

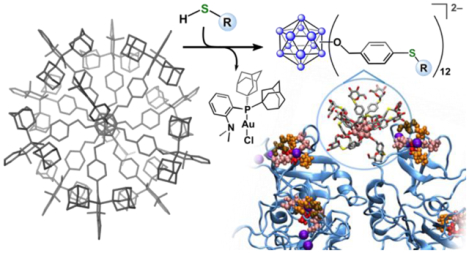
Abstract
For decades, chemists have strived to mimic the intricate design and diverse functions of naturally occurring systems through the bioinspired synthesis of programmable inorganic nanomaterials. The development of thiol-capped gold nanoparticles (AuNPs) has driven advancement in this area; however, although versatile and readily accessible, hybrid AuNPs are rarely atomically precise, which limits control over their surface topology and therefore the study of complex structure-function relationships. Here, we present a bottom-up approach to the systematic assembly of atomically precise hybrid nanoclusters employing a strategy that mimics the synthetic ease with which thiol-capped AuNPs are normally constructed, while producing well-defined covalent nanoscale assemblies with diverse surface topologies. For the first time, using a structurally characterized cluster-based organometallic building block, we demonstrate the systematic synthesis of nanoclusters with multivalent binding capabilities to complex protein targets.
Graphical Abstract
INTRODUCTION:
The rational design of surface-modified nanoscale materials using well-defined building block constituents is a powerful strategy for the bottom-up assembly of functional synthetic architectures. Drawing inspiration from nature, chemists have explored a range of platforms with programmable surface chemistry that are designed to interrogate and mimic complex biological interactions.1 These structural classes include dendrimers,2–4 polymers,5,6 coordination and supramolecular-based assemblies,7–9 and other nanoscale structures.10–12 In particular, thiol-capped gold nanoparticles (AuNPs) represent a category of hybrid materials that continue to receive considerable attention in this area for probing biological interactions of interest as they are easily accessible and tunable in terms of size and topology.13,14 Despite these attractive properties, such systems suffer from several fundamental limitations that stem from the relatively weak nature of the gold-thiolate bond (40–50 kcal mol−1, Figure 1, left).15 For example, thiol-capped AuNPs are known to undergo ligand-exchange processes with other surfactants in solution over short periods of time,16,17 contributing to limited stability profiles in biologically relevant media.18 Additionally, hybrid nanoparticle assemblies are seldom atomically precise and therefore typically possess non-uniform composition, size, and surface morphology. These drawbacks present significant challenges in rationally addressing hybrid AuNP structure-function properties on an atomic level, especially within the context of mechanistic exploration for biological applications. Such challenges have led to constant evolution of the nanochemistry field, with current efforts dedicated to addressing the areas of biological stability, atomic precision, and control over the surface topology of programmable nanostructures.19–21 Specifically, these areas include work dedicated to improving the binding of ligands to noble metal surfaces,22–25 and the development of synthetic methods that lead to the formation of gold-based nanoclusters with well-defined metal cores.26,27
Figure 1.
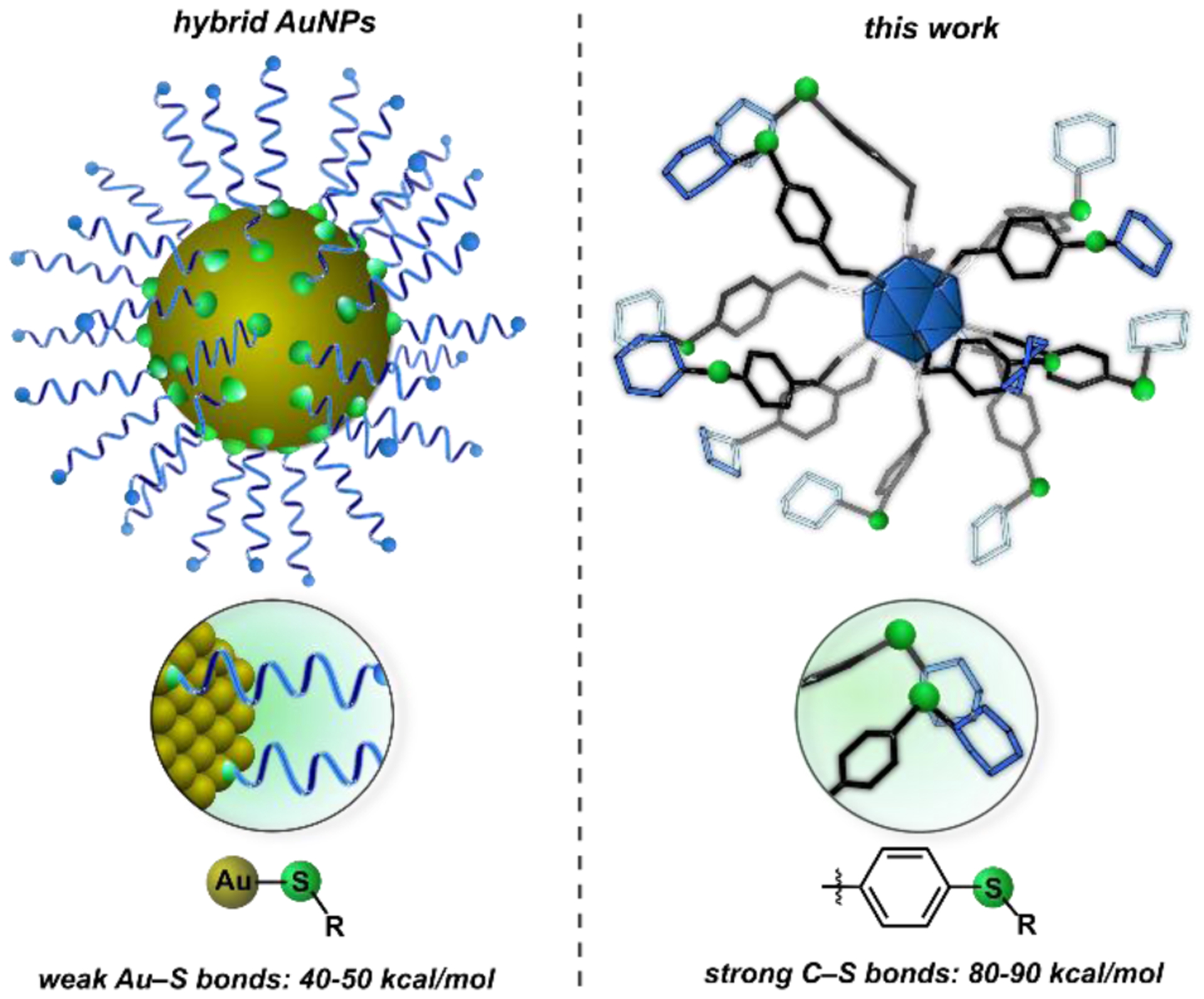
(Left) Hybrid AuNPs feature non-covalent, weak Au–S bonds (40–50 kcal/mol). (Right) This work presents a class of atomically precise nanoclusters built upon robust, and comparatively stronger C–S linkages (80–90 kcal/mol).36
Here, we present a conceptually new approach that mimics the chemistry of thiol-capped AuNP assembly processes, while producing a new method of controlling the architecture of complex abiotic molecules on an atomic scale in the context of manipulating biomolecular interactions. The presented approach incorporates, for the first time, the use of robust organometallic-based building blocks as structurally well-defined and tunable templates for assembling atomically precise and biocompatible hybrid nanoclusters. Organometallic gold-based systems have sparked interest in recent years for their use in both catalytic and stoichiometric transformations.28–31 In this work, we showcase a new class of nanostructures that contain a rigid cluster core functionalized with a dense layer of robust organometallic gold(III) sites that undergo rapid diversification with thiol-based nucleophiles via “click”-like C–S bond-forming reductive elimination. The thiol conjugation chemistry is selective and efficient, proceeding at ambient temperature under mild conditions to generate clusters with precisely controlled morphologies. This approach mimics the synthetic ease with which thiol-capped AuNPs are normally constructed,13,14,32 while providing functional advantages such as atomic precision and lack of coordinate bonds (Figure 1, right), which are properties typically associated with dendrimers and molecular systems. We investigated these hybrid nanoclusters as a new class of agents to inhibit important protein-protein and protein-sugar interactions in biological systems and report the binding affinity of saccharide-grafted clusters with plant-based concanavalin A (ConA), bacterial Shiga toxin 1, B subunit (Stx1B), and human-based DC-SIGN (Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin) targets, showcasing how this strategy allows for the rapid generation of complex, yet precise multivalency for enhancing molecular recognition.
RESULTS AND DISCUSSION:
Our approach to the programmable synthesis of hybrid nanomolecules is based upon a boron cluster core, which serves as a well-defined platform that enables atomic-level precision and control of size and surface morphology upon derivatization.33 Icosahedral dodecaborane clusters ([B12R12]) are excellent building blocks for the assembly of biologically-relevant nanoscale molecules due to their topological rigidity, appropriate size domain, low toxicity, stability under harsh biological conditions, and potential for dense surface polyfunctionalization.34–36 We envisioned access to such a class of hybrid nanoclusters would be possible through the rational evolution of synthetic handles on the cluster periphery that would ultimately lead to the incorporation of surface organometallic sites capable of facile reaction with thiol-containing substrates.
Accordingly, the synthesis of the B12(OCH2C6H4I)12 precursor was achieved through per-O-benzylation of closo-[B12(OH)12]2– with para-iodobenzyl bromide in the presence of diisopropylethylamine employing a microwave-assisted synthetic procedure.35 Following sequential two-electron chemical oxidation with Fe(III), the neutral, air-stable hypercloso-B12(OCH2C6H4I)12 cluster was isolated in 76% yield as a crystalline solid, and characterized by multinuclear NMR spectroscopy (1H, 13C, 11B), high-resolution electrospray ionization mass spectrometry (HR-ESI-MS(–)), cyclic voltammetry, UV-vis spectroscopy, and single crystal X-ray diffraction (SI Sections S2.4 and S9.1).
Bourissou et al. recently reported the oxidative addition of simple aryl iodides to Au(I) sites supported by bulky aminophosphine ligands under mild conditions.37 We envisioned that utilization of this Au(I) precursor ((Me-DalPhos)AuCl)38 with B12(OCH2C6H4I)12 under appropriate synthetic conditions could lead to the conversion of all twelve C–I sites on the cluster periphery to the corresponding C–Au(Me-DalPhos) congeners. Although conceptually simple, this strategy is far more challenging from a synthetic perspective given that successful permetalation of the cluster requires simultaneous and quantitative conversion efficiency for all twelve organometallic reactions. Furthermore, it is well documented that many organometallic-based reactions have the propensity to undergo decomposition pathways as well as generate undesired side products. These factors present additional challenges that could potentially complicate purification processes associated with assembling complex systems such as the present one.
Despite these challenges, however, treatment of B12(OCH2C6H4I)12 with excess (Me-DalPhos)AuCl in refluxing DCM (dichloromethane) in the presence of the halide scavenger AgSbF6 resulted in unprecedented 12-fold exhaustive metalation of the cluster via oxidative addition of Au(I) into each of the twelve aryl C–I bonds (Figure 2a). The permetalated product, [B12(OCH2C6H4(Me-DalPhos)AuCl)12][SbF6]11 ([1][SbF6]11), precipitated directly from the crude reaction mixture over the course of 16 h, and was isolated as a pink solid. Notably, the synthetic and purification procedures proceed cleanly when carried out under open-atmosphere conditions using unpurified solvents. The 31P{1H} NMR spectrum of [1][SbF6]11 features a single resonance at δ 75.8 ppm, which is consistent with the isolation of one, well-defined species. It is noted that during the purification procedure (SI Section S2.4), the boron cluster core undergoes a one-electron reduction from the neutral ([B12]0) to monoanionic charge state ([B12]–). This redox chemistry corresponds to an overall 11+ charge for complex 1 as opposed to the expected 12+ charge, which would be the case in the absence of any cluster-based reduction. It has been reported by our group and others that perfunctionalized dodecaborate clusters undergo redox chemistry,35,39–41 and it is likely that the DMF (dimethylformamide) used in the workup procedure serves as the reducing agent42 to result in the formation of the [1][SbF6]11 species.
Figure 2.
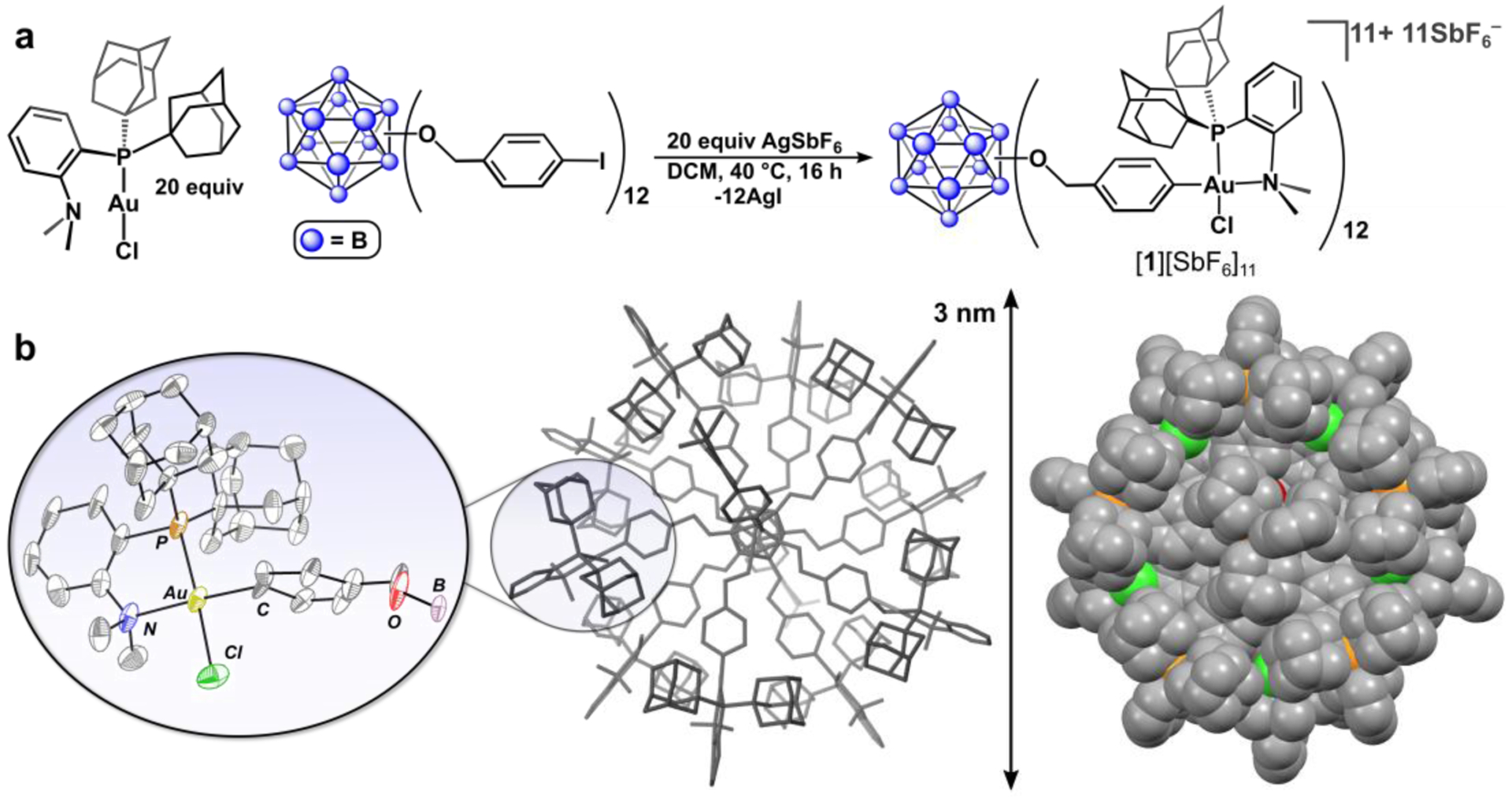
(a) Synthetic protocol for the preparation of [1][SbF6]11. (b) Solid-state structure of [1][SbF6]11 (center), and zoomed-in image of a single Au(III) organometallic fragment (left) with thermal ellipsoids rendered at the 30% probability level and with hydrogen atoms and counteranions omitted, and space-filling diagram (right). For detailed crystallographic details see SI Section S9.2.
Since we were unable to observe the intact [1]11+ complex using mass spectrometry, X-ray crystallographic analysis was the only alternative method available to directly confirm the identity of this product. Fortunately, crystallization of [1][SbF6]11 from an acetonitrile/dimethoxyethane/diethylether solution afforded single, pink crystals of suitable quality for X-ray diffraction analysis, which enabled structural determination of [1]11+ and confirmation of its identity. Diffraction data of [1][SbF6]11 were collected at a synchrotron radiation facility and revealed the complex crystallizes in the Cc monoclinic space group with the entire salt occupying the asymmetric unit. The crystallographic data were analyzed using conventional structural refinement methods and clearly show the fully-metalated framework of [1]11+ within a large cell volume of 55,900 Å3. As displayed in the solid-state structure of [1]11+ (Figure 2b), the cluster framework spans a 3 nm distance from end to end, and contains a dense layer of surface organometallic (Me-DalPhos)AuCl groups, representing a unique example of a structurally characterized atomically precise nanocluster featuring well-defined organometallic fragments.43,44
We next evaluated the ability of [1][SbF6]11 to serve as a building block for the construction of hybrid nanocluster thioether-based conjugates. Optimization of conjugation reaction conditions were carried out employing thiophenol as the model substrate. For a typical conjugation reaction, the [1][SbF6]11 species was generated according to the synthetic procedure described (vide supra) and then used in situ without its isolation as a solid. The [1][SbF6]11 complex was subsequently treated with thiophenol (30 equiv) in DMF (dimethylformamide) solution in the presence of potassium phosphate (30 equiv) according to the scheme displayed in Figure 3a. Addition of thiophenol immediately elicited a color change from deep purple to colorless due to reduction of the cluster core to the 2– charge state, which was confirmed by the presence of a singlet at δ −15.6 ppm in the 11B NMR spectrum of the reaction mixture. The location of this resonance is characteristic of reduced clusters in the 2− charge state and is consistent with the reducing capacity of the thiolate species.36 These reaction conditions afforded the twelve-fold thioether substituted conjugate, [K2][B12(OCH2C6H4SPh)12] ([K2][2]), within minutes (< 15) at 25 °C, as assayed by HR-ESI-MS(–) analysis of the crude mixture. The product was purified via silica gel column chromatography and isolated as an air-stable solid in 70% yield. The well-resolved resonances in the 1H and 11B NMR spectra in addition to the ESI-MS(–) characterization of the [K2][2] salt are consistent with its formulation as the fully functionalized thioether conjugate and the isolation of one discrete hybrid cluster species (SI Section S2.6). It is important to note that the full covalency of the nanocluster conjugate enables observation of the intact species by ESI-MS, whereas even gentle ionization techniques frequently result in fragmentation of ligands from the gold core of thiol-capped hybrid AuNPs due to cleavage of the Au–S bonds.45
Figure 3.
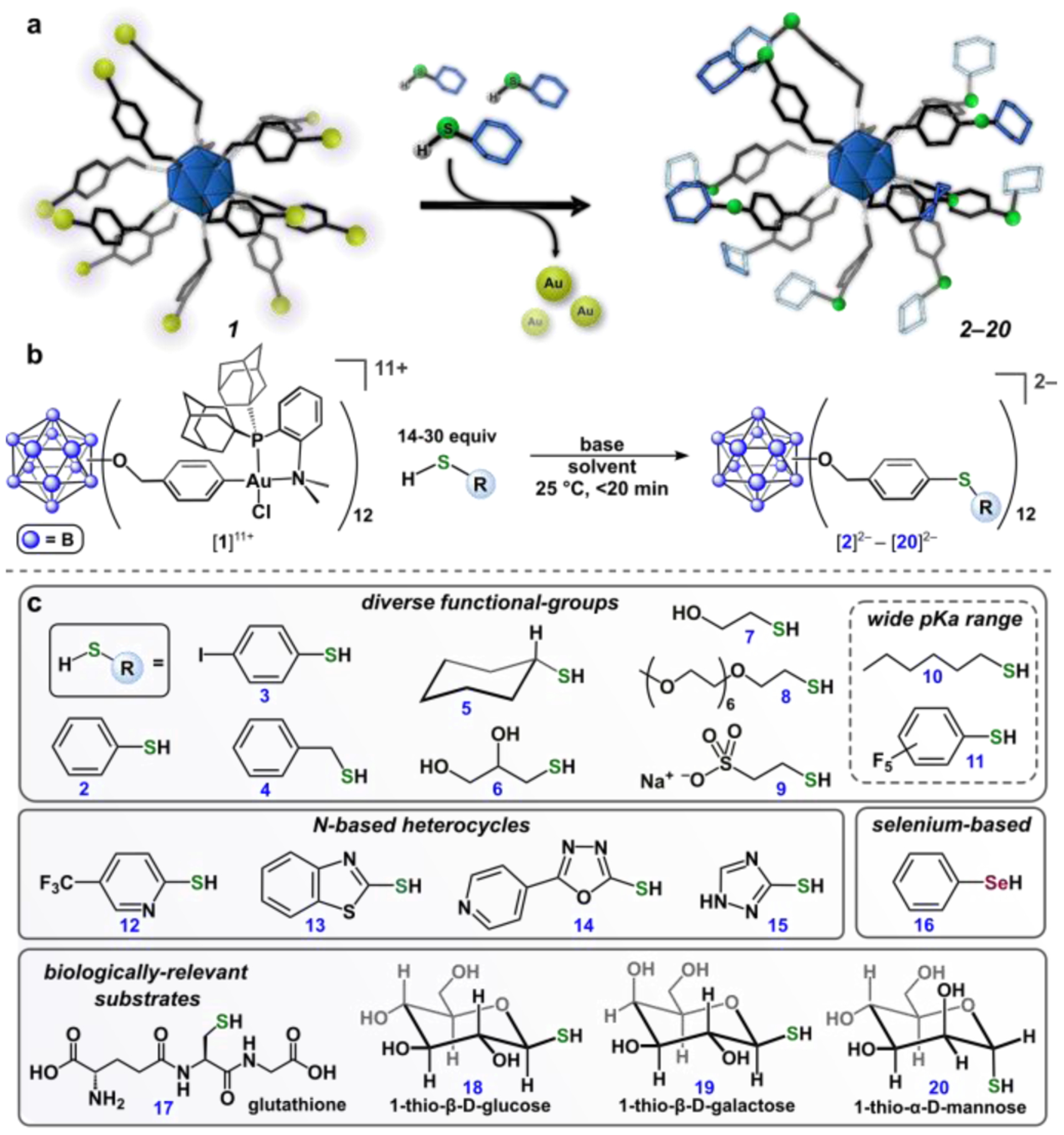
(a, b) Scheme for reactions of [1][SbF6]11 with thiol-containing substrates. (c) Substrate scope for conjugation reactions. All cluster conjugates were isolated and characterized (1H and 11B NMR spectroscopy, HR-ESI-MS(–)) after purification (SI Section S2).
The versatility of this conjugation strategy was further extended by examining the chemoselectivity of the developed chemistry through treatment of [1][SbF6]11 with thiol substrates featuring a variety of functional groups (2-11, Figure 3c). Thiol-based nucleophiles bearing aryl (2, 3, 4, 11), alkyl (5, 10), and alcohol-based groups (6, 7) cleanly generated the corresponding cluster conjugates as confirmed by ESI-MS(–) and multinuclear NMR spectroscopic analyses of the isolated products. This chemistry is also compatible with thiol nucleophiles featuring a bulky cyclohexyl group (5), a poly(ethyleneglycol) polymer (8), and an anionic sulfonate moiety (9); this wide ranging functional-group tolerance contributes to a scope of cluster conjugates with highly diverse solubility properties, surface morphologies, and electronic profiles. The conjugation chemistry is also operational with thiol-based reagents that span a broad window of nucleophilicity, as evidenced by the clean reactivity with both electron-rich (e.g. hexanethiol, 10) and electron deficient (e.g. pentafluorothiophenol, 11) substrates with pKa values ranging from ca. 3–11.46,47 Importantly, this finding expands the generality of the thiol substrate pKa scope from the previously developed organometallic-based reagents (palladium and gold) used for cysteine S-arylation.48 While N-based heterocycles are challenging coupling partners in transition-metal-mediated organic transformations due to their ability to serve as sigma-donors that can coordinate to and poison the active metal species, cluster conjugates featuring a variety of N-based heterocycles (12-15) were successfully prepared and characterized, further establishing the chemoselectivity and utility of the present approach. This conjugation strategy was even extended to include a selenium-based nucleophile (phenylselenol, 16), which further expands the versatility of the described chemistry. Finally, we extended this platform to include a variety of densely functionalized clusters bearing biologically relevant substrates such as a short peptide (17, glutathione) and three different thiol-based saccharides (18, 19, 20, vide infra). The presented organometallic-mediated approach provides a unique method of cluster-based hybrid nanomolecule synthesis,36 in which the assembly process exhibits extremely fast reaction kinetics (<15 min) under operationally simple and mild conditions and can even be conducted in mixed organic/aqueous media as necessitated by the solubility properties of the thiol substrate.
With a strategy for successful perfunctionalization of [1]11+ (vide supra), we envisioned we could establish a new generation of atomically precise nanoclusters that are modularly extended in size through a stepwise iterative synthesis. This method was accomplished by employing the radially extended iodo-aryl thioether conjugate, [3]2–, as the cluster-based precursor. Complete metalation of [3]2– to afford [B12(OCH2C6H4SC6H4(Me-DalPhos)AuCl)12][SbF6]10 ([21][SbF6]10) was accomplished using conditions similar to those described for the preparation of [1][SbF6]11. The [21][SbF6]10 cluster was characterized by 1H, 11B and 31P NMR (Figure 4b) spectroscopy, which are consistent with the formulation of [21]10+ as the fully-functionalized species (SI Section S2.24). In contrast to the neutral, hypercloso-B12(OCH2C6H4I)12 cluster that undergoes cluster-based, single electron reduction during the preparation of [1]11+ (vide supra), the [3]2– species is in its fully reduced state prior to metalation, which results in the 10+ charge of [21]10+.
Figure 4.
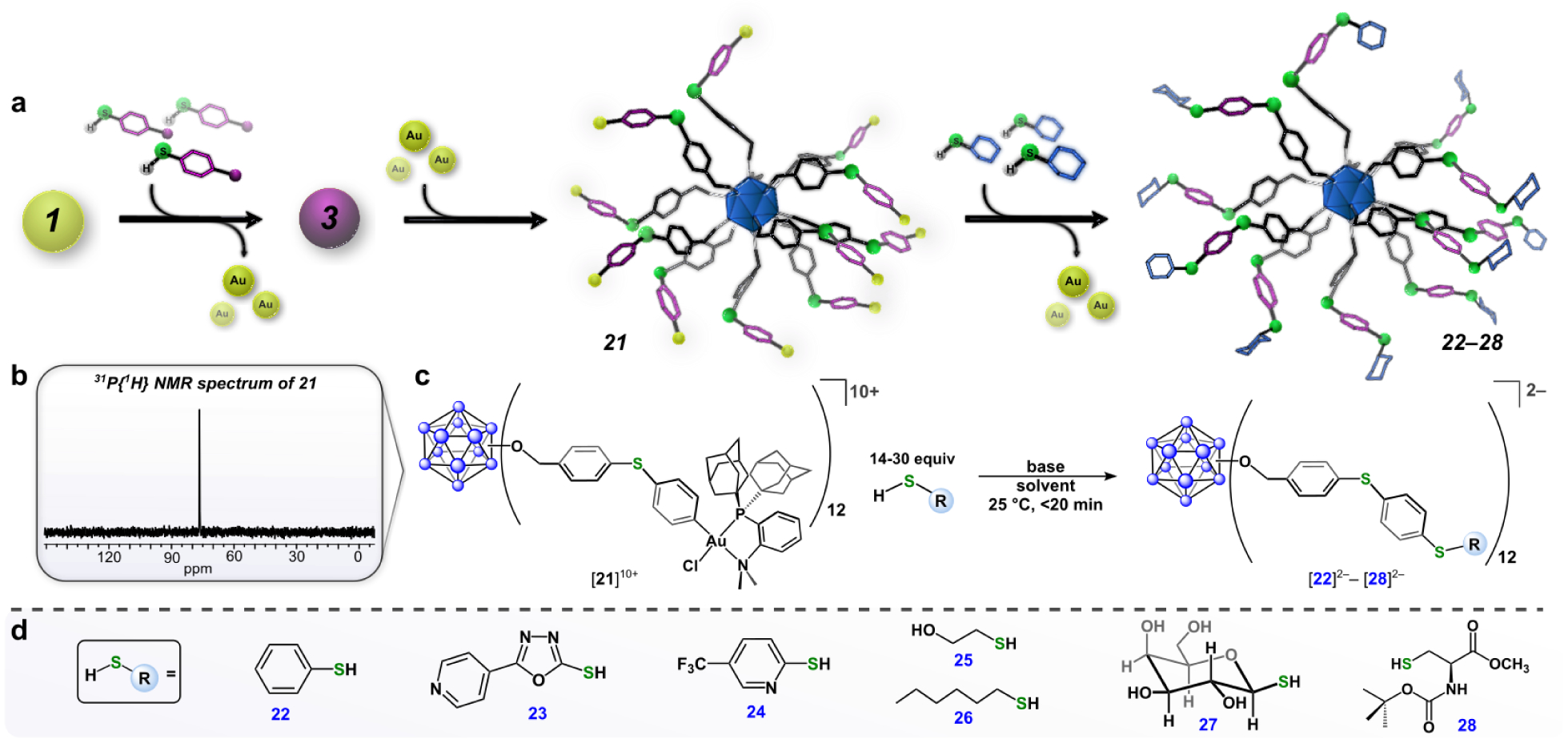
(a) “Layer-by-layer” assembly strategy for building nanoclusters that are modularly extended in size. (b) 31P{1H} NMR spectrum of [21][SbF6]10. (c) Synthetic scheme for reactions of [21][SbF6]10 and thiol-containing substrates. (d) Substrate scope for conjugation reactions. All cluster conjugates were observed in situ by ESI-MS(–) (See SI Section S2.27 for full scope and characterization data).
Complex [21][SbF6]10 undergoes facile conjugation chemistry with thiol-containing nucleophiles to generate species [22]2–-[28]2– under the same reaction conditions employed for the conjugation chemistry of [1]11+. This template-based approach to the construction of hybrid nanoclusters that are systematically extended in size is reminiscent of the layer-by-layer assembly process of building composite core-shell nanomaterials in which multiple films are layered onto colloidal particles followed by selective template removal.49,50 This method allows for rational engineering and precise control over the size, shape, composition, and surface topology of the resulting nanoclusters. Notably, we can easily recycle the (Me-DalPhos)AuCl complex that is generated as the byproduct from the conjugation reactions through purification on silica gel (SI Section S5). This recovered material can then be reused in the preparation of [1][SbF6]11 and [21][SbF6]10, thereby demonstrating the efficiency of the present approach.
We next focused on evolving the present method to rationally build a class of hybrid nanoclusters that contain specific recognition moieties capable of engaging in multivalent binding interactions with complex protein targets. Multivalency plays a vital role in numerous biological processes and especially in those involving pathogen-host relationships based on protein-glycan recognition.11,51 Naturally occurring multivalent glycoconjugates such as glycoproteins and glycolipids can bind lectins with affinities that surpass the weak binding association of the corresponding monosaccharides, making the design of high-affinity, synthetic analogues with controlled spatial and topology parameters an area of growing interest for preventing or treating pathogen-based diseases.2,7
Using commercial thiolate precursors, we prepared the glucose- ([Na2][18]), galactose- ([Na2][19]), and mannose-grafted ([Na2][20]) nanoclusters (Figure 3c) that feature precise spatial arrangements of the twelve appended saccharides on the cluster periphery. All three water-soluble glycosylated clusters were isolated after purification via size-exclusion chromatography, and characterized by heteronuclear NMR spectroscopy (1H and 11B) and HR-ESI-MS(–). We next evaluated the stability of the glycoconjugates in biologically relevant media using the glucose-functionalized cluster, [18]2–, as a model. Species [Na2][18] was exposed to fetal bovine serum cell-culture media as well as solutions of various pH environments (pH = 5 and 10) for extended periods of time (≥ 7 days) without any observable degradation as assayed by spectroscopic analyses (11B NMR, Figure 5b; ESI-MS(–), SI Section S3). The excellent stability profile of [Na2][18] in biologically relevant media is a direct consequence of its full covalency, further demonstrating the advantage of using systems built upon robust C–S linkages as opposed to the significantly weaker Au–S bonds characteristic of thiol-capped AuNPs.16,18 The excellent stability profile of [Na2][18] further motivated us to probe the use of glycosylated nanoclusters as functional agents to interrogate multivalent binding interactions with protein targets.
Figure 5.
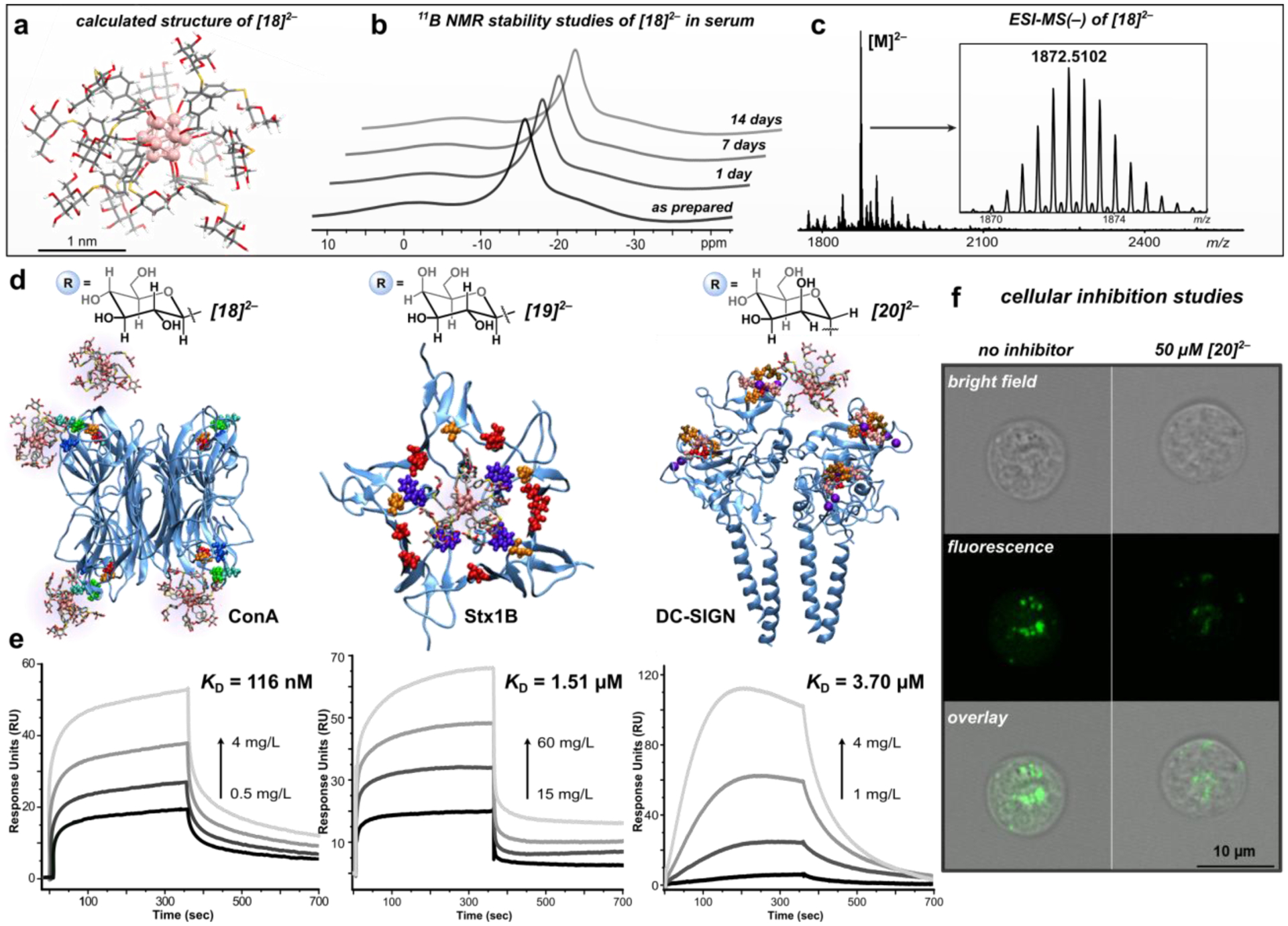
(a) Calculated structure of [18]2–. (b) 11B NMR stability studies of [18]2– in serum monitored over the course of 14 days displaying the structural integrity of the cluster. (c) ESI-MS(–) of [18]2–. (d) Snapshots of MD simulations (after 20 ns) depicting the multivalent binding interactions between [18]2– (one molecule per binding site) and ConA (left), [19]2– and Stx1B (middle), and [20]2– and DC-SIGN (right). (e) SPR sensorgrams with calculated KD values for direct binding measurements of each nanocluster and its respective protein target. (f) Confocal microscopy images of Raji DC-SIGN+ cells incubated with FITC-tagged gp120 and DMSO (control, left) and with [Na2][20] (right, 50 μM), showing significant inhibition of DC-SIGN mediated cellular entry of gp-120 in the presence of [Na2][20].
Accordingly, we evaluated and quantified the binding affinity of glycosylated clusters, [Na2][18], [Na2][19], and [Na2][20] with protein targets from three biological kingdoms (ConA, Stx1B, and DC-SIGN, respectively) by surface plasmon resonance (SPR). The PEGylated conjugate, [K2][8], was used in each case as a control to assess the extent of non-specific binding interactions for each target, and the corresponding monosaccharide was used in each case as a monovalent reference.
ConA, a protein specific for manno- and glucopyranosides,12 was initially investigated as a model target. The binding interaction (KD, dissociation constant) of the glucose-functionalized conjugate, [Na2][18], with ConA was determined to be 116 nM at pH 7.4 based on SPR binding experiments (Figure 5e). This KD value agrees well with the equilibrium constant determined for the previously developed dodecaborate perfluoroaryl analogue (54 nM)36 with ConA under similar experimental conditions. When the PEGylated cluster control ([K2][8]) was injected at the highest mass concentration of [Na2][18] evaluated (4 mgL−1, 1 μM), only minimal binding was observed (SI Section S6.1), indicating that ConA binds [18]2– with a high degree of specificity. Similarly, a negligible response was observed when a solution of the D-glucose monomer was injected at a significantly higher concentration (40 mgL−1, 200 μM) over the protein surface, suggesting that the multivalent nature of [18]2– results in a dramatically enhanced binding profile per mole of saccharide. In fact, the KD value of [Na2][18] with ConA displays a nearly 3,000-fold enhancement (250-fold increase per saccharide) when compared with the KD value reported for methyl D-glucopyranoside and ConA.3 The determined binding affinity is consistent with the reported KD value (15.8 nM) for the binding between ConA and a glycodendrimer that features over twice the number of D-glucose residues3 when compared with the functionalization of [18]2–. The comparable KD values highlight the advantages provided by the present system such as dense functionalization and core-scaffold rigidity when binding affinity is compared on a per-carbohydrate basis. To gain a better understanding of the cluster-target binding process, we performed a molecular dynamics (MD) simulation to model the dynamic nature of the multivalent interactions between [18]2– and each of the four binding sites of ConA (SI Section S8), and a snapshot of the simulation is displayed in Figure 5d.
We next probed the binding interactions of the multivalent ligand-carrier, [B12(OCH2C6H4-1-thio-D-galactose)]2– ([Na2][19]), with the B subunit of Shiga toxin 1 (Stx1B). Shiga toxin is an AB5-type cytotoxin expressed by the S. dysenteriae strain of E. coli that contains one enzymatically active A subunit, and a radially-symmetric B subunit pentamer, and represents one of the most potent bacterial poisons known.52 The B subunit features multiple sites that engage in multivalent binding interactions with the cellular receptor, globotriaosylceramide (Gb3), which is a galactose-containing glycolipid responsible for bacterial uptake into cells.52 Since cell-surface binding is a critical first step in the cytotoxicity pathway of the pathogen, building multivalent agents that block the B pentamer from binding to Gb3 is a potential method for protecting host cells from attack by the toxin.4 A KD value of 1.51 μM was determined for the binding of [19]2– with Stx1B by direct SPR studies (Figure 5e). Even at high concentrations, the free D-galactose monomer does not bind to Stx1B (100 mg/L), and a phenylene-bridged disaccharide control (gal-C6H4-gal, 100 mg/L) fails to engage in significant binding with Stx1B as displayed by its minimal SPR response. These data suggest the multivalent binding properties of [19]2– are responsible for the enhancement in its binding capacity when compared with that of its mono- and divalent controls. The proposed multivalent nature of the binding process between [19]2– and Stx1B agrees well with the MD simulation performed for this cluster-protein system (SI Section S8), and a snapshot of this dynamic interaction is displayed in Figure 5d.
Expanding our study, we investigated the multivalent binding properties of the mannosylated cluster, [20]2–, with the DC-SIGN receptor. DC-SIGN is a tetrameric receptor expressed in dendritic cells that recognizes mannose-rich glycoproteins, including the viral envelope glycoprotein of HIV-1 (gp-120) through multivalent carbohydrate-lectin interactions that facilitate receptor-mediated endocytosis.53 We envisioned [20]2– could mimic the high surface density of mannose units on gp120 and therefore potentially serve as a multivalent inhibitor of the DC-SIGN-mediated cellular uptake of the viral glycoprotein. SPR binding data reveal that [20]2– exhibits an enhanced dose-dependent avidity (KD = 3.7 μM, Figure 5e) toward DC-SIGN when compared with the negligible responses observed for the D-mannose monosaccharide and PEGylated controls ([8]2–) at significantly higher mass concentrations (SI Section 6.3). These findings support the multivalent nature of the binding interaction between [20]2– and DC-SIGN54 and agree well with the MD simulation performed for this system (Figure 5d, SI Section S8), which displays the specific binding in atomic detail. The calculated KD value is also in good agreement with that of a related mannose-coated multivalent gold nanoparticle bearing 25 surface sugars that binds DC-SIGN with high affinity (KD = 0.974 μM).53
Given the strong affinity of [20]2– towards DC-SIGN, we next conducted cell-based inhibition studies to probe the capacity of [20]2– to prevent DC-SIGN-dependent cell recognition of gp120. An assay was conducted in which Raji DC-SIGN+ cells were incubated with FITC-tagged gp120 (FITC = fluorescein isothiocyanate) and either [20]2–, [8]2–, free D-mannose, or DMSO as a control. Confocal fluorescence microscopy was used to image the cells and assess the cellular uptake of gp120, and thus the extent of glycoprotein binding inhibition. As shown in Figure 5f (left), in the absence of [20]2–, gp120-FITC readily undergoes endocytosis by DC-SIGN-expressing Raji cells. In contrast, there was a significant reduction in the gp120-FITC cellular uptake in the presence of [20]2– (Figure 5f, right), therefore indicating that the mannose-functionalized cluster can prevent the binding and cellular uptake of the gp120 viral envelope glycoprotein by blocking the extracellular receptor DC-SIGN. Consistent with the negligible binding response observed for [8]2– and free D-mannose from the SPR binding studies, there was no detectable reduction in the gp120-FITC cellular uptake when experiments were conducted in the presence of these control molecules (SI Section S7). These results highlight the excellent selectivity of the present system and the critical role the multivalent nature of the binding interactions play in effective inhibition of DC-SIGN-dependent cellular uptake of gp120.
CONCLUSION:
With this work, we have introduced a new platform that allows for the rapid generation of a library of well-defined and functional abiotic nanoclusters with protein binding capabilities. This approach is operationally reminiscent of the chemoselective assembly process associated with thiol-capped AuNP synthesis, while providing access to nanomolecules with enhanced properties such as atomic precision, and lack of coordinate bonds. Furthermore, this strategy provides exceptional control over multivalency and topological rigidity of the resulting nanoclusters, thereby expanding the underlying design principles governing the development of agents capable of engaging in the specific inhibition of complex protein targets. For the first time, combining elements of inorganic cluster chemistry, organometallic synthesis, and nanobiotechnology, we have provided a basis for the generation of robust and programmable hybrid nanoclusters with unique applications targeting molecular recognition.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH for providing the following materials: pcDNA3-DC-SIGN from Dr. S. Pöhlmann, Dr. F. Baribaud, Dr. F. Kirchhoff, and Dr. R.W. Doms, and both Raji and Raji DC-SIGN+ cells from Dr. Li Wu and Dr. Vineet N. KewalRamani. Prof. Heather Maynard is thanked for generously allowing us access to her BSL-2 laboratory space. We thank Dr. K. Sugimoto (Japan Synchrotron Radiation Research Institute/Kyoto University) and Dr. T. Sato (Rigaku Corporation) for their support and helpful advice for the crystallographic data analysis. A.M.S. thanks 3M for a Non-Tenured Faculty Award, the Alfred P. Sloan Foundation for the Sloan Research Fellowship in Chemistry, the Research Corporation for Science Advancement (RCSA) for the Cottrell Scholar Award, and the National Institutes of Health (NIH) for a Maximizing Investigators Research Award (MIRA, R35GM124746). E.A.Q. thanks the UCLA Graduate Division for the Dissertation Year Fellowship. P.K. acknowledges the NSF Division of Materials Research Grant (1506886).
Footnotes
Detailed experimental and analytical procedures and characterization data are available in the Supporting Information document. This material is available free of charge via the Internet at http://pubs.acs.org.Crystallographic data are available from the Cambridge Crystallographic Data Centre, under reference numbers CCDC 1942748 and 1949777.
UCLA has patents on several compounds reported in this work from which A.M.S. and current/former co-workers may receive royalty payments. The Cs2[B12(OH)12] salt (Catalog #902209) is commercially available through the MilliporeSigma catalog.
REFERENCES
- (1).Pieters RJ Org. Biomol. Chem 2009, 7 (10), 2013–2025. [DOI] [PubMed] [Google Scholar]
- (2).Turnbull WB; Kalovidouris SA; Stoddart JF Chem. - A Eur. J 2002, 8 (13), 2988–3000. [DOI] [PubMed] [Google Scholar]
- (3).Munoz EM; Correa J; Riguera R; Fernandez-Megia EJ Am. Chem. Soc 2013, 135 (16), 5966–5969. [DOI] [PubMed] [Google Scholar]
- (4).Nishikawa K; Matsuoka K; Kita E; Okabe N; Mizuguchi M; Hino K; Miyazawa S; Yamasaki C; Aoki J; Takashima S; Yamakawa Y; Nishijima M; Terunuma D; Kuzuhara H; Natori Y Proc. Natl. Acad. Sci 2002, 99 (11), 7669–7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Disney MD; Zheng J; Swager TM; Seeberger PH J. Am. Chem. Soc 2004, 126 (41), 13343–13346. [DOI] [PubMed] [Google Scholar]
- (6).Polizzotti BD; Kiick KL Biomacromolecules 2006, 7 (2), 483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Baldini L; Casnati A; Sansone F; Ungaro R Chem. Soc. Rev 2007, 36 (2), 254–266. [DOI] [PubMed] [Google Scholar]
- (8).Conn MM; Rebek J Chem. Rev 1997, 97 (5), 1647–1668. [DOI] [PubMed] [Google Scholar]
- (9).Kamiya N; Tominaga M; Sato S; Fujita MJ Am. Chem. Soc 2007, 129 (13), 3816–3817. [DOI] [PubMed] [Google Scholar]
- (10).Castro E; Garcia AH; Zavala G; Echegoyen LJ Mater. Chem. B 2017, 5 (32), 6523–6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kiessling LL; Gestwicki JE; Strong LE Angew. Chemie Int. Ed 2006, 45 (15), 2348–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Marra A; Staderini S; Berthet N; Dumy P; Renaudet O; Dondoni A European J. Org. Chem 2013, No. 6, 1144–1149. [Google Scholar]
- (13).Dreaden EC; Alkilany AM; Huang X; Murphy CJ; El-Sayed MA Chem. Soc. Rev 2012, 41 (7), 2740–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Daniel M; Astruc D Chem. Rev 2004, 104 (1), 293–346. [DOI] [PubMed] [Google Scholar]
- (15).Love JC; Estroff LA; Kriebel JK; Nuzzo RG; Whitesides GM Chem. Rev 2005, 105, 1103–1170. [DOI] [PubMed] [Google Scholar]
- (16).Smith AM; Marbella LE; Johnston KA; Hartmann MJ; Crawford SE; Kozycz LM; Seferos DS; Millstone JE Anal. Chem 2015, 87 (5), 2771–2778. [DOI] [PubMed] [Google Scholar]
- (17).Hostetler MJ; Templeton AC; Murray RW; Hill C; Carolina N 1999, No. 16, 3782–3789.
- (18).Häkkinen H Nat. Chem 2012, 4 (6), 443–455. [DOI] [PubMed] [Google Scholar]
- (19).Chakraborty P; Nag A; Chakraborty A; Pradeep T Acc. Chem. Res 2019, 52 (1), 2–11. [DOI] [PubMed] [Google Scholar]
- (20).Verma A; Stellacci F Small 2010, 6 (1), 12–21. [DOI] [PubMed] [Google Scholar]
- (21).Kundu PK; Samanta D; Leizrowice R; Margulis B; Zhao H; Börner M; Udayabhaskararao T; Manna D; Klajn R Nat. Chem 2015, 7 (8), 646–652. [DOI] [PubMed] [Google Scholar]
- (22).Zhukhovitskiy AV; MacLeod MJ; Johnson JA Chem. Rev 2015, 115 (20), 11503–11532. [DOI] [PubMed] [Google Scholar]
- (23).Man RWY; Li CH; MacLean MWA; Zenkina OV; Zamora MT; Saunders LN; Rousina-Webb A; Nambo M; Crudden CM J. Am. Chem. Soc 2018, 140 (5), 1576–1579. [DOI] [PubMed] [Google Scholar]
- (24).Chu Z; Han Y; Král P; Klajn R Angew. Chem. - Int. Ed 2018, 57 (24), 7023–7027. [DOI] [PubMed] [Google Scholar]
- (25).Narouz MR; Osten KM; Unsworth PJ; Man RWY; Salorinne K; Takano S; Tomihara R; Kaappa S; Malola S; Dinh CT; Padmos JD; Ayoo K; Garrett PJ; Nambo M; Horton JH; Sargent EH; Häkkinen H; Tsukuda T; Crudden CM Nat. Chem 2019, 11 (5), 419–425. [DOI] [PubMed] [Google Scholar]
- (26).Jadzinsky PD; Calero G; Ackerson CJ; Bushnell DA; Kornberg RD Science 2007, 318 (5849), 430–433. [DOI] [PubMed] [Google Scholar]
- (27).Zeng C; Chen Y; Kirschbaum K; Lambright KJ; Jin R Science 2016, 354 (6319), 1580–1584. [DOI] [PubMed] [Google Scholar]
- (28).Gorin DJ; Sherry BD; Toste FD Chem. Rev 2008, 108 (8), 3351–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yang H; Gabbaï FP J. Am. Chem. Soc 2015, 137 (41), 13425–13432. [DOI] [PubMed] [Google Scholar]
- (30).Joost M; Amgoune A; Bourissou D Angew. Chem. - Int. Ed 2015, 54, 15022–15045. [DOI] [PubMed] [Google Scholar]
- (31).Messina MS; Stauber JM; Waddington MA; Rheingold AL; Maynard HD; Spokoyny AM J. Am. Chem. Soc 2018, 140, 7065–7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Brust M; Walker M; Bethell D; Schiffrin DJ; Whyman RJ Chem. Soc. Chem. Commun 1994, No. 7, 801–802. [Google Scholar]
- (33).Fisher SP; Tomich AW; Lovera SO; Kleinsasser JF; Guo J; Asay MJ; Nelson HM; Lavallo V Chem. Rev 2019, 119, 8262–8290. [DOI] [PubMed] [Google Scholar]
- (34).Farha OK; Julius RL; Lee MW; Huertas RE; Knobler CB; Hawthorne MF J. Am. Chem. Soc 2005, 127 (51), 18243–18251. [DOI] [PubMed] [Google Scholar]
- (35).Axtell JC; Saleh LMA; Qian EA; Wixtrom AI; Spokoyny AM Inorg. Chem 2018, 57 (5), 2333–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Qian EA; Wixtrom AI; Axtell JC; Saebi A; Jung D; Rehak P; Han Y; Moully EH; Mosallaei D; Chow S; Messina MS; Wang JY; Royappa AT; Rheingold AL; Maynard HD; Král P; Spokoyny AM Nat. Chem 2017, 9 (4), 333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Zeineddine A; Estévez L; Mallet-Ladeira S; Miqueu K; Amgoune A; Bourissou D Nat. Commun 2017, 8 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hesp KD; Stradiotto MJ Am. Chem. Soc 2010, 132, 18026–18029. [DOI] [PubMed] [Google Scholar]
- (39).Wixtrom AI; Shao Y; Jung D; Machan CW; Kevork SN; Qian EA; Axtell JC; Khan SI; Kubiak CP; Spokoyny AM Inorg. Chem. Front 2016, 3, 711–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Lee MW; Farha OK; Hawthorne MF; Hansch CH Angew. Chem. - Int. Ed 2007, 46 (17), 3018–3022. [DOI] [PubMed] [Google Scholar]
- (41).Barton JL; Wixtrom AI; Kowalski JA; Qian EA; Jung D; Brushett FR; Spokoyny AM ACS Appl. Energy Mater 2019, 2 (7), 4907–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Heravi MM; Ghavidel M; Mohammadkhani L RSC Adv. 2018, 8 (49), 27832–27862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Cook AW; Jones ZR; Wu G; Scott SL; Hayton TW J. Am. Chem. Soc 2018, 140 (1), 394–400. [DOI] [PubMed] [Google Scholar]
- (44).Bai J; Virovets AV; Scheer M Science 2003, 300 (May), 781–784. [DOI] [PubMed] [Google Scholar]
- (45).Fields-zinna CA; Sampson JS; Crowe MC; Tracy JB; Parker JF; DeNey AM; Muddiman DC; Murray RW J. Am. Chem. Soc 2009, 25 (18), 13844–13851. [DOI] [PubMed] [Google Scholar]
- (46).Thapa B; Schlegel HB J. Phys. Chem. A 2016, 120 (28), 5726–5735. [DOI] [PubMed] [Google Scholar]
- (47).Jencks WP; Salvesen KJ Am. Chem. Soc 1971, 93 (18), 4433–4436. [Google Scholar]
- (48).Zhang C; Vinogradova EV; Spokoyny AM; Buchwald SL; Pentelute BL Angew. Chemie Int. Ed 2019, 58 (15), 4810–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Liu R; Priestley RD J. Mater. Chem. A 2016, 4 (18), 6680–6692. [Google Scholar]
- (50).Yan Y; Björnmalm M; Caruso F Chem. Mater 2014, 26 (1), 452–460. [Google Scholar]
- (51).Lundquist JJ; Toone EJ Chem. Rev 2002, 102 (2), 555–578. [DOI] [PubMed] [Google Scholar]
- (52).Melton-Celsa AR Microbiol. Spectr 2014, 2 (4), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Martínez-Ávila O; Hijazi K; Marradi M; Clavel C; Campion C; Kelly C; Penadés S Chem. - A Eur. J 2009, 15 (38), 9874–9888. [DOI] [PubMed] [Google Scholar]
- (54).Qian EA; Han Y; Messina MS; Maynard HD; Král P; Spokoyny AM Bioconjug. Chem 2019, 30 (10), 2594–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.