

Abstract
In aqueous solutions, the 214-residue low-complexity domain of the FUS protein (FUS-LC) is known to undergo liquid-liquid phase separation and also to self-assemble into amyloid-like fibrils. In previous work based on solid state nuclear magnetic resonance (ssNMR) methods, a structural model for the FUS-LC fibril core was developed, showing that residues 39–95 form the fibril core. Unlike fibrils formed by amyloid-β peptides, α-synuclein, and other amyloid-forming proteins, the FUS-LC core is largely devoid of purely hydrophobic amino acid sidechains. Instead, the core-forming segment contains numerous hydroxyl-bearing residues, including 18 serines, 6 threonines, and 8 tyrosines, suggesting that the FUS-LC fibril structure may be stabilized in part by inter-residue hydrogen bonds among sidechain hydroxyl groups. Here we describe ssNMR measurements, performed on 2H,15N,13C-labeled FUS-LC fibrils, that provide new information about the interactions of hydroxyl-bearing residues with one another and with water. The ssNMR data support the involvement of specific serine, threonine, and tyrosine residues in hydrogen-bonding interactions. The data also reveal differences in hydrogen exchange rates with water for different sidechain hydroxyl groups, providing information about solvent exposure and penetration of water into the FUS-LC fibril core.
Graphical Abstract
Introduction
Numerous proteins that participate in DNA transcription and RNA processing possess low-complexity (LC) domains, in addition to nucleic acid-binding and other functional domains. The LC domains have simple amino acid compositions, can have quasi-repetitive sequences, and are unstructured as monomers. Examples of proteins with relatively long LC domains include FUS, TDP43, and members of the heterogeneous nuclear ribonucleoprotein (hnRNP) family.
The self-assembly behavior of proteins with LC domains is of current interest for several reasons. These proteins typically function as multimers, with their LC domains being essential for self-association and for association of different LC-domain proteins with one another2,3. These proteins are also components of protein/nucleic acid condensates within cells, such as stress granules, P granules, and nuclear paraspeckles4,5, with their LC domains contributing to localization to such condensates6–11. In vitro, proteins containing long LC domains, or LC domains by themselves, exhibit liquid-liquid phase separation (LLPS), forming “droplets” of highly concentrated protein solution, surrounded by a dilute protein solution, at appropriate temperatures and total protein concentrations12–15. The phase-separated droplet state can be metastable, converting over time to a suspension of amyloid-like protein fibrils, which can behave as a hydrogel through entanglement of long fibrils9,16–18. Finally, fibril formation by LC-domain proteins within cells can be a pathological protein aggregation event, possibly occurring by nucleation of fibril growth within otherwise functional phase-separated condensates10,19,20. Intracellular aggregation of FUS, TDP43, and hnRNPs is associated with amyotrophic lateral sclerosis and other neurodegenerative diseases21–26.
As a step towards understanding the self-assembly behavior of LC domains at the molecular level, we recently reported a molecular structural model for fibrils formed in vitro by the 214-residue LC domain of FUS (FUS-LC)1, shown in Figs. 1a and 1b (see also Protein Data Bank file 5W3N). This model is based on a large set of solid state NMR (ssNMR) data, supplemented by information from transmission electron microscopy. The FUS-LC fibril structure has a number of intriguing properties: (i) A structurally ordered fibril core is formed by residues 39–95, while most segments outside this core remain dynamically disordered; (ii) FUS-LC fibrils are not polymorphic, with the same core structure being obtained in multiple independent sample preparations and with no evidence for detectable populations of alternative core structures or alternative core-forming segments; (iii) Although residues 39–95 form the fibril core, the amino acid composition of this segment (21.0% Gly, 31.6% Ser, 17.5% Gln, 14.0% Tyr, 10.5% Thr, 1.8% Pro, 3.5% others) is nearly the same as the overall composition of FUS-LC (24.9% Gly, 23.9% Ser, 20.2% Gln, 12.6% Tyr, 5.1% Pro, 4.7% Thr, 9.0% others); (iv) Only one residue in the core, namely Pro72, has an entirely hydrophobic sidechain. These properties make FUS-LC fibrils qualitatively different from amyloid-β27–31 and α-synuclein fibrils32–34, for example, which contain larger fractions of structurally ordered residues, are polymorphic, and have core-forming segments in which roughly 40% of the amino acids are hydrophobic (i.e., Ala, Val, Leu, Ile, Phe, or Met).
Figure 1:
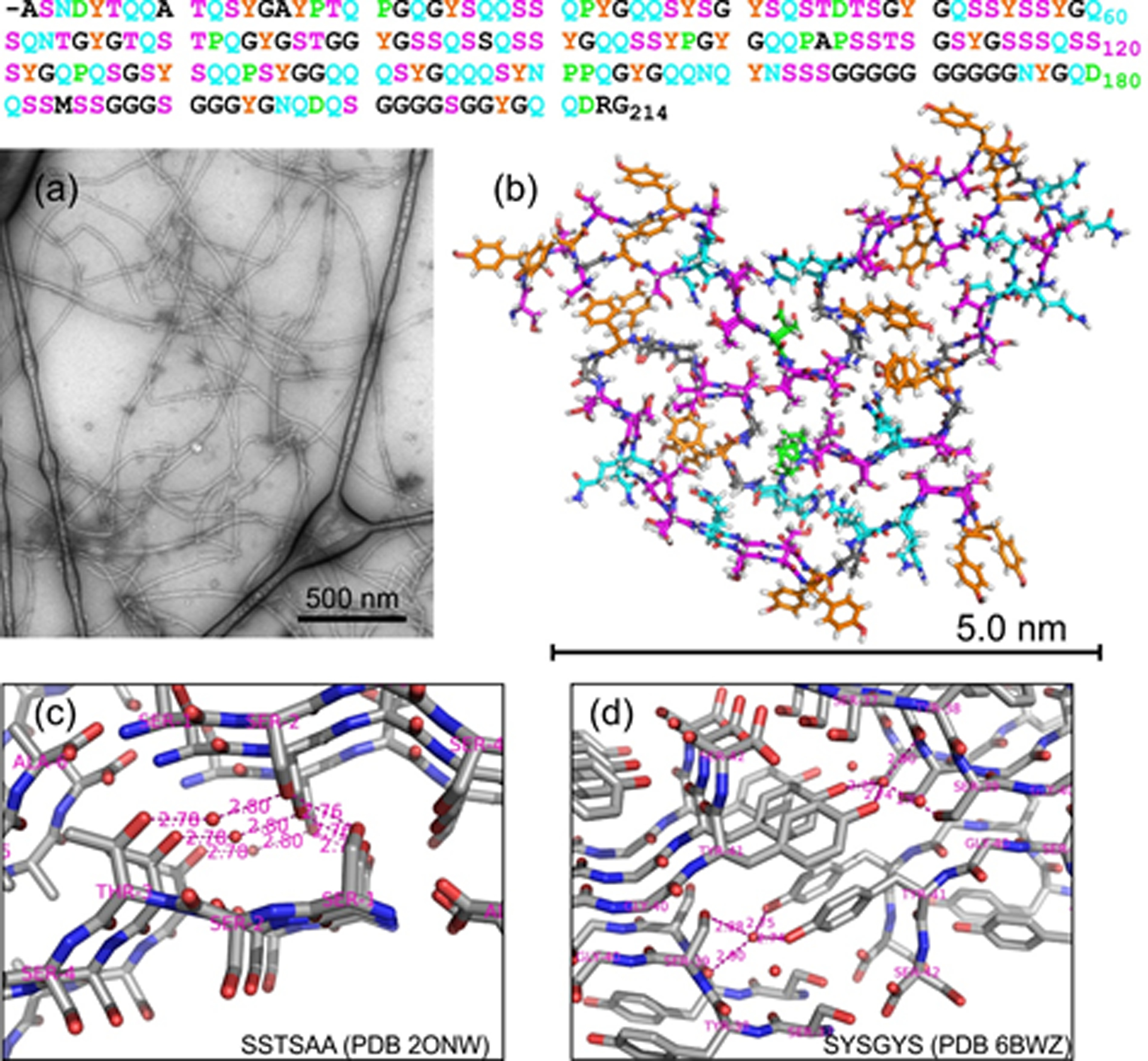
(a) TEM image of negatively stained 2H,15N,13C-FUS-LC fibrils. (b) Molecular structural model for the FUS-LC fibril core, as determined by Murray et al.1, viewed with the fibril growth axis perpendicular to the page and including residues 37–97. To illustrate the precision of the model, one molecule from model 1 of Protein Data Bank file 5W3N is superimposed on one molecule from model 16, with Ser and Thr residues in magenta, Gln and Asn residues in cyan, Tyr residues in orange, Pro and Asp residues in green, and all other residues in black. The full sequence of FUS-LC is shown above with the same color scheme. (c) Portion of the crystal structure of the hexapeptide SSTSAA, from Protein Data Bank file 2ONW. Short distances involving oxygen atoms of sidechain hydroxyl groups and water molecules are indicated in Å units. (d) Portion of the crystal structure of the hexapeptide SYSGYS, from Protein Data Bank file 6BWZ.
For FUS-LC fibrils, it is currently unclear why segments other than residues 39–95, with nearly identical amino acid compositions, do not form alternative core structures with similar thermodynamic stabilities and kinetic accessibilities, leading to polymorphism. It is also unclear why residues 39–95 adopt a unique structure in FUS-LC fibril cores, with a unique backbone conformation and a unique set of sidechain-sidechain contacts. Finally, while hydrophobic interactions clearly stabilize the structures of amyloid-β and α-synuclein fibrils, interactions that stabilize the FUS-LC fibril core structure are unclear. More precisely, intermolecular hydrogen bonds between backbone carbonyl and amide groups provide stabilization along the fibril growth direction, as in any cross-β fibril structure35. Intermolecular hydrogen bonds among sidechain amide groups of Gln and Asn residues, called polar zipper interactions36,37, may also contribute to stability along the fibril growth direction in the in-register parallel cross-β structure that was established by experimental ssNMR data and mass-per-length measurements1 (although it should be noted that residues 39–95 are not especially Gln- or Asn-rich). However, sidechain-sidechain interactions in planes perpendicular to the fibril growth direction, which ultimately determine the “fold” of the core, remain mysterious.
Insights into sidechain-sidechain interactions that may play a role in the FUS-LC core structure come from crystal structures of short peptides in cross-β motifs, including peptides from FUS-LC and other LC sequences38–41. For example, a crystal structure of the hexapeptide SSTSAA (Fig. 1c) shows inter-β-sheet hydrogen bonding between sidechain hydroxyl groups of Ser1 and Ser2, as well as water-mediated hydrogen bonding between sidechain hydroxyl groups of Ser2 and Thr3 (i.e., hydrogen bonds from Ser2 and Thr3 to the same single-file row of water molecules). A crystal structure of SYSGYS (Fig. 1d) shows water-mediated hydrogen bonding between sidechain hydroxyl groups of Ser39 and Tyr41. Additional examples of direct sidechain-sidechain, water-mediated sidechain-sidechain, and sidechain-backbone hydrogen bonding from cross-β crystal structures are shown in Fig. S2.
Do similar interactions exist in FUS-LC fibrils, thereby stabilizing the core structure in planes perpendicular to the fibril growth direction? In this paper, we present results from additional ssNMR experiments that provide information about the possible involvement of specific sidechain hydroxyl groups in stabilizing interactions within the FUS-LC fibril core, and that also indicate differential accessibilities of sidechain hydroxyl groups for hydrogen exchange with water. The ssNMR measurements were performed on a sample that was highly deuterated except at hydroxyl, amide, amine sites (i.e., exchangeable sites), thereby allowing relatively sharp 1H ssNMR signals to be observed and assigned.
Materials and Methods
Preparation of 2H,15N,13C-labeled FUS-LC fibrils
Unless indicated otherwise, chemicals were obtained from Sigma-Aldrich and solutions were 0.22 μm sterile filtered. N-terminally His6-tagged residues 2–214 of FUS-LC was expressed in BL21(DE3) pLysS E. coli cells (ThermoFisher Scientific) using a pHis-parallel vector.42 The sequence of the N-terminal tag was MSYYHHHHHHDYDIPTTENLYFQGAMDP, which was followed by the sequence in Fig. 1. (The tag is dynamically disordered in FUS-LC fibrils and does not contribute to ssNMR spectra.) Cells were incubated quiescently in 5 ml unfiltered Luria-Bertani medium at 37° C for 4 h. Then 200 μl of the culture was transferred to 2 ml of M9 medium and shaken at 37° C for 3.5 h. The 2.2 ml culture was then added to 50 ml of M9 medium and shaken overnight at 37° C. The following day, the 50 ml overnight culture was added to 950 ml of M9 medium and shaken at 37° C for 5 h. The cells were transferred to 20° C and shaken for 1 h (optical density at 600 nm equal to 1.0 ± 0.1) before adding isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM. The culture was then shaken for 21 h at 20° C before harvesting the cells, flash freezing them in liquid nitrogen, and storing them at −80° C. All culture media contained 100 μl/ml ampicillin. For isotopic labeling of FUS-LC, M9 salts were prepared in 99.9% D2O (Cambridge Isotope Labs) supplemented with 1x MEM vitamin mix (Corning), 1.0 g/l 15N ammonium chloride (Cambridge Isotope Labs), 2.0 g/l 13C6,2H7-D-glucose, and 0.3 g/l 2H,13C,15N-Isogro. The MEM vitamin mix was lyophilized and resuspended in 99.9 % D2O before use. Ampicillin and IPTG stock solutions were prepared at 1000-fold higher concentration than the working solution in 99.9% D2O. Unfiltered 100x MEM stock solutions were frozen in liquid nitrogen, lyophilized overnight, and resuspended in 99.9% D2O prior to use.
The frozen cell pellet was resuspended in 40 ml of pH 7.9 lysis buffer containing 50 mM tris(hydroxymethyl)aminomethane (KD Medical), 500 mM sodium chloride (KD Medical), 6 M guanidine HCl, 1% w/w Triton X-100 (American Bio), and 4 cOmplete Protease Inhibitor Cocktail tablets (Roche). Then 25 mg of hen egg white lysozyme powder and 4 μl of Pierce Universal Nuclease were added and the suspension was sonicated on ice with Branson 250 Sonifier using a 1/4” diameter tip, an output power of 0.3 and 10% duty cycle. The mixture was rotated at 4° C for 10 min and then centrifuged at 207,000 × g for 30 min at 4° C. The supernatant was mixed with 5 ml of NiNTA resin (Qiagen) in pH 7.4 equilibration buffer containing 20 mM sodium phosphate, 8 M urea, 500 mM sodium chloride, and 5 mM 2-mercaptoethanol before rotating at 4 °C for 30 min. The resin was loaded onto a gravity flow column (Kimble-Chase) and washed with equilibration buffer containing 0.1 mM phenylmethanesulfonyl fluoride and 20 mM imidazole until the 280 nm absorbance of the elute reached baseline. The protein was eluted using equilibration buffer with 200 mM imidazole and 7.6 M urea.
The elution fraction containing 34 μM 2H,15N,13C-FUS-LC was concentrated to 300 μM using a 0.5 ml Amicon Ultra centrifugal filter with 10 kDa cutoff (Millipore). About 175 μl of the protein solution was loaded into a 0.5 ml Slide-A-Lyzer dialysis cassette with 3.5 kDa cutoff (ThermoFisher Scientific) and dialyzed against 2.0 l of 20 mM sodium phosphate, pH 7.4. After dialysis for 19 hr about 300 μl of the protein solution was collected and allowed to sit quiescently at room temperature (approximately 22° C). After a seven-day incubation period, a mixture of fibrillar and amorphous assemblies was observed by transmission electron microscopy (TEM). The solution was then flash frozen in liquid nitrogen and thawed seven times to break up the protein aggregates, creating fragments that serve as fibril seeds. An additional 2.3 ml of 2H,15N,13C-FUS-LC protein was concentrated from 31 μM to 42 μM, then dialyzed in a 3 ml Slide-A-Lyzer dialysis cassette with 3.5 kDa cutoff against 2.0 l of 20 mM sodium phosphate, pH 7.4, for 24 h. A 1.7 ml volume of protein solution was recovered from the dialysis cassette and mixed with the seeds. The mixture was incubated quiescently at room temperature for eight days. The sample then contained almost exclusively fibrillar FUS-LC assemblies, with no significant fraction of amorphous aggregates in TEM images. Finally, the fibrils were harvested by centrifugation at 279,000 × g for 20.5 h and packed into a magic-angle spinning (MAS) NMR rotor with 1.8 mm outer diameter and 10 μl sample volume by centrifugation at 15,000 × g for 1.0 h at 4° C in a home-made centrifugal funnel device.
Electron microscopy
TEM images were obtained with a FEI Morgagni microscope, equipped with a side-mounted Advantage HR camera (Advanced Microscopy Techniques) and operating at 80 keV. Grids were home-made carbon films supported on lacey carbon on 300 mesh copper (Electron Microscopy Sciences) and were glow-discharged immediately before sample application. A 5 μl aliquot of fibril solution, diluted by a factor of 10, was adsorbed to the carbon film for two minutes, blotted, washed with 5 μl of water for 10 s, blotted, washed again, blotted, stained with 5 μl of 3% uranyl acetate for 10 s, blotted, and dried in air.
Solid state NMR
ssNMR measurements were performed at 17.5 T (746.2 MHz 1H NMR frequency) and 14.1 T (599.2 MHz 1H NMR frequency), using Varian Infinity and InfinityPlus spectrometers, respectively, and triple-channel MAS ssNMR probes with 1.8 mm rotor diameters produced by the laboratory of Prof. Ago Samoson (NMR Institute and Tallinn Institute of Technology, Estonia). Except as noted below, ssNMR data were acquired at a MAS frequency of 40.00 kHz. Sample temperatures were maintained at approximately 28° C, as indicated by the temperature-dependent 1H chemical shift of water in the sample, using −50° C nitrogen gas at the cooling gas input of the probe (17 standard liters per minute flow rate) to counteract frictional heating from high-frequency MAS. Radio-frequency (RF) pulse sequences for three-dimensional (3D) spectra are shown in Fig. S1. 1H, 13C, or 15N decoupling used the XiX technique43, with RF fields of 6–10 kHz. 13C-13C and 1H-1H polarization transfers used the finite-pulse radio-frequency-driven recoupling (fpRFDR) technique44,45. 1H-15N, 15N-1H, and 1H-13C polarization transfers used cross-polarization (CP)46, with ramped RF amplitudes as depicted in Fig. S1. A 3D 15N-13C-13C spectrum used non-selective 15N-13C CP transfers, so that NCACX and NCOCX crosspeaks were contained in the same spectrum. 3D CANH, CAN(H)H, CONH, CON(H)H, and HN(CO)CX spectra47–49 used frequency-selective 13C-15N or 15N-13C CP transfers, with the 13C carrier frequency shifted to the 13CO or 13Cα regions of the spectrum.50 In 1H-detected measurements, strong signals from water were attenuated by a train of π/2 pulses before the final CP transfer to 1H nuclei. Typically, nsat = 20 pulses separated by 15–25 ms delays were used, as depicted in Fig. S1. Other experimental parameters for individual 3D spectra are given in Table S1 and in figure captions. Two-dimensional (2D) and 3D data were processed with NMRpipe software51 and analyzed, displayed, and plotted with Sparky software (available at https://www.cgl.ucsf.edu/home/sparky/).
Water-filtered 2D 13C-13C spectra,52,53 used to characterize hydrogen exchange between water and sidechain hydroxyl groups of Ser and Thr residues, were acquired at 14. 1 T with MAS at 13.60 kHz. The sample temperature was maintained at 26° C. The RF pulse sequence for these measurements is shown in Fig. S9a. A 1H spin echo period τecho = 2.0 ms was used to suppress 1H polarization at immobilized sites on the protein, which have short T2 relaxation times at the relatively low MAS frequency, and to select 1H polarization of water, which has a much longer T2 value. The spin echo period was followed by a variable 1H-1H exchange period texch during which polarization could be transferred to sidechain hydroxyl sites (and other exchangeable sites). Hydroxyl 1H polarization was then measured through the intensities of 13Cβ/13Cα (and other) crosspeaks, which were quantified with the seriesTab program of NMRpipe. Two-pulse phase-modulated (TPPM) proton decoupling44 with an RF amplitude of 85 kHz was applied during t1 and t2 periods. A 50 ms 13C-13C mixing period with dipolar-assisted rotational resonance54 (DARR) was used.
The reference for 13C chemical shifts in this paper is nominally DSS (4,4-dimethyl-4-silapentane-1-sulfonic acid). However, the 13C chemical shift reference was adjusted to make 13Cα shifts in 2H,15N,13C-FUS-LC fibrils agree optimally with our published 13Cα shifts for 15N,13C-FUS-LC fibrils1. Due to deuterium isotope effects55, 13C chemical shifts for carbonyl, carboxyl, methylene, and methyl sites in 2H,15N,13C-FUS-LC fibrils are then expected to differ from those in 15N,13C-FUS-LC fibrils by approximately +0.3 ppm, +0.3 ppm, -0.3 ppm, and −0.6 ppm, respectively. Our 1H chemical shift reference is such that the chemical shift of water is 4.50 ppm. The nominal 15N chemical shift reference is liquid ammonia.
Chemical shift assignment calculations
After manual peak-picking of 3D solid ssNMR spectra in Sparky, assignments were generated with the Monte Carlo/simulated annealing approach described previously56. A modified version of the assignment program, called mcassign3a, was used. The modified program allows inter-residue crosspeaks with ambiguous connection directions to be used. This capability was necessary for inclusion of 3D CAN(H)H, CON(H)H, and NHH spectra in the assignment calculations, since 1H-1H spin polarization transfers can in principle occur with equal likelihood from the amide proton of residue k to the amide protons of either residue k+1 or residue k-1. The modified program also allows residue type assignments to be specified for two successive residues that are involved in an inter-residue crosspeak signal, so that the signal can only be assigned to a given residue j if both residue j and residue j+1 match the specified residue type assignments. This second modification in mcassign3a was not used in the current work but is helpful in other contexts.
Assignment calculations proceeded in an iterative fashion, as described previously1, with a gradual increase in the number of signals with definite assignments. Signal assignments were restricted to residues 31–110 of the full-length FUS-LC sequence, based on our previous identification of residues 39–95 as the structurally ordered core. The initial calculations used signal tables from 3D NCACX, NCOCX, HN(CO)CX, CANH, and CONH spectra, with 60, 52, 62, 81, and 104 signals in each table, respectively. Since 3D CANH and CONH spectra were recorded at both 14.1 T and 17.5 T, the signal tables for these spectra were generated from crosspeaks in spectra at both field strengths. The relatively large numbers of signals in these two tables is attributable to non-negligible inter-residue CA-N, non-sequential CO-N, and inter-residue N-H polarization transfers, which produce extra crosspeaks. All other signal tables were generated from spectra at 14.1 T. Signal tables were updated after each set of calculations to include any new definite assignments (defined as the same assignment for a given signal in 10 out of 10 independent mcassign3a runs). After three sets of assignment calculations, signal tables from 3D CAN(H)H, CON(H)H, and NHH spectra were added, with 28, 17, and 39 inter-residue crosspeak signals, respectively. Each of these spectra was represented by two signal tables, which for example allowed two NHH crosspeaks (one from residue k to residue k − 1, the other from residue k to residue k + 1) to be assigned to the same residue. After one set of calculations with the full set of signal tables, signals that were never assigned (in 10 out of 10 independent runs) were removed from all tables, leaving 60, 52, 59, 67, 64, 28, 17, and 39 signals from 3D NCACX, NCOCX, HN(CO)CX, CANH, CONH, CAN(H)H, CON(H)H, and NHH spectra. The final set of calculations was then performed, with 50 simulating annealing steps in each of 10 runs, with 108 Monte Carlo moves (i.e., attempted assignment changes) in each step. The final signal tables are given in Tables S2–S9.
A significant number of signals remained unassigned, specifically 9, 10, 13, 17, 10, 5, 2, and 11 signals from the final 3D NCACX, NCOCX, HN(CO)CX, CANH, CONH, CAN(H)H, CON(H)H, and NHH signal tables, respectively. We can attribute these signals to short segments or individual residues outside the main FUS-LC fibril core segment that are immobilized or partially immobilized through contacts with the core, as discussed previously1. The repetitive, low-complexity nature of the FUS-LC sequence makes signals from isolated short segments difficult to assign unambiguously.
Results
Chemical shift assignments for 2H,15N,13C-FUS-LC fibrils
Chemical shift assignments were determined from 13C-detected and 1H-detected 3D ssNMR spectra, recorded with MAS at 40.00 kHz, using a Monte Carlo/simulated annealing algorithm to assign multidimensional signals from the 3D spectra to specific amino acids (see Materials and Methods). Although 15N and 13C chemical shift assignments for 15N,13C-FUS-LC fibrils were reported previously, the assignment process was repeated independently for 2H,15N,13C-FUS-LC fibrils to obtain 1H chemical shift assignments and to determine the changes in 13C chemical shifts that are induced by deuteration.
Fig. 2 shows examples of 2D planes from 13C-detected 3D spectra to illustrate the resolution and signal-to-noise ratios of these spectra. 1H, 13C, and 15N linewidths (full widths at half-maximum, FWHM) were 0.20–0.25 ppm, 0.6–1.0 ppm, and 0.9–1.3 ppm, respectively. 3D NCACX and NCOCX spectra in Figs. 2a and 2b were obtained from a single 3D 15N-13C-13C measurement, using the RF pulse sequence in Fig. S1a with non-selective 15N-13C cross-polarization between t1 and t2 periods. These spectra primarily correlate the backbone amide 15N chemical shift of residue k with the 13Cα or 13CO chemical shift of residue k or k − 1, and then with sidechain 13C chemical shifts of residue k or k − 1. The 3D HN(CO)CX spectrum in Fig. 2c, obtained with the RF pulse sequence in Fig. S1b, primarily correlates the backbone amide 1H chemical shift of residue k with the backbone amide 15N chemical shift of residue k, and then with backbone 13CO, 13Cα, and sidechain 13C chemical shifts of residue k − 1. Additional 2D planes from these 3D spectra are shown in Fig. S3.
Figure 2:
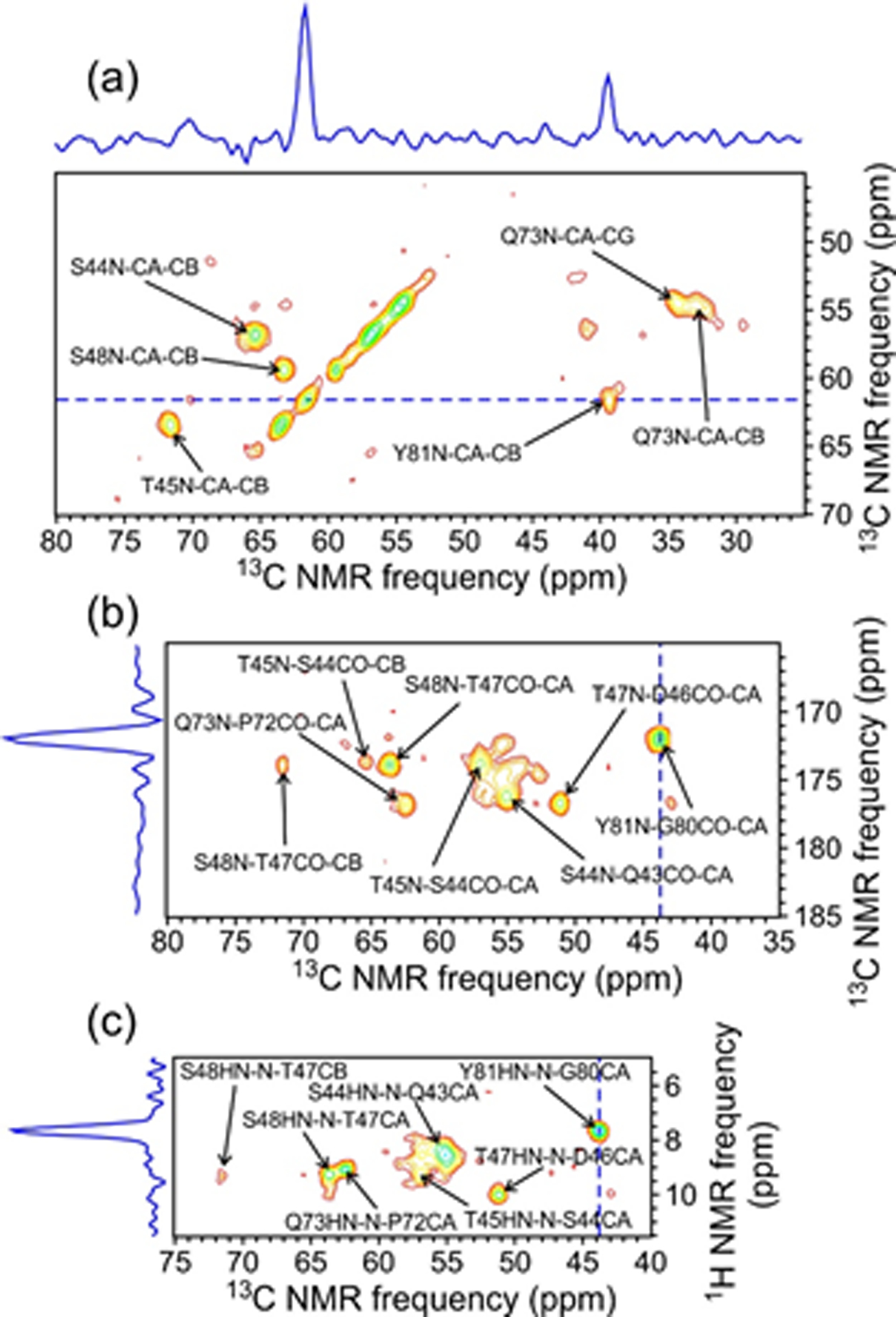
2D planes from 3D NCACX (a), NCOCX (b), and HN(CO)CX (c) spectra of 2H,15N,13C-FUS-LC fibrils, taken at a 15N chemical shift of 121.4 ppm. Assignments are shown for crosspeaks with maxima within ±0.8 ppm of 121.4 ppm in the 15N dimension. Contour levels increase by successive factors of 1.4. 1D slices shown above or on the left side of the 2D planes correspond to the dashed blue lines and illustrate the signal-to-noise ratios and ssNMR linewidths in the 3D spectra. Spectra were acquired at 14.1 T with 40.00 kHz MAS. (Note that the NCACX and NCOCX planes are different sections of the same 3D 15N-13C-13C spectrum.)
Fig. 3 shows examples of 2D planes from 1H-detected 3D spectra. 3D CANH and CONH spectra in Figs. 3a and 3b (left side) primarily correlate the 13Cα chemical shift of residue k or the 13CO chemical shift of residue k − 1 with the backbone amide 15N chemical shift of residue k, and then with the backbone amide 1H chemical shift of residue k. 3D CAN(H)H and CON(H)H spectra in Figs. 3a and 3b (right side) allow correlations to backbone amide 1H chemical shifts of residues k − 1 and k + 1 in the third dimension. These spectra were obtained with the RF pulse sequence in Fig. S1c. The 3D NHH spectrum in Fig. 3c, obtained with the RF pulse sequence in Fig. S1d, primarily correlates the backbone amide 15N chemical shift of residue k with the backbone amide 1H chemical shift of residue k, and then with the backbone amide 1H chemical shift of residues k − 1 and k + 1. Additional 2D planes from these 3D spectra are shown in Fig. S4.
Figure 3:
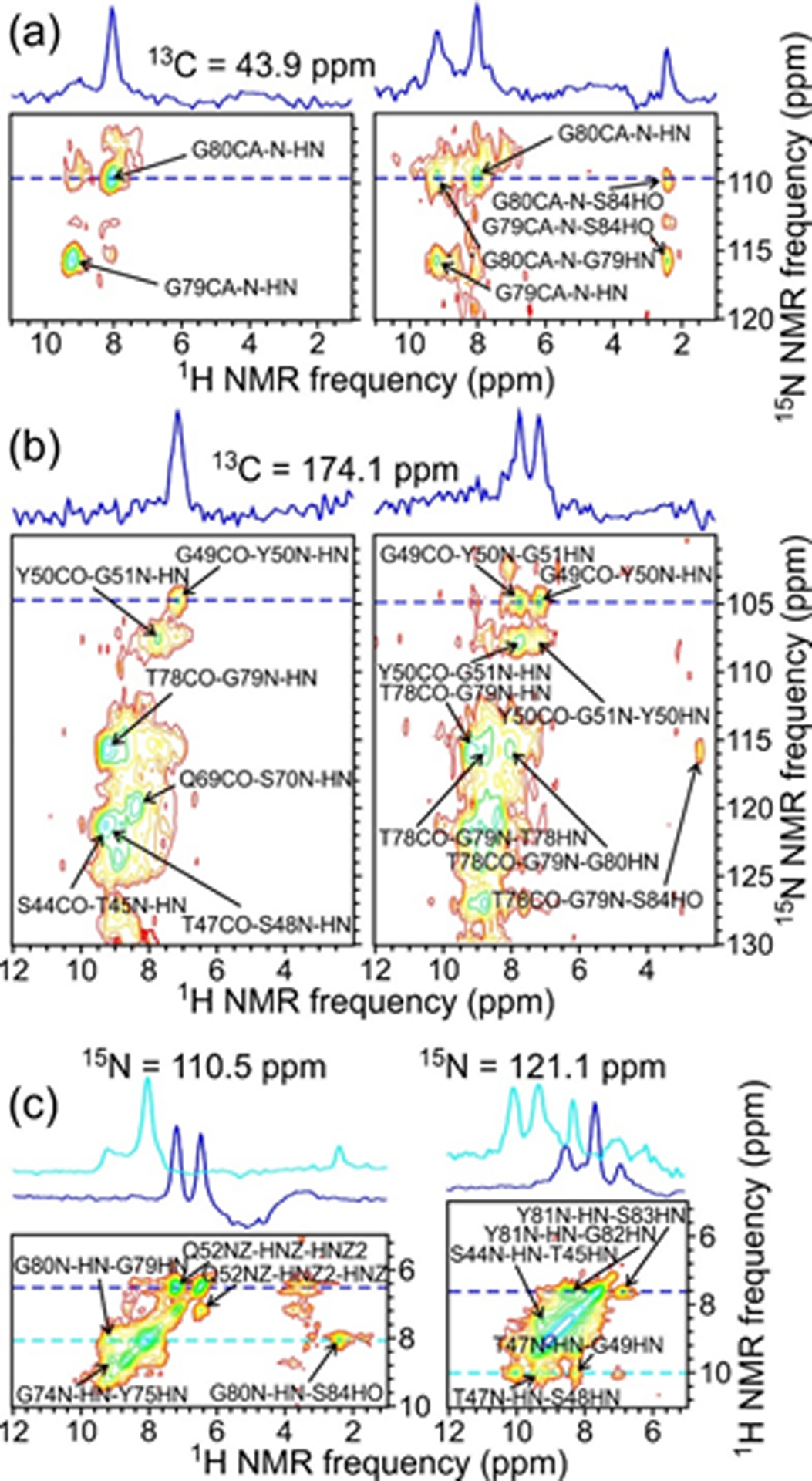
2D planes from 3D CANH and CAN(H)H spectra (a, left and right, respectively), 3D CONH and CON(H)H spectra (b, left and right, respectively), and a 3D NHH spectrum (c) of 2H,15N,13C-FUS-LC fibrils. Assignments are shown for crosspeaks with maxima within ±0.3 ppm of 43.9 ppm in the 13C dimension in panel a, within ±0.4 ppm of 174.1 ppm in the 13C dimension in panel b, and within ±0.6 ppm of 110.5 ppm (left) or 121.1 ppm (right) in the 15N dimension in panel c. Contour levels increase by successive factors of 1.4. 1D slices shown above the 2D sections correspond to the dashed blue and cyan lines. In panels a and b, these slices illustrate the development of additional inter-residue crosspeaks from 1H-1H spin polarization transfers in the CAN(H)H and CON(H)H measurements. Spectra were acquired at 14.1 T with 40.00 kHz MAS.
A simple 2D 1H-15N spectrum of 2H,15N,13C-FUS-LC fibrils is poorly resolved, showing only a small number of interpretable crosspeaks (see Fig. S5). With our fibril growth conditions, we expect all exchangeable hydrogen sites (i.e., amide, amine, and hydroxyl sites) to be protonated. This means that the overall deuteration level in residues 39–95 of FUS-LC is roughly 68%. Our MAS frequency of 40.00 kHz is then not sufficient to reduce 1H ssNMR linewidths to the point where a well-resolved 2D 1H-15N spectrum is obtained.
From the 3D spectra, definite assignments were found for residues 41–55, 63, 65–70, 72–87, 89–94, and 107–110, as given in Table S10 (Biological Magnetic Resonance Bank code 50026). For the most part, chemical shift assignments for 2H,15N,13C-FUS-LC fibrils are in good agreement with previously published assignments from spectra of 15N,13C-FUS-LC fibrils1, which included residues 39–40, 44–54, and 63–95. Differences in the identities of residues for which assignments were obtained arise from differences in the types of 3D spectra that were used and the signal-to-noise ratios of the spectra. On the average, 15N chemical shifts in Table S10 are about 0.4 ppm lower than in previously published assignments. 13C chemical shifts also differ somewhat due to isotope effects and differences in referencing.
Chemical shifts assigned to 15N of G82 and to 15N, 13Cβ, and 13CO of S89 in Table S10 differ significantly from previously published assignments1. The previously published 15N assignment for G82 is incorrect. However, S89 signals with chemical shifts in Table S10 were not present in the previously published 3D spectra of 15N,13C-FUS-LC fibrils, and S89 signals with the previously published chemical shifts are not present in 3D spectra of 2H,15N,13C-FUS-LC fibrils. Chemical shift differences for S89 apparently reflect minor structural differences.
Overall, the ssNMR spectra of 2H,15N,13C-FUS-LC fibrils support our earlier determination that the structurally ordered core of FUS-LC fibrils is formed by the segment depicted in Fig. 1b, with most of the rest of the 214-residue LC domain remaining dynamically disordered. These data indicate that residues 107–110 are rigid in the FUS-LC fibrils. Since long-range distance restraints involving residues 107–110 were not obtained, the structural role of this segment is not yet known. Secondary 13C chemical shifts do not support β-strand or α-helical secondary structure in this segment. As in our earlier work1, the ssNMR spectra include signals from outside the core-forming segment, which we can not assign definitively to specific residues. We attribute these signals to short segments (perhaps single residues or pairs of residues) that are partially ordered through interactions with the fibril core, or possibly through interactions between fibrils that are packed together densely in the MAS rotor.
Site-specific correlations with water
Correlations between the 1H chemical shift of water and certain 13C chemical shifts of 2H,15N,13C-FUS-LC are apparent in 13C-detected 2D 1H-13C and 3D 1H-13C-13C spectra. For detecting these correlations, 13C detection is preferred over 1H detection because 1H-13C crosspeaks at or near the 1H chemical shift of water are unavoidably obscured by large residual water signals in 1H-detected spectra. In the 2D spectrum (Fig. S6), correlations between the 1H chemical shift of water and 13C chemical shifts of T68 and/or T78, T45 and/or T47, S42 and/or S48, D46, G49, Y75 and/or Y81, P72, and Gln (unassigned) are observed. In 3D spectra, additional correlations with water are detected and ambiguities between T68 and T78 and between T45 and T47 are resolved. The 2D plane from a 3D spectrum with τHH = 0 (i.e., 3D HCC spectrum, pulse sequence in Fig. S1e), taken at the 1H chemical shift of water, is shown in Figs. 4a and 4c. Crosspeaks assigned to S42, S44, T45, D46, S48, T68, S83, and S110 are apparent in this 2D plane. Crosspeaks to aromatic carbons of at least one Tyr residue are also observed (Fig. S7). Crosspeak intensities are obviously not symmetric about the 13C-13C diagonal, indicating stronger nuclear spin polarization transfers from 1H of water to sidechain carbon sites, including the hydroxyl-bearing 13Cβ sites of Ser and Thr residues and the carboxyl 13Cγ site of D46, than to backbone 13Cα and 13CO sites. In the case of Tyr, the strongest polarization transfer is to the hydroxyl-bearing 13Cζ site (Fig. S7). Similar effects have been seen in previous studies of water/protein interactions by ssNMR53,57–60. We did not observe correlations to distinct water populations as reported by Wang et al. in studies of amyloid-β fibrils52, possibly because the resolution in the 1H dimension of our 3D 1H-13C-13C spectrum (about 0.2 ppm, limited by the maximum t1 value) was insufficient to distinguish signals from distinct populations.
Figure 4:
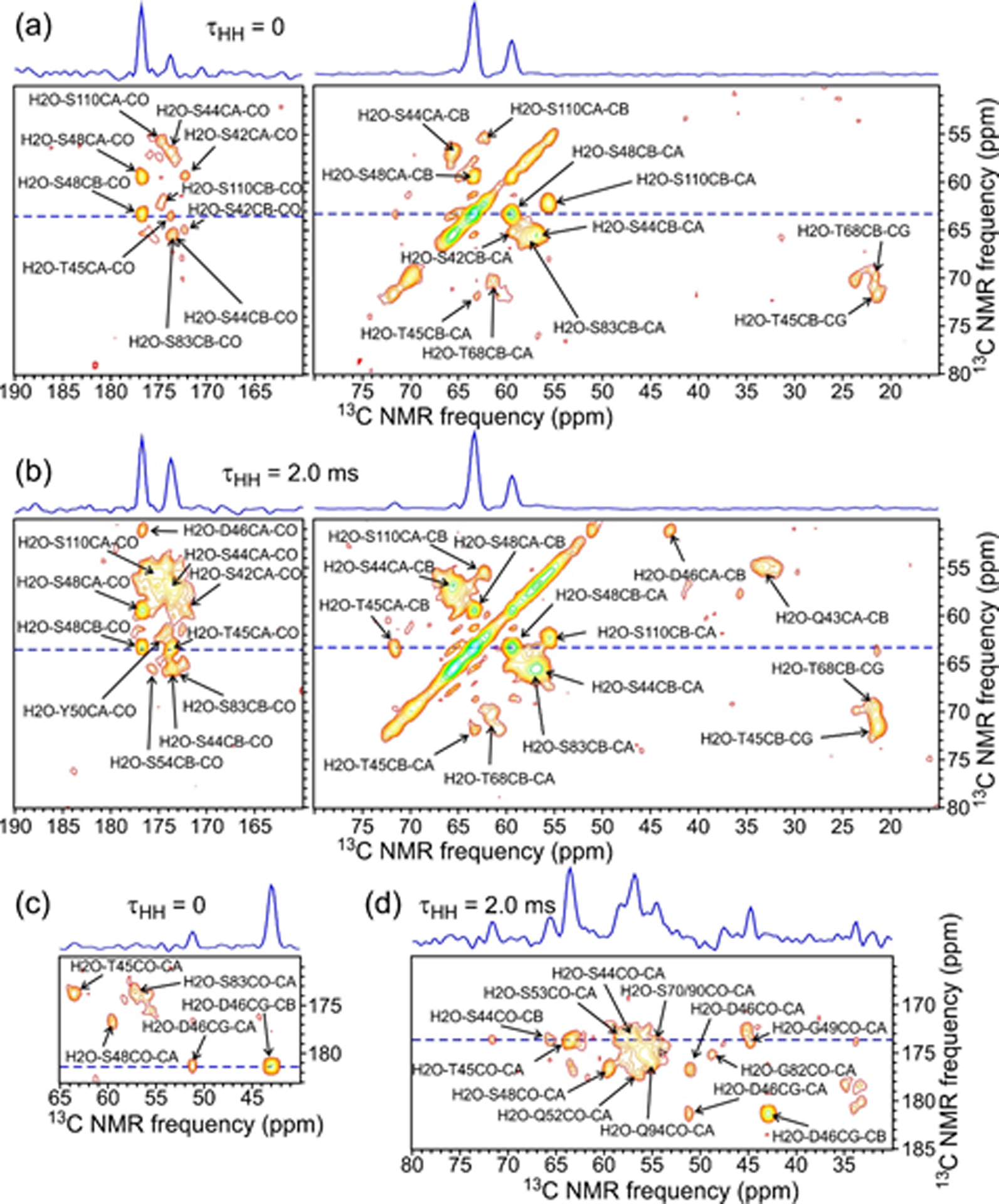
2D planes from 3D HCC (a and c; τHH = 0) and H(H)CC (b and d; τHH = 2.0 ms) spectra of 2H,15N,13C-FUS-LC fibrils at the 1H chemical shift of water (4.50 ppm). Assignments for crosspeaks within ±0.1 ppm of the water shift are shown. Contour levels increase by successive factors of 1.4. 1D slices above the 2D section correspond to dashed blue lines. Spectra were acquired at 14.1 T with 40.00 kHz MAS.
We interpret the observed correlations to water in Figs. S6, 4, and S7 as being primarily the result of hydrogen exchange between water and sidechain hydroxyl (or carboxyl) sites during the 4.0 ms τHC period of the 3D HCC pulse sequence. Near neutral pH and 30° C, intrinsic hydrogen exchange rates for sidechain hydroxyl groups of Ser and Thr are reported to be approximately 400 s−1, while the intrinsic hydrogen exchange rate for the sidechain hydroxyl group of Tyr is much greater61. Thus, we expect solvent-exposed, relatively “free” sidechain hydroxyl groups of the FUS-LC fibril core structure to exhibit correlations to water. In contrast, sidechain hydroxyl groups that are involved in hydrogen-bonding interactions within the core are expected to have slower hydrogen exchange rates, leading to weaker correlations to the water 1H chemical shift for the relevant residues57,58,62. Importantly, we do not observe correlations to water for certain Ser and Thr residues that have 13C chemical shift assignments, namely T47, S53, S54, S70, S77, T78, S84, S86, S87, S89, S90, S107, S108, and T109. Of these, S87, S89, and T109 have relatively weak crosspeaks in other types of 3D spectra, suggesting time scales or amplitudes of motion that attenuate their signals. The absence of detectable correlations to water for S87, S89, and T109 is therefore attributable to the overall weakness of signals from these three residues. On the other hand, the absence of detectable correlations to water for T47, S53, S54, S70, S77, T78, S84, S86, S90, S107, and S108 (which have relatively strong signals in other 3D spectra) is attributable to participation of these 11 residues in hydrogen-bonding interactions that reduce the rates of hydrogen exchange rates with water of their sidechain hydroxyl groups. Additional measurements of hydrogen exchange rates are described below.
Figs. 4b and 4d show the 2D plane at the 1H chemical shift of water from a 3D spectrum with τHH = 2.0 ms (i.e., 3D H(H)CC spectrum, pulse sequence in Fig. S1e). Compared to results with τHH = 0, the pattern of 13C-13C crosspeaks is changed by 1H-1H polarization transfers during the τHH period, especially polarization transfers from sidechain hydroxyl protons to backbone amide protons that lead to stronger 13Cα and 13CO signals in the t2 dimension of the 3D spectrum. Crosspeaks from additional residues, namely Q43, G49, Y50, Q52, S53, S54, G82, and Q94, appear. Crosspeaks from the water 1H chemical shift to 13C chemical shifts of Q43, G49, and G82 can be attributed to polarization transfers from the rapidly-exchanging sidechain hydroxyl protons of S42, S48, and S83 discussed above to backbone amide protons, followed by transfers from amide protons to 13Cα and 13CO sites during the τHC period. Crosspeaks in the 3D H(H)CC spectrum from the water 1H chemical shift to 13C chemical shifts of Y50, Q52, S53, S54, and Q94 can not be attributed similarly to polarization transfer pathways that involve sidechain hydroxyl sites of preceding residues. Polarization transfer pathways that produce these crosspeaks are unclear.
Correlations to sidechain hydroxyl 1H chemical shifts
Sidechain hydroxyl 1H chemical shifts of certain Ser and Thr residues in the FUS-LC fibril core can also be identified from intra-residue crosspeaks in the 13C-detected 3D 1H-13C-13C spectra. Fig. 5 shows 2D planes from these spectra at sidechain hydroxyl 1H chemical shifts of S84 (Figs 5a and 5b), S77 (Figs. 5c and 5d), S53 and T45 (Figs. 5e and 5f), and S48 (Figs. 5g and 5h). Additional 2D planes at sidechain hydroxyl 1H chemical shifts of S83, S86, T47, T78, and T68 are shown in Fig. S8. As discussed above, T45, S48, S83, and T68 also have detectable correlations with the 1H chemical shift of water, arising from hydrogen exchange between water and their sidechain hydroxyl groups, but the exchange rate is not large enough to prevent observation of the sidechain hydroxyl 1H chemical shift as a separate signal.
Figure 5:
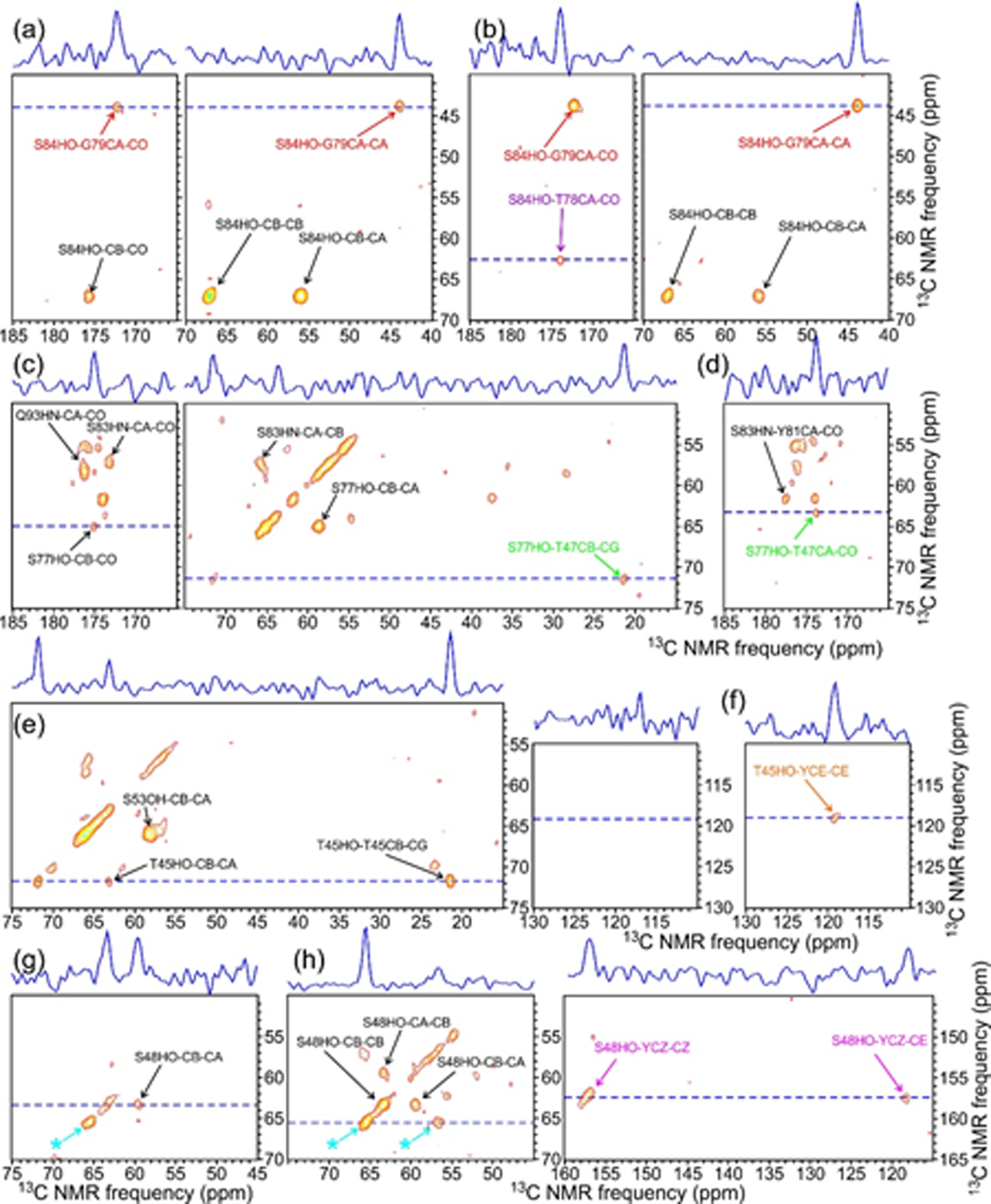
2D planes from 3D HCC (a,c,e,g) and H(H)CC (b,d,f,h) spectra of 2H,15N,13C-FUS-LC fibrils at 1H chemical shifts of 2.39 ppm (a,b), 6.91 ppm (c,d), 4.93 ppm (e,f), and 4.00 ppm (g,h), corresponding to sidechain hydroxyl protons of S84, S77, S53 and T45, and S48, respectively. Contour levels increase by successive factors of 1.4, and assignments are shown for crosspeaks within ±0.1 ppm of the nominal 1H chemical shift in each panel. (Asterisks in panels g and h indicate apparent crosspeaks arising from truncation artifacts from strong crosspeaks between the water 1H chemical shift and 13Cα and 13Cβ chemical shifts of S44.)
Inter-residue crosspeaks involving sidechain hydroxyl 1H chemical shifts can provide information about long-range contacts in the FUS-LC fibril core. With τHH = 0, correlations from the S84 hydroxyl proton to G79 and from the S77 hydroxyl proton to T47 are observed (Figs. 5a, and 5c). With τHH = 2.0 ms, additional correlations from the S84 hydroxyl to T78, from the T45 hydroxyl to a Tyr 13Cε site (unassigned), and from the S48 hydroxyl to a Tyr 13Cζ (unassigned) are observed (Figs. 5b, 5f, and 5h). These inter-residue crosspeaks are discussed further below.
Hydrogen exchange rates from water-filtered 2D 13C-13C spectra
Fig. 6a shows the region of water-filtered 2D 13C-13C spectra of 2H,15N,13C-FUS-LC fibrils that contains Cβ/Cα crosspeaks of Thr and Ser residues, with 1H-1H exchange periods texch equal to 0.1 ms and 100 ms. The RF pulse sequence in Fig. S9a was used to obtain these spectra, in which 13C ssNMR signals arise from 1H polarization that was associated with water molecules before the texch period and transferred to exchangeable sites on the protein during texch (or during the 1.5 ms 1H-13C cross-polarization period τHC).52,53 The primary mechanism for rapid build-up of Cβ/Cα crosspeaks of Thr and Ser residues during texch is expected to be hydrogen exchange from water to sidechain hydroxyl sites. Patterns of crosspeak intensities are clearly different for the two values of texch in Fig. 6a, indicating that different residues have different hydrogen exchange rates for their sidechain hydroxyl sites.
Figure 6:
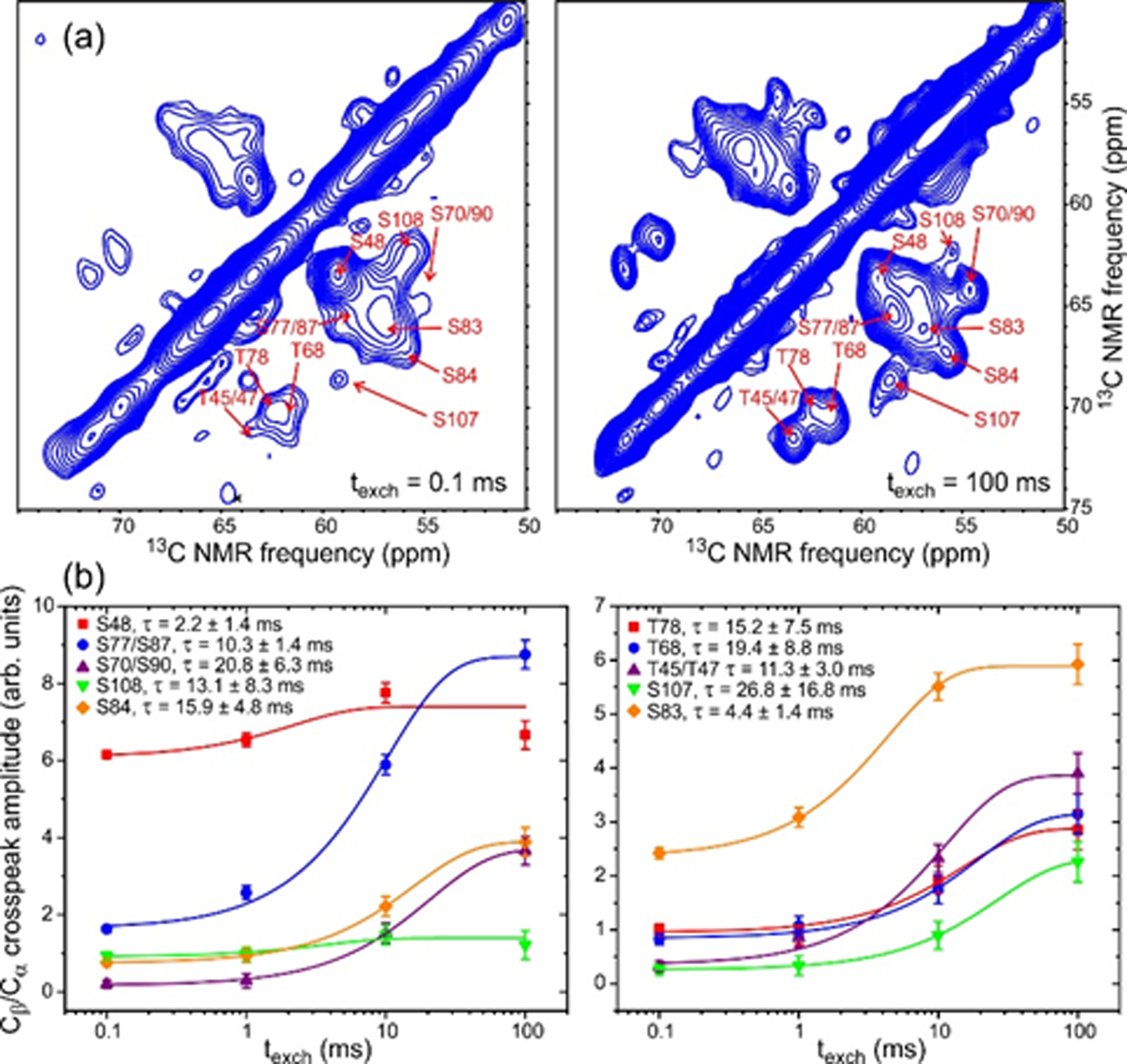
(a) Water-filtered 2D 13C-13C spectra of 2H,15N,13C-FUS-LC fibrils with 1H-1H exchange periods texch = 0.1 ms (left) and texch = 100 ms (right). Contour levels increase by successive factors of 1.2. 2D spectra were obtained with 1600 (left) and 192 (right) scans per t1 point. (b) Build-up curves for Cβ/Cα crosspeak intensities of Ser and Thr residues. Error bars represent the root-mean-squared noise in the 2D spectra. Color-coded lines are fits with single-exponential functions, yielding the indicated values of τ.
Build-up curves for resolved and partially-resolved Cβ/Cα crosspeaks are shown in Fig. 6b and are fit with single-exponential functions . Hydrogen exchange rates are then indicated by the fitted values of 1/τ. For most Thr and Ser residues, exchange rates determined in this way are 4–10 times slower than the intrinsic rates near neutral pH and 30° C,61 indicating significant protection from hydrogen exchange. Measured hydrogen exchange rates for S48 and S83 are close to intrinsic rates, consistent with an absence of stable inter-residue hydrogen bonding for sidechain hydroxyl sites of these residues.
Build-up curves for other resolved crosspeaks are shown in Fig. S9b. As expected,61 build-up of Tyr sidechain crosspeaks (unassigned) is very rapid due to the large intrinsic exchange rates for Tyr sidechain hydroxyl groups. Best-fit values of τ for most CO/Cα and CO/Cβ crosspeaks are in the 15–20 ms range, similar to the τ values for sidechain hydroxyl sites of protected Ser and Thr residues (e.g., S84 and S70/S90 in Fig. 6b). Under our experimental conditions, build-up times for 13C-13C crosspeak intensities in the 15–20 ms range may indicate an overall growth of 1H polarization within the FUS-LC fibril core, which is likely to result from hydrogen exchange at unprotected sidechain hydroxyl and backbone amide sites combined with 1H-1H spin diffusion within the fibril core. This overall growth of 1H polarization sets an upper limit on τ. Consequently, time scales for hydrogen exchange that may exceed 15–20 ms at specific, highly protected sites can not be determined accurately from our water-filtered 2D 13C-13C data.
Discussion
Fig. 7 shows two representations of our structural model for the FUS-LC fibril core, with residues color-coded to indicate the presence or absence of correlations to the 1H chemical shift of water or their involvement in inter-residue crosspeaks. In Fig. 7a, sidechains that lack correlations to water in a 3D HCC spectrum of 2H,15N,13C-FUS-LC fibrils are colored cyan. A subset of these residues, including sidechains of T47, S53, S70, S77, T78, S84, S85, and S90, are shielded within the fibril core and may participate in inter-residue hydrogen bond interactions that would reduce their rates of hydrogen exchange with water. Data in Fig. 6b also indicate protection from hydrogen exchange for T47, S70, S77, S84, and S90. (S53, T78, and S85 do not have sufficiently well-resolved Cβ/Cα crosspeaks to be included in Fig. 6b.) However, sidechains of other residues that lack correlations to water, including sidechains of S54, S87, and S89, are apparently exposed on the FUS-LC fibril surface. As mentioned above, crosspeak signals of S87 and S89 are relatively weak in all 3D spectra, so the absence of detectable correlations to water for these residues may be due to the overall weakness of their 13C ssNMR signals, which is presumably a consequence of molecular motions in residues 87–89.
Figure 7:
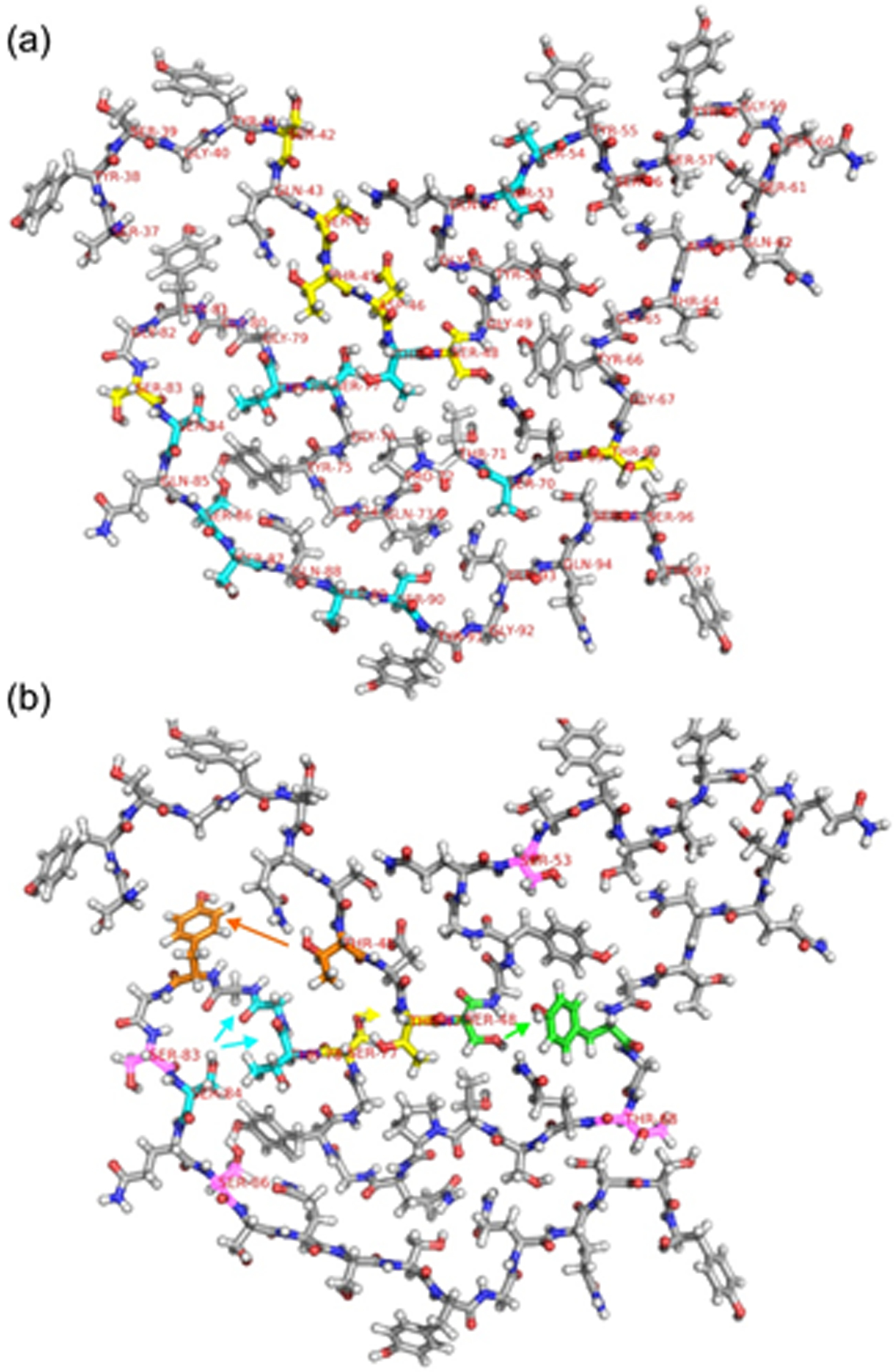
(a) One molecule from Protein Data Bank file 5W3N, with carbons of Ser, Thr, Asp residues that have correlations to the 1H chemical shift of water in the 3D HCC spectrum of 2H,15N,13C-FUS-LC fibrils (and also have 13C chemical shift assignments) shown in yellow. Carbons of Ser and Thr residues that have 13C chemical shift assignments, but do not have correlations to water, are shown in cyan. (b) One molecule from Protein Data Bank file 5W3N, shown with residue number labels on all Ser and Thr residues that have 1H chemical shift assignments for sidechain hydroxyl protons. Carbons of residues that are involved in inter-residue crosspeaks in 3D HCC or 3D H(H)CC spectra are colored orange, cyan, yellow, and green, with arrows indicating the direction of these crosspeaks from hydroxyl 1H chemical shifts to 13C chemical shifts. Based on the structural model, crosspeaks from hydroxyl 1H chemical shifts of T48 and S48 to aromatic 13C chemical shifts of Tyr residues are depicted as T48-Y81 and S48- Y66 crosspeaks. Carbons of Ser and Thr residues that have hydroxyl 1H chemical shift assignments but do not exhibit detectable inter-residue crosspeaks are colored pink.
For the most part, sidechains of residues that do exhibit correlations to water (yellow in Fig. 7a) are exposed on the FUS-LC fibril surface or would be exposed after minor adjustments of the structural model depicted in Fig. 7. In this context, it should be noted that sidechains of S44, T45, and D46 are fully accessible to solvent in some models within the bundle of FUS-LC fibril core structures that were found to be consistent with all ssNMR restraints (see Protein Data Bank file 5W3N). Moreover, in simple molecular dynamic simulations with explicit solvent63 that start with the structure in Fig. 7, sidechains of S44 and D46 become solvated within 20 ns (see Fig. S10). Water molecules also enter a cavity that develops between S48 and T71, accounting for the correlations to water exhibited by S48. In addition, data in Fig. 6b indicate a lack of protection from hydrogen exchange for S48. Thus, the observed correlations to water (and absence thereof) for Ser and Thr residues that have assigned 13C ssNMR signals are consistent with the structural model in Fig. 7 after including adjustments that occur upon immersion of this model in water.
In Fig. 7b, carbon atoms of S84, T78, and G79 are colored cyan to illustrate the observed correlations from the S84 hydroxyl 1H signal to 13C signals of T78 and G79 in Figs. 5a, 5b, and S6a. The greater intensity of S84-G79 crosspeaks and the fact that these crosspeaks are observed with τHH = 0 (Fig. 5a) suggests the presence of a hydrogen bond between the S84 sidechain and the backbone carbonyl group of G79. Data in Fig. 6b also indicate protection from hydrogen exchange for S84. The S84-T78 crosspeak with τHH = 2.0 ms (Fig. 5b) may result from 1H-1H polarization transfers from G79 to T78 during the τHH period. Interestingly, two hydroxyl 1H chemical shifts (5.89 ppm and 6.24 ppm) can be assigned to T78, since both 1H chemical shifts correlate with the 13Cα and 13Cβ chemical shifts of T78, as shown in Fig. S8. Neither T78 hydroxyl 1H signal has detectable correlations with 13C signals of S84. We interpret these observations as evidence that the T78 hydroxyl group does not form hydrogen bonds with S84, but may interact with other sidechains, such as that of Y75, with two distinct configurations that are in slow exchange. As discussed above, T78 does not show correlations to water in the 3D HCC spectrum, and therefore is apparently protected from rapid hydrogen exchange with water.
Carbon atoms of S77 and T47 are colored yellow in Fig. 7b to illustrate the observed correlations from the S77 hydroxyl 1H signal to 13C signals of T47 (Figs. 5c and 5d). We attribute these crosspeaks to hydrogen bonding between S77 and T47 sidechains. In water-filtered 2D 13C-13C spectra, crosspeaks from S77 and T47 are not resolved from those of S87 and T45. Nonetheless, values of the apparent hydrogen exchange time τ for these crosspeaks are approximately 10 ms, greater than the intrinsic hydrogen exchange time of an unprotected Ser or Thr hydroxyl group.61 Although the T47 hydroxyl 1H signal is relatively strong, crosspeaks from the T47 hydroxyl 1H signal to 13C signals of S77 can not be observed conclusively in our 3D HCC and H(H)CC spectra, as such inter-residue crosspeaks are only partially resolved from intra-residue crosspeaks of S87 (see Table S10).
Carbon atoms of T45 and Y81 are colored orange and carbon atoms of S48 and Y66 are colored green in Fig. 7b to illustrate the observed correlations from the T45 and S48 hydroxyl 1H signals to 13C signals of Tyr sidechains (Figs. 5f and 5h). Assignments to Y81 and Y66 are based tentatively on the structural model itself, because the Tyr 13C chemical shifts do not have site-specific assignments. These correlations may result from T45-Y81 and S48-Y66 hydrogen bonds.
As described in the Introduction above, evidence for the importance of sidechain hydrogen bonding in self-assembly of low complexity sequences was provided previously by structural studies of peptides in amyloid-like crystalline states.38–41 In particular, Luo et al. identified segments of FUS-LC that form amyloid fibrils in a thermally reversible manner, which they call RAC1 (residues 37–42 of FUS-LC, sequence SYSGYS) and RAC2 (residues 54–59 of FUS-LC, sequence SYSSYG). High-resolution structures of crystalline RAC1 and RAC2 were solved by micro-electron diffraction (PDB 5XSG) and x-ray diffraction (PDB 5XRR, see Fig. S2d), respectively.38 A similar crystal structure of RAC1 (PDB 6BWZ) was also reported by Hughes et al.39 For RAC1, stabilizing interactions include hydrogen bonding of S39 and Y41 sidechains with an ordered row of water molecules (Fig. 1d), as well as intermolecular hydrogen bonding between sidechains of Y38 and S42 within each cross-β layer. For RAC2, stabilizing interactions include water-mediated hydrogen bonding among Y58 sidechains in different cross-β layers, as well as hydrogen bonding of Y55 and S57 sidechains with ordered rows of water molecules.
The specific sidechain interactions observed in structures of RAC1 and RAC2 are not directly transferable to the FUS-LC fibril structure for several reasons: (i) FUS-LC fibrils are noncrystalline; (ii) FUS-LC fibrils contain the full low-complexity sequence, so that a given segment of the sequence interacts both with the same segment in neighboring molecules and with different segments in the same and neighboring molecules; (iii) residues 37–42 and 54–59 are not fully structurally ordered in FUS-LC fibrils, as indicated by the ssNMR data of Murray et al.1 as well as the ssNMR data for 2H,15N,13C-FUS-LC fibrils presented above (see Table S10). Nonetheless, the data for 2H,15N,13C-FUS-LC fibrils indicate that similar interactions are present in FUS-LC fibrils.
Conclusions
Data presented above provide evidence that the FUS-LC fibril core structure is stabilized in part by hydrogen bonds involving sidechains of hydroxyl-bearing residues, or more precisely that such hydrogen bonds exist within the core structure. In particular, the sidechain hydroxyl group of S84 appears to form a hydrogen bond to the backbone carbonyl group of G79, bridging a β-turn structure in residues 79–84. The S84 sidechain may also interact with the sidechain of T78, although evidence for a S84-T78 hydrogen bond is not clear. In addition, the sidechain hydroxyl group of S77 appears to form a hydrogen bond to the sidechain of T47, contributing to energetically favorable stacking of a cross-β layer formed by residues 44–47 on a layer formed by residues 76–79. Data presented above also provide information about interactions of Thr and Ser hydroxyl groups with water. In particular, sidechain hydroxyl groups of specific residues (i.e., T47, S53, S70, S77, T78, S84, S86, and S90) are relatively protected from hydrogen exchange with water, consistent with their involvement in hydrogen bonds that may contribute to structural stabilization. Protection from hydrogen exchange with water is indicated by the absence of correlations with water in 3D 1H-13C-13C spectra, as well as by crosspeak build-up rates in water-filtered 2D 13C-13C spectra that are less than intrinsic hydrogen exchange rates.
Additionally, data presented above demonstrate how multidimensional ssNMR measurements on deuterated, back-exchanged samples can provide site-specific insights into hydrogen bonding, hydrogen exchange, and internal water locations within protein assemblies.
Supplementary Material
Acknowledgements
This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health. D.T.M. was supported by a Postdoctoral Research Associate Training (PRAT) fellowship from the National Institute of General Medical Sciences, award number 1Fi2GM117604-01.
Footnotes
References
- (1).Murray DT, Kato M, Lin Y, Thurber KR, Hung I, McKnight SL, and Tycko R (2017) Structure of FUS protein fibrils and its relevance to self-assembly and phase separation of low-complexity domains, Cell 171, 615–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Zinszner H, Sok J, Immanuel D, Yin Y, and Ron D (1997) TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling, J. Cell Sci 110, 1741–1750. [DOI] [PubMed] [Google Scholar]
- (3).Schwartz JC, Wang XY, Podell ER, and Cech TR (2013) RNA seeds higher-order assembly of FUS protein, Cell Reports 5, 918–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Fox AH, Nakagawa S, Hirose T, and Bond CS (2018) Paraspeckles: Where long noncoding RNA meets phase separation, Trends Biochem. Sci 43, 124–135. [DOI] [PubMed] [Google Scholar]
- (5).Seydoux G (2018) The P granules of C. elegans: A genetic model for the study of RNA-protein condensates, J. Mol. Biol 430, 4702–4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bosco DA, Lemay N, Ko HK, Zhou HR, Burke C, Kwiatkowski TJ, Sapp P, McKenna-Yasek D, Brown RH, and Hayward LJ (2010) Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules, Hum. Mol. Genet 19, 4160–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Julicher F, and Hyman AA (2009) Germline P granules are liquid droplets that localize by controlled dissolution/condensation, Science 324, 1729–1732. [DOI] [PubMed] [Google Scholar]
- (8).Han TNW, Kato M, Xie SH, Wu LC, Mirzaei H, Pei JM, Chen M, Xie Y, Allen J, Xiao GH, and McKnight SL (2012) Cell-free formation of RNA granules: Bound RNAs identify features and components of cellular assemblies, Cell 149, 768–779. [DOI] [PubMed] [Google Scholar]
- (9).Kato M, Han TNW, Xie SH, Shi K, Du XL, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei JM, Grishin NV, Frantz DE, Schneider JW, Chen S, Li L, Sawaya MR, Eisenberg D, Tycko R, and McKnight SL (2012) Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels, Cell 149, 753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, and Taylor JP (2015) Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization, Cell 163, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mittag T, and Parker R (2018) Multiple modes of protein-protein interactions promote RNP granule assembly, J. Mol. Biol 430, 4636–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Brady JP, Farber PJ, Sekhar A, Lin YH, Huang R, Bah A, Nott TJ, Chan HS, Baldwin AJ, Forman-Kay JD, and Kay LE (2017) Structural and hydrodynamic properties of an intrinsically disordered region of a germ cell-specific protein on phase separation, Proc. Natl. Acad. Sci. U. S. A 114, E8194–E8203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Nott TJ, Petsalaki E, Farber P, Jervis D, Fussner E, Plochowietz A, Craggs TD, Bazett-Jones DP, Pawson T, Forman-Kay JD, and Baldwin AJ (2015) Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles, Mol. Cell 57, 936–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ryan VH, Dignon GL, Zerze GH, Chabata CV, Silva R, Conicella AE, Amaya J, Burke KA, Mittal J, and Fawzi NL (2018) Mechanistic view of hnRNPA2 low-complexity domain structure, interactions, and phase separation altered by mutation and arginine methylation, Mol. Cell 69, 465–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Amaya J, Ryan VH, and Fawzi NL (2018) The SH3 domain of Fyn kinase interacts with and induces liquid-liquid phase separation of the low-complexity domain of hnRNPA2, J. Biol. Chem 293, 19522–19531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lin Y, Protter DSW, Rosen MK, and Parker R (2015) Formation and maturation of phase-separated liquid droplets by RNA-binding proteins, Mol. Cell 60, 208–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Xiang SH, Kato M, Wu LC, Lin Y, Ding M, Zhang YJ, Yu YH, and McKnight SL (2015) The LC domain of hnRNPA2 adopts similar conformations in hydrogel polymers, liquid-like droplets, and nuclei, Cell 163, 829–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Murakami T, Qamar S, Lin JQ, Schierle GSK, Rees E, Miyashita A, Costa AR, Dodd RB, Chan FTS, Michel CH, Kronenberg-Versteeg D, Li Y, Yang SP, Wakutani Y, Meadows W, Ferry RR, Dong L, Tartaglia GG, Favrin G, Lin WL, Dickson DW, Zhen M, Ron D, Schmitt-Ulms G, Fraser PE, Shneider NA, Holt C, Vendruscolo M, Kaminski CF, and St George-Hyslop P (2015) ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function, Neuron 88, 678–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Li YR, King OD, Shorter J, and Gitler AD (2013) Stress granules as crucibles of ALS pathogenesis, J. Cell Biol 201, 361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bentmann E, Haass C, and Dormann D (2013) Stress granules in neurodegeneration: Lessons learnt from TAR DNA binding protein of 43 kDa and fused in sarcoma, FEBS J. 280, 4348–4370. [DOI] [PubMed] [Google Scholar]
- (21).Ling SC, Polymenidou M, and Cleveland DW (2013) Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis, Neuron 79, 416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C, Annu K, Baker M, Perkerson RB, Kurti A, Matchett BJ, Mittag T, Temirov J, Hsiung GYR, Krieger C, Murray ME, Kato M, Fryer JD, Petrucelli L, Zinman L, Weintraub S, Mesulam M, Keith J, Zivkovic SA, Hirsch-Reinshagen V, Roos RP, Zuchner S, Graff-Radford NR, Petersen RC, Caselli RJ, Wszolek ZK, Finger E, Lippa C, Lacomis D, Stewart H, Dickson DW, Kim HJ, Rogaeva E, Bigio E, Boylan KB, Taylor JP, and Rademakers R (2017) TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics, Neuron 95, 808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mackenzie IRA, Rademakers R, and Neumann M (2010) TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia, Lancet Neurol. 9, 995–1007. [DOI] [PubMed] [Google Scholar]
- (24).Deng HX, Zhai H, Bigio EH, Yan JH, Fecto F, Ajroud K, Mishra M, Ajroud-Driss S, Heller S, Sufit R, Siddique N, Mugnaini E, and Siddique T (2010) FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis, Ann. Neurol 67, 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li SQ, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, Topp S, Gkazi AS, Miller J, Shaw CE, Kottlors M, Kirschner J, Pestronk A, Li YR, Ford AF, Gitler AD, Benatar M, King OD, Kimonis VE, Ross ED, Weihl CC, Shorter J, and Taylor JP (2013) Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS, Nature 495, 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Murray DT, Zhou XM, Kato M, Xiang SH, Tycko R, and McKnight SL (2018) Structural characterization of the D290V mutation site in hnRNPA2 low-complexity-domain polymers, Proc. Natl. Acad. Sci. U. S. A 115, E9782–E9791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Walti MA, Ravotti F, Arai H, Glabe CG, Wall JS, Bockmann A, Guntert P, Meier BH, and Riek R (2016) Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril, Proc. Natl. Acad. Sci. U. S. A 113, E4976–E4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I, Rosay M, Donovan KJ, Michael B, Wall J, Linse S, and Griffin RG (2016) Atomic resolution structure of monomorphic Aβ(42) amyloid fibrils, J. Am. Chem. Soc 138, 9663–9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Xiao YL, Ma BY, McElheny D, Parthasarathy S, Long F, Hoshi M, Nussinov R, and Ishii Y (2015) Aβ(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease, Nat. Struct. Mol. Biol 22, 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Schutz AK, Vagt T, Huber M, Ovchinnikova OY, Cadalbert R, Wall J, Guntert P, Bockmann A, Glockshuber R, and Meier BH (2015) Atomic-resolution three-dimensional structure of amyloid-β fibrils bearing the Osaka mutation, Angew. Chem.-Int. Edit 54, 331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, and Tycko R (2013) Molecular structure of β-amyloid fibrils in Alzheimer’s disease brain tissue, Cell 154, 1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, Courtney JM, Kim JK, Barclay AM, Kendall A, Wan W, Stubbs G, Schwieters CD, Lee VMY, George JM, and Rienstra CM (2016) Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein, Nat. Struct. Mol. Biol 23, 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Li YW, Zhao CY, Luo F, Liu ZY, Gui XR, Luo ZP, Zhang X, Li D, Liu C, and Li XM (2018) Amyloid fibril structure of α-synuclein determined by cryoelectron microscopy, Cell Res. 28, 897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Li BS, Ge P, Murray KA, Sheth P, Zhang M, Nair G, Sawaya MR, Shin WS, Boyer DR, Ye SL, Eisenberg DS, Zhou ZH, and Jiang L (2018) Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel, Nat. Commun 9, article number 3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Tycko R (2014) Physical and structural basis for polymorphism in amyloid fibrils, Protein Sci. 23, 1528–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Perutz MF, Johnson T, Suzuki M, and Finch JT (1994) Glutamine repeats as polar zippers: Their possible role in inherited neurodegenerative diseases, Proc. Natl. Acad. Sci. U. S. A 91, 5355–5358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, and Eisenberg D (2005) Structure of the cross-β spine of amyloid-like fibrils, Nature 435, 773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Luo F, Gui XR, Zhou H, Gu JG, Li YC, Liu XY, Zhao ML, Li D, Li XM, and Liu C (2018) Atomic structures of FUS LC domain segments reveal bases for reversible amyloid fibril formation, Nat. Struct. Mol. Biol 25, 341–346. [DOI] [PubMed] [Google Scholar]
- (39).Hughes MP, Sawaya MR, Boyer DR, Goldschmidt L, Rodriguez JA, Cascio D, Chong L, Gonen T, and Eisenberg DS (2018) Atomic structures of low-complexity protein segments reveal kinked β-sheets that assemble networks, Science 359, 698–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Wiltzius JJW, Sievers SA, Sawaya MR, Cascio D, Popov D, Riekel C, and Eisenberg D (2008) Atomic structure of the cross-β spine of islet amyloid polypeptide (amylin), Protein Sci. 17, 1467–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJW, McFarlane HT, Madsen AO, Riekel C, and Eisenberg D (2007) Atomic structures of amyloid cross-β spines reveal varied steric zippers, Nature 447, 453–457. [DOI] [PubMed] [Google Scholar]
- (42).Sheffield P, Garrard S, and Derewenda Z (1999) Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors, Protein Expr. Purif 15, 34–39. [DOI] [PubMed] [Google Scholar]
- (43).Ernst M, Samoson A, and Meier BH (2003) Low-power XIX decoupling in MAS NMR experiments, J. Magn. Reson 163, 332–339. [DOI] [PubMed] [Google Scholar]
- (44).Bennett AE, Rienstra CM, Griffiths JM, Zhen WG, Lansbury PT, and Griffin RG (1998) Homonuclear radio frequency-driven recoupling in rotating solids, J. Chem. Phys 108, 9463–9479. [Google Scholar]
- (45).Ishii Y (2001) 13C-13C dipolar recoupling under very fast magic angle spinning in solid-state nuclear magnetic resonance: Applications to distance measurements, spectral assignments, and high-throughput secondary-structure determination, J. Chem. Phys 114, 8473–8483. [Google Scholar]
- (46).Pines A, Gibby MG, and Waugh JS (1973) Proton-enhanced NMR of dilute spins in solids, J. Chem. Phys 59, 569–590. [Google Scholar]
- (47).Knight MJ, Webber AL, Pell AJ, Guerry P, Barbet-Massin E, Bertini I, Felli IC, Gonnelli L, Pierattelli R, Emsley L, Lesage A, Herrmann T, and Pintacuda G (2011) Fast resonance assignment and fold determination of human superoxide dismutase by high-resolution proton-detected solid-state MAS NMR spectroscopy, Angew. Chem.-Int. Edit 50, 11697–11701. [DOI] [PubMed] [Google Scholar]
- (48).Linser R, Dasari M, Hiller M, Higman V, Fink U, del Amo JML, Markovic S, Handel L, Kessler B, Schmieder P, Oesterhelt D, Oschkinat H, and Reif B (2011) Proton-detected solid-state NMR spectroscopy of fibrillar and membrane proteins, Angew. Chem.-Int. Edit 50, 4508–4512. [DOI] [PubMed] [Google Scholar]
- (49).Zhou DH, Shah G, Cormos M, Mullen C, Sandoz D, and Rienstra CM (2007) Proton-detected solid-state NMR spectroscopy of fully protonated proteins at 40 kHz magic-angle spinning, J. Am. Chem. Soc 129, 11791–11801. [DOI] [PubMed] [Google Scholar]
- (50).Baldus M, Petkova AT, Herzfeld J, and Griffin RG (1998) Cross polarization in the tilted frame: Assignment and spectral simplification in heteronuclear spin systems, Mol. Phys 95, 1197–1207. [Google Scholar]
- (51).Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, and Bax A (1995) NMRpipe: A multidimensional spectral processing system based on Unix pipes, J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- (52).Wang T, Jo H, DeGrado WF, and Hong M (2017) Water distribution, dynamics, and interactions with Alzheimer’s β-amyloid fibrils investigated by solid-state NMR, J. Am. Chem. Soc 139, 6242–6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Williams JK, and Hong M (2014) Probing membrane protein structure using water polarization transfer solid-state NMR, J. Magn. Reson 247, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Takegoshi K, Nakamura S, and Terao T (2001) 13C-1H dipolar-assisted rotational resonance in magic-angle spinning NMR, Chem. Phys. Lett 344, 631–637. [Google Scholar]
- (55).Hansen PE (2000) Isotope effects on chemical shifts of proteins and peptides, Magn. Reson. Chem 38, 1–10. [Google Scholar]
- (56).Tycko R (2015) On the problem of resonance assignments in solid state NMR of uniformly 15N,13C-labeled proteins, J. Magn. Reson 253, 166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Bockmann A, Juy M, Bettler E, Emsley L, Galinier A, Penin F, and Lesage A (2005) Water-protein hydrogen exchange in the micro-crystalline protein Crh as observed by solid state NMR spectroscopy, J. Biomol. NMR 32, 195–207. [DOI] [PubMed] [Google Scholar]
- (58).Agarwal V, Linser R, Fink U, Faelber K, and Reif B (2010) Identification of hydroxyl protons, determination of their exchange dynamics, and characterization of hydrogen bonding in a microcrystallin protein, J. Am. Chem. Soc 132, 3187–3195. [DOI] [PubMed] [Google Scholar]
- (59).Van Melckebeke H, Schanda P, Gath J, Wasmer C, Verel R, Lange A, Meier BH, and Bockmann A (2011) Probing water accessibility in HET-s(218–289) amyloid fibrils by solid-state NMR, J. Mol. Biol 405, 765–772. [DOI] [PubMed] [Google Scholar]
- (60).Dicke A, Gopinath T, Wang YJ, and Veglia G (2016) Probing residue-specific water-protein interactions in oriented lipid membranes via solid-state NMR spectroscopy, J. Phys. Chem. B 120, 10959–10968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Liepinsh E, and Otting G (1996) Proton exchange rates from amino acid side chains: Implications for image contrast, Magn.Reson.Med 35, 30–42. [DOI] [PubMed] [Google Scholar]
- (62).Takeda M, Jee J, Ono AM, Terauchi T, and Kainosho M (2011) Hydrogen exchange study on the hydroxyl groups of serine and threonine residues in proteins and structure refinement using NOE restraints with polar side-chain groups, J. Am. Chem. Soc 133, 17420–17427. [DOI] [PubMed] [Google Scholar]
- (63).Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, and Schulten K (2005) Scalable molecular dynamics with NAMD, J. Comput. Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.