

Abstract
A novel coronavirus [severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), or 2019 novel coronavirus] has been identified as the pathogen of coronavirus disease 2019. The main protease (Mpro, also called 3‐chymotrypsin‐like protease) of SARS‐CoV‐2 is a potential target for treatment of COVID‐19. A Mpro homodimer structure suitable for docking simulations was prepared using a crystal structure (PDB ID: https://doi.org/10.2210/pdb6Y2G/pdb; resolution 2.20 Å). Structural refinement was performed in the presence of peptidomimetic α‐ketoamide inhibitors, which were previously disconnected from each Cys145 of the Mpro homodimer, and energy calculations were performed. Structure‐based virtual screenings were performed using the ChEMBL database. Through a total of 1 485 144 screenings, 64 potential drugs (11 approved, 14 clinical, and 39 preclinical drugs) were predicted to show high binding affinity with Mpro. Additional docking simulations for predicted compounds with high binding affinity with Mpro suggested that 28 bioactive compounds may have potential as effective anti‐SARS‐CoV‐2 drug candidates. The procedure used in this study is a possible strategy for discovering anti‐SARS‐CoV‐2 drugs from drug libraries that may significantly shorten the clinical development period with regard to drug repositioning.
Keywords: 2019 novel coronavirus, COVID‐19, drug repositioning, Mpro, SARS‐CoV‐2, virtual screening
Here, I describe virtual screenings for compounds targeting main protease of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) to predict potential anti‐SARS‐CoV‐2 drugs and lead compounds. The procedure described herein may be a suitable drug repositioning strategy for the discovery of anti‐SARS‐CoV‐2 drugs from drug libraries.

Abbreviations
- Mpro
main protease
- SARS‐CoV‐2
severe acute respiratory syndrome coronavirus 2
A novel coronavirus (severe acute respiratory syndrome coronavirus 2 [SARS‐CoV‐2], or 2019 novel coronavirus [2019‐nCoV]) has been identified as the pathogen of coronavirus disease 2019. The coronavirus has spread worldwide and exhibits strong contagious and infective characteristics. There are no effective anti‐SARS‐CoV‐2 drugs at the time of writing. The discovery of potential anti‐SARS‐CoV‐2 drugs from known drug libraries is thought to be an effective drug repositioning strategy for shortening the clinical development period.
Severe acute respiratory syndrome coronavirus 2 belongs to the betacoronavirus group. One of the best‐characterized drug targets among viral constituents is the main protease (Mpro, also called 3‐chymotrypsin‐like protease) [1]. Crystal structures of the Mpro dimer, the biological active form, have been resolved with or without synthetic inhibitors, some of which covalently bind to Cys145 at the catalytic dyad (i.e., Cys145 and His41) or have been designed to covalently bind to Cys145 (Fig. 1) [2, 3]. However, side effects, toxicity, and lower potency often cause covalent inhibitors to drop out. Therefore, noncovalent inhibitors with high binding affinity are more suited for treatment of such viral infections.
Fig. 1.
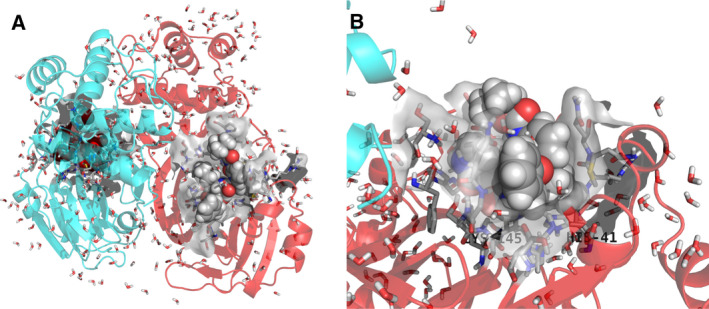
Refined crystal structure (PDB ID: https://doi.org/10.2210/pdb6Y2G/pdb) of a SARS‐CoV‐2 Mpro homodimer with peptidomimetic α‐ketoamide inhibitors. (A) Whole structure and (B) enlarged structure of the active site. Chains A and B of the Mpro homodimer are shown as red and cyan ribbons, respectively. Peptidomimetic α‐ketoamide inhibitors are shown in CPK color using space‐filling models. Water molecules are shown using tubes. Residues located 3 Å from the inhibitors are shown in CPK color using tubes without nonpolar hydrogen atoms. Van der Waals surfaces of the active sites are shown in gray.
In this study, I performed stepwise structure‐based virtual screenings using two different docking simulations in order to discover potential drugs that target Mpro using the ChEMBL database [4], which mainly lists drugs and known bioactive compounds. I present these potential anti‐SARS‐CoV‐2 drug candidates here. The structural information of the potential drugs will be useful for improving their pharmacokinetic properties more effectively and for developing specific anti‐SARS‐CoV‐2 drugs.
Methods
Refinement of the Mpro crystal structure
I prepared the Mpro homodimer structure suitable for docking simulations using a crystal structure (PDB ID: https://doi.org/10.2210/pdb6Y2G/pdb [2]; resolution 2.20 Å). Structural refinement was performed using Homology Modeling Professional for hyperchem (HMHC) software [5, 6], and energy calculations were performed under the AMBER99 force field with the following parameters: root‐mean‐square gradient, 1.0 kcal·mol−1·Å−1; algorithm, Polak–Ribière; cutoffs, none; 1–4 van der Waals scale factor, 0.5; 1–4 electrostatic scale factor, 0.833; dielectric scale factor, 1.0; and distance‐dependent dielectric condition. Structural refinement was conducted in the presence of peptidomimetic α‐ketoamide inhibitors, which were previously disconnected from each Cys145 of the Mpro homodimer. After adding hydrogen atoms automatically, I assigned Mulliken atomic charges of inhibitors; their α‐carbonyl moiety (disconnected from Cys145) was treated as a hydroxy carbocation using single‐point calculations of the semiempirical MNDO/d method. Mulliken atomic charges obtained from the MNDO/d calculation showed empirically good correlation with AMBER charges [7]. In addition, AMBER99 atom types were assigned. N‐ and C‐terminals of the Mpro homodimer were treated as zwitterions, aspartic and glutamic acid residues were treated as anions, while lysine, arginine, and histidine residues were treated as cations under physiological conditions. Next, a free glycine included in the crystal structure was removed, and the initial coordinates of hydrogen atoms of crystal waters (one water, WAT620, was removed, since the initial coordinate of the hydrogen atoms could not be assigned) were predicted using HMHC. Subsequently, partial optimization by Belly calculations for all components, except heavy atoms, was performed, and distance restraint conditions (7.0 kcal·mol−1·Å−2) were applied to all heavy atoms of the above structure; next, geometry optimization calculations were performed. The resulting structure was subjected to low‐temperature molecular dynamics annealing (starting temperature 0 K; heat time 30 ps; simulation temperature 300 K; run time 100 ps; final temperature 0 K; cooling time 30 ps; step size 0.001 ps; and temperature step 0.01 K). Finally, all distance restraint conditions were removed, and the structure was further optimized to obtain the final structure. Precision of the final structure was confirmed using a Ramachandran plot program of HMHC.
Preparation of 3D structures from the ChEMBL database
Planar structures of the ChEMBL database (ChEMBL26; 1 950 760 distinct compounds, including 13 308 drugs) [4] were downloaded from the ChEMBL website in SDF file format. MayaChemTools (2019) [8] was used to remove counterions and inorganic compounds from the database. Then, 3D structures were obtained using balloon version 1.6.9 [9] under an MMFF94 force field. The resulting 3D structure database was treated using babel version 2.4.1 [10]; the compounds’ protonation state was prepared under physiological conditions (pH = 7.4) and filtered by molecular weight (MW ≥ 100 and ≤ 500) to reduce the database to a more drug‐like library. A total of 1 485 144 compounds were used for subsequent virtual screenings.
Structure‐based virtual screenings
Structure‐based virtual screenings were performed using rdock (2013) [11] and autodock vina version 1.1.2 [12]; both interfaces are available in Docking Study with hyperchem (DSHC) software [5, 13], and the resulting docking modes filtered by the rdock score threshold were more precisely simulated using autodock vina.
Prior to docking simulations, the inhibitor of the A chain of the Mpro homodimer was removed from the system. Then, docking simulations for the 3D structure database (1 485 144 compounds) were performed using the relatively reliable, high‐speed docking simulation program rdock under the default condition. I expected that the concentrated potential drugs would consist of ~ 70 distinct drugs, the others being bioactive compounds. On the basis of this, I determined the rdock score threshold to be ≤ −50 kcal·mol−1. The cavity condition for rdock docking simulations was prepared using peptidomimetic α‐ketoamide inhibitor of the A chain under a default condition. Docking simulations output the top three docking modes per trial compound in SDF file format. As a result, 4 455 432 docking modes were obtained. These docking modes were filtered by the rdock score threshold of ≤ −50 kcal·mol−1 to obtain 27 561 distinct compounds (57 649 docking modes) in SDF file format. The ChEMBL IDs of these distinct compounds were subjected to knime version 4.1.2 [14] to collect compound information from the ChEMBL web server; some information was manually collected from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database [15].
From the 57 649 docking modes obtained by virtual screenings, the 27 561 distinct hit compounds had two docking modes on average. The hit compounds, including the 64 drugs I found, could be more precisely investigated using autodock vina docking simulations with these docking modes as the initial structure. Subsequently, the resulting 57 649 docking modes were separated and converted into individual PDBQT files using DSHC. Then, more precise docking simulations were performed using the autodock vina In Silico Screenings interface integrated into DSHC. The Mpro homodimer system prepared above in PDB file format was also converted to a PDBQT file using DSHC. A configuration file with cavity information was prepared using DSHC, and other docking conditions were set to default values (the top nine docking modes per trial compound were maximally outputted). Docking simulations with autodock vina produced 513 597 docking modes, which were filtered by the autodock vina score (empirical binding free energy) threshold of −10 kcal·mol−1. Since the autodock vina score is an empirical binding free energy, I expected that −9 kcal·mol−1 of a score would theoretically show an nM order of binding affinity with Mpro. When the threshold for screening was set to less than this value, I obtained 659 distinct compounds (1216 docking modes) as hit compounds. To more realistically concentrate the number of hit compounds, I determined the threshold value to be ≤ −10 kcal·mol−1. As a result, I obtained 29 distinct compounds (total 41 docking modes). The ChEMBL IDs of these distinct compounds were subjected to KNIME to collect compound information from the ChEMBL web server.
Results and Discussion
Structure‐based virtual screenings of the ChEMBL database
In the ChEMBL database, drugs, including approved, clinical, and preclinical drugs, constitute ~ 0.7% of the total number of compounds; the others are mainly bioactive compounds, whose synthesis is, therefore, promising. The advantage for using the ChEMBL database is that it covers all types of drugs, from preclinical to approved stages. I expected that the hit compounds would largely differ from candidates obtained from virtual screenings using focused and targeted libraries [16, 17]. With regard to drug repositioning, the ChEMBL database is more suitable for searching for effective known drugs or bioactive compounds when urgent therapy is necessary and effective drugs are not known. The rdock score threshold of ≤ −50 kcal·mol−1 showed relatively high binding affinity with Mpro.
Table 1 shows the 64 potential drugs that showed high binding affinity with Mpro, with some drug information collected from the ChEMBL web server using KNIME. I found 11 approved, 14 clinical, and 39 preclinical drugs from the hit compounds (27 561 distinct compounds with 57 649 docking modes); the other 27 497 were bioactive compounds. The 64 drugs were largely classified into antibacterial, antidiabetic, anti‐infective, anti‐inflammatory, antineoplastic, cardiovascular, gastrointestinal, human immunodeficiency virus, and neuropsychiatric drugs. Interestingly, the potential drugs obtained contained sepimostat and curcumin, which are recommended as potential anti‐SARS‐CoV‐2 drugs by researchers [18, 19].
Table 1.
Potential anti‐SARS‐CoV‐2 drugs obtained from rdock virtual screenings of the ChEMBL database.
| CHEMBL ID | Drug synonym | Stage | Action | Target | rdock Score (kcal·mol−1) | Vina Score (kcal·mol−1) |
|---|---|---|---|---|---|---|
| CHEMBL2105088 | LOBENDAZOLE | Anthelmintic | −52.1429 | −6.5 | ||
| CHEMBL2105653 | SETILEUTON | Antiasthmatic | 5‐Lipoxygenase inhibitor | −60.4636 | −8.3 | |
| CHEMBL1191 | SULFAMETHIZOLE | Approved | Antibacterial | Dihydropteroate synthase inhibitor | −79.7939 | −6.6 |
| CHEMBL437 | SULFATHIAZOLE | Approved | Antibacterial | Dihydropteroate synthase inhibitor | −72.0537 | −6.5 |
| CHEMBL1384 | KANAMYCIN | Approved | Antibacterial | 30S ribosomal subunit inhibitor | −71.2391 | −7.5 |
| CHEMBL1747 | TOBRAMYCIN | Approved | Antibacterial | 50S ribosomal subunit inhibitor | −56.0916 | −6.6 |
| CHEMBL1524273 | PHTHALYLSULFATHIAZOLE | Approved | Antibacterial | Cytochrome P450 3A4, dihydropteroate synthase inhibitor | −51.7695 | −7.3 |
| CHEMBL2105399 | SULFAMOXOLE | Antibacterial | Dihydropteroate synthase inhibitor | −87.8995 | −7.2 | |
| CHEMBL1355299 | SULFAETHIDOLE | Antibacterial | Putative fructose‐1,6‐bisphosphate aldolase | −84.7512 | −7.0 | |
| CHEMBL2105398 | SULFAMETROLE | Antibacterial | −69.6628 | −6.6 | ||
| CHEMBL2105403 | PENTISOMICIN | Antibacterial | −59.2134 | −7.3 | ||
| CHEMBL2110604 | BETAMICIN | Antibacterial | −54.6510 | −7.7 | ||
| CHEMBL2107073 | SANFETRINEM CILEXETIL | Antibacterial | −52.6940 | −7.8 | ||
| CHEMBL94087 | GLYBUTHIAZOL | Antidiabetic | −83.8342 | −6.8 | ||
| CHEMBL490070 | BENAXIBINE | Antidiabetic | Monoamine oxidase A | −52.5382 | −6.9 | |
| CHEMBL2107408 | GLYBUZOLE | Antidiabetic, Anti‐Hyperglycemic, | −73.5918 | −6.6 | ||
| CHEMBL2104694 | ACEFLURANOL | Antiestrogen | −57.3375 | −7.4 | ||
| CHEMBL1950289 | TANZISERTIB | Phase2 | Antifibrotic | c‐Jun N‐terminal kinase inhibitor | −60.6067 | −8.5 |
| CHEMBL2107669 | VIPROSTOL | Antihypertensive | Prostaglandin analogue | −52.3341 | −6.5 | |
| CHEMBL2106914 | PHTHALYLSULFAMETHIZOLE | Anti‐infective | −84.7500 | −7.9 | ||
| CHEMBL2106807 | MALEYLSULFATHIAZOLE | Anti‐infective | −66.6682 | −7.0 | ||
| CHEMBL157337 | RAMIFENAZONE | Anti‐Inflammatory | Adrenergic receptor beta | −79.4409 | −6.3 | |
| CHEMBL2104561 | ELTENAC | Anti‐Inflammatory | COX2 | −72.5029 | −6.1 | |
| CHEMBL114586 | SEPIMOSTAT | Anti‐Inflammatory | Serine protease inhibitor | −58.1205 | −7.9 | |
| CHEMBL2110642 | DIBUPYRONE | Anti‐Inflammatory | −57.8675 | −6.1 | ||
| CHEMBL2104226 | ETERSALATE | Anti‐Inflammatory | −53.3912 | −7.0 | ||
| CHEMBL2058833 | GANAPLACIDE | Phase2 | Antimalarial | −70.6688 | −7.7 | |
| CHEMBL2396661 | ALPELISIB | Approved | Antineoplastic | Serine‐protein kinase ATM | −67.1970 | −8.3 |
| CHEMBL25336 | BISANTRENE | Phase3 | Antineoplastic | −54.2373 | −8.5 | |
| CHEMBL2103842 | VARLITINIB | Phase2 | Antineoplastic | EGFR‐HER2 inhibitor | −69.1763 | −8.1 |
| CHEMBL2180604 | TAK‐593 | Phase1 | Antineoplastic | Vascular endothelial growth factor receptor 3 | −65.4614 | −8.1 |
| CHEMBL3182444 | MK‐5108 | Phase1 | Antineoplastic | Aurora‐A kinase inhibitor | −52.9359 | −6.7 |
| CHEMBL1079 | TIZANIDINE | Approved | Cardiovascular | Adrenergic receptor alpha agonist | −78.7516 | −6.3 |
| CHEMBL259223 | MENATETRENONE | Phase3 | Cardiovascular | Vitamin K‐dependent gamma‐carboxylase | −75.9905 | −6.3 |
| CHEMBL321582 | BUCINDOLOL | Phase2 | Cardiovascular | Adrenergic receptor beta antagonist | −50.6285 | −7.0 |
| CHEMBL12552 | BIMAKALIM | Cardiovascular | Potassium channel opener | −67.8339 | −7.1 | |
| CHEMBL2106134 | DALBRAMINOL | Cardiovascular | Beta blocker | −67.3284 | −6.3 | |
| CHEMBL358373 | INDANIDINE | Cardiovascular | Adrenergic receptor alpha agonist | −66.5682 | −6.2 | |
| CHEMBL297362 | XYLAZINE | Cardiovascular | Adrenergic receptor alpha agonist | −53.0909 | −5.7 | |
| CHEMBL689 | MANNITOL | Approved | Gastrointestinal | −51.6980 | −5.3 | |
| CHEMBL70209 | ZALTIDINE | Gastrointestinal | Histamine receptor H2 antagonist | −57.8372 | −6.3 | |
| CHEMBL1742413 | PIBUTIDINE | Gastrointestinal | Histamine 2 receptor antagonist | −53.1955 | −7.7 | |
| CHEMBL116438 | CURCUMIN | Phase3 | HIV | HIV‐1 integrase | −55.7724 | −7.3 |
| CHEMBL2360841 | RO‐24‐7429 | Phase2 | HIV | Tyrosyl‐DNA phosphodiesterase 1 | −58.6922 | −6.7 |
| CHEMBL2105488 | THYMOTRINAN | Immunostimulant | −50.6933 | −7.1 | ||
| CHEMBL593262 | PARA‐NITROSULFATHIAZOLE | Leishmania Infantum | −80.0130 | −7.0 | ||
| CHEMBL2107425 | GLUCUROLACTONE | Liver function improving | −50.5937 | −5.8 | ||
| CHEMBL1108 | DROPERIDOL | Approved | Neuropsychiatric | Dopamine D2‐receptor antagonist | −59.2556 | −7.5 |
| CHEMBL1522 | ESZOPICLONE | Approved | Neuropsychiatric | GABA‐A receptor agonist | −54.5048 | −10.0 |
| CHEMBL1618018 | HOMATROPINE | Approved | Neuropsychiatric | Muscarinic cholinergic receptor antagonist | −50.4433 | −6.7 |
| CHEMBL1394756 | ESOXYBUTYNIN | Neuropsychiatric | NF‐Kappa‐B, muscarinic cholinergic receptor antagonist | −51.7716 | −5.9 | |
| CHEMBL2110912 | DIHEXYVERINE | Neuropsychiatric | Muscarinic cholinergic receptor antagonist | −51.2083 | −6.8 | |
| CHEMBL55214 | NERIDRONIC ACID | Phase3 | Osteogenesis Imperfecta | −52.9425 | −5.6 | |
| CHEMBL2106834 | METOXEPIN | Psychotropic | −53.3412 | −7.4 | ||
| CHEMBL1231124 | AZD‐1480 | Phase2 | Tyrosine‐protein kinase JAK2 inhibitor | −56.3449 | −8.0 | |
| CHEMBL10188 | TALNETANT | Phase2 | Neurokinin 3 receptor antagonist | −52.4637 | −7.7 | |
| CHEMBL563646 | EVATANEPAG | Phase2 | Prostanoid EP2 receptor | −50.5628 | −8.0 | |
| CHEMBL2105528 | BISFENAZONE | Carboxylesterase | −66.3130 | −7.9 | ||
| CHEMBL2105110 | LAMTIDINE | Histamine 2 receptor antagonist | −65.9473 | −6.9 | ||
| CHEMBL67654 | CAREBASTINE | Histamine H1 receptor antagonist | −55.9690 | −7.7 | ||
| CHEMBL155674 | ASOBAMAST | TNF receptor 2 | −52.7795 | −7.1 | ||
| CHEMBL1603949 | BITHIONOLOXIDE | Menin/histone‐lysine N‐methyltransferase MLL | −52.4736 | −6.9 | ||
| CHEMBL2105536 | SULFACECOLE | −52.0995 | −7.0 | |||
| CHEMBL2104446 | VANYLDISULFAMIDE | −50.1930 | −8.3 |
Additional docking simulations for hit compounds
Table 2 shows the 29 hit compounds obtained using autodock vina virtual screenings with ≤ −10 kcal·mol−1 of binding free energy for Mpro. For the 64 drugs, autodock vina scores of the most stable docking modes are also shown in Table 1.
Table 2.
Hit compounds obtained by combining autodock vina and rdock virtual screenings of the ChEMBL database.
| CHEMBL ID | Structure | Target | Vina score (kcal·mol−1) |
|---|---|---|---|
| CHEMBL1559003 |
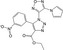
|
Survival motor neuron protein | −10.6 |
| CHEMBL2237553 |
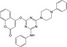
|
Aspergillus niger | −10.5 |
| CHEMBL1511674 |
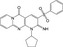
|
Histone‐lysine N‐methyltransferase MLL | −10.5 |
| CHEMBL3260476 |
|
Heat shock protein HSP 90‐alpha | −10.4 |
| CHEMBL1170272 |
|
Serotonin 6(5‐HT6) receptor | −10.4 |
| CHEMBL1335000 |
|
−10.4 | |
| CHEMBL2235580 |
|
Mus musculus | −10.3 |
| CHEMBL3264032 |
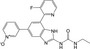
|
Staphylococcus aureus | −10.3 |
| CHEMBL1447105 |
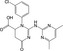
|
4′‐phosphopantetheinyl transferase FFP | −10.2 |
| CHEMBL589899 |
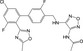
|
Bradykinin B1 receptor | −10.2 |
| CHEMBL1539803 |
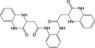
|
Lysine‐specific demethylase 4D‐like | −10.2 |
| CHEMBL2216842 |
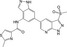
|
PI3‐kinase p110‐delta subunit | −10.2 |
| CHEMBL427787 |
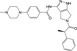
|
Serine threonine‐protein kinase aurora‐A | −10.2 |
| CHEMBL1339675 |
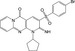
|
−10.2 | |
| CHEMBL3126648 |
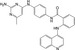
|
DNA(Cytosine‐5)‐methyltransferase 1 | −10.1 |
| CHEMBL1302388 |
|
Prelamin‐A/C | −10.1 |
| CHEMBL3234783 |
|
Staphylococcus aureus | −10.1 |
| CHEMBL1807774 |
|
Tyrosine‐protein kinase receptor RET | −10.1 |
| CHEMBL2087984 |
|
−10.1 | |
| CHEMBL2387487 |
|
ACHN | −10.0 |
| CHEMBL2113271 |
|
Adenosine A1 receptor | −10.0 |
| CHEMBL476947 |
|
Cannabinoid CB2 receptor | −10.0 |
| CHEMBL399042 |
|
Cyclin‐dependent kinase 1 | −10.0 |
| CHEMBL2000247 |
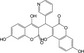
|
Integrase | −10.0 |
| CHEMBL3236740 |
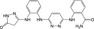
|
Mus musculus | −10.0 |
| CHEMBL1447944 |
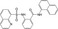
|
Nonstructural protein 1 | −10.0 |
| CHEMBL1760165 |
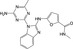
|
Serine threonine‐protein kinase mTOR | −10.0 |
| CHEMBL2087965 |
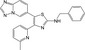
|
−10.0 | |
| CHEMBL1522 |
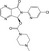
|
GABA‐A receptor agonist | −10.0 |
Almost all the 29 hit compounds were bioactive compounds registered to the ChEMBL database, except for eszopiclone (CHEMBL1522; approved drug for neuropsychiatric disease). I believe that these hit compounds were not developed for common targets, although the structural feature could be categorized into some mother skeletons, such as diazole, azine, and sulfone derivatives (Table 2).
Figure 2 shows the most stable docking modes of sepimostat (Fig. 2B; autodock vina score −7.9 kcal·mol−1), curcumin (Fig. 2C; autodock vina score −7.3 kcal·mol−1), and eszopiclone (Fig. 2D; autodock vina score −10.0 kcal·mol−1) obtained from autodock vina docking simulations, in addition to the binding mode of peptidomimetic α‐ketoamide inhibitor in the crystal structure (Fig. 2A). Researchers consider sepimostat, curcumin, and the α‐ketoamide inhibitor to be potential anti‐SARS‐CoV‐2 drugs. In this study, eszopiclone was also the only approved drug with the highest score on autodock vina docking simulations (Tables 1 and 2). The carbonyl moiety of these compounds was close to the catalytic site of the Cys145 and His41 catalytic dyad, and docking modes were similar to each other. These results suggest that these compounds may function through the same underlying mechanism.
Fig. 2.
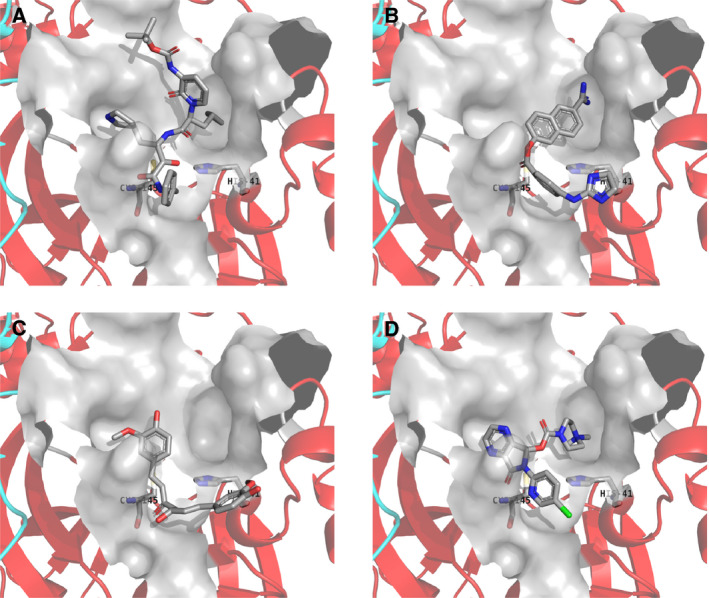
The most stable docking mode obtained from autodock vina docking simulations. (A) Binding mode of peptidomimetic α‐ketoamide inhibitor in the crystal structure. (B) Sepimostat, (C) curcumin, and (D) eszopiclone. Chains A and B of the Mpro homodimer are shown as red and cyan ribbons, respectively. The compound, Cys145, and His41 are shown as tubes. Van der Waals surface of the active site is shown in gray color. Water molecules and hydrogen atoms are neglected.
Conclusions
This study was performed to rapidly identify potential anti‐SARS‐CoV‐2 drug candidates from a known drug library on the basis of drug repositioning. The drug candidates presented in this study could be further examined for their anti‐SARS‐CoV‐2 activities, together with those of earlier studies using more limited drug libraries [20, 21, 22]. Bioactive compounds with high binding affinity for SARS‐CoV‐2 Mpro could be used as a basis for improving pharmacokinetic properties and for developing specific anti‐SARS‐CoV‐2 drugs. Combinations of structure‐based docking simulations are valuable for high‐throughput virtual screenings to identify urgently needed therapies for viral infections. Determination of the effect of potential anti‐SARS‐CoV‐2 drugs obtained in this study is in progress, and the results will be published in the near future.
Conflict of interest
The author declares no conflict of interest.
Author contributions
MT designed the experiments, prepared the infrastructure of the experiments, developed the software, performed the experiments, analyzed the data, and wrote the paper.
References
- 1. Yang H, Yang M, Ding Y, Liu Y, Lou Z, Zhou Z, Sun L, Mo L, Ye S, Pang H et al (2003) The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. PNAS 100, 13190–13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang L, Lin D, Sun X, Rox K and Hilgenfeld R (2020) X‐ray structure of main protease of the novel coronavirus SARS‐CoV‐2 enables design of α‐ketoamide inhibitors. bioRxiv [Preprint]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Peng C et al (2020) Structure of Mpro from COVID‐19 virus and discovery of its inhibitors. bioRxiv [Preprint]. [Google Scholar]
- 4. Davies M, Nowotka M, Papadatos G, Dedman N, Gaulton A, Atkinson F, Bellis L and Overington JP (2015) ChEMBL web services: streamlining access to drug discovery data and utilities. Nucleic Acids Res 43, W612–W620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tsuji M (2007) Development of the structure‐based drug design systems, HMHC and DSHC. Mol Sci 1, NP004. [Google Scholar]
- 6. Tsuji M (2018) Homology Modeling Professional for HyperChem, Revision H1. Institute of Molecular Function, Saitama, Japan. [Google Scholar]
- 7. Tsuji M, Shudo K and Kagechika H (2015) Docking simulations suggest that all‐trans retinoic acid could bind to retinoid X receptors. J Comput Aided Mol Des 29, 975–988. [DOI] [PubMed] [Google Scholar]
- 8. Sud M (2016) MayaChemTools: an open source package for computational drug discovery. J Chem Inf Model 56, 2292–2297. [DOI] [PubMed] [Google Scholar]
- 9. Vainio MJ and Johnson MS (2007) Generating conformer ensembles using a multiobjective genetic algorithm. J Chem Inf Model 47, 2462–2474. [DOI] [PubMed] [Google Scholar]
- 10. O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T and Hutchison GR (2011) Open babel: an open chemical toolbox. J Cheminform 3, 33 10.1186/1758-2946-3-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ruiz‐Carmona S, Alvarez‐Garcia D, Foloppe N, Garmendia‐Doval AB, Juhos S, Schmidtke P, Barril X, Hubbard RE and Morley SD (2014) rDock: a fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLoS Comput Biol 10, e1003571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Trott O and Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comp Chem 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tsuji M (2018) Docking Study with HyperChem, Revision H1. Institute of Molecular Function, Saitama, Japan. [Google Scholar]
- 14. Berthold MR, Cebron N, Dill F, Gabriel TR, Kötter T, Meinl T, Ohl P, Thiel K and Wiswedel B (2009) KNIME: The konstanz information miner. ACM SIGKDD Explorations Newslett 11, 26. [Google Scholar]
- 15. Kanehisa M and Sato Y (2020) KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci 29, 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, Wang Q, Xu Y, Li M, Li X et al (2020) Analysis of therapeutic targets for SARS‐CoV‐2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 10.1016/j.apsb.2020.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gentile D, Patamia V, Scala A, Sciortino MT, Piperno A and Rescifina A (2020) Inhibitors of SARS‐CoV‐2 main protease from a library of marine natural products: a virtual screening and molecular modeling study. Preprints, 2020030372. 10.20944/preprints202003.0372.v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Khaerunnisa S, Kurniawan H, Awaluddin R, Suhartati S and Soetjipto S (2020) Potential inhibitor of COVID‐19 main protease (Mpro) from several medicinal plant compounds by molecular docking study. Preprints 2020030226. 10.20944/preprints202003.0226.v1 [DOI] [Google Scholar]
- 19. Hoffmann M, Kleine‐Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N, Nitsche A et al (2020) SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Talluri S (2020) Virtual screening based prediction of potential drugs for COVID‐19. Preprints, 2020020418. 10.20944/preprints202002.0418.v2 [DOI] [PubMed] [Google Scholar]
- 21. Wang Q, Zhao Y, Chen X and Hong A (2020) Virtual screening of approved clinic drugs with main protease (3CLpro) reveals potential inhibitory effects on SARS‐CoV‐2. Preprints, 2020030144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen YW, Yiu CB and Wong K. (2020) Prediction of the SARS‐CoV‐2 (2019‐nCoV) 3C‐like protease (3CLpro) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research 9, 2020030226 10.12688/f1000research.22457.1 [DOI] [PMC free article] [PubMed] [Google Scholar]