 | 383 | 531 | 4,446 |
| D1 | NA | NA | >10,000 | Sigma 2 | 2,750 | N/A | NA |
| D2 | NA | 3,902 | 743.7 | ||||
Ki (nM) values for the indicated compounds were determined as described in the Experimental Section.
NA indicates inhibition of binding was <50% in the primary assay conducted using a single 10 μM concentration.
Notably, in the PDSP screen, 20 exhibited a Ki for the D3R of 1.24 μM. Thus, as was initially observed with compound 1, when assessed using [3H]-methylspiperone binding in membrane preparations, 20 exhibits an affinity for the D3R that is much weaker than its potency observed using any of the functional assays, even those that lack amplification. In contrast, the agonists pramipexole and CJ-1639 both display similar D3R binding affinities and functional potencies. For instance, when we examined pramipexole and CJ-1639 in the D3R-mediated β-arrestin recruitment assay, we found EC50 values of 5.4 ± 1.5 nM (mean ± SEM, n = 3), and 9.3 ± 1.5 nM (mean ± SEM, n = 3), respectively, in good agreement with their affinities observed in the binding assays (see above).
Given the above observations, we decided to investigate the effects of assay buffer conditions on the radioligand binding results (Table 7). Our standard binding buffer is EBSS, which is an isotonic bicarbonate-phosphate buffer containing 117 mM NaCl, 5.37 mM KCl, 1.8 mM CaCl2, 0.8 mM MgSO4, and 5.55 mM glucose. In addition to EBSS, we utilized Tris buffer with no additional salts, as well as Tris plus 140 mM NaCl, and performed [3H]-methylspiperone competition binding with 20 and pramipexole (Table 7). Interestingly, 20 was ~10-fold more potent using the Tris buffer compared to EBSS whereas 20’s potency decreased by ~4-fold when 140 mM NaCl was added to the Tris buffer (Tris + Na+). Notably, the [3H]-methylspiperone binding buffer used by the PDSP is HEPES plus 50 mM NaCl and 5 mM MgCl2, more similar to that of EBSS with respect to the concentrations of Na+ and divalent cations. Not surprisingly, the potency of 20 is similar when comparing the EBSS (Ki = 3.7 μM) and HEPES (Ki = 1.24 μM) buffer systems. In contrast to 20, the potency of pramipexole appeared to be relatively unaffected by the assay buffer conditions (Table 7), as is [3H]-methylspiperone (data not shown).
Table 7.
Affinities1 of 20 and pramipexole for inhibiting either [3H]-methylspiperone or [3H]-7OH-DPAT binding to the D3R in various buffer systems.
| [3H]-Methylspiperone Ki (nM) | [3H]-7-OH-DPAT Ki (nM) | |||||||
|---|---|---|---|---|---|---|---|---|
| EBSS | Tris | Tris + Na+ | EBSS | Tris | Tris + Na+ | Tris + Mg2+ | ||
| 20 | 3,700 ± 900 | 350 ± 130 | 1,500 ± 370 | 720 ± 31 | 22 ± 3.7 | 94 ± 11 | 12.5 ± 3.8 | |
| Pramipexole | 8.2 ± 2.0 | 13 ± 0.8 | 17 ± 0.6 | 1.1 ± 0.3 | 0.94 ± 0.24 | 5.6 ± 1.3 | ND | |
Ki (nM) values for the indicated compounds were determined as described in the Experimental Section.
Data are expressed as mean ± S.E.M. of individual experiments performed 3–16 times.
ND indicates that the value was not determined.
Typically, agonist ligands appear more potent when competing for radiolabeled agonist binding as the binding sites represent the high-affinity active G protein-coupled state of the GPCR.68–70 As such, we repeated our competition binding experiments using [3H]-7OH-DPAT, which is an agonist of the D3R (Table 7). The same potency trends were observed with the various buffer systems, although both 20 and pramipexole appeared more potent using [3H]-7-OH-DPAT compared with [3H]-methylspiperone, as was expected. In addition, we noted that the addition of 5 mM MgCl2 to Tris buffer (Tris + Mg2+) increased the potency of 20 by ~2-fold. These observations are in line with previous results showing that Na+ typically decreases agonist binding affinity, likely through stabilizing the inactive state of the GPCR,71, 72 while Mg2+ increases agonist binding affinity through promoting GPCR-G protein interactions.72, 73 Taken together, these results suggest that the apparent binding affinity of 20 for the D3R in membrane preparations can vary by as much as 300-fold depending on the buffer constituents and radioligand utilized. Moreover, the potency of 20 using intact cell functional assays (EC50s = 0.18–86 nM) is more closely aligned with its affinity for the active signaling state of the D3R as detected using [3H]-7OH-DPAT membrane binding, as previously observed for multiple GPCRs.68–70
We extended our selectivity screening to the DiscoverX gpcrMAX™ functional assay, which measures agonist-stimulated β-arrestin recruitment to 168 known GPCRs (http://www.DiscoverX.com). We chose to perform this screen using a high concentration (10 μM) of 20 in order to maximize the detection of off-target activities. Figure 6A shows the results of this screen performed in agonist mode where each GPCR is stimulated with 20 and β-arrestin recruitment is then measured. Notably, the D3R is the only GPCR in this array that was fully activated, although small, but measurable agonist responses were observed with the short and long isoforms of the D2R. These latter activities were comparable to the low activity of 20 at the D2R observed in Figure 4B using 10 μM and higher concentrations. Figure 6B shows the results of this screen in antagonist mode and that 20 exhibits low potency partial antagonist activity at several GPCRs. These include the Epstein-Barr virus-induced GPCR 2 (EBI2), the sphingosine-1-phosphate 5 receptor (EDG8), the cholecystokinin A receptor (CCKAR), the alpha2C-adrenergic receptor (ADRA2C), and the 5-HT2A serotonergic receptor (HTR2A). Antagonism of these receptors is not associated with any known clinical side-effects and, in any case, such interactions would not occur at the nM concentrations employed to selectively activate the D3R. Importantly, 20 was identified as exhibiting submicromolar affinity for the 5-HT2B receptor in the PDSP screen (Table 6) which is a potential liability as drugs that activate the 5-HT2B receptor have been associated with cardiac valvulopathy.74, 75 Unfortunately, the DiscoverX gpcrMAX™ panel does not include the 5-HT2B receptor, so this screen did not provide us with a functional profile of 20 at this receptor subtype. Thus, in separate experiments, we assessed the effects of 20 (10 μM) on 5-HT2B receptor stimulation of inositol 1-phosphate (IP1) accumulation (see the Experimental Section). Fortunately, no agonist activity was observed, but this concentration of 20 produced an 80% inhibition of the response elicited by 30 nM serotonin (data not shown) suggesting that 20 may be a low potency antagonist of the 5-HT2B receptor at high micromolar concentrations. Taken together, these findings indicate that 20 is a selective D3R agonist with very limited cross-reactivity at other GPCRs.
Figure 6. Functional profiling of compound 20 against an array of 168 known GPCRs.

A single high concentration (10 μM) of 20 was screened against the DiscoverX gpcrMAX™ assay panel in both agonist (A) or antagonist (B) modes as described in the Experimental Section. Data represent the percent maximum stimulation observed by a reference agonist for each GPCR (agonist mode) or the percent inhibition of a response generated by an EC80 concentration of a reference agonist (antagonist mode). A complete key to the GPCR array and numerical results are provided in Supplemental Table S1. In B, partial antagonist activity was observed at the ADRA2C (36%), CCKAR (63%), EDG8 (77%), EBI2 (88%), and HTR2A (37%) receptors using a 10 μM concentration of 20.
For comparison with 20, the D3R-preferring agonists CJ-1639 and pramipexole were also screened in the DiscoverX gpcrMAX™ panel. Figure S1A shows that, at 10 μM, CJ-1639 is a full agonist of both the D2R and D3R, with partial agonist activities at the D4R and the chemokine CXCR7 receptor. Conversely, at 10 μM, CJ-1639 displays >50% antagonist activity at the alpha1B-adrenergic receptor, H1, H2, and H3 histamine receptors, and the μ opioid receptor (Figure S1B). Pramipexole also exhibits full agonist activity at the D2R and D3R as well as partial agonist activity at the alpha2A- and alpha2B-adrenergic receptors (Figure S2A). Interestingly, pramipexole did not exhibit antagonist activity at any of the GPCRs tested, even at the 10 μM concentration (Figure S2A). Overall, using these assays, CJ-1639 and pramipexole appear less globally selective than 20, although they may exhibit greater D3R selectivity if lower screening concentrations are employed.
Molecular Modeling Predicts Unique Interactions of Compound 20 with the D3R.
To characterize the binding pose of 20, we carried out a computational modeling and simulation study of a D3R model in complex with 20. We first docked 20 into a D3R model that we have equilibrated previously,76 and found the majority of the resulting poses of 20 are with the indole amide moiety pointing away from the orthosteric binding site (OBS, defined in77), similar to other indole amide D3R ligands that we have characterized previously.6, 78 Using three subtly different poses in such an orientation with the lowest docking scores, we collected three molecular dynamics (MD) simulation trajectories. The simulations converged to a 20 pose in which at one end its anisole moiety binds in the OBS and protrudes into the interface between TM5 and TM6 and interacts with Tyr1985.48 (superscripts denote Ballesteros and Weinstein numbering),79 while at the other end its indole amide moiety interacts with Tyr3657.35 (Figure 7B). Further, the positively charged piperazine nitrogen of 20 closest to the anisole moiety was found to interact with Asp1103.32 (Figure 7B), the highly conserved aspartic acid residue found in TM3 of all biogenic amine GPCRs77. Given this observation, it is not surprising that compounds either lacking this nitrogen (50) or with immediately adjacent methyl groups (46 and 48) that could produce steric hindrance exhibited greatly reduced or no activity for interacting with the D3R (Table 3).
Figure 7. Molecular modeling identified residues that uniquely interact with 20.

(A) a side view of a representative D3R model in complex with 20 resulting from our MD simulations. The model was based on the D3R crystal structure (PDB: 3PBL).6 (B) A zoom-in view of the ligand binding pocket showing that two Tyr residues, Tyr1985.48 and Tyr3657.35, uniquely interact with the two ends of the ligand. The conserved TM3 aspartate residue, Asp1103.32, in contact with the basic piperazine nitrogen is also depicted.
We noticed that the two tyrosine residues, Tyr1985.48 and Tyr3657.35, in contact with 20 cannot directly interact with dopamine.57 Thus, to validate the predicted binding pose of 20 at D3R, we mutated Tyr1985.48 and Tyr3657.35 to alanine residues either individually or in combination, and compared the pharmacological profiles of dopamine and 20 at these mutants. The mutant D3R constructs were found to express similarly to D3R-WT as determined using[3H]-methylspiperone saturation binding assays (Bmax values (fmol/mg): WT = 3,700 ± 650, Y198A = 4,300 ± 1,000, Y365A = 3,000 ± 500, Y198A/Y365A = 2,700 ± 930; Kd values (nM): WT = 0.51 ± 0.08, Y198A = 0.53 ± 0.06, Y365A = 0.49 ± 0.11, Y198A/Y365A = 0.35 ± 0.1; mean ± SEM from 3 independent experiments). We then compared the ability of compound 20 and dopamine to stimulate β-arrestin recruitment to these constructs. Figure 8A shows that they stimulate β-arrestin recruitment to the D3R-WT with similar potencies (EC50 = 1.4 nM and 3.7 nM for 20 and dopamine, respectively). The Y198A mutation resulted in a reduction in efficacy (Emax = 38%) and a 13-fold reduction in the potency (EC50 = 17 nM) of 20 for stimulating β-arrestin recruitment (Figure 8B). Because the mutation similarly reduced the potency of dopamine by 14-fold (EC50 = 55 nM) but not its efficacy, the drastically reduced efficacy of 20 is a specific effect of the mutation on this ligand. The Y365A mutation resulted in a pronounced 400-fold reduction in the potency (EC50 = 520 nM) and decrease in efficacy (Emax = 26%) for 20, whereas dopamine’s potency was reduced by only 9-fold (EC50 = 33 nM) without any reduction of its efficacy (Figure 8C). Notably, the double Y198A/Y365A mutation completely abolished the efficacy of 20 for stimulating β-arrestin recruitment to the D3R (Figure 8D), whereas the effects on dopamine’s potency (EC50 = 820 nM) appeared to be additive (215-fold) compared to the effects of the single mutants. The unique dependence of 20 on these two residues for full activation of the receptor, supports our computational results that show they are in direct contact with 20 but not dopamine. Interestingly, Tyr3657.35 has previously been suggested as an interaction site for a DAR agonist with D3R>D2R selectivity.51
Figure 8. Mutagenesis studies support D3R binding site model for compound 20.

HEK293 cells were transiently transfected with the following Rluc8-fused receptor constructs: D3R-WT (A), D3R-Y198A (B), D3R-Y365A (C), or D3R-Y198A/Y365A (D), as described in the Experimental Section. Cells were stimulated as indicated and analyzed using the BRET-based β-arrestin recruitment assay (see the Experimental Section). Data are expressed as a percentage of the maximum dopamine (DA) response. (A) 20 is a full agonist at the D3R-WT; EC50 = 1.4 ± 0.7 nM, Emax = 92 ± 10% (mean ± SEM, n = 8); DA EC50 = 3.7 ± 1.2 nM, Emax = 100 ± 0.3% (mean ± SEM, n = 8). (B) 20 has reduced potency and efficacy at the D3R-Y198A mutant; EC50 = 17 ± 7.6 nM*, Emax = 38 ± 6.2%** (mean ± SEM, n = 4); DA EC50 = 55 ± 16 nM***, Emax = 100 ± 0.2% (mean ± SEM, n = 4). (C) 20 has reduced potency and efficacy at the D3RY365A; EC50 = 520 ± 150 nM**; Emax 26 ± 11%** (mean ± SEM, n = 4); DA EC50 = 33 ± 3.3 nM****; Emax = 100 ± 0.1% (mean ± SEM, n = 4). (D) The D3R-Y198A/Y365A mutation abolishes agonist activity of 20. DA EC50 = 820 ± 56 nM****; Emax = 100 ± 0.1% (mean ± SEM, n = 3). Statistical comparisons between WT and mutant parameters were made using a two-tailed t-test: *p<0.05, **p<0.005, ***p<0.0.001, ****p<0.0001.
We thought it would be of interest to also characterize and compare the effects of the Y198A and Y365A mutations on the signaling properties of pramipexole. Figure S3A shows that pramipexole and dopamine have similar potencies in stimulating β-arrestin recruitment to the D3R-WT. Introduction of the Y198A, Y365A, or double Y198A/Y365A mutations into the D3R decreased the potency of pramipexole to a similar extent as that seen with dopamine, while, in contrast to that seen with compound 20, there was no effect on the efficacy of pramipexole for stimulating β-arrestin recruitment (Figure S3). These results suggest that different efficacy determinants exist for activation of the D3R by pramipexole and 20. Taken together, the mutational data lend support to the molecular modeling results that describe unique interactions of 20 with the D3R.
Neuroprotective properties of compound 20.
As D3R-preferring agonists, such as pramipexole and related compounds, have shown neuroprotective properties in several models of neurodegeneration,11, 13–16 we evaluated the effects of compound 20, using pramipexole as a comparator, in a cellular model of neuroprotection. We genetically engineered a human iPSC cell line so that it stably expresses the human D3R (3–5 pmol/mg) and differentiated these cells into dopaminergic neurons in culture (see Experimental Section). The cells were then treated with the dopaminergic neurotoxin 6-OHDA to induce cell death. As shown in Figure 9, 6-OHDA treatment reduced cell viability by 62 ± 8% after 24 hr. Both compound 20 and pramipexole demonstrated a dose-dependent reduction in 6-OHDA-induced cell death. This effect was significant at 50 nM of 20 and maximal at 500 nM. In contrast, pramipexole was somewhat less potent in not achieving a significant level of protection until a concentration of 500 nM was employed (Figure 9). These data indicate that compound 20 simulates the previously described neuroprotective properties of pramipexole and highlights the utility of pursuing this compound for in vivo development, including investigations of additional models of neurodegeneration and as a probe compound to understand the role of the D3R in neuroprotection.
Figure 9. Compound 20 protects D3R-expressing dopaminergic neurons from 6-OHDA-induced cell death.

Human iPSCs expressing the D3R were differentiated into dopaminergic neurons as described in the Experimental Section. Cells were treated with the indicated concentrations of vehicle, 20, or pramipexole for 24 hours and then incubated with 30 μM of 6-hydroxydopamine (6-OHDA) for 24 hours to induce cell death (the control cell group did not receive 6-OHDA). MTT cell viability assays were performed as described in the Experimental Section. Data represent the means ± SEM from six experiments performed in quadruplicate. In the absence of drugs (vehicle), 6-OHDA treatment reduced cell viability by 62 ± 8%. Pretreatment with 20 and pramipexole protected against 6-OHDA-induced cell death in a dose-dependent fashion. Statistical significance for differences between the drug and vehicle treated groups was assessed using two-way ANOVA followed by Dunnett’s multiple comparison post-hoc test: *p < 0.05, **p < 0.01, #p < 0.001, & p < 0.0001.
Pharmacokinetics and Toxicology.
As we are interested in the use of 20 as an in vivo probe and as a lead compound for therapeutics development, we performed a number of preliminary toxicology, ADME and pharmacokinetics experiments. Compound 20 showed no liability (AC50s >50 μM) using a cytotoxicity screening panel that measures changes in nuclear size, DNA structure, cell membrane permeability, mitochondrial mass, mitochondrial membrane potential, and cytochrome C release (see the Experimental Section). Further, no toxicity was observed in the AMES reverse mutation assay (data not shown). Compound 20 displays reasonable liver microsomal stability (t1/2 = 21.2 min) and excellent permeability using the PAMPA assay (541 (10−6 cm/sec)), however, its aqueous solubility is low (1.1 μg/ml). In vivo pharmacokinetics experiments in mice (using 20 mg/kg IP) reveals that compound 20 is brain penetrant and exhibits a plasma half-life of 3.44 h and a brain half-life of 4.23 h (Figure S4 and Tables S2 and S3). Further, compound 20 displays a plasma Tmax of 0.5 h and Cmax of 6,500 ng/ml, and a brain T max of 0.25 h and Cmax of 28,000 ng/ml (Tables S2 and S3). Taken together, these findings are very encouraging for future in vivo studies using compound 20 and the potential therapeutic development of this scaffold.
CONCLUSIONS
In summary, we have identified a novel and highly selective D3R agonist scaffold originating from a high-throughput screening campaign. Focused optimization led to the potent and uniquely selective D3R agonist, 20. Its exquisite receptor selectivity suggests that 20 will prove to be a valuable pharmacological tool to interrogate the physiological functions of the D3R in normal and pathological conditions. 20’s demonstrated neuroprotective effects along with promising toxicology and pharmacokinetic profiles further suggest that it may show utility as a therapeutic lead for the treatment of neurodegenerative or other disorders.
EXPERIMENTAL SECTION
Materials.
[3H]-methylspiperone (80 Ci/mmol) was obtained from PerkinElmer Life Sciences (Waltham, MA). D1R, D2R, D3R, D4R, and D5R expressing CHO-K1 cells and CP2 media were purchased from DiscoverX (Fremont, CA). Other cell culture media and reagents were purchased from MediaTech/Cellgro (Manassas, VA). Cell culture flasks, materials and all assay plates were purchased from ThermoFisher Scientific (Waltham, MA), and Greiner Bio-One (Monroe, NC). DNA constructs for the BRET assays were kind gifts from Dr. Jonathan A. Javitch. Compound CJ-1639 was a kind gift from Dr. Shaomeng Wang. Receptor mutants were prepared at Bioinnovatise (Rockville, MD), and all mutations were verified by DNA sequencing. Chemicals and buffer components were purchased from Sigma-Aldrich (St Louis, MO), except where indicated. All tested analogs were synthesized as described below and were characterized as being >95% pure.
Chemistry.
General synthesis and analysis experimental details:
All reagents were used as received from the following suppliers: Alfa Aesar, Aldrich, Ark Pharm, Combi-Blocks, Fisher Scientific Oakwood, Matrix and 1 Click Chemistry. Acetonitrile and THF were purified using the Innovative Technology PureSolv solvent purification system using two alumina columns. The 1H and 13C spectra were recorded on a 400 MHz Bruker Avance spectrometer equipped with a broadband observe probe and a 500 MHz Bruker AVIII spectrometer equipped with a dual cryoprobe, respectively. Chemical shifts are reported in parts per million and were referenced to residual proton solvent signals. 13C multiplicities were determined with the aid of an APT pulse sequence, differentiating the signals for methyl (CH3) and methyne (CH) carbons as “d” from methylene (CH2) and quarternary (C) carbons as “u”. The infrared (IR) spectra were acquired as thin films using a universal ATR sampling accessory on a Thermo Fisher Nicolet iS5 FT-IR spectrometer and the absorption frequencies are reported in cm−1. Melting points were determined on a Stanford Research Systems Optimelt automated melting point system interfaced through a PC and are uncorrected. Microwave syntheses were conducted in a Biotage Initiator constant temperature microwave synthesizer. Flash column chromatography separations were performed using the Teledyne Isco CombiFlash Rf using RediSep Rf silica gel columns. TLC was performed on Analtech UNIPLATE silica gel GHLF plates (gypsum inorganic hard layer with fluorescence). TLC plates were developed using iodine vapor or ceric ammonium molybdate stain, as required. Automated preparative RP HPLC purification was performed using an Agilent 1200 Mass-Directed Fractionation system (Prep Pump G1361 with gradient extension, make-up pump G1311A, pH modification pump G1311A, HTS PAL autosampler, UV-DAD detection G1315D, fraction collector G1364B, and Agilent 6120 quadrapole spectrometer G6120A). HRMS determinations for compounds 106–109 were analyzed with a ThermoFisher Q Exactive HF-X (ThermoFisher, Bremen, Germany) mass spectrometer coupled with a Waters Acquity H-class liquid chromatograph system. Samples were introduced via a heated electrospray source (HESI) at a flow rate of 0.6 mL/min. Electrospray source conditions were set as: spray voltage 3.0 kV, sheath gas (nitrogen) 60 arb, auxillary gas (nitrogen) 20 arb, sweep gas (nitrogen) 0 arb, nebulizer temperature 375 degrees C, capillary temperature 380 degrees C, RF funnel 45 V. The mass range was set to 150–2000 m/z. All measurements were recorded at a resolution setting of 120,000. The preparative chromatography conditions included a Waters X-Bridge C18 column (19 × 150 mm, 5 μm, with 19 × 10-mm guard column), elution with a water and acetonitrile gradient, which increases 20% in acetonitrile content over 4 min at a flow rate of 20 mL/min (modified to pH 9.8 through addition of NH4OH by auxiliary pump), and sample dilution in DMSO. The preparative gradient, triggering thresholds, and UV wavelength were selected according to the analytical RP HPLC analysis of each crude sample. The analytical method used an Agilent 1200 RRLC system with UV detection (Agilent 1200 DAD SL) and mass detection (Agilent 6224 TOF). The analytical method conditions included a Waters Aquity BEH C18 column (2.1 × 50 mm, 1.7 μm) and elution with a linear gradient of 5% acetonitrile in pH 9.8 buffered aqueous ammonium formate to 100% acetonitrile at 0.4 mL/min flow rate. Compound purity was measured on the basis of peak integration (area under the curve) from UV/Vis absorbance (at 214 nm), and compound identity was determined on the basis of mass analysis. All compounds used for assays or biological studies possessed HPLC purity >95%. The analytical HPLC system used is a dedicated instrument for assessing compound purity and routinely detects impurities as low as 0.1%. Any compounds with a measured HPLC purity of 100% were thus conservatively assigned a purity of “> 99.5%”. Any compounds purified by automated preparative RP HPLC purification utilized the same solvent gradient and column material in the analytical conditions to minimize the possibility of undetected impurities carrying over from the purification run.
All final compounds were inspected for functional groups known to contribute PAINS liabilities, and none were found.
General Procedure A: piperazine coupling with alkyl bromides.
The monocarboxamide piperazine substrate, alkyl bromide (1.1–1.2 equiv), potassium carbonate (3.0 equiv) and potassium iodide (1.0 equiv) were charged in a reaction vial and slurried with MeCN (50 mL/mmol substrate) and stirred at 60 °C for 14–20 h. The reaction was filtered and the solids washed with CH2Cl2 (2 × 5 mL). The combined filtrates were evaporated and the residue purified by silica gel chromatography to afford the alkylated piperazine product.
General Procedure B: piperazine coupling with alkyl bromides: high-throughput synthesis protocol.
The monocarboxamide piperazine substrate as a solution in DMF (0.2 M) and alkyl bromide (1.0 equiv) as a solution in DMF (0.2 M) were added to a reaction tube (Bohdan MiniBlock) containing potassium carbonate (3.0 equiv) and potassium iodide (0.1 equiv). The vial was sealed and the reaction stirred at 60 °C for 14–20 h. The reaction was filtered and the solids washed with CH2Cl2 (2 × 5 mL). The combined filtrates were evaporated and the residue purified by mass-directed, reverse-phase HPLC to afford the alkylated piperazine product.
General Procedure C: Mitsunobu reaction route to aryl ether analogs.
A solution of (2-hydroxylethyl)piperazine substrate, phenol (1.0 equiv) and triphenyl phosphine (1.0 equiv) in THF or CH2Cl2 (10 mL/mmol substrate) was cooled in an ice/water bath and DIAD (1.25 equiv) added in a single portion. After 1 min, the ice/water bath was removed and the reaction was stirred at rt for an additional 5 min then stirred at 60 °C for 16–40 h. The reaction was monitored for conversion by LC-MS and declared complete when no further change was observed in the chromatogram. For reactions in THF, the solvent was removed in vacuo and the residue dissolved in CH2Cl2 (5 mL). For reactions in CH2Cl2, additional CH2Cl2 was added to adjust the volume to 5 mL. The CH2Cl2 solution was washed with aqueous 1 N NaOH (2 × 1 mL) then water (1 mL) and the organic layer was purified by silica gel chromatography to afford the aryl ether product.
General Procedure D: late-stage acylation route to disubstituted piperazine analogs.
A solution of piperazine 114 and triethyl amine (1.5 equiv) in CH2Cl2 (20 mL/mmol substrate) was cooled in an ice/water bath and the appropriate acid chloride (1.0–1.3 equiv) added in a single portion. The reaction was capped and stirred for 16–20 h, slowly warming to rt. The reaction was washed with water (2 × 5 mL) then dried over Na2SO4 and the organic layer was purified by silica gel chromatography to afford the aryl ether product.
General Procedure E: PyBOP-mediated coupling route to disubstituted piperazine analogs.
To a solution of arylcarboxylic acid (1.0–1.3 equiv) in DMF (10 mL/mmol substrate) was added PyBOP (1.2 equiv) and the reaction stirred at rt for 10 mins. A solution of 1-(2-(aryloxy)ethyl)piperazine 114 (1.0 equiv) and diisopropylethyl amine (3.0 equiv) in DMF (10 mL/mmol substrate) was added and the reaction stirred at rt for 16–20 h. The reaction solvent was removed in vacuo and the residue partitioned between saturated, aqueous NaHCO3 and CH2Cl2 (2 × 5 mL). The combined organic layers were dried over Na2SO4, concentrated in vacuo and purified by silica gel chromatography or reverse-phase, mass-directed preparative HPLC to afford the disubstituted piperazine product.
General Procedure F: Piperazine alkyl chloride displacement route to aryl ether piperazine analogs.
A solution of alkyl chloride substrate, phenol nucleophile (1.6–1.9 equiv) and potassium carbonate (2.5 equiv) in DMF (10 mL/mmol substrate) was heated at 60 °C for the stated reaction time and cooled to rt. The reaction was diluted with water (10 mL) and extracted with CH2Cl2 (3 × 2 mL). The combined organic layers were dried over Na2SO4, concentrated in vacuo and purified by silica gel chromatography or reverse-phase, mass-directed preparative HPLC to afford the disubstituted piperazine product.
General Procedure G: Piperazine alkyl chloride displacement route to aryl ether piperazine analogues, parallel synthesis protocol.
A solution of piperazine alkyl chloride substrate in DMF (0.528 M, 1.0 equiv) was added to a 24-position Bohdan MiniBlock reaction tube containing a mixture of phenol, aniline or thiol nucleophile (1.6–1.9 equiv) and potassium carbonate (3.9 equiv) in DMF (800 μL). The reaction was heated at 60 °C for 16 h, concentrated on a Genevac centrifugal evaporator for 3 h at 35 °C. The residue was partitioned between water (2 mL) and CH2Cl2 (3 mL), filtered through a Biotage phase separator tube into a 16 × 100 mm tube. The organic layer was concentrated on a Genevac centrifugal evaporator and purified by preparative, mass-directed, reverse-phase HPLC to afford the aryl ether product.
General Procedure H: Alkylation of phenols with 1,2-dibromoethane.
To a solution of phenol 112 and potassium hydroxide (2.0 equiv) in water (3 mL/mmol of phenol) was added sequentially tetrabutylammonium bromide (0.2 equiv) then 1,2-dibromoethane (5 equiv). The reaction was heated at reflux for 19 h, cooled to rt and extracted with CH2Cl2 (2 × 50 mL). The combined organic layers were dried with Na2SO4 and concentrated in vacuo. The residue was purified by silica chromatography to afford the bromide product 112.
(4-(2-(4-Methoxyphenoxy)ethyl)piperazin-1-yl)(4-methoxyphenyl)methanone (1).
(4-Methoxyphenyl)(piperazin-1-yl)methanone (49 mg, 0.22 mmol) and 1-(2-bromoethoxy)-4-methoxybenzene (61 mg, 0.27 mmol, 1.2 equiv) were reacted according to General Procedure A to afford the alkylated piperazine as a white solid (66 mg, 0.18 mmol, 81% yield). Rf = 0.29 (EtOAc); mp = 87–89 °C. 1H NMR (400 MHz, CDCl3) δ 2.51–2.68 (complex, 4H), 2.81 (t, J = 5.6 Hz, 2H), 3.51–3.74 (complex, 4H), 3.76 (s, 3H), 3.82 (s, 3H), 4.06 (t, J = 5.6 Hz, 2H), 6.80–6.86 (complex, 4H), 6.89–6.92 (m, 2H), 7.36–7.40 (m, 2H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d: 55.3, 55.7, 113.7, 114.6, 115.6, 129.1; u: 53.5, 53.7, 57.3, 66.5, 127.8, 152.8, 154.0, 160.7, 170.2; FTIR (neat): 1623, 1607, 1506 cm−1; HRMS (m/z): calcd for C21H27N2O4 [M + H]+ 371.1965; found 371.1973; HPLC purity = 99.2%.
(4-(2-(2-Methoxyphenoxy)ethyl)piperazin-1-yl)(4-methoxyphenyl)methanone (2).
(4-Methoxyphenyl)(piperazin-1-yl)methanone (44 mg, 0.20 mmol) and 1-(2-bromoethoxy)-2-methoxybenzene (46 mg, 0.20 mmol, 1.0 equiv) were reacted according to General Procedure B to afford the alkylated piperazine as a light yellow solid (28 mg, 0.076 mmol, 38% yield). Rf = 0.14 (EtOAc); mp = 119–121 °C. 1H NMR (400 MHz, CDCl3) δ 2.52–2.68 (complex, 4H), 2.86 (t, J = 5.9 Hz, 2H), 3.45–3.88 (complex, 4H), 3.81 (s, 3H), 3.83 (s, 3H), 4.14 (t, J = 5.9 Hz, 2H), 6.85–6.94 (complex, 6H), 7.35–7.39 (m, 2H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d: 55.3, 55.9, 111.9, 113.7, 113.9, 120.8, 121.6, 129.1; u: 53.5, 53.7, 57.0, 67.0, 127.8, 148.1, 149.6, 160.7, 170.2; FTIR (neat): 1608, 1504 cm−1; HRMS (m/z): calcd for C21H27N2O4 [M + H]+ 371.1965; found 371.1985; HPLC purity = 96%.
(4-(2-(3-Methoxyphenoxy)ethyl)piperazin-1-yl)(4-methoxyphenyl)methanone (3).
(4-Methoxyphenyl)(piperazin-1-yl)methanone (44 mg, 0.20 mmol) and 1-(2-bromoethoxy)-3-methoxybenzene (46 mg, 0.20 mmol, 1.0 equiv) were reacted according to General Procedure B to afford the alkylated piperazine as a pale yellow oil (47 mg, 0.13 mmol, 63% yield). Rf = 0.24 (EtOAc). 1H NMR (400 MHz, CDCl3) δ 2.51–2.66 (complex, 4H), 2.81 (t, J = 5.6 Hz, 2H), 3.47–3.82 (complex, 4H), 3.76 (s, 3H), 3.80 (s, 3H), 4.08 (t, J = 5.6 Hz, 2H), 6.44–6.51 (complex, 3H), 6.87–6.91 (m, 2H), 7.17 (t, J = 8.2 Hz, 1H), 7.36–7.39 (m, 2H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d: 55.25, 55.32, 101.1, 106.5, 106.6, 113.7, 129.1, 129.9; u: 53.57, 53.63, 57.1, 65.8, 127.8, 159.8, 160.7, 160.8, 170.2; FTIR (neat): 1603, 1492 cm−1; HRMS (m/z): calcd for C21H27N2O4 [M + H]+ 371.1965; found 371.1988; HPLC purity = 99%.
(4-(2-(4-Ethylphenoxy)ethyl)piperazin-1-yl)(4-methoxyphenyl)methanone (4).
(4-Methoxyphenyl)(piperazin-1-yl)methanone (44 mg, 0.20 mmol) and 1-(2-bromoethoxy)-4-ethylbenzene (46 mg, 0.20 mmol, 1.0 equiv) were reacted according to General Procedure B to afford the alkylated piperazine as a waxy white solid (44 mg, 0.12 mmol, 59% yield). Rf = 0.19 (EtOAc); mp = 57–64 °C. 1H NMR (400 MHz, CDCl3) δ 1.20 (t, J = 7.6 Hz, 3H), 2.52–2.65 (complex, 4H), 2.58 (q, J = 5.6 Hz, 2H), 2.82 (t, J = 5.6 Hz, 2H), 3.43–3.87 (complex, 4H), 3.81 (s, 3H), 4.08 (t, J = 5.6 Hz, 2H), 6.79–6.84 (m, 2H), 6.88–6.92 (m, 2H), 7.07–7.12 (m, 2H), 7.36–7.39 (m, 2H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d: 15.9, 55.3, 113.7, 114.4, 128.7, 129.1; u: 28.0, 53.5, 53.6, 57.2, 65.9, 127.8, 136.7, 156.6, 160.7, 170.2; FTIR (neat): 1625, 1607, 1510 cm−1; HRMS (m/z): calcd for C22H29N2O3 [M + H]+ 369.2173; found 369.2195; HPLC purity = 98%.
(4-(2-(4-Chlorophenoxy)ethyl)piperazin-1-yl)(4-methoxyphenyl)methanone (5).
(4-Methoxyphenyl)(piperazin-1-yl)methanone (44 mg, 0.20 mmol) and 1-(2-bromoethoxy)-4-chlorobenzene (46 mg, 0.20 mmol, 1.0 equiv) were reacted according to General Procedure B to afford the alkylated piperazine as a white solid (28 mg, 0. 08 mmol, 38% yield). Rf = 0.18 (EtOAc); mp = 69–72 °C. 1H NMR (400 MHz, CDCl3) δ 2.47–2.64 (complex, 4H), 2.81 (t, J = 5.6 Hz, 2H), 3.48–3.79 (complex, 4H), 3.81 (s, 3H), 4.06 (t, J = 5.6 Hz, 2H), 6.79–6.82 (m, 2H), 6.87–6.91 (m, 2H), 7.18–7.23 (m, 2H), 7.35–7.38 (m, 2H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d: 55.3, 113.7, 115.8, 129.1, 129.3; u: 53.6, 53.7, 57.0, 66.2, 125.8, 127.7, 157.2, 160.8, 170.2; FTIR (neat): 1623, 1607 cm−1; HRMS (m/z): calcd for C20H24ClN2O3 [M + H]+ 375.1470; found 375.1501; HPLC purity = 99%.
(4-(2-(4-(Trifluoromethoxy)phenoxy)ethyl)piperazin-1-yl)(4-methoxyphenyl)methanone (6).
(4-(2-Hydroxyethyl)piperazin-1-yl)(4-methoxyphenyl)methanone (96 mg, 0.36 mmol) and 4-(trifluoromethoxy)phenol (65 mg, 0.36 mmol, 1.0 equiv) in CH2Cl2 were reacted according to General Procedure C for 46 h to afford the aryl ether as a light orange oil (56 mg, 0.13 mmol, 36% yield). Rf = 0.27 (EtOAc). 1H NMR (400 MHz, CDCl3) δ 2.50–2.68 (complex, 4H), 2.84 (t, J = 5.6 Hz, 2H), 3.47–3.82 (complex, 4H), 3.83 (s, 3H), 4.10 (t, J = 5.6 Hz, 2H), 6.86–6.92 (complex, 4H), 7.11–7.16 (m, 2H), 7.37–7.41 (m, 2H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d: 55.4, 114.0, 115.5, 122.7, 129.5; u: 52.7, 56.4, 63.26, 63.30, 120.4 (q, J = 257.9 Hz), 125.6, 143.6, 155.5, 161.6, 170.4; 19F NMR (376 MHz, CDCl3) δ –58.4; FTIR (neat): 1634, 1607, 1507 cm−1; HRMS (m/z): calcd for C21H24F3N2O4 [M + H]+ 425.1683; found 425.1696; HPLC purity > 99.5%.
(4-(2-(3,5-Dimethoxyphenoxy)ethyl)piperazin-1-yl)(4-methoxyphenyl)methanone (7).
(4-Methoxyphenyl)(piperazin-1-yl)methanone (44 mg, 0.20 mmol) and 1-(2-bromoethoxy)-3,5-dimethoxybenzene (52 mg, 0.20 mmol, 1.0 equiv) were reacted according to General Procedure B to afford the alkylated piperazine as a yellow oil (57 mg, 0.14 mmol, 71% yield). Rf = 0.17 (EtOAc). 1H NMR (400 MHz, CDCl3) δ 2.48–2.67 (complex, 4H), 2.81 (t, J = 5.5 Hz, 2H), 3.74 (s, 6H), 3.81 (s, 3H), 3.45–3.79 (complex, 4H), 4.05 (t, J = 5.5 Hz, 2H), 6.04–6.08 (complex, 3H), 6.89 (d, J = 8.7 Hz, 2H), 7.37 (d, J = 8.7 Hz, 2H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d: 55.3, 93.1, 93.5, 113.7, 129.1; u: 53.5, 53.6, 57.0, 65.8, 127.8, 160.5, 160.7, 161.5, 170.2; FTIR (neat): 1592, 1456, 1425 cm−1; HRMS (m/z): calcd for C22H29N2O5 [M + H]+ 401.2071; found 401.2102; HPLC purity = 98%.
(4-Methoxyphenyl)(4-(2-phenoxyethyl)piperazin-1-yl)methanone (8).
(4-(2-Hydroxyethyl)piperazin-1-yl)(4-methoxyphenyl)methanone (76 mg, 0.29 mmol) and phenol (27 mg, 0.29 mmol, 1.0 equiv) in THF were reacted according to General Procedure C for 22 h to afford the aryl ether as an orange oil (31 mg, 0.091 mmol, 32% yield). Rf = 0.17 (EtOAc). 1H NMR (400 MHz, CDCl3) δ 2.53–2.72 (complex, 4H), 2.85 (t, J = 5.6 Hz, 2H), 3.43–3.82 (complex, 4H), 3.83 (s, 3H), 4.12 (t, J = 5.6 Hz, 2H), 6.86–6.93 (complex, 4H), 6.95 (tt, J = 1.0, 7.4 Hz, 1H), 7.25–7.31 (m, 2H), 7.37–7.40 (m, 2H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d: 55.3, 113.7, 114.5, 120.9, 129.1, 129.4; u: 53.57, 53.60, 57.1, 65.7, 127.8, 158.5, 160.7, 170.2; FTIR (neat): 1622, 1599, 1456, 1427 cm−1; HRMS (m/z): calcd for C20H25N2O3 [M + H]+ 341.1860; found 341.1872; HPLC purity = 99%.
(4-(2-(Benzo[d][1,3]dioxol-5-yloxy)ethyl)piperazin-1-yl)(4-methoxyphenyl)methanone (9).
(4-Methoxyphenyl)(piperazin-1-yl)methanone (44 mg, 0.20 mmol) and 5-(2-bromoethoxy)benzo[d][1,3]dioxole (49 mg, 0.20 mmol, 1.0 equiv) were reacted according to General Procedure B to afford the alkylated piperazine as a tan solid (50 mg, 0.13 mmol, 65% yield). Rf = 0.16 (EtOAc); mp = 97–101 °C. 1H NMR (400 MHz, CDCl3) δ 2.48–2.64 (complex, 4H), 2.77 (t, J = 5.6 Hz, 2H), 3.44–3.86 (complex, 4H), 3.80 (s, 3H), 4.01 (t, J = 5.6 Hz, 2H), 5.87 (s, 2H), 6.29 (dd, J = 2.5, 8.5 Hz, 1H), 6.47 (d, J = 2.4 Hz, 1H), 6.67 (d, J = 8.4 Hz, 1H), 6.88 (d, J = 8.3 Hz, 1H), 7.36 (d, J = 8.8 Hz, 1H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d: 55.3, 98.2, 105.7, 107.9, 113.7, 129.1; u: 53.5, 53.6, 57.1, 66.8, 127.8, 141.8, 148.2, 154.1, 160.7, 170.2; FTIR (neat): 1622, 1607, 1486 cm−1; HRMS (m/z): calcd for C21H25N2O5 [M + H]+ 385.1758; found 385.1789; HPLC purity = 99%.
(4-Methoxyphenyl)(4-(2-(pyridin-3-yloxy)ethyl)piperazin-1-yl)methanone (10).
(4-(2-Hydroxyethyl)piperazin-1-yl)(4-methoxyphenyl)methanone (81 mg, 0.31 mmol) and 3-hydroxypyridine (29 mg, 0.31 mmol, 1.0 equiv) in CH2Cl2 were reacted according to General Procedure C for 22 h to afford the aryl ether as a colorless oil (16 mg, 0.047 mmol, 15% yield). 1H NMR (400 MHz, CDCl3) δ 2.53–2.65 (complex, 4H), 2.86 (t, J = 5.6 Hz, 2H), 3.45–3.80 (complex, 4H), 3.83 (s, 3H), 4.16 (t, J = 5.6 Hz, 2H), 6.89–6.93 (m, 2H), 7.19–7.23 (m, 2H), 7.37–7.41 (m, 2H), 8.23 (dd, J = 2.0, 4.0 Hz, 1H), 8.32 (dd, J = 1.0, 2.4 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 53.6, 53.7, 55.4, 57.0, 66.2, 113.7, 121.2, 123.8, 127.7, 129.2, 138.0, 142.4, 154.8, 160.8, 170.3; HRMS (m/z): calcd for C19H24N3O3 [M + H]+ 342.1812; found 342.1822; HPLC purity = 99%.
(4-(2-(4-Methoxyphenoxy)ethyl)piperazin-1-yl)(phenyl)methanone (11).
1-(2-(4-Methoxyphenoxy)ethyl)piperazine (107 mg, 0.45 mmol) and benzoyl chloride (83 mg, 0.59 mmol, 1.3 equiv) were reacted according to General Procedure D for 16 h to afford the acylated product as a viscous, pale yellow oil (106 mg, 0.31 mmol, 69% yield). Rf = 0.28 (EtOAc). 1H NMR (500 MHz, CDCl3) δ 2.49–2.70 (complex, 4H), 2.82 (t, J = 5.5 Hz, 2H), 3.39–3.50 (m, 2H), 3.75–3.86 (m, 2H), 3.77 (s, 3H), 4.07 (t, J = 5.6 Hz, 2H), 6.79–6.86 (complex, 4H), 7.37–7.44 (complex, 5H); 13C NMR (126 MHz, CDCl3) δ 53.3, 53.9, 55.7, 57.2, 66.5, 114.6, 115.6, 127.0, 128.5, 129.7, 135.8, 152.7, 154.0, 170.3; FTIR (neat): 1626, 1506, 1431 cm−1; HRMS (m/z): calcd for C20H25N2O3 [M + H]+ 341.1860; found 341.1874; HPLC purity = 98%.
(4-(2-(4-Methoxyphenoxy)ethyl)piperazin-1-yl)(2-methoxyphenyl)methanone (12).
(2-Methoxyphenyl)(piperazin-1-yl)methanone (44 mg, 0.20 mmol) and 1-(2-bromoethoxy)-4-methoxybenzene (46 mg, 0.20 mmol, 1.0 equiv) were reacted according to General Procedure B to afford the alkylated piperazine as an orange oil (22 mg, 0.059 mmol, 30% yield). Rf = 0.21 (EtOAc). 1H NMR (400 MHz, CDCl3) δ 2.37–2.71 (complex, 4H), 2.79 (t, J = 5.6 Hz, 2H), 3.19–3.33 (m, 2H), 3.74 (s, 3H), 3.77–3.92 (m, 2H), 3.80 (s, 3H), 4.04 (t, J = 5.6 Hz, 2H), 6.78–6.84 (complex, 4H), 6.88 (d, J = 6.4 Hz, 1H), 6.96 (t, J = 7.5 Hz, 1H), 7.20–7.25 (m, 1H), 7.32 (tt, J = 1.1, 7.9 Hz, 1H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d: 55.5, 55.7, 110.8, 114.6, 115.5, 120.9, 128.0, 130.3; u: 41.5, 46.7, 53.3, 53.8, 57.3, 66.5, 125.7, 152.8, 153.9, 155.3, 167.7; FTIR (neat): 1628, 1600, 1506 cm−1; HRMS (m/z): calcd for C21H27N2O4 [M + H]+ 371.1965; found 371.1969; HPLC purity = 97%.
(4-(2-(4-Methoxyphenoxy)ethyl)piperazin-1-yl)(3-methoxyphenyl)methanone (13).
(3-Methoxyphenyl)(piperazin-1-yl)methanone (44 mg, 0.20 mmol) and 1-(2-bromoethoxy)-4-methoxybenzene (46 mg, 0.20 mmol, 1.0 equiv) were reacted according to General Procedure B to afford the alkylated piperazine as an orange oil (44 mg, 0.12 mmol, 59% yield). Rf = 0.23 (EtOAc). 1H NMR (400 MHz, CDCl3) δ 2.45–2.66 (complex, 4H), 2.78 (t, J = 5.6 Hz, 2H), 3.36–3.49 (m, 2H), 3.71–3.84 (m, 2H), 3.73 (s, 3H), 3.79 (s, 3H), 4.03 (t, J = 5.6 Hz, 2H), 6.76–6.83 (complex, 4H), 6.89–6.95 (complex, 3H), 7.23–7.30 (m, 1H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d: 55.3, 55.7, 112.4, 114.6, 115.5, 115.5, 119.0, 129.5; u: 53.2, 53.8, 57.2, 66.5, 137.0, 152.7, 153.9, 159.6, 170.0; FTIR (neat): 1628, 1578, 1506 cm−1; HRMS (m/z): calcd for C21H27N2O4 [M + H]+ 371.1965; found 371.1987; HPLC purity = 99%.
(4-Chlorophenyl)(4-(2-(4-methoxyphenoxy)ethyl)piperazin-1-yl)methanone (14).
(4-Chlorophenyl)(piperazin-1-yl)methanone (45 mg, 0.20 mmol) and 1-(2-bromoethoxy)-4-methoxybenzene (46 mg, 0.20 mmol, 1.0 equiv) were reacted according to General Procedure B to afford the alkylated piperazine as a light yellow solid (27 mg, 0.073 mmol, 37% yield). Rf = 0.29 (EtOAc); mp = 78–80 °C. 1H NMR (400 MHz, CDCl3) δ 2.47–2.67 (complex, 4H), 2.80 (t, J = 5.6 Hz, 2H), 3.36–3.49 (m, 2H), 3.72–3.84 (m, 2H), 3.74 (s, 3H), 4.04 (t, J = 5.6 Hz, 2H), 6.78–6.85 (complex, 4H), 7.32–7.38 (complex, 4H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d: 55.7, 114.6, 115.5, 128.6, 128.7; u: 53.2, 53.8, 57.2, 66.5, 134.1, 135.7, 152.7, 154.0, 169.1; FTIR (neat): 1628, 1594, 1505 cm−1; HRMS (m/z): calcd for C20H24ClN2O3 [M + H]+ 375.1470; found 375.1478; HPLC purity > 99.5%.
(4-Ethylphenyl)(4-(2-(4-methoxyphenoxy)ethyl)piperazin-1-yl)methanone (15).
(4-Ethylphenyl)(piperazin-1-yl)methanone (44 mg, 0.20 mmol) and 1-(2-bromoethoxy)-4-methoxybenzene (46 mg, 0.20 mmol, 1.0 equiv) were reacted according to General Procedure B to afford the alkylated piperazine as a light yellow oil (40 mg, 0.11 mmol, 54% yield). Rf = 0.29 (EtOAc). 1H NMR (400 MHz, CDCl3) δ 1.21 (t, J = 7.6 Hz, 3H), 2.46–2.68 (complex, 4H), 2.64 (q, J = 7.6 Hz, 2H), 2.79 (t, J = 5.6 Hz, 2H), 3.39–3.54 (m, 2H), 3.67–3.84 (m, 2H), 3.73 (s, 3H), 4.04 (t, J = 5.6 Hz, 2H), 6.77–6.84 (complex, 4H), 7.18–7.22 (m, 2H), 7.28–7.32 (m, 2H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d: 15.4, 55.7, 114.6, 115.5, 127.2, 127.9; u: 28.7, 53.7, 53.9, 57.2, 66.5, 133.0, 146.1, 152.7, 153.9, 170.5; FTIR (neat): 1627, 1506, 1426 cm−1; HRMS (m/z): calcd for C22H29N2O3 [M + H]+ 369.2173; found 369.2196; HPLC purity = 99%.
(4-(2-(4-Methoxyphenoxy)ethyl)piperazin-1-yl)(pyridine-4-yl)methanone (16).
1-(2-(4-Methoxyphenoxy)ethyl)piperazine (108 mg, 0.46 mmol) and isonicotinoyl chloride hydrocholoride (99 mg, 0.56 mmol, 1.2 equiv) were reacted according to General Procedure D for 16 h to afford the acylated product as an orange-brown oil (95 mg, 0.28 mmol, 61% yield). Rf = 0.11 (EtOAc). 1H NMR (400 MHz, CDCl3) δ 2.51–2.56 (m, 2H), 2.65–2.71 (m, 2H), 2.83 (t, J = 5.5 Hz, 2H), 3.36–3.41 (m, 4H), 3.77 (s, 3H), 3.79–3.85 (m, 2H), 4.06 (t, J = 5.5 Hz, 2H), 6.81–6.87 (complex, 4H), 7.28 (d, J = 6.0 Hz, 2H), 8.70 (d, J = 6.0 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ 42.0, 47.4, 53.1, 53.7, 55.7, 57.2, 66.5, 114.6, 115.5, 121.2, 143.4, 150.3, 152.7, 154.0, 167.6; FTIR (neat): 1632, 1506, 1436 cm−1; HRMS (m/z): calcd for C19H24N3O3 [M + H]+ 342.1812; found 342.1827; HPLC purity > 99.5%.
(4-(2-(4-Methoxyphenoxy)ethyl)piperazin-1-yl)(5-methoxypyridin-2-yl)methanone (17).
5-Methoxypicolinic acid (19 mg, 0.12 mmol, 1.2 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as a light yellow oil (38 mg, 0.10 mmol, 93% yield). Rf = 0.13 (EtOAc). 1H NMR (400 MHz, DMSO-d6) δ 2.42–2.51 (m, 2H), 2.52–2.58 (m, 2H), 2.70 (t, J = 5.8 Hz, 2H), 3.48–3.53 (m, 2H), 3.60–3.66 (m, 2H), 3.69 (s, 3H), 3.87 (s, 3H), 4.02 (t, J = 5.7 Hz, 2H), 6.82–6.89 (complex, 4H), 7.49 (dd, J = 2.9, 8.7 Hz, 1H), 7.59 (d, J = 8.6 Hz, 1H), 8.27 (dd, J = 0.6, 2.9 Hz, 1H); 13C NMR (126 MHz, DMSO-d6) δ 41.8, 46.7, 52.9, 53.4, 55.4, 55.8, 56.6, 65.8, 114.6, 115.4, 121.2, 124.8, 135.7, 146.0, 152.4, 153.4, 155.9, 166.3; FTIR (neat): 1628, 1507, 1458 cm−1; HRMS (m/z): calcd for C20H26N3O4 [M + H]+ 372.1918; found 372.1926; HPLC purity > 99.5%.
(1H-Indol-5-yl)(4-(2-(4-methoxyphenoxy)ethyl)piperazin-1-yl)methanone (18).
1H-Indole-5-carboxylic acid (21 mg, 0.13 mmol, 1.2 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as a white solid (29 mg, 0.08 mmol, 69% yield). Rf = 0.17 (EtOAc); mp = 123–127 °C; 1H NMR (500 MHz, CDCl3) δ 2.50–2.69 (complex, 4H), 2.83 (t, J = 5.6 Hz, 2H), 3.50–3.85 (complex, 4H), 3.77 (s, 3H), 4.07 (t, J = 5.7 Hz, 2H), 6.57–6.60 (m, 1H), 6.81–6.85 (complex, 4H), 7.24–7.28 (m, 2H), 7.38 (d, J = 8.4 Hz, 1H), 7.73 (d, J = 1.6 Hz, 1H), 8.52 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 46.5, 53.8, 55.9, 57.4, 66.6, 103.3, 111.1, 114.8, 115.7, 120.4, 121.6, 125.5, 127.3, 127.5, 136.5, 152.9, 154.1, 171.9; FTIR (neat): 1599, 1506, 1431 cm−1; HRMS (m/z): calcd for C22H26N3O3 [M + H]+ 380.1969; found 380.1971; HPLC purity = 96%.
(1H-Indol-3-yl)(4-(2-(4-methoxyphenoxy)ethyl)piperazin-1-yl)methanone (19).
1H-Indole-3-carboxylic acid (21 mg, 0.13 mmol, 1.2 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as an off-white solid (6.4 mg, 0.017mmol, 15% yield). 1H NMR (500 MHz, CDCl3) δ 2.59–2.66 (complex, 4H), 2.84 (t, J = 5.6 Hz, 2H), 3.72–3.81 (complex, 4H), 3.76 (s, 3H), 4.08 (t, J = 5.6 Hz, 2H), 6.79–6.86 (complex, 4H), 7.19–7.25 (m, 2H), 7.40–7.43 (m, 1H), 7.55 (d, J = 2.7 Hz, 1H), 7.71 (d, J = 7.7 Hz, 1H), 8.47 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 46.4, 54.0, 55.9, 57.5, 66.7, 111.7, 112.4, 114.8, 115.7, 120.6, 121.3, 123.1, 125.5, 127.0, 135.6, 152.9, 154.1, 166.6; HRMS (m/z): calcd for C22H26N3O3 [M + H]+ 380.1969; found 380.1973; HPLC purity = 99%.
(1H-Indol-2-yl)(4-(2-(4-methoxyphenoxy)ethyl)piperazin-1-yl)methanone (20).
To a mixture of 1-(2-(4-methoxyphenoxy)ethyl)piperazine (146 mg, 0.618 mmol), indole-2-carboxylic acid (119 mg, 0.741 mmol, 1.2 equiv) and DMAP (8 mg, 0.062 mmol, 0.1 equiv) in THF (10 mL) was added diisopropylcarbodiimide (0.29 mL, 234 mg, 1.85 mmol, 3.0 equiv). The reaction was stirred at rt for 15 h and the solvents removed under vacuum. The residue was purified via silica gel chromatography to afford the acylated product as an off-white solid (172 mg, 0.453 mmol, 73% yield). Rf = 0.54 (MeOH (10%) and NH4OH (2%) in CH2Cl2); mp = 163–165 °C. 1H NMR (400 MHz, CDCl3) δ 2.68 (t, J = 5.1 Hz, 4H), 2.85 (t, J = 5.6 Hz, 2H), 3.77 (s, 3H), 3.90–4.05 (m, 4H), 4.10 (t, J = 5.5 Hz, 2H), 6.78 (dd, J = 1.0, 2.1 Hz, 1H), 6.82–6.89 (complex, 4H), 7.13 (ddd, J = 1.0, 7.0, 8.0 Hz, 1H), 7.24–7.32 (m, 1H), 7.43 (dd, J = 1.1, 8.3 Hz, 1H), 7.65 (dd, J = 1.2, 8.0 Hz, 1H), 9.65 (br s, 1H) 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d: 55.7, 105.2, 111.8, 114.7, 115.6, 120.5, 121.8, 124.4; u: 53.6, 57.3, 66.6, 127.4, 129.2, 135.7, 152.8, 154.0, 162.3; FTIR (neat): 3258, 1597, 1506, 1437 cm−1; HRMS (m/z): calcd for C22H26N3O3 [M + H]+ 380.1969; found 380.1995; HPLC purity = 96% (for LC/MS chromatogram and spectrum see the Supporting Information Figures S5 and S6).
(7-Methoxy-1H-indol-2-yl)(4-(2-(4-methoxyphenoxy)ethyl)piperazin-1-yl)methanone (21).
7-Methoxy-1H-indole-2-carboxylic acid (22 mg, 0.11 mmol, 1.1 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as a tan solid (31.6 mg, 0.077 mmol, 71% yield). 1H NMR (500 MHz, CDCl3) δ 2.67 (t, J = 5.1 Hz, 4H), 2.84 (t, J = 5.6 Hz, 2H), 3.77 (s, 3H), 3.89–4.01 (complex, 4H), 3.96 (s, 3H), 4.09 (t, J = 5.6 Hz, 2H), 6.69 (dd, J = 0.8, 7.7 Hz, 1H), 6.74 (d, J = 2.3 Hz, 1H), 6.82–6.87 (complex, 4H), 7.05 (t, J = 7.9 Hz, 1H), 7.21–7.25 (m, 1H), 9.22 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 41.1, 53.7, 55.6, 55.9, 57.4, 66.6, 103.6, 105.5, 114.2, 114.8, 115.7, 121.2, 126.8, 128.7, 129.1, 146.6, 152.9, 154.1, 162.3; HRMS (m/z): calcd for C23H28N3O4 [M + H]+ 410.2074; found 410.2073; HPLC purity = 99%.
(6-Methoxy-1H-indol-2-yl)(4-(2-(4-methoxyphenoxy)ethyl)piperazin-1-yl)methanone (22).
6-Methoxy-1H-indole-2-carboxylic acid (22 mg, 0.11 mmol, 1.1 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as an off-white solid (30 mg, 0.073 mmol, 67% yield). 1H NMR (500 MHz, CDCl3) δ 2.67 (t, J = 5.1 Hz, 4H), 2.84 (t, J = 5.6 Hz, 2H), 3.77 (s, 3H), 3.86 (s, 3H), 3.90–4.01 (complex, 4H), 4.09 (t, J = 5.6 Hz, 2H), 6.72 (dd, J = 0.9, 2.2 Hz, 1H), 6.81 (dd, J = 2.3, 8.7 Hz, 1H), 6.82–6.88 (complex, 5H), 7.44–7.62 (m, 1H), 9.10 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 53.7, 55.7, 55.9, 57.4, 66.7, 93.8, 105.8, 112.1, 114.8, 115.7, 122.0, 122.8, 128.3, 136.7, 152.9, 154.1, 158.4, 162.3; FTIR (neat): 1596, 1505 cm−1; HRMS (m/z): calcd for C23H28N3O4 [M + H]+ 410.2074; found 410.2074; HPLC purity = 98%.
(5-Methoxy-1H-indol-2-yl)(4-(2-(4-methoxyphenoxy)ethyl)piperazin-1-yl)methanone (23).
5-Methoxy-1H-indole-2-carboxylic acid (22 mg, 0.11 mmol, 1.1 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as a white solid (32 mg, 0.078 mmol, 72% yield). Rf = 0.20 (EtOAc); mp = 154–155 °C. 1H NMR (500 MHz, CDCl3) δ 2.67 (t, J = 5.1 Hz, 4H), 2.85 (t, J = 5.6 Hz, 2H), 3.77 (s, 3H), 3.84 (s, 3H), 3.89–4.04 (complex, 4H), 4.09 (t, J = 5.6 Hz, 2H), 6.70 (dd, J = 0.9, 2.2 Hz, 1H), 6.82–6.89 (complex, 4H), 6.95 (dd, J = 2.4, 8.9 Hz, 1H), 7.05 (d, J = 2.4 Hz, 1H), 7.32 (td, J = 0.8, 9.1 Hz, 1H), 9.22 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 41.1, 53.7, 55.9, 57.4, 66.7, 102.4, 105.1, 112.8, 114.8, 115.7, 115.8, 127.9, 129.8, 131.1, 152.9, 154.1, 154.7, 162.4; FTIR (neat): 1595, 1525, 1506, 1437 cm−1; HRMS (m/z): calcd for C23H28N3O4 [M + H]+ 410.2074; found 410.2076; HPLC purity > 99.5%.
(4-Methoxy-1H-indol-2-yl)(4-(2-(4-methoxyphenoxy)ethyl)piperazin-1-yl)methanone (24).
4-Methoxy-1H-indole-2-carboxylic acid (22 mg, 0.11 mmol, 1.1 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as a white solid (30 mg, 0.072 mmol, 67% yield). Rf = 0.64 (10% MeOH in CH2Cl2); mp = 127–129 °C. 1H NMR (500 MHz, CDCl3) δ 2.67 (t, J = 5.1 Hz, 4H), 2.84 (t, J = 5.6 Hz, 2H), 3.77 (s, 3H), 3.90–4.05 (complex, 4H), 3.96 (s, 3H), 4.09 (t, J = 5.6 Hz, 2H), 6.51 (dd, J = 0.6, 7.8 Hz, 1H), 6.90 (dd, J = 0.9, 2.3 Hz, 1H), 7.04 (ddd, J = 1.8, 8.0, 79.5 Hz, 1H), 7.20 (t, J = 7.9 Hz, 1H), 9.29 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 41.1, 53.7, 55.4, 55.9, 57.4, 66.7, 99.7, 102.8, 105.0, 114.8, 115.7, 118.8, 125.5, 128.1, 137.1, 152.9, 154.1, 154.2, 162.2; FTIR (neat): 1595, 1580, 1505, 1433 cm−1; HRMS (m/z): calcd for C23H28N3O4 [M + H]+ 410.2074; found 410.2076; HPLC purity > 99.5%.
4-(2-(4-Methoxyphenoxy)ethyl)piperazin-1-yl)(7-methyl-1H-indol-2-yl)methanone (25).
7-Methyl-1H-indole-2-carboxylic acid (20 mg, 0.11 mmol, 1.1 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as an off-white solid (28.0 mg, 0.071 mmol, 66% yield). Rf = 0.64 (10% MeOH in CH2Cl2); mp = 134–138 °C. 1H NMR (400 MHz, CDCl3) δ 2.50 (s, 3H), 2.68 (t, J = 5.1 Hz, 4H), 2.85 (t, J = 5.6 Hz, 2H), 3.77 (s, 3H), 3.87–4.05 (complex, 4H), 4.10 (t, J = 5.5 Hz, 2H), 6.78 (d, J = 2.1 Hz, 1H), 6.82–6.91 (complex, 4H), 7.01–7.11 (m, 2H), 7.49 (dd, J = 2.2, 6.7 Hz, 1H), 9.12 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 16.9, 41.1, 53.7, 55.4, 57.2, 66.0, 101.2, 105.9, 106.7, 106.8, 119.6, 121.0, 121.2, 124.8, 127.1, 129.0, 130.1, 135.5, 160.0, 161.0, 162.5; FTIR (neat): 1598, 1534, 1505, 1436 cm−1; HRMS (m/z): calcd for C23H28N3O3 [M + H]+ 394.2125; found 394.2124; HPLC purity > 99.5%.
4-(2-(4-Methoxyphenoxy)ethyl)piperazin-1-yl)(6-methyl-1H-indol-2-yl)methanone (26).
6-Methyl-1H-indole-2-carboxylic acid (19 mg, 0.11 mmol, 1.0 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as a white solid (26.7 mg, 0.068 mmol, 63% yield). Rf = 0.60 (10% MeOH in CH2Cl2); mp = 130–131 °C. 1H NMR (500 MHz, CDCl3) δ 2.46 (s, 3H), 2.67 (t, J = 5.1 Hz, 4H), 2.85 (t, J = 5.6 Hz, 2H), 3.77 (s, 3H), 3.90–4.03 (complex, 4H), 4.09 (t, J = 5.6 Hz, 2H), 6.73 (dd, J = 0.9, 2.1 Hz, 1H), 6.81–6.89 (complex, 4H), 6.97 (ddd, J = 0.6, 1.4, 8.2 Hz, 1H), 7.21 (m, 1H), 7.52 (dd, J = 0.9, 8.2 Hz, 1H), 9.08 (br s, 1H); 13C NMR (101 MHz, CDCl3) δ 22.1, 53.7, 55.9, 57.4, 66.7, 105.4, 111.6, 114.8, 115.7, 121.6, 122.8, 125.5, 128.8, 134.6, 136.2, 152.9, 154.1, 162.5; FTIR (neat): 1596, 1523, 1507, 1438 cm−1; HRMS (m/z): calcd for C23H28N3O4 [M + H]+ 394.2125; found 394.2128; HPLC purity = 99%.
4-(2-(4-Methoxyphenoxy)ethyl)piperazin-1-yl)(5-methyl-1H-indol-2-yl)methanone (27).
5-Methyl-1H-indole-2-carboxylic acid (20 mg, 0.11 mmol, 1.1 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as a white solid (29 mg, 0.074 mmol, 68% yield). Rf = 0.60 (10% MeOH in CH2Cl2); mp = 175–179 °C. 1H NMR (500 MHz, CDCl3) δ 2.44 (s, 3H), 2.67 (t, J = 5.1 Hz, 4H), 2.85 (t, J = 5.6 Hz, 2H), 3.77 (s, 3H), 3.91–4.02 (complex, 4H), 4.09 (t, J = 5.5 Hz, 2H), 6.69 (dd, J = 0.9, 2.2 Hz, 1H), 6.81–6.89 (complex, 4H), 7.11 (dd, J = 1.3, 8.4 Hz, 1H), 7.31 (dt, J = 0.8, 8.4 Hz, 1H), 7.41 (dd, J = 0.8, 1.7 Hz, 1H), 9.10 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 53.7, 55.7, 55.9, 57.4, 66.7, 93.8, 105.8, 112.1, 114.8, 115.7, 122.0, 122.8, 128.3, 136.7, 152.9, 154.1, 158.4, 162.3; FTIR (neat): 1592, 1536, 1506, 1434 cm−1; HRMS (m/z): calcd for C23H28N3O4 [M + H]+ 394.2125; found 394.2127; HPLC purity = 99%.
4-(2-(4-Methoxyphenoxy)ethyl)piperazin-1-yl)(4-methyl-1H-indol-2-yl)methanone (28).
4-Methyl-1H-indole-2-carboxylic acid (21 mg, 0.12 mmol, 1.1 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as an off-white solid (31.3 mg, 0.080 mmol, 74% yield). Rf = 0.68 (10% MeOH in CH2Cl2); mp = 134–136 °C. 1H NMR (500 MHz, CDCl3) δ 2.56 (s, 2H), 2.69 (t, J = 5.1 Hz, 4H), 2.85 (t, J = 5.5 Hz, 2H), 3.77 (s, 3H), 3.91–4.06 (complex, 4H), 4.10 (t, J = 5.5 Hz, 2H), 6.78 (dd, J = 1.0, 2.2 Hz, 1H), 6.81–6.89 (complex, 4H), 6.93 (td, J = 0.9, 7.1 Hz, 1H), 7.19 (dd, J = 7.0, 8.3 Hz, 1H), 7.24–7.27 (m, 1H), 9.20 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 18.9, 41.1, 53.7, 55.9, 57.4, 66.7, 103.9, 109.4, 114.8, 115.7, 120.7, 124.7, 127.7, 128.8, 131.6, 135.5, 152.9, 154.1, 162.4; FTIR (neat): 1597, 1506, 1456, 1437 cm−1; HRMS (m/z): calcd for C23H28N3O4 [M + H]+ 394.2125; found 394.2125; HPLC purity = 99%.
(7-Chloro-1H-indol-2-yl)(4-(2-(4-methoxyphenoxy)ethyl)piperazin-1-yl)methanone (29).
7-Chloro-1H-indole-2-carboxylic acid (23 mg, 0.12 mmol, 1.1 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as a yellow solid (31.8 mg, 0.077 mmol, 71% yield). Rf = 0.68 (10% MeOH in CH2Cl2); mp = 125–130 °C. 1H NMR (400 MHz, CDCl3) δ 2.68 (t, J = 5.1 Hz, 4H), 2.85 (t, J = 5.5 Hz, 2H), 3.77 (s, 3H), 3.87–4.04 (complex, 4H), 4.09 (t, J = 5.5 Hz, 2H), 6.79 (d, J = 2.2 Hz, 1H), 6.83–6.90 (m, 4H), 7.08 (t, J = 7.8 Hz, 1H), 7.28 (dd, J = 0.9, 7.6 Hz, 1H), 7.54 (td, J = 0.9, 8.0 Hz, 1H), 9.23 (br s, 1H) 13C NMR (126 MHz, CDCl3) δ 41.1, 53.7, 55.9, 57.4, 66.7, 105.9, 114.8, 115.7, 117.3, 120.6, 121.5, 123.7, 128.8, 130.1, 133.2, 152.9, 154.1, 161.8; FTIR (neat): 1604, 1505, 1435 cm−1; HRMS (m/z): calcd for C22H25ClN3O3 [M + H]+ 414.1579; found 414.1578; HPLC purity > 99.5%.
(6-Chloro-1H-indol-2-yl)(4-(2-(4-methoxyphenoxy)ethyl)piperazin-1-yl)methanone (30).
6-Chloro-1H-indole-2-carboxylic acid (24 mg, 0.12 mmol, 1.1 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as a white solid (31.2 mg, 0.075 mmol, 70% yield). Rf = 0.61 (10% MeOH in CH2Cl2); mp = 165–168 °C. 1H NMR (500 MHz, CDCl3) δ 2.69 (t, J = 5.1 Hz, 4H), 2.85 (t, J = 5.5 Hz, 2H), 3.77 (s, 3H), 3.89–4.04 (m, 4H), 4.10 (t, J = 5.5 Hz, 2H), 6.75 (dd, J = 1.0, 2.1 Hz, 1H), 6.82–6.88 (complex, 4H), 7.10 (dd, J = 1.8, 8.5 Hz, 1H), 7.40–7.44 (m, 1H), 7.55 (d, J = 8.7 Hz, 1H), 9.46 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 53.7 (× 2 C), 55.9, 57.4, 66.7, 105.4, 111.7, 114.8, 115.7, 121.7, 122.9, 126.1, 130.1, 130.4, 136.0, 152.9, 154.1, 162.0; FTIR (neat): 1591, 1506, 1456, 1437 cm−1; HRMS (m/z): calcd for C22H25ClN3O3 [M + H]+ 414.1579; found 414.1575; HPLC purity = 95%.
(5-Chloro-1H-indol-2-yl)(4-(2-(4-methoxyphenoxy)ethyl)piperazin-1-yl)methanone (31).
5-Chloro-1H-indole-2-carboxylic acid (22 mg, 0.11 mmol, 1.1 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as an off-white solid (30.6 mg, 0.074 mmol, 68% yield). 1H NMR (400 MHz, CDCl3) δ 2.68 (t, J = 5.1 Hz, 4H), 2.85 (t, J = 5.5 Hz, 2H), 3.77 (s, 3H), 3.88–4.02 (m, 4H), 4.09 (t, J = 5.5 Hz, 2H), 6.70 (dd, J = 0.9, 2.2 Hz, 1H), 6.81–6.88 (complex, 4H), 7.23 (dd, J = 2.0, 8.8 Hz, 1H), 7.35 (d, J = 8.7 Hz, 1H), 7.60 (d, J = 2.0 Hz, 1H), 9.41 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 53.7 (× 2 C), 55.9, 57.4, 66.7, 104.7, 113.0, 114.8, 115.7, 121.2, 125.0, 126.3, 128.5, 130.6, 134.1, 152.9, 154.1, 162.0; HRMS (m/z): calcd for C22H25ClN3O3 [M + H]+ 414.1579; found 414.1578; HPLC purity = 99.5%.
(4-Chloro-1H-indol-2-yl)(4-(2-(4-methoxyphenoxy)ethyl)piperazin-1-yl)methanone (32).
4-Chloro-1H-indole-2-carboxylic acid (21 mg, 0.11 mmol, 1.1 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as a white solid (29.9 mg, 0.072 mmol, 67% yield). Rf = 0.64 (10% MeOH in CH2Cl2); mp = 139–141 °C. 1H NMR (400 MHz, DMSO-d6) δ 2.57 (t, J = 5.1 Hz, 4H), 2.73 (t, J = 5.7 Hz, 2H), 3.69 (s, 3H), 3.71–3.82 (m, 4H), 4.04 (t, J = 5.7 Hz, 2H), 6.76 (s, 1H), 6.82–6.91 (complex, 4H), 7.13 (dd, J = 1.0, 7.6 Hz, 1H), 7.16–7.23 (m, 1H), 7.40 (d, J = 8.0 Hz, 1H), 11.97 (s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 53.1 (× 2 C), 55.4, 56.6, 65.9, 101.6, 111.3, 114.6, 115.4, 119.4, 124.0, 125.2, 125.4, 131.0, 136.6, 152.5, 153.4, 161.3; FTIR (neat): 1599, 1505, 1458, 1437 cm−1; HRMS (m/z): calcd for C22H25ClN3O3 [M + H]+ 414.1579; found 414.1581; HPLC purity > 99.5%.
(4-(2-(4-Methoxyphenoxy)ethyl)piperazin-1-yl)(1-methyl-1H-indol-2-yl)methanone (33).
1-(2-(4-Methoxyphenoxy)ethyl)piperazine (109 mg, 0.461 mmol) and 1-methyl-1H-indole-2-carbonyl chloride (107 mg, 0.554 mmol, 1.2 equiv) were reacted according to General Procedure D to afford the acylated piperazine product as a tan oil (117 mg, 0.297 mmol, 65% yield). Rf = 0.81 (10% MeOH in CH2Cl2c). 1H NMR (500 MHz, CDCl3) δ 2.53–2.72 (m, 4H), 2.84 (t, J = 5.5 Hz, 2H), 3.77 (s, 3H), 3.84 (s, 3H), 3.74–3.87 (m, 4H), 4.08 (t, J = 5.5 Hz, 2H), 6.60 (d, J = 0.9 Hz, 1H), 6.80–6.88 (complex, 4H), 7.14 (ddd, J = 1.0, 6.9, 8.0 Hz, 1H), 7.30 (ddd, J = 1.2, 6.9, 8.3 Hz, 1H), 7.36 (td, J = 0.9, 8.4 Hz, 1H), 7.62 (d, J = 8.0 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 31.1, 53.7 (× 2 C), 55.7, 57.2, 66.5, 103.6, 109.8, 114.7, 115.6, 120.3, 121.5, 123.3, 126.4, 131.6, 137.9, 152.7, 154.0, 163.0; FTIR (neat): 1624, 1506, 1462, 1438 cm−1; HRMS (m/z): calcd for C23H28N3O3 [M + H]+ 394.2125; found 394.2146; HPLC purity = 98.2%.
(4-(2-(4-Methoxyphenoxy)ethyl)piperazin-1-yl)(3-methyl-1H-indol-2-yl)methanone (34).
3-Methyl-1H-indole-2-carboxylic acid (19 mg, 0.11 mmol, 1.0 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as a light yellow solid (20.4 mg, 0.052 mmol, 48% yield). Rf = 0.73 (10% MeOH in CH2Cl2); mp = 105–123 °C. 1H NMR (400 MHz, DMSO-d6) δ 2.26 (s, 3H), 2.48–2.55 (m, 4H), 2.72 (t, J = 5.7 Hz, 2H), 3.52–3.61 (m, 4H), 3.69 (s, 3H), 4.02 (t, J = 5.7 Hz, 2H), 6.84–6.89 (complex, 4H), 7.03 (ddd, J = 1.0, 6.9, 7.9 Hz, 1H), 7.16 (ddd, J = 1.2, 7.0, 8.2 Hz, 1H), 7.33 (d, J = 8.2 Hz, 1H), 7.51–7.58 (m, 1H), 11.19 (s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 9.3, 53.2 (× 2 C), 55.3, 56.5, 65.8, 109.9, 111.6, 114.5, 115.3, 118.9, 119.2, 122.8, 127.4, 127.7, 135.6, 152.4, 153.3, 163.3; FTIR (neat): 1603, 1506, 1451, 1440 cm−1; HRMS (m/z): calcd for C23H28N3O3 [M + H]+ 394.2125; found 394.2126; HPLC purity = 99.2%.
(4-(2-(4-Methoxyphenoxy)ethyl)piperazin-1-yl)(1H-pyrrolo[2,3-b]pyridin-2-yl)methanone (35).
1H-Pyrrolo[2,3-b]pyridine-2-carboxylic acid (20 mg, 0.12 mmol, 1.1 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as a white solid (27.3 mg, 0.072 mmol, 66% yield). Rf = 0.71 (10% MeOH in CH2Cl2); mp = 167–176 °C; 1H NMR (500 MHz, CDCl3) δ 2.69 (t, J = 5.1 Hz, 4H), 2.85 (t, J = 5.5 Hz, 2H), 3.77 (s, 3H), 3.90–4.03 (m, 4H), 4.09 (t, J = 5.5 Hz, 2H), 6.72 (d, J = 1.4 Hz, 1H), 6.81–6.92 (complex, 4H), 7.13 (dd, J = 4.7, 7.9 Hz, 1H), 7.98 (dd, J = 1.6, 7.9 Hz, 1H), 8.50 (dd, J = 1.6, 4.7 Hz, 1H), 10.73 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 53.6, 55.7, 57.2, 66.5, 103.4, 114.7, 115.6, 116.9, 119.8, 129.9, 130.3, 146.0, 147.7, 152.8, 154.0, 162.0; FTIR (neat): 1618, 1579, 1506, 1436 cm−1; HRMS (m/z): calcd for C21H25N4O3 [M + H]+ 381.1921; found 381.1921; HPLC purity > 99.5%.
(4-(2-(4-Methoxyphenoxy)ethyl)piperazin-1-yl)(1H-pyrrolo[2,3-c]pyridin-2-yl)methanone (36).
1H-Pyrrolo[2,3-c]pyridine-2-carboxylic acid (20 mg, 0.12 mmol, 1.1 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as a white solid (30.5 mg, 0.080 mmol, 74% yield). Rf = 0.28 (10% MeOH in CH2Cl2); mp = 163–165 °C. 1H NMR (500 MHz, CDCl3) δ 2.69–2.74 (m, 4H), 2.86 (t, J = 5.4 Hz, 2H), 3.77 (s, 3H), 3.90–4.03 (m, 4H), 4.10 (t, J = 5.5 Hz, 2H), 6.70–6.97 (complex, 5H), 7.34 (d, J = 5.8 Hz, 1H), 8.37 (d, J = 5.8 Hz, 1H), 8.99 (d, J = 1.1 Hz, 1H), 10.02 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 53.6, 55.7, 57.2, 66.5, 104.3, 106.8, 114.7, 115.6, 124.6, 130.3, 139.1, 142.7, 145.5, 152.7, 154.0, 161.7; FTIR (neat): 1609, 1578, 1506 cm−1; HRMS (m/z): calcd for C21H25N4O3 [M + H]+ 381.1921; found 381.1926; HPLC purity = 99.5%.
(4-(2-(4-Methoxyphenoxy)ethyl)piperazin-1-yl)(1H-pyrrolo[3,2-c]pyridin-2-yl)methanone (37).
1H-Pyrrolo[3,2-c]pyridine-2-carboxylic acid (23 mg, 0.14 mmol, 1.3 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as a light yellow solid (30.3 mg, 0.080 mmol, 74% yield). Rf = 0.28 (10% MeOH in CH2Cl2); mp = 160–164 °C. 1H NMR (400 MHz, DMSO-d6) δ 2.57 (t, J = 5.1 Hz, 4H), 2.74 (t, J = 5.7 Hz, 2H), 3.69 (s, 3H), 3.70–3.83 (m, 4H), 4.04 (t, J = 5.7 Hz, 2H), 6.82–6.91 (complex, 4H), 6.92–7.01 (m, 1H), 7.36 (d, J = 5.8 Hz, 1H), 8.22 (d, J = 5.8 Hz, 1H), 8.90 (d, J = 1.2 Hz, 1H), 11.99 (s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 53.1, 55.3, 56.5, 65.8, 103.1, 107.0, 114.5, 115.3, 124.0, 131.0, 138.9, 141.5, 144.7, 152.4, 153.3, 161.3; FTIR (neat): 1608, 1575, 1505 cm−1; HRMS (m/z): calcd for C21H25N4O3 [M + H]+ 381.1921; found 381.1926; HPLC purity = 99.7%.
(4-(2-(4-Methoxyphenoxy)ethyl)piperazin-1-yl)(1H-pyrrolo[3,2-b]pyridin-2-yl)methanone (38).
1H-Pyrrolo[3,2-b]pyridine-2-carboxylic acid (19 mg, 0.12 mmol, 1.1 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as a white solid (26.9 mg, 0.071 mmol, 65% yield). Rf = 0.34 (10% MeOH in CH2Cl2); mp = 165–166 °C. 1H NMR (500 MHz, CDCl3) δ 2.70 (t, J = 5.1 Hz, 4H), 2.86 (t, J = 5.5 Hz, 2H), 3.77 (s, 3H), 3.85–4.14 (m, 4H), 4.10 (t, J = 5.6 Hz, 2H), 6.75–6.92 (complex, 4H), 6.98 (d, J = 1.1 Hz, 1H), 7.20 (dd, J = 4.6, 8.3 Hz, 1H), 7.74 (d, J = 8.2 Hz, 1H), 8.53 (dd, J = 1.4, 4.6 Hz, 1H), 9.69 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 53.6, 55.7, 57.2, 66.5, 105.7, 114.7, 115.6, 119.1, 119.2, 128.8, 131.7, 144.9, 145.3, 152.7, 154.0, 161.9; FTIR (neat): 1601, 1505, 1437, 1407 cm−1; HRMS (m/z): calcd for C21H25N4O3 [M + H]+ 381.1921; found 381.1929; HPLC purity = 99.0%.
(1H-Benzo[d]imidazol-2-yl)(4-(2-(4-methoxyphenoxy)ethyl)piperazin-1-yl)methanone (39).
1H-Benzo[d]imidazole-2-carboxylic acid (20 mg, 0.12 mmol, 1.1 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (26 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as a white solid (29.3 mg, 0.077 mmol, 71% yield). Rf = 0.78 (10% MeOH in CH2Cl2); mp = 139–141 °C. 1H NMR (500 MHz, CDCl3) δ 2.70–2.81 (m, 4H), 2.86 (t, J = 5.6 Hz, 2H), 3.77 (s, 3H), 3.94 (t, J = 5.0 Hz, 2H), 4.11 (t, J = 5.6 Hz, 2H), 4.82 (t, J = 5.2 Hz, 2H), 6.75–6.95 (complex, 4H), 7.30–7.43 (m, 2H), 7.48–7.58 (m, 1H), 7.82 (d, J = 8.0 Hz, 1H), 11.00 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 43.2, 46.7, 53.4, 54.2, 55.7, 57.2, 66.5, 111.7, 114.6, 115.6, 121.0, 123.1, 125.1, 132.5, 143.2, 145.3, 152.8, 154.0, 158.2; FTIR (neat): 1608, 1506, 1440, 1406 cm−1; HRMS (m/z): calcd for C21H25N4O3 [M + H]+ 381.1921; found 381.1919; HPLC purity = 99.8%.
Benzofuran-2-yl(4-(2-(4-methoxyphenoxy)ethyl)piperazin-1-yl)methanone (40).
Benzofuran-2-carboxylic acid (21.0 mg, 0.13 mmol, 1.2 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (25.7 mg, 0.11 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as a white solid (25.3 mg, 0.067 mmol, 61% yield). Rf = 0.68 (10% MeOH in CH2Cl2); mp = 113–116 °C. 1H NMR (500 MHz, CDCl3) δ 2.70–2.81 (m, 4H), 2.86 (t, J = 5.6 Hz, 2H), 3.77 (s, 3H), 3.94 (t, J = 5.0 Hz, 2H), 4.11 (t, J = 5.6 Hz, 2H), 4.82 (t, J = 5.2 Hz, 2H), 6.75–6.95 (complex, 4H), 7.30–7.43 (m, 2H), 7.48–7.58 (m, 1H), 7.82 (d, J = 8.0 Hz, 1H), 11.00 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 53.4, 54.0, 55.7, 57.2, 66.5, 111.9, 112.0, 114.6, 115.6, 122.2, 123.6, 126.4, 127.0, 149.0, 152.8, 154.0, 154.6, 159.7; FTIR (neat): 1630, 1561, 1506, 1433 cm−1; HRMS (m/z): calcd for C22H25N2O4 [M + H]+ 381.1809; found 381.1814; HPLC purity = 98.6%.
Benzo[b]thiophen-2-yl(4-(2-(4-methoxyphenoxy)ethyl)piperazin-1-yl)methanone (41).
Benzo[b]thiophen-2-carboxylic acid (25 mg, 0.15 mmol, 1.2 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (30 mg, 0.13 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as a white solid (27.0 mg, 0.077 mmol, 60% yield). Rf = 0.68 (10% MeOH in CH2Cl2); mp = 57–59 °C. 1H NMR (500 MHz, CDCl3) δ 2.62–2.69 (m, 4H), 2.84 (t, J = 5.5 Hz, 2H), 3.77 (s, 3H), 3.78–3.88 (m, 4H), 4.08 (t, J = 5.6 Hz, 2H), 6.80–6.89 (m, 4H), 7.37–7.42 (m, 2H), 7.48 (d, J = 0.8 Hz, 1H), 7.79–7.83 (m, 1H), 7.84–7.87 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 55.7, 57.2, 66.5, 114.6, 115.6, 122.4, 124.6, 124.8, 125.2, 125.8, 136.6, 138.6, 140.2, 152.7, 154.0, 163.8; FTIR (neat): 1619, 1505, 1458, 1437 cm−1; HRMS (m/z): calcd. for C22H25N2O3S [M + H]+ 397.1580; found 397.1587; HPLC purity = 99.5%.
Cyclohexyl(4-(2-(4-methoxyphenoxy)ethyl)piperazin-1-yl)methanone (42).
Cyclohexanecarbonyl chloride (75 μL, 0.56 mmol, 1.2 equiv) and 1-(2-(4-methoxyphenoxy)ethyl)piperazine (110 mg, 0.47 mmol) were reacted according to General Procedure D to afford the acylated piperazine product as a yellow solid (101.3 mg, 0.293 mmol, 63% yield); Rf = 0.58 (10% MeOH in CH2Cl2); mp = 54–58 °C. 1H NMR (500 MHz, CDCl3) δ 1.17–1.32 (m, 4H), 1.44–1.59 (m, 2H), 1.69–1.87 (m, 4H), 2.39–2.61 (m, 5H), 2.80 (t, J = 5.6 Hz, 2H), 3.58 (dt, J = 5.2, 60.8 Hz, 4H), 3.77 (s, 3H), 4.07 (t, J = 5.6 Hz, 2H), 6.78–6.88 (m, 4H); 13C NMR (126 MHz, CDCl3) δ 25.8, 25.9, 29.4, 40.4, 41.5, 45.3, 53.4, 54.1, 55.7, 57.3, 66.5, 114.6, 115.6, 152.8, 154.0, 174.5; FTIR (neat): 1634, 1506, 1433 cm−1; HRMS (m/z): calcd. for C20H31N2O3 [M + H]+ 347.2329; found 347.2349; HPLC purity = 99.2%.
-(4-(2-(4-Methoxyphenoxy)ethyl)piperazin-1-yl)ethan-1-one (43).
Acetyl chloride (32.8 μL, 0.461 mmol, 1.0 equiv), 1-(2-(4-methoxyphenoxy)ethyl)piperazine (109 mg, 0.461 mmol) and triethylamine (100 μL, 0.717 mmol, 1.5 equiv) were reacted according to General Procedure D to afford the acylated piperazine product as a brown oil (79.6 mg, 0.287 mmol, 62% yield). Rf = 0.57 (10% MeOH in CH2Cl2). 1H NMR (500 MHz, CDCl3) δ 2.09 (s, 3H), 2.54 (t, J = 5.2 Hz, 2H), 2.58 (t, J = 5.0 Hz, 2H), 2.81 (t, J = 5.6 Hz, 2H), 3.49 (t, J = 5.3 Hz, 2H), 3.64 (t, J = 4.6 Hz, 2H), 3.77 (s, 3H), 4.06 (t, J = 5.6 Hz, 2H), 6.77–6.87 (m, 4H); 13C NMR (126 MHz, CDCl3) δ 21.3, 41.4, 46.3, 53.2, 53.7, 55.7, 57.2, 66.5, 114.6, 115.6, 152.8, 154.0, 168.9; FTIR (neat): 1637, 1505, 1461, 1426 cm−1; HRMS (m/z): calcd. for C15H23N2O3 [M + H]+ 279.1703; found 279.1716; HPLC purity > 99.5%.
1-(2-(4-Methoxyphenoxy)ethyl)-4-((4-methoxyphenyl)sulfonyl)piperazine (44).
To a solution of 1-(2-(4-methoxyphenoxy)ethyl)piperazine (76 mg, 0.32 mmol) and triethylamine (90 μL, 0.64 mmol, 2.0 equiv) in toluene (25 mL) at rt was added 4-methoxybenzene-1-sulfonyl chloride (67 mg, 0.32 mmol, 1.0 equiv). The reaction was capped and stirred for 19 h at rt. The reaction solvents were removed in vacuo and the residue partitioned between saturated, aqueous NaHCO3 (40 mL) and CH2Cl2 (2 × 20 mL). The combined organic phases were dried over Na2SO4 and purified by silica gel chromatography to afford the sulfonamide product as an off-white solid (71.4 mg, 0.176 mmol, 55% yield). 1H NMR (400 MHz, CDCl3) δ 2.65 (t, J = 5.0 Hz, 4H), 2.76 (t, J = 5.5 Hz, 2H), 3.03 (t, J = 4.9 Hz, 2H), 3.75 (s, 3H), 3.86 (s, 3H), 3.98 (t, J = 5.5 Hz, 2H), 6.77–6.81 (m, 4H), 6.98 (d, J = 8.9 Hz, 2H), 7.68 (d, J = 9.0 Hz, 2H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d: 55.7, 55.8, 114.3, 114.7, 115.7, 130.1; u: 46.1, 52.7, 57.0, 66.6, 127.0, 152.8, 154.1; HRMS (m/z): calcd for C20H27N2O5S [M + H]+ 407.1635; found 407.1635; HPLC purity > 99.5%.
1-(2-(4-Methoxyphenoxy)ethyl)-4-(4-methoxyphenyl)piperazine (45).
To a solution of 1-(4-methoxyphenyl)piperazine (39 mg, 0.20 mmol) and 1-(2-bromoethoxy)-4-methoxybenzene (52 mg, 0.22 mmol, 1.1 equiv) in MeCN (10 mL) was added triethylamine (40 μL, 0.29 mmol, 1.4 equiv) and the reaction stirred at 60 °C for 18 h. The reaction was filtered and the solids washed with CH2Cl2 (2 × 5 mL). The combined filtrates were evaporated and the residue purified by silica gel chromatography to afford the alkylated piperazine product as a tan solid (47.5 mg, 0.139 mmol, 68% yield). Rf = 0.45 (5% MeOH/CH2Cl2); mp = 156–159 °C. 1H NMR (500 MHz, CDCl3) δ 2.72–2.78 (m, 4H), 2.85 (t, J = 5.8 Hz, 2H), 3.08–3.16 (m, 4H), 3.76 (s, 3H), 3.78 (s, 3H), 4.10 (t, J = 5.8 Hz, 2H), 6.81–6.88 (m, 6H), 6.89–6.92 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 50.6, 53.8, 55.6, 55.7, 57.4, 66.6, 114.4, 114.6, 115.6, 118.2, 145.7, 152.9, 153.8, 153.9; FTIR (neat): 1508, 1457, 1441 cm−1; HRMS (m/z): calcd. for C20H27N2O3 [M + H]+ 343.2016; found 343.2032; HPLC purity = 97.1%.
(4-(1-(4-Methoxyphenoxy)propan-2-yl)piperazin-1-yl)(4-methoxyphenyl)methanone (46).
A solution of (4-methoxyphenyl)(piperazin-1-yl)methanone2,2,2-trifluoroacetate (78 mg, 0.23 mmol, 1.0 equiv), 1-(4-methoxyphenoxy)propan-2-one (44 mg, 0.24 mmol, 1.0 equiv), acetic acid (4 μL, 0.07 mmol, 0.3 equiv) and sodium triacetoxyborohydride (76 mg, 0.36 mmol, 1.5 equiv) in dichloroethane (2 mL) was heated at 50°C for 7 days and cooled to rt. The reaction was diluted with CH2Cl2 (4 mL) and washed with saturated aqueous NaHCO3 (2 × 2 mL) and water (1 × 2 mL). The combined organic layers were dried over MgSO4, concentrated in vacuo and purified by silica gel chromatography to afford the reductive amination product as a tan solid (51.1 mg, 0.132 mmol, 57% yield). 1H NMR (400 MHz, CDCl3) δ 1.17 (d, J = 6.8 Hz, 3H), 2.54–2.75 (m, 4H), 3.03 (dtd, J = 5.6, 6.8, 12.4 Hz, 1H), 3.41–3.75 (br m, 4H), 3.77 (s, 3H), 3.83 (s, 3H), 3.83–3.86 (m, 1H), 4.00 (dd, J = 5.6, 9.6 Hz, 1H), 6.83 (m, 4H), 6.87–6.95 (m, 2H), 7.35–7.42 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 13.3, 55.3, 55.7, 58.4, 70.6, 113.7, 114.6, 115.5, 127.9, 129.2, 152.9, 153.9, 160.7, 170.2; HRMS (m/z): calcd. for C22H29N2O4 [M + H]+ 385.2122; found 385.2135; HPLC purity = 99.6%.
(4-(2-(4-methoxyphenoxy)propyl)piperazin-1-yl)(4-methoxyphenyl)methanone (47).
4-Methoxybenzoic acid (22 mg, 0.14 mmol, 1.2 equiv) and 1-(2-(4-methoxyphenoxy)propyl)piperazine 114d (29 mg, 0.12 mmol, 1.0 equiv) were reacted according to General Procedure E to afford the acylated piperazine product 47 as a yellow oil (30.3 mg, 0.0789 mmol, 68% yield). Rf = 0.41 (5% MeOH/CH2Cl2). 1H NMR (500 MHz, CDCl3) δ 1.28 (d, J = 6.1 Hz, 3H), 2.50 (dd, J = 4.5, 13.3 Hz, 1H), 2.53–2.63 (m, 4H), 2.71 (dd, J = 6.8, 13.3 Hz, 1H), 3.28–3.74 (m, 4H), 3.77 (s, 3H), 3.83 (s, 3H), 4.42 (td, J = 4.6, 6.5 Hz, 1H), 6.83 (qt, J = 2.8, 9.4 Hz, 4H), 6.88–6.92 (m, 2H), 7.34–7.39 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 18.6, 55.3, 55.7, 63.5, 73.3, 113.7, 114.6, 117.6, 127.9, 129.1, 151.7, 154.1, 160.7, 170.2; FTIR (neat): 1625, 1606, 1503, 1427 cm−1; HRMS (m/z): calcd. for C22H29N2O4 [M + H]+ 385.2122; found 385.2121; HPLC purity = 98.4%.
(4-(2-(4-Methoxyphenoxy)ethyl)-3-methylpiperazin-1-yl)(4-methoxyphenyl)methanone (48).
(4-Methoxyphenyl)(3-methylpiperazin-1-yl)methanone (45.0 mg, 0.19 mmol) and 1-(2-bromoethoxy)-4-methoxybenzene (48.8 mg, 0.21 mmol, 1.1 equiv) were reacted according to General Procedure A then purified by preparative, reverse-phase HPLC to afford the alkylated piperazine as a sticky, golden solid (20.1 mg, 0.052 mmol, 27% yield). Rf = 0.38 (5% MeOH/CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 1.01–1.19 (m, 3H), 2.44–2.64 (m, 4H), 2.78 (dt, J = 5.8, 13.9 Hz, 1H), 2.90–3.02 (m, 2H), 3.10 (dt, J = 5.9, 12.7 Hz, 1H), 3.32 (ddd, J = 3.1, 9.9, 13.0 Hz, 1H), 3.76 (s, 3H), 3.83 (s, 3H), 4.03 (t, J = 5.9 Hz, 2H), 6.80–6.83 (m, 4H), 6.90 (d, J = 8.7 Hz, 2H), 7.37 (d, J = 8.7 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 52.4, 55.1, 55.3, 55.7, 66.4, 113.7, 114.6, 115.5, 127.8, 129.1, 152.8, 153.9, 160.7, 170.2; FTIR (neat): 1627, 1608, 1507, 1429 cm−1; HRMS (m/z): calcd for C22H29N2O4 [M + H]+ 385.2122; found 385.2140; HPLC purity = 95.1%.
(4-(2-(4-Methoxyphenoxy)ethyl)-2-methylpiperazin-1-yl)(4-methoxyphenyl)methanone (49).
A mixture of 1-(2-bromoethoxy)-4-methoxybenzene (51.0 mg, 0.221 mmol, 1.1 equiv), (4-methoxyphenyl)(2-methylpiperazin-1-yl)methanone 111f (47.0 mg, 0.201 mmol, 1.0 equiv) and potassium carbonate (56.0 mg, 0.405 mmol, 2.0 equiv) in DMF (1 mL) was heated at 100 °C for 5 h and cooled to rt. The reaction was diluted with water (10 mL) and extracted with CH2Cl2 (4 × 2 mL). The combined organic layers were dried over Na2SO4, concentrated in vacuo and purified by silica gel chromatography to afford the aryl ether product as a light yellow oil (28.4 mg, 0.0741 mmol, 37% yield). Rf = 0.48 (5% MeOH/CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 1.35 (d, J = 6.8 Hz, 3H), 2.14–2.24 (m, 1H), 2.34 (dd, J = 3.8, 11.3 Hz, 1H), 2.67–2.84 (m, 3H), 2.90 (d, J = 11.3 Hz, 1H), 3.29 (t, J = 12.5 Hz, 1H), 3.77 (s, 3H), 3.83 (s, 3H), 3.93–4.15 (m, 3H), 4.24–4.60 (m, 1H), 6.80–6.87 (m, 4H), 6.88–6.94 (m, 2H), 7.31–7.38 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 16.6, 54.1, 55.3, 55.7, 57.2, 58.1, 66.8, 113.7, 114.6, 115.6, 128.5, 128.7, 152.8, 153.9, 160.5, 170.3; FTIR (neat): 1624, 1607, 1506, 1421 cm−1; HRMS (m/z): calcd. for C22H29N2O4 [M + H]+ 385.2122; found 385.2121; HPLC purity = 99.7%.
(4-(2-(4-Methoxyphenoxy)ethyl)piperidin-1-yl)(4-methoxyphenyl)methanone (50).
A mixture of 4-(2-chloroethyl)piperidin-1-yl)(4-methoxyphenyl)methanone (64.9 mg, 0.230 mmol, 1.0 equiv), 4-methoxyphenol (32.0 mg, 0.258 mmol, 1.1 equiv) and potassium carbonate (57.0 mg, 0.412 mmol, 1.8 equiv) in DMF (1 mL) was heated at 55 °C for 19 h, followed by 80 °C for 45 h and then cooled to rt. The reaction was diluted with CH2Cl2 (10 mL) and washed with 1 M NaOH (3 × 2 mL) and water (4 × 2 mL). The organic layer was dried over Na2SO4, concentrated in vacuo and purified by silica gel chromatography to afford the aryl ether product as an off-white solid (12.2 mg, 0.033 mmol, 14% yield). 1H NMR (400 MHz, CDCl3) δ 1.12–1.35 (br m, 3H), 1.74 (q, J = 6.3 Hz, 3H), 1.78–1.90 (m, 2H), 2.69–3.07 (br m, 2H), 3.77 (s, 3H), 3.83 (s, 3H), 3.97 (t, J = 6.2 Hz, 2H), 4.47–4.79 (m, 1H), 6.79–6.85 (m, 4H), 6.88–6.93 (m, 2H), 7.35–7.40 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 33.2, 35.8, 55.3, 55.7, 65.8, 113.6, 114.7, 115.3, 128.4, 128.9, 153.0, 153.8, 160.6, 170.3; HRMS (m/z): calcd. for C22H28NO4 [M + H]+ 370.2013; found 370.2004; HPLC purity = 98.1%.
4-Methoxy-N-(2-((2-(4-methoxyphenoxy)ethyl)(methyl)amino)ethyl)-N-methylbenzamide (51).
N-(2-((2-Chloroethyl)(methyl)amino)ethyl)-4-methoxy-N-methylbenzamide hydrochloride (70.7 mg, 0.220 mmol, 1.0 equiv), 4-methoxyphenol (38.0 mg, 0.306 mmol, 1.4 equiv) and potassium carbonate (101 mg, 0.731 mmol, 3.3 equiv) in DMF (1 mL) was heated at 50 °C for 7 h and cooled to rt. The reaction was diluted with CH2Cl2 (10 mL) and washed with water (4 × 2 mL). The organic layer was dried over Na2SO4, concentrated in vacuo and purified by silica gel chromatography to afford the aryl ether product as a colorless oil (56.5 mg, 0.161 mmol, 73% yield). 1H NMR (400 MHz, CDCl3) δ 2.11–2.52 (br m, 3H), 2.58–2.92 (br m, 4H), 3.05 (s, 3H), 3.36–3.69 (br m, 2H), 3.77 (s, 3H), 3.81 (s, 3H), 3.97 (d, J = 17.9 Hz, 2H), 6.78–6.84 (m, 4H), 6.84–6.90 (m, 2H), 7.32–7.41 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 43.0, 55.3, 55.7, 56.7, 66.8, 113.6, 114.6, 115.4, 128.8, 128.9, 152.9, 153.8, 160.4; HRMS (m/z): calcd. for C21H29N2O4 [M + H]+ 373.2122; found 373.2132; HPLC purity = 97.9%.
(4-(2-(3-Methoxyphenoxy)ethyl)piperazin-1-yl)(1H-pyrrolo[2,3-b]pyridin-2-yl)methanone (52).
1H-Pyrrolo[2,3-b]pyridine-2-carboxylic acid (19 mg, 0.12 mmol, 1.2 equiv) and 1-(2-(3-methoxyphenoxy)ethyl)piperazine (200 μL, 0.488 M, 0.0980 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as a white solid (24.8 mg, 0.0653 mmol, 67% yield). Rf = 0.54 (10% MeOH/CH2Cl2); mp = 136–139 °C. 1H NMR (400 MHz, CDCl3) δ 2.68 (t, J = 5.1 Hz, 4H), 2.86 (t, J = 5.5 Hz, 2H), 3.78 (s, 3H), 3.85–4.03 (m, 4H), 4.12 (t, J = 5.5 Hz, 2H), 6.43–6.55 (m, 3H), 6.68–6.74 (m, 1H), 7.09–7.22 (m, 2H), 7.98 (dd, J = 1.5, 7.9 Hz, 1H), 8.54 (dd, J = 1.5, 4.7 Hz, 1H), 11.57 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 26.4, 26.5, 46.3, 46.3, 55.3, 57.1, 65.8, 101.1, 105.7, 106.58, 106.63, 119.1, 119.2, 128.8, 129.9, 131.7, 144.9, 145.3, 159.8, 160.9, 161.9; FTIR (neat): 1602, 1530, 1492 cm−1; HRMS (m/z): calcd. for C21H25N4O3 [M + H]+ 381.1921; found 381.1928; HPLC purity = 99.4%.
(4-(2-(3-Methoxyphenoxy)ethyl)piperazin-1-yl)(1H-pyrrolo[3,2-c]pyridin-2-yl)methanone (53).
1H-Pyrrolo[3,2-c]pyridine-2-carboxylic acid (18 mg, 0.11 mmol, 1.1 equiv) and 1-(2-(3-methoxyphenoxy)ethyl)piperazine (200 μL, 0.488 M, 0.0980 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as a sticky, off-white solid (22.9 mg, 0.0598 mmol, 61% yield); Rf = 0.11 (5% MeOH/CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 2.70 (t, J = 5.1 Hz, 4H), 2.87 (t, J = 5.4 Hz, 2H), 3.78 (s, 3H), 3.85–4.06 (m, 4H), 4.12 (t, J = 5.4 Hz, 2H), 6.45–6.56 (m, 3H), 6.82–6.89 (m, 1H), 7.18 (t, J = 8.2 Hz, 1H), 7.31–7.37 (m, 1H), 8.36 (d, J = 5.9 Hz, 1H), 8.96–9.02 (m, 1H), 10.32 (br s, 1H); 13C NMR (101 MHz, CDCl3) δ 55.3, 57.0, 65.9, 101.2, 104.3, 106.5, 106.7, 106.9, 124.5, 129.9, 130.4, 139.2, 142.5, 145.4, 159.8, 160.9, 161.8; FTIR (neat): 1603, 1534, 1492, 1451, 1435 cm−1; HRMS (m/z): calcd. for C21H25N4O3 [M + H]+ 381.1921; found 381.1928; HPLC purity = 96.4%.
(1H-Indol-2-yl)(4-(2-(3-methoxyphenoxy)ethyl)piperazin-1-yl)methanone (54).
4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and 4-methoxyphenol (19 μL, 0.17 mmol, 1.6 equiv) were reacted according to General Procedure G to afford the aryl ether product as a light yellow solid (24.1 mg, 0.0638 mmol, 60% yield). Rf = 0.59 (10% MeOH/CH2Cl2); mp = 99–102 °C. 1H NMR (500 MHz, CDCl3) δ 2.64–2.71 (m, 4H), 2.87 (t, J = 5.5 Hz, 2H), 3.79 (s, 3H), 3.88–4.04 (m, 4H), 4.13 (t, J = 5.5 Hz, 2H), 6.49 (t, J = 2.4 Hz, 1H), 6.50–6.54 (m, 2H), 6.78 (dd, J = 0.9, 2.1 Hz, 1H), 7.14 (ddd, J = 1.0, 7.0, 8.0 Hz, 1H), 7.19 (t, J = 8.2 Hz, 1H), 7.28 (ddd, J = 1.2, 7.0, 8.2 Hz, 1H), 7.43 (dq, J = 0.9, 8.2 Hz, 1H), 7.65 (dq, J = 0.9, 8.1 Hz, 1H), 9.25 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 41.0, 53.6, 55.3, 57.1, 65.8, 101.1, 105.3, 106.5, 106.7, 111.7, 120.6, 121.9, 124.4, 127.5, 129.2, 129.9, 135.6, 159.9, 160.8, 162.2; FTIR (neat): 1592, 1526, 1491 cm−1; HRMS (m/z): calcd. for C22H26N3O3 [M + H]+ 380.1969; found 380.1980; HPLC purity = 99.2%.
(5-Methoxy-1H-indol-2-yl)(4-(2-(3-methoxyphenoxy)ethyl)piperazin-1-yl)methanone (55).
5-Methoxy-1H-indole-2-carboxylic acid (20 mg, 0.10 mmol, 1.0 equiv) and 1-(2-(3-methoxyphenoxy)ethyl)piperazine (200 μL, 0.488 M, 0.0980 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as an off-white solid (24.0 mg, 0.0588 mmol, 60% yield). Rf = 0.64 (10% MeOH/CH2Cl2); mp = 116–118 °C. 1H NMR (500 MHz, CDCl3) δ 2.63–2.73 (m, 4H), 2.86 (t, J = 5.6 Hz, 2H), 3.79 (s, 3H), 3.85 (s, 3H), 3.90–4.02 (m, 4H), 4.13 (t, J = 5.5 Hz, 2H), 6.48 (t, J = 2.4 Hz, 1H), 6.50–6.55 (m, 2H), 6.70 (dd, J = 0.9, 2.2 Hz, 1H), 6.96 (dd, J = 2.5, 8.9 Hz, 1H), 7.05 (d, J = 2.5 Hz, 1H), 7.19 (t, J = 8.2 Hz, 1H), 7.32 (dt, J = 0.8, 8.9 Hz, 1H), 9.21 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 53.6, 55.3, 55.7, 57.1, 65.8, 101.1, 102.2, 104.9, 106.5, 106.7, 112.6, 115.7, 127.8, 129.7, 129.9, 130.9, 154.6, 159.9, 160.8, 162.2; FTIR (neat): 1591, 1525, 1492 cm−1; HRMS (m/z): calcd. for C23H28N3O4 [M + H]+ 410.2074; found 410.2071; HPLC purity > 99.5%.
(1H-Benzo[d]imidazol-2-yl)(4-(2-(3-methoxyphenoxy)ethyl)piperazin-1-yl)methanone (56).
1H-Benzo[d]imidazole-2-carboxylic acid (17 mg, 0.11 mmol, 1.1 equiv) and 1-(2-(3-methoxyphenoxy)ethyl)piperazine (200 μL, 0.488 M, 0.0980 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as a white solid (22.7 mg, 0.0598 mmol, 61% yield). Rf = 0.38 (5% MeOH/CH2Cl2) mp = 50–65 °C. 1H NMR (400 MHz, CDCl3) δ 2.75 (q, J = 4.6 Hz, 4H), 2.88 (t, J = 5.6 Hz, 2H), 3.79 (s, 3H), 3.96 (t, J = 5.1 Hz, 2H), 4.14 (t, J = 5.6 Hz, 2H), 4.82 (t, J = 5.1 Hz, 2H), 6.46–6.55 (m, 3H), 7.18 (t, J = 8.2 Hz, 1H), 7.27–7.39 (m, 2H), 7.52 (d, J = 7.6 Hz, 1H), 7.82 (d, J = 8.0 Hz, 1H), 11.53 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 43.1, 46.6, 53.3, 54.1, 55.2, 57.0, 65.7, 101.0, 106.4, 106.5, 111.6, 121.0, 123.0, 125.0, 129.8, 132.5, 143.1, 145.2, 158.2, 159.8, 160.7; FTIR (neat): 1604, 1588, 1491, 1447 cm−1; HRMS (m/z): calcd. for C21H25N4O3 [M + H]+ 381.1921; found 381.1924; HPLC purity = 99.4%.
(4-Methoxy-2-methylphenyl)(4-(2-(pyridin-3-yloxy)ethyl)piperazin-1-yl)methanone (57).
4-Methoxy-2-methyl-benzoic acid (26 mg, 0.15 mmol, 1.2 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (545 μL, 0.235 M, 0.128 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as a light yellow solid (29.5 mg, 0.083 mmol, 65% yield). Rf = 0.29 (10% MeOH/CH2Cl2); mp = 91–93 °C. 1H NMR (500 MHz, CDCl3) δ 2.30 (s, 3H), 2.48 (t, J = 5.1 Hz, 2H), 2.65 (t, J = 5.2 Hz, 2H), 2.85 (t, J = 5.5 Hz, 2H), 3.26–3.34 (m, 2H), 3.78–3.89 (m, 2H), 3.80 (s, 3H), 4.15 (t, J = 5.5 Hz, 2H), 6.71–6.75 (m, 2H), 7.08–7.13 (m, 1H), 7.17–7.24 (m, 2H), 8.23 (dd, J = 1.7, 4.3 Hz, 1H), 8.31–8.34 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 19.4, 41.5, 46.9, 53.4, 54.1, 55.3, 57.0, 66.3, 111.2, 115.8, 121.2, 123.8, 127.4, 128.5, 136.3, 138.0, 142.4, 154.8, 159.8, 170.1; FTIR (neat): 1625, 1606, 1574, 1459 cm−1; HRMS (m/z): calcd for C20H26N3O3 [M + H]+ 356.1969; found 356.1970; HPLC purity = 98.6%.
(6-Methoxy-1H-indol-2-yl)(4-(2-(pyridin-3-yloxy)ethyl)piperazin-1-yl)methanone (58).
6-Methoxy-1H-indole-2-carboxylic acid (26 mg, 0.14 mmol, 1.1 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (200 μL, 0.645 M, 0.129 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as a white solid (42.2 mg, 0.111 mmol, 86% yield). Rf = 0.40 (10% MeOH/CH2Cl2); mp = 142–144 °C. 1H NMR (400 MHz, CDCl3) δ 2.67 (t, J = 5.1 Hz, 4H), 2.87 (t, J = 5.5 Hz, 2H), 3.83 (s, 3H), 3.88–4.04 (m, 4H), 4.17 (t, J = 5.5 Hz, 2H), 6.68–6.75 (m, 1H), 6.79 (dd, J = 1.4, 8.8 Hz, 1H), 6.83–6.88 (m, 1H), 7.17–7.24 (m, 2H), 7.49 (d, J = 8.7 Hz, 1H), 8.19–8.28 (m, 1H), 8.30–8.37 (m, 1H), 9.44 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 26.4, 26.5, 46.2, 46.3, 55.7, 57.1, 66.3, 93.7, 106.1, 111.9, 121.3, 121.8, 122.6, 123.9, 128.1, 136.6, 138.0, 142.4, 154.8, 158.3, 162.2; FTIR (neat): 1623, 1607, 1506 cm−1; HRMS (m/z): calcd. for C23H28N3O3 [M + H]+ 381.1921; found 381.1905; HPLC purity = 95%.
(5-Methoxy-1H-indol-2-yl)(4-(2-(pyridin-3-yloxy)ethyl)piperazin-1-yl)methanone (59).
5-Methoxy-1H-indole-2-carboxylic acid (26 mg, 0.14 mmol, 1.1 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (200 μL, 0.645 M, 0.129 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as a pale yellow solid (36.6 mg, 0.0963 mmol, 75% yield). 1H NMR (500 MHz, CDCl3) δ 2.67 (t, J = 5.1 Hz, 4H), 2.87 (t, J = 5.5 Hz, 2H), 3.83 (s, 3H), 3.88–4.03 (m, 4H), 4.17 (t, J = 5.5 Hz, 2H), 6.67–6.72 (m, 1H), 6.94 (dd, J = 2.4, 8.9 Hz, 1H), 7.04 (d, J = 2.4 Hz, 1H), 7.19–7.23 (m, 2H), 7.31 (d, J = 8.9 Hz, 1H), 8.23 (dd, J = 2.2, 3.7 Hz, 1H), 8.30–8.37 (m, 1H), 9.57 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 53.7, 55.7, 57.1, 66.3, 102.3, 104.9, 112.7, 115.7, 121.3, 123.9, 127.8, 129.7, 131.1, 138.0, 142.4, 154.6, 154.9, 162.4; HRMS (m/z): calcd. for C21H25N4O3 [M + H]+ 381.1921; found 381.1922; HPLC purity = 98.7%.
(7-Methyl-1H-indol-2-yl)(4-(2-(pyridin-3-yloxy)ethyl)piperazin-1-yl)methanone (60).
7-Methyl-1H-indole-2-carboxylic acid (24 mg, 0.14 mmol, 1.1 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (200 μL, 0.645 M, 0.129 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as an orange solid (33.5 mg, 0.0919 mmol, 71% yield). Rf = 0.26 (5% MeOH/CH2Cl2); mp = 49–60 °C. 1H NMR (400 MHz, CDCl3) δ 2.49 (s, 3H), 2.67 (t, J = 4.8 Hz, 4H), 2.88 (t, J = 5.5 Hz, 2H), 3.83–4.06 (m, 4H), 4.18 (t, J = 5.5 Hz, 2H), 6.78 (d, J = 2.0 Hz, 1H), 6.99–7.10 (m, 2H), 7.18–7.24 (m, 2H), 7.44–7.51 (m, 1H), 8.24 (q, J = 2.5, 3.7 Hz, 1H), 8.29–8.36 (m, 1H), 9.24 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 16.7, 53.7, 57.1, 66.3, 105.8, 119.5, 120.9, 121.2, 121.4, 123.9, 124.7, 127.0, 128.9, 135.4, 138.0, 142.4, 154.9, 162.5; FTIR (neat): 1601, 1574, 1536, 1429 cm−1; HRMS (m/z): calcd. for C21H25N4O2 [M + H]+ 365.1972; found 365.1974; HPLC purity = 98.2%.
(6-Methyl-1H-indol-2-yl)(4-(2-(pyridin-3-yloxy)ethyl)piperazin-1-yl)methanone (61).
6-Methyl-1H-indole-2-carboxylic acid (24 mg, 0.14 mmol, 1.1 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (200 μL, 0.645 M, 0.129 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as a white solid (31.8 mg, 0.0872 mmol, 68% yield). Rf = 0.29 (10% MeOH/CH2Cl2); mp = 163–166 °C. 1H NMR (500 MHz, CDCl3) δ 2.47 (s, 3H), 2.62–2.72 (m, 4H), 2.89 (t, J = 5.5 Hz, 2H), 3.85–4.02 (m, 4H), 4.19 (t, J = 5.5 Hz, 2H), 6.74 (dd, J = 0.9, 2.2 Hz, 1H), 6.86–7.01 (m, 1H), 7.19–7.24 (m, 3H), 7.46–7.64 (m, 1H), 8.23–8.25 (m, 1H), 8.33–8.35 (m, 1H), 9.07 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 21.9, 53.6, 57.1, 66.3, 105.3, 111.4, 121.3, 121.5, 122.7, 123.9, 125.3, 128.6, 134.6, 136.0, 138.0, 142.4, 154.8, 162.3; FTIR (neat): 1596, 1573, 1522, 1427 cm−1; HRMS (m/z): calcd. for C21H25N4O2 [M + H]+ 365.1972; found 365.1974; HPLC purity > 99.5%.
(5-Methyl-1H-indol-2-yl)(4-(2-(pyridin-3-yloxy)ethyl)piperazin-1-yl)methanone (62).
5-Methyl-1H-indole-2-carboxylic acid (24 mg, 0.14 mmol, 1.1 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (200 μL, 0.645 M, 0.129 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as an off-white solid (38.2 mg, 0.105 mmol, 81% yield). Rf = 0.29 (10% MeOH/CH2Cl2); mp = 145–147 °C. 1H NMR (400 MHz, CDCl3) δ 2.44 (s, 3H), 2.64–2.73 (m, 4H), 2.89 (t, J = 5.5 Hz, 2H), 3.82–4.02 (m, 4H), 4.19 (t, J = 5.5 Hz, 2H), 6.69 (dd, J = 0.9, 2.2 Hz, 1H), 7.11 (dd, J = 1.6, 7.1 Hz, 1H), 7.21–7.24 (m, 2H), 7.29–7.33 (m, 1H), 7.40–7.42 (m, 1H), 8.22–8.26 (m, 1H), 8.33–8.35 (m, 1H), 9.14 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 21.5, 53.6, 57.0, 66.2, 104.8, 111.4, 121.2, 121.3, 123.9, 126.4, 127.7, 129.2, 129.9, 134.0, 138.0, 142.4, 154.8, 162.4; FTIR (neat): 1597, 1574, 1529, 1427 cm−1; HRMS (m/z): calcd. for C21H25N4O2 [M + H]+ 365.1972; found 365.1977; HPLC purity = 99.5%.
(4-Methyl-1H-indol-2-yl)(4-(2-(pyridin-3-yloxy)ethyl)piperazin-1-yl)methanone (63).
4-Methyl-1H-indole-2-carboxylic acid (24 mg, 0.14 mmol, 1.1 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (200 μL, 0.645 M, 0.129 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as a tan solid (38.4 mg, 0.105 mmol, 82% yield). Rf = 0.28 (5% MeOH/CH2Cl2); mp = 52–58 °C. 1H NMR (400 MHz, CDCl3) δ 2.55 (s, 3H), 2.68 (t, J = 5.3 Hz, 4H), 2.87 (t, J = 5.5 Hz, 2H), 3.91–4.09 (m, 4H), 4.17 (t, J = 5.5 Hz, 2H), 6.77 (d, J = 1.4 Hz, 1H), 6.91 (d, J = 7.0 Hz, 1H), 7.13–7.22 (m, 3H), 7.24–7.29 (m, 1H), 8.21–8.26 (m, 1H), 8.32–8.37 (m, 1H), 9.99 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 18.7, 53.6, 57.0, 66.3, 103.7, 109.4, 120.5, 121.3, 123.9, 124.4, 127.5, 128.6, 131.3, 135.7, 138.0, 142.4, 154.8, 162.6; FTIR (neat): 1600, 1585, 1519, 1427 cm−1; HRMS (m/z): calcd. for C21H25N4O2 [M + H]+ 365.1972; found 365.1976; HPLC purity = 98.1%.
(3-Methyl-1H-indol-2-yl)(4-(2-(pyridin-3-yloxy)ethyl)piperazin-1-yl)methanone (64).
3-Methyl-1H-indole-2-carboxylic acid (24 mg, 0.14 mmol, 1.1 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (200 μL, 0.645 M, 0.129 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as a golden oil (40.9 mg, 0.112 mmol, 87% yield). Rf = 0.17 (5% MeOH/CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 2.37 (s, 3H), 2.57–2.69 (m, 4H), 2.85 (t, J = 5.5 Hz, 2H), 3.69–3.81 (m, 4H), 4.15 (t, J = 5.5 Hz, 2H), 7.10–7.16 (m, 1H), 7.17–7.28 (m, 3H), 7.36 (d, J = 8.2 Hz, 1H), 7.59 (d, J = 7.9 Hz, 1H), 8.23 (dd, J = 1.8, 4.1 Hz, 1H), 8.32 (d, J = 2.5 Hz, 1H), 8.86 (br s, 1H); 13C NMR (101 MHz, CDCl3) δ 10.2, 53.8, 57.0, 66.2, 111.5, 112.1, 119.8, 119.8, 121.3, 123.9, 124.0, 127.0, 128.2, 135.9, 138.0, 142.4, 154.8, 164.7; FTIR (neat): 1607, 1574, 1472, 1450, 1424 cm−1; HRMS (m/z): calcd. for C21H25N4O2 [M + H]+ 365.1972; found 365.1973; HPLC purity = 98.1%.
(1-Methyl-1H-indol-2-yl)(4-(2-(pyridin-3-yloxy)ethyl)piperazin-1-yl)methanone (65).
1-Methyl-1H-indole-2-carboxylic acid (24 mg, 0.14 mmol, 1.1 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (200 μL, 0.645 M, 0.129 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as a yellow oil (41.6 mg, 0.114 mmol, 88% yield). Rf = 0.34 (5% MeOH/CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 2.54–2.72 (m, 4H), 2.87 (t, J = 5.5 Hz, 2H), 3.77–3.82 (m, 4H), 3.83 (s, 3H), 4.16 (t, J = 5.5 Hz, 2H), 6.60 (d, J = 0.7 Hz, 1H), 7.10–7.17 (m, 1H), 7.20–7.22 (m, 1H), 7.24–7.39 (m, 3H), 7.62 (d, J = 7.9 Hz, 1H), 8.23 (dd, J = 2.1, 3.8 Hz, 1H), 8.28–8.35 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 26.4, 31.1, 46.3, 57.0, 66.3, 103.7, 109.8, 120.3, 121.4, 121.5, 123.4, 123.9, 126.4, 131.5, 137.9, 142.3, 154.8, 163.1; FTIR (neat): 1625, 1574, 1522, 1463, 1422 cm−1 HRMS (m/z): calcd. for C21H25N4O2 [M + H]+ 365.1972; found 365.1972; HPLC purity = 98.7%.
(1-Methyl-1H-indol-3-yl)(4-(2-(pyridin-3-yloxy)ethyl)piperazin-1-yl)methanone (66).
1-Methyl-1H-indole-3-carboxylic acid (27 mg, 0.15 mmol, 1.2 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (545 μL, 0.235 M, 0.128 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as a golden oil (29.5 mg, 0.0803 mmol, 63% yield). Rf = 0.23 (5% MeOH/CH2Cl2). 1H NMR (500 MHz, CDCl3) δ 2.63 (t, J = 5.1 Hz, 4H), 2.87 (t, J = 5.6 Hz, 2H), 3.77 (t, J = 4.9 Hz, 4H), 3.82 (s, 3H), 4.17 (t, J = 5.6 Hz, 2H), 7.18–7.24 (m, 3H), 7.27–7.31 (m, 1H), 7.35 (dt, J = 1.0, 8.2 Hz, 1H), 7.43 (s, 1H), 7.69 (dt, J = 1.0, 7.9 Hz, 1H), 8.23 (dd, J = 2.0, 4.0 Hz, 1H), 8.32 (dd, J = 1.1, 2.5 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 33.2, 41.0, 53.9, 57.1, 66.2, 109.8, 110.4, 120.5, 120.9, 121.2, 122.4, 123.8, 125.9, 131.7, 136.4, 138.0, 142.4, 154.8, 166.6; FTIR (neat): 1604, 1532, 1472, 1424 cm−1; HRMS (m/z): calcd. for C21H25N4O2 [M + H]+ 365.1972; found 365.1972; HPLC purity = 99.0%.
(7-Chloro-1H-indol-2-yl)(4-(2-(pyridin-3-yloxy)ethyl)piperazin-1-yl)methanone (67).
7-Chloro-1H-indole-2-carboxylic acid (27 mg, 0.14 mmol, 1.1 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (200 μL, 0.645 M, 0.129 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as a golden oil (37.9 mg, 0.0978 mmol, 76% yield). Rf = 0.29 (5% MeOH/CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 2.69 (t, J = 5.1 Hz, 4H), 2.89 (t, J = 5.5 Hz, 2H), 3.87–4.03 (m, 4H), 4.19 (t, J = 5.5 Hz, 2H), 6.80 (d, J = 2.2 Hz, 1H), 7.08 (t, J = 7.8 Hz, 1H), 7.20–7.24 (m, 2H), 7.28 (dd, J = 0.9, 7.6 Hz, 1H), 7.55 (dt, J = 0.9, 8.0 Hz, 1H), 8.24 (dd, J = 2.3, 3.7 Hz, 1H), 8.34 (dd, J = 1.3, 2.4 Hz, 1H), 9.24 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 41.0, 57.0, 66.3, 105.8, 117.2, 120.4, 121.3, 121.4, 123.6, 123.9, 128.7, 129.9, 133.0, 138.0, 142.4, 154.8, 161.7; FTIR (neat): 1614, 1574, 1532, 1430 cm−1; HRMS (m/z): calcd. for C20H22ClN4O2 [M + H]+ 386.1426; found 386.1431; HPLC purity = 98.7%.
(6-Chloro-1H-indol-2-yl)(4-(2-(pyridin-3-yloxy)ethyl)piperazin-1-yl)methanone (68).
6-Chloro-1H-indole-2-carboxylic acid (28 mg, 0.14 mmol, 1.1 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (200 μL, 0.645 M, 0.129 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as an off-white solid (39.1 mg, 0.101 mmol, 78% yield). Rf = 0.25 (5% MeOH/CH2Cl2); mp = 161–163 °C. 1H NMR (400 MHz, CDCl3) δ 2.70 (t, J = 5.2 Hz, 4H), 2.89 (t, J = 5.5 Hz, 2H), 3.83–4.09 (m, 4H), 4.19 (t, J = 5.5 Hz, 2H), 6.75 (dd, J = 0.9, 2.2 Hz, 1H), 7.10 (dd, J = 1.8, 8.5 Hz, 1H), 7.19–7.24 (m, 2H), 7.42 (dt, J = 0.8, 1.7 Hz, 1H), 7.55 (dt, J = 0.7, 8.5 Hz, 1H), 8.25 (dd, J = 2.3, 3.7 Hz, 1H), 8.34 (dd, J = 1.2, 2.5 Hz, 1H), 9.52 (br s, 1H).; 13C NMR (126 MHz, CDCl3) δ 41.0, 53.6, 57.0, 66.3, 105.3, 111.6, 121.3, 121.6, 122.8, 123.9, 126.0, 129.9, 130.3, 136.0, 138.0, 142.4, 154.8, 162.0; FTIR (neat): 1606, 1574, 1522, 1430 cm−1; HRMS (m/z): calcd. for C20H22ClN4O2 [M + H]+ 386.1426; found 386.1423; HPLC purity = 99.7%.
(5-Chloro-1H-indol-2-yl)(4-(2-(pyridin-3-yloxy)ethyl)piperazin-1-yl)methanone (69).
5-Chloro-1H-indole-2-carboxylic acid (25.2 mg, 0.129 mmol, 1.0 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (200 μL, 0.645 M, 0.129 mmol) were reacted according to General Procedure E and purified by mass-directed, preparative HPLC to afford the acylated product as a white solid (33.6 mg, 0.087 mmol, 67% yield). 1H NMR (400 MHz, CDCl3) δ 2.69 (t, J = 5.1 Hz, 4H), 2.89 (t, J = 5.5 Hz, 2H), 3.89–4.04 (m, 4H), 4.19 (t, J = 5.5 Hz, 2H), 6.71 (dd, J = 0.8, 2.1 Hz, 1H), 7.21–7.24 (m, 3H), 7.35 (td, J = 0.6, 8.8 Hz, 1H),7.60–7.62 (m, 1H), 8.24–8.25 (m, 1H), 8.32–8.35 (m, 1H), 9.39 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 53.6 (br, 2C), 57.0, 66.2, 104.6, 112.8, 121.0, 121.3, 123.9, 124.9, 126.1, 128.4, 130.4, 133.9, 138.0, 142.4, 154.8, 161.9; HRMS (m/z): calcd. for C20H22ClN4O2 [M + H]+ 385.1426; found 385.1418; HPLC purity = 98.8%.
(4-Chloro-1H-indol-2-yl)(4-(2-(pyridin-3-yloxy)ethyl)piperazin-1-yl)methanone (70).
4-Chloro-1H-indole-2-carboxylic acid (27 mg, 0.14 mmol, 1.1 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (200 μL, 0.645 M, 0.129 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as an off-white solid (36.4 mg, 0.0939 mmol, 73% yield). Rf = 0.22 (5% MeOH/CH2Cl2); mp = 155–158 °C. 1H NMR (400 MHz, CDCl3) δ 2.65–2.76 (m, 4H), 2.90 (t, J = 5.5 Hz, 2H), 3.81–4.10 (m, 4H), 4.19 (t, J = 5.5 Hz, 2H), 6.86 (dd, J = 0.9, 2.4 Hz, 1H), 7.11–7.25 (m, 4H), 7.33 (dt, J = 1.0, 8.0 Hz, 1H), 8.20–8.28 (m, 1H), 8.35 (dd, J = 1.3, 2.4 Hz, 1H), 9.61 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 41.0, 53.6, 57.0, 66.3, 103.6, 110.4, 120.2, 121.3, 123.9, 124.9, 126.4, 127.0, 129.7, 136.2, 138.0, 142.4, 154.8, 161.8; FTIR (neat): 1604, 1572, 1527, 1427 cm−1; HRMS (m/z): calcd. for C20H22ClN4O2 [M + H]+ 386.1426; found 386.1424; HPLC purity = 99.4%.
(4-(2-(Pyridin-3-yloxy)ethyl)piperazin-1-yl)(1H-pyrrolo[2,3-b]pyridin-2-yl)methanone (71).
1H-Pyrrolo[2,3-b]pyridine-2-carboxylic acid (25 mg, 0.15 mmol, 1.2 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (200 μL, 0.645 M, 0.129 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as a white solid (39.9 mg, 0.113 mmol, 88% yield). Rf = 0.13 (5% MeOH/CH2Cl2); mp = 133–135 °C. 1H NMR (500 MHz, CDCl3) δ 2.70 (t, J = 5.1 Hz, 4H), 2.90 (t, J = 5.4 Hz, 2H), 3.85–4.08 (m, 4H), 4.19 (t, J = 5.5 Hz, 2H), 6.73 (d, J = 1.7 Hz, 1H), 7.14 (dd, J = 4.7, 7.9 Hz, 1H), 7.22 (dt, J = 1.5, 4.5 Hz, 2H), 7.99 (dd, J = 1.6, 7.9 Hz, 1H), 8.24 (dd, J = 2.0, 3.9 Hz, 1H), 8.34 (dd, J = 1.2, 2.5 Hz, 1H), 8.51 (dd, J = 1.6, 4.7 Hz, 1H), 10.81 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 41.0, 53.6, 57.0, 66.3, 103.4, 116.9, 119.8, 121.3, 123.9, 129.9, 130.4, 138.0, 142.4, 146.0, 147.7, 154.8, 162.0; FTIR (neat): 1618, 1575, 1521, 1430 cm−1; HRMS (m/z): calcd. for C19H22N5O2 [M + H]+ 352.1768; found 352.1769; HPLC purity > 99.5%.
(4-(2-(Pyridin-3-yloxy)ethyl)piperazin-1-yl)(1H-pyrrolo[2,3-c]pyridin-2-yl)methanone (72).
1H-Pyrrolo[2,3-c]pyridine-2-carboxylic acid (24 mg, 0.15 mmol, 1.1 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (200 μL, 0.645 M, 0.129 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as a light yellow solid (35.7 mg, 0.101 mmol, 79% yield). Rf = 0.07 (5% MeOH/CH2Cl2); mp = 166–172 °C. 1H NMR (400 MHz, CDCl3) δ 2.71 (t, J = 5.2 Hz, 4H), 2.90 (t, J = 5.4 Hz, 2H), 3.86–4.07 (m, 4H), 4.20 (t, J = 5.4 Hz, 2H), 6.87 (dd, J = 1.0, 1.9 Hz, 1H), 7.20–7.25 (m, 2H), 7.34 (dt, J = 1.1, 5.8 Hz, 1H), 8.24–8.26 (m, 1H), 8.35 (dd, J = 1.4, 2.3 Hz, 1H), 8.38 (d, J = 5.8 Hz, 1H), 8.96–9.01 (m, 1H), 9.75 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 41.0, 53.6, 57.0, 66.3, 104.3, 106.8, 121.3, 123.9, 124.6, 130.3, 138.0, 139.0, 142.5, 142.8, 145.5, 154.8, 161.6; FTIR (neat): 1611, 1573, 1534, 1428 cm−1; HRMS (m/z): calcd. for C19H22N5O2 [M + H]+ 352.1768; found 352.1766; HPLC purity > 99.5%.
(4-(2-(Pyridin-3-yloxy)ethyl)piperazin-1-yl)(1H-pyrrolo[3,2-c]pyridin-2-yl)methanone (73).
1H-Pyrrolo[3,2-c]pyridine-2-carboxylic acid (22 mg, 0.14 mmol, 1.0 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (200 μL, 0.645 M, 0.129 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as an off-white solid (35.9 mg, 0.103 mmol, 80% yield). Rf = 0.07 (5% MeOH/CH2Cl2); mp = 163–170 °C. 1H NMR (500 MHz, CDCl3) δ 2.71 (t, J = 5.1 Hz, 4H), 2.91 (t, J = 5.5 Hz, 2H), 3.80–4.13 (m, 4H), 4.20 (t, J = 5.5 Hz, 2H), 6.87 (d, J = 1.4 Hz, 1H), 7.20–7.26 (m, 2H), 7.34 (dt, J = 1.1, 5.9 Hz, 1H), 8.24 (dd, J = 1.2, 1.7 Hz, 1H), 8.35 (dd, J = 1.2, 2.5 Hz, 1H), 8.38 (d, J = 5.8 Hz, 1H), 8.99 (d, J = 1.1 Hz, 1H), 9.90 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 41.0, 53.6, 57.0, 66.3, 104.3, 106.8, 121.3, 123.9, 124.6, 130.3, 138.0, 139.0, 142.5, 142.8, 145.5, 154.8, 161.7; FTIR (neat): 1610, 1573, 1534, 1427 cm−1; HRMS (m/z): calcd. for C19H22N5O2 [M + H]+ 352.1768; found 352.1768; HPLC purity > 99.5%.
(4-(2-(Pyridin-3-yloxy)ethyl)piperazin-1-yl)(1H-pyrrolo[3,2-b]pyridin-2-yl)methanone (74).
1H-Pyrrolo[3,2-b]pyridine-2-carboxylic acid (25 mg, 0.15 mmol, 1.2 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (200 μL, 0.645 M, 0.129 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as a white solid (35.8 mg, 0.102 mmol, 79% yield). Rf = 0.11 (5% MeOH/CH2Cl2); mp = 173–177 °C. 1H NMR (500 MHz, CDCl3) δ 2.71 (t, J = 5.1 Hz, 4H), 2.90 (t, J = 5.5 Hz, 2H), 3.76–4.13 (m, 4H), 4.19 (t, J = 5.5 Hz, 2H), 6.98 (dd, J = 0.9, 2.1 Hz, 1H), 7.18–7.25 (m, 3H), 7.74 (dt, J = 1.2, 8.2 Hz, 1H), 8.24 (dd, J = 1.9, 4.2 Hz, 1H), 8.35 (dd, J = 1.1, 2.5 Hz, 1H), 8.53 (dd, J = 1.4, 4.6 Hz, 1H), 9.72–9.82 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 53.5, 57.0, 66.2, 105.7, 119.1, 119.2, 121.3, 123.9, 128.8, 131.7, 137.9, 142.4, 145.0, 145.2, 154.8, 161.9; FTIR (neat): 1617, 1574, 1526, 1428 cm−1; HRMS (m/z): calcd. for C19H22N5O2 [M + H]+ 352.1768; found 352.1766; HPLC purity = 99.5%.
(1H-Benzo[d]imidazol-2-yl)(4-(2-(pyridin-3-yloxy)ethyl)piperazin-1-yl)methanone (75).
1HBenzo[d]imidazole-2-carboxylic acid (23 mg, 0.14 mmol, 1.1 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (200 μL, 0.645 M, 0.129 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as an off-white solid (34.2 mg, 0.0972 mmol, 75% yield). Rf = 0.14 (5% MeOH/CH2Cl2); mp = 163–164 °C. 1H NMR (400 MHz, CDCl3) δ 2.75 (dt, J = 5.1, 10.1 Hz, 4H), 2.90 (t, J = 5.5 Hz, 2H), 3.93 (t, J = 5.1 Hz, 2H), 4.20 (t, J = 5.5 Hz, 2H), 4.82 (t, J = 5.1 Hz, 2H), 7.23 (dd, J = 1.5, 3.5 Hz, 2H), 7.29–7.40 (m, 2H), 7.53 (d, J = 8.0 Hz, 1H), 7.82 (d, J = 8.0 Hz, 1H), 8.25 (dd, J = 2.5, 3.6 Hz, 1H), 8.35 (dd, J = 1.4, 2.3 Hz, 1H), 10.84 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 41.0, 43.2, 46.6, 53.4, 54.2, 57.0, 66.2, 111.7, 121.1, 121.3, 123.2, 123.9, 125.1, 132.5, 138.0, 142.4, 143.2, 145.3, 154.8, 158.2; FTIR (neat): 1618, 1574, 1489, 1428 cm−1; HRMS (m/z): calcd. for C19H22N5O2 [M + H]+ 352.1768; found 352.1764; HPLC purity = 99.6%.
(1H-Indazol-3-yl)(4-(2-(pyridin-3-yloxy)ethyl)piperazin-1-yl)methanone (76).
1H-Indazole-3-carboxylic acid (25 mg, 0.15 mmol, 1.2 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (545 μL, 0.235 M, 0.128 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as a tan solid (25.8 mg, 0.0739 mmol, 57% yield). Rf = 0.11 (5% MeOH/CH2Cl2). mp = 121–123 °C. 1H NMR (500 MHz, CDCl3) δ 2.64–2.76 (m, 4H), 2.89 (t, J = 5.6 Hz, 2H), 3.86–4.15 (m, 4H), 4.19 (t, J = 5.6 Hz, 2H), 7.20–7.23 (m, 2H), 7.25 (dd, J = 1.1, 6.9 Hz, 1H), 7.39–7.44 (m, 1H), 7.49 (dt, J = 0.9, 8.5 Hz, 1H), 8.13 (dt, J = 1.0, 8.2 Hz, 1H), 8.24 (dd, J = 1.9, 4.1 Hz, 1H), 8.34 (dd, J = 1.2, 2.5 Hz, 1H), 10.86 (br s, 1H).; 13C NMR (126 MHz, CDCl3) δ 42.4, 46.9, 53.5, 54.2, 57.0, 66.2, 109.7, 121.3, 122.2, 122.4, 123.4, 123.9, 127.3, 138.0, 140.0, 140.5, 142.3, 154.9, 162.7; FTIR (neat): 1613, 1574, 1486, 1429 cm−1; HRMS (m/z): calcd. for C19H22N5O2 [M + H]+ 352.1768; found 352.1768; HPLC purity = 97.5%.
Benzofuran-2-yl(4-(2-(pyridin-3-yloxy)ethyl)piperazin-1-yl)methanone (77).
Benzofuran-2-carboxylic acid (25 mg, 0.15 mmol, 1.2 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (545 μL, 0.235 M, 0.128 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as a sticky yellow solid (40.1 mg, 0.114 mmol, 89% yield). Rf = 0.27 (5% MeOH/CH2Cl2). 1H NMR (500 MHz, CDCl3) δ 2.63–2.70 (m, 4H), 2.86 (t, J = 5.5 Hz, 2H), 3.76–3.96 (m, 4H), 4.16 (t, J = 5.5 Hz, 2H), 7.18–7.21 (m, 2H), 7.24–7.28 (m, 1H), 7.34–7.41 (m, 1H), 7.50 (d, J = 8.3 Hz, 1H), 7.63 (d, J = 7.8 Hz, 1H), 8.17–8.24 (m, 1H), 8.31 (br s, 1H); 13C NMR (101 MHz, CDCl3) δ 26.3, 26.4, 46.2, 46.3, 57.0, 66.2, 111.8, 112.1, 121.3, 122.2, 123.6, 123.8, 126.5, 126.9, 138.0, 142.4, 148.9, 154.6, 154.8, 159.7; FTIR (neat): 1630, 1573, 1475, 1428 cm−1; HRMS (m/z): calcd. for C20H22N3O3 [M + H]+ 352.1656; found 352.1663; HPLC purity = 99.2%.
Benzo[b]thiophen-2-yl(4-(2-(pyridin-3-yloxy)ethyl)piperazin-1-yl)methanone (78).
Benzo[b]thiophene-2-carboxylic acid (27 mg, 0.15 mmol, 1.2 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (545 μL, 0.235 M, 0.128 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as a light yellow oil (31.4 mg, 0.0855 mmol, 67% yield). Rf = 0.38 (5% MeOH/CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 2.59–2.73 (m, 4H), 2.86 (t, J = 5.5 Hz, 2H), 3.75–3.86 (m, 4H), 4.15 (t, J = 5.5 Hz, 2H), 7.20 (d, J = 2.7 Hz, 2H), 7.34–7.41 (m, 2H), 7.45–7.49 (m, 1H), 7.76–7.87 (m, 2H), 8.18–8.26 (m, 1H), 8.27–8.36 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 46.26, 46.30, 53.6, 57.0, 66.3, 121.3, 122.4, 123.8, 124.6, 124.8, 125.2, 125.8, 136.5, 138.0, 138.6, 140.1, 142.4, 154.8, 163.8; FTIR (neat): 1617, 1573, 1521, 1421 cm−1; HRMS (m/z): calcd. for C20H22N3O2S [M + H]+ 368.1427; found 368.1427; HPLC purity = 99.0%.
(4-(2-(Pyridin-3-yloxy)ethyl)piperazin-1-yl)(1H-pyrrol-2-yl)methanone (79).
1H-Pyrrole-2-carboxylic acid (17 mg, 0.15 mmol, 1.2 equiv) and 1-(2-(pyridin-3-yloxy)ethyl)piperazine (545 μL, 0.235 M, 0.128 mmol) were reacted according to General Procedure E to afford the acylated piperazine product as an off-white solid (19.7 mg, 0.0646 mmol, 50% yield). Rf = 0.17 (5% MeOH/CH2Cl2); mp = 131–133 °C. 1H NMR (500 MHz, CDCl3) δ 2.64 (t, J = 5.2 Hz, 4H), 2.87 (t, J = 5.5 Hz, 2H), 3.80–3.97 (m, 4H), 4.18 (t, J = 5.6 Hz, 2H), 6.25 (dt, J = 2.7, 3.8 Hz, 1H), 6.52 (ddd, J = 1.3, 2.5, 3.8 Hz, 1H), 6.92 (td, J = 1.2, 2.7 Hz, 1H), 7.16–7.25 (m, 2H), 8.24 (dd, J = 2.1, 3.7 Hz, 1H), 8.34 (dd, J = 1.3, 2.3 Hz, 1H), 9.53 (br s, 1H);13C NMR (126 MHz, CDCl3) δ 53.6, 57.1, 66.2, 109.6, 112.1, 120.9, 121.3, 123.8, 124.5, 138.0, 142.4, 154.8, 161.6; FTIR (neat): 1584, 1573, 1465, 1426 cm−1; HRMS (m/z): calcd. for C16H21N4O2 [M + H]+ 301.1659; found 301.1667; HPLC purity = 98.3%.
(1H-Indol-2-yl)(4-(2-phenoxyethyl)piperazin-1-yl)methanone (80).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (37.1 mg, 0.128 mmol) and phenol (19.3 mg, 0.205 mmol, 1.6 equiv) were reacted according to General Procedure F and purified by silica gel chromatography to afford the aryl ether product as a light yellow solid (26.3 mg, 0.075 mmol, 59% yield). 1H NMR (400 MHz, CDCl3) δ 2.69 (t, J = 5.1 Hz, 4H), 2.88 (t, J = 5.6 Hz, 2H), 3.89–4.04 (m, 4H), 4.15 (t, J = 5.6 Hz, 2H), 6.78 (dd, J = 0.9, 2.1 Hz, 1H), 6.90–6.98 (m, 3H), 7.12–7.15 (m, 1H), 7.25–7.32 (m, 3H), 7.43 (dd, J = 0.8, 8.3 Hz, 1H), 7.65 (dd, J = 0.9, 8.0 Hz, 1H), 9.22 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 53.6 (br, 2C), 57.2, 65.8, 105.3, 111.7, 114.6, 120.6, 121.0, 121.8, 124.4, 127.4, 129.2, 129.5, 135.5, 158.6, 162.2; HRMS (m/z): calcd. for C21H24N3O2 [M + H]+ 350.1863; found 350.1860; HPLC purity = 99.0%.
(4-(2-(Benzo[d][1,3]dioxol-5-yloxy)ethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (81).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and benzo[d][1,3]dioxol-5-ol (25 mg, 0.18 mmol, 1.7 equiv) were reacted according to General Procedure G to afford the aryl ether product as a white solid (20.5 mg, 0.052 mmol, 49% yield). Rf = 0.34 (5% MeOH/CH2Cl2); mp = 142–146 °C. 1H NMR (400 MHz, DMSO-d6) δ 2.56 (t, J = 5.0 Hz, 4H), 2.72 (t, J = 5.7 Hz, 2H), 3.69–3.83 (m, 4H), 4.03 (t, J = 5.7 Hz, 2H), 5.95 (s, 2H), 6.38 (dd, J = 2.5, 8.5 Hz, 1H), 6.65 (d, J = 2.5 Hz, 1H), 6.76 – 6.85 (m, 2H), 7.04 (ddd, J = 1.0, 6.9, 8.0 Hz, 1H), 7.18 (ddd, J = 1.2, 7.0, 8.2 Hz, 1H), 7.41 (dd, J = 0.9, 8.2 Hz, 1H), 7.60 (dd, J = 1.0, 8.0 Hz, 1H), 11.56 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 53.11, 53.13, 56.4, 66.1, 97.8, 100.9, 103.9, 105.7, 108.0, 112.0, 119.7, 121.3, 123.1, 126.7, 129.8, 135.8, 141.1, 147.9, 153.8, 161.9; FTIR 1600, 1527, 1487, 1437 cm−1; HRMS (m/z): calcd. for C22H24N3O4 [M + H]+ 394.1761; found 394.1782; HPLC purity = 99.4%.
(1H-Indol-2-yl)(4-(2-((4-methoxyphenyl)thio)ethyl)piperazin-1-yl)methanone (82).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and 4-methoxybenzenethiol (21 μL, 0.17 mmol, 1.6 equiv) were reacted according to General Procedure G to afford the aryl thioether product as a tan solid (19.3 mg, 0.0487 mmol, 46% yield). Rf = 0.39 (5% MeOH/CH2Cl2); mp = 114–122 °C. 1H NMR (400 MHz, DMSO-d6) δ 2.46 (t, J = 5.1 Hz, 4H), 2.51–2.56 (m, 2H), 2.96–3.03 (m, 2H), 3.62–3.81 (m, 7H), 6.76 (dd, J = 0.9, 2.2 Hz, 1H), 6.88–6.94 (m, 2H), 7.04 (ddd, J = 1.0, 7.0, 8.0 Hz, 1H), 7.18 (ddd, J = 1.2, 7.0, 8.2 Hz, 1H), 7.30–7.37 (m, 2H), 7.41 (dq, J = 1.0, 8.3 Hz, 1H), 7.59 (dq, J = 0.9, 8.0 Hz, 1H), 11.55 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 30.3, 51.4, 54.0, 55.9, 102.8, 110.9, 113.6, 118.6, 120.1, 122.0, 124.8, 125.6, 128.7, 130.8, 134.7, 157.1, 160.7; FTIR (neat) 1595, 1526, 1492, 1437 cm−1; HRMS (m/z): calcd for C22H26N3O2S [M + H]+ 396.1740; found 396.1739; HPLC purity > 99.5%.
(1H-Indol-2-yl)(4-(2-(4-(methylthio)phenoxy)ethyl)piperazin-1-yl)methanone (83).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and 4-(methylthio)phenol (24 mg, 0.17 mmol, 1.6 equiv) were reacted according to General Procedure G to afford the aryl ether product as a tan residue (10.3 mg, 0.0265 mmol, 25% yield). Rf = 0.38 (5% MeOH/CH2Cl2). 1H NMR (500 MHz, DMSO-d6) δ 2.40–2.42 (s, 3H), 2.57 (t, J = 5.1 Hz, 4H), 2.75 (t, J = 5.7 Hz, 2H), 3.72–3.82 (m, 4H), 4.09 (t, J = 5.7 Hz, 2H), 6.78 (dd, J J = 0.9, 2.2 Hz, 1H), 6.92–6.96 (m, 2H), 7.04 (ddd, J = 1.0, 6.9, 7.9 Hz, 1H), 7.18 (ddd, J = 1.2, 6.9, 8.2 Hz, 1H), 7.22–7.25 (m, 2H), 7.41 (dq, J = 1.0, 8.2 Hz, 1H), 7.60 (dq, J = 0.9, 8.0 Hz, 1H), 11.57 (br s, 1H). 13C NMR (126 MHz, DMSO-d6) δ 16.5, 53.1, 56.4, 65.5, 103.9, 112.0, 115.3, 119.7, 121.3, 123.1, 126.7, 128.3, 129.0, 129.8, 135.8, 156.7, 161.9; FTIR (neat) 3247, 1598, 1574, 1525, 1427 cm−1; HRMS (m/z): calcd for C22H26N3O2S [M + H]+ 396.1740; found 396.1754; HPLC purity = 98%.
(4-(2-(4-Ethoxyphenoxy)ethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (84).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and 4-ethoxyphenol (25 mg, 0.18 mmol, 1.7 equiv) were reacted according to General Procedure G to afford the aryl ether product as a white residue (18.6 mg, 0.0477 mmol, 45% yield). Rf = 0.35 (5% MeOH/CH2Cl2). 1H NMR (400 MHz, DMSO-d6) δ 1.29 (t, J = 7.0 Hz, 3H), 2.56 (t, J = 5.1 Hz, 4H), 2.73 (t, J = 5.7 Hz, 2H), 3.67–3.82 (m, 4H), 3.94 (q, J = 7.0 Hz, 2H), 4.04 (t, J = 5.7 Hz, 2H), 6.78 (dd, J = 0.9, 2.2 Hz, 1H), 6.80–6.90 (m, 4H), 7.04 (ddd, J = 1.0, 6.9, 8.0 Hz, 1H), 7.18 (ddd, J = 1.2, 7.0, 8.2 Hz, 1H), 7.41 (dq, J = 1.0, 8.3 Hz, 1H), 7.60 (dq, J = 0.9, 8.0 Hz, 1H), 11.56 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 14.6, 53.1, 56.4, 63.2, 65.7, 103.8, 111.9, 115.1, 115.2, 119.6, 121.2, 123.1, 126.7, 129.8, 135.8, 152.2, 152.5, 161.8; FTIR (neat) 1598, 1526, 1506, 1437 cm−1; HRMS (m/z): calcd. for C23H28N3O3 [M + H]+ 394.2125; found 394.2139; HPLC purity > 99.5%.
(1H-Indol-2-yl)(4-(2-(4-(trifluoromethoxy)phenoxy)ethyl)piperazin-1-yl)methanone (85).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and 4-(trifluoromethoxy)phenol (30.3 mg, 0.170 mmol, 1.6 equiv) were reacted according to General Procedure G to afford the aryl ether product as an off-white solid (27.7 mg, 0.0647 mmol, 61% yield). Rf = 0.35 (5% MeOH/CH2Cl2); mp = 147–150 °C. 1H NMR (500 MHz, CDCl3) δ 2.65–2.70 (m, 4H), 2.87 (t, J = 5.5 Hz, 2H), 3.85–4.02 (m, 4H), 4.13 (t, J = 5.5 Hz, 2H), 6.78 (dd, J = 0.9, 2.2 Hz, 1H), 6.88–6.93 (m, 2H), 7.12–7.17 (m, 3H), 7.29 (ddd, J = 1.2, 7.0, 8.2 Hz, 1H), 7.43 (dq, J = 0.9, 8.2 Hz, 1H), 7.65 (dq, J = 0.9, 8.0 Hz, 1H), 9.23 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 53.6, 57.0, 66.4, 105.3, 111.7, 115.3, 117.5, 119.5, 120.6, 121.6, 121.9, 122.5, 123.6, 124.5, 127.5, 129.2, 135.6, 142.9 (q, J = 1.6, 2.5 Hz), 157.1, 162.2; FTIR (neat) 1598, 1526, 1506, 1437 cm−1; HRMS (m/z): calcd. for C22H23F3N3O3 [M + H]+ 434.1686; found 434.1685; HPLC purity > 99.5%.
(1H-Indol-2-yl)(4-(2-(p-tolyloxy)ethyl)piperazin-1-yl)methanone (86).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and 4-methylphenol (18.6 mg, 0.172 mmol, 1.6 equiv) were reacted according to General Procedure G to afford the aryl ether product as a white solid (19.3 mg, 0.0535 mmol, 50% yield). Rf = 0.36 (5% MeOH/CH2Cl2); mp = 151–153 °C. 1H NMR (400 MHz, CDCl3) δ 2.29 (s, 3H), 2.65–2.71 (m, 4H), 2.86 (t, J = 5.6 Hz, 2H), 3.85–4.04 (m, 4H), 4.12 (t, J = 5.6 Hz, 2H), 6.78 (dd, J = 1.0, 2.2 Hz, 1H), 6.79–6.84 (m, 2H), 7.06–7.11 (m, 2H), 7.14 (ddd, J = 1.0, 7.0, 8.0 Hz, 1H), 7.26–7.31 (m, 1H), 7.42 (dq, J = 0.9, 8.2 Hz, 1H), 7.65 (dq, J = 1.0, 8.0 Hz, 1H), 9.20 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 20.5, 41.0, 53.6, 57.2, 65.9, 105.3, 111.7, 114.4, 120.6, 121.9, 124.4, 127.5, 129.2, 129.9, 130.2, 135.5, 156.5, 162.2; FTIR (neat) 1599, 1527, 1510, 1437 cm−1; HRMS (m/z): calcd. for C22H26N3O2 [M + H]+ 364.2020; found 364.2038; HPLC purity = 99.5%.
(4-(2-(4-Ethylphenoxy)ethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (87).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and 4-ethylphenol (22 mg, 0.18 mmol, 1.7 equiv) were reacted according to General Procedure G to afford the aryl ether product as a white solid (20.1 mg, 0.0535 mmol, 50% yield). Rf = 0.32 (5% MeOH/CH2Cl2); mp = 135–138 °C. 1H NMR (500 MHz, DMSO-d6) δ 1.14 (t, J = 7.6 Hz, 3H), 2.52–2.59 (m, 6H), 2.75 (t, J = 5.7 Hz, 2H), 3.70–3.82 (m, 4H), 4.07 (t, J = 5.7 Hz, 2H), 6.78 (dd, J = 0.9, 2.2 Hz, 1H), 6.84–6.88 (m, 2H), 7.04 (ddd, J = 1.0, 6.9, 7.9 Hz, 1H), 7.07–7.13 (m, 2H), 7.18 (ddd, J = 1.2, 6.9, 8.2 Hz, 1H), 7.42 (dq, J = 0.9, 8.3 Hz, 1H), 7.60 (dq, J = 0.9, 7.8 Hz, 1H), 11.57 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 15.9, 27.2, 53.1, 56.5, 65.3, 103.9, 112.0, 114.3, 119.7, 121.3, 123.1, 126.7, 128.6, 129.8, 135.7, 135.8, 156.4, 161.9; FTIR (neat) 1598, 1526, 1510, 1436 cm−1; HRMS (m/z): calcd. for C23H28N3O2 [M + H]+ 378.2176; found 378.2183; HPLC purity = 99.6%.
(4-(2-(4-(tert-Butyl)phenoxy)ethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (88).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and 4-(tert-butyl)phenol (27 mg, 0.18 mmol, 1.7 equiv) were reacted according to General Procedure G to afford the aryl ether product as a white solid (18.2 mg, 0.0477 mmol, 45% yield). Rf = 0.34 (5% MeOH/CH2Cl2); mp = 171–175 °C. 1H NMR (400 MHz, DMSO-d6) δ 1.25 (s, 9H), 2.57 (t, J = 5.1 Hz, 4H), 2.75 (t, J = 5.7 Hz, 2H), 3.70–3.83 (m, 4H), 4.08 (t, J = 5.7 Hz, 2H), 6.78 (dd, J = 0.9, 2.2 Hz, 1H), 6.84–6.90 (m, 2H), 7.04 (ddd, J = 1.0, 7.0, 8.0 Hz, 1H), 7.18 (ddd, J = 1.2, 7.0, 8.3 Hz, 1H), 7.25–7.30 (m, 2H), 7.41 (dq, J = 0.9, 8.3 Hz, 1H), 7.60 (dq, J = 0.9, 8.1 Hz, 1H), 11.57 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 31.2, 33.6, 53.0, 56.4, 65.2, 103.8, 111.9, 113.8, 119.6, 121.2, 123.1, 125.9, 126.7, 129.8, 135.8, 142.6, 156.0, 161.8; FTIR (neat) 1599, 1512, 1459, 1436 cm−1; HRMS (m/z): calcd. for C25H32N3O2 [M + H]+ 406.2489; found 406.2488; HPLC purity = 99.6%.
(4-(2-(4-Fluorophenoxy)ethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (89).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and 4-fluorophenol (22 mg, 0.19 mmol, 1.8 equiv) were reacted according to General Procedure G to afford the aryl ether product as an off-white solid (24.3 mg, 0.0668 mmol, 63% yield). Rf = 0.44 (5% MeOH/CH2Cl2); mp = 162–164 °C. 1H NMR (500 MHz, DMSO-d6) δ 2.57 (t, J = 5.1 Hz, 4H), 2.75 (t, J = 5.7 Hz, 2H), 3.72–3.80 (m, 4H), 4.09 (t, J = 5.7 Hz, 2H), 6.78 (dd, J = 0.9, 2.2 Hz, 1H), 6.95–6.99 (m, 2H), 7.04 (ddd, J = 1.0, 7.0, 8.0 Hz, 1H), 7.09–7.13 (m, 2H), 7.18 (ddd, J = 1.2, 6.9, 8.2 Hz, 1H), 7.42 (dq, J = 0.9, 8.3 Hz, 1H), 7.60 (dq, J = 0.9, 8.0 Hz, 1H), 11.58 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 52.8, 56.4, 65.9, 103.9, 112.0, 115.7 (d, J = 7.3 Hz), 115.9, 119.7, 121.3, 123.1, 126.7, 129.8, 135.8, 154.7 (d, J = 1.6 Hz), 161.9; FTIR (neat): 1596, 1527, 1504, 1436 cm−1; HRMS (m/z): calcd. for C21H23FN3O2 [M + H]+ 368.1769; found 368.1780; HPLC purity = 99.7%.
(4-(2-(4-Chlorophenoxy)ethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (90).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and 4-chlorophenol (25 mg, 0.19 mmol, 1.8 equiv) were reacted according to General Procedure G to afford the aryl ether product as a light yellow solid (24.1 mg, 0.0625 mmol, 59% yield). Rf = 0.43 (5% MeOH/CH2Cl2); mp = 170–173 °C. 1H NMR (500 MHz, DMSO-d6) δ 2.56 (t, J = 5.1 Hz, 4H), 2.75 (t, J = 5.6 Hz, 2H), 3.64–3.86 (m, 4H), 4.11 (t, J = 5.7 Hz, 2H), 6.78 (dd, J = 0.9, 2.2 Hz, 1H), 6.96–7.01 (m, 2H), 7.04 (ddd, J = 1.0, 7.0, 8.0 Hz, 1H), 7.18 (ddd, J = 1.2, 7.0, 8.3 Hz, 1H), 7.30–7.34 (m, 2H), 7.41 (dq, J = 0.9, 8.2 Hz, 1H), 7.60 (dq, J = 1.0, 8.0 Hz, 1H), 11.57 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 53.0, 56.2, 65.6, 103.8, 111.9, 116.2, 119.6, 121.2, 123.1, 124.1, 126.6, 129.1, 129.7, 135.8, 157.2, 161.8; FTIR (neat) 1596, 1526, 1490, 1436 cm−1; HRMS (m/z): calcd. for C21H23ClN3O2 [M + H]+ 384.1473; found 384.1474; HPLC purity = 99.7%.
(4-(2-(4-Bromophenoxy)ethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (91).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and 4-bromophenol (31 mg, 0.18 mmol, 1.7 equiv) were reacted according to General Procedure G to afford the aryl ether product as a light yellow solid (27.4 mg, 0.0646 mmol, 61% yield). Rf = 0.41 (5% MeOH/CH2Cl2); mp = 165–168 °C. 1H NMR (500 MHz, DMSO-d6) δ 2.56 (q, J = 5.2 Hz, 4H), 2.75 (t, J = 5.7 Hz, 2H), 3.68–3.82 (m, 4H), 4.10 (t, J = 5.7 Hz, 2H), 6.78 (dd, J = 0.9, 2.2 Hz, 1H), 6.92–6.96 (m, 2H), 7.04 (ddd, J = 1.0, 6.9, 7.9 Hz, 1H), 7.18 (ddd, J = 1.2, 6.9, 8.2 Hz, 1H), 7.40–7.46 (m, 3H), 7.60 (dq, J = 0.9, 8.1 Hz, 1H), 11.57 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 53.1, 56.3, 65.7, 103.9, 111.9, 112.0, 116.8, 119.7, 121.3, 123.1, 126.7, 129.8, 132.1, 135.8.; FTIR (neat) 1597, 1526, 1487, 1436 cm−1; HRMS (m/z): calcd. for C21H23BrN3O2 [M + H]+ 429.0968; found 430.0954; HPLC purity = 99.6%.
(1H-Indol-2-yl)(4-(2-(4-nitrophenoxy)ethyl)piperazin-1-yl)methanone (92).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and 4-nitrophenol (25 mg, 0.18 mmol, 1.7 equiv) were reacted according to General Procedure G to afford the aryl ether product as an off-white solid (5.3 mg, 0.0138 mmol, 13% yield). Rf = 0.40 (5% MeOH/CH2Cl2); mp = 188–204 °C. 1H NMR (500 MHz, DMSO-d6) δ 2.55–2.60 (m, 4H), 2.81 (t, J = 5.6 Hz, 2H), 3.68–3.82 (m, 4H), 4.28 (t, J = 5.6 Hz, 2H), 6.78 (dd, J = 0.9, 2.2 Hz, 1H), 7.04 (ddd, J = 1.0, 6.9, 7.9 Hz, 1H), 7.16–7.21 (m, 3H), 7.42 (dq, J = 1.0, 8.2 Hz, 1H), 7.60 (dq, J = 0.9, 8.0 Hz, 1H), 8.19–8.24 (m, 2H), 11.57 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 53.0, 56.1, 66.4, 103.9, 112.0, 115.1, 119.7, 121.3, 123.2, 125.9, 126.7, 129.8, 135.8, 140.8, 161.9, 163.8; FTIR (neat) 1592, 1509, 1437 cm−1; HRMS (m/z): calcd. for C21H23N4O4 [M + H]+ 395.1714; found 395.1712; HPLC purity > 99.5%.
(4-(2-(3,4-Dimethoxyphenoxy)ethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (93).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and 3,4-dimethoxyphenol (28 mg, 0.18 mmol, 1.7 equiv) were reacted according to General Procedure G to afford the aryl ether product as a colorless, sticky solid (25.5 mg, 0.0625 mmol, 59% yield). Rf = 0.36 (5% MeOH/CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 2.63–2.73 (m, 4H), 2.85 (t, J = 5.5 Hz, 2H), 3.84 (s, 3H), 3.85 (s, 3H), 3.92–4.05 (m, 4H), 4.09 (t, J = 5.5 Hz, 2H), 6.40 (td, J = 2.8, 7.2, 8.0 Hz, 1H), 6.54 (d, J = 2.8 Hz, 1H), 6.75–6.81 (m, 2H), 7.13 (t, J = 7.5 Hz, 1H), 7.27 (t, J = 7.6 Hz, 1H), 7.43 (d, J = 8.3 Hz, 1H), 7.64 (d, J = 8.0 Hz, 1H), 9.59 (br s, 1H); 13C NMR (101 MHz, CDCl3) δ 53.6, 55.9, 56.40, 56.44, 57.2, 66.3, 69.4, 101.0, 101.4, 103.7, 104.4, 105.2, 111.7, 111.8, 120.5, 121.8, 124.3, 127.4, 129.2, 143.7, 149.9, 153.2; FTIR (neat) 1596, 1510, 1437 cm−1; HRMS (m/z): calcd. for C23H28N3O4 [M + H]+ 410.2074; found 410.2088; HPLC purity > 99.5%.
(4-(2-(3,4-Dimethylphenoxy)ethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (94).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and 3,4-dimethylphenol (24 mg, 0.20 mmol, 1.9 equiv) were reacted according to General Procedure G to afford the aryl ether product as a white solid (20.7 mg, 0.0548 mmol, 52% yield). Rf = 0.42 (5% MeOH/CH2Cl2); mp = 129–137 °C. 1H NMR (400 MHz, DMSO-d6) δ 2.13 (s, 3H), 2.18 (s, 3H), 2.56 (t, J = 5.0 Hz, 4H), 2.73 (t, J = 5.7 Hz, 2H), 3.65–3.85 (m, 4H), 4.05 (t, J = 5.7 Hz, 2H), 6.66 (dd, J = 2.7, 8.3 Hz, 1H), 6.74–6.81 (m, 2H), 6.98–7.09 (m, 2H), 7.18 (ddd, J = 1.2, 7.0, 8.2 Hz, 1H), 7.41 (dd, J = 1.0, 8.2 Hz, 1H), 7.56–7.70 (m, 1H), 11.56 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 18.4, 19.6, 53.1, 56.5, 65.2, 103.9, 111.4, 112.0, 115.8, 119.7, 121.3, 123.1, 126.7, 127.9, 129.8, 130.1, 135.8, 137.2, 156.5, 161.9; FTIR (neat) 1600, 1526, 1502, 1437 cm−1; HRMS (m/z): calcd. for C23H28N3O2 [M + H]+ 378.2176; found 378.2181; HPLC purity = 99.2%.
(4-(2-(3-Chloro-4-methylphenoxy)ethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (95).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and 3-chloro-4-methylphenol (25 mg, 0.17 mmol, 1.7 equiv) were reacted according to General Procedure G to afford the aryl ether product as a white solid (25.0 mg, 0.0636 mmol, 60% yield). Rf = 0.53 (5% MeOH/CH2Cl2); mp = 156–158 °C. 1H NMR (500 MHz, DMSO-d6) δ 2.24 (s, 3H), 2.26 (t, J = 5.1 Hz, 4H), 2.74 (t, J = 5.6 Hz, 2H), 3.68–3.83 (m, 4H), 4.10 (t, J = 5.7 Hz, 2H), 6.78 (dd, J = 0.9, 2.2 Hz, 1H), 6.86 (dd, J = 2.6, 8.4 Hz, 1H), 7.02–7.06 (m, 2H), 7.18 (ddd, J = 1.2, 6.9, 8.2 Hz, 1H), 7.24 (dd, J = 0.8, 8.4 Hz, 1H), 7.41 (dq, J = 0.9, 8.2 Hz, 1H), 7.60 (dq, J = 0.9, 8.0 Hz, 1H), 11.57 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 18.6, 53.1, 56.3, 65.8, 103.9, 112.0, 113.8, 114.7, 119.7, 121.3, 123.1, 126.7, 127.0, 129.8, 131.6, 133.5, 135.8, 157.3, 161.9; FTIR (neat) 1599, 1526, 1495, 1437 cm−1; HRMS (m/z): calcd. for C22H25ClN3O2 [M + H]+ 398.1630; found 398.1640; HPLC purity = 99.3%.
(4-(2-(4-Chloro-3-methylphenoxy)ethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (96).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and 4-chloro-3-methylphenol (26 mg, 0.18 mmol, 1.7 equiv) were reacted according to General Procedure G to afford the aryl ether product as a white solid (25.1 mg, 0.0636 mmol, 60% yield). Rf = 0.49 (5% MeOH/CH2Cl2); mp = 146–148 °C. 1H NMR (500 MHz, DMSO-d6) δ 2.29 (s, 3H), 2.56 (t, J = 5.1 Hz, 4H), 2.75 (t, J = 5.7 Hz, 2H), 3.68–3.83 (m, 4H), 4.09 (t, J = 5.7 Hz, 2H), 6.78 (dd, J = 0.9, 2.2 Hz, 1H), 6.79–6.83 (m, 1H), 6.98 (dd, J = 0.8, 3.1 Hz, 1H), 7.04 (ddd, J = 1.0, 6.9, 8.0 Hz, 1H), 7.18 (ddd, J = 1.1, 6.9, 8.2 Hz, 1H), 7.29 (d, J = 8.8 Hz, 1H), 7.41 (dq, J = 1.0, 8.4 Hz, 1H), 7.60 (dq, J = 0.9, 8.0 Hz, 1H), 11.57 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 19.7, 53.1, 56.3, 65.6, 103.9, 112.0, 113.6, 117.2, 119.7, 121.3, 123.1, 124.5, 126.7, 129.4, 129.8, 135.8, 136.4, 157.2, 161.9; FTIR (neat) 1597, 1526, 1480, 1436 cm−1; HRMS (m/z): calcd. for C22H25ClN3O2 [M + H]+ 398.1630; found 398.1630; HPLC purity > 99.5%.
(4-(2-(2-Chloro-4-methoxyphenoxy)ethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (97).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and 2-chloro-4-methoxyphenol (30 mg, 0.19 mmol, 1.8 equiv) were reacted according to General Procedure G to afford the aryl ether product as a white solid (26.6 mg, 0.0647 mmol, 61% yield). Rf = 0.46 (5% MeOH/CH2Cl2); mp = 157–159 °C. 1H NMR (400 MHz, DMSO-d6) δ 2.60 (t, J = 5.1 Hz, 4H), 2.78 (t, J = 5.7 Hz, 2H), 3.72 (s, 3H) 3.73–3.82 (m, 4H), 4.13 (t, J = 5.7 Hz, 2H), 6.78 (dd, J = 0.9, 2.2 Hz, 1H), 6.87 (dd, J = 3.0, 9.0 Hz, 1H), 7.01–7.07 (m, 2H), 7.12 (d, J = 9.1 Hz, 1H), 7.18 (ddd, J = 1.2, 7.0, 8.2 Hz, 1H), 7.41 (dq, J = 0.9, 8.3 Hz, 1H), 7.60 (dq, J = 0.9, 8.0 Hz, 1H), 11.56 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 52.8, 55.7, 56.3, 67.5, 103.9, 112.0, 113.4, 115.3, 115.5, 119.7, 121.3, 122.0, 123.1, 126.7, 129.8, 135.8, 148.0, 153.5, 161.9; FTIR (neat) 1596, 1526, 1496, 1437 cm−1; HRMS (m/z): calcd. for C22H25ClN3O3 [M + H]+ 414.1579; found 414.1594; HPLC purity = 99.5%.
(1H-Indol-2-yl)(4-(2-(pyridin-2-yloxy)ethyl)piperazin-1-yl)methanone (98).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and pyridine-2-ol (16 mg, 0.17 mmol, 1.6 equiv) were reacted according to General Procedure G to afford the pyridyl ether product as a light yellow, sticky solid (21.4 mg, 0.0615 mmol, 58% yield). Rf = 0.27 (5% MeOH/CH2Cl2). 1H NMR (500 MHz, DMSO-d6) δ 2.49–2.54 (m, 4H), 2.61 (t, J = 6.4 Hz, 2H), 3.68–3.78 (m, 4H), 4.01 (t, J = 6.4 Hz, 2H), 6.21 (td, J = 1.4, 6.6 Hz, 1H), 6.37 (ddd, J = 0.6, 1.4, 9.1 Hz, 1H), 6.77 (dd, J = 0.9, 2.2 Hz, 1H), 7.04 (ddd, J = 1.1, 6.9, 8.0 Hz, 1H), 7.18 (ddd, J = 1.2, 6.9, 8.2 Hz, 1H), 7.37–7.43 (m, 2H), 7.59 (dq, J = 0.9, 8.0 Hz, 1H), 7.67 (ddd, J = 0.7, 2.1, 6.8 Hz, 1H), 11.57 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 45.5, 52.8, 55.9, 103.9, 104.8, 112.0, 119.3, 119.7, 121.3, 123.2, 126.7, 129.8, 135.8, 139.7, 139.9, 161.4, 161.9; FTIR (neat) 1652, 1572, 1538, 1435 cm−1; HRMS (m/z): calcd. for C20H23N4O2 [M + H]+ 351.1816; found 351.1844; HPLC purity = 99.4%.
(1H-Indol-2-yl)(4-(2-(pyridin-3-yloxy)ethyl)piperazin-1-yl)methanone (99).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and pyridine-2-ol (19 mg, 0.20 mmol, 1.9 equiv) were reacted according to General Procedure G to afford the pyridyl ether product as a tan solid (18.1 mg, 0.0517 mmol, 49% yield). Rf = 0.50 (10% MeOH/CH2Cl2); mp = 129–132 °C. 1H NMR (500 MHz, DMSO-d6) δ 2.58 (t, J = 5.1 Hz, 5H), 2.78 (t, J = 5.6 Hz, 2H), 3.69–3.85 (m, 4H), 4.19 (t, J = 5.6 Hz, 2H), 6.78 (dd, J = 0.9, 2.2 Hz, 1H), 7.04 (ddd, J = 1.0, 6.9, 7.9 Hz, 1H), 7.18 (ddd, J = 1.2, 6.9, 8.2 Hz, 1H), 7.33 (ddd, J = 0.7, 4.6, 8.4 Hz, 1H), 7.41 (dd, J = 1.2, 2.8 Hz, 1H), 7.43 (dd, J = 1.2, 2.9 Hz, 1H), 7.60 (dd, J = 1.0, 8.0 Hz, 1H), 8.17 (dd, J = 1.3, 4.6 Hz, 1H), 8.31 (dd, J = 0.7, 3.0 Hz, 1H), 11.58 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 53.1, 56.3, 65.6, 103.9, 112.0, 119.7, 121.0, 121.3, 123.1, 124.1, 126.7, 129.8, 135.8, 137.8, 141.7, 154.7, 161.9; FTIR (neat) 1598, 1574, 1525, 1427 cm−1; HRMS (m/z): calcd. for C20H23N4O2 [M + H]+ 351.1816; found 351.1815; HPLC purity > 99.5%.
(1H-Indol-2-yl)(4-(2-(pyridin-4-yloxy)ethyl)piperazin-1-yl)methanone (100).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and pyridin-4-ol (19 mg, 0.20 mmol, 1.9 equiv) were reacted according to General Procedure G to afford the pyridyl ether product as an off-white residue (9.0 mg, 0.025 mmol, 24% yield). Rf = 0.29 (5% MeOH/CH2Cl2). 1H NMR (400 MHz, DMSO-d6) δ 2.57 (t, J = 5.1 Hz, 4H), 2.78 (t, J = 5.6 Hz, 2H), 3.67–3.85 (m, 4H), 4.20 (t, J = 5.6 Hz, 2H), 6.78 (dd, J = 0.9, 2.2 Hz, 1H), 6.96–7.01 (m, 2H), 7.04 (ddd, J = 1.0, 6.9, 8.0 Hz, 1H), 7.18 (ddd, J = 1.2, 6.9, 8.2 Hz, 1H), 7.41 (dq, J = 1.0, 8.2 Hz, 1H), 7.58–7.62 (m, 1H), 8.34–8.41 (m, 2H), 11.56 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 53.0, 56.1, 65.3, 103.9, 110.4, 112.0, 119.7, 121.3, 123.1, 126.7, 129.8, 135.8, 150.9, 161.9, 164.2; FTIR (neat) 1638, 1608, 1549, 1436 cm−1; HRMS (m/z): calcd. for C20H23N4O2 [M + H]+ 351.1816; found 351.1808; HPLC purity > 99.5%.
(4-(2-((5-Chloropyridin-3-yl)oxy)ethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (101).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.9 mg, 0.106 mmol) and 5-chloropyridin-3-ol (26 mg, 0.20 mmol, 1.9 equiv) were reacted according to General Procedure G to afford the pyridyl ether product as a white solid (23.1 mg, 0.0604 mmol, 57% yield). Rf = 0.38 (5% MeOH/CH2Cl2); mp = 184–186 °C. 1H NMR (500 MHz, DMSO-d6) δ 2.57 (t, J = 5.0 Hz, 4H), 2.78 (t, J = 5.6 Hz, 2H), 3.68–3.83 (m, 4H), 4.24 (t, J = 5.6 Hz, 2H), 6.78 (dd, J = 0.9, 2.3 Hz, 1H), 7.04 (ddd, J = 1.0, 6.9, 8.0 Hz, 1H), 7.18 (ddd, J = 1.1, 6.9, 8.2 Hz, 1H), 7.42 (dq, J = 1.0, 8.2 Hz, 1H), 7.59–7.62 (m, 1H), 7.64–7.67 (m, 1H), 8.22 (d, J = 1.9 Hz, 1H), 8.29 (d, J = 2.5 Hz, 1H), 11.57 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 51.9, 55.0, 65.1, 102.8, 110.9, 118.6, 119.9, 120.2, 122.0, 125.6, 128.7, 130.0, 134.7, 135.7, 138.8, 154.1, 160.7; FTIR (neat) 1611, 1576, 1535, 1429 cm−1; HRMS (m/z): calcd. for C20H22ClN4O2 [M + H]+ 385.1426; found 385.1422; HPLC purity > 99.5%.
(1H-Indol-2-yl)(4-(2-(4-methoxyphenoxy)propyl)piperazin-1-yl)methanone (102).
1H-Indole-2-carboxylic acid (24 mg, 0.15 mmol, 1.3 equiv) and 1-(2-(4-methoxyphenoxy)propyl)piperazine 114d (29 mg, 0.11 mmol, 1.0 equiv) were reacted according to General Procedure E to afford the aryl ether product as an off-white solid (27.1 mg, 0.0684 mmol, 59% yield). Rf = 0.54 (5% MeOH/CH2Cl2); mp = 136–138 °C. 1H NMR (500 MHz, CDCl3) δ 1.30 (d, J = 6.1 Hz, 3H), 2.53 (dd, J = 4.4, 13.3 Hz, 1H), 2.65 (tq, J = 5.6, 6.3, 11.5 Hz, 4H), 2.74 (dd, J = 6.9, 13.3 Hz, 1H), 3.77 (s, 3H), 3.86–3.98 (m, 4H), 4.45 (pd, J = 4.3, 6.2 Hz, 1H), 6.76 (dd, J = 0.9, 2.2 Hz, 1H), 6.81–6.90 (m, 4H), 7.13 (ddd, J = 0.9, 6.9, 7.9 Hz, 1H), 7.26–7.30 (m, 1H), 7.42 (dq, J = 0.9, 8.4 Hz, 1H), 7.64 (dq, J = 0.9, 8.1 Hz, 1H), 9.23 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 18.6, 29.7, 53.8, 55.7, 63.5, 73.4, 105.2, 111.7, 114.7, 117.6, 120.6, 121.8, 124.4, 127.5, 129.3, 135.5, 151.8, 154.1, 162.2; FTIR (neat) 1597, 1526, 1504, 1438 cm−1; HRMS (m/z): calcd. for C23H28N3O3 [M + H]+ 394.2125; found 394.2124; HPLC purity > 99.5%.
(4-(2-(4-Chlorophenoxy)propyl)piperazin-1-yl)(1H-indol-2-yl)methanone (103).
1H-Indole-2-carboxylic acid (22.0 mg, 0.137 mmol, 1.2 equiv) and 1-(2-(4-chlorophenoxy)propyl)piperazine 114e (29.0 mg, 0.116 mmol, 1.0 equiv) were reacted according to General Procedure E to afford the acylated product 103 as a white solid (22.3 mg, 0.0558 mmol, 49% yield). Rf = 0.46 (5% MeOH/CH2Cl2); mp = 149–151 °C. 1H NMR (500 MHz, CDCl3) δ 1.32 (d, J = 6.2 Hz, 3H), 2.55 (dd, J = 4.4, 13.4 Hz, 1H), 2.64 (tq, J = 5.6, 6.4, 11.5 Hz, 4H), 2.74 (dd, J = 6.8, 13.4 Hz, 1H), 3.82–4.01 (m, 4H), 4.53 (pd, J = 4.3, 6.2 Hz, 1H), 6.76 (dd, J = 0.9, 2.2 Hz, 1H), 6.83–6.87 (m, 2H), 7.13 (ddd, J = 1.0, 7.0, 8.0 Hz, 1H), 7.21–7.25 (m, 2H), 7.26–7.30 (m, 1H), 7.42 (dq, J = 0.9, 8.3 Hz, 1H), 7.64 (dq, J = 0.9, 8.0 Hz, 1H), 9.26 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 18.4, 29.7, 53.8, 63.3, 72.8, 105.2, 111.7, 117.3, 120.6, 121.8, 124.4, 125.7, 127.4, 129.2, 129.4, 135.5, 156.4, 162.2; FTIR (neat) 1595, 1526, 1487, 1436 cm−1; HRMS (m/z): calcd. for C22H25ClN3O2 [M + H]+ 398.1630; found 398.1633; HPLC purity = 99.7%.
(1H-Indol-2-yl)(4-(2-((4-methoxyphenyl)(methyl)amino)ethyl)piperazin-1-yl)methanone (104).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone in DMF (200 μL, 0.528 M, 0.106 mmol) and 4-methoxy-N-methylaniline (24 mg, 0.18 mmol, 1.6 equiv) were reacted according to General Procedure G to afford the tertiary aniline product as a white solid (1.9 mg, 0.0048 mmol, 5% yield). HRMS (m/z): calcd. for C23H29N4O2 [M + H]+ 393.2285; found 393.2283; HPLC purity > 99.5%.
(4-(2-((1H-Indol-5-yl)oxy)ethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (105).
(4-(2-Chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (30.8 mg, 0.106 mmol) and 5-hydroxyindole (22.6 mg, 0.170 mmol, 1.6 equiv) were reacted according to General Procedure F and purified by mass-directed, preparative HPLC to afford the aryl ether product as a colorless oil (9.8 mg, 0.025 mmol, 24% yield). 1H NMR (500 MHz, DMSO-d6) δ 2.59 (t, J = 5.1 Hz, 4H), 2.77 (t, J = 5.8 Hz, 2H), 3.70–3.85 (m, 4H), 4.10 (t, J = 5.7 Hz, 2H), 6.31–6.33 (m, 1H), 6.74 (dd, J = 2.4, 8.7 Hz, 1H), 6.79 (dd, J = 0.8, 2.2 Hz, 1H), 7.03–7.07 (m, 2H), 7.16–7.20 (m, 1H), 7.26–7.28 (m, 2H), 7.41 (dd, J = 0.9, 8.3 Hz, 1H), 7.61 (dd, J = 0.6, 8.0 Hz, 1H), 10.90 (br s, 1H), 11.57 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 53.17, 53.19, 56.7, 65.9, 100.8, 102.8, 103.9, 111.6, 111.9, 112.0, 119.7, 121.3, 123.1, 125.7, 126.7, 127.9, 129.8, 131.0, 135.8, 152.2, 161.9; HRMS (m/z): calcd. for C23H25N4O2 [M + H]+ 389.1972; found 389.1971; HPLC purity = 96.8%.
(1H-Indol-2-yl)(4-(3-(4-methoxyphenoxy)propyl)piperazin-1-yl)methanone (106).
(1H-Indol-2-yl)(piperazin-1-yl)methanone (43 mg, 0.19 mmol) and 1-(3-bromopropoxy)-4-methoxybenzene (55 mg, 0.23 mmol, 1.2 equiv) were reacted according to General Procedure A to afford the alkylated piperazine as a tan solid (61 mg, 0.16 mmol, 83% yield). Rf = 0.36 (5% MeOH/CH2Cl2); mp = 122–127 °C. 1H NMR (400 MHz, CDCl3) δ 1.95–2.02 (m, 2H), 2.53–2.61 (complex, 6H), 3.77 (s, 3H), 3.94–4.09 (complex, 6H), 6.78 (d, J = 1.9 Hz, 1H), 6.83–6.88 (complex, 4H), 7.13 (t, J = 7.5 Hz, 1H), 7.25–7.29 (m, 1H), 7.45 (d, J = 8.3 Hz, 1H), 7.65 (d, J = 8.0 Hz, 1H), 10.02 (br s, 1H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d 55.7, 105.2, 111.9, 114.7, 115.5, 120.4, 121.7, 124.2; u 26.8, 53.19, 53.23, 55.0, 66.6, 127.4, 129.3, 135.9, 153.1, 153.8, 162.5; FTIR (neat) 1595, 1526, 1505, 1436 cm−1; HRMS (m/z): calcd. for C23H28N3O3 [M + H]+ 394.2125; found 394.2117; HPLC purity = 97.2 %.
(1H-Indol-2-yl)(4-(4-(4-methoxyphenoxy)butyl)piperazin-1-yl)methanone (107).
(1H-Indol-2-yl)(piperazin-1-yl)methanone (38 mg, 0.17 mmol) and 1-(4-bromobutoxy)-4-methoxybenzene (52 mg, 0.20 mmol, 1.2 equiv) were reacted according to General Procedure A to afford the alkylated piperazine as a tan solid (51 mg, 0.13 mmol, 76% yield). Rf = 0.36 (5% MeOH/CH2Cl2); mp = 111–113 °C. 1H NMR (400 MHz, CDCl3) δ 1.64–1.72 (m, 2H), 1.77–1.84 (m, 2H), 2.44 (t, J = 7.4 Hz, 2H), 2.53 (t, J = 5.0 Hz, 4H), 3.75 (s, 3H), 3.91–3.99 (complex, 6H), 6.75–6.77 (m, 1H), 6.80–6.85 (m, 1H), 7.09–7.13 (m, 1H), 7.23–7.27 (m, 1H), 7.42 (dd, J = 0.7, 8.3 Hz, 1H), 7.63 (dd, J = 0.6, 7.4 Hz, 1H), 9.92 (br s, 1H); 13C NMR (101 MHz, CDCl3) δ 23.3, 27.3, 53.2 (br, 2 C), 55.7, 58.1, 68.2, 105.2, 111.9, 114.6, 115.4, 120.4, 121.7, 124.2, 127.4, 129.3, 135.8, 153.1, 153.7, 162.5; FTIR (neat) 1593, 1506, 1463, 1437 cm−1; HRMS (m/z): calcd. for C24H30N3O3 [M + H]+ 408.2282; found 408.2265; HPLC purity = 98.1%.
(1H-Indol-2-yl)(4-(2-(4-methoxyphenoxy)ethyl)piperidin-1-yl)methanone (108).
To a mixture of 2-(1-(1H-indole-2-carbonyl)piperidin-4-yl)ethyl 4-methylbenzenesulfonate 122 (82 mg, 0.19 mmol) and potassium carbonate (80 mg, 0.58 mmol, 3.0 equiv) in MeCN (3 mL) was added 4-methoxyphenol (72 mg, 0.58 mmol, 3 equiv) in THF (5 mL) and the reaction was stirred at 60 °C for 17 h. After cooling to rt, the reaction was filtered and the solids washed with CH2Cl2 (2 × 5 mL). The combined filtrates were evaporated and the residue purified by silica gel chromatography to afford the aryl ether product 108 as an off-white solid (51 mg, 0.14 mmol, 70% yield). Rf = 0.58 (5% MeOH/CH2Cl2); mp = 176–179 °C. 1H NMR (400 MHz, CDCl3) δ 1.29–1.41 (m, 2H), 1.74–1.80 (m, 2H), 1.87–1.95 (complex, 3H), 2.97–3.17 (br m, 2H), 3.78 (s, 3H), 4.00 (t, J = 6.2 Hz, 2H), 4.70–4.4.76 (m, 2H), 6.77–6.79 (m, 1H), 6.84–6.85 (m, 4H), 7.11–7.15 (m, 1H), 7.25–7.30 (m, 1H), 7.43 (dd, J = 0.7, 8.3 Hz, 1H), 7.65 (dd, J = 0.6, 8.0 Hz, 1H), 9.35 (br s, 1H); 13C NMR (101 MHz, CDCl3) δ 32.4 (br, 2 C), 33.2, 35.8, 55.7, 65.8, 104.9, 111.7, 114.7, 115.4, 120.5, 121.8, 124.2, 127.5, 129.6, 135.6, 153.0, 153.8, 162.2; FTIR (neat) 1596, 1534, 1505, 1440 cm−1; HRMS (m/z): calcd. for C23H27N2O3 [M + H]+ 379.2016; found 379.2020; HPLC purity = 97.8%.
1-(1H-Indol-2-yl)-2-(4-(2-(4-methoxyphenoxy)ethyl)piperazin-1-yl)ethan-1-one (109).
1-(2-(4-Methoxyphenoxy)ethyl)piperazine (60 mg, 0.25 mmol), 2-chloro-1-(1H-indol-2-yl)ethan-1-one (49 mg, 0.25 mmol, 1.0 equiv), potassium carbonate (140 mg, 1.02 mmol, 4.0 equiv) and potassium iodide (42 mg, 0.25 mmol, 1.0 equiv) were charged in a reaction vial and slurried with MeCN (4 mL) and stirred at 65 °C for 18 h. The reaction was filtered and the solids washed with CH2Cl2 (2 × 5 mL). The combined filtrates were evaporated and the residue purified by silica gel chromatography to afford the aryl ether product as a tan residue (51 mg, 0.13 mmol, 51% yield). Rf = 0.30 (5% MeOH/CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 2.64–2.75 (complex, 8H), 2.82 (t, J = 5.8 Hz, 2H), 3.75–3.77 (complex, 5H), 4.06 (t, J = 5.8 Hz, 2H), 6.79–6.85 (complex, 4H), 7.12–7.17 (m, 1H), 7.32–7.38 (m, 2H), 7.43 (d, J = 8.3 Hz, 1H), 7.71 (d, J = 8.1 Hz, 1H), 9.78 (br s, 1H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d 55.7, 109.4, 112.3, 114.6, 115.6, 121.0, 123.1, 126.4; u 53.4, 53.6, 57.3, 64.8, 66.5, 127.4, 134.4, 137.1, 152.9, 153.9, 189.6; HRMS (m/z): calcd. for C23H28N3O3 [M + H]+ 394.2125; found 394.2132; HPLC purity = 96.9%.
(4-Methoxyphenyl)(piperazin-1-yl)methanone (111a).
This material was purchased from Oakwood Chemical.
(2-Methoxyphenyl)(piperazin-1-yl)methanone (111b).
This material was purchased from 1Click Chemistry Inc.
(3-Methoxyphenyl)(piperazin-1-yl)methanone (111c).
This material was purchased from Combi-Blocks Inc.
(4-Chlorophenyl)(piperazin-1-yl)methanone (111d).
This material was synthesized according to the protocol of Wang and coworkers.80 Thus, to a solution of piperazine (1.26 g, 14.6 mmol) in THF (100 ml) was cooled in a rt water bath and butyllithium (13.8 ml, 30.7 mmol) was added. The reaction was stirred at rt for 30 min and 4-chlorobenzoyl chloride (1.9 ml, 14.6 mmol) was added as a solution in THF (10 mL). The reaction stirred for 10 additional mins and quenched with MeOH (10 mL). All solvents were removed in vacuo and the residue was partitioned between saturated, aqueous NaHCO3 (50 mL) and EtOAc (25 mL). The aqueous layer was extracted with EtOAc (2 × 25 mL) and the combined organic layers dried (Na2SO4), concentrated in vacuo and purified by silica chromatography to afford the previously reported acylated product.81 (818 mg, 2.72 mmol, 68% yield). 1H NMR (400 MHz, MeOD-d4) δ 2.68–2.96 (m, 4H), 3.34–3.78 (m, 4H), 7.36–7.44 (m, 2H), 7.44–7.51 (m, 2H); 13C NMR (101 MHz, MeOD-d4) δ 42.1, 45.6, 128.4, 128.5, 134.0, 135.6, 169.9.
(4-Ethylphenyl)(piperazin-1-yl)methanone (111e).
To a solution of tert-butyl piperazine-1-carboxylate (1.44 g, 7.73 mmol) and triethylamine (2.69 mL, 19.33 mmol, 2.5 equiv) in THF (120 mL) at 0 °C was added 4-ethylbenzoyl chloride (1.25 mL, 8.50 mmol, 1.1 equiv). The reaction was stirred overnight (19 h), slowly warming to rt. The reaction was quenched with saturated aqueous NaHCO3 and the aqueous layer extracted with EtOAc (2 × 30 mL). The combined organic layers were dried (Na2SO4), evaporated and purified by silica chromatography to afford the acylated product, tert-butyl 4-(4-ethylbenzoyl)piperazine-1-carboxylate, as a white solid (2.14 g, 6.72 mmol, 87% yield) 1H NMR (400 MHz, CDCl3) δ 1.23 (t, J = 7.6 Hz, 3H), 1.45 (s, 9H), 2.66 (q, J = 7.6 Hz, 2H), 3.31–3.74 (m, 8H), 7.19–7.27 (m, 2H), 7.31 (d, J = 8.1 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 15.2, 15.4, 28.4, 28.7, 29.0, 80.3, 127.2, 127.8, 128.0, 130.2, 132.7, 146.4, 154.6, 170.9.
To a solution of tert-butyl 4-(4-ethylbenzoyl)piperazine-1-carboxylate (1.90 g, 5.97 mmol) in CH2Cl2 (60 mL) was added trifluoroacetic acid (9.19 mL, 119 mmol, 20 equiv). The reaction was stirred at rt for 4 h and concentrated under vacuum. The residue was partitioned between aqueous, saturated sodium bicarbonate (100 mL) and CH2Cl2 (3 × 30 mL). The combined organic layers were concentrated under vacuum to afford the piperazine product 111e as a white solid (1.87 g, 5.63 mmol, 94% yield). 1H NMR (400 MHz, MeOD-d4) δ 1.24 (t, J = 7.6 Hz, 3H), 2.68 (q, J = 7.6 Hz, 2H), 2.72–2.94 (m, 4H), 3.35–3.78 (m, 4H), 7.25–7.35 (m, 4H); 13C NMR (101 MHz, MeOD-d4) δ 14.5, 28.3, 42.3, 45.4, 126.8, 127.7, 132.6, 146.5, 171.3.
(4-Methoxyphenyl)(2-methylpiperazin-1-yl)methanone (111f).
To a solution of 2-methylpiperazine (0.99g, 9.88 mmol) in THF (100 mL) was added butyllithium (2.22 M in hexanes, 9.35 mL, 20.76 mmol, 2.1 equiv) and the reaction stirred at rt for 30 mins. This preformed anion solution was cooled to 0 °C and a solution of 4-methoxybenzoyl chloride (1.68 g, 9.88 mmol, 1.0 equiv) in THF (10 mL) was added. After stirring at 0 °C for 10 mins, the reaction was quenched by the addition of MeOH (10 mL) and the solvents removed in vacuo. The residue was partitioned between saturated, aqueous NaHCO3 (50 mL) and EtOAc (3 × 25 mL). The combined organic layers were dried (Na2SO4), concentrated in vacuo and purified on C-18 functionalized silica gel chromatography to afford the acylated product 111f as a tan solid (427.9 mg, 1.83 mmol, 18% yield).
(4-Methoxyphenyl)(2-methylpiperazin-1-yl)methanone (111g).
To a solution of tert-butyl 3-methylpiperazine-1-carboxylate (1.20 g, 6.01 mmol) and triethylamine (1.0 mL, 7.17 mmol, 1.2 equiv) in CH2Cl2 (25 mL) was added 4-methoxybenzoyl chloride (1.12 g, 6.54 mmol, 1.1 equiv). The reaction was stirred at rt for 4 h and quenched with aqueous citric acid (1 M, 15 mL). The organic layer was washed with water (5 mL) then aqueous NaOH (1M, 5 mL) and dried with MgSO4. The solvent was removed in vacuo and the residue purified by silica chromatography to afford the acylated product, tert-butyl 4-(4-methoxybenzoyl)-3-methylpiperazine-1-carboxylate, as a colorless oil (1.98 g, 5.92 mmol, 98% yield). 1H NMR (400 MHz, CDCl3) δ 1.25 (d, J = 6.7 Hz, 3H), 1.47 (s, 9H), 2.69–3.27 (complex, 5H), 3.84 (s, 3H), 3.96–4.15 (m, 2H), 6.92 (d, J = 8.8 Hz, 2H), 7.35 (d, J = 8.8 Hz, 2H).
To a solution of tert-butyl 4-(4-methoxybenzoyl)-3-methylpiperazine-1-carboxylate (1.98 g, 5.91 mmol) in CH2Cl2 (10 mL) was added trifluoroacetic acid (10 mL, 129 mmol, 22 equiv). The reaction was stirred at rt for 5 h and concentrated under vacuum. The residue was partitioned between aqueous sodium hydroxide (1 M, 50 mL) and CH2Cl2 (4 × 10 mL). The combined organic layers were dried with Na2SO4, concentrated under vacuum and purified by silica chromatography to afford the piperazine product 111g as a viscous, tan oil (1.30 g, 5.56 mmol, 94% yield). 1H NMR (400 MHz, CDCl3) δ 1.36 (d, J = 6.9 Hz, 3H), 2.76 (dt, J = 3.5, 12.3 Hz, 1H), 2.82–2.87 (m, 1H), 2.97 (dd, J = 4.0, 12.4 Hz, 1H), 3.00–3.05 (m, 1H), 3.16–3.27 (m, 1H), 3.83 (s, 3H), 3.91–4.05 (m, 1H), 4.34–4.48 (m, 1H), 6.88–6.96 (m, 2H), 7.32–7.38 (m, 2H); 13C NMR (126 MHz, DMSO-d6) δ 15.5, 40.7, 46.1, 47.6, 50.2, 55.4, 113.8, 128.3, 128.6, 160.6, 170.6.
(1H-Indol-2-yl)(piperazin-1-yl)methanone (111h).
1H-Indole-2-carboxylic acid (1.55 g, 9.62 mmol) and HATU (4.39 g, 11.55 mmol, 1.2 equiv) were dissolved in EtOAc (80 mL) and stirred at rt for 10 mins followed by the addition of a tert-butyl piperazine-1-carboxylate (1.79 g, 9.62 mmol, 1.0 equiv) solution in THF (80 mL). After stirring at rt for an additional 10 mins, Et3N (4.0 mL, 28.87 mmol, 3.0 equiv) was added and the reaction stirred at rt for 43 h. The solvents were removed in vacuo and the residue washed with water (4 × 50 mL). The crude solid product was further purified by silica chromatography to afford the amide piperazine product, tert-butyl 4-(1H-indole-2-carbonyl)piperazine-1-carboxylate, as a white solid (2.65 g, 9.62 mmol, 84% yield). 1H NMR (400 MHz, CDCl3) δ 1.49 (s, 9H), 3.56 (dd, J = 4.2, 6.4 Hz, 4H), 3.91 (s, 4H), 6.76–6.81 (m, 1H), 7.14 (ddd, J = 1.0, 7.0, 8.0 Hz, 1H), 7.25–7.34 (m, 1H), 7.43 (dd, J = 1.0, 8.4 Hz, 1H), 7.62–7.69 (m, 1H), 9.29 (br s, 1H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d 28.4, 105.4, 111.7, 120.7, 121.9, 124.6; u 37.2, 41.9, 80.4, 127.4, 128.9, 135.6, 154.6, 162.6.
A solution of tert-butyl 4-(1H-indole-2-carbonyl)piperazine-1-carboxylate (1.66 g, 5.03 mmol) in hexafluoroisopropanol (15 mL) was heated under microwave irradiation at 145 °C for 2.5 h and cooled to rt. The solvent was removed in vacuo and the residue recrystallized from methyl tert-butyl ether to afford the free base piperazine product 111h as a white solid (998 mg, 4.35 mmol, 87% yield). Mp 185–187 °C. 1H NMR (400 MHz, CDCl3) δ 1.49 (s, 9H), 3.56 (dd, J = 4.2, 6.4 Hz, 4H), 3.91 (s, 4H), 6.76–6.81 (m, 1H), 7.14 (ddd, J = 1.0, 7.0, 8.0 Hz, 1H), 7.25–7.34 (m, 1H), 7.43 (dd, J = 1.0, 8.4 Hz, 1H), 7.62–7.69 (m, 1H), 9.29 (br s, 1H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d 105.1, 111.7, 120.2, 121.5, 124.0; u 45.5 (2C), 127.0, 128.7, 136.0, 163.1.
1-(2-Bromoethoxy)-4-methoxybenzene (112a).
This material was synthesized as previously described.82
1-(2-Bromoethoxy)-2-methoxybenzene (112b).
2-Methoxyphenol (2.84 g, 22.88 mmol) was reacted according to General Procedure H to afford the bromide as a white solid (2.0 g, 8.65 mmol, 38% yield). Rf = 0.70 (25% EtOAc in hexanes); 1H NMR (400 MHz, CDCl3) δ 3.62 (t, J = 6.6 Hz, 2H), 3.87 (s, 3H), 4.33 (t, J = 6.9 Hz, 2H), 6.88–7.03 (complex, 4H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d 56.0, 112.3, 115.0, 120.9, 122.4; u 28.9, 69.3, 147.5, 149.9. These data are in agreement with those previously reported.83
1-(2-Bromoethoxy)-3-methoxybenzene (112c).
3-Methoxyphenol (2.05 g, 16.51 mmol) was reacted according to General Procedure H to afford the bromide as a colorless oil (1.90 g, 8.22 mmol, 50% yield). Rf = 0.71 (25% EtOAc in hexanes); 1H NMR (400 MHz, CDCl3) δ 3.62 (t, J = 6.3 Hz, 2H), 3.78 (s, 3H), 4.26 (t, J = 6.3 Hz, 2H), 6.47–6.55 (complex, 3H), 7.16–7.21 (m, 1H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d 55.3, 101.3, 106.7, 107.1, 130.0; u 29.2, 67.9, 159.3, 160.9; IR (thin film): 1589, 1491, 1450 cm−1.
1-(2-Bromoethoxy)-4-ethylbenzene (112d).
4-Ethylphenol (3.55 g, 29.10 mmol) was reacted according to General Procedure H to afford the bromide as a dark golden oil (2.10 g, 9.17 mmol, 31% yield). Rf = 0.79 (25% EtOAc in hexanes); 1H NMR (400 MHz, CDCl3) δ 1.19 (t, J = 7.6 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.56 (t, J = 6.3 Hz, 2H), 4.20 (t, J = 6.3 Hz, 2H), 6.78–6.86 (m, 2H), 7.07–7.10 (m, 2H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d 16.0, 114.8, 128.9; u 28.1, 29.4, 68.1, 137.3, 156.2. These data are in agreement with those previously reported.84
1-(2-Bromoethoxy)-4-chlorobenzene (112e).
4-Chlorophenol (3.18 g, 24.74 mmol) was reacted according to General Procedure H to afford the bromide as a white solid (2.80 g, 11.89 mmol, 48% yield). Rf = 0.73 (25% EtOAc in hexanes); 1H NMR (400 MHz, CDCl3) δ 3.61 (t, J = 6.2 Hz, 2H), 4.23 (t, J = 6.2 Hz, 2H), 6.80–6.87 (m, 2H), 7.21–7.25 (m, 2H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d 116.1, 129.5; u 29.0, 68.2, 126.4, 156.7; IR (neat): 1594, 1581, 1488, 1457 cm−1.These data are in agreement with those previously reported.85
1-(2-Bromoethoxy)-3,5-dimethoxybenzene (112f).
3,5-Dimethoxyphenol (3.02 g, 19.6 mmol) was reacted according to General Procedure H to afford the bromide as a colorless oil (2.70 g, 10.3 mmol, 53% yield). Rf = 0.43 (25% EtOAc in hexanes); 1H NMR (400 MHz, CDCl3) 3.61 (t, J = 6.2 Hz, 2H), 3.76 (s, 6H), 4.24 (t, J = 6.2 Hz, 2H), 6.08 (d, J = 2.1 Hz, 2H), 6.10–6.12 (m, 1H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d 55.4, 93.61, 93.62; u 29.1, 67.9, 160.0, 161.6; IR (thin film): 1590, 1474, 1456 cm−1. These data are in agreement with those previously reported.86
5-(2-Bromoethoxy)benzo[d][1,3]dioxole (112g).
3,4-(Methylenedioxy)phenol (2.31 g, 16.72 mmol) was reacted according to General Procedure H to afford the bromide as a faintly purple solid (2.70 g, 11.0 mmol, 66% yield). Rf = 0.55 (25% EtOAc in hexanes); 1H NMR (400 MHz, CDCl3) δ 3.50 (t, J = 6.3 Hz, 2H), 4.11 (t, J = 6.2 Hz, 2H), 5.82 (s, 2H), 6.24 (dd, J = 2.5, 8.4 Hz, 1H), 6.42 (d, J = 2.5 Hz, 1H), 6.61 (d, J = 8.4 Hz, 1H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d 98.6, 106.3, 108.0; u 29.3, 69.0, 101.3, 142.3, 148.4, 153.5; IR (thin film): 1632, 1494, 1481 cm−1.
3-(2-Chloroethoxy)pyridine (112h).
To a mixture of 3-hydroxypyridine (872 mg, 9.17 mmol) and potassium carbonate (2.54 mg, 18.35 mmol, 2.0 equiv) in DMF (15 mL) was added 1-dibromo-2-chloroethane (3.0 mL, 5.19 g, 36.2 mmol, 4.0 equiv). The reaction was heated at 60 °C for 22 h, cooled to rt and partitioned between water (150 mL) and CH2Cl2 (5 × 10 mL). The combined organic layers were washed with aqueous sodium hydroxide (1 M, 2 × 10 mL) then water (2 × 10 mL) and dried with Na2SO4. The organic layer was adsorbed onto silica and purified by silica chromatography to afford the ether product as a dark orange oil (284 mg, 1.80 mmol, 20% yield). 1H NMR (400 MHz, CDCl3) δ 3.84 (t, J = 5.8 Hz, 2H), 4.29 (t, J = 5.8 Hz, 2H), 7.22–7.25 (m, 2H), 8.27 (dd, J = 2.2, 3.8 Hz, 1H), 8.34 (dd, J = 1.3, 2.4 Hz, 1H).
1-(3-Bromopropoxy)-4-methoxybenzene (112i).
This material was purchased from 1Click Chemistry Inc.
1-(4-Bromobutoxy)-4-methoxybenzene (112j).
This material was purchased from 1Click Chemistry Inc.
1-(4-Methoxyphenyl)piperazine (113).
This material was purchased from Combi-Blocks Inc.
1-(2-(4-Methoxyphenoxy)ethyl)piperazine (114a).
A mixture of tert-butyl piperazine-1-carboxylate (490 mg, 2.63 mmol), 1-(2-bromoethoxy)-4-methoxybenzene 112a (608 mg, 2.63 mmol), potassium carbonate (727 mg, 5.26 mmol, 2.0 equiv) and potassium iodide (44 mg, 0.263 mmol) in acetonitrile (35 mL) was heated at 70 °C for 16 h. The reaction was cooled to rt, filtered and the solids washed with acetonitrile (2 × 15 mL). The combined organics were adsorbed onto celite and purified by silica chromatography to afford the ether product, tert-butyl 4-(2-(4-methoxyphenoxy)ethyl)piperazine-1-carboxylate, as a colorless oil (622 mg, 1.85 mmol, 70% yield). Rf = 0.57 (EtOAc); 1H NMR (400 MHz, CDCl3) δ 1.46 (s, 9 H), 2.51 (t, J = 4.8 Hz, 4 H), 2.79 (t, J = 6.0 Hz, 2 H), 3.45 (t, J = 4.8 Hz, 4 H), 3.76 (s, 3 H), 4.05 (t, J = 6.0 Hz, 2 H), 6.83 (d, J = 2.4 Hz, 4 H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d 28.5, 55.8, 114.7, 115.7; u 53.5, 57.5, 66.7, 79.6, 153.0, 154.1, 154.8; HRMS (m/z): calcd. for C18H29N2O4 ([M]++H) 337.2122; found 337.2122; HPLC purity = 99.0%.
To a solution of tert-butyl 4-(2-(4-methoxyphenoxy)ethyl)piperazine-1-carboxylate (185 mg, 0.55 mmol) and triethylsilane (96 mg, 0.825 mmol) in CH2Cl2 (5 mL) was added trifluoroacetic acid (0.85 mL, 1,254 mg, 11.00 mmol). The reaction was stirred at rt for 4 h and concentrated under vacuum. The residue was partitioned between aqueous sodium bicarbonate (10 mL) and CH2Cl2 (3 × 10 mL). The combined organic layers were concentrated under vacuum to afford the piperazine product 114a as a colorless oil (118 mg, 0.499 mmol, 91% yield). 1H NMR (400 MHz, CDCl3) δ 2.54–2.60 (m, 4 H), 2.78 (t, J = 6.0 Hz, 2 H), 2.94 (t, J = 4.8 Hz, 4 H), 3.76 (s, 3 H), 4.05 (t, J = 6.0 Hz, 2 H), 6.83 (d, J = 2.8 Hz, 4 H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d 55.8, 114.8, 115.7; u 45.9, 54.6, 58.0, 66.6, 153.0, 154.0; HRMS (m/z): calcd. for C13H21N2O2 ([M]++H) 237.1598; found 237.1573; HPLC purity = 94.1%.
1-(2-(3-Methoxyphenoxy)ethyl)piperazine (114b).
This material was purchased from 1Click Chemistry Inc.
1-(2-(Pyridin-3-yloxy)ethyl)piperazine (114c).
A slurry of tert-butyl piperazine-1-carboxylate (699 mg, 3.59 mmol), 3-(2-chloroethoxy)pyridine 112h (283 mg, 1.80 mmol) and potassium carbonate (499 mg, 3.61 mmol, 2.0 equiv) in DMF (2 mL) was heated at 90 °C for 21 h. The reaction was cooled to rt, diluted with water (20 mL) and extracted with CH2Cl2 (4 × 5 mL). The combined organics were dried with Na2SO4, concentrated under vacuum and purified by silica chromatography to afford the alkylated product, tert-butyl 4-(2-(4 pyridin-3-yloxy)ethyl)piperazine-1-carboxylate, as a golden oil (434 mg, 1.41 mmol, 79% yield). 1H NMR (400 MHz, CDCl3) δ 2.53 (t, J = 5.0 Hz, 4H), 2.83 (t, J = 5.6 Hz, 2H), 3.46 (t, J = 5.1 Hz, 4H), 4.15 (t, J = 5.7 Hz, 2H), 7.19–7.23 (m, 2H), 8.20–8.26 (m, 1H), 8.32–8.34 (m, 1H).
To a solution of tert-butyl 4-(2-(4 pyridin-3-yloxy)ethyl)piperazine-1-carboxylate (1.54 g, 5.02 mmol) and triethylsilane (0.874 g, 7.51 mmol, 1.5 equiv) in CH2Cl2 (6 mL) was added trifluoroacetic acid (6.0 mL, 8.94 g, 78.4 mmol). The reaction was stirred at rt for 19 h and concentrated under vacuum. The residue was partitioned between aqueous sodium hyroxide (5 M, 11 mL) and CH2Cl2 (5 × 10 mL). The combined organic layers were dried with Na2SO4, concentrated under vacuum and purified by chromatography on basic alumina (50 g) to afford the piperazine product 114c as a light orange oil (915 mg, 4.41 mmol, 88% yield). 1H NMR (400 MHz, CDCl3) δ 2.49–2.61 (m, 4H), 2.81 (t, J = 5.8 Hz, 2H), 2.92 (t, J = 4.9 Hz, 4H), 4.15 (t, J = 5.8 Hz, 2H), 7.18–7.24 (m, 2H), 8.22 (dd, J = 2.4, 3.6 Hz, 1H), 8.32 (dd, J = 1.4, 2.3 Hz, 1H).
1-(2-(4-Methoxyphenoxy)propyl)piperazine (114d).
To a mixture of tert-butyl piperazine-1-carboxylate (931 mg, 5.00 mmol) and potassium carbonate (1.73 g, 12.5 mmol, 2.5 equiv) in DMF (5 mL) was added 2-chloropropanoyl chloride (698 mg, 5.50 mmol, 1.1 equiv) and the reaction stirred at rt for 20 min. 4-Methoxyphenol (746 mg, 6.01 mmol, 1.2 equiv) was added and the reaction stirred at rt for 5 min then at 80 °C for 4 h. The reaction was partitioned between water (50 mL) and CH2Cl2 (3 × 10 mL). The combined organic layers were sequentially washed with saturated aqueous NaHCO3 (3 mL), water (3 mL), aqueous citric acid (1 M, 3 mL) and water (2 × 3 mL). The organic layer was dried over Na2SO4, adsorbed onto silica gel (4 g) and purified by silica gel chromatography to afford the acylated ether product, tert-butyl 4-(2-(4-methoxyphenoxy)propyl)piperazine-1-carboxylate, as a colorless oil (430 mg, 1.18 mmol, 24% yield). ). 1H NMR (400 MHz, CDCl3) δ 1.44 (s, 9H), 1.54–1.64 (complex, 5H), 3.06–3.27 (m, 2H), 3.39–3.69 (m, 4H), 3.76 (s, 3H), 4.88 (q, J = 6.8 Hz, 1H), 6.72–6.93 (m, 4H); HRMS (m/z): calcd. for C19H29N2O5 [M + H]+ 365.2071; found 365.2082.
To a solution of tert-butyl 4-(2-(4-methoxyphenoxy)propyl)piperazine-1-carboxylate (376 mg, 1.03 mmol) in CH2Cl2 (4 mL) was added sequentially triethylsilane (240 mg, 2.07 mmol, 2.0 equiv) then trifluoroacetic acid (4 mL) and the reaction stirred at rt for 26 h. The reaction was concentrated in vacuo and partitioned between aqueous NaOH (2 M, 6 mL) and CH2Cl2 (3 × 5 mL). The combined organic layers were washed with water (2 mL), dried over Na2SO4, evaporated and purified by silica gel chromatography to afford the deprotected product, 2-(4-methoxyphenoxy)-1-(piperazin-1-yl)propan-1-one, as a colorless oil (219 mg, 0.829 mmol, 80% yield). 1H NMR (400 MHz, CDCl3) δ 1.59 (d, J = 6.9 Hz, 3H), 2.58–2.91 (m, 4H), 3.47–3.73 (m, 4H), 3.76 (s, 3H), 4.88 (q, J = 6.8 Hz, 1H), 6.77–6.88 (m, 4H).
To a solution of 2-(4-methoxyphenoxy)-1-(piperazin-1-yl)propan-1-one (219 mg, 0.829 mmol) in THF (2 mL) at 0 °C was added a THF solution of borane (2.8 mL, 2.8 mmol, 3.2 equiv). After bubbling had ceased (6 min), the reaction was stirred at 65 °C for 4 h. MeOH (2 mL) was added and the reaction concentrated in vacuo. The residue was dissolved in MeOH (3 mL) and concentrated HCl (0.25 mL) added and the solution stirred at 65 °C for 30 min. The solution was concentrated in vacuo and the residue partitioned between aqueous HCl (1 M, 4 mL) and CH2Cl2 (4 mL). Aqueous sodium hydroxide (10 M, 3 mL) was added to raise the pH to >9 and the aqueous layer was extracted with CH2Cl2 (4 × 2 mL). The combined organic layers were dried over Na2SO4, evaporated and purified by silica gel chromatography to afford the reduced product 114d as a colorless oil (178 mg, 0.711 mmol, 86% yield). 1H NMR (400 MHz, CDCl3) δ 1.27 (d, J = 6.2 Hz, 3H), 2.38–2.57 (complex, 5H), 2.68 (dd, J = 6.7, 13.1 Hz, 1H), 2.88 (t, J = 4.9 Hz, 4H), 3.77 (s, 3H), 4.35–4.48 (m, 1H), 6.75–6.92 (m, 4H).
1-(2-(4-Chlorophenoxy)propyl)piperazine (114e).
To a mixture of tert-butyl piperazine-1-carboxylate (931 mg, 5.00 mmol) and potassium carbonate (1.73 g, 12.5 mmol, 2.5 equiv) in DMF (5 mL) was added 2-chloropropanoyl chloride (698 mg, 5.50 mmol, 1.1 equiv) and the reaction stirred at rt for 20 min. 4-chlorophenol (771 mg, 6.00 mmol, 1.2 equiv) was added and the reaction stirred at rt for 5 min then at 80 °C for 4 h. The reaction was partitioned between water (50 mL) and CH2Cl2 (3 × 10 mL). The combined organic layers were sequentially washed with saturated aqueous NaHCO3 (3 mL), water (3 mL), aqueous citric acid (1 M, 3 mL) and water (2 × 3 mL). The organic layer was dried over MgSO4, adsorbed onto silica gel (4 g) and purified by silica gel chromatography to afford the acylated ether product, tert-butyl 4-(2-(4-chlorophenoxy)propyl)piperazine-1-carboxylate, as a white solid (515 mg, 1.40 mmol, 28% yield). 1H NMR (400 MHz, CDCl3) δ 1.45 (s, 9H), 1.62 (d, J = 6.9 Hz, 3H), 3.09–3.26 (m, 3H), 3.41–3.79 (m, 5H), 4.91 (q, J = 6.8 Hz, 1H), 6.83 (d, J = 9.1 Hz, 2H), 7.23 (d, J = 9.0 Hz, 2H); HRMS (m/z): calcd. for C14H18ClN2O4 [M – tert-butyl + 2H]+ 313.0950; found 313.0944.
To a solution of tert-butyl 4-(2-(4-chlorophenoxy)propyl)piperazine-1-carboxylate (458 mg, 1.24 mmol) in CH2Cl2 (4 mL) was added sequentially triethylsilane (291 mg, 2.50 mmol, 2.0 equiv) then trifluoroacetic acid (4 mL) and the reaction stirred at rt for 19 h. The reaction was concentrated in vacuo and partitioned between aqueous NaOH (2 M, 6 mL) and CH2Cl2 (3 × 5 mL). The combined organic layers were washed with water (2 mL), dried over Na2SO4, evaporated and purified by silica gel chromatography to afford the deprotected product, 2-(4-chlorophenoxy)-1-(piperazin-1-yl)propan-1-one, as a colorless oil (299 mg, 1.11 mmol, 90% yield). 1H NMR (400 MHz, CDCl3) δ 1.61 (d, J = 6.8 Hz, 3H), 2.56–2.91 (m, 5H), 3.45–3.76 (m, 3H), 4.91 (q, J = 6.8 Hz, 1H), 6.83 (d, J = 9.0 Hz, 2H), 7.23 (d, J = 9.1 Hz, 2H).
To a solution of 2-(4-chlorophenoxy)-1-(piperazin-1-yl)propan-1-one (308 mg, 1.15 mmol) in THF (2 mL) at 0 °C was added a THF solution of borane (1 M, 3.5 mL, 3.5 mmol, 3.1 equiv). After bubbling had ceased (6 min), the reaction was stirred at 65 °C for 4 h. MeOH (2 mL) was added and the reaction concentrated in vacuo. The residue was dissolved in MeOH (3 mL) and concentrated HCl (0.25 mL) added and the solution stirred at 65 °C for 30 min. The solution was concentrated in vacuo and the residue partitioned between aqueous HCl (1 M, 4 mL) and CH2Cl2 (4 mL). Aqueous sodium hydroxide (10 M, 3 mL) was added to raise the pH to >9 and the aqueous extracted with CH2Cl2 (4 × 2 mL). The combined organic layers were dried over Na2SO4, evaporated and purified by silica gel chromatography to afford the amide reduction product 114e as a colorless oil (257 mg, 1.10 mmol, 88% yield). 1H NMR (400 MHz, CDCl3) δ 1.29 (d, J = 6.2 Hz, 3H), 2.39–2.55 (complex, 5H), 2.68 (dd, J = 6.6, 13.2 Hz, 1H), 2.86 (t, J = 4.9 Hz, 4H), 4.49 (td, J = 4.8, 6.3 Hz, 1H), 6.84 (d, J = 9.0 Hz, 2H), 7.21 (d, J = 9.0 Hz, 2H).
(4-(2-chloroethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (116).
To a solution of 2-(piperazin-1-yl)ethanol (904 mg, 6.94 mmol) and triethylamine (843 mg, 8.33 mmol, 1.2 equiv) in CH2Cl2 (18 mL) was added 1H-indole-2-carbonyl chloride (1,367 mg, 7.61 mmol, 1.1 equiv) while cooling the reaction solution in an ice/water bath. The reaction was stirred for 20 h, slowly warming to rt. The reaction was concentrated in vacuo and the residue suspended in 50% saturated, aqueous NaHCO3 (40 mL). The solids were collected by filtration and washed with water (2 × 20 mL) and dried under vacuum to afford the acylated product, (4-(2-hydroxyethyl)piperazin-1-yl)(1H-indol-2-yl)methanone, as a tan solid (1.41 g, 5.17 mmol, 75% yield), which was used without further purification. 1H NMR (400 MHz, CDCl3) δ 2.59 (t, J = 5.3 Hz, 2H), 3.65 (t, J = 5.4 Hz, 2H), 3.84 (s, 3H), 6.91 (d, J = 8.7 Hz, 2H), 7.39 (d, J = 8.8 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ 53.0 (br), 55.4, 57.8, 59.3, 113.7, 127.7, 129.2, 160.8, 170.3; HRMS (m/z): calcd. for C14H21N2O3 [M + H]+ 265.1547; found 265.1575.
Chlorination of the primary alcohol was accomplished utilizing the protocol of Kartika and coworkers.87 Thus, a solution of (4-(2-hydroxyethyl)piperazin-1-yl)(1H-indol-2-yl)methanone (1.96 g, 7.18 mmol) and triethylamine (2.50 mL, 1.82 g, 17.94 mmol, 2.5 equiv) in CH2Cl2 (65 mL) was cooled to 0 °C. Triphosgene (1.07 g, 3.60 mmol, 0.5 equiv) was added as a solid and the reaction stirred for 5 min at 0 °C and then at rt for 3 h. The reaction was quenched with saturated, aqueous NaHCO3 (50 mL) and extracted with CH2Cl2 (2 × 10 mL). The combined organic layers were washed sequentially with saturated, aqueous NaHCO3 (25 mL) then water (25 mL), dried with Na2SO4 and concentrated in vacuo. The residue was purified by silica chromatography to afford the chloride 116 as a tan solid (1.10 g, 3.77 mmol, 53% yield). 1H NMR (400 MHz, DMSO-d6) δ 2.44 (t, J = 6.2 Hz, 2H), 2.46–2.53 (m, 4H), 3.53 (q, J = 6.1 Hz, 2H), 3.64–3.83 (m, 4H), 4.47 (t, J = 5.3 Hz, 1H), 6.77 (dd, J = 0.9, 2.2 Hz, 1H), 7.04 (ddd, J = 1.0, 6.9, 7.9 Hz, 1H), 7.18 (ddd, J = 1.2, 7.0, 8.2 Hz, 1H), 7.41 (dd, J = 1.0, 8.2 Hz, 1H), 7.60 (dd, J = 1.0, 8.0 Hz, 1H), 11.56 (br s, 1H).
(4-(2-Chloroxyethyl)piperidin-1-yl)(4-methoxyphenyl)methanone (118).
To a solution of 2-(piperidin-4-yl)ethanol (518 mg, 4.01 mmol) and triethylamine (508 mg, 5.02 mmol, 1.25 equiv) in CH2Cl2 (20 mL) was added 4-methoxybenzoyl chloride (0.60 mL, 4.36 mmol, 1.1 equiv) while cooling the solution in an ice/water bath. The reaction was stirred for 16 h, slowly warming to rt, then washed sequentially with aqueous HCl (1 M, 2 × 5 mL), water (1 × 5 mL) and aqueous NaHCO3 (1 × 4 mL). The organic layer was adsorbed onto silica gel (1.5 g) and purified by silica chromatography to afford the acylated product, (4-(2-hydroxyethyl)piperidin-1-yl)(4-methoxyphenyl)methanone, as a colorless oil (845 mg, 3.21 mmol, 80% yield). 1H NMR (400 MHz, CDCl3) δ 1.12–1.30 m, 1H), 1.50–1.59 (m, 2H), 1.64–1.86 (m, 4H), 2.69–3.06 (m, 4H), 3.71 (t, J = 6.5 Hz, 3H), 3.83 (s, 5H), 6.90 (d, J = 8.7 Hz, 2H), 7.37 (d, J = 8.8 Hz, 2H); HRMS (m/z): calcd. for C15H22NO3 [M + H]+ 264.1594; found 264.1605.
To a solution of (4-(2-hydroxyethyl)piperidin-1-yl)(4-methoxyphenyl)methanone (144 mg, 0.547 mmol) in CHCl3 (3 mL) at 0 °C was added thionyl chloride (65.1 mg, 0.547 mmol, 1.0 equiv). The reaction was stirred at 0 °C for 18 min, rt for 2 h and heated at 50 °C for 46 h, then cooled to rt. The reaction mixture was diluted with CH2Cl2 (4 mL) and washed sequentially aqueous NaHCO3 (2 × 2 mL) then water (1 × 2 mL). The organic layer was dried with MgSO4 and purified by silica gel chromatography to afford the chlorinated product 118 as a beige oil (141 mg, 0.470 mmol, 86% yield). 1H NMR (400 MHz, CDCl3) δ 1.10–1.29 (m, 1H), 1.68–1.88 (complex, 6H), 2.72–3.09 (m, 4H), 3.59 (t, J = 6.6 Hz, 2H), 3.83 (s, 3H), 6.90 (d, J = 8.7 Hz, 2H), 7.37 (d, J = 8.7 Hz, 2H).
N-(2-((2-Chloroethyl)(methyl)amino)ethyl)-4-methoxy-N-methylbenzamide hydrochloride (120).
To a solution of N-(2-hydroxyethyl)-4-methoxy-N-methylbenzamide88 119 (572 mg, 2.73 mmol) in CHCl3 (6 mL) at 0 °C was added thionyl chloride (489 mg, 4.11 mmol, 1.5 equiv). The reaction was stirred at 0 °C for 3 min and heated at 55 °C for 35 min, then cooled to rt. The reaction mixture was adsorbed onto silica gel (1.0 g) and purified by silica gel chromatography to afford the chlorinated product, N-(2-chloroethyl)-4-methoxy-N-methylbenzamide, as a colorless oil (336 mg, 1.48 mmol, 54% yield). 1H NMR (400 MHz, CDCl3) δ 1.53–1.69 (m, 2H), 3.12 (s, 3H), 3.68–3.88 (m, 2H), 3.84 (s, 3H), 6.92 (d, J = 8.7 Hz, 2H), 7.41 (d, J = 8.2 Hz, 2H).
To a solution of N-(2-chloroethyl)-4-methoxy-N-methylbenzamide (332 mg, 1.45 mmol) in MeCN (6 mL) was added 2-(methylamino)ethanol (563 mg, 7.50 mmol, 5.1 equiv). The reaction was stirred at 80 °C for 25 h, then cooled to rt. The reaction mixture was adsorbed onto silica gel (1.4 g) and purified by silica gel chromatography to afford the alkylated product, N-(2-((2-hydroxyethyl)(methyl)amino)ethyl)-4-methoxy-N-methylbenzamide, as a colorless oil (259 mg, 0.972 mmol, 67% yield). 1H NMR (400 MHz, CDCl3) δ 2.29–2.74 (complex, 7H), 3.04 (s, 3H), 3.39–3.68 (m, 4H), 3.83 (s, 3H), 6.90 (d, J = 8.7 Hz, 2H), 7.39 (d, J = 8.7 Hz, 2H); HRMS (m/z): calcd. for C14H23N2O3 [M + H]+ 267.1703; found 267.1726.
To a solution of N-(2-((2-hydroxyethyl)(methyl)amino)ethyl)-4-methoxy-N-methylbenzamide (121 mg, 0.454 mmol) in CHCl3 (3 mL) at 0 °C was added thionyl chloride (82.0 mg, 0.685 mmol, 1.5 equiv). The reaction was stirred at 0 °C for 3 min, rt for 30 min and heated at 50 °C for 25 min, then cooled to rt and concentrated in vacuo. The residue was partitioned between aqueous sodium hydroxide (1 M, 3 mL) and CH2Cl2 (2 × 10 mL). The combined organic layers were dried over Na2SO4, evaporated and purified by silica gel chromatography to afford the hydroxy product 120 as a beige oil (109 mg, 0.38 mmol, 84% yield). 1H NMR (400 MHz, CDCl3) δ 3.00 (s, 3H), 3.17 (s, 3H), 3.38–3.54 (m, 2H), 3.60–3.71 (m, 2H), 3.84 (s, 3H), 3.92–4.23 (m, 4H), 6.93 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.7 Hz, 2H).
(4-(2-Hydroxyethyl)piperazin-1-yl)(4-methoxyphenyl)methanone (121).
To a solution of 2-(piperazin-1-yl)ethanol (521 mg, 4.00 mmol) and triethylamine (508 mg, 5.02 mmol, 1.3 equiv) in CH2Cl2 (20 mL) was added 4-methoxybenzoyl chloride (0.60 mL, 4.36 mmol, 1.1 equiv) while cooling the solution in an ice/water bath. The reaction was stirred for 24 h, slowly warming to rt. The reaction was adsorbed onto silica gel (2 g) and purified by silica chromatography to afford the previously reported acylated product89 as a white solid (818 mg, 2.72 mmol, 68% yield). 1H NMR (400 MHz, CDCl3) δ 2.42–2.66 (m, 4H), 2.59 (t, J = 5.3 Hz, 2H), 3.46–3.79 (m, 4H), 3.65 (t, J = 5.4 Hz, 2H), 3.84 (s, 3H), 6.91 (d, J = 8.7 Hz, 2H), 7.39 (d, J = 8.8 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ 53.0 (br), 55.4, 57.8, 59.3, 113.7, 127.7, 129.2, 160.8, 170.3; HRMS (m/z): calcd. for C14H21N2O3 [M + H]+ 265.1547; found 265.1575.
2-(1-(1H-Indole-2-carbonyl)piperidin-4-yl)ethyl 4-methylbenzenesulfonate (122).
To a solution of 1H-indole-2-carboxylic acid (374 mg, 2.32 mmol, 1.0 equiv) in CH2Cl2 (40 mL) was added HOBt (314 mg, 2.32 mmol, 1.0 equiv) and EDC•HCl (445 mg, 2.32 mmol, 1.0 equiv) and the reaction stirred at rt for 10 mins. 2-(Piperidin-4-yl)ethan-1-ol (300 mg, 2.32 mmol) was added and the reaction stirred at rt for 18 h. The reaction was partitioned between water (40 mL) and CH2Cl2 (3 × 10 mL). The combined organic layers were dried over Na2SO4, evaporated and the residue purified by silica gel chromatography to afford the previously reported amide product, (4-(2-hydroxyethyl)piperidin-1-yl)(1H-indol-2-yl)methanone, as a white solid (310 mg, 1.14 mmol, 49% yield).90 1H NMR (400 MHz, CDCl3) δ 1.13–1.32 (m, 2H), 1.45–1.52 (m, 2H), 1.67–1.85 (m, 3H), 2.68–3.21 (complex, 4H), 3.60 (t, J = 6.6 Hz, 2H), 4.56 (d, J = 13.4 Hz, 1H), 6.68 (s, 1H), 7.05 (t, J = 7.5 Hz, 1H), 7.19 (t, J = 7.7 Hz, 1H), 7.37 (d, J = 8.3 Hz, 1H), 7.56 (d, J = 8.0 Hz, 1H), 10.00 (br s, 1H); 13C NMR (101 MHz, CDCl3) δ 32.2, 32.7, 38.7, 43.5, 59.5, 104.9, 111.8, 120.3, 121.5, 124.0, 127.1, 129.2, 135.8, 162.9.
To (4-(2-hydroxyethyl)piperidin-1-yl)(1H-indol-2-yl)methanone (166 mg, 0.61 mmol) in CH2Cl2 (13 mL) was added Et3N (0.34 mL, 2.44 mmol, 4.0 equiv) then p-toluenesulfonyl chloride (232 mg, 1.22 mmol, 2.0 equiv). The reaction was stirred at rt until TLC indicated conversion of alcohol (2 h). The reaction was quenched with saturated, aqueous ammonium chloride (10 mL) and the layers separated. The aqueous layer was extracted with CH2Cl2 (2 × 10 mL) and the combined organic layers were dried over Na2SO4, evaporated and the residue purified by silica gel chromatography to afford the tosylate 122 as a white solid (120 mg, 0.28 mmol, 46% yield). 1H NMR (400 MHz, CDCl3) δ 1.16–1.28 (m, 2H), 1.61–1.66 (m, 2H), 1.70–1.83 (complex, 3H), 2.45 (s, 3H), 2.86–3.12 (m, 2H), 4.10 (t, J = 6.2 Hz, 2H), 4.66–4.73 (m, 2H), 6.73–6.75 (m, 1H), 7.10–7.14 (m, 1H), 7.24–7.28 (m, 1H), 7.36(d, J = 8.0 Hz, 2H), 7.42 (dd, J = 0.8, 8.3 Hz, 1H), 7.64 (dd, J = 0.7, 8.0 Hz, 1H), 7.81 (d, J = 8.3 Hz, 1H), 9.66 (s, 1H); 13C NMR (101 MHz, APT pulse sequence, CDCl3) δ d 21.7, 32.4, 104.9, 111.8, 120.4, 121.7, 124.2, 127.9, 129.9; u 32.1, 35.1, 67.8, 127.4, 129.5, 132.9, 135.7, 144.9, 162.3.
Transient transfections.
HEK293 cells were grown in DMEM supplemented with 10% fetal bovine serum and maintained at 37°C in a humidified incubator containing 5% CO2. The day before transfection, cells were plated in 100 mm tissue culture dishes at 4 × 106 cells/dish in serum-free DMEM. The cells were transfected with the indicated DNA constructs using polyethylenimine (PEI) as the transfection reagent at a ratio of 3:1 (μl PEI: μg DNA). Twenty-four hours after transfection, the media was changed to DMEM supplemented with 10% fetal bovine serum, and cells were used for experiments the next day.
Radioligand binding assays.
HEK293 cells that were stably transfected with the human D2RL or D3R (Codex Biosolutions, Inc., Gaithersburg, MD) or parental HEK293 cells that were transiently transfected with either the D2RL, D3R, or indicated point mutants, as described above were removed mechanically using calcium-free Earle’s Balanced Salt Solution (EBSS-). Intact cells were collected by centrifugation and then lysed with 5 mM Tris-HCl and 5 mM MgCl2 at pH 7.4. Homogenates were centrifuged at 30,000 × g for 30 minutes. The membranes were re-suspended in EBSS pH 7.4. For competition binding studies, membrane preparations were incubated for 90 minutes at room temperature with various concentrations of compound and a single or various concentration(s) of [3H]-methylspiperone in a reaction volume of 250 μL. Non-specific binding was determined in the presence of 4 μM (+)-butaclamol. Bound ligand was separated from unbound by filtration through GF/C filters using a PerkinElmer cell harvester and quantified using a TopCount instrument (PerkinElmer). Ki values were calculated using the Cheng-Prusoff equation91 from observed IC50 values and the radioligand Kd value determined from separate saturation binding experiments.
β-arrestin recruitment DiscoverX PathHunter® assay.
In this assay, a CHO-K1 cell line expressing the human D3R fused with a small 42 amino acid fragment of β-galactosidase called ProLink™ and also expressing a fusion protein consisting of β-arrestin and a larger N-terminal deletion mutant of β-galactosidase were employed. Upon receptor activation, β-arrestin is recruited to the ProLink™-tagged D3R, resulting in complementation of β-galactosidase to form a functional enzyme; addition of the substrate (PathHunter® Detection Reagent) generates a chemiluminescent signal. Assays were conducted as previously described by our laboratory.53, 92 Briefly, CHO-K1 DiscoverX cells stably expressing various dopamine receptors were plated in CP2 media (DiscoverX, Fremont, CA) in 384-well black, clear-bottom plates at a density of 2625 cells/well and incubated overnight at 37°C. Cells were then incubated with indicated compounds for 90 min at 37 °C for agonist mode or for 120 min in the presence of an EC80 concentration of dopamine for antagonist mode. Cells were then incubated with DiscoverX detection reagent for 30 min at room temperature and luminescence was read on a FDSS μCell (Hamamatsu, Bridgewater, NJ). Data were collected as relative luminescence units (RLUs) and normalized as a percentage of the luminescence produced by a maximum concentration of dopamine.
β-arrestin recruitment BRET assay.
HEK293 cells were transiently transfected with D3RRluc8, β-arrestin2-mVenus, and G protein-coupled receptor kinase 3. GRK3 was used because it provides the greatest enhancement of β-arrestin recruitment for the D3R compared to other GRKs (unpublished observations). This GRK preference has also been noted by other labs.56 Cells were harvested with EBSS-, plated in 96-well white plates at 20,000 cells/well in Dulbecco’s phosphate-buffered saline (DPBS) and incubated at room temperature for 45 min. Cells were incubated with 5 μM coelenterazine h (Nanolight Technology, Pinetop, AZ) for 5 min, then stimulated with the indicated concentrations of test compound for 5 min. The BRET signal was determined by quantifying and calculating the ratio of the light emitted by mVenus (525 nm) over that emitted by Rluc8 (485 nm) using a PHERAstar FSX Microplate Reader (BMG Labtech, Cary, NC).
Go BRET activation assay.
HEK293 cells transiently expressing the D3R and GαoA-Rluc8, untagged-β1, and mVenus-γ2 were harvested with EBSS-, plated in 96-well white plates at 20,000 cells/well in DPBS and incubated at room temperature for 45 min. Cells were incubated with 5 μM coelenterazine h (Nanolight Technology, Pinetop, AZ) for 5 min, then stimulated with the indicated concentrations of test compound for 5 min. The BRET signal was determined by quantifying and calculating the ratio of the light emitted by mVenus (525 nm) over that emitted by RLuc8 (485 nm) using a PHERAstar FSX Microplate Reader (BMG Labtech, Cary, NC).
ERK1/2 phosphorylation assay.
ERK1/2 phosphorylation was measured using the AlphaScreen SureFire Ultra ERK kit (PerkinElmer, Waltham, USA). CHO-K1 DiscoverX cells stably expressing the D3R were seeded into 384-well small volume white plates at a density of 40,000 cells/well in serum-free Ham’s F12 media overnight. Cells were stimulated with the indicated concentration of test compound for 15 min, followed by cell lysis as specified by manufacture’s protocol. The plate was shaken for 10 min at room temperature, followed by the addition of Surefire activation buffer, Surefire reaction buffer, AlphaScreen acceptor beads, and AlphaScreen donor beads in ratios specified by the manufacturer. The plate was incubated in the dark for 2 h, then read using a PHERAstar FSX Microplate Reader (BMG Labtech, Cary, NC).
cAMP CAMYEL biosensor assay.
HEK293 cells transiently expressing the D3R and the CAMYEL cAMP biosensor (yellow fluorescence protein-Epac-Rluc)93 were harvested with EBSS-, plated in 96-well white plates at 20,000 cells/well in DPBS and incubated at room temperature for 45 min. Cells were pretreated for 5 min with 10 μM forskolin and 10 μM propranolol (to block endogenous β-adrenergic receptors), then incubated with 5 μM coelenterazine h (Nanolight Technology, Pinetop, AZ) for 5 min, followed by stimulation with the indicated concentrations of test compound for 5 min. The BRET signal was determined by quantifying and calculating the ratio of the light emitted by mVenus (525 nm) over that emitted by Rluc8 (485 nm) using a PHERAstar FSX Microplate Reader (BMG Labtech, Cary, NC).
DiscoverX gpcrMAX™ GPCR Panel.
To determine the selectivity profiles of ML417, CJ-1639, and pramipexole, these compounds were screened using the DiscoverX gpcrMAX™ GPCR panel which measures GPCR activation of β-arrestin recruitment to different GPCRs in either agonist or antagonist modes using a single high concentration (10 μM) of test compound. This study was conducted by DiscoverX, Inc. (Fremont, CA). Reference standards were run for each GPCR in the panel as an integral part of each assay to ensure the validity of the results. Assay results are presented as the mean percent activation or inhibition of the indicated GPCRs (for n = 2 replicates) for each compound tested. For a full description of the DiscoverX gpcrMAX™GPCR panel and the Experimental Section see: http://www.DiscoverX.com.
Psychoactive Drug Screening Program (PDSP) radioligand binding panel.
20, CJ-1639, and pramipexole were screened using the National Institute of Mental Health (NIMH) Psychoactive Drug Screening Program (PDSP) directed by Dr. Bryan L. Roth (University of North Carolina, Chapel Hill, NC). For experimental details including radioligands used and associated Kd values for each individual target, please refer to the PDSP website http://pdsp.med.unc.edu/. Primary screening is performed using 10 μM of the test compound and, if >50% inhibition of radioligand binding is observed, then secondary screening is performed in full concentration-response format in order to derive affinity values.
Inositol 1-phosphate (IP1) Accumulation Assay.
Compound 20 was tested as both an agonist and antagonist of the 5-HT2B receptor using the Eurofins Cerep service (Celle l’Evescault, France). Briefly, recombinant CHO cells expressing the human 5-HT2B were used to measure IP1 accumulation in response to agonist. Serotonin was used as the control agonist while SB20655394 was used as the control antagonist. In these assays, serotonin had a an EC50 of 4.5 nM while SB206553 had an IC50 of 13 nM. Antagonist assays were conducted in the presence of an EC80 concentration (30 nM) of serotonin. The results obtained were the means of two separate assays.
Molecular Dynamics Simulations.
The binding mode of 20 at the D3R was predicted by computational docking and molecular dynamics (MD) simulations. The ligand was docked to an equilibrated model of the D3R, which was built76 based on the D3R crystal structure (PDB: 3PBL).6 Briefly, the missing N terminus was predicted de novo, and a truncated poly-Gly segment was used in place of the ICL3.76 The initial poses of the ligand were acquired by using an induced-fit docking protocol95 in the Schrodinger software (release 2015–3; Schrodinger Inc., New York, NY). The MD simulations were performed in the explicit water-POPC lipid bilayer environment using Desmond MD System (version 3.8; D. E. Shaw Research, New York, NY) with the CHARMM36 protein and lipid force field96, 97 and the TIP3P water model. The ligand parameters were acquired from the GAAMP server.98
As the predicted pKa of 20 is ambiguous using both empirical and quantum mechanics approaches, we experimentally titrated the molecule using a Sirius T3 instrument with a pH electrode and onboard titration tubes (Sirius Analytical Inc, Beverly, MA). Reference spectra without compound and with controls containing either piroxicam or sulphathiazole were collected prior to the assays (data not shown). Samples were titrated using onboard robotics via addition of HCl or KOH to generate spectra and pKa values were calculated using the onboard Sirius T3 software. The pKa of 20 was determined to be 8.62 and thus the basic piperazine nitrogen is predicted to be largely protonated at physiological pH. Consequently, we used the protonated, i.e., the charged form of the ligand in our simulations. The system charges were neutralized, and a solvent concentration of 0.15 M NaCl was added. The system was initially minimized and equilibrated with restraints on the ligand heavy atoms and protein backbone atoms, followed by a production stage in an isothermal–isobaric (NPT) ensemble at 310 K and 1 atm with all atoms unrestrained, as described previously.71, 76 We used Langevin constant pressure and temperature dynamical system99 to maintain the pressure and the temperature, on an anisotropic flexible periodic cell with a constant-ratio constraint applied on the lipid bilayer in the X-Y plane.
Insertion of human D3R into NSCs.
The human D3R open reading frame (ORF) was cloned into a donor vector containing homologous arms for the CLYBL safe harbor site and a neomycin resistance selection cassette. The ORF was synthesized as a codon optimized gBlock (Integrated DNA Technologies) and inserted into the BsrGI site of the donor vector via Gibson assembly. The donor vector with the DRD3 knock-in and two TALEN pairs targeting the CLYBL locus were delivered by nucleofection (2 μg of each vector) into human neural stem cells (NSC) derived from the NCRM-1 iPSC line. Forty-eight hours after transfection, the drug selection was initiated in media containing 600 μg/mL of G418. Selection was continued for seven days at which point the surviving NSCs were expanded in media without G418. Details about the TALEN-based safe harbor targeting approach are described by Cerbini et al. (2015).100 Targeted cell lines were characterized for D3R expression by [3H]-methyspiperone binding assays.
iPSC cell culture and differentiation into dopaminergic neurons.
Neural stem cells (NSCs) were maintained in a growth medium comprised of Neurobasal medium (Gibco), B-27 (Gibco), non-essential amino acid (Sigma-Aldrich), GlutaMAX (Gibco), and 10 ng/mL of fibroblast growth factor (FGF). Differentiation into a dopaminergic phenotype was carried out over 10 days in medium comprised of Neurobasal medium, 1X B27 supplements, 1X non-essential amino acids, 20 ng/mL brain-derived neurotrophic factor (BDNF), 20 ng/mL glial-derived neurotrophic factor (GDNF), and 20 ng/mL TGFb3.101 All growth factors were purchased from Peprotech (Rocky Hill, NJ). NSCs were grown in plates/flasks coated with Geltrex ™ (ThermoFisher Scientific, Carlsbad, CA). 50% of the differentiation media was replaced with fresh media every 72 hours.
MTT assay.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from ThermoFisher Scientific (Carlsbad, CA) (catalog number: M6494). MTT was dissolved in PBS at a concentration of 5 mg/mL (12 mM) and filtered. NSCs were plated at 10,000 cells per well in a 96-well plate and differentiation as described above for 10 days. On day 10 of the differentiation protocol, the cells were treated with the indicated concentrations of DMSO vehicle, 20, or pramipexole. 24 hours later, the cells were treated with 30 μM of 6-hydroxydopamine (6-OHDA). MTT assays were performed 24 h after 6-OHDA treatment. Each dilution was done in quadruplicates and the experiments performed at least six times. MTT assays were performed per manufacturer’s protocol (ThermoFisher Scientific, Waltham, MA). Briefly, all media were pipetted out of the wells and fresh 100 μl of media was added. 10% (10 μl) of 5 mg/mL MTT solution was added to each well and incubated at 37°C for 4 hours. All but 25 μl of the MTT/media solution was pipetted out of each well. 50 μl of DMSO was added to dissolve the resultant formazan crystals. The plate was agitated on a shaker for 10 min. Optical density (OD) was read at 540 nm wavelength on PHERAstar FX (BMG Labtech, Cary, NC) with a reference wavelength at 650 nm.
Ames assay:
The mutagenicity potential of 20 was tested using the Ames reverse mutation assay by Cyprotex Inc (Watertown, MA). Briefly, 10 million bacteria were exposed in triplicate to 20 for 90 min in medium containing a low concentration of histidine. The cultures were then diluted into an indicator medium lacking histidine, dispensed into a 384-well plate, and incubated for 48 h at 37 °C. Cells that have undergone a reversion will grow, resulting in a color change. The number of wells showing growth were counted and compared to the vehicle control. An increase in the number of colonies of at least 2-fold over baseline and a dose response indicated a positive response. Data were analyzed using an unpaired, one-sided Student’s t-test.
Cytotoxicity screening panel:
HepG2 cells were plated on 384-well tissue culture treated black walled clear bottomed polystyrene plates. The cells were treated with test compound using a range of concentrations. At the end of the incubation period, the cells were loaded with the relevant dye/antibody for each cell health marker. The plates were then scanned using an automated fluorescent cellular imager, ArrayScan® (Thermo Scientific Cellomics, Waltham, MA). Cytotoxicity was assessed using a multi-parametric approach using High Content Screening (HCS).
Mouse plasma and brain tissue sampling:
All animal studies were conducted by Cyprotex Inc (Watertown, MA). The levels of 20 in mouse plasma and brain tissue samples were assessed as follows. A single IP dose (20 mg/kg) of 20 was administered to 6–8 week old male C57BL/6 mice. The formulation consisted of 10% dimethylacetamide (DMA) and 60% PEG400, balanced with 30% saline. Plasma and brain samples were collected across 8 time points (5, 15, 30, 60, 120, 240, 480, and 1440 minutes). Brain samples were homogenized in two volumes (1:2 w/v dilution) of phosphate buffer solution (PBS). Once homogenized, the samples were precipitated with three volumes of acetonitrile containing an analytical internal standard (bucetin). Samples were then centrifuged to remove precipitated protein, and the supernatant was analyzed by LC-MS/MS. All brain samples were compared to a calibration curve prepared from a control mouse brain. Plasma samples (5, 15, 30, 60, and 120 min time points) were diluted ten-fold with control plasma. No dilutions were made for the 240, 480, and 1440 min plasma samples. All plasma samples were precipitated with three volumes of acetonitrile containing an analytical internal standard (bucetin). Samples were then centrifuged to remove precipitated protein, and the supernatant was analyzed by LC-MS/MS. All plasma samples were compared to a calibration curve prepared from control mouse plasma. Samples were analyzed by LC-MS/MS using Waters Xevo TQ mass spectrometer coupled with an Acquity UPLC and a CTC PAL chilled autosampler, all controlled by MassLynx software (Waters). After separation on a C18 reverse phase HPLC column (Waters Acquity HSS T3 2.1×50mm 1.8 μM) using an acetonitrile-water gradient system, peaks were analyzed by mass spectrometry (MS) using ESI ionization in MRM mode. All work was performed with appropriate local health regulations and ethical approval. Three mice were used for each timepoint collected and the data represent means ± SEM.
Statistical analyses.
Nonlinear regression analyses were conducted using GraphPad Prism version 8.0.1 (GraphPad Software, Inc., La Jolla, CA). Results are expressed as means ± SEM. EC50, IC50, Emax, and Imax values were calculated from individual concentration response curves and then averaged to generate means and SEM values. Other statistical tests were performed as described in the legends.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Benjamin Neuenswander for preparative and analytical HPLC and Patrick Porubsky for compound management. We thank the University of North Carolina’s Department of Chemistry Mass Spectrometry Core Laboratory for their assistance with mass spectrometry analysis. Q Exactive HF-X system HRMS determinations were supported by the National Science Foundation under Grant No. (CHE-1726291). We thank Shaomeng Wang, PhD at the University of Michigan for the kind gift of Compound CJ-1639. Receptor binding profiles were generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, contract # HHSN-271-2018-00023-C (NIMH PDSP). The NIMH PDSP is directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at NIMH, Bethesda MD, USA. Support for this research was provided by the National Institute of Neurological Disorders and Stroke-Intramural Research Program, National Institute on Drug Abuse-Intramural Research Program (Z1A DA000609), Molecular Libraries Initiative funding to the University of Kansas Specialized Chemistry Center (U54HG005031) and generous support provided by the Eshelman Institute for Innovation at the UNC Eshelman School of Pharmacy.
ABBREVIATIONS USED
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- HTR2A
5-HT2A serotonergic receptor
- 6-OHDA
6-hydroxydopamine
- ADRA2C
alpha2C-adrenergic receptor
- BRET
bioluminescence resonance energy transfer
- BDNF
brain-derived neurotrophic factor
- CCKAR
cholecystokininA receptor
- DA
Dopamine
- DAR
Dopamine Receptor
- DPBS
Dulbecco’s Phosphate-buffered Saline
- EBSS
Earle’s Balanced Salt Solution
- EBI2
Epstein-Barr virus-induced GPCR 2
- pERK
ERK1/2 phosphorylation
- FGF
fibroblast growth factor
- GRKs
G protein-coupled receptor kinases
- GDNF
glial-derived neurotrophic factor
- GPCR
G-protein coupled receptor
- HTS
high-throughput screen
- IP1
inositol 1-phosphate
- MD
Molecular Dynamics
- MLPCN
Molecular Libraries Probe Production Centers Network
- OBS
orthosteric binding site
- PD
Parkinson’s disease
- PEI
polyethylenimine
- PTGER2
Prostaglandin E Receptor 2
- PDSP
Psychoactive Drug Screening Program
- RLU
Relative Luminescence Unit
- RLS
Restless Legs Syndrome
- EDG8
sphingosine-1-phosphate 5 receptor
- SAR
structure–activity relationship
- TM
transmembrane domain
Footnotes
The authors declare no competing financial interests
Supporting Information
Supporting Tables, Figures, HPLC and NMR spectra (PDF)
Molecular formula strings and SAR data (.csv file)
Protein data bank for D3R homology model (.pdb file)
The supporting information is available free of charge on the ACS website.
REFERENCES
- 1.Sibley DR; Monsma FJ Jr., Molecular biology of dopamine receptors. Trends Pharmacol. Sci 1992, 13, 61–69. [DOI] [PubMed] [Google Scholar]
- 2.Money KM; Stanwood GD, Developmental origins of brain disorders: roles for dopamine. Front. Cell. Neurosci 2013, 7:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beaulieu JM; Espinoza S; Gainetdinov RR, Dopamine receptors - IUPHAR Review 13. Br. J. Pharmacol 2015, 172, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaar SJ; Natesan S; McCutcheon R; Howes OD, Antipsychotics: mechanisms underlying clinical response and side-effects and novel treatment approaches based on pathophysiology. Neuropharmacology 2019, 107704. [DOI] [PubMed] [Google Scholar]
- 5.Klein MO; Battagello DS; Cardoso AR; Hauser DN; Bittencourt JC; Correa RG, Dopamine: functions, signaling, and association with neurological diseases. Cell Mol Neurobiol 2019, 39, 31–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chien EYT; Liu W; Zhao Q; Katritch V; Won Han G; Hanson MA; Shi L; Newman AH; Javitch JA; Cherezov V; Stevens RC, Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho DI; Zheng M; Kim K-M, Current perspectives on the selective regulation of dopamine D2 and D3 receptors. Arch. Pharm. Res 2010, 33, 1521–1538. [DOI] [PubMed] [Google Scholar]
- 8.Moritz AE; Benjamin Free R; Sibley DR, Advances and challenges in the search for D2 and D3 dopamine receptor-selective compounds. Cell. Signal 2018, 41, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joyce JN, Dopamine D3 receptor as a therapeutic target for antipsychotic and antiparkinsonian drugs. Pharmacol. Ther 2001, 90, 231–259. [DOI] [PubMed] [Google Scholar]
- 10.Parkinson Study Group CCI, Long-term effect of initiating pramipexole vs levodopa in early Parkinson disease. Arch. Neurol 2009, 66, 563–570. [DOI] [PubMed] [Google Scholar]
- 11.Joyce JN; Presgraves S; Renish L; Borwege S; Osredkar T; Hagner D; Replogle M; PazSoldan M; Millan MJ, Neuroprotective effects of the novel D3/D2 receptor agonist and antiparkinson agent, S32504, in vitro against 1-methyl-4-phenylpyridinium (MPP+) and in vivo against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): a comparison to ropinirole. Exp. Neurol 2003, 184, 393–407. [DOI] [PubMed] [Google Scholar]
- 12.Joyce JN; Der TC; Renish L; Osredkar T; Hagner D; Reploge M; Sakakibara S; Ueda S, Loss of D3 receptors in the zitter mutant rat is not reversed by L-dopa treatment. Exp. Neurol 2004, 187, 178–189. [DOI] [PubMed] [Google Scholar]
- 13.Li C; Biswas S; Li X; Dutta AK; Le W, Novel D3 dopamine receptor-preferring agonist D-264: evidence of neuroprotective property in Parkinson’s disease animal models induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and lactacystin. J. Neurosci. Res 2010, 88, 2513–2523. [DOI] [PubMed] [Google Scholar]
- 14.Joyce JN; Woolsey C; Ryoo H; Borwege S; Hagner D, Low dose pramipexole is neuroprotective in the MPTP mouse model of Parkinson’s disease, and downregulates the dopamine transporter via the D3 receptor. BMC Biol 2004, 2, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iravani MM; Haddon CO; Cooper JM; Jenner P; Schapira AH, Pramipexole protects against MPTP toxicity in non-human primates. J. Neurochem 2006, 96, 1315–1321. [DOI] [PubMed] [Google Scholar]
- 16.Vu TQ; Ling ZD; Ma SY; Robie HC; Tong CW; Chen EY; Lipton JW; Carvey PM, Pramipexole attenuates the dopaminergic cell loss induced by intraventricular 6-hydroxydopamine. J. Neural Transm. (Vienna) 2000, 107, 159–176. [DOI] [PubMed] [Google Scholar]
- 17.Ramirez AD; Wong SK; Menniti FS, Pramipexole inhibits MPTP toxicity in mice by dopamine D3 receptor dependent and independent mechanisms. Eur. J. Pharmacol 2003, 475, 29–35. [DOI] [PubMed] [Google Scholar]
- 18.Lao CL; Kuo YH; Hsieh YT; Chen JC, Intranasal and subcutaneous administration of dopamine D3 receptor agonists functionally restores nigrostriatal dopamine in MPTP-treated mice. Neurotox. Res 2013, 24, 523–531. [DOI] [PubMed] [Google Scholar]
- 19.Weintraub D, Dopamine and impulse control disorders in Parkinson’s disease. Ann. Neurol 2008, 64, S93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balarajah S; Cavanna AE, The pathophysiology of impulse control disorders in Parkinson disease. Behav. Neurol 2013, 26, 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newman AH; Blaylock BL; Nader MA; Bergman J; Sibley DR; Skolnick P, Medication discovery for addiction: translating the dopamine D3 receptor hypothesis. Biochem. Pharmacol 2012, 84, 882–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Das B; Modi G; Dutta A, Dopamine D3 agonists in the treatment of Parkinson’s disease. Curr. Top. Med. Chem 2015, 15, 908–926. [DOI] [PubMed] [Google Scholar]
- 23.Keck TM; Burzynski C; Shi L; Newman AH, Beyond small-molecule SAR: using the dopamine D3 receptor crystal structure to guide drug design. Adv. Pharmacol 2014, 69, 267–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luedtke RR; Rangel-Barajas C; Malik M; Reichert DE; Mach RH, Bitropic D3 dopamine receptor selective compounds as potential antipsychotics. Curr. Pharm 2015, 21, 3700–3724. [DOI] [PubMed] [Google Scholar]
- 25.Grundt P; Carlson EE; Cao J; Bennett CJ; McElveen E; Taylor M; Luedtke RR; Newman AH, Novel heterocyclic trans olefin analogues of N-{4-[4-(2,3-dichlorophenyl)piperazin-1-yl]butyl}arylcarboxamides as selective probes with high affinity for the dopamine D3 receptor. J. Med. Chem 2005, 48, 839–848. [DOI] [PubMed] [Google Scholar]
- 26.Chen J; Levant B; Jiang C; Keck TM; Newman AH; Wang S, Tranylcypromine substituted cis-hydroxycyclobutylnaphthamides as potent and selective dopamine D(3) receptor antagonists. J. Med. Chem 2014, 57, 4962–4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar V; Banala AK; Garcia EG; Cao J; Keck TM; Bonifazi A; Deschamps JR; Newman AH, Chiral resolution and serendipitous fluorination reaction for the selective dopamine D3 receptor antagonist BAK2–66. ACS Med. Chem. Lett 2014, 5, 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boateng CA; Bakare OM; Zhan J; Banala AK; Burzynski C; Pommier E; Keck TM; Donthamsetti P; Javitch JA; Rais R; Slusher BS; Xi ZX; Newman AH, High affinity dopamine D3 receptor (D3R)-selective antagonists attenuate heroin self-administration in wild-type but not D3R knockout mice. J. Med. Chem 2015, 58, 6195–6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar V; Bonifazi A; Ellenberger MP; Keck TM; Pommier E; Rais R; Slusher BS; Gardner E; You ZB; Xi ZX; Newman AH, Highly selective dopamine D3 receptor (D3R) antagonists and partial agonists based on eticlopride and the D3R crystal structure: new leads for opioid dependence treatment. J. Med. Chem 2016, 59, 7634–7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shaik AB; Kumar V; Bonifazi A; Guerrero AM; Cemaj SL; Gadiano A; Lam J; Xi ZX; Rais R; Slusher BS; Newman AH, Investigation of novel primary and secondary pharmacophores and 3-substitution in the linking chain of a series of highly selective and bitopic dopamine D3 receptor antagonists and partial agonists. J. Med. Chem 2019, 62, 9061–9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valant C; Robert Lane J; Sexton PM; Christopoulos A, The best of both worlds? Bitopic orthosteric/allosteric ligands of g protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol 2012, 52, 153–178. [DOI] [PubMed] [Google Scholar]
- 32.Lane JR; Sexton PM; Christopoulos A, Bridging the gap: bitopic ligands of G-protein-coupled receptors. Trends Pharmacol. Sci 2013, 34, 59–66. [DOI] [PubMed] [Google Scholar]
- 33.Kumar V; Moritz AE; Keck TM; Bonifazi A; Ellenberger MP; Sibley CD; Free RB; Shi L; Lane JR; Sibley DR; Newman AH, Synthesis and pharmacological characterization of novel trans-cyclopropylmethyl-linked bivalent ligands that exhibit selectivity and allosteric pharmacology at the dopamine D3 receptor (D3R). J. Med. Chem 2017, 60, 1478–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Battiti FO; Cemaj SL; Guerrero AM; Shaik AB; Lam J; Rais R; Slusher BS; Deschamps JR; Imler GH; Newman AH; Bonifazi A, The significance of chirality in drug design and synthesis of bitopic ligands as D3 receptor (D3R) selective agonists. J. Med. Chem 2019, 62, 6287–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Newman AH; Battiti FO; Bonifazi A, 2016 Philip S. Portoghese medicinal chemistry lectureship: designing bivalent or bitopic molecules for G-protein coupled receptors. The whole is greater than the sum of its parts. J. Med. Chem 2019, 63, 1779–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy A; Nestor LJ; McGonigle J; Paterson L; Boyapati V; Ersche KD; Flechais R; Kuchibatla S; Metastasio A; Orban C; Passetti F; Reed L; Smith D; Suckling J; Taylor E; Robbins TW; Lingford-Hughes A; Nutt DJ; Deakin JF; Elliott R, Acute D3 antagonist GSK598809 selectively enhances neural response during monetary reward anticipation in drug and alcohol dependence. Neuropsychopharmacology 2017, 42, 1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Di Ciano P; Mansouri E; Tong J; Wilson AA; Houle S; Boileau I; Duvauchelle T; Robert P; Schwartz JC; Le Foll B, Occupancy of dopamine D2 and D3 receptors by a novel D3 partial agonist BP1.4979: a [(11)C]-(+)-PHNO PET study in humans. Neuropsychopharmacology 2019, 44, 1284–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.You ZB; Gao JT; Bi GH; He Y; Boateng C; Cao J; Gardner EL; Newman AH; Xi ZX, The novel dopamine D3 receptor antagonists/partial agonists CAB2–015 and BAK4–54 inhibit oxycodone-taking and oxycodone-seeking behavior in rats. Neuropharmacology 2017, 126, 190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.You ZB; Bi GH; Galaj E; Kumar V; Cao J; Gadiano A; Rais R; Slusher BS; Gardner EL; Xi ZX; Newman AH, Dopamine D3R antagonist VK4–116 attenuates oxycodone self-administration and reinstatement without compromising its antinociceptive effects. Neuropsychopharmacology 2019, 44, 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jordan CJ; Humburg BA; Thorndike EB; Shaik AB; Xi ZX; Baumann MH; Newman AH; Schindler CW, Newly developed dopamine D3 receptor antagonists, R-VK4–40 and R-VK4–116, do not potentiate cardiovascular effects of cocaine or oxycodone in rats. J. Pharmacol. Exp. Ther 2019, 371, 602–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jordan CJ; Humburg B; Rice M; Bi GH; You ZB; Shaik AB; Cao J; Bonifazi A; Gadiano A; Rais R; Slusher B; Newman AH; Xi ZX, The highly selective dopamine D3R antagonist, R-VK4–40 attenuates oxycodone reward and augments analgesia in rodents. Neuropharmacology 2019, 158, 107597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carlsson J; Coleman RG; Setola V; Irwin JJ; Fan H; Schlessinger A; Sali A; Roth BL; Shoichet BK, Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nat. Chem. Biol 2011, 7, 769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lane JR; Chubukov P; Liu W; Canals M; Cherezov V; Abagyan R; Stevens RC; Katritch V, Structure-based ligand discovery targeting orthosteric and allosteric pockets of dopamine receptors. Mol. Pharmacol 2013, 84, 794–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen J; Levant B; Wang S, High-affinity and selective dopamine D(3) receptor full agonists. Bioorg. Med. Chem. Lett 2012, 22, 5612–5617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen J; Collins GT; Levant B; Woods J; Deschamps JR; Wang S, CJ-1639: a potent and highly selective dopamine D3 receptor full agonist. ACS Med. Chem. Lett 2011, 2, 620–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen J; Jiang C; Levant B; Li X; Zhao T; Wen B; Luo R; Sun D; Wang S, Pramipexole derivatives as potent and selective dopamine D(3) receptor agonists with improved human microsomal stability. ChemMedChem 2014, 9, 2653–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Biswas S; Hazeldine S; Ghosh B; Parrington I; Kuzhikandathil E; Reith ME; Dutta AK, Bioisosteric heterocyclic versions of 7-{[2-(4-phenyl-piperazin-1-yl)ethyl]propylamino}-5,6,7,8-tetrahydronaphthalen-2- ol: identification of highly potent and selective agonists for dopamine D3 receptor with potent in vivo activity. J. Med. Chem 2008, 51, 3005–3019. [DOI] [PubMed] [Google Scholar]
- 48.Ghosh B; Antonio T; Zhen J; Kharkar P; Reith ME; Dutta AK, Development of (S)-N6-(2-(4-(isoquinolin-1-yl)piperazin-1-yl)ethyl)-N6-propyl-4,5,6,7-tetrahydro benzo[d]-thiazole-2,6-diamine and its analogue as a D3 receptor preferring agonist: potent in vivo activity in Parkinson’s disease animal models. J. Med. Chem 2010, 53, 1023–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnson M; Antonio T; Reith ME; Dutta AK, Structure-activity relationship study of N(6)-(2-(4-(1H-Indol-5-yl)piperazin-1-yl)ethyl)-N(6)-propyl-4,5,6,7-tetrahydroben zo[d]thiazole-2,6-diamine analogues: development of highly selective D3 dopamine receptor agonists along with a highly potent D2/D3 agonist and their pharmacological characterization. J. Med. Chem 2012, 55, 5826–5840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhen J; Antonio T; Jacob JC; Grandy DK; Reith ME; Dutta AK; Selley DE, Efficacy of hybrid tetrahydrobenzo[d]thiazole based aryl piperazines D-264 and D-301 at D(2) and D(3) receptors. Neurochem. Res 2016, 41, 328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cao Y; Sun N; Zhang J; Liu Z; Tang YZ; Wu Z; Kim KM; Cheon SH, Correction: design, synthesis, and evaluation of bitopic arylpiperazine-phthalimides as selective dopamine D3 receptor agonists. Medchemcomm 2018, 9, 1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schreiber SL; Kotz JD; Li M; Aube J; Austin CP; Reed JC; Rosen H; White EL; Sklar LA; Lindsley CW; Alexander BR; Bittker JA; Clemons PA; de Souza A; Foley MA; Palmer M; Shamji AF; Wawer MJ; McManus O; Wu M; Zou B; Yu H; Golden JE; Schoenen FJ; Simeonov A; Jadhav A; Jackson MR; Pinkerton AB; Chung TD; Griffin PR; Cravatt BF; Hodder PS; Roush WR; Roberts E; Chung DH; Jonsson CB; Noah JW; Severson WE; Ananthan S; Edwards B; Oprea TI; Conn PJ; Hopkins CR; Wood MR; Stauffer SR; Emmitte KA, Advancing biological understanding and therapeutics discovery with small-molecule probes. Cell 2015, 161, 1252–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Free RB; Chun LS; Moritz AE; Miller BN; Doyle TB; Conroy JL; Padron A; Meade JA; Xiao J; Hu X; Dulcey AE; Han Y; Duan L; Titus S; Bryant-Genevier M; Barnaeva E; Ferrer M; Javitch JA; Beuming T; Shi L; Southall NT; Marugan JJ; Sibley DR, Discovery and characterization of a G protein–biased agonist that inhibits ß-arrestin recruitment to the D2 dopamine receptor. Mol. Pharmacol 2014, 86, 96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiao J; Free RB; Barnaeva E; Conroy JL; Doyle T; Miller B; Bryant-Genevier M; Taylor MK; Hu X; Dulcey AE; Southall N; Ferrer M; Titus S; Zheng W; Sibley DR; Marugan JJ, Discovery, optimization, and characterization of novel D2 dopamine receptor selective antagonists. J. Med. Chem 2014, 57, 3450–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Conn PJ; Lindsley CW; Meiler J; Niswender CM, Opportunities and challenges in the discovery of allosteric modulators of GPCRs for treating CNS disorders. Nat. Rev. Drug Discov 2014, 13, 692–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim KM; Valenzano KJ; Robinson SR; Yao WD; Barak LS; Caron MG, Differential regulation of the dopamine D2 and D3 receptors by G protein-coupled receptor kinases and beta-arrestins. J. Biol. Chem 2001, 276, 37409–37414. [DOI] [PubMed] [Google Scholar]
- 57.Newman AH; Beuming T; Banala AK; Donthamsetti P; Pongetti K; LaBounty A; Levy B; Cao J; Michino M; Luedtke RR; Javitch JA; Shi L, Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J. Med. Chem 2012, 55, 6689–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zou MF; Keck TM; Kumar V; Donthamsetti P; Michino M; Burzynski C; Schweppe C; Bonifazi A; Free RB; Sibley DR; Janowsky A; Shi L; Javitch JA; Newman AH, Novel analogues of (R)-5-(methylamino)-5,6-dihydro-4H-imidazo[4,5,1-ij]quinolin-2(1H)-one (sumanirole) provide clues to dopamine D2/D3 receptor agonist selectivity. J. Med. Chem 2016, 59, 2973–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sanchez-Soto M; Verma RK; Willette BKA; Gonye EC; Moore AM; Moritz AE; Boateng CA; Yano H; Free RB; Shi L; Sibley DR, A structural basis for how ligand binding site changes can allosterically regulate GPCR signaling and engender functional selectivity. Sci. Signal 2020, 13, 617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Salahpour A; Espinoza S; Masri B; Lam V; Barak LS; Gainetdinov RR, BRET biosensors to study GPCR biology, pharmacology, and signal transduction. Front. Endocrinol 2012, 3, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sanchez-Soto M; Bonifazi A; Cai NS; Ellenberger MP; Newman AH; Ferre S; Yano H, Evidence for noncanonical neurotransmitter activation: norepinephrine as a dopamine D2-like receptor agonist. Mol. Pharmacol 2016, 89, 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bonifazi A; Yano H; Ellenberger MP; Muller L; Kumar V; Zou MF; Cai NS; Guerrero AM; Woods AS; Shi L; Newman AH, Novel bivalent ligands based on the sumanirole pharmacophore reveal dopamine D2 receptor (D2R) biased agonism. J. Med. Chem 2017, 60, 2890–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jain R; Watson U; Vasudevan L; Saini DK, ERK activation pathways downstream of GPCRs. Int. Rev. Cell. Mol. Biol 2018, 338, 79–109. [DOI] [PubMed] [Google Scholar]
- 64.Gurevich VV; Gurevich EV, Arrestin-mediated signaling: is there a controversy? World J. Biol. Chem 2018, 9, 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Locht C; Coutte L; Mielcarek N, The ins and outs of pertussis toxin. FEBS J 2011, 278, 4668–4682. [DOI] [PubMed] [Google Scholar]
- 66.Mangmool S; Kurose H, G(i/o) protein-dependent and -independent actions of pertussis toxin (PTX). Toxins 2011, 3, 884–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Besnard J; Ruda GF; Setola V; Abecassis K; Rodriguiz RM; Huang XP; Norval S; Sassano MF; Shin AI; Webster LA; Simeons FR; Stojanovski L; Prat A; Seidah NG; Constam DB; Bickerton GR; Read KD; Wetsel WC; Gilbert IH; Roth BL; Hopkins AL, Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sibley DR; De Lean A; Creese I, Anterior pituitary dopamine receptors. Demonstration of interconvertible high and low affinity states of the D-2 dopamine receptor. J. Biol. Chem 1982, 257, 6351–6361. [PubMed] [Google Scholar]
- 69.Sibley DR; Mahan LC; Creese I, Dopamine receptor binding on intact cells. Absence of a high-affinity agonist-receptor binding state. Mol. Pharmacol 1983, 23, 295–302. [PubMed] [Google Scholar]
- 70.Narendran R; Hwang DR; Slifstein M; Talbot PS; Erritzoe D; Huang Y; Cooper TB; Martinez D; Kegeles LS; Abi-Dargham A; Laruelle M, In vivo vulnerability to competition by endogenous dopamine: comparison of the D2 receptor agonist radiotracer (−)-N-[11C]propyl-norapomorphine ([11C]NPA) with the D2 receptor antagonist radiotracer [11C]-raclopride. Synapse 2004, 52, 188–208. [DOI] [PubMed] [Google Scholar]
- 71.Michino M; Free RB; Doyle TB; Sibley DR; Shi L, Structural basis for Na(+)-sensitivity in dopamine D2 and D3 receptors. Chem. Commun. (Camb.) 2015, 51, 8618–8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zarzycka B; Zaidi SA; Roth BL; Katritch V, Harnessing ion-binding sites for GPCR pharmacology. Pharmacol. Rev 2019, 71, 571–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Watanabe M; George SR; Seeman P, Regulation of anterior pituitary D2 dopamine receptors by magnesium and sodium ions. J. Neurochem 1985, 45, 1842–1849. [DOI] [PubMed] [Google Scholar]
- 74.Roth BL, Drugs and valvular heart disease. N. Engl. J. Med 2007, 356, 6–9. [DOI] [PubMed] [Google Scholar]
- 75.Zanettini R; Antonini A; Gatto G; Gentile R; Tesei S; Pezzoli G, Valvular heart disease and the use of dopamine agonists for Parkinson’s disease. N. Engl. J. Med 2007, 356, 39–46. [DOI] [PubMed] [Google Scholar]
- 76.Michino M; Boateng CA; Donthamsetti P; Yano H; Bakare OM; Bonifazi A; Ellenberger MP; Keck TM; Kumar V; Zhu C; Verma R; Deschamps JR; Javitch JA; Newman AH; Shi L, Toward understanding the structural basis of partial agonism at the dopamine D3 receptor. J. Med. Chem 2017, 60, 580–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Michino M; Beuming T; Donthamsetti P; Newman AH; Javitch JA; Shi L, What can crystal structures of aminergic receptors tell us about designing subtype-selective ligands? Pharmacol. Rev 2015, 67, 198–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Michino M; Donthamsetti P; Beuming T; Banala A; Duan L; Roux T; Han Y; Trinquet E; Newman AH; Javitch JA; Shi L, A single glycine in extracellular loop 1 is the critical determinant for pharmacological specificity of dopamine D2 and D3 receptors. Mol. Pharmacol 2013, 84, 854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ballesteros JA, Weinstein H, Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci 1995, 25, 366–428. [Google Scholar]
- 80.Wang TZ, Z.; Meanwell, N. A., Benzoylation of dianions: preparation of monobenzoylated derivatives of symmetrical secondary diamines. J. Org. Chem 1999, 64, 7661–7662. [Google Scholar]
- 81.Wang LX; Zhou XB; Xiao ML; Jiang N; Liu F; Zhou WX; Wang XK; Zheng ZB; Li S, Synthesis and biological evaluation of substituted 4-(thiophen-2-ylmethyl)-2H-phthalazin-1-ones as potent PARP-1 inhibitors. Bioorg. Med. Chem. Lett 2014, 24, 3739–3743. [DOI] [PubMed] [Google Scholar]
- 82.Schultz DM; Prescher JA; Kidd S; Marona-Lewicka D; Nichols DE; Monte A, ‘Hybrid’ benzofuran-benzopyran congeners as rigid analogs of hallucinogenic phenethylamines. Bioorg. Med. Chem 2008, 16, 6242–6251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Canale V; Kurczab R; Partyka A; Satala G; Lenda T; Jastrzebska-Wiesek M; Wesolowska A; Bojarski AJ; Zajdel P, Towards new 5-HT7 antagonists among arylsulfonamide derivatives of (aryloxy)ethyl-alkyl amines: multiobjective based design, synthesis, and antidepressant and anxiolytic properties. Eur. J. Med. Chem 2016, 108, 334–346. [DOI] [PubMed] [Google Scholar]
- 84.Valhondo M; Marco I; Martin-Fontecha M; Vazquez-Villa H; Ramos JA; Berkels R; Lauterbach T; Benhamu B; Lopez-Rodriguez ML, New serotonin 5-HT1A receptor agonists endowed with antinociceptive activity in vivo. J. Med. Chem 2013, 56, 7851–7861. [DOI] [PubMed] [Google Scholar]
- 85.Kim MS; Buisson LA; Heathcote DA; Hu H; Braddock DC; Barrett AG; Ashton-Rickardt PG; Snyder JP, Approaches to design non-covalent inhibitors for human granzyme B (hGrB). Org. Biomol. Chem 2014, 12, 8952–8965. [DOI] [PubMed] [Google Scholar]
- 86.Malik HB, W.; Schmidt, R. R., Maltose and maltotriose derivatives as potential inhibitors of the maltose-binding protein. Eur. J. Org. Chem 2008, 2084–2099. [Google Scholar]
- 87.Ayala CE; Villalpando A; Nguyen AL; McCandless GT; Kartika R, Chlorination of aliphatic primary alcohols via triphosgene-triethylamine activation. Org. Lett 2012, 14, 3676–3679. [DOI] [PubMed] [Google Scholar]
- 88.Nishii YH,T; Fernandez S; Knochel P; Mashima K, Zinc-catalyzed esterification of N-β-hydroxyethylamides: removal of directing groups under mild conditions. Eur. J. Org. Chem 2017, 5010–5014. [Google Scholar]
- 89.Kuder KJS,M; Schunack W; Szymanska E; Kiec-Kononowicz K, Discovery of novel nead in the group of N-substituted piperazine ether derivatives with potential histamine H3 receptor activity. Med. Chem 2014, 10, 588–599. [DOI] [PubMed] [Google Scholar]
- 90.Watanabe K; Kakefuda A; Yasuda M; Enjo K; Kikuchi A; Furutani T; Naritomi Y; Otsuka Y; Okada M; Ohta M, Discovery of 2-methyl-1-{1-[(5-methyl-1H-indol-2-yl)carbonyl]piperidin-4-yl}propan-2-ol: a novel, potent and selective type 5 17beta-hydroxysteroid dehydrogenase inhibitor. Bioorg. Med. Chem 2013, 21, 5261–5270. [DOI] [PubMed] [Google Scholar]
- 91.Cheng Y; Prusoff WH, Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- 92.Banala AK; Levy BA; Khatri SS; Furman CA; Roof RA; Mishra Y; Griffin SA; Sibley DR; Luedtke RR; Newman AH, N-(3-fluoro-4-(4-(2-methoxy or 2,3-dichlorophenyl)piperazine-1-yl)butyl)arylcarboxamides as selective dopamine D3 receptor ligands: critical role of the carboxamide linker for D3 receptor selectivity. J. Med. Chem 2011, 54, 3581–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jiang LI; Collins J; Davis R; Lin KM; DeCamp D; Roach T; Hsueh R; Rebres RA; Ross EM; Taussig R; Fraser I; Sternweis PC, Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J. Biol. Chem 2007, 282, 10576–10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kennett GA; Wood MD; Bright F; Cilia J; Piper DC; Gager T; Thomas D; Baxter GS; Forbes IT; Ham P; Blackburn TP, In vitro and in vivo profile of SB 206553, a potent 5-HT2C/5-HT2B receptor antagonist with anxiolytic-like properties. Br. J. Pharmacol 1996, 117, 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sherman W; Day T; Jacobson MP; Friesner RA; Farid R, Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem 2006, 49, 534–553. [DOI] [PubMed] [Google Scholar]
- 96.Pastor RW; Mackerell AD Jr., Development of the CHARMM Force Field for Lipids. J. Phys. Chem. Lett 2011, 2, 1526–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Best RB; Zhu X; Shim J; Lopes PE; Mittal J; Feig M; Mackerell AD Jr., Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput 2012, 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Huang L; Roux B, Automated force field parameterization for non-polarizable and polarizable atomic models based on ab initio target data. J. Chem. Theory Comput 2013, 9, 3543–3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Feller SEZ,Y; Pastor RW; Brooks BR, Constant pressure molecular dynamics simulation: the langevin piston method. J. Chem. Phys 1995, 103, 4613–4621. [Google Scholar]
- 100.Cerbini T; Luo Y; Rao MS; Zou J, Transfection, selection, and colony-picking of human induced pluripotent stem cells TALEN-targeted with a GFP gene into the AAVS1 safe harbor. J. Vis. Exp 2015, (96). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Swistowski A; Peng J; Han Y; Swistowska AM; Rao MS; Zeng X, Xeno-free defined conditions for culture of human embryonic stem cells, neural stem cells and dopaminergic neurons derived from them. PLoS One 2009, 4 (7), e6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.