

Abstract
Megalencephaly-capillary malformation syndrome (MCAP) is an overgrowth disorder characterized by cerebrocortical malformations, vascular anomalies, and segmental overgrowth secondary to somatic activating mutations in the PI3K-AKT-MTOR pathway (PIK3CA). Cases of growth failure and hypoglycemia have been reported in patients with MCAP, raising the suspicion for unappreciated growth hormone (GH) deficiency. Here we report an observational multi-center study of children with MCAP and GH deficiency. Eleven participants were confirmed to have GH deficiency, all with very low or undetectable circulating concentrations of insulin-like growth factor-1 and insulin-like growth factor binding protein-3. Seven underwent GH stimulation testing and all had insufficient responses with a median GH peak of 3.7 ng/ml (range 1.1-8.6). Growth patterns revealed a drastic decline in length z-scores within the first year of life but then stabilized afterward. Five were treated with GH; one discontinued due to inconsolability. The other four participants continued on GH with improvement in linear growth velocity. Other endocrinopathies were identified in seven of the 11 participants in this cohort. This study indicates that GH deficiency is associated with MCAP and that children with MCAP and hypoglycemia and/or postnatal growth failure should be evaluated for GH deficiency and other endocrinopathies.
Keywords: MCAP, megalencephaly-capillary malformation syndrome, growth hormone deficiency, hypoglycemia, PIK3CA
INTRODUCTION
The megalencephaly-capillary malformation syndrome (MCAP; OMIM#602501) is a pediatric overgrowth disorder characterized by brain overgrowth (i.e. megalencephaly), macrosomia, cutaneous vascular malformations, digital anomalies (polydactyly, syndactyly) and cortical brain malformations (typically bilateral perisylvian polymicrogyria) (Clayton-Smith et al., 1997; Mirzaa et al., 2012; Moore et al., 1997). As an overgrowth disorder, MCAP is characterized by several distinctive features that are typically recognizable at birth including macrosomia, megalencephaly with or without overlying cortical malformations, and cutaneous vascular malformations (Mirzaa, Riviere, & Dobyns, 2013). MCAP is most commonly caused by mosaic (i.e., postzygotic) gain-of-function mutations in the PIK3CA gene on the long arm of chromosome 3. These mutations lead to activation of the PI3K-AKT-MTOR pathway, a critical cell-signaling pathway that regulates various cellular functions including growth, proliferation, and metabolism (Cantley, 2002; Engelman, Mukohara, et al., 2006; Mirzaa et al., 2013; Riviere et al., 2012).
While growth failure has not been reported as a key finding in classic MCAP, we have identified a subset of children with MCAP and postnatal growth failure. Furthermore, several children with this syndrome have been reported to have hypoglycemia, usually of unknown etiology (Kinross et al., 2015; McDermott, Byers, & Clayton-Smith, 2016). Both postnatal growth failure and hypoglycemia can be presenting signs of GH deficiency, yet there is only one published report to date of a child with MCAP and confirmed growth hormone (GH) deficiency (Minagawa et al., 2005). This prompted us to launch this observational study describing the phenotypes, growth trajectories, and endocrinological manifestations of children with both MCAP and GH deficiency to facilitate early identification and better management approaches for children with MCAP and endocrinopathies.
METHODS
Participants were enrolled at one of two tertiary care referral centers under the appropriate institutional research protocols. The first site is the Center for Integrative Brain Research (CIBR) at the Seattle Children’s Research Institute (SCRI) that recruited nationally and internationally for a study of megalencephaly and other developmental brain disorders. The second site is the University of Colorado/Children’s Hospital Colorado that recruited participants with MCAP and growth failure seen in the pediatric endocrinology clinic. To avoid duplicate enrollment, initials and birth year were cross-compared between sites. One participant was identified to have participated in studies at both sites, and data were therefore merged for analysis. The local institutional review boards at both institutions approved these studies with written informed consent provided from all enrolled families.
Children with clinically and/or molecularly confirmed MCAP who also had confirmed GH deficiency were included in this study. The diagnosis of MCAP was confirmed by clinical geneticists, and when genetic testing results were available, by molecular diagnostic testing (i.e., the identification of a disease-causing PIK3CA variant). Relevant data regarding clinical phenotypes, comorbidities, and genetic findings were collected from medical records. Growth parameters were obtained from clinical growth charts. To standardize growth assessments, growth data were converted to age and sex-specific z-scores based on the Fenton curves for birth parameters and Center for Disease Control (CDC) for postnatal growth parameters (Fenton & Kim, 2013; Grummer-Strawn, Reinold, & Krebs, 2010). Annual growth velocity was calculated by dividing the change in linear growth in centimeters by the time between visits and normalizing it to 12 months. Due to expected age differences in growth velocity, calculations were analyzed separately for age 0-1 years, 1-2 years, 2-4 years, 5-10 years, and 10+ years. Data on thyroid function, insulin-like growth factor-1 (IGF-1), insulin-like factor binding protein-3 (IGFBP-3) concentrations, GH response to hypoglycemia and/or stimulation tests, pituitary structure on brain magnetic resonance imaging (MRI), and management of GH deficiency were collected, when available. All laboratory data were measured clinically using different assays. Therefore, the normal ranges for these assays vary and the data were not statistically compared. All records were reviewed by a pediatric endocrinologist who confirmed the diagnosis of GH deficiency (clinical symptoms plus laboratory criteria diagnostic of GH deficiency). Descriptive statistics were used to summarize outcomes of interest using mean and standard deviation unless otherwise specified.
RESULTS
Eleven children had confirmed GH deficiency. The clinical characteristics of each participant are presented in Table 1. All participants with GH deficiency who underwent genetic testing (N=9) had a pathogenic variant in PIK3CA; for the other two, the diagnosis of MCAP was made clinically but genetic testing was not pursued.
Table 1.
Summary of the clinical and genetic data of patients with MCAP and confirmed GH deficiency (N=11).
| ID | PIK3CA variant | Sex | Race/Ethnicity | Phenotype | Neurodevelopment† | Birth weight (kg, z-score‡) | Birth length (cm, z-score‡) | Age at diagnosis of GH deficiency (yrs) | IGF-1 (ng/mL, normal range) | IGFBP-3 (mg/L, normal range) | GH stimulation maximum peak (ng/mL) | Free T4 (ng/dL) Ref: 0.8-2.0 ng/dL | Pituitary gland on MRI | Other endocrine diagnoses |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 103 | p.Asn345Ile | F | White/Hispanic | H+, M, CCM, HEMI | Moderate delays | 4.6, 3.2 | 54.0, 2.6 | 0.8 | <25 (55-327) | 0.6 (0.7-3.6) | 7.3 | 1.2 | Normal | Hypoglycemia |
| LR11-418 | p.Cys378Tyr | M | White/Caucasian | H-, M, CCM, PM, DA, ChM | Mild delays | 3.6, 2.7 | - | 7.4 | 49 (18-307) | 1.8 (1.8-5.0) | 5.3 | - | Normal | Type 1 diabetes |
| LR12-037 | p.Glu726Lys | M | White | H+, M, DA | Severe delays, seizures | 3.8, 3.1 | - | 4 | 32 (49-351) | 0.8 (1.8-7.1) | 1.1 | 1.0 | Normal | |
| LR11-230 | p.Gly1049Ser | M | Unknown | H+, M, DA | Moderate delays | 4.0, 3.0 | 54.5, 3.1 | 3.7 | 24 (30-155) | 0.8 (0.9-4.3) | 8.6 | - | Normal | Hypoglycemia; cryptorchidism |
| 102 | p.Gly914Arg | M | White | H-§, M, CCM | Moderate delays, seizures | 2.5, 0.5 | 49.5, 1.9 | 0.6 | <10 (30-22) | <0.5 (0.7-3.6) | - | 0.8 | Normal | Central hypothyroidism; cryptorchidism |
| LR15-337 | p.Gly914Arg | M | White/Caucasian | M, HEMI, ChM | Moderate delays | 3.4, 1.7 | 49.0, 0.8 | 2.2 | 16 (18-176) | N/A | 3.4 | - | Normal | |
| 105 | p.Met1043Ile | F | White | H+, M, CCM | Moderate delays | 3.0, 0.3 | 53.3, 2.3 | 1.8 | <25 (55-327) | 0.6 (0.7-3.6) | 2.5 | 0.8 | Normal | Hypoglycemia |
| LR14-100 | p.Met1043Ile | M | White/Caucasian | M, CCM, DA, PM | Severe delays | 4.0, 3.7 | 52.7, 2.8 | - | <10 (16-233) | <0.5 (1.8-5.0) | - | - | Normal | |
| LR18-218 | p.Met1043Ile | M | White/Ashkenazi Jewish | M, PM, DA, CCM, CA, HEMI | Moderate delays | 4.8, 3.8 | - | 3.5 | 18 (49-283) | 0.78 (1.6-4.6) | 3.7 | 1.5 | Pars intermedia cyst | Hypoglycemia; central adrenal insufficiency |
| 101 | Not performed | F | White/Hispanic | H+, M, CCM | Mild delays | 4.4, 2.1 | 53.5, 1.7 | 3.8 | <25 (49-283) | <0.5 (0.8-0.3) | - | - | Normal | |
| 104 | Not performed | F | White | H+, M, CCM | Profound delays, seizures | 4.2, 3.2 | 52.7, 2.4 | 9.2¶ | <25 (88-452) | <0.5 (1.8-7.1) | - | 1.1 | N/A | Hypoglycemia; osteoporosis |
Abbreviations: M = megalencephaly; H+ = hydrocephalus with shunt; H− = hydrocephalus without a shunt; CCM = cutaneous capillary malformation; PM = polymicrogyria; DA = digit abnormalities (syndactyly or polydactyly); CA = cardiac arrhythmia; HEMI = hemihypertrophy; ChM = Chiari malformation.
Based on clinical review.
Based on Fenton growth charts.
Participant underwent a ventriculostomy.
Growth failure occurred by age 6 but was not evaluated due to more significant health concerns.
In our cohort, the mean gestational age was 36.0±1.4 weeks, mean birth weight z-score was 2.7±1.1 and mean birth length z-score was 2.3±0.8. Length or height z-scores from birth through 5 years of age and growth velocities through 10 years of age are presented in Figure 1 and Figure 2, respectively. For most participants, there was a sharp decline in height z-scores in the first year of life. After the first year of life, growth velocity remained stable and in the low-normal range. However, there were individuals who did not follow this pattern and seemed to grow normally until later in childhood. One participant (LR11-418) did not have growth faltering at all and a diagnostic evaluation was only pursued due to anecdotal knowledge of other cases of GH deficiency associated with MCAP (see Table 1 for laboratory results).
Figure 1. Growth trajectory (length or height z-scores) from birth to five years for each participant with MCAP and confirmed GH deficiency.
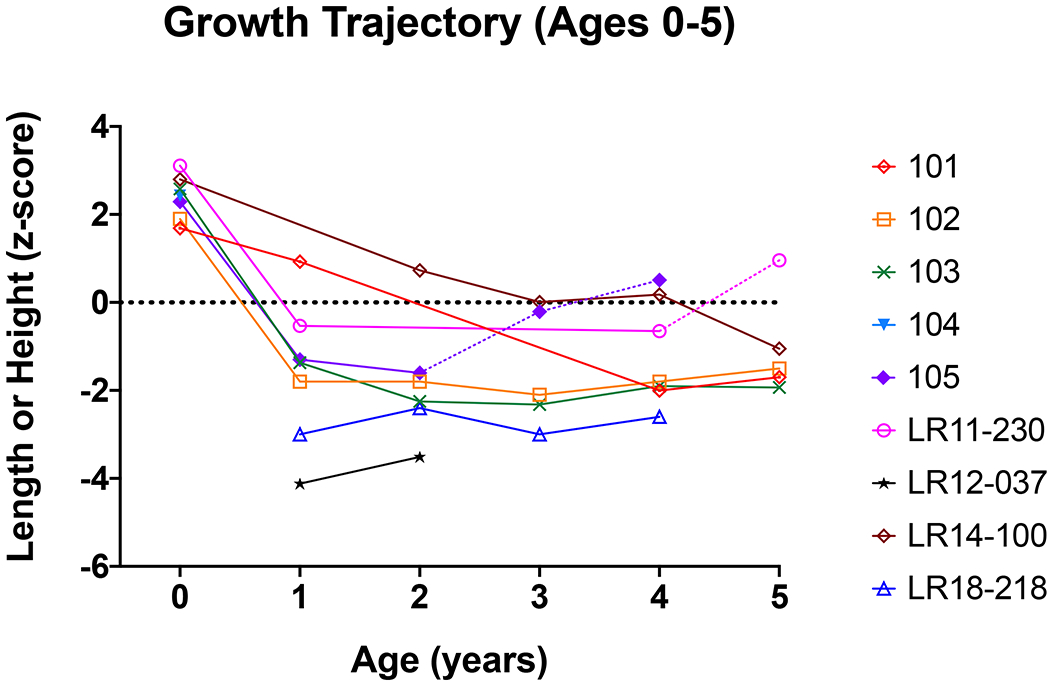
Dotted lines for participants 105 (purple diamond) and LR11-230 (pink circle) indicate growth trajectory following the initiation of GH treatment.
Figure 2. Growth velocity from birth to ten years of age for participants with MCAP and confirmed GH deficiency.
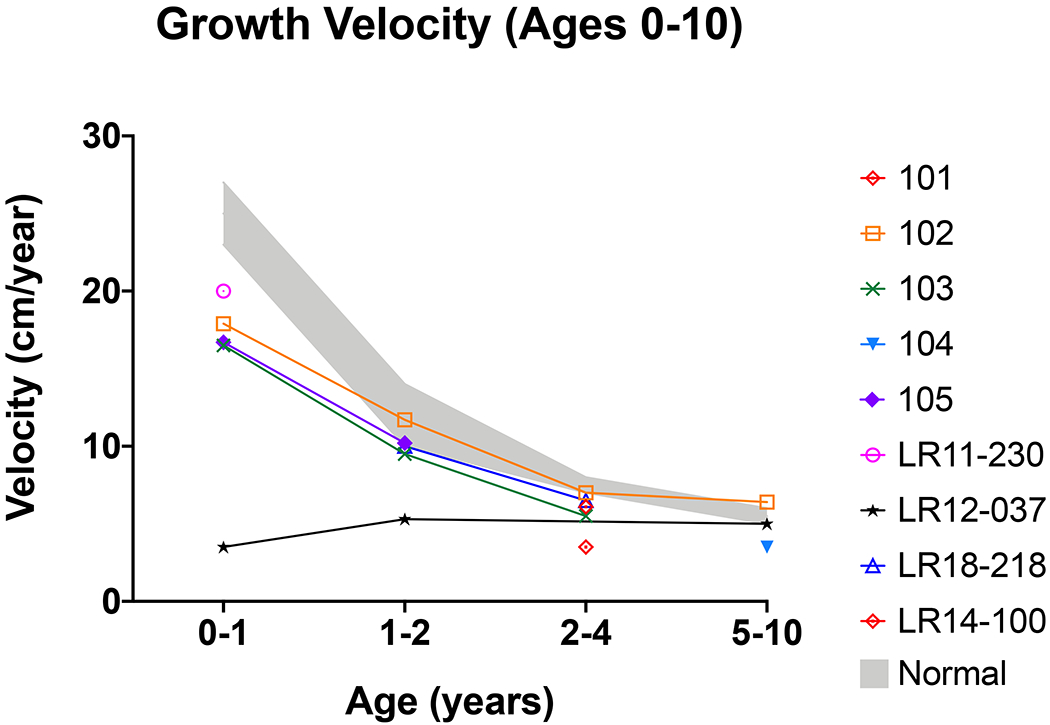
Normal growth velocity is shown in grey. Growth velocity is notably lower in the first year of life.
The mean age at the time of GH deficiency diagnosis was 4.2±3.0 years. At the time of diagnosis, circulating IGF-1 and IGFBP-3 concentrations were low or undetectable in all participants. Four underwent GH stimulation testing and/or had GH measured at the time of hypoglycemia, and all had insufficient responses with mean peak GH concentration of 3.7 ng/ml (normal response >10 ng/ml). Of the 10 participants with brain MRIs, nine had structurally normal pituitary glands and one had an incidental finding of a pars intermedia cyst. Five participants (102, 105, LR11-230, LR12-037, and LR15-337) were treated with subcutaneous recombinant human GH. Participant 102 discontinued GH after one week of treatment due to symptoms of headache and inconsolability, although the etiology of his irritability was not confirmed (more specifically, no papilledema or MRI changes were noted). This child’s irritability resolved after discontinuing GH treatment. The other four participants continued on GH with robust increases in linear growth velocity and no reported side effects, including no clinical worsening of macrocephaly or hemihypertrophy. Five (45%) participants with GH deficiency also had confirmed hypoglycemia. In four, the occurrence of hypoglycemia preceded the diagnosis of GH deficiency. Notably, hypoglycemia was attributed to several etiologies among these children, including hypoglycemia secondary to GH deficiency (N=2), hyperinsulinism (N=1), and ketotic hypoglycemia (N=2). Additional endocrine diagnoses observed in the participants with GH deficiency included cryptorchidism (N=2), central hypothyroidism (N=1), central adrenal insufficiency (N=1), antibody-positive type 1 diabetes (N=1), and osteoporosis with pathologic fractures (N=1).
DISCUSSION
In this study, we describe the clinical presentation and management of 11 children with MCAP and GH deficiency. Most participants presented with growth failure leading to a diagnostic evaluation for GH deficiency. Four of the five participants treated with recombinant human GH had an excellent response. This report of GH deficiency as well as other endocrinopathies, including hypoglycemia and other pituitary deficiencies, should raise clinicians’ suspicion of these disorders in children with MCAP.
GH is secreted by the pituitary gland and has systemic effects, especially on bone and muscle growth, as well as indirect effects through hepatic production of IGF-1 (De Luca, 2018). GH deficiency can result in poor linear growth, increased adiposity, decreased muscle bulk, decreased bone mineralization, unfavorable lipid profiles, and occasionally fasting hypoglycemia due to an inadequate counter-regulatory response (Chinoy & Murray, 2016). While low IGF-1 is a sensitive but not specific screening test for GH deficiency, low IGFBP-3 is recognized as the more specific but less sensitive biomarker of GH deficiency (Trivin et al., 2006). One of the unique features found in this cohort of children with MCAP is the universally low IGFBP-3 concentrations. Whether this is reflective of the severity of GH deficiency or due to a unique mechanism in MCAP is unknown.
We also suspect that postnatal growth retardation may be more common than previously appreciated in MCAP. In one of the first papers describing 13 children with MCAP, macrosomia at birth was nearly universal (Moore et al., 1997). However, at the time of follow up, seven of these individuals had lengths <10th%ile and all but one had a decrease in their length percentile from birth to the most recent follow up. The first published case of MCAP and GH deficiency demonstrated postnatal growth retardation and developed hypoglycemic seizures at seven months of age (Minagawa et al., 2005). His IGF-1 concentration was very low, and he had a low GH (1.1 ng/ml) at the time of hypoglycemia. However, the patient had a normal response to GH releasing hormone (GHRH) stimulation testing with a peak GH of 21 ng/dl. The management and clinical course of this patient were not further described. Another report of a child with MCAP (megalencephaly, polymicrogyria, hydrocephalus requiring shunt, and a mosaic gain-of-function mutation p.Met1043Ile found in 16% in blood and 37% in saliva) also may have had GH deficiency. He had hypoglycemia and low GH, IGF-1, and IGFBP-3 but there was no mention of growth; he was briefly treated with GH but this did not resolve the hypoglycemia and he was not given a diagnosis of GH deficiency (Stutterd et al., 2018). Finally, another child with megalencephaly due to a germline mutation in AKT3 (p.Glu40Lys) has also been reported to have GH deficiency and hypoglycemia. He was treated with recombinant GH around 15 months of age and hypoglycemia resolved. He did not have polymicrogyria, seizures, or developmental delays (Takagi et al., 2017). This large case series substantially adds to the literature describing GH deficiency among children with MCAP with PIK3CA mutations.
The underlying mechanisms of GH deficiency in MCAP are not well-understood. Classic childhood GH deficiency has been attributed to inadequate GH production by the anterior pituitary due to genetic mutations involved in pituitary gland development or signaling, tumors, or brain malformations (especially optic nerve hypoplasia sequence) (Chinoy & Murray, 2016). Given that a few of the children in our series had multiple pituitary hormone deficiencies, congenital or acquired pituitary dysfunction is a potential etiology of GH deficiency in MCAP as well. While the pituitary glands in our participants appeared structurally normal on MRI, this does not exclude anterior pituitary dysfunction. Furthermore, hydrocephalus is a known feature of MCAP and was present in 73% of our cohort, and hydrocephalus has been associated with anterior pituitary deficiencies including GH deficiency (Cholley et al., 2001; Mirzaa et al., 2012; Moin, Bergsneider, Vespa, & Heaney, 2012).
The normal physiologic role of the PI3K-AKT pathway in downstream signaling for the IGF-1, insulin and to a lesser extent, GH receptors, suggests alternative possible mechanisms for the findings in our patients (Grimberg & DiMeglio, 2018). Gain-of-function mutations in PIK3CA may mimic over- or constitutive activity of these 3 hormone receptors and depending on the location of the post-zygotic mutations in patients with MCAP, determine the pathophysiologic mechanisms and clinical features involved. If such gain-of-function mutations occur centrally, they can mimic increased negative feedback on the hypothalamus and pituitary, leading to reduced GH production and secretion. If the PIK3CA mutation is not present in hypothalamic or pituitary tissues, individuals would be expected to have normal GH releasing hormone (GHRH) and GH production and not exhibit symptoms of GH deficiency. If instead the post-zygotic gain-of-function mutation occurs in target tissues, direct negative feedback may inhibit growth factors through any number of known or unknown regulatory feedback loops (Engelman, Luo, & Cantley, 2006). With GH and IGF-1 production downregulated, tissues that do not harbor the PIK3CA gain-of-function mutation would not have this pathway activated, leading to symptoms of GH deficiency including poor linear growth and hypoglycemia. Therefore, knowing which tissues the PIK3CA mutation is present in would determine the phenotype. Finally, augmented clearance of GH, IGF-1 and IGFBP-3 could be a mechanism of these low serum concentrations found in our cohort. Given the complexity of the interactions that the PI3K-AKT-MTOR pathway has in various tissues, further investigation of the underlying molecular mechanisms leading to GH deficiency in MCAP are needed.
The management of GH deficiency in MCAP is particularly challenging. Treatment of GH deficiency in several reported cases appeared to improve hypoglycemia and normalize linear growth velocity. However, it potentially could lead to worsened asymmetric overgrowth in tissues with the activating PIK3CA mutation, particularly the brain. Although our sample size is small, four of five children treated with GH had a positive response with no apparent side effects, however one participant developed irritability and a concern for possible intracranial hypertension, a known side effect of GH treatment. An additional consideration with GH therapy is the theoretical concern for tumorigenesis. According to Pediatric Endocrine Society, while data are insufficient to suggest that GH causes tumorigenesis, children on GH treatment should be monitored closely if they have an increase risk of cancer (Allen et al., 2016; Grimberg & Allen, 2017; Raman et al., 2015). Unlike several other overgrowth syndromes, the risk of tumorigenesis in MCAP does not appear to be significantly elevated (Mirzaa, Conway, Graham, & Dobyns, 1993; Swerdlow et al., 2017). Overall, given the risk for death and neurodevelopmental issues with untreated hypoglycemia that can be associated with GH deficiency, we recommend treatment with conservative doses of GH in children with MCAP confirmed to have GH deficiency and hypoglycemia. In the absence of hypoglycemia, the risks and benefits of management are less clear and need to be evaluated on an individual basis. As there is little urgency to treat for the purpose of increasing linear growth, waiting until after a child is two years of age when the most rapid period of brain growth has ended is reasonable. Additional longitudinal studies are needed to better delineate the safety and efficacy of GH in this context.
This study had several limitations. First, this is an observational study based on clinical data, therefore, not all relevant data were uniformly available for review. Furthermore, detailed endocrine data were specifically collected from those children with suspected or confirmed endocrine dysfunction. Therefore, the prevalence of GH deficiency and other endocrinopathies in all individuals with MCAP is difficult to determine with certainty, due to ascertainment bias of this cohort. Finally, although it would be useful to fully understand the mechanisms underlying GH deficiency in MCAP to enable an optimal therapeutic approach, the role of PIK3CA mutations in various tissues to cause GH deficiency is currently unclear, as mentioned above.
In summary, we report a series of children with MCAP and GH deficiency, suggesting that GH deficiency is an underappreciated and clinically relevant feature of this megalencephaly overgrowth disorder. Therefore, we recommend that children with MCAP and either hypoglycemia and/or postnatal growth failure be evaluated for GH deficiency and treatment considered on a case-by-case basis. This study includes a large sample size for this rare genetic syndrome, however additional research is needed to accurately determine the overall prevalence, mechanisms, and best management for GH deficiency in children with MCAP.
Figure 3. Possible mechanisms for growth hormone (GH) deficiency in children with mosaic gain-of-function mutations in the PI3K-AKT-MTOR pathway:
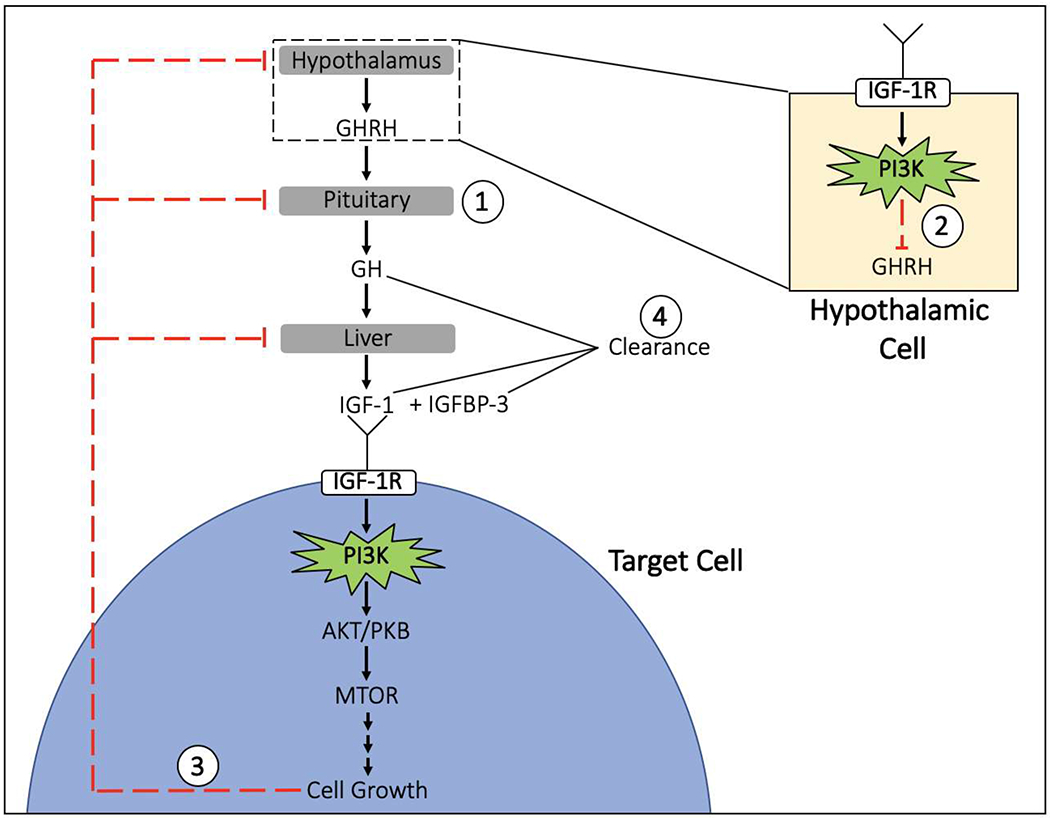
1) Hypopituitarism associated with other central nervous system abnormalities (classic GH deficiency); 2) negative inhibition of GHRH from the IGF-1R signaling pathway being constitutively activated at the level of the hypothalamus; 3) negative feedback from cell growth signaling products of the constituently active PI3K pathway in target cells; and 4) increased clearance of growth factors. GHRH = growth hormone releasing hormone; GH = growth hormone; IGF-1 = insulin-like growth factor 1; R=receptor; IGFBP-3 = insulin-like binding protein 3
ACKNOWLEDGEMENTS
The authors would like to thank the supporting organizations and the patients and families who contributed to this work. This work was supported by NINDS K08NS092898 and Jordan’s Guardian Angels (G.M.) and NICHD K23HD092588 (S.D.).
Funding: NINDS K08NS092898 (G.M.), Jordan’s Guardian Angels (G.M.), NICHD K23HD092588 (S.D.)
Footnotes
CONFLICT OF INTEREST
A.G. serves on the Steering Committee of the Pfizer International Growth Study Database. S.D., M.A.W., J.Z., M.A.D., K.G., S.H., M.K., S.M., R.P.M., M.P. N.N., C.P., A.C.R., M.L.M., & G.M. have nothing to disclose.
DATA AVAILABILITY
The data from this study are available from the corresponding author upon reasonable request.
REFERENCES
- Allen DB, Backeljauw P, Bidlingmaier M, Biller BM, Boguszewski M, Burman P, … Thorner M (2016). GH safety workshop position paper: a critical appraisal of recombinant human GH therapy in children and adults. Eur J Endocrinol, 174(2), P1–9. doi: 10.1530/eje-15-0873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley LC (2002). The phosphoinositide 3-kinase pathway. Science, 296(5573), 1655–1657. doi: 10.1126/science.296.5573.1655 [DOI] [PubMed] [Google Scholar]
- Chinoy A, & Murray PG (2016). Diagnosis of growth hormone deficiency in the paediatric and transitional age. Best Pract Res Clin Endocrinol Metab, 30(6), 737–747. doi: 10.1016/j.beem.2016.11.002 [DOI] [PubMed] [Google Scholar]
- Cholley F, Trivin C, Sainte-Rose C, Souberbielle JC, Cinalli G, & Brauner R (2001). Disorders of growth and puberty in children with non-tumoral hydrocephalus. J Pediatr Endocrinol Metab, 14(3), 319–327. [DOI] [PubMed] [Google Scholar]
- Clayton-Smith J, Kerr B, Brunner H, Tranebjaerg L, Magee A, Hennekam RC, … Donnai D (1997). Macrocephaly with cutis marmorata, haemangioma and syndactyly--a distinctive overgrowth syndrome. Clin Dysmorphol, 6(4), 291–302. [DOI] [PubMed] [Google Scholar]
- De Luca F (2018). Regulatory Role for Growth Hormone in Statural Growth: IGF-Dependent and IGF-Independent Effects on Growth Plate Chondrogenesis and Longitudinal Bone Growth. Pediatr Endocrinol Rev, 16(Suppl 1), 33–38. doi: 10.17458/per.vol16.2018.l.igfeffectschondrogenesis [DOI] [PubMed] [Google Scholar]
- Engelman JA, Luo J, & Cantley LC (2006). The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet, 7(8), 606–619. doi: 10.1038/nrg1879 [DOI] [PubMed] [Google Scholar]
- Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borras AM, Gale CM, … Janne PA (2006). Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest, 116(10), 2695–2706. doi: 10.1172/jci28656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton TR, & Kim JH (2013). A systematic review and meta-analysis to revise the Fenton growth chart for preterm infants. BMC Pediatr, 13, 59. doi: 10.1186/1471-2431-13-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimberg A, & Allen DB (2017). Growth hormone treatment for growth hormone deficiency and idiopathic short stature: new guidelines shaped by the presence and absence of evidence. Curr Opin Pediatr, 29(4), 466–471. doi: 10.1097/mop.0000000000000505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimberg A, & DiMeglio L (2018). Mechanisms of Hormone Action In Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemian LS, Rudolph AM, Rudolph CD, Kushner J, & Lefkothea KP (Eds.), Rudolph’s Pediatrics (23rd ed., Vol. 1, pp. 2481–2490). New York: McGraw-Hill. [Google Scholar]
- Grummer-Strawn LM, Reinold C, & Krebs NF (2010). Use of World Health Organization and CDC growth charts for children aged 0–59 months in the United States. MMWR Recomm Rep, 59(Rr-9), 1–15. [PubMed] [Google Scholar]
- Kinross KM, Montgomery KG, Mangiafico SP, Hare LM, Kleinschmidt M, Bywater MJ, … Phillips WA (2015). Ubiquitous expression of the Pik3caH1047R mutation promotes hypoglycemia, hypoinsulinemia, and organomegaly. Faseb j, 29(4), 1426–1434. doi: 10.1096/fj.14-262782 [DOI] [PubMed] [Google Scholar]
- Lapunzina P, Gairi A, Delicado A, Mori MA, Torres ML, Goma A, … Pajares IL (2004). Macrocephaly-cutis marmorata telangiectatica congenita: report of six new patients and a review. Am J Med Genet A, 130A(1), 45–51. doi: 10.1002/ajmg.a.30235 [DOI] [PubMed] [Google Scholar]
- McDermott JH, Byers H, & Clayton-Smith J (2016). Detection of a mosaic PIK3CA mutation in dental DNA from a child with megalencephaly capillary malformation syndrome. Clin Dysmorphol, 25(1), 16–18. doi: 10.1097/mcd.0000000000000099 [DOI] [PubMed] [Google Scholar]
- Minagawa M, Nagai F, Kanazawa M, Sato Y, Shimohashi K, I K, & Kohno Y (2005). A Case of Macrocephaly-Cutis Marmorata Telangiectatica Congenita Complicated with Growth Hormone (GH)-Deficiency and Hypothyroidism. Clin Pediatr Endocrinol, 14(Suppl 22), 53–56. [Google Scholar]
- Mirzaa G, Conway R, Graham JM Jr., & Dobyns WB (1993). PIK3CA-Related Segmental Overgrowth In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, & Amemiya A (Eds.), GeneReviews((R)). Seattle (WA): University of Washington, Seattle [Google Scholar]
- University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle: All rights reserved. [Google Scholar]
- Mirzaa G, Timms AE, Conti V, Boyle EA, Girisha KM, Martin B, … Dobyns WB (2016). PIK3CA-associated developmental disorders exhibit distinct classes of mutations with variable expression and tissue distribution. JCI Insight, 1(9). doi: 10.1172/jci.insight.87623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzaa GM, Conway RL, Gripp KW, Lerman-Sagie T, Siegel DH, deVries LS, … Dobyns WB (2012). Megalencephaly-capillary malformation (MCAP) and megalencephaly-polydactyly-polymicrogyria-hydrocephalus (MPPH) syndromes: two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis. Am J Med Genet A, 158a(2), 269–291. doi: 10.1002/ajmg.a.34402 [DOI] [PubMed] [Google Scholar]
- Mirzaa GM, Riviere JB, & Dobyns WB (2013). Megalencephaly syndromes and activating mutations in the PI3K-AKT pathway: MPPH and MCAP. Am J Med Genet C Semin Med Genet, 163c(2), 122–130. doi: 10.1002/ajmg.c.31361 [DOI] [PubMed] [Google Scholar]
- Moin T, Bergsneider M, Vespa P, & Heaney AP (2012). Pituitary function in patients with normal pressure hydrocephalus before and after neurosurgical correction. J Clin Endocrinol Metab, 97(10), 3545–3549. doi: 10.1210/jc.2012-1978 [DOI] [PubMed] [Google Scholar]
- Moore CA, Toriello HV, Abuelo DN, Bull MJ, Curry CJ, Hall BD, … Dobyns WB (1997). Macrocephaly-cutis marmorata telangiectatica congenita: a distinct disorder with developmental delay and connective tissue abnormalities. Am J Med Genet, 70(1), 67–73. [PubMed] [Google Scholar]
- Raman S, Grimberg A, Waguespack SG, Miller BS, Sklar CA, Meacham LR, & Patterson BC (2015). Risk of Neoplasia in Pediatric Patients Receiving Growth Hormone Therapy--A Report From the Pediatric Endocrine Society Drug and Therapeutics Committee. J Clin Endocrinol Metab, 100(6), 2192–2203. doi: 10.1210/jc.2015-1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riviere JB, Mirzaa GM, O’Roak BJ, Beddaoui M, Alcantara D, Conway RL, … Dobyns WB (2012). De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet, 44(8), 934–940. doi: 10.1038/ng.2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutterd C, McGillivray G, Stark Z, Messazos B, Cameron F, White S, … Leventer R (2018). Polymicrogyria in association with hypoglycemia points to mutation in the mTOR pathway. Eur J Med Genet, 61(12), 738–740. doi: 10.1016/j.ejmg.2018.06.002 [DOI] [PubMed] [Google Scholar]
- Swerdlow AJ, Cooke R, Beckers D, Borgstrom B, Butler G, Carel JC, … Zandwijken GRJ (2017). Cancer Risks in Patients Treated With Growth Hormone in Childhood: The SAGhE European Cohort Study. J Clin Endocrinol Metab, 102(5), 1661–1672. doi: 10.1210/jc.2016-2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi M, Dobashi K, Nagahara K, Kato M, Nishimura G, Fukuzawa R, … Hasegawa T (2017). A novel de novo germline mutation Glu40Lys in AKT3 causes megalencephaly with growth hormone deficiency. Am J Med Genet A, 173(4), 1071–1076. doi: 10.1002/ajmg.a.38099 [DOI] [PubMed] [Google Scholar]
- Trivin C, Souberbielle JC, Aubertin G, Lawson-Body E, Adan L, & Brauner R (2006). Diagnosis of idiopathic growth hormone deficiency: contributions of data on the acid-labile subunit, insulin-like growth factor (IGF)-I and-II, and IGF binding protein-3. J Pediatr Endocrinol Metab, 19(4), 481–489. [PubMed] [Google Scholar]
- Yano S, & Watanabe Y (2001). Association of arrhythmia and sudden death in macrocephaly-cutis marmorata telangiectatica congenita syndrome. Am J Med Genet, 102(2), 149–152. [DOI] [PubMed] [Google Scholar]