

Abstract
Background
CM082 is a novel angiogenesis inhibitor targeting vascular endothelial growth factor receptor (VEGFR) and platelet‐derived growth factor receptor (PDGFR). The purpose of this research was to evaluate the antitumor activity of CM082 combined with gefitinib on epidermal growth factor receptor (EGFR) mutant non‐small cell lung cancer (NSCLC) in vitro and in vivo.
Methods
The effect of CM082 on human umbilical vein endothelial cells (HUVECs) was assessed. In vitro and in vivo efficacy of CM082 combined with gefitinib on EGFR NSCLC cell lines (HCC827 harboring E746_A750 deletion and H3255 harboring L858R) and a xenograft model was evaluated.
Results
CM082 inhibited VEGF‐induced cell growth, phosphorylation of VEGFR and downstream signaling molecules, tube formation, and cell migration of HUVECs. Furthermore, CM082 combined with gefitinib was more effective in inhibiting growth and colony formation and inducing apoptosis of H3255 and HCC827 cells in vitro than monotherapy. Moreover, tumor growth inhibition by the combination in a H3255 xenograft model was 107.7% more than that by gefitinib (93.6%) (P < 0.0001) and CM082 (51.4%) (P < 0.0001) alone. In addition, coadministration had a better effect on inhibiting cell proliferation, inducing apoptosis, and inhibiting the expression of CD31 and VEGF‐A. The combination therapy had a stronger inhibition effect on STAT3 phosphorylation than monotherapy.
Conclusions
CM082, a novel angiogenesis inhibitor, enhances the antitumor activity of gefitinib on EGFR mutant NSCLC by inhibiting proliferation and angiogenesis and promoting apoptosis of tumor cells.
Key points
Significant findings of the study CM082, a novel angiogenesis inhibitor, enhances the antitumor activity of gefitinib on EGFR mutant NSCLC by inhibiting proliferation and angiogenesis and promoting apoptosis of tumor cells. What this study adds These findings justify evaluation of the efficacy of CM082 combined with gefitinib in patients with EGFR mutant advanced NSCLC in clinical trials.
Keywords: Angiogenesis inhibitor, combination therapy, gefitinib, non‐small cell lung cancer
Introduction
Lung cancer is a malignant tumor with the highest morbidity and mortality worldwide. Most patients are diagnosed at the advanced or metastatic stage, and non‐small cell lung cancer (NSCLC) accounts for nearly 85% of primary lung cancer cases.1, 2 The main driver gene in NSCLC is epidermal growth factor receptor (EGFR).3, 4 First‐generation EGFR tyrosine kinase inhibitors (EGFR‐TKIs) are effective in the treatment of advanced EGFR mutant NSCLC, but most patients have disease progression in approximately one year.5, 6, 7, 8, 9 Many approaches including combinations of EGFR‐TKIs with angiogenesis inhibitors have been evaluated to delay the progression.
Angiogenesis plays an important role in tumor development and progression.10, 11 Blockade of the VEGF/VEGFR signaling pathway of angiogenesis is a key method in inhibiting tumor growth.12 Several angiogenesis inhibitors targeting the VEGF/VEGFR signaling pathway have been approved for the treatment of advanced lung cancer.13, 14, 15 Angiogenesis inhibitors combined with EGFR‐TKIs prolong the progression‐free survival of patients with EGFR mutant advanced NSCLC while increasing the incidence of side effects compared to EGFR‐TKI monotherapy.16, 17 Regardless of the drug type (whether antibody‐based drugs or small‐molecule tyrosine kinase inhibitors), a long half‐life can pose a challenge in managing toxicity especially in combination therapy. Murine studies on the pharmacokinetics and pharmacodynamics of sunitinib showed that the continuous inhibition of VEGFR2 phosphorylation was not required to achieve a therapeutic effect. However, clinical studies have suggested that sunitinib has a longer half‐life (>40 hours) and accumulates in various tissues, which leads to the drug holiday of sunitinib for metastatic renal cell carcinoma treatment at 50 mg once daily with four weeks on and two weeks off.18, 19 CM082 is a novel angiogenesis inhibitor targeting VEGFR and platelet‐derived growth factor receptor (PDGFR), developed on the same chemical framework as sunitinib (Fig 1). In a phase I clinical study, CM082 had a shorter half‐life (<6.5 hours) in humans, meeting the pharmacokinetics/pharmacodynamics requirement of intermittent inhibition, and exhibiting no dose‐limiting toxicity at all the doses administered daily.20 Our research aimed to evaluate the antitumor effect of CM082 combined with gefitinib on EGFR mutant NSCLC via preclinical experiments.
Figure 1.
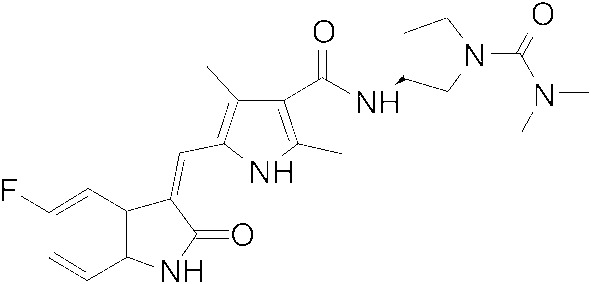
Chemical structure of CM082 (provided by Betta Pharmaceutica Co., Ltd). CAS number: 1013920‐15‐4. Molecular formula: C23H26FN5O3.
Methods
Cell lines and reagents
Human umbilical vein endothelial cells (HUVECs) were purchased from Sciencell (CA, USA), and human lung cancer cell lines, HCC827 and H3255, were purchased from American Type Culture Collection (VA, USA). HUVECs were cultured in endothelial cell medium (ECM) kit (Sciencell) containing 5% fetal bovine serum (FBS) and 1% endothelial cell growth supplement. The human lung cancer cell lines were cultured in medium 1640 (Thermo Fisher Scientific, MA, USA) containing 10% FBS (Thermo Fisher Scientific), and 100 U/mL penicillin and 0.1 mg/mL streptomycin were added to the medium. All cells were cultured in an incubator at 37°C with 5% CO2.
Gefitinib and sunitinib were purchased from Selleck (TX, USA). CM082 was provided by Betta Pharmaceutica Co., Ltd. (Hangzhou, China). Recombinant human VEGF was purchased from R&D Systems (MN, USA). Antibodies for VEGFR2, p‐VEGFR2, ERK, p‐ERK, AKT, p‐AKT, p‐STAT3, and STAT3 were purchased from Cell Signaling Technology (MA, USA). Antibodies for VEGF‐A, CD31, Ki‐67, β‐actin, and β‐tubulin, and horseradish peroxidase‐conjugated antirabbit/mouse IgG antibodies, were purchased from ABclonal Technology (Wuhan, China).
Cell viability assay
Cell counting kit‐8 (CCK‐8) was used to detect cell viability according to the manufacturer's instructions. In brief, cells (2 × 103/well) were placed onto 96‐well plates, cultured overnight at 37°C, and then incubated with different concentrations of the drugs for 72 hours. The CCK‐8 reagent was then added, and the mixture was incubated at 37°C for 0.5–4 hours. The absorbance value at 450 nm was detected using a microplate reader. The cell growth rate was evaluated in comparison with that of the control, and the 50% inhibitory concentration (IC) value was calculated using the GraphPad Prism 7.0 software. Each experiment was conducted in triplicate. For the detection of VEGF‐induced HUVEC growth, the cells were cultured in ECM medium containing 0.1% FBS and 40 ng/mL VEGF and incubated with different concentrations of the drugs.
Drug combination research
To evaluate the combined index (CI) of the effect of CM082 with gefitinib on the growth of HCC827 and H3255 cells, the inhibitory effect of a fixed concentration of CM082 combined with varying concentrations of gefitinib on the growth of HCC827 and H3255 cells was analyzed. Administration concentrations were selected with similar inhibitory rates, with two fixed concentrations of CM082 and five gradient concentrations of gefitinib. CalcuSyn software (version 2.0) was used to calculate the CI of CM082 combined with gefitinib. CI > 1 indicated antagonism, CI = 1 indicated an additive effect, and CI < 1 indicated synergism.
Colony formation
For colony formation assay, H3255 and HCC827 cells (2 × 103 per well) were seeded in 6‐well plates and incubated with gefitinib, CM082, or both at IC25. The culture medium containing the drugs was changed every three days. After 10–14 days, the cells were fixed in 4% formaldehyde for 15 minutes, stained with 0.1% crystal violet for 10 minutes at room temperature, and the colonies were then visualized and counted.
Cell apoptosis
Cell apoptosis was detected using flow cytometry. Briefly, H3255 and HCC827 cells (2 × 105 per well) were placed into 6‐well plates and incubated with gefitinib, CM082, or both at IC25. After 48 hours, apoptotic cells were marked using the Annexin V/FITC‐propidium iodide apoptosis detection kit (BD Biosciences, CA, USA) and were detected using flow cytometry.
Tube formation and migration of endothelial cells
To evaluate the effect of the drugs on microvascular formation, Matrigel (50 μL per well) (Corning, New York, USA) was plated in precooled 96‐well plates and incubated at 37°C for 30 minutes. HUVECs (3 × 104 per well) incubated with ECM containing 10% FBS and the drugs were then placed in 96‐well plates. After incubation for 4–6 hours at 37°C, the microvessels were imaged using a microscope.
To detect endothelial cell migration, HUVECs (1 × 105 per well) were resuspended in ECM containing 1% FBS and different concentrations of the drugs, and then the mixture was placed in the top Transwell chamber (Corning). ECM containing 10% FBS was added into the lower chamber. After incubation at 37°C for 24 hours, cells that moved to the bottom of the membrane were stained with Hochest33342 (Sanofi, Paris, France) for 10–15 minutes, and then imaged using a fluorescence microscope.
Western blot
Sodium dodecyl sulfate lysis buffer containing protease and phosphatase inhibitor was used to extract proteins from the cells and tissues. The extracted proteins were separated using sodium dodecyl sulfate‐polyacrylamide gel electrophoresis, transferred onto polyvinylidene difluoride membranes, and incubated with primary antibodies and horseradish peroxidase‐conjugated secondary antibodies. Electrochemiluminescence detection reagent (Thermo Fisher Scientific, MA, USA) was used to detect the target bands.
Tumor xenograft and immunohistochemistry
The animal experiments were approved by the Ethics Committee of the Academy of Military Medical Sciences of the People's Liberation Army. Female BALB/c nude mice, aged five weeks, were purchased from Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). The mice were raised in specific pathogen‐free conditions with a 12 hour light‐dark cycle. Next, 2 × 106 H3255 cells were subcutaneously injected into the right flank of the mice. When the tumor volume reached approximately 100 mm3, the mice were randomly assigned to six groups (n = 6) and treated with dimethyl sulfoxide (DMSO), CM082 (80 mg/kg b.i.d.), CM082 (160 mg/kg b.i.d.), sunitinib (50 mg/kg q.d.), gefitinib (10 mg/kg q.d.), and gefitinib (10 mg/kg q.d.) combined with CM082 (80 mg/kg b.i.d.), respectively. Bodyweight and tumor volume of the mice were measured every three days. After 21 days, mice were killed, and the xenograft tumors were weighed and imaged. The tumors were used for western blotting and immunohistochemistry (IHC). Tumor volume was calculated as (length × width2)/2. Tumor growth inhibition (TGI) was calculated from the start of treatment by comparing changes in tumor volumes for control and treatment groups.
IHC was used to detect the expression of Ki‐67, CD31, and VEGF‐A in the tumor tissues according to the guidelines provided by Zhongshan Golden Bridge Biotechnology Co., Ltd. (Beijing, China). Meanwhile, apoptotic cells in the tumor tissues were detected using the TUNEL method.
Statistical analysis
All data are expressed as mean ± standard deviation. Statistical analyses were carried out using an unpaired, two‐tailed Student's t‐test. * represented P < 0.05 and ** represented P < 0.01.
Results
CM082 inhibited the growth of HUVECs in vitro
VEGF/VEGFR pathway is an important proangiogenic signaling pathway that promotes the growth of endothelial cells.10 CM082 inhibited the growth of VEGF‐stimulated HUVECs with an IC50 of 0.031 ± 0.005 μmol/L, while the IC50 of sunitinib was 0.012 ± 0.003 μmol/L. Notably, the IC50 values of CM082 and sunitinib for inhibiting FBS‐stimulated HUVEC growth were 29.9 ± 3.9 μmol/L and 4.4 ± 0.5 μmol/L, respectively (Fig 2a). The IC50 of CM082 for inhibiting VEGF‐induced HUVEC proliferation was 1/1000 of its IC50 for inhibiting FBS‐induced HUVEC proliferation, while the ratio of sunitinib was 3/1000, suggesting that CM082 had higher selectivity for VEGFR2 than did sunitinib.
Figure 2.
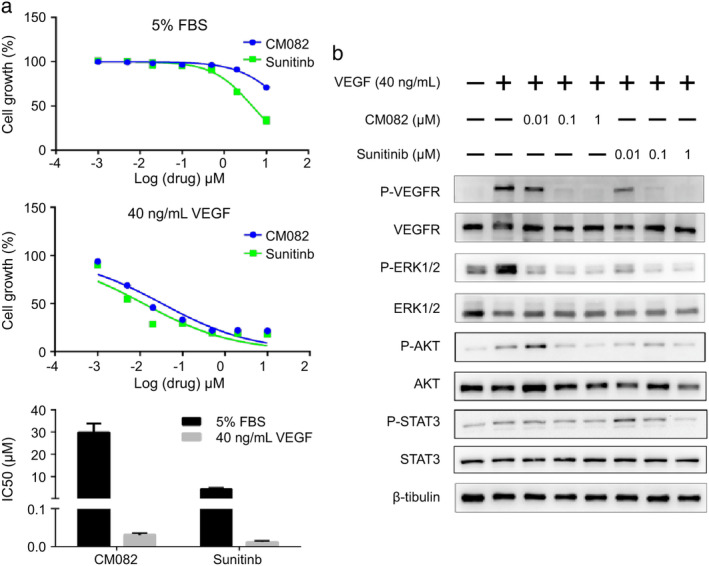
CM082 inhibited VEGF‐induced cell growth and signaling pathway molecule activation of HUVECs. (a) Dose‐dependent response curves of the effects of CM082 or sunitinib on the growth of HUVECs under different culture conditions (5% FBS, 40 ng/mL VEGF). The histogram represents the mean ± standard deviation of three experiments. (b) Effects of the different concentrations of CM082 or sunitinib on phosphorylation of VEGF‐stimulated HUVEC receptors.
To further study the selectivity of CM082 towards VEGFR2, we examined the effect of CM082 on VEGF‐induced receptor phosphorylation. As shown in Fig 2b, CM082 and sunitinib exhibited similar concentration‐dependent inhibition on VEGF‐induced phosphorylation of VEGFR2 and its downstream signaling molecules ERK1/2, AKT, and STAT3. CM082 and sunitinib were able to obviously block the activation of VEGFR2 and AKT at a concentration of 0.1 μmol/L, and significantly inhibited the phosphorylation of ERK1/2 at 0.01 μmol/L. However, CM082 slightly inhibited the phosphorylation of STAT3 at 1 μmol/L while sunitinib significantly inhibited the phosphorylation of STAT3 at 1 μmol/L. These results indicated that CM082 and sunitinib exhibited similar effect on VEGF induced phosphorylation of VEGFR2, but the molecular mechanisms were not exactly the same. Interestingly, although the inhibition of AKT phosphorylation by CM082 and the inhibition of STAT3 phosphorylation by sunitinib were concentration‐dependent, CM082 activated AKT and sunitinib activated STAT3 at a low concentration (0.01 μmol/L). There may be a compensation mechanism here. In a previous study, VEGF‐stimulated phosphorylation of VEGFR2 was decreased by apatinib in a concentration‐dependent manner and apatinib also activated the phosphorylation of VEGFR2 at a low concentration (0.0001, 0.001 μmol/L), but the reason for this is not clear.21 Besides, the maximum plasma drug concentration of CM082 or apatinib at the maximum tolerated dose in human is more than 1 μmol/L.20, 22
CM082 inhibited tube formation and cell migration of HUVECs in vitro
Angiogenesis is the formation of new capillaries from existing blood vessels upon the migration of endothelial cells, which are key to this process.23 Previous reports have shown that VEGF could not induce tube formation by HUVECs on Matrigel.21 Therefore, 10% FBS was chosen as a stimulant to evaluate the effect of CM082 on HUVEC tube formation. As shown in Fig 3a, CM082 inhibited tube formation by HUVECs in a concentration‐dependent manner. At 1 μmol/L, very few tubes were detected. The inhibitory effect of CM082 was similar to that of sunitinib.
Figure 3.
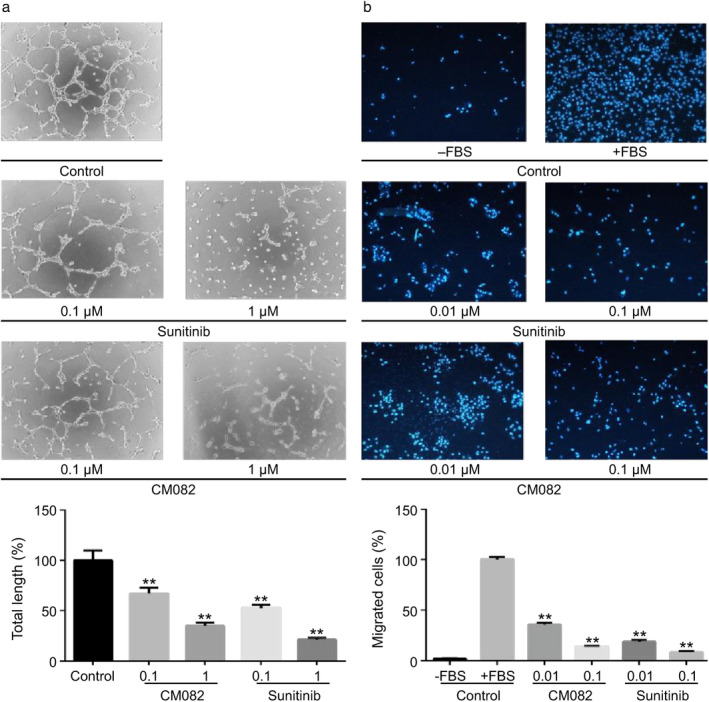
CM082 inhibited tube formation and cell migration of HUVECs. (a) Effects of the different concentrations of CM082 or sunitinib on FBS‐stimulated tube formation in HUVECs. (b) Effects of the different concentrations of CM082 or sunitinib on FBS‐stimulated HUVEC migration. The histogram represents the mean ± standard deviation of three experiments, **P < 0.01.
We next tested the effect of CM082 on the migration of HUVECs using Transwell assay. As shown in Fig 3b, CM082 inhibited FBS‐induced HUVEC migration in a concentration‐dependent manner. At a concentration of 0.1 μmol/L of CM082, minimal cell migration was detected. Similarly, HUVEC migration was almost completely inhibited by sunitinib at 0.1 μmol/L.
CM082 combined with gefitinib inhibited the growth of EGFR mutant NSCLC cells in vitro
To study the inhibition of tumor growth by CM082 combined with gefitinib in EGFR mutant lung cancer cells, HCC827 (harboring EGFR exon 19 deletion) and H3255 (harboring EGFR L858R mutation) were selected for the experiments. As shown in Fig 4a, CM082 evidently inhibited the growth of both cell lines; the inhibitory effect of CM082 was weaker than that of gefitinib. Even at the highest concentration, 50% of the cells were still able to grow with CM082, while cell growth was almost completely inhibited by gefitinib. The IC50 values of CM082 for HCC827 and H3255 cell growth inhibition were 35.65 ± 4.23 μmol/L and 43.2 ± 4.31 μmol/L respectively, while the IC50 values of gefitinib were 0.029 ± 0.003 μmol/L and 0.18 ± 0.03 μmol/L, respectively.
Figure 4.
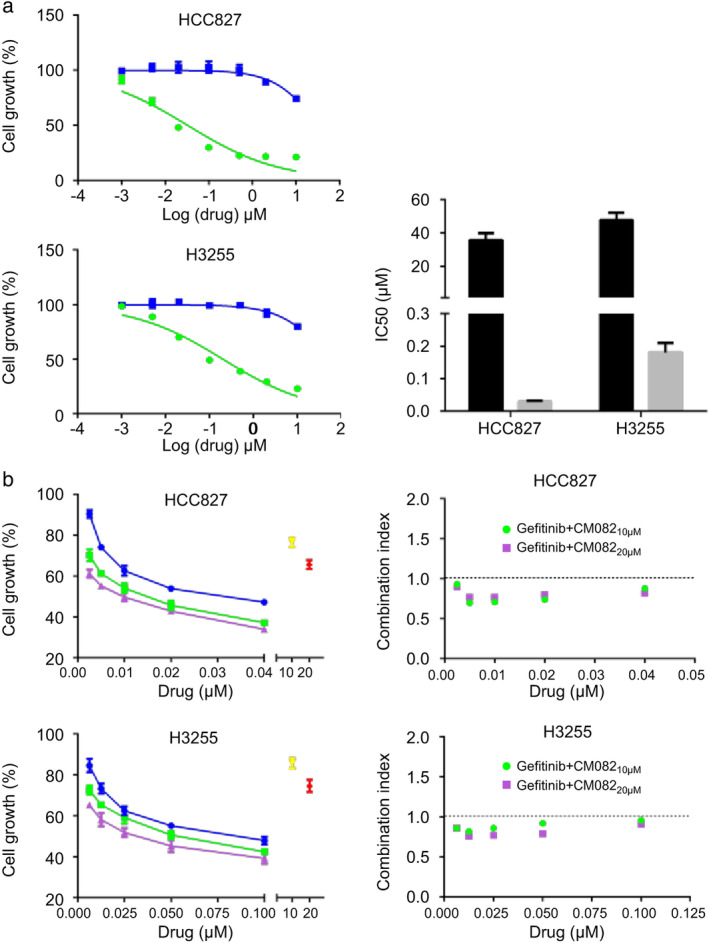
CM082 combined with gefitinib inhibited HCC827 and H3255 cell growth. (a) Dose‐dependent response curves of the effects of CM082 or gefitinib on the growth of HCC827 and H3255 cells. The histogram represents the mean ± standard deviation of three experiments. (b) Dose‐dependent response curves and combination index of CM082 plus gefitinib on the growth of HCC827 and H3255 cells.
We then investigated the inhibitory effect of CM082 combined with gefitinib on the growth of HCC827 and H3255 cells. As shown in Fig 4b, coadministration enhanced the inhibitory effect more than monotherapy. CM082, administered at 10 or 20 μmol/L, showed a synergistic inhibitory effect in combination with gefitinib on the growth of HCC827 and H3255 cells.
CM082 combined with gefitinib inhibited proliferation of EGFR mutant NSCLC cells in vitro
The colony formation assay further verified the effect of CM082 combined with gefitinib on cell proliferation. As shown in Fig 5, both CM082 and gefitinib inhibited the formation of HCC827 and H3255 cell colonies at IC25. The inhibitory effect of the coadministration on H3255 colony formation was stronger than that of monotherapy (P = 0.0079 vs. gefitinib, P = 0.0015 vs. CM082). Surprisingly, coadministration completely inhibited colony formation by HCC827 cells (P = 0.0002 vs. gefitinib, P = 0.0011 vs. CM082).
Figure 5.
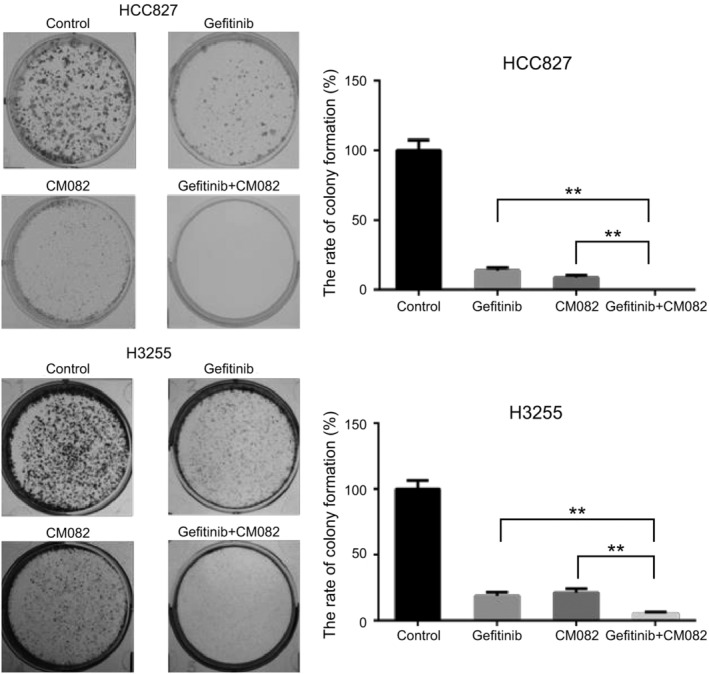
CM082 combined with gefitinib inhibited HCC827 and H3255 colony formation. The histogram represents the mean ± standard deviation of three experiments, **P < 0.01.
CM082 combined with gefitinib promoted apoptosis of EGFR mutant NSCLC cells in vitro
Flow cytometry was used to evaluate the effect of CM082 combined with gefitinib on apoptosis of EGFR mutant NSCLC cells. As shown in Fig 6, CM082 and gefitinib individually promoted the apoptosis of HCC827 and H3255 cells. However, CM082 combined with gefitinib was superior to monotherapy in promoting the apoptosis of HCC827 cells (P = 0.018 vs. gefitinib, P = 0.005 vs. CM082) and H3255 cells (P = 0.003 vs. gefitinib, P = 0.0007 vs. CM082).
Figure 6.
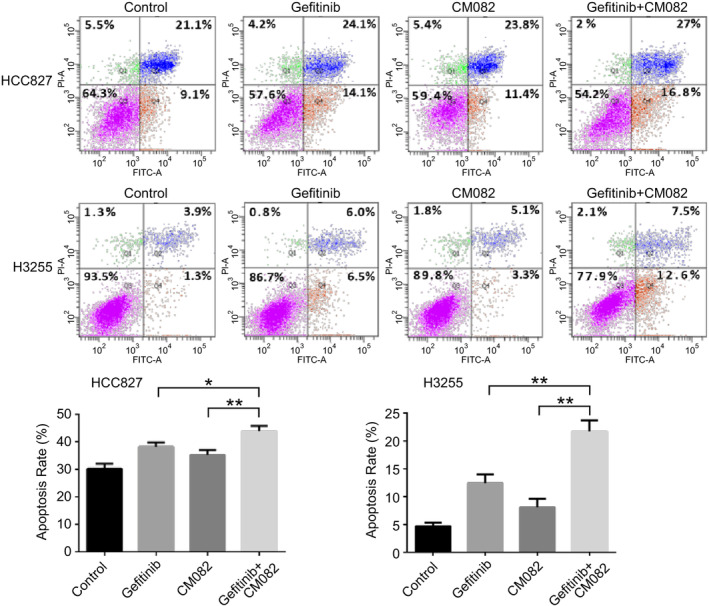
The effect of CM082 combined with gefitinib on apoptosis of HCC827 and H3255 cells. The histogram represents the mean ± standard deviation of three experiments, *P < 0.05, **P < 0.01.
Antitumor effect of CM082 combined with gefitinib in xenograft models
Based on the in vitro antiangiogenic activity of CM082 and antitumor activity of its combination with gefitinib, we evaluated the in vivo antitumor potential of CM082 combined with gefitinib in H3255 tumor xenograft model. As shown in Fig 7a and b, CM082, administered at 80 mg/kg and 160 mg/kg twice daily, had a similar inhibitory effect on tumor growth compared to that in the control group. The TGI was 51.4% (80 mg/kg) and 53.6% (160 mg/kg), which was lower than that of sunitinib (66.8%), but there was no statistically significant difference (the P‐values were 0.22 and 0.29, respectively). The TGI by CM082 (80 mg/kg twice daily) combined with gefitinib was 107.7% and was significantly higher than that by monotherapy with gefitinib (93.6%) (P < 0.0001) and CM082 (51.4%) (P < 0.0001) (Fig. 7c). At the same time, the administration of CM082 combined with gefitinib had no significant effect on the weight of the mice, suggesting that coadministration had no significant toxic or side effects (Fig. 7d).
Figure 7.
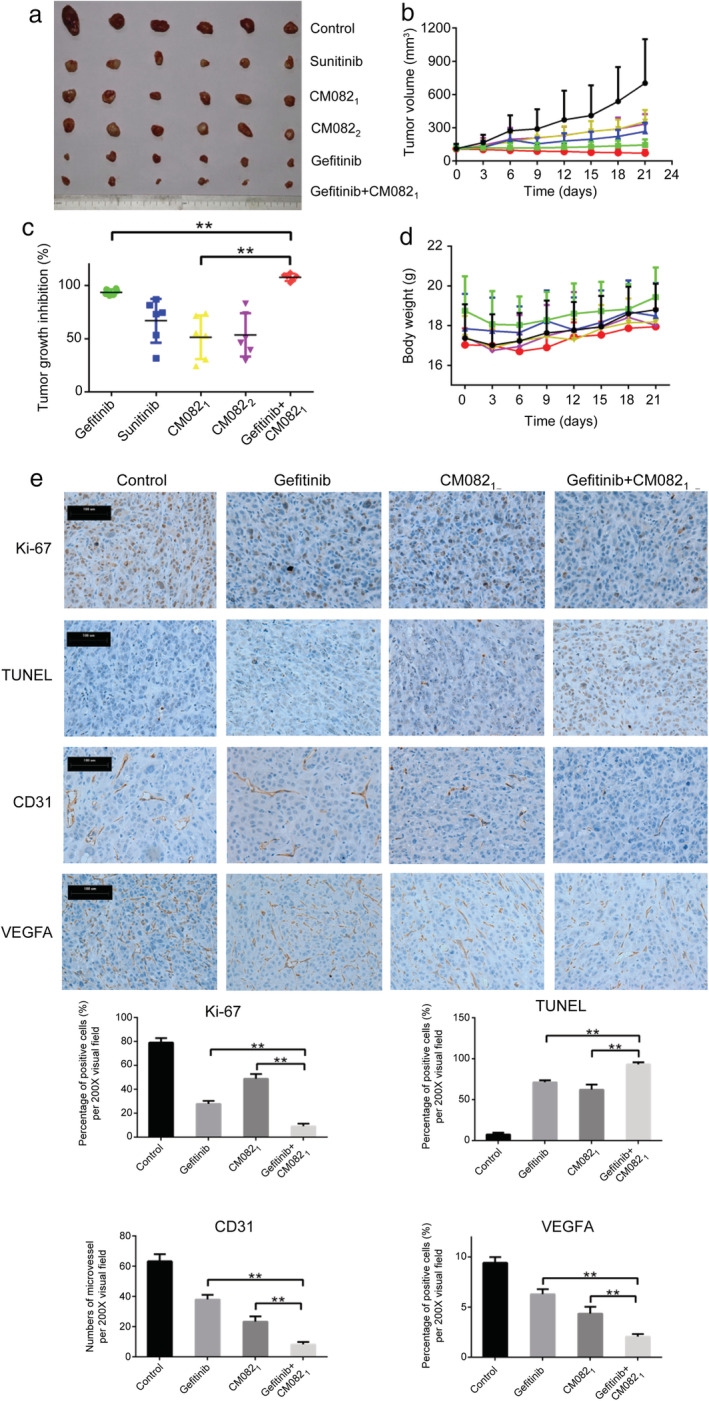
The antitumor activity of CM082 combined with gefitinib on H3255 tumor xenograft. (a) Images of the tumors after 21 days. (b) The tumor volumes of the mice. (c) The tumor growth inhibition rate of different drugs. (d) The bodyweights of the mice. (e) Immunohistochemistry was used to detect the proliferation marker Ki‐67, apoptotic cells (TUNEL method), endothelial marker CD31, and VEGF‐A. CM0821: 80 mg b.i.d., CM0822: 160 mg b.i.d. The data are expressed as mean ± standard deviation, **P < 0.01. Scale bar = 100 μm.
We then assessed the effects of CM082 combined with gefitinib on tumor proliferation, apoptosis, and angiogenesis. Apoptotic cells were detected by the TUNEL method. Ki‐67 and CD31 were used as proliferation and vascular endothelial cell markers, respectively. As shown in Fig 7e, CM082 combined with gefitinib inhibited tumor proliferation (P = 0.0005 vs. gefitinib, P = 0.0001 vs. CM082), promoted tumor apoptosis (P = 0.01 vs. gefitinib, P = 0.008 vs. CM082), and inhibited angiogenesis (P = 0.0001 vs. gefitinib, P = 0.0025 vs. CM082) compared to monotherapy. At the same time, both CM082 and gefitinib could inhibit the expression of VEGF‐A, and combination therapy exhibited a synergistic inhibitory effect on VEGF‐A compared to monotherapy (P = 0.0002 vs. gefitinib, P = 0.0054 vs. CM082).
Finally, we analyzed the effects of CM082 combined with gefitinib on the related downstream signaling molecules ERK1/2, AKT, and STAT3. As shown in Fig 8, CM082 and gefitinib both inhibited the phosphorylation of ERK1/2, AKT, and STAT3 in H3255 tumor xenograft model. However, CM082 significantly inhibited the phosphorylation of ERK1/2 and AKT, and gefitinib obviously inhibited the phosphorylation of ERK1/2 and STAT3. Most importantly, coadministration was superior to monotherapy in inhibiting the phosphorylation of STAT3 (P = 0.0015 vs. gefitinib, P = 0.0002 vs. CM082).
Figure 8.
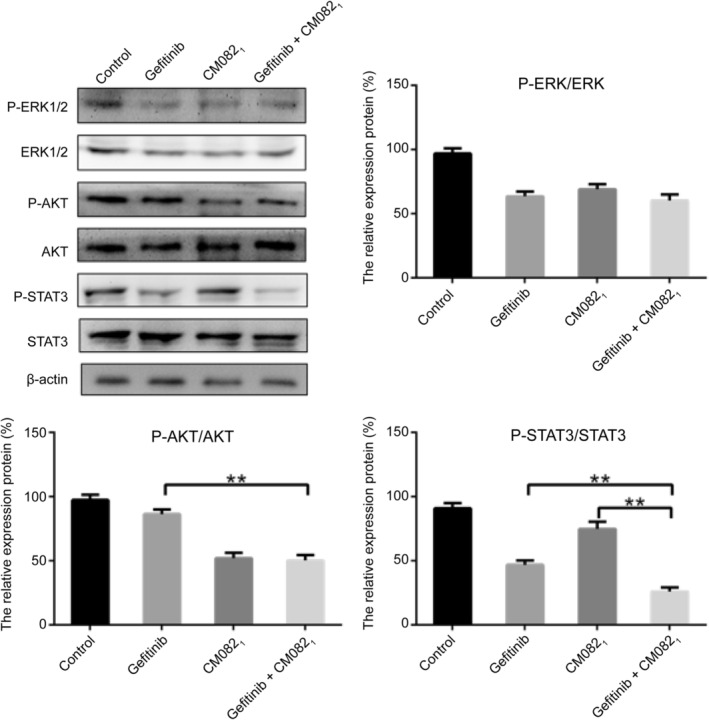
The effect of CM082 combined with gefitinib on the signaling pathway molecules in H3255 tumor xenograft model. The data are expressed as mean ± standard deviation, **P < 0.01.
Discussion
Angiogenesis plays an important role in tumor development and progression.10, 11 VEGF/VEGFR is a key signaling pathway taking part in angiogenesis.12 In this study, CM082, a novel angiogenesis inhibitor, showed promising antiangiogenic and antitumor activities. CM082 inhibited VEGF‐induced cell growth, tube formation and migration of HUVECs at nanomolar levels. Besides, CM082 had higher selectivity for VEGFR than did sunitinib. Further, CM082 (80 mg/kg or 160 mg/kg b.i.d.) had similar inhibitory effects on tumor growth to sunitinib (50 mg/kg q.d.) in tumor xenograft models. There is an interaction between the EGFR and VEGFR pathways, and simultaneous inhibition of EGFR and VEGFR pathways may have a synergistic antitumor effect.24, 25 In our study, CM082 combined with gefitinib inhibited the growth of tumor cells more effectively than monotherapy in vitro and in vivo. Interestingly, monotherapy slowed tumor growth, while coadministration stopped tumor growth and shrunk the tumor volume in vivo.
It is well‐known that sustaining proliferation and resisting apoptosis are the two hallmarks of cancer.26 Cancer cells sustain proliferation by producing growth factors themselves or stimulating normal cells to supply various growth factors.27, 28 Apoptosis serves as a natural barrier to cancer development and cancer cells have evolved a number of methods to limit apoptosis.29, 30 Our results showed that coadministration had a greater inhibitory effect on proliferation and significantly induced apoptosis of the tumor cells in vitro and in vivo. Inducing angiogenesis is another important hallmark of cancer.26 In our study, gefitinib and CM082 both inhibited the expression VEGF‐A and angiogenesis, and coadministration exhibited superior inhibitory activity. These results suggested that CM082 combined with gefitinib inhibited tumor growth by inhibiting proliferation, promoting apoptosis of tumor cells and inhibiting angiogenesis.
ERK1/2, AKT, and STAT3 are important downstream signaling molecules of EGFR and VEGFR, which support tumor cell proliferation, survival and angiogenesis.12, 31, 32, 33 Our results showed that CM082 significantly inhibited the phosphorylation of ERK1/2 and AKT, but slightly inhibited the phosphorylation of STAT3 in H3255 xenograft model, which is consistent with the effect of CM082 on VEGF‐induced HUVECs. In addition, gefitinib obviously inhibited the phosphorylation of ERK1/2 and STAT3 in H3255 xenograft model. Previous results showed that STAT3 could be activated by EGFR to participate in oncogenesis and was simultaneously involved in angiogenesis, especially the expression of VEGF‐A.34, 35 In our study, gefitinib also inhibited the expression VEGF‐A in H3255 xenograft model. All the results indicated that gefitinib showed antivascular effect through STAT3/VEGFA. Moreover, only STAT3 phosphorylation was inhibited to a greater extent by the combined administration than by the monotherapy in H3255 xenograft model. Tumor cell intrinsic activation of STAT3 is frequently observed in solid malignancies and is mainly caused by an oversupply of growth factors, they ultimately accelerate proliferation, increase resistance to apoptosis, and promote angiogenesis and metastatic potential.33 In H3255 xenograft tumor cells, STAT3 may be an important downstream signaling molecule for the synergistic antitumor effect of CM082 combined with gefitinib.
In conclusion, CM082, a novel angiogenesis inhibitor, enhances the antitumor activity of gefitinib on EGFR mutant NSCLC by inhibiting proliferation and angiogenesis and promoting apoptosis of tumor cells. These findings support evaluation of the efficacy of CM082 combined with gefitinib on patients with EGFR mutant advanced NSCLC in clinical trials.
Disclosure
All the authors declare that there are no conflicts of interest.
Acknowledgments
This study was funded by the Beijing Municipal Science & Technology Commission, PR China (No. Z181100001718074) and Chinese National Instrumentation Program (2011YQ170067). We would like to thank Betta Pharmaceutical Co., Ltd. for providing CM082 and its chemical structure. Study concept, design, and supervision: Liu Xiaoqing, Zhang Kun, and Wang Lili; technical assistant: Wang Lili; experimental work: Zhang Kun, Wei Aili, and Jia Xinfei; data analysis and manuscript writing: Zhang Kun; manuscript editing: Liu Xiaoqing.
References
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018; 68: 394–424. [DOI] [PubMed] [Google Scholar]
- 2. Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non‐small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc 2008; 83: 584–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hsu WH, Yang JC, Mok TS, Loong HH. Overview of current systemic management of EGFR‐mutant NSCLC. Ann Oncol 2018; 29 (suppl_1): i3–9. [DOI] [PubMed] [Google Scholar]
- 4. Midha A, Dearden S, McCormack R. EGFR mutation incidence in non‐small‐cell lung cancer of adenocarcinoma histology: A systematic review and global map by ethnicity (mutMapII). Am J Cancer Res 2015; 5: 2892–911. [PMC free article] [PubMed] [Google Scholar]
- 5. Mitsudomi T, Morita S, Yatabe Y et al Gefitinib versus cisplatin plus docetaxel in patients with non‐small‐cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol 2010; 11: 121–8. [DOI] [PubMed] [Google Scholar]
- 6. Rosell R, Carcereny E, Gervais R et al Erlotinib versus standard chemotherapy as first‐line treatment for European patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (EURTAC): A multicentre, open‐label, randomised phase 3 trial. Lancet Oncol 2012; 13: 239–46. [DOI] [PubMed] [Google Scholar]
- 7. Shi Y, Zhang L, Liu X et al Icotinib versus gefitinib in previously treated advanced non‐small‐cell lung cancer (ICOGEN): A randomised, double‐blind phase 3 non‐inferiority trial. Lancet Oncol 2013; 14: 953–61. [DOI] [PubMed] [Google Scholar]
- 8. Wu YL, Zhou C, Liam CK et al First‐line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer: Analyses from the phase III, randomized, open‐label, ENSURE study. Ann Oncol 2015; 26: 1883–9. [DOI] [PubMed] [Google Scholar]
- 9. Zhou C, Wu Y‐L, Chen G et al Erlotinib versus chemotherapy as first‐line treatment for patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (OPTIMAL, CTONG‐0802): A multicentre, open‐label, randomised, phase 3 study. Lancet Oncol 2011; 12: 735–42. [DOI] [PubMed] [Google Scholar]
- 10. Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature 2005; 438: 967–74. [DOI] [PubMed] [Google Scholar]
- 11. Manzo A, Montanino A, Carillio G et al Angiogenesis inhibitors in NSCLC. Int J Mol Sci 2017. 10.3390/ijms18102021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kerbel RS. Tumor angiogenesis. N Engl J Med 2008; 358: 2039–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ferrara N, Adamis AP. Ten years of anti‐vascular endothelial growth factor therapy. Nat Rev Drug Discov 2016; 15: 385–403. [DOI] [PubMed] [Google Scholar]
- 14. Han B, Li K, Wang Q et al Effect of anlotinib as a third‐line or further treatment on overall survival of patients with advanced non‐small cell lung cancer: The ALTER 0303 phase 3 randomized clinical trial. JAMA Oncol 2018; 4: 1569–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kurzrock R, Stewart DJ. Exploring the benefit/risk associated with antiangiogenic agents for the treatment of non‐small cell lung cancer patients. Clin Cancer Res 2017; 23: 1137–48. [DOI] [PubMed] [Google Scholar]
- 16. Nakagawa K, Garon EB, Seto T et al Ramucirumab plus erlotinib in patients with untreated, EGFR‐mutated, advanced non‐small‐cell lung cancer (RELAY): A randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol 2019; 20: 1655–69. [DOI] [PubMed] [Google Scholar]
- 17. Saito H, Fukuhara T, Furuya N et al Erlotinib plus bevacizumab versus erlotinib alone in patients with EGFR‐positive advanced non‐squamous non‐small‐cell lung cancer (NEJ026): Interim analysis of an open‐label, randomised, multicentre, phase 3 trial. Lancet Oncol 2019; 20: 625–35. [DOI] [PubMed] [Google Scholar]
- 18. Faivre S, Delbaldo C, Vera K et al Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J Clin Oncol 2006; 24: 25–35. [DOI] [PubMed] [Google Scholar]
- 19. Mendel DB, Laird AD, Xin X et al In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet‐derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res 2003; 9: 327–37. [PubMed] [Google Scholar]
- 20. Bendell JC, Patel MR, Moore KN et al Phase I, first‐in‐human, dose‐escalation study to evaluate the safety, tolerability, and pharmacokinetics of vorolanib in patients with advanced solid tumors. Oncologist 2019; 24: 455–e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tian S, Quan H, Xie C et al YN968D1 is a novel and selective inhibitor of vascular endothelial growth factor receptor‐2 tyrosine kinase with potent activity in vitro and in vivo. Cancer Sci 2011; 102: 1374–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li J, Zhao X, Chen L et al Safety and pharmacokinetics of novel selective vascular endothelial growth factor receptor‐2 inhibitor YN968D1 in patients with advanced malignancies. BMC Cancer 2010; 10: 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chung AS, Lee J, Ferrara N. Targeting the tumour vasculature: Insights from physiological angiogenesis. Nat Rev Cancer 2010; 10: 505–14. [DOI] [PubMed] [Google Scholar]
- 24. Tortora G, Ciardiello F, Gasparini G. Combined targeting of EGFR‐dependent and VEGF‐dependent pathways: Rationale, preclinical studies and clinical applications. Nat Clin Pract Oncol 2008; 5: 521–30. [DOI] [PubMed] [Google Scholar]
- 25. van Cruijsen H, Giaccone G, Hoekman K. Epidermal growth factor receptor and angiogenesis: Opportunities for combined anticancer strategies. Int J Cancer 2005; 117: 883–8. [DOI] [PubMed] [Google Scholar]
- 26. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell 2011; 144: 646–74. [DOI] [PubMed] [Google Scholar]
- 27. Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature 2004; 432: 332–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cheng N, Chytil A, Shyr Y, Joly A, Moses HL. Transforming growth factor‐beta signaling‐deficient fibroblasts enhance hepatocyte growth factor signaling in mammary carcinoma cells to promote scattering and invasion. Mol Cancer Res 2008; 6: 1521–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Adams JM, Cory S. The Bcl‐2 apoptotic switch in cancer development and therapy. Oncogene 2007; 26: 1324–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature 2004; 432: 307–15. [DOI] [PubMed] [Google Scholar]
- 31. Burotto M, Chiou VL, Lee JM, Kohn EC. The MAPK pathway across different malignancies: A new perspective. Cancer 2014; 120: 3446–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fumarola C, Bonelli MA, Petronini PG, Alfieri RR. Targeting PI3K/AKT/mTOR pathway in non small cell lung cancer. Biochem Pharmacol 2014; 90: 197–207. [DOI] [PubMed] [Google Scholar]
- 33. Huynh J, Etemadi N, Hollande F, Ernst M, Buchert M. The JAK/STAT3 axis: A comprehensive drug target for solid malignancies. Semin Cancer Biol 2017; 45: 13–22. [DOI] [PubMed] [Google Scholar]
- 34. Harris TJ, McCormick F. The molecular pathology of cancer. Nat Rev Clin Oncol 2010; 7: 251–65. [DOI] [PubMed] [Google Scholar]
- 35. Zhang Q, Xu F, Shi Y et al C‐X‐C motif chemokine receptor 4 promotes tumor angiogenesis in gastric cancer via activation of JAK2/STAT3. Cell Biol Int 2017; 41: 854–62. [DOI] [PubMed] [Google Scholar]