

Abstract
Tumor cells employ various immune suppressive strategies to overcome anti-tumor immunity. One such method is by their expression of programmed death ligand-1 (PDL1), which triggers apoptotic death or anergy upon binding programmed death-1 (PD1) on T cells. Our previous in vitro cellular studies with human and mouse PDL1+ tumor cells demonstrated that a soluble form of the costimulatory molecule CD80 prevented PDL1-mediated immune suppression and restored T cell activation by binding PDL1 and blocking interaction with PD1. We now report that in vivo treatment of established syngeneic PDL1+ CT26 colon carcinoma and B16F10 melanoma tumors with CD80-Fc delays tumor growth and promotes tumor-infiltrating T cells. Studies with PD1−/− and CD28−/− mice demonstrate that soluble CD80 acts in vivo by simultaneously neutralizing PD1 suppression and activating through CD28. We also report that soluble CD80 mediates its effects by activating transcription factors EGR1–4, NF-κB and MAPK, downstream signaling components of the CD28 and T cell receptor pathways. Soluble CD80 binds to CTLA4 on activated human PBMC; however, increasing quantities of CTLA4 antagonist antibodies do not increase T cell activation. These results indicate that soluble CD80 does not suppress T cell function through CTLA4 and suggest that CTLA4 acts as a decoy receptor for CD80, rather than functioning as a suppressive signaling receptor. Collectively, these studies demonstrate that soluble CD80 has therapeutic efficacy in vivo in mouse tumor systems and that its effects are due to its ability to inhibit PD1-mediated suppression while concurrently activating T cells through CD28.
Keywords: Immune suppression, T cell activation, CD28, CTLA4
Introduction
Tumor cells employ various methods of immune suppression to overcome anti-tumor immunity. One such method is mediated by programmed death ligand-1 (PDL1). PDL1 is an inhibitory molecule of the B7 family which triggers apoptotic death or anergy upon binding programmed death-1 (PD1) on T cells. Activated human and mouse T cells upregulate surface PD1 to limit their effector functions in peripheral tissue as a mechanism to terminate an immune response and minimize damage of healthy surrounding tissue(1). Human and mouse cells such as T cells, B cells, dendritic cells, macrophages, vascular endothelial cells, pancreatic islet cells, and tumor cells may constitutively express PDL1, and PDL1 levels are up-regulated in response to IFNγ (reviewed in (2)).
PDL1 triggers immune suppression through numerous mechanisms including tolerization of T cells through the PD1 pathway and reverse signaling through CD80, and by promotion of T regulatory cells (3–7). PDL1 expression on many human and mouse tumors including non-small cell lung carcinoma, melanoma, renal carcinoma, and Hodgkin’s lymphoma has made PDL1 a major target for tumor immunotherapies (8–13). Therefore, many antagonist monoclonal antibodies to both PD1 and PDL1 have been developed and tested as monotherapies in clinical trials (14–16). Immune checkpoint antagonist monoclonal antibody therapies to CTLA4, PD1, and PDL1 have proven to be successful treatments in late stage cancers and in cancers that were once thought to be non-immunogenic. Several of these therapies are now FDA approved for cancer patients (16–23).
Nevertheless, 60% or more of patients do not respond to these therapies which only utilize one function: blocking a ligand/receptor interaction. In both mouse and human systems combination of checkpoint blockade antibodies with other treatments such as radiotherapy, stimulatory cytokines, vaccines, CpG ODN, or other checkpoint blockade antibodies have resulted in higher response rates (20, 24–30). We recently described a soluble form of CD80 (CD80-Fc) as a novel potential immunotherapeutic (31–34). We studied soluble CD80 as a potential immunotherapeutic based on its binding affinity for PDL1(35, 36), and proposed that soluble CD80 could simultaneously act as an antagonist and agonist by (i) binding to PDL1 and blocking PD1 signaling, and (ii) co-stimulating T cells through CD28. Our previous studies have shown that CD80-Fc maintains the activation of human and mouse T cells in vitro in the presence of suppressive human and mouse PDL1+ tumor cells, respectively. These studies further demonstrated that at the cellular level, soluble CD80 prevents PDL1-PD1 binding and suggested that CD80 also activates T cells through a CD28 dependent mechanism(32). While these in vitro results confirmed soluble CD80 as a potential immunotherapeutic, animal studies are needed to determine if the soluble form of CD80 has therapeutic efficacy in vivo. Likewise, additional studies are needed to clarify the mechanistic action of soluble CD80 and to determine if it acts by signaling through CD28. Given the known suppressive function of CD80 through CTLA4, studies are also needed to ascertain if CD80-Fc minimizes T cell function by suppressing through CTLA4.
Using CD80-Fc, we have now tested the ability of soluble CD80 to control progression of PDL1+ tumors in mice, and we report that in vivo treatment of established CT26 and B16F10 tumors with CD80-Fc delays tumor growth and promotes the influx of T cells into solid tumors. We clarify the mechanism by which soluble CD80 mediates its effects and report it stimulates downstream signaling components of the CD28 and T cell receptor pathways. We also demonstrate that CD80-Fc binds to CTLA4; however, the binding does not impact T cell function, suggesting that CTLA4 serves as a competitive inhibitor for CD80 and does not in itself have suppressive activity.
Materials and Methods
Cell Lines and mice
Human melanoma cell line C8161 and mouse C57BL/6-derived melanoma B16F10 and BALB/c-derived colon carcinoma CT26 cells were cultured as previously described (31, 37–39). Cell lines were authenticated by the original suppliers. C57BL/6 CD28-deficient, C57BL/6 PD1-deficient, and breeding stocks of BALB/c and C57BL/6 mice were purchased from The Jackson Laboratory. Mice were bred (BALB/c and C57BL/6) and maintained (all strains) in the UMBC animal facility and all animal procedures were approved by the UMBC Institutional Animal Care and Use Committee.
Tumor inoculation and tumor growth
Female 6 week old BALB/c or C57BL/6 mice were inoculated s.c. in the flank on day ~−7 with 5×105 CT26 colon carcinoma or 1×106 B16F10 melanoma cells, respectively. Intra-tumoral (i.t.) injections of mouse CD80-Fc (20μg/50μl/mouse, R&D Systems), rat IgG2a clone 2A3 (20μg/50μl/mouse, BioXCell), or CpG ODN 1555 (100μg/50μl/mouse) were started on day 0 when tumors were approximately 5mm in diameter and administered twice a week for three weeks (7). For some experiments mice were inoculated on ~day −7 with B16F10 cells on both flanks and tumors on only one flank were treated. Intraperitoneal (i.p.) injections of mouse CD80-Fc (200ug/mouse, R&D Systems), rat IgG2a clone 2A3 (200ug/mouse BioXCell), or anti-mouse PDL1 clone 10F.9G2 (200ug/mouse BioXCell) were administered on days 3, 6, 9, and 22 following tumor inoculation. Mice were sacrificed when they became moribund and their tumors were cryopreserved and analyzed by immunohistochemistry. Tumors were measured every 2–3 days in two perpendicular diameters. Tumor volume = 4/3πr3 where r = (diameter 1 + diameter 2)/4.
Reagents, antibodies and flow cytometry
Soluble CD80 (CD80-Fc) and TROY-Fc were from R&D Systems (Biotechne). Human CD3-APC (clone OKT3) and CD28-FITC (clone CD28.2) were from BioLegend (San Diego, CA). Cells were stained for cell-surface expression and subjected to flow cytometry as described, and analyzed using a Beckman Coulter Cyan ADP flow cytometer and Summit V4.3 software (40, 41).
For intracellular staining, cells were fixed with 2% paraformaldehyde and permeabilized with 0.2% saponin, and subsequently stained with fluorescent antibodies. For assessment of phosphorylated signal transduction factors, 2×105 cells/ml (RPMI, 10% FBS, 1% Glutamax, 5 × 10–5 M β-mercaptoethanol, and 1% pen-strep) were stimulated with anti-human CD28 agonist antibody (clone 28.2, BioLegend or BD, 2μg/ml), CD80-Fc (10μg/ml), TROY-Fc (10μg/ml), or mouse IgG1 isotype antibody (BD, 2μg/ml) for 30 minutes at 37oC, transferred to ice, fixed and permeabilized, and then stained with rabbit mAbs for phosphor-p44 MAPK + p42 MAPK pTyr204 (clone B.742.5; ThermoScientific, Rockford, IL), pNF-κB p65 (S536, clone 93H1, Cell Signaling Technologies, Danvers, MA), or rabbit IgG isotype control (ThermoFisher Scientific, 02–6102), followed by F(ab’)2 goat-anti-rabbit IgG-AF488 mAb (ThermoFisher; A-11070).
In vitro PBMC and T cell activation
Cryopreserved PBMC from healthy donors were provided by Dr. Dean Mann (UMB Medical School). Tumor cells (50 Gy-irradiated, 3×104) and PBMC (6×104) were co-cultured in triplicate in 96 well plates as described (32) with the following modifications. In some experiments, human CD80-Fc, human TROY-Fc, human IgG1 (R&D Systems), antagonist anti-CTLA4 mAb L3D10 (BioLegend), Ipilimumab (Bristol-Meyers Squibb), or CTLA4-Fc was added at 10μg/ml or the specified concentration. IFNγ was quantified by ELISA according to the manufacturer’s protocol (R&D Systems) as previously described (32). Percent T activation = [(IFNγExperimental)/(IFNγActivated PBMC + C8161 + CD80-Fc)] × 100 %. PBMC (1×107) were stimulated for 72 hours with PHA (5 μg/ml), treated for 30 minutes with 10 μg/ml of human CD80-Fc or human TROY-Fc, or 2 μg/ml agonist CD28 antibody clone 28.2 (BioLegend).
Cryopreservation and immunohistochemistry
Tumors were cryopreserved in OCT by freezing in a slurry of dry ice plus 2-methylbutane. Frozen tumors were stored at −80°C until they were sectioned, fixed in cold acetone, dried, and stained with H&E or hematoxylin and/or stained with anti-CD3 mAb (5μg/ml; BioLegend).
Western blots
2×106 Jurkat cells were left untreated or incubated at 37°C for 30 minutes with 20ug/ml CD80-Fc or anti-CD28 mAb. Cells were harvested, resuspended in M-PER Mammalian Protein Extraction Reagent with Halt Protease and Phosphatase Inhibitor Cocktail (ThermoFisher Scientific), and lysed in gentleMACS M tubes (Miltenyi Biotec) using program Protein_01according to the manufacturer’s recommendation. Protein concentration was assed via Bradford Assay, and protein was electrophoresed on 15% SDS-PAGE gels in SDS running buffer (BioRad) at 100 volts for 90 minutes, and transferred overnight in transfer buffer (BioRad) at 30 volts to PVDF membranes (GE Healthcare). Membranes were blocked with 5% milk in TBST. pNF-κB and pMAPK were detected with anti-pNF-KB antibody (clone 93H1; 1:1000 in 10ml of 2.5% milk/TBST) or anti-pMAPK antibody (clone D13.14.4E; 1:2000 in 10ml of 2.5% milk/TBST) (both from Cell Signaling) followed by goat-anti-rabbit-HRP (Biolegend; 1:10000 in 10ml of 2.5% milk/TBST). Beta-actin was detected with anti-β-actin antibody (Sigma-Aldrich; clone AC-15, ascites fluid; 1:10000 in 10ml of 2.5% milk/TBST) followed by sheep-anti-mouse-HRP (GE Healthcare Life Sciences). Protein was visualized using an HRP detection kit (Denville Scientific, Inc).
qRT-PCR
CD3+ T cells from the blood of healthy human donors were isolated via negative selection using a Miltenyi Human T cell Isolation Kit according to the manufacturer’s protocol and were >85% CD3+ T cells. 1×107 cells were incubated for 72 hours with 5 μg/ml PHA followed by a 2 hour treatment with LEAF purified mouse IgG1K; isotype control antibody (2 μ/ml BioLegend), agonist CD28 mAb clone CD28.2 (2 μg/ml), soluble TROY-Fc (10 μg/ml, R&D Systems), or CD80-Fc (10 μg/ml, R&D Systems). RNA was isolated from T cells according to the manufacturer’s protocol and quantified using a BioTek Synergy 2 microplate reader. cDNA was created from each RNA sample using the Maxima cDNA kit (Thermo Scientific) per the manufacturer’s protocol. Specific transcripts were amplified using the KiCqStart SYBR Green qPCR ReadyMix, iQ (Sigma Life Science) following the manufacturer’s protocol and detected using a CFX96 Real-Time PCR Detection System (Bio-Rad). Each sample was analyzed in triplicate and each primer set was tested by a melting curve. Relative expression of each experimental transcript was normalized to β-actin. Primers used were EGR1
forward: ACCGCAGAGTCTTTTCCTGA; EGR1
reverse: GTGGGTTGGTCATGCTCACT; EGR2 forward:
ACGGCTTTTCTGACACTCCA; EGR2 reverse:
AATGTTGATCATGCCATCTCCG, EGR3 forward:
GGTGACCATGAGCAGTTTGC; EGR3 reverse:
ACCGATGTCCATTACATTCTCTGT; EGR4 forward: TAGCGAGTTTTCCGAACCCG; EGR4
reverse: TCAAGAAGTCGCCTGCTCC; β actin forward: CCTCGCCTTTGCCGATCC; β actin
reverse: AATCCTTCTGACCCATGCCC.
Statistical analysis
Statistical analysis of tumor growth rates was conducted using the compare Growth Curves function of the Statmod software package (http://bioinf.wehi.edu.au/software/compareCurves/). Statistical analysis of Kaplan-Meier graphs was conducted using the Log Rank Test function of the Statmod software package (http://bioinf.wehi.edu.au/software/russell/logrank/). Student’s t test was utilized to determine statistical significance between two sets of data using Microsoft Excel Version 2010. p values <0.05 were considered statistically significant.
Results
CD80-Fc slows tumor growth and extends survival of tumor-bearing mice
We previously reported that in vitro CD80-Fc potentially acts simultaneously as an antagonist and agonist by binding to PDL1 to block PD1 signaling and by co-stimulating T cells through CD28, respectively (31–33). To ascertain if CD80-Fc is therapeutic in vivo, BALB/c and C57BL/6 mice were injected with CT26 colon carcinoma cells and B16F10 melanoma cells, respectively. Tumors were allowed to grow to approximately 4.5mm in diameter. Studies in mice showed that intratumoral (i.t.) drug delivery requires less drug than systemic delivery (42), and i.t. injections are now being used clinically (43, 44). Therefore, when tumors were established, the mice were given i.t. injections of CD80-Fc or control IgG antibody twice a week for three weeks and monitored for tumor progression (Fig. 1A and 1B). Day 0 on the graphs is the day therapy was started. CD80-Fc treatment slowed tumor growth in both CT26 (p = 0.029) and B16F10 (p = 0.045) tumor bearing mice in comparison to control IgG treatment.
Figure 1. Monotherapy with CD80-Fc delays tumor growth in mice with CT26 tumors.
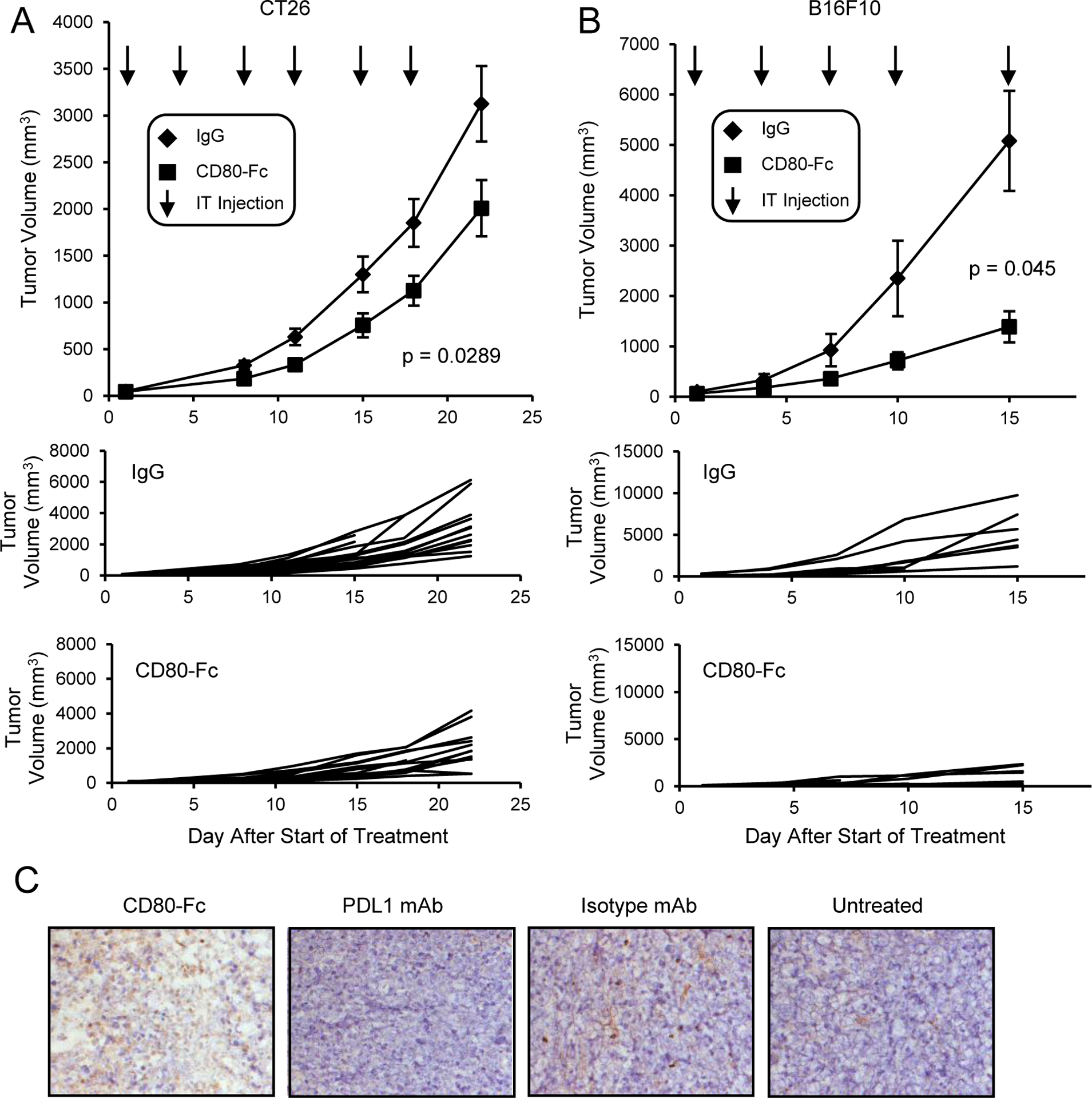
A, BALB/c or B, C57BL/6 mice were inoculated s.c. on day 0 with 1×105 CT26 colon carcinoma or B16F10 melanoma cells. When the average tumor diameter was 4.5mm, mice were started on twice weekly i.t. injections of 20μg IgG or CD80-Fc/mouse. I.t. injections were continued for three weeks. Graphs show average tumor volumes ± SD, survival time, and tumor growth in individual mice. Day 0 is the first day of therapy. n= 14 (CD80-Fc) or 15 (IgG) mice/group and 8 (CD80-Fc and IgG) mice/group for A and B, respectively, and are pooled from multiple experiments. Statistical analyses for A and B were performed using the compare Growth Curves function of the Statmod software package. C, BALB/c mice were inoculated s.c. on day 0 with 5×105 CT26 colon carcinoma cells. I.p. injections of 200μg of mouse CD80-Fc, rat IgG2a, or anti-mouse PDL1 were administered on days 3, 6, 9, and 22 following tumor inoculation. Surviving mice were sacrificed on day 42. Tumors were cryopreserved and analyzed by immunohistochemistry. Images are representative of 5 mice/group.
CD80-Fc therapy has also been administered intraperitoneally (i.p.) since this route has routinely been used in many mouse studies. BALB/c mice were inoculated s.c. with 5×105 CT26 cells and either untreated or treated with 200μg/mouse CD80-Fc, anti-PDL1 mAb or isotype mAb (rat IgG2a) on days 3, 6, 9, and 22 following tumor inoculation (Fig. 1C). At day 42 after tumor inoculation, 100% of the CD80-Fc-treated mice were still alive, while only 65% of the PDL1 mAb-treated, 50% of the untreated, and none of the isotype mAb-treated mice survived. To determine if CD80-Fc promotes an influx of TIL, tumors were harvested at the time of euthanization according to IACUC guidelines (day 42 for all groups except isotype-treated mice which were euthanized on day 39), and frozen sections were stained for CD3+ cells. CD80-Fc therapy was more effective in increasing TIL in comparison to the IgG control, untreated, and PDL1 mAb-treated mice.
Treatment of both mice and cancer patients with a combination of immune monotherapies has in some cases produced greater reduction in tumor growth than either monotherapy alone (reviewed in (45, 46)). To determine if CD80-Fc has the potential to amplify the effect of another immune-based therapeutic, we combined i.t. CD80-Fc therapy with CpG ODN 1555 therapy. CpG was selected for the combination therapy for several reasons. It facilitates both adaptive and innate immunity by enhancing the activation of professional APC (dendritic cells) through the engagement of TLR9 and it reprograms myeloid-derived suppressor cells (MDSC) to become non-suppressive macrophages and thus eliminates another potent immune suppressive mechanism in the tumor microenvironment (7). However, CpG induces PDL1 expression on human B cells (47), a drawback that should be neutralized by soluble CD80.
BALB/c mice were inoculated on day 1 with 5×105 CT26 tumor cells. On day 14 when tumors were an average of 6.5 ± 1.1 mm in diameter, mice were started on twice weekly i.t. injections of either control IgG, CpG, or CD80-Fc + CpG (Fig. 2). Monotherapy with CpG alone slowed primary tumor growth; however, combination therapy of CpG plus CD80-Fc was more effective and significantly delayed tumor progression.
Figure 2. Combination i.t. therapy with CD80-Fc plus CpG is more effective than CpG monotherapy.
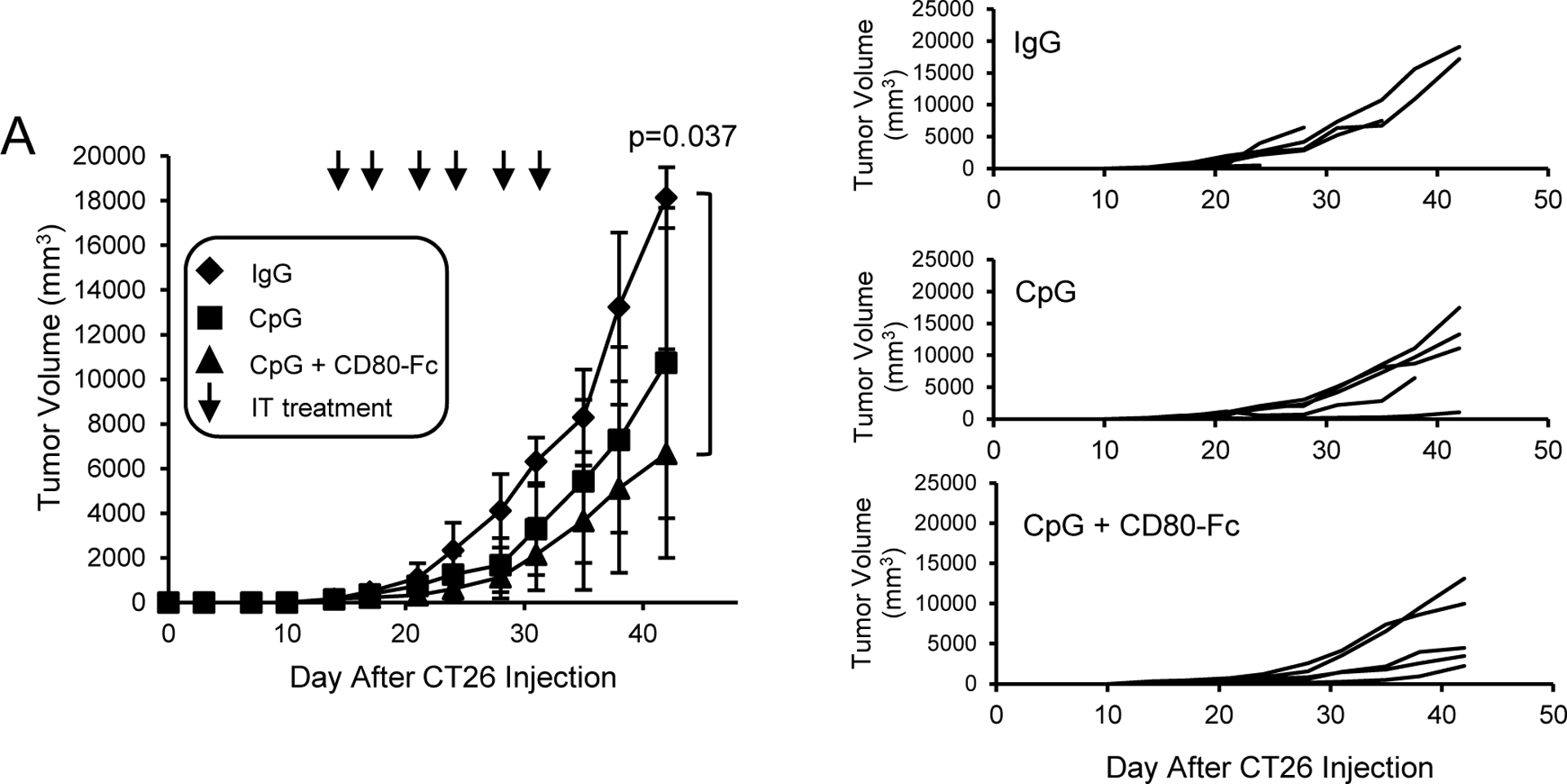
A, BALB/c mice were inoculated s.c. on day 0 with 1×105 CT26 colon carcinoma cells. On day 14 when the average tumor diameters were 6.4 – 6.7 mm, the mice were started on twice weekly i.t. injections of 20μg IgG, 200 μg CpG/mouse, or 200 μg CpG + 20μg CD80-Fc/mouse. I.t. injections were continued for three weeks. Graphs show average tumor volumes ± SD, percent surviving mice, and tumor growth in individual mice. Day 0 is the day of tumor inoculation. n= 5 mice/group. Statistical analysis was performed using pairwise comparison test.
Collectively, the in vivo studies demonstrate that CD80-Fc delays tumor progression of established tumors, enhances the therapeutic efficacy of other immune monotherapies, and promotes TIL.
CD80-Fc activates the CD28 pathway and downstream targets of the TCR
CD80 is an established costimulatory molecule and second signal for T cell activation(48). Our previous studies using wild type and CD28-deficient T cells demonstrated that CD80-Fc binds to CD28 and suggested that in addition to preventing PD1 suppression, CD80-Fc costimulates through CD28. If CD80-Fc uses the CD28 pathway, then CD80-Fc binding to CD28 will activate EGR1–4 downstream components of the CD28 signaling pathway (49). To test this hypothesis, T cells were purified from PBMC of healthy human donors, treated with PHA for 72 hours, and then treated with either a CD28 agonist antibody, CD80-Fc, or the irrelevant recombinant protein TROY-Fc. RNA was then isolated and screened by qRT-PCR (Fig. 3A). mRNA levels in the CD80-Fc-treated cells were upregulated approximately 3 fold for EGR1, EGR2, EGR3, and EGR4 in comparison to the TROY-Fc-treated T cells (p = 0.032, 0.041, 0.029, 0.035 respectively). T cells treated with the agonist CD28 antibody had upregulated levels of EGR1, EGR2, EGR3, and EGR4 similar to that of CD80-Fc-treated cells. EGR1–4 were similarly activated when human Jurkat T lymphoma cells were used (Supplementary Fig. S1).
Figure 3. CD80-Fc activates transcription factors downstream of CD28 and the TcR.
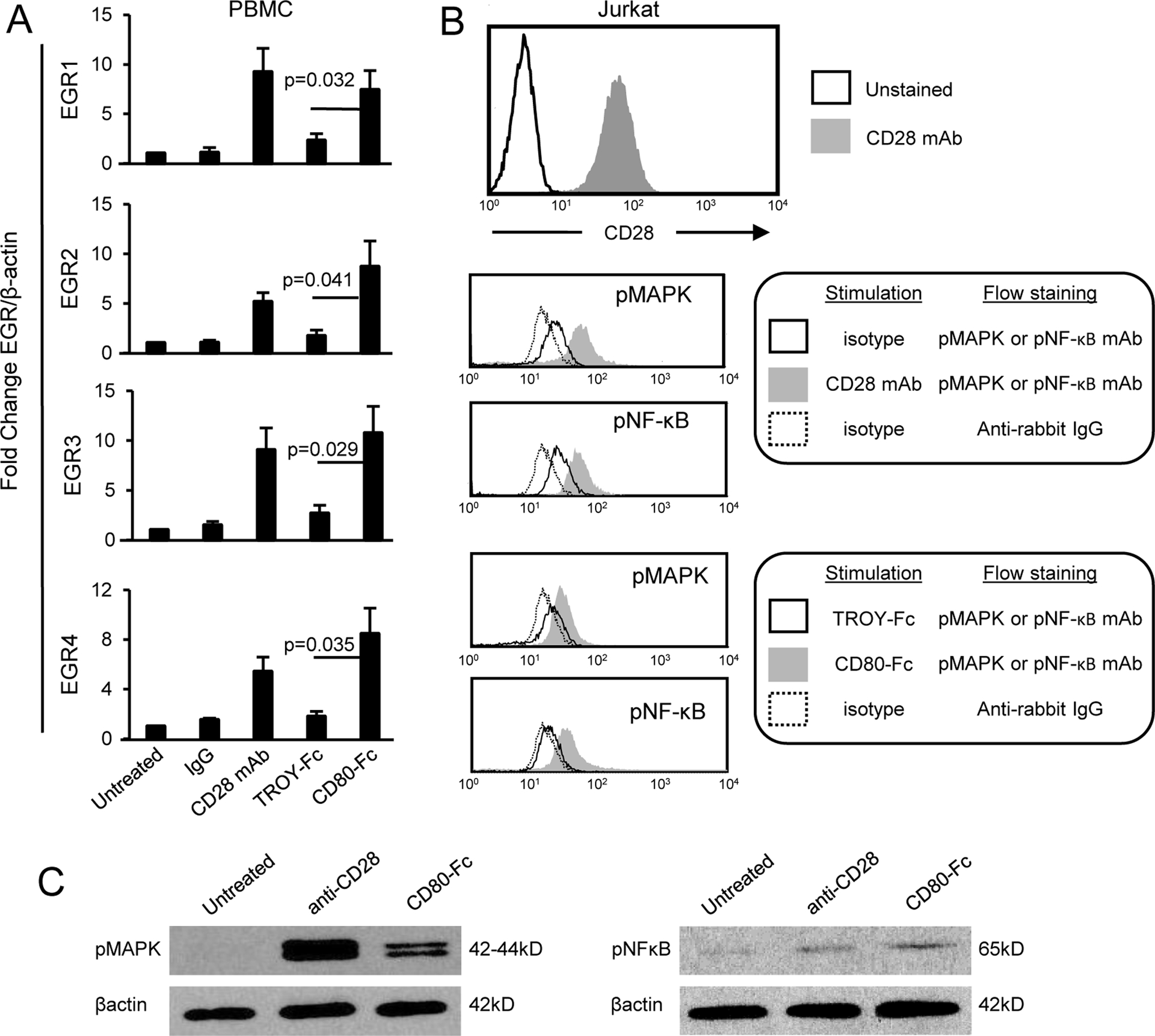
A, CD80-Fc induces transcription of EGR1/2/3/4. CD3+ cells were isolated by negative selection from the blood of healthy human donors, activated with PHA for 72 hours, and then either untreated or incubated for two hours with isotype control mAb, agonist CD28 mAb, TROY-Fc, or CD80-Fc. RNA was then isolated, converted into cDNA, and the DNA analyzed by qRT-PCR for EGR1/2/3/4. Values were normalized to β-actin expression. Results are shown as fold change in expression level compared to untreated T cells. Statistical analysis was performed using Student’s t test. Data are from one of three independent experiments. B, CD3+CD28+ Jurkat cells were treated for 30 min at 37OC with isotype mAb, agonist CD28 mAb, TROY-Fc, or CD80-Fc. Cells were then fixed and permeabilized, and stained with mAbs to pMAPK or pNF-κB followed by anti-rabbit IgG-FITC. C, Jurkat cells were untreated or treated for 30 min with agonist anti-CD28 mAb and then lysed and analyzed by western blot for phosphorylated MAPK and NFκB. Data for B and C are representative of one of two and one of three independent experiments, respectively.
MAPK and NF-κB are established downstream signal transducers of the TCR and CD28 pathways (50–52). To confirm that CD80-Fc activates T cells through these pathways, CD3+CD28+Jurkat cells were stimulated for 30 min at 37°C with agonist anti-CD28 mAb, TROY-Fc, or CD80-Fc. The resulting cells were then fixed and permeabilized and stained with mAbs to CD3, pMAPK, and pNF-κB, and the CD3+ cells were gated and analyzed (Fig. 3B). To confirm the flow cytometry findings, Jurkat cells were analyzed by Western blotting for pMAPK and pNF-κB following stimulation with agonist anti-CD28 mAb or CD80-Fc (Fig. 3C). CD3+ cells treated with either agonist CD28 or CD80-Fc contained elevated levels of phosphorylated MAPK and NF-κB. These results demonstrate that CD80-Fc activates transcription factors downstream of CD28 and the TCR.
CD80-Fc does not affect T cell activation by interacting with CTLA4.
CD80 binding to CTLA4 drives T cell tolerance (53). Our cellular studies indicate that CD80-Fc sustains T cell activation; however, there is the potential for CD80-Fc to decrease T cell function by interacting with CTLA4 on activated T cells. If CD80-Fc facilitates T cell suppression through CTLA4, then antibody blocking of CTLA4 will release the suppression and increase T cell activation. To test this hypothesis, PBMC from healthy human donors were activated with PHA and co-cultured for three days with PDL1+ human melanoma cells (C8161) in the presence or absence of CD80-Fc, a CTLA4 antagonist mAb, TROY-Fc, or irrelevant antibody. T cell activation was assessed by IFNγ production. PHA activated T cells express CTLA4 (Fig. 4A) and blocking CTLA4 with either Ipilimumab (Ipi) or L3D10 anti-CTLA4 mAb did not increase T cell activation (Fig. 4B). These results indicate that CD80-Fc does not suppress T cell function through CTLA4.
Figure 4. CD80-Fc does not affect T cell activation by interacting with CTLA4.
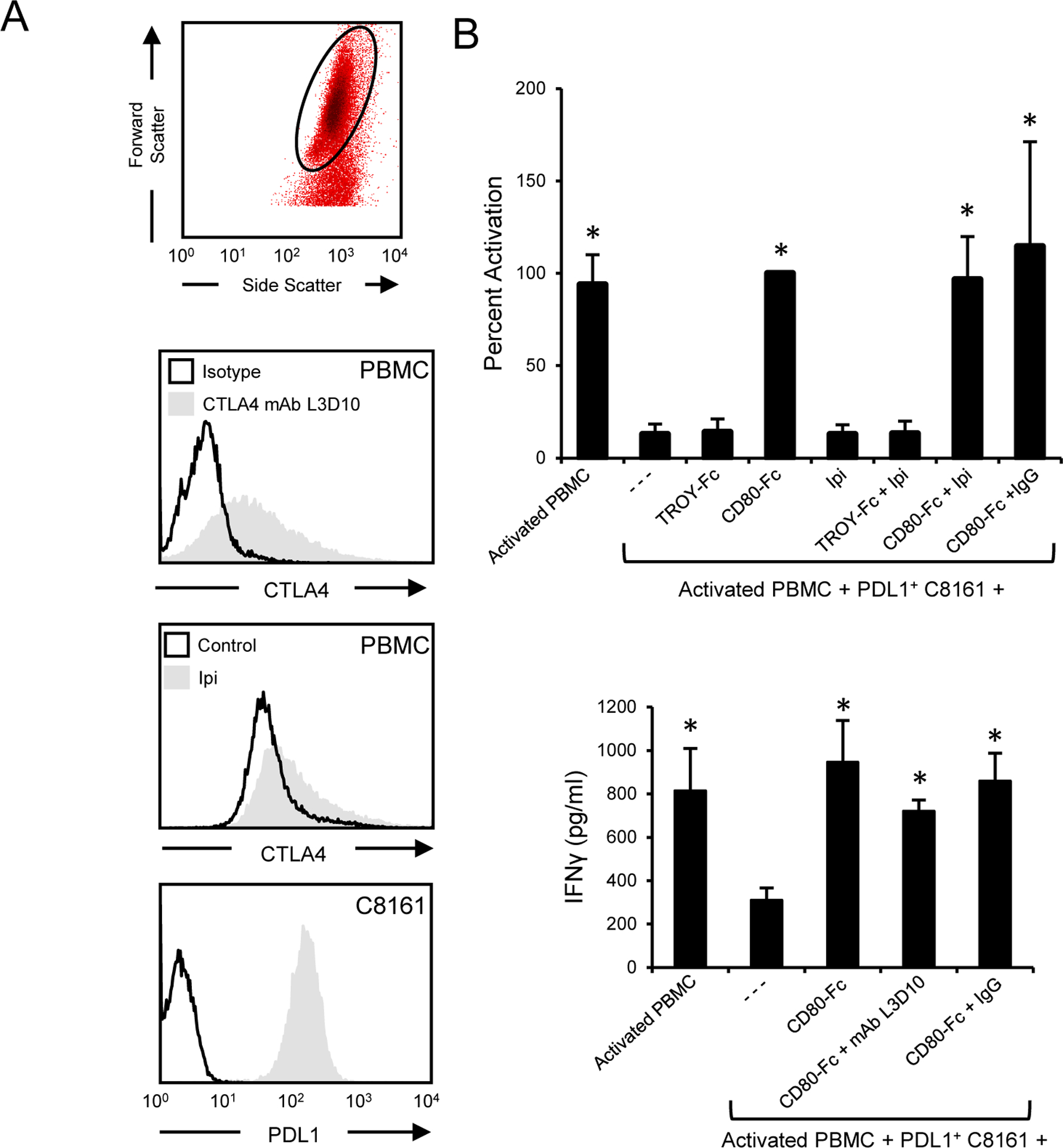
A, PBMC from a healthy human donor were activated with PHA for 72 hours and subsequently stained with antagonist anti-CTLA4 mAb L3D10 or Ipilimumab. Human melanoma C8161 cells were stained for PDL1 expression (29E.2A3 mAb). B, PHA-treated CTLA4+ T cells were co-cultured with PDL1+ C8161 human melanoma cells in the presence or absence of CD80-Fc, Ipilimumab, control recombinant protein TROY-Fc, antagonist CTLA4 mAb L3D10, or irrelevant isotype antibody. After three days of culture T cell activation was assessed by measuring IFNγ in the supernatants. Top panel shows one of three independent experiments; bottom panel shows the average of three independent experiments. Error bars indicate SD. Bars with * are statistically different from all other values and are not statistically different from each other at p < 0.05.
CD80-Fc prevents CTLA4 suppression by saturating CTLA4
Since the inhibitory receptor CTLA4 is an established receptor for CD80, it was surprising that blocking CTLA4 when CD80 was present did not increase T cell activation. Early studies suggested that CTLA4-CD80 interactions mediated T cell suppression by CTLA4 signaling into the T cell (53, 54). However, downstream signaling molecules have not been identified for CTLA4, and more recent publications speculate that CTLA4 limits T cell function because it is a decoy receptor for CD80 (reviewed in (55)). If CTLA4 is a decoy receptor, then CTLA4 will bind CD80-Fc and prevent CD80-Fc from binding to PDL1 and CD28. To test this hypothesis, we first determined the minimum dose of CD80-Fc that maintains T cell activation in the presence of PDL1+ tumor cells (Supplementary Fig. S2). We then co-cultured PHA-activated human PBMC for three days with PDL1+ human C8161 melanoma cells in the presence of 2μg/ml CD80-Fc. Increasing quantities of CTLA4-Fc or irrelevant control recombinant protein TROY-Fc were included in the cultures to determine if CTLA4 competed with CD80-Fc and suppressed T cell production of IFNγ (Fig. 5). Higher concentrations of CTLA4-Fc, but not irrelevant TROY-Fc, prevented CD80-Fc from restoring T cell activation in the presence of PDL1+ tumor cells, consistent with the concept that CTLA4 suppresses T cell function by binding CD80 and preventing it from activating T cells through CD28.
Figure 5. CD80-Fc prevents CTLA4-mediated suppression by saturating CTLA4.
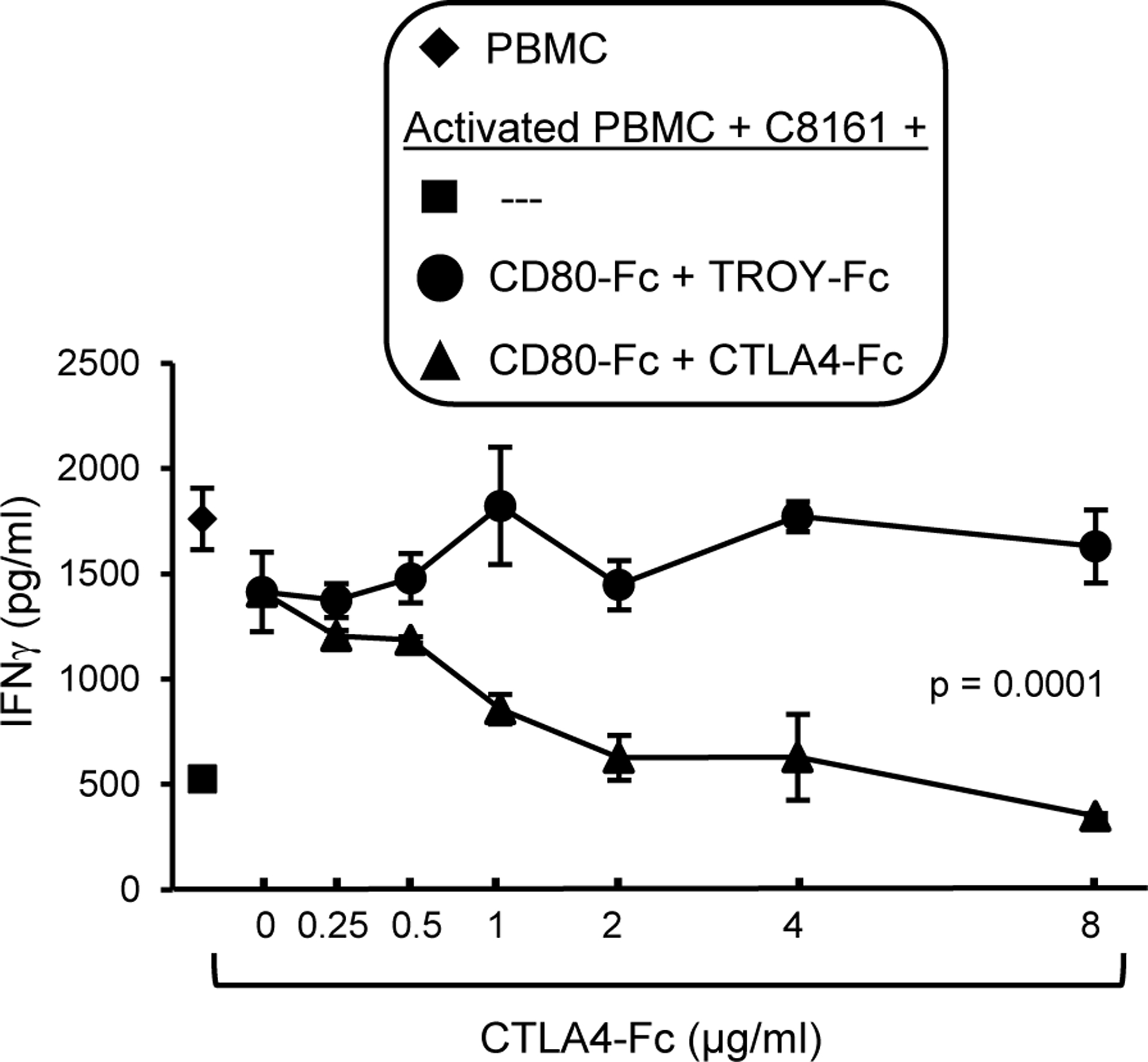
PHA-activated human PBMC, PDL1+ C8161 cells, and CD80-Fc (2 μg/ml) were incubated with increasing amounts of soluble CTLA4-Fc. or control irrelevant protein TROY-Fc. T cell activation was assessed by measuring IFNγ in the supernatants. Statistical analyses were performed using Student’s t test. Data are from one of two independent experiments.
In vivo therapeutic effect of CD80-Fc requires neutralization of PD1 suppression and activation through CD28
Our in vitro studies indicate that CD80-Fc functions by simultaneously blocking PD1 suppression while activating through CD28. To determine if these functions occur in vivo, C57BL/6 wild type, CD28−/−, and PD1−/− mice were inoculated s.c. with B16F10 tumor cells and treated with either CD80-Fc or PBS starting when tumors were ~5mm in diameter. We reasoned that if CD28 activation was essential for a therapeutic effect, then CD80-Fc therapy would not benefit CD28−/− mice. However, if neutralizing PD1 suppression while activating through CD28 was essential, then the therapeutic effect of CD80-Fc would be the same in PD1+/+ and PD1−/− mice. To control for the impact of genetic background on tumor progression, the mice were also injected in their contralateral flank on day 0 with tumor cells, and these tumors were not treated. Treated tumors progressed significantly more rapidly in PBS-treated wild type and CD80-Fc-treated CD28−/− mice as compared to CD80-Fc-treated wild type and CD80-Fc-treated PD1−/− mice (Fig. 6A), demonstrating that the CD80-Fc therapeutic effect involves both PD1 and CD28. Progression of the contralateral untreated tumors in the CD28−/− and PD1−/− mice was not significantly different from progression of the injected tumors, further demonstrating that CD80-Fc had no effect unless CD28 was present and the PD1 pathway was inhibited (Fig. 6B).
Figure 6. The in vivo therapeutic effect of CD80-Fc involves neutralization of PD1 suppression and activation through CD28.
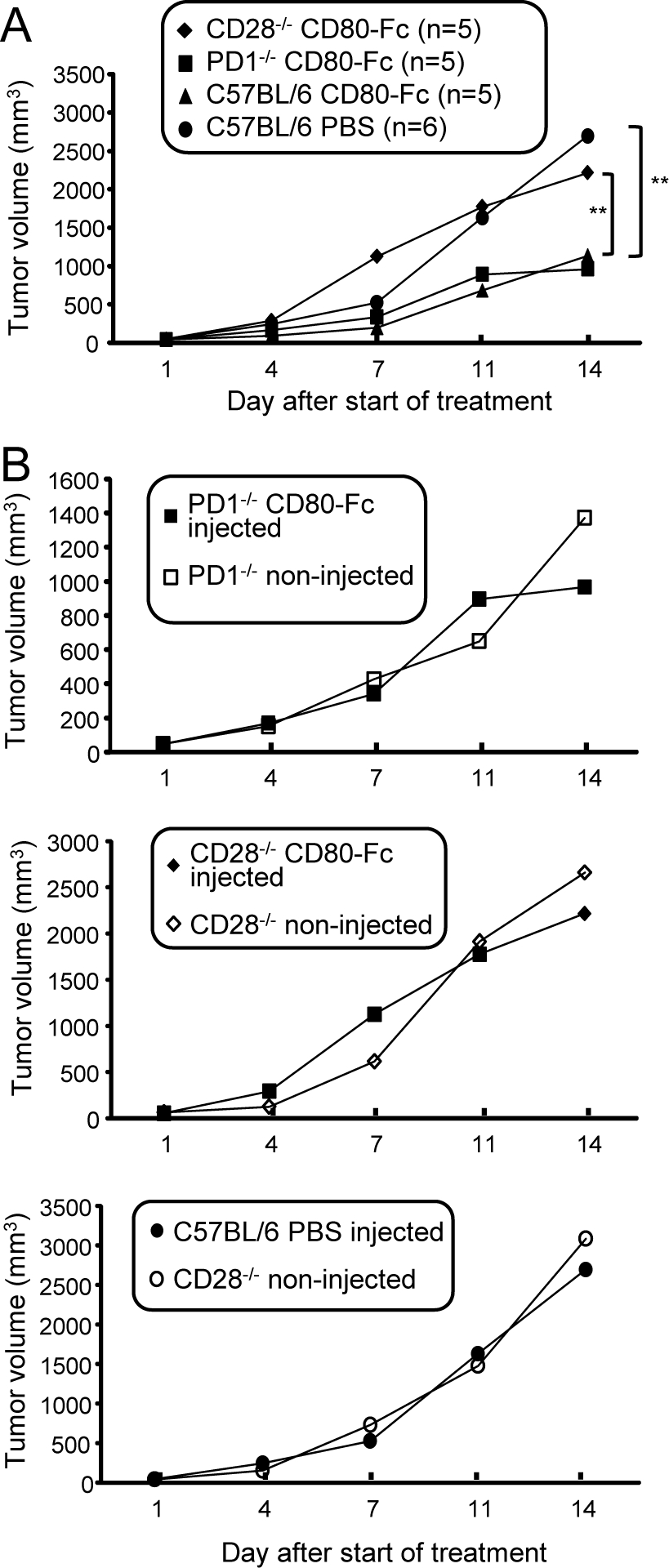
C57BL/6 wild type, CD28-deficient, and PD1-deficient mice were inoculated with B16F10 cells in both flanks. When tumors were ~5 mm in diameter (~day 7), tumors on one flank were treated with CD80-Fc (wild type, CD28−/−, and LPD1−/− mice) or PBS (wild type mice). Mice were followed for tumor progression. A, Progression of treated tumors. B, Comparison of progression of treated and contralateral untreated tumors.
Discussion
This present study demonstrates that CD80-Fc enhances T cell activation in the tumor site and extends survival time either as a monotherapy or in conjunction with another immunotherapy, and that the effects occur in mice with relatively large, established tumors. These in vivo results complement earlier in vitro studies showing that soluble CD80 activates tumor-reactive T cells and is a potential therapeutic for facilitating anti-tumor immunity (31–33). The present studies also reveal that soluble CD80 functions by activating the CD28 and TcR signal transduction pathways. A concern of our earlier studies was that soluble CD80 might limit T cell-mediated anti-tumor activity by interacting with CTLA4. However, the current studies demonstrate that CTLA4 does not impact the therapeutic effect of soluble CD80.
Until very recently it was unclear how PD1 anergized T cells and inhibited T cell function. However, elegant studies using a biochemical reconstitution system and intact cells have demonstrated that PDL1 signaling through PD1 results in the dephosphorylation of CD28 by PD1-recruited Shp2 phosphatase (56). Therefore, PD1 suppresses T cell function by inhibiting costimulation through CD28. Our initial hypothesis that CD80-Fc acts by inhibiting PD1 signaling while concurrently activating through CD28 is therefore not only supported by the studies presented here, but is also consistent with this newly identified mechanism explaining PD1-mediated suppression.
The literature does not conclusively identify the mechanism by which CTLA4 suppresses T cells. Several mechanisms have been proposed including (i) CTLA4 initiates a negative signal transduction pathway; (ii) CTLA4 increases T cell motility that limits antigen presenting cell/T cell interactions; or (iii) CTLA4 is a decoy receptor that sequesters CD80 and CD86 away from CD28 (reviewed in (55)). There are no known signaling molecules activated by CTLA4, so we have not explored if soluble CD80 activates a CTLA4 negative signal transduction pathway. However, we have
demonstrated that CTLA4 can suppress T cells by competing for CD80-Fc and preventing it from binding to PDL1 and CD28. Therefore, therapy with soluble CD80 will depend on the saturation of CTLA4 at both the tumor site and the tumor draining lymph node, and excess soluble CD80 will be necessary to allow costimulation through CD28 and neutralization of PDL1. Saturating quantities of soluble CD80 may have the added benefit of preventing CTLA4-mediated suppression and thereby keeping exhausted T cells activated.
Therapeutic antibodies and recombinant proteins have been delivered via multiple routes including intravenous (i.v.), i.p., and i.t. Antibody accumulation within solid tumors does not differ between the i.v. and i.p delivery methods (57). Although i.v. and i.p. delivery have been the traditional routes clinically and in mice, respectively, i.t. delivery is increasingly being used in both species. Intratumoral injection has been utilized for the delivery of bacteria, oncolytic viruses, immunoagonists, STING agonist, cytokines, activated immune cells, and antibodies in mice (reviewed in(42)). Additionally, i.t. injection of ipilimumab in combination with radiotherapy (44) and i.t. injection of an IL-12 plasmid in combination with pembrolizumab (43) are being tested in clinical trials. Studies using i.t. injections of anti-CTLA4 mAbs in mice have resulted in systemic anti-tumor immune responses without the serious adverse side effects of a systemic injection (58, 59). Other studies have shown that some immunotherapeutic drugs only elicited a therapeutic response when delivered i.t. and not i.v. (60). Intratumoral injection makes it likely that the drug arrives at the tumor site in high concentrations where it can immediately bind to PDL1 and costimulate tumor-specific T cells. Intratumoral injection may also be beneficial because it decreases the potential for off target side effects.
A 2006 clinical trial of the superagonist anti-CD28 mAb TGN1412 rendered six healthy recipients critically ill with a systemic inflammatory response and cytokine release syndrome (“cytokine storm”) (61). CD28 superagonist antibodies fully activate naïve T cells in the absence of TcR ligation and downstream phosphorylation of ZAP70 and the TcRζ. Their effects are due to their binding to the exposed C”D loop of the CD28 Ig-like domain, in contrast to conventional anti-CD28 antibodies that bind to a region close to the binding site for CD80 and CD86 (62). In addition, superagonist anti-CD28 mAbs induce T regulatory cells (63). In contrast to superagonist antibodies, soluble CD80 binds to the natural binding site of CD28 and has a binding constant five orders of magnitude lower than an antibody. Therefore, it is unlikely that soluble CD80 will have the same adverse effects as TGN1412.
The mouse studies of Fig. 1C indicate that CD80-Fc therapy is more effective than monotherapy with PDL1 antibody at promoting TIL. The specific PDL1 antibody used in these experiments was selected because it is known to have therapeutic efficacy in mice. Since the binding constants for mAbs are much higher than the binding constant for CD80 to PDL1 (10−6–10−9 for antibodies vs. ~1.4 μM for CD80, (35)), the difference in function is not due to tighter binding. Several mechanisms could be involved: (i) The ability to costimulate through CD28 allows soluble CD80 to deliver additional activating signals to T cells while concurrently preventing T cell anergy through PD1; (ii) Unlike antibody to PDL1 which only prevents suppression via PDL1, soluble CD80 may also prevent tolerance by reverse signaling of PDL1 through T cell-expressed CD80; (iii) Since PD1 is predominantly expressed on activated T cells, antibody therapy will likely only be effective in individuals who already have activated T cells. In contrast, soluble CD80 may also facilitate the activation of naïve tumor-specific T cells that are in the tumor microenvironment.
Immune cells including activated T cells, dendritic cells, macrophages, and B cells, as well as non-immune endothelial cells are not constitutively PDL1+, but have the potential to express PDL1 when exposed to IFNγ and/or inflammation (64, 65). Tumor microenvironments can be inflamed and may contain activated T cells producing IFNγ, so soluble CD80 may protect against PDL1 expression on these cells as well. There is the potential that CD80 could activate autoreactive T cells against these non-malignant PDL1+ cells. However, this possibility is unlikely since it would require the presence of T cells with an autoreactive TcR in an IFNγ-rich local environment.
Collectively, these findings identify soluble CD80 as a therapeutic agent for mice with large, established tumors, and suggest it may have potential for the treatment of cancer patients.
Supplementary Material
Acknowledgments
The authors thank Dr. Dean Mann (University of Maryland School of Medicine) for human PBMC and Dr. Dennis Klinman (NIH, National Cancer Institute) for CpG ODN1555.
Financial Support: NIH RO1GM021248, RO1CA84232, TEDCO MII
Footnotes
Conflict of Interest statement: The authors declare no conflicts of interest
References
- 1.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.He J, Hu Y, Hu M, Li B. Development of PD-1/PD-L1 Pathway in Tumor Immune Microenvironment and Treatment for Non-Small Cell Lung Cancer. Scientific reports 2015;5:13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azuma T, Yao S, Zhu G, Flies AS, Flies SJ, Chen L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood 2008;111:3635–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nature Med 2002;8:793–800. [DOI] [PubMed] [Google Scholar]
- 5.Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med 2009;206:3015–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, et al. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci USA 2004;101:10691–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shirota Y, Shirota H, Klinman DM. Intratumoral injection of CpG oligonucleotides induces the differentiation and reduces the immunosuppressive activity of myeloid-derived suppressor cells. J Immunol 2012;188:1592–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 2010;28:3167–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen BJ, Chapuy B, Ouyang J, Sun HH, Roemer MG, Xu ML, et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin Canc Res 2013;19:3462–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest 2015;125:3384–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamanishi J, Mandai M, Matsumura N, Abiko K, Baba T, Konishi I. PD-1/PD-L1 blockade in cancer treatment: perspectives and issues. Int J Clin Oncol 2016;21:462–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thompson RH, Gillett MD, Cheville JC, Lohse CM, Dong H, Webster WS, et al. Costimulatory B7-H1 in renal cell carcinoma patients: Indicator of tumor aggressiveness and potential therapeutic target. Proc Natl Acad Sci USA 2004;101:17174–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Velcheti V, Schalper KA, Carvajal DE, Anagnostou VK, Syrigos KN, Sznol M, et al. Programmed death ligand-1 expression in non-small cell lung cancer. Lab Invest 2014;94:107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gettinger SN, Horn L, Gandhi L, Spigel DR, Antonia SJ, Rizvi NA, et al. Overall Survival and Long-Term Safety of Nivolumab (Anti-Programmed Death 1 Antibody, BMS-936558, ONO-4538) in Patients With Previously Treated Advanced Non-Small-Cell Lung Cancer. J Clin Oncol 2015;33:2004–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013;369:134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolchok JD, Hodi FS, Weber JS, Allison JP, Urba WJ, Robert C, et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci 2013;1291:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Antonia S, Goldberg SB, Balmanoukian A, Chaft JE, Sanborn RE, Gupta A, et al. Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: a multicentre, phase 1b study. Lancet Oncol 2016;17:299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 2015;373:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 2016;387:1540–50. [DOI] [PubMed] [Google Scholar]
- 23.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 2015;372:2018–28. [DOI] [PubMed] [Google Scholar]
- 24.Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest 2014;124:687–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.West EE, Jin HT, Rasheed AU, Penaloza-Macmaster P, Ha SJ, Tan WG, et al. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J Clin Invest 2013;123:2604–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, Chai Y, et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J immunother 2015;38:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mangsbo SM, Sandin LC, Anger K, Korman AJ, Loskog A, Totterman TH. Enhanced tumor eradication by combining CTLA-4 or PD-1 blockade with CpG therapy. J Immunother 2010;33:225–35. [DOI] [PubMed] [Google Scholar]
- 28.Spain L, Larkin J. Combination immune checkpoint blockade with ipilimumab and nivolumab in the management of advanced melanoma. Exp Opin Biol Ther 2016;16:389–96. [DOI] [PubMed] [Google Scholar]
- 29.Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med 2013;368:1365–6. [DOI] [PubMed] [Google Scholar]
- 30.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haile ST, Bosch JJ, Agu NI, Zeender AM, Somasundaram P, Srivastava MK, et al. Tumor cell programmed death ligand 1-mediated T cell suppression is overcome by coexpression of CD80. J Immunol 2011;186:6822–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haile ST, Dalal SP, Clements V, Tamada K, Ostrand-Rosenberg S. Soluble CD80 restores T cell activation and overcomes tumor cell programmed death ligand 1-mediated immune suppression. J Immunol 2013;191:2829–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haile ST, Horn LA, Ostrand-Rosenberg S. A soluble form of CD80 enhances antitumor immunity by neutralizing programmed death ligand-1 and simultaneously providing costimulation. Cancer Immunol Res 2014;2:610–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ostrand-Rosenberg S, Horn LA, Alvarez JA. Novel strategies for inhibiting PD-1 pathway-mediated immune suppression while simultaneously delivering activating signals to tumor-reactive T cells. Cancer Immunol Immunother 2015;64:1287–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7–1 costimulatory molecule to inhibit T cell responses. Immunity 2007;27:111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Butte MJ, Pena-Cruz V, Kim MJ, Freeman GJ, Sharpe AH. Interaction of human PD-L1 and B7–1. Molec Immunol 2008;45:3567–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fidler IJ, Hart IR. Biological diversity in metastatic neoplasms: origins and implications. Science 1982;217:998–1003. [DOI] [PubMed] [Google Scholar]
- 38.Pulaski BA, Ostrand-Rosenberg S. Reduction of established spontaneous mammary carcinoma metastases following immunotherapy with major histocompatibility complex class II and B7.1 cell-based tumor vaccines. Cancer Res 1998;58:1486–93. [PubMed] [Google Scholar]
- 39.Srivastava MK, Bosch JJ, Wilson AL, Edelman MJ, Ostrand-Rosenberg S. MHC II lung cancer vaccines prime and boost tumor-specific CD4+ T cells that cross-react with multiple histologic subtypes of nonsmall cell lung cancer cells. Int J Cancer 2010;127:2612–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dissanayake SK, Thompson JA, Bosch JJ, Clements VK, Chen PW, Ksander BR, et al. Activation of tumor-specific CD4(+) T lymphocytes by major histocompatibility complex class II tumor cell vaccines: a novel cell-based immunotherapy. Cancer Res 2004;64:1867–74. [DOI] [PubMed] [Google Scholar]
- 41.Thompson JA, Srivastava MK, Bosch JJ, Clements VK, Ksander BR, Ostrand-Rosenberg S. The absence of invariant chain in MHC II cancer vaccines enhances the activation of tumor-reactive type 1 CD4+ T lymphocytes. Cancer Immunol Immunother 2008;57:389–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh M, Overwijk WW. Intratumoral immunotherapy for melanoma. Cancer Immunol Immunther 2015;64:911–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.https://clinicaltrials.gov/ct2/show/NCT02493361. Trial of pIL-12/MK-3475 in Metastatic Melanoma. 2017
- 44.https://clinicaltrials.gov/ct2/show/results/NCT01769222?sect=X370156#evnt. Ipilimumab and Local Radiation Therapy in Treating Patients With Recurrent Melanoma, Non-Hodgkin Lymphoma, Colon, or Rectal Cancer. 2017. [Google Scholar]
- 45.Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Disc 2015;14:561–84. [DOI] [PubMed] [Google Scholar]
- 46.Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nature Rev Clin Oncol 2016;13:273–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kubo S, Yamada T, Osawa Y, Ito Y, Narita N, Fujieda S. Cytosine-phosphate-guanosine-DNA induces CD274 expression in human B cells and suppresses T helper type 2 cytokine production in pollen antigen-stimulated CD4-positive cells. Clin Exp Immunol 2012;169:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harris NL, Ronchese F. The role of B7 costimulation in T-cell immunity. Immunol Cell Biol 1999;77:304–11. [DOI] [PubMed] [Google Scholar]
- 49.Riley JL, Mao M, Kobayashi S, Biery M, Burchard J, Cavet G, et al. Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc Natl Acad Sci U S A 2002;99:11790–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Appleman LJ, van Puijenbroek AA, Shu KM, Nadler LM, Boussiotis VA. CD28 costimulation mediates down-regulation of p27kip1 and cell cycle progression by activation of the PI3K/PKB signaling pathway in primary human T cells. J Immunol 2002;168:2729–36. [DOI] [PubMed] [Google Scholar]
- 51.Marinari B, Costanzo A, Marzano V, Piccolella E, Tuosto L. CD28 delivers a unique signal leading to the selective recruitment of RelA and p52 NF-kappaB subunits on IL-8 and Bcl-xL gene promoters. Proc Natl Acad Sci USA 2004;101:6098–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.August A, Dupont B. Activation of extracellular signal-regulated protein kinase (ERK/MAP kinase) following CD28 cross-linking: activation in cells lacking p56lck. Tissue Antigens 1995;46:155–62. [DOI] [PubMed] [Google Scholar]
- 53.Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994;1:405–13. [DOI] [PubMed] [Google Scholar]
- 54.Walunas TL, Bakker CY, Bluestone JA. CTLA-4 ligation blocks CD28-dependent T cell activation. J Exp Med 1996;183:2541–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Walker LS, Sansom DM. Confusing signals: recent progress in CTLA-4 biology. Trends Immunol 2015;36:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017;355:1428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dou S, Smith M, Wang Y, Rusckowski M, Liu G. Intraperitoneal injection is not always a suitable alternative to intravenous injection for radiotherapy. Cancer Biother Radiopharm 2013;28:335–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fransen MF, van der Sluis TC, Ossendorp F, Arens R, Melief CJ. Controlled local delivery of CTLA-4 blocking antibody induces CD8+ T-cell-dependent tumor eradication and decreases risk of toxic side effects. Clin Canc Res 2013;19:5381–9. [DOI] [PubMed] [Google Scholar]
- 59.Marabelle A, Kohrt H, Levy R. Intratumoral anti-CTLA-4 therapy: enhancing efficacy while avoiding toxicity. Clin Canc Res 2013;19:5261–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rommelfanger DM, Compte M, Diaz RM, Ilett E, Alvarez-Vallina L, Thompson JM, et al. The efficacy versus toxicity profile of combination virotherapy and TLR immunotherapy highlights the danger of administering TLR agonists to oncolytic virus-treated mice. Molec Ther 2013;21:348–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med 2006;355:1018–28. [DOI] [PubMed] [Google Scholar]
- 62.Luhder F, Huang Y, Dennehy KM, Guntermann C, Muller I, Winkler E, et al. Topological requirements and signaling properties of T cell-activating, anti-CD28 antibody superagonists. J Exp Med 2003;197:955–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beyersdorf N, Hanke T, Kerkau T, Hunig T. CD28 superagonists put a break on autoimmunity by preferentially activating CD4+CD25+ regulatory T cells. Autoimmunity reviews 2006;5:40–5. [DOI] [PubMed] [Google Scholar]
- 64.Mazanet MM, Hughes CC. B7-H1 is expressed by human endothelial cells and suppresses T cell cytokine synthesis. J Immunol 2002;169:3581–8. [DOI] [PubMed] [Google Scholar]
- 65.Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 2002;169:5538–45. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.