

Abstract
Background
Tyrosine kinase domain (TKD) mutation and particularly exon 20 insertion mutations of ERBB2 have been extensively reported in non‐small cell lung cancer (NSCLC). Due to the increased accessibility of next‐generation sequencing, more ERBB2 mutations within the non‐TKD can be detected in clinical practice. Nevertheless, the clinical significance of non‐TKD mutations remains unknown. Hence, this study was designed to comprehensively outline the landscape and characteristics of ERBB2 mutations in NSCLC.
Methods
A total of 1934 patients with NSCLC from cBioPortal were included in the study. An ERBB2 mutation cohort was identified, while subsequent analyses revealed clinical and genomic characteristics.
Results
The frequency of ERBB2 mutation was 4.5%, and it was determined to be more likely to occur in never‐smokers. ERBB2 mutations occurring in the non‐TKD accounted for 57.5% of ERBB2 mutations. In the non‐TKD, missense mutation was the most recurrent mutation type, and S310F was the most recurrent mutation variant. ERBB2 mutations within non‐TKD also had a strong oncogenic ability where up to 37.5% of ERBB2 oncogenic mutations were within non‐TKD. The co‐mutation of EGFR or KRAS was higher in the non‐TKD mutation compared to the TKD mutation. Shorter overall survival was observed in ERBB2‐mutant patients compared with ERBB2 wild‐type patients. There was no significant difference in overall survival between patients with non‐TKD mutations and TKD mutations.
Conclusions
The present study showed that a considerable portion of non‐TKD mutations were oncogenic. ERBB2 mutation was a poor prognostic factor. The non‐TKD mutation might also be used as a therapeutic target in ERBB2‐directed target therapy.
Key points
• Significant findings of the study
ERBB2 mutations were more abundant within a nontyrosine domain than those within the tyrosine domain. Up to 37.5% of ERBB2 oncogenic mutations were within the nontyrosine domain. ERBB2 mutation was a poor prognostic factor.
• What this study adds
The frequency of EGFR or KRAS co‐mutations were significantly higher in ERBB2 mutations within the nontyrosine kinase domain compared to ERBB2 mutations within the tyrosine kinase domain. Nontyrosine domain mutations confer equal overall survival to tyrosine domain mutations.
Keywords: Co‐mutation, ERBB2 mutation, non‐small cell lung cancer, oncogenic function, prognosis
Introduction
The epidermal growth factor receptor (EGFR) target therapy has been the cornerstone for the precise treatment of NSCLC. Nowadays, the classification of NSCLC is not just built on the histology but is also based on tumor driver mutations. A driver mutation leads to abnormal activation of cellular signaling pathways, thus resulting in abnormal proliferation and survival of cancer cells.1 Treatments that target driver gene mutations improve the prognosis in patients with NSCLC compared with conventional chemotherapy.2 A previous study reported that in over 60% of patients with lung adenocarcinomas with detected driver mutations, 9%–14% were rare driver mutations, including erb‐b2 receptor tyrosine kinase 2 (ERBB2).3
ERBB2 has been extensively studied in breast cancer. Its amplification or overexpression was a biomarker of anti‐ERBB2 target therapy in breast cancer. Instead, the mutation is predominant in lung cancer, so conventional ERBB2‐targeting drugs are not effective against ERBB2 mutations in lung cancer. Therefore, a thorough analysis of the ERBB2 mutation spectrum in NSCLC is necessary for the future study of targeted drugs. ERBB2 is composed of an extracellular domain that contains two receptor‐L domain and furin‐like cysteine‐rich domain, a transmembrane domain (TMD), and an intracellular structure that contains a tyrosine kinase domain (TKD) and a carboxyl‐terminal tail.4 ERBB2 TKD mutations and particularly exon 20 insertion mutations are classical driver mutations that have been extensively reported in NSCLC. However, ERBB2 non‐TKD mutation, such as V659E and G660D mutations within the TMD, can also act as driver mutations in NSCLC.5 It has been reported that ERBB2 V659E has shown sensitivity to afatinib and lapatinib in in vitro models.6, 7 In addition, Pahuja et al. found multiple oncogenic mutations in the TMD and the juxtamembrane domain in human tumors.8 They reported that small molecule inhibitors and ERBB2 inhibiting antibodies could efficiently inhibit non‐TKD oncogenic mutations. Some recurring extracellular domain mutations of ERBB2, such as S310F, are also potently oncogenic but can be inhibited by treatment with small‐molecule inhibitors of ERBB2.9 All these preclinical studies indicated that the non‐TKD mutations could be used as candidates for targeted anti‐ERBB2 therapy.
Thanks to easier accessibility to next‐generation sequencing, it is possible to detect more ERBB2 mutations that occur within the non‐TKD in clinical practice; yet, the clinical significance remains unknown in most of these mutations. Hence, this study was designed to comprehensively outline the landscape and characteristics of ERBB2 mutations in NSCLC.
Methods
Patient cohorts
A total of 5222 patients with NSCLC pooled from The Cancer Genome Atlas cohort and other available studies10, 11, 12, 13, 14, 15 via a public database cBioPortal for Cancer Genomics (https://www.cbioportal.org/), were initially screened.16, 17 Briefly, 2725 duplicated patients and 563 patients without ERBB2 sequencing were excluded. Finally, 1934 patients were included in the analysis.
Mutation analyses
The next‐generation sequencing was applied in the present study.10, 11, 12, 13, 14, 15 The mutation domain was defined as the region where ERBB2 mutation occurs. Mutation domain was referred to the Pfam database (http://pfam.xfam.org/), including receptor‐L domain (amino acid position: 52–173 and 366–486), furin‐like cysteine‐rich domain (183–343), growth factor receptor domain IV (510–643), transmembrane domain (654–675), and tyrosine kinase domain (TKD) (720–976). Nontyrosine kinase domain (non‐TKD) was defined as ERBB2 domains mentioned above, except for the TKD. The oncogenic function of mutation was first referred to the OncoKB (https://oncokb.org/), a precision oncology knowledge base containing information on the biological effects and treatment implications of specific cancer gene alterations.18 Mutations with unknown oncogenic function in the OncoKB, including missense mutation and splice site mutation, were analyzed using the Polyphen‐2 (http://genetics.bwh.harvard.edu/pph2/) and Human Splicing Finder (http://www.umd.be/HSF/), respectively, to predict whether a given mutation had an impact on the ERBB2 protein. The oncogenic function was defined as the ability to induce tumor of specific ERBB2 mutations, catalogued as oncogenic, benign, and unknown function. ERBB2 synonymous mutations were generally excluded from the ERBB2 mutation cohort, but synonymous mutations in splice sites were included due to their potential impact on alternative splicing. Splice site was defined as a region near the intron/exon junction or two base pairs into an intron adjacent to the intron/exon junction, referring to Sequence Ontology (http://www.sequenceontology.org/).
Clinical characteristics
Age at diagnosis, sex, smoking history, tumor pathology, and stage was summarized after identifying patients with ERBB2 mutation. Overall survival (OS) was defined as the time from initial diagnosis until death. Survival analysis was performed between ERBB2‐mutant patients and ERBB2 wild‐type patients.
Statistical analysis
Measurement data were tested using Student's t‐test. Categorical data were analyzed using the chi‐square test or Fisher's exact test, and the odds ratio or risk ratio was assessed for the association. The Kaplan‐Meier method was used to estimate the event‐time distribution, and the log‐rank test was used to compare OS between ERBB2‐mutant patients and ERBB2 wild‐type patients. Multivariate survival analysis was performed using the Cox regression model. To balance confounding factors, ERBB2‐mutant patients with survival data were matched to ERBB2 wild‐type patients at a ratio of 1:3 using the propensity score matching method. Matched factors included age at diagnosis, sex, smoking history, pathology, stage, and common oncogenes (EGFR and KRAS). Propensity score matching was performed using R software (version 3.6.1) with matchit package. All statistical tests were two‐sided, and the P‐value < 0.05 was considered statistically significant. Statistical analyses were performed using IBM SPSS, version 23.0 (Armonk, NY).
Results
Prevalence and clinical characteristics
As shown in Table 1, a total of 87 ERBB2 mutations (4.5%, 87/1934) were found, and three patients carried double ERBB2 mutations. Exon 20 insertion mutations accounted for 34.4% (30/87) of ERBB2 mutations; all mutation variants are summarized in Fig 1. A total of 53 ERBB2 mutation variants were defined in 84 patients. The most recurrent mutation variant was Y772_A775dup (25.0%, 21/84), followed by S310F (6.0%, 5/84), G776delinsVC (4.8%, 4/84), and G778_P780dup (4.8%, 4/84). All other mutations occurred in two or fewer patients, with frequency ranging from 1.2% to 2.4%. ERBB2 mutation was associated with smoking history, but not with age, sex, stage, and pathology (never‐smokers vs. smokers, odds ratio = 2.2, 95% confidence interval [CI], 1.3–3.6; P = 0.002).
Table 1.
Clinical characteristics of patients included in the study
| Variables | ERBB2 mutation n = 84 | ERBB2 wild‐type n = 1850 | P‐value |
|---|---|---|---|
| Age, mean (SD) | 65.5 (9.1) | 66.9 (8.6) | 0.159 |
| Sex (%) | 0.14 | ||
| Female | 47 (60.3) | 847 (51.7) | |
| Male | 31 (39.7) | 791 (48.3) | |
| Unknown | 6 | 212 | |
| Stage (%) | 0.937 | ||
| I | 22 (30.6) | 417 (28.3) | |
| II | 8 (11.1) | 188 (12.7) | |
| III | 12 (16.7) | 226 (15.3) | |
| IV | 30 (41.7) | 644 (43.7) | |
| Unknown | 12 | 375 | |
| Pathology (%) | 0.062 | ||
| LUAD | 65 (79.3) | 1554 (86.2) | |
| LUSC | 17 (20.7) | 242 (13.4) | |
| LUNE | 0 (0.0) | 6 (0.3) | |
| NSCLCa | 2 | 48 | |
| Smoker (%) | 0.002 | ||
| Yes | 43 (62.3) | 1119 (78.5) | |
| No | 26 (37.7) | 307 (21.5) | |
| Unknown | 15 | 424 |
a Specific pathological type was unknown.
LAUD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; LUNE, lung neuroendocrine carcinoma; NSCLC, non‐small cell lung cancer.
Figure 1.
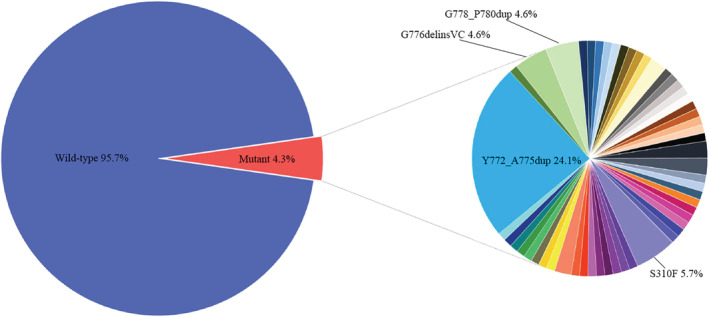
Mutational landscape of ERBB2 in 1934 NSCLC patients.  Wild‐type,
Wild‐type,  R47H,
R47H,  V94I,
V94I,  P122L,
P122L,  G152V,
G152V,  K200N,
K200N,  G222C,
G222C,  D277Y,
D277Y,  G292C,
G292C,  A293P,
A293P,  S310F,
S310F,  N302K,
N302K,  V308M,
V308M,  Q329L,
Q329L,  S335C,
S335C,  R340P,
R340P,  Q396K,
Q396K,  S418T,
S418T,  L651V,
L651V,  V659E,
V659E,  I661V,
I661V,  Q680H,
Q680H,  V697L,
V697L,  Q711H,
Q711H,  G727A,
G727A,  L755A,
L755A,  L755P,
L755P,  V777M,
V777M,  Y772_A775dup,
Y772_A775dup,  G776delinsAVGC,
G776delinsAVGC,  G776delinsVC,
G776delinsVC,  G778_P780dup,
G778_P780dup,  R840W,
R840W,  W906*,
W906*,  Q943*,
Q943*,  G1015E,
G1015E,  E1021Q,
E1021Q,  G1057V,
G1057V,  G1188W,
G1188W,  P1233S,
P1233S,  A1232Gfs*45,
A1232Gfs*45,  ERBB2‐CTTN,
ERBB2‐CTTN,  ERBB2‐PPP1R1B,
ERBB2‐PPP1R1B,  ERBB2‐TCAP,
ERBB2‐TCAP,  SHC1‐ERBB2,
SHC1‐ERBB2,  CASC3‐ERBB2,
CASC3‐ERBB2,  ST14‐ERBB2,
ST14‐ERBB2,  L215=,
L215=,  P300=,
P300=,  X192_splice,
X192_splice,  X254_splice,
X254_splice,  X408_splice,
X408_splice,  X1054_splice
X1054_splice
Mutational characteristics
A total of 42.5% (37/87) and 57.5% (50/87) of ERBB2 mutations occurred within the TKD and the non‐TKD, respectively. Within the non‐TKD, mutation rate was ranked by furin‐like cysteine‐rich region (17.2%, 15/87), splice site (10.3%, 9/87), receptor‐L domain (5.7%, 5/87), and TMD (4.6%, 4/87) (shown in Fig 2a). Missense mutation (43.7%, 38/87) was the most frequent mutation type, followed by in‐frame insertion, splice site mutation, rearrangement, nonsense mutation, and frameshift insertion (34.5%, 10.3%, 8.0%, 2.3%, and 1.1%, respectively). All splice site mutations were predicted to have an impact on alternative splicing except for X192_splice mutation. ERBB2 rearrangements were discovered in seven patients, and five were concurrent with ERBB2 copy number amplification.
Figure 2.

Clinical and molecular characteristics in ERBB2 mutations. (a) An overview of the ERBB2 mutation region; mutation region is referred to as the Pfam database. (b) Concurrent mutations of oncogenes and tumor suppressor genes in patients with ERBB2 mutations. aThree patients carried double ERBB2 mutations illustrated by longer bars based on the mutation types: S310F and D277Y; G727A and Q711H; X254_splice and W906*.  No data,
No data,  Female,
Female,  Male No data,
Male No data,  Yes,
Yes,  No No Data,
No No Data,  LUAD,
LUAD,  LUSC,
LUSC,  NSCLC,
NSCLC,  IA,
IA,  IB,
IB,  IIA,
IIA,  IIB,
IIB,  IIIA,
IIIA,  IIIB,
IIIB,  IV No data,
IV No data,  TKD,
TKD,  non‐TKD,
non‐TKD,  Oncogenic,
Oncogenic,  Benign,
Benign,  Unknow. Mutation type:
Unknow. Mutation type:  Inframe mutation,
Inframe mutation,  Missense mutation,
Missense mutation,  Frame shift mutation,
Frame shift mutation,  No sense mutation,
No sense mutation,  Splice site,
Splice site,  Rearrangement,
Rearrangement,  Amplification,
Amplification,  Deep deletion,
Deep deletion,  No alterations,
No alterations,  Not profiled. Abbreviations: LAUD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; NSCLC, non‐small cell lung cancer. TKD, tyrosine kinase domain; non‐TKD, nontyrosine kinase domain.
Not profiled. Abbreviations: LAUD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; NSCLC, non‐small cell lung cancer. TKD, tyrosine kinase domain; non‐TKD, nontyrosine kinase domain.
Human Splicing Finder and Polyphen‐2 were used to predict the biological effects of ERBB2 variants. Moreover, 47.2% (25/53) of mutation variants were oncogenic (Table 2). Oncogenic function was significantly stronger in TKD mutation compared with non‐TKD mutation (risk ratio = 1.9, 95% CI, 1.3–2.6; P = 0.03), although up to 37.5% of ERBB2 oncogenic mutations were within the non‐TKD.
Table 2.
Oncogenic function of ERBB2 mutation variants identified in the present study
| Variants | Mutation region | Exon | Oncogenic function | Source |
|---|---|---|---|---|
| R47H | ‐ | 2 | Benign | Polyphen‐2 |
| V94I | Receptor‐L domain | 3 | Benign | Polyphen‐2 |
| P122L | Receptor‐L domain | 3 | Benign | Polyphen‐2 |
| G152V | Receptor‐L domain | 4 | Oncogenic | Polyphen‐2 |
| K200N | Furin‐like cysteine rich region | 5 | Benign | Polyphen‐2 |
| G222C | Furin‐like cysteine rich region | 6 | Oncogenic | OncoKB |
| D277Y | Furin‐like cysteine rich region | 7 | Oncogenic | OncoKB |
| G292C | Furin‐like cysteine rich region | 7 | Oncogenic | OncoKB |
| A293P | Furin‐like cysteine rich region | 7 | Oncogenic | OncoKB |
| N302K | Furin‐like cysteine rich region | 8 | Oncogenic | Polyphen‐2 |
| V308M | Furin‐like cysteine rich region | 8 | Oncogenic | Polyphen‐2 |
| S310F | Furin‐like cysteine rich region | 8 | Oncogenic | OncoKB |
| Q329L | Furin‐like cysteine rich region | 8 | Oncogenic | Polyphen‐2 |
| S335C | Furin‐like cysteine rich region | 8 | Oncogenic | OncoKB |
| R340P | Furin‐like cysteine rich region | 8 | Benign | Polyphen‐2 |
| Q396K | Receptor‐L domain | 10 | Benign | Polyphen‐2 |
| S418T | Receptor‐L domain | 11 | Benign | Polyphen‐2 |
| L651V | Transmembrane domain | 17 | Oncogenic | OncoKB |
| V659E | Transmembrane domain | 17 | Oncogenic | OncoKB |
| I661V | Transmembrane domain | 17 | Benign | Polyphen‐2 |
| Q680H | ‐ | 17 | Benign | Polyphen‐2 |
| V697L | ‐ | 18 | Oncogenic | OncoKB |
| Q711H | ‐ | 18 | Benign | Polyphen‐2 |
| G727A | Tyrosine kinase domain | 18 | Oncogenic | Polyphen‐2 |
| L755A | Tyrosine kinase domain | 19 | Oncogenic | OncoKB |
| L755P | Tyrosine kinase domain | 19 | Oncogenic | OncoKB |
| Y772_A775dup | Tyrosine kinase domain | 20 | Oncogenic | OncoKB |
| G776delinsAVGC | Tyrosine kinase domain | 20 | Oncogenic | OncoKB |
| G776delinsVC | Tyrosine kinase domain | 20 | Oncogenic | OncoKB |
| G778_P780dup | Tyrosine kinase domain | 20 | Oncogenic | OncoKB |
| V777M | Tyrosine kinase domain | 20 | Oncogenic | OncoKB |
| R840W | Tyrosine kinase domain | 21 | Oncogenic | Polyphen‐2 |
| W906* | Tyrosine kinase domain | 22 | Unknown | ‐ |
| Q943* | Tyrosine kinase domain | 23 | Unknown | ‐ |
| E1021Q | ‐ | 25 | Oncogenic | Polyphen‐2 |
| G1015E | ‐ | 25 | Benign | Polyphen‐2 |
| G1057V | ‐ | 26 | Benign | Polyphen‐2 |
| G1188W | ‐ | 27 | Oncogenic | Polyphen‐2 |
| A1232Gfs*45 | ‐ | 27 | Oncogenic | OncoKB |
| P1233S | ‐ | 27 | Benign | Polyphen‐2 |
| ERBB2‐CTTN | ‐ | ‐ | Unknown | ‐ |
| ERBB2‐PPP1R1B | ‐ | ‐ | Unknown | ‐ |
| ERBB2‐TCAP | ‐ | ‐ | Unknown | ‐ |
| CASC3‐ERBB2 | ‐ | ‐ | Unknown | ‐ |
| SHC1‐ERBB2 | ‐ | ‐ | Unknown | ‐ |
| ST14‐ERBB2 | ‐ | ‐ | Unknown | ‐ |
| L215= | Splice site | 6 | Unknown, affecting splicing | Human Splicing Finder |
| P300= | Splice site | 7 | Unknown, affecting splicing | Human Splicing Finder |
| X192_splice | Splice site | 5 | Benign | Human Splicing Finder |
| X254_splice | Splice site | 7 | Unknown, Affecting splicing | Human Splicing Finder |
| X408_splice | Splice site | 11 | Unknown, affecting splicing | Human Splicing Finder |
| X633_splice | Splice site | 16 | Unknown, affecting splicing | Human Splicing Finder |
| X1054_splice | Splice site | 26 | Unknown, affecting splicing | Human Splicing Finder |
Concurrent mutations of cancer gene and tumor suppressor gene
Among ERBB2‐mutant patients, 17 cancer genes or tumor suppressor genes were observed with a co‐mutation rate not less than 10%, including TP53, EGFR, KRAS, STK11, and KEAP1. Concurrent mutations of the aforementioned five genes and other well‐known oncogenes in NSCLC (ALK, BRAF, MET, ROS1, and RET) were analyzed in ERBB2‐mutant patients.
TP53 was the most frequently co‐mutated gene in ERBB2 mutations (69.0%) (Fig 2b). No concurrent oncogene mutations were found in the TKD mutation cohort except for one EGFR co‐mutation. Further, a comparison of concurrent oncogene mutations between TKD mutation and non‐TKD mutation was performed. The frequency of EGFR and KRAS mutations was higher in non‐TKD mutation (EGFR: 19.1% vs. 2.9%, P = 0.038; KRAS: 19.1% vs. 0.0%, P = 0.017), but no difference was observed for ALK, BRAF, MET, ROS1, and RET. Tumor suppressor genes KEAP1 and STK11 concurrently mutated with 10.0% and 10.7% of ERBB2 mutations, respectively, similar to ERBB2 wild‐type patients (ERBB2 wild‐type: KEAP1: 16.3%, P = 0.134; STK11: 15.1%, P = 0.272). Likewise, a comparison of KEAP1 or STK11 comutation between TKD mutation and non‐TKD mutation was performed. Although the frequency of STK11 and KEAP1 mutations was higher in non‐TKD mutation cohort, no statistical difference was observed for both of them (non‐TKD mutation vs. TKD mutation: STK11: 14.9% vs. 2.9%, P = 0.094; KEAP1: 12.8% vs. 2.9%, P = 0.164).
Prognostic values of ERBB2 mutation in patients with NSCLC
Overall survival data were available for 31 ERBB2‐mutant patients and 478 ERBB2 wild‐type patients (stage I–IV). The median OS was 28.4 months (95% CI, 24.1–32.7) and 50.3 months (95% CI, 41.1–59.5) for ERBB2‐mutant patients and ERBB2 wild‐type patients, respectively (Fig 3, P = 0.059). The multivariate survival analysis showed that OS was significantly associated with stage (P = 0.005), but not with ERBB2 mutation (P = 0.098) (Table 3). Considering that confounding factors and sample size varied between two cohorts, a propensity score matching method was performed to match ERBB2‐mutant patients and ERBB2 wild‐type patients. Age, sex, smoking, pathology, stage, and matched oncogenes were balanced between two cohorts (Table 4). The multivariate survival analysis after propensity score matching showed that both ERBB2 mutation and advanced stage were poor prognostic factors (ERBB2 mutation vs. wild‐type: hazard ratio = 2.54, 95% CI, 1.25–5.18, P = 0.010; IIIB–IV vs. IA–IIIA: hazard ratio = 3.54, 95% CI, 1.07–11.71, P = 0.038) (Table 3).
Figure 3.

Overall survival in patients from ERBB2 mutation and wild‐type cohorts. (a) Survival curve before PSM.  ERBB2 mutation, median = 28.4 months,
ERBB2 mutation, median = 28.4 months,  ERBB2 wild‐type, median = 50.3 months. P = 0.059. (b) Survival curve after PSM.
ERBB2 wild‐type, median = 50.3 months. P = 0.059. (b) Survival curve after PSM.  ERBB2 mutation, median = 28.4 months,
ERBB2 mutation, median = 28.4 months,  ERBB2 wild‐type, median = 62.8 months. P = 0.005. Abbreviations: PSM, propensity score matching.
ERBB2 wild‐type, median = 62.8 months. P = 0.005. Abbreviations: PSM, propensity score matching.
Table 3.
Multivariate Cox regression analysis of overall survival in patients with NSCLC
| Before PSM | After PSM | |||||||
|---|---|---|---|---|---|---|---|---|
| Univariate | Multivariate | Univariate | Multivariate | |||||
| Variables | HR (95% CI) | P‐value | HR (95% CI) | P‐value | HR (95% CI) | P‐value | HR (95% CI) | P‐value |
| Age (<65 years vs. ≥65 years) | 0.80 (0.59–1.07) | 0.135 | 0.85 (0.47–1.54) | 0.587 | ||||
| Gender (male vs. female) | 1.14 (0.86–1.51) | 0.363 | 0.84 (0.45–1.55) | 0.569 | ||||
| Pathology (LUSC vs. LUAD) | 0.93 (0.81–1.07) | 0.288 | 1.00 (0.51–1.96) | 0.996 | ||||
| Smoker (no vs. yes) | 0.87 (0.27–2.74) | 0.806 | 0.76 (0.17–3.35) | 0.719 | ||||
| Stage (IIIB‐IV vs. IA–IIIA) | 1.88 (1.21–2.93) | 0.005 | 1.88 (1.21–2.93) | 0.005 | 3.43 (1.04–11.30) | 0.043 | 3.54 (1.07–11.71) | 0.038 |
| ERBB2 (mutation vs. wild‐type) | 1.75 (0.97–3.14) | 0.063 | ‐ | 0.098 | 2.69 (1.35–5.35) | 0.005 | 2.54 (1.25–5.18) | 0.010 |
NSCLC, non‐small cell lung cancer; LAUD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; PSM, propensity score matching; HR, hazard ratio.
Table 4.
Clinical characteristics of patients included in the survival analysis
| Variable | ERBB2 mutation n = 31 | ERBB2 wild‐type | |||
|---|---|---|---|---|---|
| Before PSM n = 478 | P‐value | After PSM n = 93 | P‐value | ||
| Age (%) | 0.234 | 0.678 | |||
| <65 years | 15 (48.4) | 180 (37.7) | 49 (52.7) | ||
| ≥65 years | 16 (51.6) | 298 (62.3) | 44 (47.3) | ||
| Gender (%) | 0.086 | 0.852 | |||
| Female | 18 (60.0) | 210 (43.9) | 54 (58.1) | ||
| Male | 12 (40.0) | 268 (56.1) | 39 (41.9) | ||
| Unknown | 1 | 0 | 0 | ||
| Stagea(%) | 0.138 | 0.663 | |||
| IA | 10 (33.3) | 105 (22.0) | 35 (37.6) | ||
| IB | 10 (33.3) | 150 (31.4) | 31 (33.3) | ||
| IIA | 1 (3.3) | 40 (8.4) | 2 (2.2) | ||
| IIB | 5 (16.7) | 73 (15.3) | 12 (12.9) | ||
| IIIA | 3 (10.0) | 64 (13.4) | 11 (11.8) | ||
| IIIB | 0 (0.0) | 21 (4.4) | 0 (0.0) | ||
| IV | 1 (3.3) | 15 (3.1) | 2 (2.2) | ||
| Unknown | 1 | 0 | 0 | ||
| Pathology (%) | 0.534 | 0.533 | |||
| LUSC | 15 (48.4) | 204 (42.7) | 51 (54.8) | ||
| LUAD | 16 (51.6) | 274 (57.3) | 42 (45.2) | ||
| Smoker (%) | 0.013 | 0.489 | |||
| Yes | 17 (81.0) | 280 (95.9) | 54 (87.1) | ||
| No | 4 (19.0) | 12 (4.1) | 8 (12.9) | ||
| Unknown | 10 | 31 | 31 | ||
| KRAS wild‐type (%) | 26 (83.9) | 390 (79.6) | 0.75 | 74 (79.6) | 0.793 |
| EGFR wild‐type (%) | 31 (100.0) | 424 (88.7) | 0.093 | 93 (100.0) | ‐ |
a Difference between stage was tested by Mann‐Whitney Wilcoxon test.
LAUD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma. PSM, propensity score matching.
Next, we compared the OS between patients with TKD and non‐TKD mutations. Subgroup analysis showed that OS was longer in patients with non‐TKD mutations; the observed difference was not statistically significant (OS: non‐TKD vs. TKD, 30.1 months vs. 15.0 months; P = 0.475). After excluding patients with benign ERBB2 mutation, the OS was longer in patients with non‐TKD mutations (OS: non‐TKD vs. TKD, 28.4 months vs. 15.0 months; P = 0.177).
Discussion
In this study, the prevalence of ERBB2 mutation was 4.5%, which was relatively high in comparison to previous studies.19, 20, 21, 22 This discrepancy is reasonable if we take into account that these studies were limited to the detection of TKD mutations, particularly exon 20 mutation in ERBB2. Our results showed that a considerable number of non‐TKD mutations may have significant oncogenic capacity.
In the present study, Y772_A775dup, G776delinsVC, G778_P780dup, and S310F were the most recurrent ERBB2 mutations in NSCLC, which is consistent with a previous report.22 In colorectal cancer, the most recurrent ERBB2 mutations are I655V, V842I, and R678Q.23 The difference indicated different preferred mutant variants among different types of cancer with the same oncogene. In the present study, smoking status was the only factor associated with ERBB2 mutation, which was not in line with previous studies reporting that ERBB2 mutation was associated with lung adenocarcinoma, female sex, and never‐smokers.24, 25 Previous studies have mostly focused on TKD mutations, particularly exon 20‐insertion mutation, barely detecting ERBB2 non‐TKD mutations. Excluding two mutations that occurred in NSCLC (specific histology not defined), all TKD mutations identified in the present study occurred in lung adenocarcinoma. Hence, the results of the present study do not contradict previous studies but provide more comprehensive information regarding mutations throughout ERBB2. Smoking status is usually related to sex. Two previous studies showed that sex was not associated with EGFR mutations after balancing the smoking status of participants, considering that sex‐related smoking was a confounding factor.26, 27
As shown in the present study, the oncogenic function varied among mutations in different ERBB2 domains. Most of the oncogenic mutations occurred in TKD. The extracellular domain of ERBB2 included four parts: two receptor‐L domains, a furin‐like cysteine‐rich domain, and a growth factor receptor domain IV. The receptor‐l domain is related to leucine‐rich segments that participate in ligand binding in EGFR, but ligand binding to ERBB2 has not been discovered.4 This might explain why 80% (4/5) of mutations in the receptor‐L domain found in the present study were benign. Furin‐like cysteine‐rich domain and growth factor receptor domain IV contain numerous cysteine residues that participate in disulfide bond formation, and in homodimer and heterodimer formation with other ErbB family members.4 A high frequency of oncogenic mutations (86.7%) was observed in the furin‐like cysteine‐rich domain. A total of 15n mutations were found in furin‐like cysteine‐rich domain, including a recurrent mutation S310F (n = 5). Two oncogenic mechanisms were found in the extracellular domain mutation. The oncogenic mechanism of S310F implied an elevation of C‐terminal phosphorylation,9 and then again, cysteine substitution in the furin‐like cysteine‐rich domain was identified as another oncogenic mechanism mediated by the formation of disulfide‐linked dimers.9 The TMD not only serves as a membrane anchor but also has a significant role in receptor dimerization.28 In the present study, we identified three mutation variants in TMD (V659E, L651V, and I661V). V659E and G660D are oncogenic mutations that respond to afatinib treatment.7 All these results implied that the oncogenic function of a specific mutation depended on the biological function of the mutation domain.
Our findings indicated that concurrent driver mutation was excluded by ERBB2 TKD mutation. However, a significantly higher frequency of co‐mutation with EGFR and KRAS was observed in non‐TKD mutation. This could be explained by the weaker oncogenic function of non‐TKD mutation compared with TKD mutation. Similarly, the mutation count was much higher in non‐TKD mutation compared with TKD mutation (P < 0.001), but it should be carefully interpreted for different gene test methods applied in the present study. KEAP1 and STK11 mutations were frequently observed in our patient population (16.0% for KEAP1 and 14.9% for STK11). KEAP1 mutation is a poor prognostic factor, while STK11 mutation is a negative predictor of immune checkpoint inhibitors.29, 30 STK11 regulates cellular metabolism/energy homeostasis, growth, and polarity.31 Inactivation of STK11 mediated by mutation is associated with a “cold” tumor immune microenvironment and a decreased density of infiltrating cytotoxic CD8+ T lymphocytes in both genetically engineered murine models and human tumors.32 Both KEAP1 and STK11 mutations rarely coexisted with ERBB2 TKD mutations. Interestingly, a greater portion of these poor‐prognosis and immune‐negative genes concurrently mutated in the non‐TKD mutation cohort with a higher mutation count, although without statistical significance.
Previous studies have also shown a tendency toward shorter OS in both ERBB2‐mutant NSCLC and ERBB2‐mutant colorectal cancer.23, 25 In the present study, the survival of ERBB2‐mutant patients was significantly shorter compared to ERBB2 wild‐type patients after propensity score matching. This might indicate that ERBB2 mutation was a poor prognostic factor. Although the previous case report showed that ERBB2 mutated NSCLC patient could benefit from afatinib treatment,33 a retrospective study in China suggested that compared with chemotherapy, afatinib was not more beneficial to ERBB2 mutated NSCLC patients.34 Also, most of the clinical trials, including afatinib, dacomitinib, and neratinib, focused on ERBB2 exon 20 insertion mutations and failed with a low objective response rate of 11%–19%.35, 36, 37 Two‐phase II studies showed a promising response of poziotinib and pyrotinib in advanced NSCLC with ERBB2 exon 20 mutation. The objective response rate and median PFS were 50% and 5.1 months for poziotinib, and 55% and 6.2 months for pyrotinib, respectively.38, 39 None of these clinical trials included ERBB2 mutations that occurred in non‐TKD. However, basket trials verified the efficacy of tyrosine kinase inhibitor as well as ado‐trastuzumab emtansine on non‐TKD mutations in patients with NSCLC.40, 41 This indicated that non‐TKD mutation was targetable and could be considered as a target during the management of ERBB2‐mutant patients. It is necessary to identify mutations that can benefit from such treatment, which should be initiated by defining a subset of oncogenic mutations.
The present study has certain limitations. First, the patients included in the present analysis were from different cohorts. Also, different gene test methods were used, leading to a mutation detection bias. The aim of the present study was mainly to characterize mutations throughout the entire ERBB2 and emphasize that they might also be important in carcinogenesis and have the potential to be used as therapeutic targets. Second, survival data were only available for a small sample of ERBB2‐mutant patients. Hence, a larger population of ERBB2‐mutant patients is needed to validate the prognostic value of ERBB2 mutation. Finally, the oncogenic function of specific ERBB2 mutation predicted in this study still needs further validation.
Above all, the present study demonstrated that the non‐TKD mutation accounted for over half of ERBB2 mutations. A considerable portion of non‐TKD mutations were oncogenic, while ERBB2 mutation resulted in a poor prognostic factor. The non‐TKD mutation might also be used as a therapeutic target in ERBB2‐directed target therapy.
Disclosure
The authors declare that they have no competing interests.
Acknowledgments
The study was supported by the National Key R&D Program of China (Grant No. 2016YFC1303800), the National Natural Science Foundation of China (Grant No. 81871891), the High‐level Hospital Construction Project (Grant No. DFJH201810), and the Guangdong Provincial Key Laboratory of Lung Cancer Translational Medicine (Grant No. 2017B030314120).
References
- 1. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature 2009; 458: 719–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fukuoka M, Wu YL, Thongprasert S et al Biomarker analyses and final overall survival results from a phase III, randomized, open‐label, first‐line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non‐small‐cell lung cancer in Asia (IPASS). J Clin Oncol 2011; 29: 2866–74. [DOI] [PubMed] [Google Scholar]
- 3. Oxnard GR, Binder A, Jänne PA. New targetable oncogenes in non–small‐cell lung cancer. J Clin Oncol 2013; 31: 1097–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roskoski RJ. The ErbB/HER family of protein‐tyrosine kinases and cancer. Pharmacol Res 2014; 79: 34–74. [DOI] [PubMed] [Google Scholar]
- 5. Ou SI, Schrock AB, Bocharov EV et al HER2 transmembrane domain (TMD) mutations (V659/G660) that stabilize homo‐ and heterodimerization are rare oncogenic drivers in lung adenocarcinoma that respond to afatinib. J Thorac Oncol 2017; 12: 446–57. [DOI] [PubMed] [Google Scholar]
- 6. Serra V, Vivancos A, Puente XS et al Clinical response to a lapatinib‐based therapy for a Li‐Fraumeni syndrome patient with a novel HER2V659E mutation. Cancer Discov 2013; 3: 1238–44. [DOI] [PubMed] [Google Scholar]
- 7. Yamamoto H, Toyooka S, Ninomiya T et al Therapeutic potential of afatinib for cancers with ERBB2 (HER2) transmembrane domain mutations G660D and V659E. Oncologist 2018; 23: 150–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pahuja KB, Nguyen TT, Jaiswal BS et al Actionable activating oncogenic ERBB2/HER2 transmembrane and juxtamembrane domain mutations. Cancer Cell 2018; 34: 792–806.e5. 10.1016/j.ccell.2018.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Greulich H, Bethany K, Mertins P et al Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of ERBB2. Proc Natl Acad Sci U S A 2012; 109: 14476–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ding L, Getz G, Wheeler DA et al Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008; 455: 1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jordan EJ, Kim HR, Arcila ME et al Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov 2017; 7: 596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Imielinski M, Berger AH, Hammerman PS et al Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012; 150: 1107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vavala T, Monica V, Lo Iacono M et al Precision medicine in age‐specific non‐small‐cell‐lung‐cancer patients: Integrating biomolecular results into clinical practice‐A new approach to improve personalized translational research. Lung Cancer 2017; 107: 84–90. [DOI] [PubMed] [Google Scholar]
- 14. Rizvi H, Sanchez‐Vega F, La K et al Molecular determinants of response to anti‐programmed cell death (PD)‐1 and anti‐programmed death‐ligand 1 (PD‐L1) blockade in patients with non‐Small‐cell lung cancer profiled with targeted next‐generation sequencing. J Clin Oncol 2018; 36: 633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rizvi NA, Hellmann MD, Snyder A et al Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science 2015; 348: 124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gao J, Aksoy BA, Dogrusoz U et al Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cerami E, Gao J, Dogrusoz U et al The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2: 401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chakravarty D, Gao J, Phillips S et al OncoKB: A precision oncology Knowledge Base. JCO Precis Oncol 2017; 1: 16 10.1200/PO.17.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buttitta F, Barassi F, Fresu G et al Mutational analysis of the HER2 gene in lung tumors from Caucasian patients: Mutations are mainly present in adenocarcinomas with bronchioloalveolar features. Int J Cancer 2006; 119: 2586–91. [DOI] [PubMed] [Google Scholar]
- 20. Suzuki M, Shiraishi K, Yoshida A et al HER2 gene mutations in non‐small cell lung carcinomas: Concurrence with Her2 gene amplification and Her2 protein expression and phosphorylation. Lung Cancer 2015; 87: 14–22. [DOI] [PubMed] [Google Scholar]
- 21. Stephens P, Hunter C, Bignell G et al Lung cancer: Intragenic ERBB2 kinase mutations in tumours. Nature 2004; 431: 525–6. [DOI] [PubMed] [Google Scholar]
- 22. Arcila ME, Chaft JE, Nafa K et al Prevalence, clinicopathologic associations, and molecular spectrum of ERBB2 (HER2) tyrosine kinase mutations in lung adenocarcinomas. Clin Cancer Res 2012; 18: 4910–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Loree JM, Bailey AM, Johnson AM et al Molecular landscape of ERBB2/ERBB3 mutated colorectal cancer. J Natl Cancer Inst 2018; 110: 1409–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mazieres J, Peters S, Lepage B et al Lung cancer that harbors an HER2 mutation: Epidemiologic characteristics and therapeutic perspectives. J Clin Oncol 2013; 31: 1997–2003. [DOI] [PubMed] [Google Scholar]
- 25. Tomizawa K, Suda K, Onozato R et al Prognostic and predictive implications of HER2/ERBB2/neu gene mutations in lung cancers. Lung Cancer 2011; 74: 139–44. [DOI] [PubMed] [Google Scholar]
- 26. Tanaka T, Matsuoka M, Sutani A et al Frequency of and variables associated with the EGFR mutation and its subtypes. Int J Cancer 2010; 126: 651–5. [DOI] [PubMed] [Google Scholar]
- 27. Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: Biological and clinical implications. Cancer Res 2004; 64: 8919–23. [DOI] [PubMed] [Google Scholar]
- 28. Mineev KS, Bocharov EV, Pustovalova YE, Bocharova OV, Chupin VV, Arseniev AS. Spatial structure of the transmembrane domain heterodimer of ErbB1 and ErbB2 receptor tyrosine kinases. J Mol Biol 2010; 400: 231–43. [DOI] [PubMed] [Google Scholar]
- 29. Skoulidis F, Goldberg ME, Greenawalt DM et al STK11/LKB1 mutations and PD‐1 inhibitor resistance in KRAS‐mutant lung adenocarcinoma. Cancer Discov 2018; 8: 822–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Arbour KC, Jordan E, Kim HR et al Effects of co‐occurring genomic alterations on outcomes in patients with KRAS‐mutant non‐Small cell lung cancer. Clin Cancer Res 2018; 24: 334–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shackelford DB, Shaw RJ. The LKB1‐AMPK pathway: Metabolism and growth control in tumour suppression. Nat Rev Cancer 2009; 9: 563–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koyama S, Akbay EA, Li YY et al STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T‐cell activity in the lung tumor microenvironment. Cancer Res 2016; 76: 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shi Y, Wang M. Afatinib as first‐line treatment for advanced lung adenocarcinoma patients harboring HER2 mutation: A case report and review of the literature. Thorac Cancer 2018; 9: 1788–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu F, Yang G, Xu H, Yang L, Qiu W, Wang Y. Treatment outcome and clinical characteristics of HER2 mutated advanced non‐small cell lung cancer patients in China. Thorac Cancer 2020; 23: 1759–7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kris MG, Camidge DR, Giaccone G et al Targeting HER2 aberrations as actionable drivers in lung cancers: Phase II trial of the pan‐HER tyrosine kinase inhibitor dacomitinib in patients with HER2‐mutant or amplified tumors. Ann Oncol 2015; 26: 1421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gandhi L, Besse B, Mazieres J et al MA04.02 Neratinib ± temsirolimus in HER2‐mutant lung cancers: An international, randomized phase II study. J Thorac Oncol 2017; 12: S358–S9. [Google Scholar]
- 37. Dziadziuszko R, Smit EF, Dafni U et al Afatinib in NSCLC with HER2 mutations: Results of the prospective, open‐label phase II NICHE trial of European thoracic oncology platform (ETOP). J Thorac Oncol 2019; 14: 1086–94. [DOI] [PubMed] [Google Scholar]
- 38. Wang Y, Jiang T, Qin Z et al HER2 exon 20 insertions in non‐small‐cell lung cancer are sensitive to the irreversible pan‐HER receptor tyrosine kinase inhibitor pyrotinib. Ann Oncol 2019; 30: 447–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Robichaux JP, Elamin YY, Tan Z et al Mechanisms and clinical activity of an EGFR and HER2 exon 20‐selective kinase inhibitor in non‐small cell lung cancer. Nat Med 2018; 24: 638–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hyman DM, Piha‐Paul SA, Won H et al HER kinase inhibition in patients with HER2‐ and HER3‐mutant cancers. Nature 2018; 554: 189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li BT, Shen R, Buonocore D et al Ado‐trastuzumab emtansine for patients with HER2‐mutant lung cancers: Results from a phase II basket trial. J Clin Oncol 2018; 36: 2532–7. [DOI] [PMC free article] [PubMed] [Google Scholar]