

Abstract
Chronic rhinosinusitis (CRS) is one of the most common chronic diseases worldwide. It is a heterogeneous disease, and geographical or ethnic differences in inflammatory pattern in nasal mucosa are major issues. Tissue eosinophilia in CRS is highly associated with extensive sinus disease, recalcitrance, and a higher nasal polyp (NP) recurrence rate after surgery. The prevalence of eosinophilic CRS (ECRS) is increasing in Asian countries within the last 2 decades, and this trend appears to be occurring across the world. International consensus criteria for ECRS are required for the accurate understanding of disease pathology and precision medicine. In a multicenter large-scale epidemiological survey, the “Japanese Epidemiological Survey of Refractory Eosinophilic Chronic Rhinosinusitis study,” ECRS was definitively defined when the eosinophil count in nasal mucosa is greater than or equal to 70 eosinophils/hpf (magnification, ×400), and this study proposed an algorithm that classifies CRS into 4 groups according to disease severity. The main therapeutic goal with ECRS is to eliminate or diminish the bulk of NP tissue. NPs are unique abnormal lesions that grow from the lining of the nasal and paranasal sinuses, and type 2 inflammation plays a critical role in NP development in patients with ECRS. An imbalance between protease and endogenous protease inhibitors might play a pivotal role in the initiation and exacerbation of type 2 inflammation in ECRS. Intraepithelial mast cells in NPs, showing a tryptase1, chymase2 phenotype, may also enhance type 2 inflammation. Intense edema and reduced fibrosis are important histological features of eosinophilic NPs. Mucosal edema mainly consists of exuded plasma protein, and excessive fibrin deposition would be expected to contribute to the retention of proteins from capillaries and thereby perpetuate mucosal edema that may play an etiological role in NPs. Upregulation of the coagulation cascade and downregulation of fibrinolysis strongly induce abnormal fibrin deposition in nasal mucosa, and type 2 inflammation plays a central role in the imbalance of coagulation and fibrinolysis.
Keywords: Chronic rhinosinusitis, nasal polyps, mast cell, tryptase, fibrin, coagulation cascade, fibrinolysis, tissue plasminogen activator
Chronic rhinosinusitis (CRS) is one of the most common chronic diseases worldwide, with a prevalence of approximately 5% to 12% in the adult population. It is characterized by inflammation of the nasal mucosa and paranasal sinuses, and by definition, patients with CRS must report the presence of anterior or posterior rhinorrhea, nasal congestion, hyposmia, and/or facial pressure or pain that lasts for more than 12 weeks.1,2 Although CRS is not life-threatening, symptoms such as nasal congestion, nasal discharge, pain or facial pressure, loss of smell, and cough impair the patient’s quality of life and economic activity.3,4 CRS is heterogeneous and is generally classified into 2 phenotypes, namely, CRS with nasal polyps (CRSwNP) and CRS without nasal polyps. In Western countries, the sinonasal tissue of patients with CRS without nasal polyps can be either eosinophilic or neutrophilic, whereas polyp tissue from patients with CRSwNP is mostly characterized by extreme eosinophilic infiltration. Consequently, identifying whether nasal polyps (NPs) are present or absent is useful for estimating the likelihood of eosinophilic inflammation and can influence treatment strategies. In Asian countries, NP tissues from patients with CRS have fewer eosinophils than do those from patients with CRS in Europe and the United States.5,6 However, the degree of eosinophilia in NPs has increased in Asia in the past 2 decades.7–9 The geographical differences and changes in histological characteristics of Asian NPs may be related to genetic, nutritional, or environmental factors.10,11 Therefore, compared with phenotyping by presence of NPs, classifying CRS by inflammatory endotypes can improve the accuracy of identifying the underlying pathophysiology and provide guidance for specific treatment. The most common CRS classification by endotype divides CRS into 2 subtypes, namely, eosinophilic CRS (ECRS) and noneosinophilic CRS (non-ECRS). ECRS is characterized by severe type 2 inflammation and is difficult to treat. In addition, NP recurrence after medical treatment or surgical intervention is frequent in ECRS. Comorbid asthma, including aspirin/ nonsteroidal anti-inflammatory drug intolerance, is also common in ECRS.12–15 The main therapeutic goal with patients with ECRS is to eliminate or diminish the bulk of NP tissue and reduce the sinonasal inflammation, and treatment of CRSwNP can not only alleviate upper airway symptoms such as persistent stuffiness, facial pain, headache, and impairment or loss of smell but also sometimes suppress comorbid asthma.16,17 Polypoid sinus mucosa, including NP tissue, may be a primary source of cysteinyl leukotrienes (CysLTs), which may contribute to comorbid asthma.18,19 Consequently, precise understanding of the pathogenesis of NP formation is particularly significant for improving the efficacy of ECRS therapy.
This review article highlights the recent information on CRS endotypes and phenotypes and clarifies the role of imbalances of coagulation and fibrinolysis in the pathophysiology of eosinophilic NP development.
HETEROGENEITY AND DISEASE SEVERITY OF CRS
Tissue eosinophilia in CRS is highly associated with extensive sinus disease, recalcitrance, and a higher NP recurrence rate after endoscopic sinus surgery.20,21 To our knowledge, Okuda5 was the first to report the differences in the inflammation pattern of CRS nasal mucosa between European and Asian (in this case, Japanese) patients. The prevalence of eosinophilic CRSwNP is increasing in Asian countries within the last 2 decades however, and this trend appears to be occurring across the world, possibly due to the rapid global adoption of a Western lifestyle.7,8,10,22 Therefore, intractable CRSwNP, which exhibits eosinophilia in NPs, has been referred as ECRS in Japan and we will use that terminology here.23 However, no international consensus criteria exist for ECRS, and the variable use of criteria in different studies can cloud understanding of disease pathology. Globally, mucosal eosinophilia is defined by determining the mucosal eosinophil count or eosinophil percentage in the total inflammatory cells, an end point that can provide prognostic information about disease severity or outcome.24,25 In a multicenter large-scale epidemiological study of 1716 patients with CRS titled “Japanese Epidemiological Survey of Refractory Eosinophilic Chronic Rhinosinusitis (JESREC) study,” ECRS was definitively defined as whether the eosinophil count in the nasal mucosa was greater than or equal to 70 eosinophils/hpf (magnification, ×400). Although this number is rather high and may not be applicable internationally, this cutoff value provided the most significant assessment of the likelihood of CRS recurrence in Japan.26 In addition, this study created an algorithm that classifies CRS according to blood eosinophilia, ethmoid sinus-dominant shadow in computed tomography, and comorbidity (bronchial asthma and aspirin/nonsteroidal anti-inflammatory drug intolerance). Eventually, CRS was classified into the following 4 groups: non-ECRS, mild ECRS, moderate ECRS, and severe ECRS. These 4 groups varied significantly in the rate of recurrence and treatment refractoriness (Fig 1).26 In this classification scheme, moderate and severe ECRS were considered refractory. Molecular and cellular identification of diverse inflammatory pathological mechanisms and associated immunologic endotyping of CRS will likely be important for establishing the prognosis of disease severity and recurrence after surgery, and appropriate use of biologics in the near future.4,13,27,28 Two recent studies investigated whether the JESREC algorithm is appropriate for classifying CRS subgroups into endotypes by unsupervised statistical analysis. The moderate/severe ECRS groups showed type 2–dominant inflammation, whereas non-ECRS/mild ECRS groups showed type 1/type 3 mixed inflammation, suggesting that the CRS clinical scoring system from the JESREC study to some extent reflects the underlying inflammatory endotype.29,30 However, the applicability of the JESREC algorithm, which was developed in a Japanese population and validated by 2 Asian studies, to other regions outside of Asia remains unknown. So far, for definition of nasal mucosal eosinophilia, various cutoff values of tissue eosinophil counts (5–350/hpf) and eosinophil percentage (5%−50%) were proposed by different researchers.25 Besides tissue eosinophil counts, inflammatory endotype markers including IFN-γ (type 1), Charcot-Leyden crystal galectin (type 2), IL-5 (type 2), and IL-17A (type 3) are useful. Genetics, lifestyle, and environmental factors might affect the relationship between endotype and phenotype of CRS, and global studies are required to address this issue.
FIG 1.
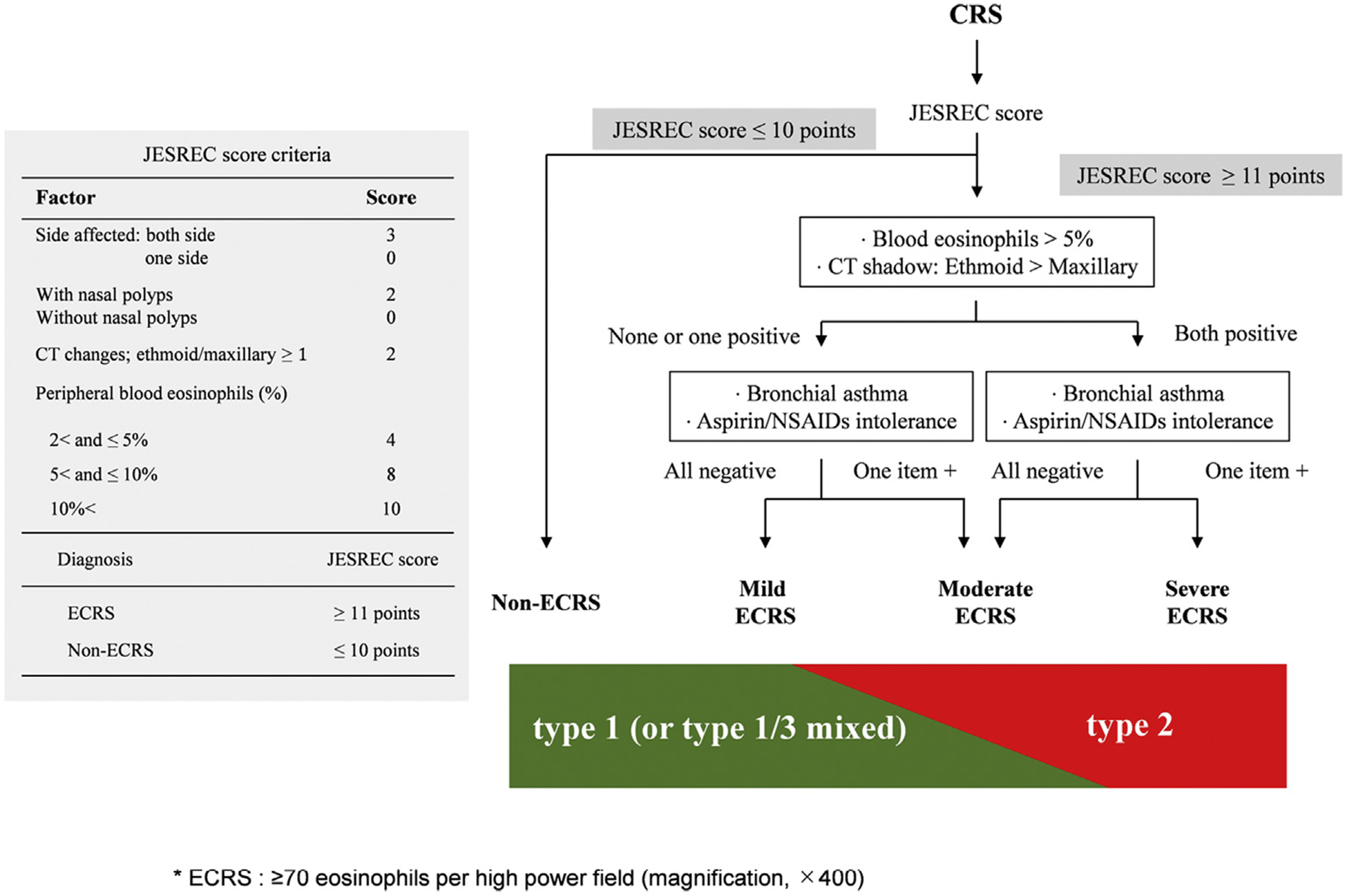
JESREC score criteria for the diagnosis of ECRS (left). Diagnostic algorithm for ECRS severity. Patients with CRS would be classified into 4 groups by JESREC score: blood eosinophilia, ethmoid dominant shadow in CT, and comorbidity (bronchial asthma, aspirin/NSAID intolerance). The moderate/severe ECRS groups showed type 2–dominant inflammation, and non-ECRS/mild ECRS groups showed type 1/type 3 mixed inflammation (right). CT, Computed tomography; NSAID, nonsteroidal anti-inflammatory drug.
ESTABLISHMENT OF TYPE 2 INFLAMMATION IN ECRS
Regardless of ethnicity and geographic region, eosinophilia in patients with CRS strongly correlates with type 2 inflammatory response and generally demonstrates severe symptoms and high recurrence rate, and high prevalence and severity of asthma.6 Therefore, understanding the mechanisms of establishing type 2 inflammation in nasal and paranasal sinus mucosa is therapeutically important. It is clear that type 2 cytokines, especially IL-5 and IL-13 but possibly also IL-4, play important roles mediating inflammation in ECRS and NP development.31 In NPs, these cytokines are produced by mast cells, group 2 innate lymphoid cells (ILC2s), and TH2 cells. Current thinking suggests that an innate response to exogeneous proteases from allergens, such as pollen, mite, fungi, and microorganisms, can induce the epithelial-derived cytokines IL-33, IL-25, and thymic stromal lymphopoietin (TSLP), which drive the activation of ILC2s to release type 2 cytokines in an antigen-independent manner (Fig 2).32 The protease activity of allergens is critical for the secretion of epithelial-derived cytokines, which are involved in the initiation and development of type 2 inflammation. Endogenous protease inhibitors (EPIs) of both endogenous and exogenous proteases are present in various organs to control the adverse effects of proteases, and EPIs may prevent the initiation or exacerbation of type 2 inflammation.33,34 Kouzaki et al35 have recently reported that the endogenous cysteine and serine protease inhibitors, namely, cystatin A and serine protease inhibitor Kazal-type 5 (SPINK5), are less expressed in the NP tissues of patients with ECRS than in the NP tissues of patients with non-ECRS or healthy control subjects. In cell culture and animal experiments, these EPIs decreased the allergen-induced secretion of IL-33, IL-25, and TSLP.35 Taken together, an imbalance between allergen proteases and EPIs might play a role in the initiation and exacerbation of type 2 inflammation in ECRS (Fig 2). The expression of cystatin SN, which is a cysteine protease inhibitor, is significantly increased in the epithelium of NPs from patients with ECRS versus non-ECRS. In addition, the expression of cystatin SN was positively correlated with the eosinophil count and the levels of TSLP, CCL11 (eotaxin-1), and periostin in NPs.36 In vitro experiments revealed that treatment with cystatin SN further increased TSLP expression stimulated by double-stranded RNA plus IL-4 and that stimulation with TSLP or IL-33 increased cystatin SN expression in nasal epithelial cells. Cystatin SN may thus participate in an innate immune type 2 inflammation positive-feedback loop with epithelial-derived cytokines in ECRS. Fibroblasts are also increased in NPs and are associated with the disease severity of CRSwNP.37 The release of CCL11, which belongs to the CC chemokine family, leads to the selective recruitment of eosinophils through binding with CCR3. Hulse et al38 reported that CCL11 expression is increased in NPs and is closely associated with eosinophil infiltration in CRSwNP. Periostin is an extracellular matrix protein that is produced by fibroblasts and is associated with the activation and migration of eosinophils.39,40 Reportedly, periostin levels were increased in ECRS, and cystatin SN stimulation increased periostin expression in cultured fibroblasts isolated from NPs.36,41 These results further reinforce the possible role of cystatin SN in eosinophilic inflammation (Fig 2). According to a report from China, cystatin SN promotes the activation and recruitment of eosinophils through IL-5 induction, further supporting this hypothesis.42 Extracellular endogenous proteases, proteases from allergens, and mast cell proteases may all play key roles in allergic inflammation.43 Mast cells are significantly increased in NPs and are primarily accumulated in the epithelium.44,45 Interestingly, mast cell levels are higher in NPs from patients with ECRS than in NPs from patients with non-ECRS.46 Furthermore, intraepithelial mast cells in NPs showed a mast cell-tryptase (MC-T) but not MC-T/chymase phenotype.44,45 Similar mast cells (MC-T) were increased in a subset of patients with asthma and eosinophilic esophagitis that displayed type 2–dominant inflammation.47,48 Although the mechanisms underlying how these differences in mast cell phenotype influence ECRS pathogenesis remain unclear, several possibilities are noted. Proteinase-activated receptor 2 (PAR-2) contributes to the development of type 2 immunity and eosinophilic inflammation of the airways, and PAR-2 levels were increased in the epithelium of patients with asthma, allergic rhinitis, and ECRS.49,50 Among epithelial-derived cytokines, TSLP is most highly expressed in ECRS, and it strongly enhances the expression of substantial quantities of type 2 cytokines, especially IL-5 and IL-13 in both ILC2s and mast cells, independent of TH2 cells and adaptive immunity.30,51,52 The production of TSLP is induced by airway epithelial cells through PAR-2 signaling.49 Considering that tryptase is a potent activator of PAR-2,53 tryptase/PAR-2 signaling might be involved in the production of TSLP in ECRS. Although mast cells are generally associated with proinflammatory functions, some anti-inflammatory effects of chymase were observed in airway allergic inflammation.54,55 Chymase, but not tryptase, can degrade cytokines such as IL-6, IL-13, and IL-33,55–57 and further supporting a protective role of chymase, clinical evidence showed a positive correlation between chymase-positive mast cells and lung function in severe asthma.58,59 According to these findings, the absence of mast cell chymase in NP epithelium may lead to an excessive accumulation of IL-33, thereby exaggerating type 2 inflammation (Fig 2). It has been reported that IL-33 plays a pivotal role in chronic inflammation of CRSwNP by enhancing TH2-type cytokine production, which could contribute to a further increase of an established TH2 profile in CRSwNP.60 Baba et al61 and Cao et al62 reported that local IgE production and IgE class-switch recombination were increased in ECRS. Because polyclonal IgE antibodies can activate mast cells in NPs,63 increased levels of local IgE production might contribute to eosinophilic inflammation in the patients with CRS. The net impact of mast cells may be a result of a balance between detrimental and protective effects, mediated by distinct mast cell proteases in the main 2 mast cell phenotypes. Further studies are necessary to determine the precise mechanisms by which the interaction between mast cells and NP epithelium influences mucosal immunity. It is also important to point out at this juncture that the relative importance of adaptive (ie, antigen-driven) and innate (eg, protease and epithelial cytokine-driven) activation of mast cells, ILC2s, and TH2 cells is currently unknown and under active investigation.
FIG 2.
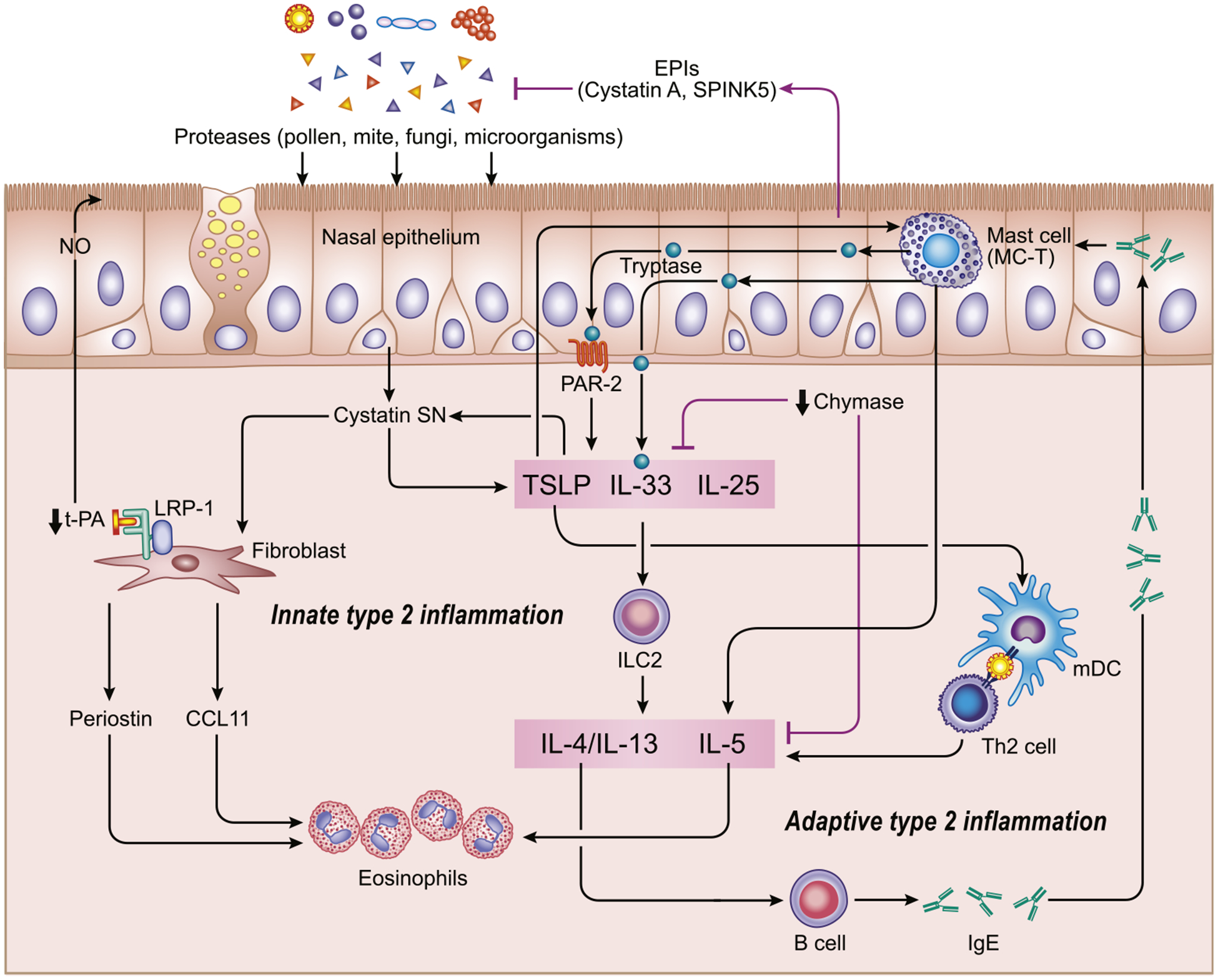
Establishment of type 2 inflammation in ECRS. Innate response to exogeneous proteases from allergens, such as pollen, mite, fungi, and microorganisms, can induce the epithelial-derived cytokines IL-33, IL-25, and TSLP, which drive the activation of ILC2s to release type 2 cytokines in an antigen-independent manner. An imbalance between allergen proteases and EPIs might play a role in the initiation and exacerbation of type 2 inflammation in ECRS. Intraepithelial mast cells, showing a tryptase+, chymase− phenotype (MC-T), enhance type 2 inflammation. Cystatin SN enhances TSLP expression, and that stimulation with TSLP or IL-33 increased cystatin SN expression in nasal epithelial cells. Cystatin SN plays a role in eosinophilic inflammation in ECRS through inducing CCL11 and periostin expression in NPs fibroblast. Downregulation of the tPA/LRP-1 pathway reduces NO production. NO contributes to the local host defense through regulation of ciliary motility. mDC, Myeloid dendritic cell.
NATURE OF NP AND FIBRIN DEPOSITION
NPs are painless benign lesions that originate from around the middle nasal meatus or paranasal sinus cavity, and unlike polyps in the digestive tract or bladder, malignant transformation is extremely rare. NPs have a semitranslucent pale gray appearance, resembling a cystic lesion; nonetheless, the content is mostly a jelly-like solid. Striking histological features of NPs include intense edematous stroma filled with plasma proteins, mainly albumin, and less collagen production and fibrosis.64,65 Shi et al66 compared the remodeling pattern between ECRS and non-ECRS and found more edematous and less fibrotic nasal mucosa in ECRS.21 TGF-β promotes fibroblast proliferation and collagen production, may participate in various pathological inflammation in the upper and lower airway, and is involved in the initiation and maintenance of tissue fibrosis.67 Although conflicting reports exist, TGF-β is reportedly downregulated in ECRS.67,68 Some mechanisms of NP development overlap with asthma remodeling. However, NP-like lesions or intense mucosal edema have not been reported in the lower respiratory tract mucosa in asthma. Interestingly, Nonaka et al69 demonstrated that TGF-β stimulation induced procollagen type 1 synthesis in the lungs but not in nasal fibroblasts, indicating organ specificity of fibroblast responses.
Most studies of ECRS focus on the mechanisms underlying type 2 skewing in nasal mucosa. Although conflicting reports exist, previous clinical trials revealed that biologics directed against IL-5 reduce eosinophils and shrink NPs.70 An important recent clinical trial demonstrated that a biologic directed against IL-4/IL-13 receptor alpha chain significantly decreased NP size.71 These data support a role of type 2 cytokines in NP pathogenesis. IL-5 is a key activation and survival factor for eosinophils in NPs. In contrast, IL-4 and IL-13 are factors that control IgE responses in B cells and plasma cells, enhance mucus and chemokine production in epithelial cells, and activate the endothelium for recruitment of VLA4+ cells such as eosinophils and basophils. A deeper understanding of the mechanisms by which type 2 inflammation drives the formation of NPs will be indispensable for precision medicine. Various inflammatory mediators such as eosinophil granule proteins, mast cell tryptase, and CysLTs increase plasma exudation from capillaries and cause mucosal edema. A high level of CysLTs is a hallmark of aspirin-exacerbated respiratory disease (also recently referred to as nonsteroidal anti-inflammatory drug–exacerbated respiratory disease [N-ERD]), which has very recalcitrant NPs. Recent studies have reported that mutual activation of platelets and eosinophils enhances the production of CysLTs via transcellular metabolism of arachidonic acid in eosinophils.72,73 However, since plasma proteins also pass through the airway epithelial layer, plasma may not only induce and perpetuate edema. Antigen stimulation induces the influx of plasma proteins into the nasal lumen in allergic rhinitis, and substantial levels of plasma proteins also exist in the airway lumen in asthma.74,75 We previously reported excessive fibrin deposition in NP tissue from the patients with CRSwNP.65 Excessive fibrin deposition would be expected to contribute to the retention of exuded plasma proteins from capillaries and thereby perpetuate mucosal edema and play an etiological role in NPs. Our recent study demonstrated that nattokinase, a serine protease possessing strong fibrinolytic activity, effectively shrunk surgically removed NP tissue in vitro through degradation of fibrin in the tissue, whereas papain, a well-known representative of the cysteine protease family, did not.76 Interestingly, fibrin mesh was also shown to be responsible for the viscosity of mucus in these studies. These results reinforce the view that profound fibrin deposition might be involved in the retention of plasma proteins, apparent tissue remodeling, intense edema, and mucosal adherence of mucus in NPs. Fibrin is an end product of the coagulation cascade and plays a major role in blood clotting at the site of vascular injury. In addition, because components of the coagulation factors reside in, or are transported to, tissue and can stimulate extravascular fibrin formation, fibrin production in response to inflammation can be integral to normal repair and restoration of tissues.75 This is believed to play a role in the confinement of toxic agents or microbial organisms to a limited area and in the formation of a provisional matrix for the influx of fibroblasts, endothelial cells, and monocytes.77 Suppression of fibrin turnover facilitates fibrin deposition and can have proinflammatory properties.78 Indeed, several studies demonstrated that removal of fibrin can ameliorate disease development and symptoms.75,78–80
INCREASES IN LEVELS OF COAGULATION FACTORS IN NPs
Tissue factor (TF) initiates the extrinsic coagulation cascade and subsequent fibrin deposition. Leakage of coagulation factors contained in the exuded plasma into tissues facilitates factor VIIa binding to TF on cell surfaces, and the formed complex activates factor Xa, which in turn leads to thrombin generation and subsequent fibrin clot formation. Eosinophils were found to express TF, which initiates the extrinsic coagulation cascade and subsequent fibrin deposition, and significant positive correlations were observed between eosinophil numbers and fibrin deposition levels in NP tissue.81,82 It is reasonable to propose that TF expressed in eosinophils has a role in the formation of excessive fibrin deposition, which might be involved in tissue remodeling in ECRS. Although binding of TF to factor VIIa on cell surfaces is necessary for the initiation of the extrinsic coagulation cascade, preformed TF is stored in the intracellular compartment of eosinophils under resting conditions.75 Proteomic analysis revealed that L-plastin, a leukocyte-specific actin-binding protein, was upregulated in NPs from patients with aspirin-exacerbated respiratory disease, which is the most severe type of ECRS. Immunohistochemistry data showed that TF was most prominently located on the periphery of eosinophils and colocalized with L-plastin in NPs. In a cell culture experiment using Eol-1 cells, a human eosinophil cell line, silencing L-plastin disrupted TF translocation to the cell surface.81 Therefore, it is reasonable to speculate that L-plastin might be involved in the translocation of TF to the eosinophil cell surface, which in turn initiates the extrinsic coagulation cascade by binding to factor VIIa and induces subsequent excessive fibrin deposition in the nasal mucosa of patient with ECRS (Fig 3).
FIG 3.
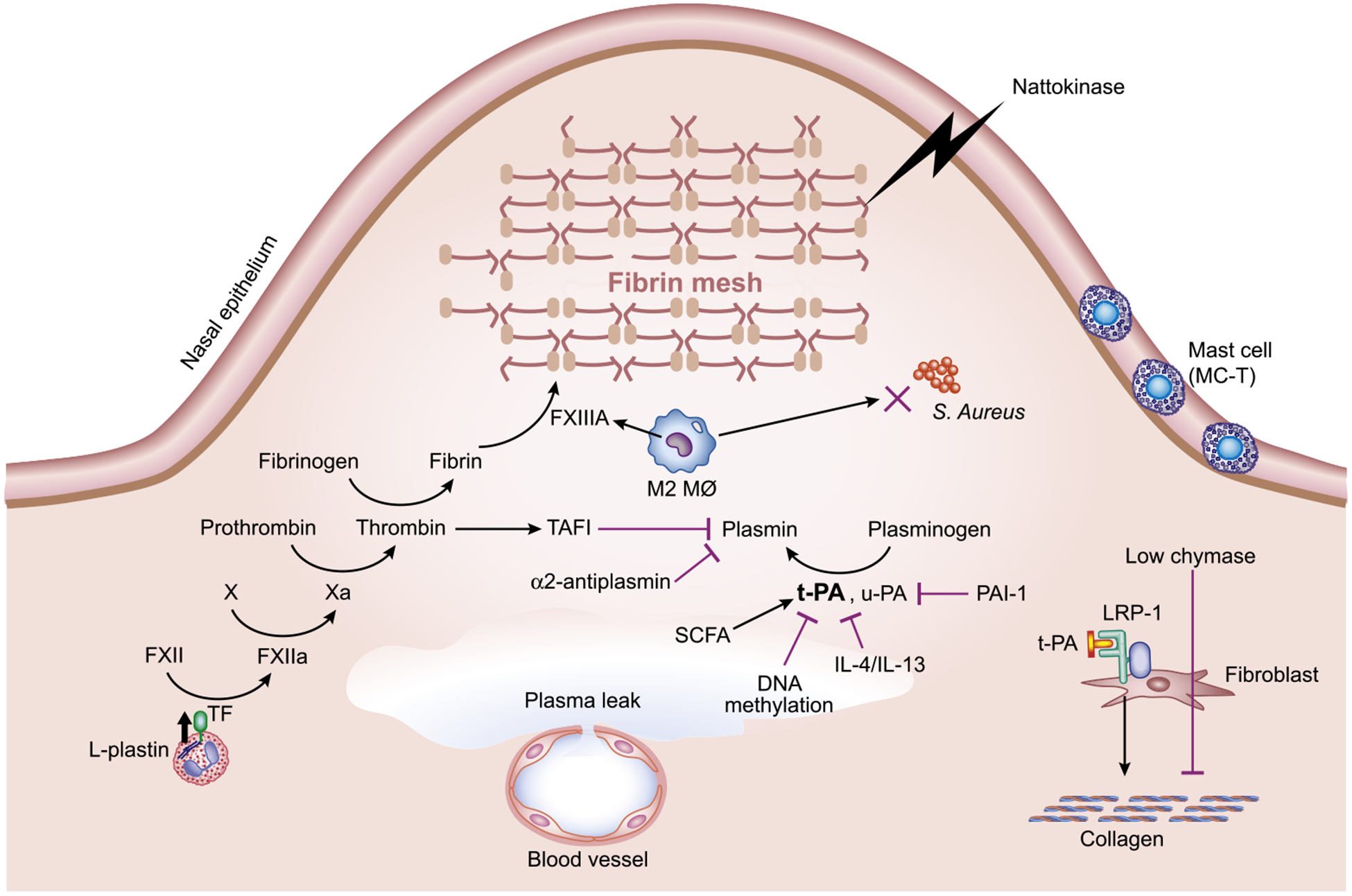
Hypothetical models to explain the role of coagulation and fibrinolysis in excessive fibrin deposition and low fibrosis in eosinophilic NPs. L-plastin might be involved in the translocation of TF to the eosinophil cell surface, which in turn initiates coagulation cascade and induces subsequent fibrin deposition in nasal mucosa. Increased expression of TAFI downregulates plasmin activity. Type 2 inflammation leads to the recruitment of M2 macrophages and the subsequent production of FXIII-A, which induces fibrin cross-linking. Type 2 cytokine and DNA methylation attenuates tPA expression, causing impaired plasmin generation, which in turn decreased fibrinolysis. Nattokinase shrinks NPs through strong fibrinolytic activity. SCFAs mediate tPA expression in nasal epithelial cells.
Thrombin, as a component of the coagulation cascade, also regulates fibrinolysis. In the fibrinolysis cascade, the initial cleavage of fibrin by plasmin generates C-terminal lysine residues on fibrin that are capable of binding both plasminogen and plasminogen activator. Generation of C-terminal lysine residues stimulates plasminogen activator–mediated plasminogen activation and further accelerates fibrinolysis. Thrombin activatable fibrinolysis inhibitor (TAFI) regulates this feedback by cleaving the newly formed C-terminal lysine residues from the fibrin surface and decreasing cofactor activity. TAFI is a carboxypeptidase zymogen originally identified in plasma, which is synthesized mainly in the liver and by megakaryocytes. Increased levels of TAFI have been reported in several inflammatory diseases and reflect the degree of inflammation.83 Imoto et al84 recently reported that the levels of TAFI were significantly increased in NPs from patients with CRSwNP. They also found that the levels of TAFI in all subjects with CRS were significantly elevated in subjects with asthma compared with those without asthma. Taken together, thrombin might play a central role in forming a vicious cycle, leading to activation of the coagulation and attenuation of fibrinolysis, that may result in tissue remodeling in NP (Fig 3).
We previously reported that coagulation factor XIII (FXIII) A levels are increased in NP tissue from patients with CRSwNP and that M2 macrophages are the sole or major FXIII-A–producing cells in NPs.74 FXIII is a transglutaminase that participates in the final stage of the coagulation cascade. There are 2 forms of FXIII. Plasma FXIII consists of 2 enzymatically active A subunits and 2 inhibitor/carrier B subunits, whereas cellular FXIII is a dimer of FXIII-A, present in platelets, monocytes, and macrophages. FXIII catalyzes the formation of covalent cross-links between γ-glutamyl and ε-lysyl residues on adjacent fibrin chains in polymerized fibrin to yield the mature clot and cross-links α2-antiplasmin with fibrin. The cross-linking of fibrin enhances its stiffness and rigidity, which allows it to retain plasma proteins. Cross-linking of α2-antiplasmin to fibrin in the matrix has the predominant role of protecting the newly formed fibrin from degradation by the fibrinolytic enzyme plasmin.85 Macrophages are now widely recognized to be polarized by their microenvironment, especially by TH cytokines and pathogens. Classically activated macrophages (also known as M1 macrophages) develop in response to proinflammatory stimuli, such as type 1 cytokines (IFN-γ) or bacterial products (LPS). In contrast, alternatively activated macrophages are induced by exposure to type 2 cytokines, including IL-4 and IL-13, and are therefore called M2 macrophages. The numbers of M2 macrophages, but not M1 macrophages, were dramatically increased in NPs.74,86 It is possible that the increased numbers of M2 macrophages could be explained by either the type 2 inflammatory milieu of CRSwNP or the presence of mast cells and ILC2s, as demonstrated in recent reports.3 A strong role for colonization with Staphylococcus aureus in driving ECRS has been advocated by several researchers. Krysko et al86 reported that the phagocytic capacity of M2 macrophages is impaired in NP tissue, indicating that decreased phagocytosis of S aureus and predominance of M2 macrophages in CRSwNP could potentially contribute to the persistence of chronic inflammation in CRSwNP (Fig 3).86
FIBRINOLYTIC IMPAIRMENT CAUSES ABNORMAL FIBRIN DEPOSITION IN NPs
The serine protease plasmin is responsible for the degradation of cross-linked fibrin (ie, fibrinolysis) to prevent or reverse excess fibrin deposition in tissue. Plasmin is generated through cleavage of the proenzyme plasminogen by 2 physiological plasminogen activators, urokinase plasminogen activator (uPA) and tissue plasminogen activator (tPA). The activities of uPA and tPA are inhibited by plasminogen activator inihibitor-1.
We have previously found that NPs from patients with CRSwNP have significantly decreased levels of tPA, but not uPA, which could diminish fibrinolysis and in turn contribute to excessive fibrin deposition in this disease.65 In addition, Stevens et al87 reported further reduction of tPA gene expression in NPs from patients with aspirin-exacerbated respiratory disease. Such profound reduction of tPA in nasal mucosa could nearly completely eliminate the fibrinolytic mechanism and potentially lead to greatly enhanced fibrin deposition in nasal mucosa, forming NPs (Fig 3).
It has long been known that type 2 inflammation with predominant mucosal eosinophilia significantly correlates with disease severity of CRS. Levels of tPA, but not uPA, were significantly negatively correlated with eosinophil cationic protein as a marker of type 2 inflammation, and immunohistochemical analysis revealed that tPA expression was mainly observed in epithelial cells in nasal mucosa. We have also found that nasal epithelial cells constitutively express tPA and that stimulation with type 2 cytokines IL-4 and IL-13 significantly downregulated tPA expression while leaving uPA expression unaltered.65 These findings suggest that type 2 inflammation in NPs might downregulate the expression of tPA and play a role in the induction of excessive fibrin deposition through suppression of fibrinolysis (Fig 3). A recent large phase 3 clinical trial demonstrated that dupilumab, a fully human mAb that inhibits signaling of IL-4 and IL-13, significantly decreased NP size.71 Although it was not tested, it is reasonable to speculate that dupilumab blocks IL-4/IL-13 signaling and may subsequently restore tPA production in nasal epithelial cells, which would then activate fibrinolysis and result in NP shrinkage. We do note that fibrin deposition is not restricted to NPs associated with type 2 inflammation, and are aware of studies by Z. Liu et al showing that fibrin deposition is a feature of noneosinophilic polyps in Wuhan, China (personal communication, 2020). This suggests that fibrin deposition can be a feature of more than just the type 2 endotype of CRSwNP.
Gene expression patterns are strongly influenced by epigenetic changes. Hypermethylation of a promoter region can result in silencing gene expression by folding the chromatin structure and inhibiting transcription initiation. Conversely, hypomethylation of an inactive gene promoter region can increase gene expression levels by spreading chromatin compaction.88 It has been reported that DNA methylation changes were induced by type 2 cytokines in asthmatic airway tissues and might contribute to asthma phenotype.89 Kidoguchi et al90 have reported that the proximal promoter, especially at the −618 CpG site of PLAT (tPA; gene name PLAT), was hypermethylated in NPs, and the degree of methylation was negatively correlated with expression levels of PLAT.90 They concluded that hypermethylation of the proximal PLAT promoter may lead to decreased gene expression in NPs, leading to abnormal fibrin deposition by impairment of fibrinolysis.
If it is the case that regulation of tPA expression can be a key factor for NP development, particularly in type 2 inflammatory milieu, then it may be feasible to diminish NP formation by inducing tPA, which would cause fibrinolysis and NP dissolution. The underlying mechanisms of the induction of tPA gene and tPA activity in various cells were reported.91 Short-chain fatty acids (SCFAs) are carboxylic acids with 1 to 6 carbon atoms. Acetic acid, propionic acid, and butyric acid are important species of SCFAs and produced by microbiota in the distal small intestine and colon. Beneficial effects of SCFAs on human health have been reported, and SCFAs exhibit their function via specific SCFA receptors, including G protein–coupled receptor (GPR) 41/free fatty acid receptor 3 and GPR43/free fatty acid receptor 2.92 Imoto et al93 recently showed that SCFAs, especially propionic acid, butyric acid, and valeric acid, induced the expression and release of tPA by airway epithelial cells and that the tPA released was in the active form. They also found that the SCFA receptors GPR41 and GPR43 are expressed by nasal epithelial cells and can mediate the induction of tPA after stimulation with SCFAs.93 These findings support the rationale of topical administration of SCFAs or other agonists of these receptors in patients with NPs to induce tPA and potentially shrink NPs.
A growing body of evidence suggests that tPA can function as a cytokine and bind to the cell membrane receptor low-density-lipoprotein receptor–related protein-1 (LRP-1). Independent of its proteolytic effect, binding by t-PA to LRP-1 induces receptor tyrosine phosphorylation, triggers intracellular signaling, and induces collagen production by fibroblasts.94 LRP-1 is ubiquitously expressed, and it can be detected in nasal mucosa (our unpublished data, 2019). Thus, the reduction in levels of tPA that has been reported in CRSwNP may also explain the reduction in collagen production that has been reported in NPs.65 In addition, it has been reported that the t-PA/LRP-1 pathway facilitates nitric oxide (NO) production in the central nervous system.95 We have recently demonstrated that nasal NO levels were significantly decreased in patients with ECRS compared with patients without ECRS and that nasal NO may be useful as a marker of ECRS severity.96 It is not clear whether downregulation of the t-PA/LRP-1 pathway is responsible for the decreased levels of nasal NO in ECRS however. The upper airway, especially the paranasal sinuses, is the major source of NO in the respiratory tract, and studies have demonstrated the multifunctional role of NO in homeostasis. NO contributes to local host defenses against bacterial, viral, and fungal infection and helps to maintain a bacteriostatic state in the nasal and paranasal sinuses.97 It has been reported that low NO levels impair mucociliary function in the upper airways.98 Indeed, low nasal NO levels in patients with cystic fibrosis and primary dyskinesia tend to increase the susceptibility of these patient groups to airway infection. It has been well documented that the nasal or paranasal sinuses of patients with CRSwNP are often chronically colonized with fungi and bacteria and that these microbes may play an important role in skewing toward type 2 inflammatory condition in CRS.38 S aureus colonization is also common in NPs, and many studies have demonstrated that S aureus and its superantigens play an important role in the pathogenesis of ECRS. Wang et al99 have shown that the presence of Staphylococcal enterotoxin–specific IgE within the NP tissue varied in parallel with the type 2 inflammation signature. Because NO contributes to the local host defense against bacterial infection and because it regulates ciliary motility for adequate clearance of foreign material from the respiratory tract, decreased nasal NO production might be involved in the pathogenesis of ECRS.
Area specificity of NP formation is one of the most intriguing questions in the study of CRS. The mechanisms explaining why NPs arise from only in and around the middle nasal meatus and paranasal sinuses but not inferior turbinate (IT) have so far not been elucidated. We previously reported that protein levels of plasminogen activator, uPA and tPA, were lower in uncinate process tissue in comparison with those seen in IT in disease and control.65 This suggests that lower levels of plasminogen activators might confer an increased susceptibility to excess fibrin deposition in uncinate tissue and explain why NPs arise from the mucous membrane in and around the middle nasal meatus but not in the IT. Our previous studies demonstrated that IT and uncinate tissue differ dramatically in levels of host defense molecules, so such a regional difference might also influence the area specification of NP formation.100,101
SUMMARY AND CONCLUSIONS
CRS is a heterogeneous disease, and geographical or racial differences in inflammatory pattern in nasal mucosa are major issues. Although it was believed that eosinophilic NPs are most common in Western countries and neutrophilic NPs are most common in Asian countries, recent studies reported that the prevalence of eosinophilic NPs is increasing in Asian countries within the last 2 decades, and this trend will possibly be seen throughout the world. International consensus criteria for CRS are required for the accurate understanding of disease pathology and precision medicine. In a multicenter large-scale epidemiological survey, the “JESREC study,” ECRS was definitively defined when the eosinophil count in nasal mucosa is greater than or equal to 70/hpf. In addition, this study proposed an algorithm that classifies CRS into 4 groups according to disease severity. Further detailed studies are required to verify the CRS endotypes that are valid internationally and allow comparison between patient groups and populations around the world. Eosinophilia in patients with CRS correlates strongly with type 2 inflammation response and demonstrates severe symptoms and high recurrence rate and greater prevalence of comorbid asthma. An imbalance between proteases and EPI might play a pivotal role in the initiation and exacerbation of type 2 inflammation in ECRS. Intraepithelial mast cells in NPs, showing a tryptase+, chymase− phenotype (MC-T), may enhance type 2 inflammation. Intense edema and reduced fibrosis are important histological features of eosinophilic NPs, and the main therapeutic target for ECRS is to control NP development. Mucosal edema mainly consists of exuded plasma protein, and excessive fibrin deposition would be expected to contribute to the retention of proteins from capillaries and thereby perpetuate mucosal edema that may play an etiological role in NPs. Upregulation of the coagulation cascade and downregulation of fibrinolysis strongly induce abnormal fibrin deposition in nasal mucosa, and type 2 inflammation plays a central role in the imbalance of coagulation and fibrinolysis. This strong promotion of the formation of a large fibrin mesh is likely to be a primary driver of the formation of NP tissue.
INFORMATION FOR CATEGORY 1 CME CREDIT.
Credit can now be obtained, free for a limited time, by reading the review articles in this issue. Please note the following instructions.
Method of Physician Participation in Learning Process:
The core material for these activities can be read in this issue of the Journal or online at the JACI Web site: www.jacionline.org. The accompanying tests may only be submitted online at www.jacionline.org. Fax or other copies will not be accepted.
Copyright Statement:
Copyright © 2020–2021. All rights reserved.
Overall Purpose/Goal:
To provide excellent reviews on key aspects of allergic disease to those who research, treat, or manage allergic disease.
Target Audience:
Physicians and researchers within the field of allergic disease.
Accreditation/Provider Statements and Credit Designation:
The American Academy of Allergy, Asthma & Immunology (AAAAI) is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to provide continuing medical education for physicians. The AAAAI designates this journal-based CME activity for a maximum of 1.00 AMA PRA Category 1 CreditTM. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
List of Design Committee Members:
Tetsuji Takabayashi, MD, and Robert P. Schleimer, PhD (authors); Zuhair K. Ballas, MD (editor)
Disclosure of Significant Relationships with Relevant Commercial Companies/Organizations:
R. P. Schleimer reports National Institutes of Health grant money paid to the institution and to himself, and consulting fees and/or stock ownership from Allakos, BioMarck, Sanofi, Genentech, Exicure Inc, Aqualung Therapeutics Corp, ActoBio Therapeutics, Lyra Therapeutics, Astellas Pharm Inc, and Genzyme/Sanofi Corp; and has Siglec-8 and Siglec-8 ligand-related patents licensed to Allakos Inc. T. Takabayashi declares no relevant conflicts of interest. Z. K. Ballas (editor) disclosed no relevant financial relationships.
Activity Objectives:
To understand the key protease and endogenous protease inhibitors that contribute to type 2 inflammation within the epithelium of nasal polyps (NPs).
To understand how phenotypic differences in intraepithelial mast cells likely affect NP type 2 inflammation and its clinical implications in allergic disease.
To become familiar with the key mediators of type 2 inflammation in eosinophilic chronic rhinosinusitis (ECRS).
To understand the connection between the coagulation cascade and fibrinolysis and type 2 inflammation.
Recognition of Commercial Support:
This CME activity has not received external commercial support.
List of CME Exam Authors:
Victoria Eng, MD, Amie Nguyen, MD, Javed Sheikh, MD, Shefali A. Samant, MD, and Michael S. Kaplan, MD.
Disclosure of Significant Relationships with Relevant Commercial Companies/Organizations:
The examination authors declare no relevant conflicts of interest.
Acknowledgments
The authors would like to thank Ms Jacqueline Schaffer for the illustrations included in this review.
This research was supported in part by a Grant-in-Aid for Scientific Research (KAKENHI) (C) grant number 16K11207. R. P. Schleimer was supported in part by grants AI137174 on AERD and PO1AI145818 (Chronic Rhinosinusitis Integrative Studies Program 2 [CRISP2]) from the NIH and by The Ernest S. Bazley Charitable Fund.
Abbreviations used
- CRS
Chronic rhinosinusitis
- CRSwNP
CRS with nasal polyps
- CysLT
Cysteinyl leukotriene
- ECRS
Eosinophilic CRS
- EPI
Endogenous protease inhibitor
- FXIII
Factor XIII
- GPR
G protein–coupled receptor
- ILC2
Group 2 innate lymphoid cell
- IT
Inferior turbinate
- JESREC
Japanese Epidemiological Survey of Refractory Eosinophilic Chronic Rhinosinusitis
- LRP-1
Lipoprotein receptor–related protein-1
- MC-T
Mast cell-tryptase
- NO
Nitric oxide
- NP
Nasal polyp
- PAR-2
Proteinase-activated receptor 2
- SCFA
Short-chain fatty acid
- TAFI
Thrombin activatable fibrinolysis inhibitor
- tPA
Tissue plasminogen activator
- TSLP
Thymic stromal lymphopoietin
- uPA
Urokinase plasminogen activator
GLOSSARY
- CCL11 (EOTAXIN-1)
A CC (2 adjacent cysteine residues near the amino terminus) chemokine that is a strong eosinophil and basophil chemoattractant. Binds to CCR3
- CYSTEINYL LEUKOTRIENES
Inflammatory mediators produced from arachidonic acid by 5-lipoxygenase in mast cells, eosinophils, and other inflammatory cells. Cysteinyl leukotrienes include leukotriene C4, leukotriene D4, and leukotriene E4
- FIBROBLASTS
Cells that synthesize most of the extracellular matrix of connective tissue via secretion of collagen and elastin
- GROUP 2 INNATE LYMPHOID CELLS (ILC2s)
Cells possessing lymphoid morphology, but without antigen receptors. They can quickly respond to multiple tissue-derived factors, such as cytokines, eicosanoids, and alarmins, by producing multiple proinflammatory and immunoregulatory cytokines. ILCs’ classification into groups based on their transcription factors and cytokines parallels helper T-cell subsets
- hpf
An area visible by light microscopy under a slide at a magnification of approximately 400 to 500 times
- IL-33
A member of the IL-1 superfamily that functions as an endogenous alarmin. IL-33 helps promote TH2 immune deviation and is a chemoattractant for TH2 cells. IL-33 exists in both a nuclear form and a secreted form. The nuclear form is believed to function as a transcription factor
- MC-T
The “tryptase-only” phenotype of mast cells. MC-T cells are distinguished from MC-T chymase cells by a “scroll” granule pattern under electron microscopy after antitryptase staining. MC-T cells have a relatively low IL-4 and IL-13 cytokine profile in comparison to MC-T chymase cells
- SPINK5
Serine protease inhibitor Kazal type 5. Loss-of-function mutations in SPINK5 result in Netherton syndrome, an autosomal-recessive disorder characterized by profound ichthyosis, as well as atopy
- SUPERANTIGENS
A class of T-cell receptor ligands that have the ability to activate large fractions of the T-cell population. They do not require processing to bind to MHC class II molecules. They bind to polymorphic residues on the periphery of the class II molecule and interact with the T-cell receptor via their Vβ domain
- THROMBIN
One of the key enzymes in blood coagulation. Thrombin was first discovered in the 19th century. Prothrombin (factor II) is converted to thrombin (factor IIa). Thrombin can factor as both a procoagulant and an anticoagulant. Anticoagulant activity occurs when thrombin is bound to thrombomodulin. During coagulation, thrombin cleaves fibrinogen, which activates factor XIII to form a cross-linked fibrin clot
- TISSUE FACTOR
A transmembrane protein important in the extrinsic pathway of coagulation. Tissue factor is usually not expressed on surfaces of cells in contact with blood, but can be upregulated on cell surfaces by proinflammatory mediators. Exposed tissue factor binds to circulating factor VIIa and the complex activates both factor IX and factor X
- UNCINATE PROCESS
A thin, hook-shaped portion of the ethmoid bone that usually overlies the ostium of the maxillary sinus in the lateral wall of the nasal cavity
- VLA41 CELLS
Very late antigen 4. Also known as α4β1 integrin. VLA-4 expression is thought to help promote cellular attachment to the endothelium, a prerequisite for transmigration
REFERENCES
- 1.Fokkens W, Desrosiers M, Harvey R, Hopkins C, Mullol J, Philpott C, et al. EPOS2020: development strategy and goals for the latest European Position Paper on Rhinosinusitis. Rhinology 2019;57:162–8. [DOI] [PubMed] [Google Scholar]
- 2.Orlandi RR, Kingdom TT, Hwang PH, Smith TL, Alt JA, Baroody FM, et al. International Consensus Statement on Allergy and Rhinology: Rhinosinusitis. Int Forum Allergy Rhinol 2016;6:S22–209. [DOI] [PubMed] [Google Scholar]
- 3.Schleimer RP. Immunopathogenesis of chronic rhinosinusitis and nasal polyposis. Annu Rev Pathol 2017;12:331–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bachert C, Zhang N, Hellings PW, Bousquet J. Endotype-driven care pathways in patients with chronic rhinosinusitis. J Allergy Clin Immunol 2018;141: 1543–51. [DOI] [PubMed] [Google Scholar]
- 5.Okuda M [Differences in chronic rhinitis with reference to its incidence and type in Chiba and Vienna]. Monatsschr Ohrenheilkd Laryngorhinol 1969;103: 56–71. [PubMed] [Google Scholar]
- 6.Wang ET, Zheng Y, Liu PF, Guo LJ. Eosinophilic chronic rhinosinusitis in East Asians. World J Clin Cases 2014;2:873–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang W, Gao Y, Zhu Z, Zha Y, Wang X, Qi F, et al. Changes in the clinical and histological characteristics of Chinese chronic rhinosinusitis with nasal polyps over 11 years. Int Forum Allergy Rhinol 2019;9:149–57. [DOI] [PubMed] [Google Scholar]
- 8.Kim SJ, Lee KH, Kim SW, Cho JS, Park YK, Shin SY. Changes in histological features of nasal polyps in a Korean population over a 17-year period. Otolaryngol Head Neck Surg 2013;149:431–7. [DOI] [PubMed] [Google Scholar]
- 9.Fujieda S, Imoto Y, Kato Y, Ninomiya T, Tokunaga T, Tsutsumiuchi T, et al. Eosinophilic chronic rhinosinusitis. Allergol Int 2019;68:403–12. [DOI] [PubMed] [Google Scholar]
- 10.Katotomichelakis M, Tantilipikorn P, Holtappels G, De Ruyck N, Feng L, Van Zele T, et al. Inflammatory patterns in upper airway disease in the same geographical area may change over time. Am J Rhinol Allergy 2013;27: 354–60. [DOI] [PubMed] [Google Scholar]
- 11.Mahdavinia M, Suh LA, Carter RG, Stevens WW, Norton JE, Kato A, et al. Increased noneosinophilic nasal polyps in chronic rhinosinusitis in US second-generation Asians suggest genetic regulation of eosinophilia. J Allergy Clin Immunol 2015;135:576–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Batra PS, Tong L, Citardi MJ. Analysis of comorbidities and objective parameters in refractory chronic rhinosinusitis. Laryngoscope 2013;123:S1–11. [DOI] [PubMed] [Google Scholar]
- 13.Tomassen P, Vandeplas G, Van Zele T, Cardell LO, Arebro J, Olze H, et al. Inflammatory endotypes of chronic rhinosinusitis based on cluster analysis of biomarkers. J Allergy Clin Immunol 2016;137:1449–56.e4. [DOI] [PubMed] [Google Scholar]
- 14.Langdon C, Mullol J. Nasal polyps in patients with asthma: prevalence, impact, and management challenges. J Asthma Allergy 2016;9:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kowalski ML, Agache I, Bavbek S, Bakirtas A, Blanca M, Bochenek G, et al. Diagnosis and management of NSAID-exacerbated respiratory disease (N-ERD)—a EAACI position paper. Allergy 2019;74:28–39. [DOI] [PubMed] [Google Scholar]
- 16.Proimos E, Papadakis CE, Chimona TS, Kiagiadaki D, Ferekidis E, Yiotakis J. The effect of functional endoscopic sinus surgery on patients with asthma and CRS with nasal polyps. Rhinology 2010;48:331–8. [DOI] [PubMed] [Google Scholar]
- 17.Dejima K, Hama T, Miyazaki M, Yasuda S, Fukushima K, Oshima A, et al. A clinical study of endoscopic sinus surgery for sinusitis in patients with bronchial asthma. Int Arch Allergy Immunol 2005;138:97–104. [DOI] [PubMed] [Google Scholar]
- 18.Okano M, Kariya S, Ohta N, Imoto Y, Fujieda S, Nishizaki K. Association and management of eosinophilic inflammation in upper and lower airways. Allergol Int 2015;64:131–8. [DOI] [PubMed] [Google Scholar]
- 19.Choi JH, Kim MA, Park HS. An update on the pathogenesis of the upper airways in aspirin-exacerbated respiratory disease. Curr Opin Allergy Clin Immunol 2014; 14:1–6. [DOI] [PubMed] [Google Scholar]
- 20.Szucs E, Ravandi S, Goossens A, Beel M, Clement PA. Eosinophilia in the ethmoid mucosa and its relationship to the severity of inflammation in chronic rhinosinusitis. Am J Rhinol 2002;16:131–4. [PubMed] [Google Scholar]
- 21.Kountakis SE, Arango P, Bradley D, Wade ZK, Borish L. Molecular and cellular staging for the severity of chronic rhinosinusitis. Laryngoscope 2004;114: 1895–905. [DOI] [PubMed] [Google Scholar]
- 22.Sejima T, Holtappels G, Kikuchi H, Imayoshi S, Ichimura K, Bachert C. Cytokine profiles in Japanese patients with chronic rhinosinusitis. Allergol Int 2012;61: 115–22. [DOI] [PubMed] [Google Scholar]
- 23.Takeno S, Hirakawa K, Ishino T. Pathological mechanisms and clinical features of eosinophilic chronic rhinosinusitis in the Japanese population. Allergol Int 2010;59:247–56. [DOI] [PubMed] [Google Scholar]
- 24.Jiang N, Kern RC, Altman KW. Histopathological evaluation of chronic rhinosinusitis: a critical review. Am J Rhinol Allergy 2013;27:396–402. [DOI] [PubMed] [Google Scholar]
- 25.Lou H, Zhang N, Bachert C, Zhang L. Highlights of eosinophilic chronic rhinosinusitis with nasal polyps in definition, prognosis, and advancement. Int Forum Allergy Rhinol 2018;8:1218–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tokunaga T, Sakashita M, Haruna T, Asaka D, Takeno S, Ikeda H, et al. Novel scoring system and algorithm for classifying chronic rhinosinusitis: the JESREC study. Allergy 2015;70:995–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao PP, Wang ZC, Schleimer RP, Liu Z. Pathophysiologic mechanisms of chronic rhinosinusitis and their roles in emerging disease endotypes. Ann Allergy Asthma Immunol 2019;122:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liao B, Liu JX, Li ZY, Zhen Z, Cao PP, Yao Y, et al. Multidimensional endotypes of chronic rhinosinusitis and their association with treatment outcomes. Allergy 2018;73:1459–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakayama T, Sugimoto N, Okada N, Tsurumoto T, Mitsuyoshi R, Takaishi S, et al. JESREC score and mucosal eosinophilia can predict endotypes of chronic rhinosinusitis with nasal polyps. Auris Nasus Larynx 2019;46:374–83. [DOI] [PubMed] [Google Scholar]
- 30.Kim DK, Kang SI, Kong IG, Cho YH, Song SK, Hyun SJ, et al. Two-track medical treatment strategy according to the clinical scoring system for chronic rhinosinusitis. Allergy Asthma Immunol Res 2018;10:490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kato A Immunopathology of chronic rhinosinusitis. Allergol Int 2015;64:121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ebbo M, Crinier A, Vély F, Vivier E. Innate lymphoid cells: major players in inflammatory diseases. Nat Rev Immunol 2017;17:665–78. [DOI] [PubMed] [Google Scholar]
- 33.Ogawa T, Takai T, Kato T, Kikuchi Y, Niyonsaba F, Ikeda S, et al. Upregulation of the release of granulocyte-macrophage colony-stimulating factor from keratinocytes stimulated with cysteine protease activity of recombinant major mite allergens, Der f 1 and Der p 1. Int Arch Allergy Immunol 2008; 146:27–35. [DOI] [PubMed] [Google Scholar]
- 34.Richer SL, Truong-Tran AQ, Conley DB, Carter R, Vermylen D, Grammer LC, et al. Epithelial genes in chronic rhinosinusitis with and without nasal polyps. Am J Rhinol 2008;22:228–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kouzaki H, Matsumoto K, Kikuoka H, Kato T, Tojima I, Shimizu S, et al. Endogenous protease inhibitors in airway epithelial cells contribute to eosinophilic chronic rhinosinusitis. Am J Respir Crit Care Med 2017;195:737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kato Y, Takabayashi T, Sakashita M, Imoto Y, Tokunaga T, Ninomiya T, et al. Expression and functional analysis of CST1 in intractable nasal polyps. Am J Respir Cell Mol Biol 2018;59:448–57. [DOI] [PubMed] [Google Scholar]
- 37.Park SK, Jin YD, Park YK, Yeon SH, Xu J, Han RN, et al. IL-25-induced activation of nasal fibroblast and its association with the remodeling of chronic rhinosinusitis with nasal polyposis. PLoS One 2017;12:e0181806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hulse KE, Stevens WW, Tan BK, Schleimer RP. Pathogenesis of nasal polyposis. Clin Exp Allergy 2015;45:328–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jia G, Erickson RW, Choy DF, Mosesova S, Wu LC, Solberg OD, et al. Periostin is a systemic biomarker of eosinophilic airway inflammation in asthmatic patients. J Allergy Clin Immunol 2012;130:647–54.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noguchi T, Nakagome K, Kobayashi T, Uchida Y, Soma T, Nakamoto H, et al. Periostin upregulates the effector functions of eosinophils. J Allergy Clin Immunol 2016;138:1449–52.e5. [DOI] [PubMed] [Google Scholar]
- 41.Ninomiya T, Noguchi E, Haruna T, Hasegawa M, Yoshida T, Yamashita Y, et al. Periostin as a novel biomarker for postoperative recurrence of chronic rhinosinitis with nasal polyps. Sci Rep 2018;8:11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan B, Lou H, Wang Y, Li Y, Meng Y, Qi S, et al. Epithelium-derived cystatin SN enhances eosinophil activation and infiltration through IL-5 in patients with chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol 2019;144: 455–69. [DOI] [PubMed] [Google Scholar]
- 43.Amin K The role of mast cells in allergic inflammation. Respir Med 2012;106: 9–14. [DOI] [PubMed] [Google Scholar]
- 44.Takabayashi T, Kato A, Peters AT, Suh LA, Carter R, Norton J, et al. Glandular mast cells with distinct phenotype are highly elevated in chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol 2012;130:410–20.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shaw JL, Ashoori F, Fakhri S, Citardi MJ, Luong A. Increased percentage of mast cells within sinonasal mucosa of chronic rhinosinusitis with nasal polyp patients independent of atopy. Int Forum Allergy Rhinol 2012;2:233–40. [DOI] [PubMed] [Google Scholar]
- 46.Baba S, Kondo K, Suzukawa M, Ohta K, Yamasoba T. Distribution, subtype population, and IgE positivity of mast cells in chronic rhinosinusitis with nasal polyps. Ann Allergy Asthma Immunol 2017;119:120–8. [DOI] [PubMed] [Google Scholar]
- 47.Dougherty RH, Sidhu SS, Raman K, Solon M, Solberg OD, Caughey GH, et al. Accumulation of intraepithelial mast cells with a unique protease phenotype in T(H)2-high asthma. J Allergy Clin Immunol 2010;125:1046–53.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abonia JP, Blanchard C, Butz BB, Rainey HF, Collins MH, Stringer K, et al. Involvement of mast cells in eosinophilic esophagitis. J Allergy Clin Immunol 2010;126:140–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kouzaki H, O’Grady SM, Lawrence CB, Kita H. Proteases induce production of thymic stromal lymphopoietin by airway epithelial cells through protease-activated receptor-2. J Immunol 2009;183:1427–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dinh QT, Cryer A, Trevisani M, Dinh S, Wu S, Cifuentes LB, et al. Gene and protein expression of protease-activated receptor 2 in structural and inflammatory cells in the nasal mucosa in seasonal allergic rhinitis. Clin Exp Allergy 2006;36: 1039–48. [DOI] [PubMed] [Google Scholar]
- 51.Nagarkar DR, Poposki JA, Tan BK, Comeau MR, Peters AT, Hulse KE, et al. Thymic stromal lymphopoietin activity is increased in nasal polyps of patients with chronic rhinosinusitis. J Allergy Clin Immunol 2013;132: 593–600.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ogasawara N, Klingler AI, Tan BK, Poposki JA, Hulse KE, Stevens WW, et al. Epithelial activators of type 2 inflammation: elevation of thymic stromal lymphopoietin, but not IL-25 or IL-33, in chronic rhinosinusitis with nasal polyps in Chicago, Illinois. Allergy 2018;73:2251–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Levi-Schaffer F, Piliponsky AM. Tryptase, a novel link between allergic inflammation and fibrosis. Trends Immunol 2003;24:158–61. [DOI] [PubMed] [Google Scholar]
- 54.Waern I, Jonasson S, Hjoberg J, Bucht A, Abrink M, Pejler G, et al. Mouse mast cell protease 4 is the major chymase in murine airways and has a protective role in allergic airway inflammation. J Immunol 2009;183:6369–76. [DOI] [PubMed] [Google Scholar]
- 55.Waern I, Lundequist A, Pejler G, Wernersson S. Mast cell chymase modulates IL-33 levels and controls allergic sensitization in dust-mite induced airway inflammation. Mucosal Immunol 2013;6:911–20. [DOI] [PubMed] [Google Scholar]
- 56.Roy A, Ganesh G, Sippola H, Bolin S, Sawesi O, Dag€alv A, et al. Mast cell chymase degrades the alarmins heat shock protein 70, biglycan, HMGB1, and interleukin-33 (IL-33) and limits danger-induced inflammation. J Biol Chem 2014;289:237–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao W, Oskeritzian CA, Pozez AL, Schwartz LB. Cytokine production by skin-derived mast cells: endogenous proteases are responsible for degradation of cytokines. J Immunol 2005;175:2635–42. [DOI] [PubMed] [Google Scholar]
- 58.Balzar S, Chu HW, Strand M, Wenzel S. Relationship of small airway chymase-positive mast cells and lung function in severe asthma. Am J Respir Crit Care Med 2005;171:431–9. [DOI] [PubMed] [Google Scholar]
- 59.Zanini A, Chetta A, Saetta M, Baraldo S, D’Ippolito R, Castagnaro A, et al. Chymase-positive mast cells play a role in the vascular component of airway remodeling in asthma. J Allergy Clin Immunol 2007;120:329–33. [DOI] [PubMed] [Google Scholar]
- 60.Soyka MB, Holzmann D, Basinski TM, Wawrzyniak M, Bannert C, B€urgler S, et al. The induction of IL-33 in the sinus epithelium and its influence on T-helper cell responses. PLoS One 2015;10:e0123163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baba S, Kondo K, Toma-Hirano M, Kanaya K, Suzukawa K, Ushio M, et al. Local increase in IgE and class switch recombination to IgE in nasal polyps in chronic rhinosinusitis. Clin Exp Allergy 2014;44:701–12. [DOI] [PubMed] [Google Scholar]
- 62.Cao PP, Zhang YN, Liao B, Ma J, Wang BF, Wang H, et al. Increased local IgE production induced by common aeroallergens and phenotypic alteration of mast cells in Chinese eosinophilic, but not non-eosinophilic, chronic rhinosinusitis with nasal polyps. Clin Exp Allergy 2014;44:690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang N, Holtappels G, Gevaert P, Patou J, Dhaliwal B, Gould H, et al. Mucosal tissue polyclonal IgE is functional in response to allergen and SEB. Allergy 2011; 66:141–8. [DOI] [PubMed] [Google Scholar]
- 64.Bachert C, Gevaert P, Holtappels G, Cuvelier C, van Cauwenberge P. Nasal polyposis: from cytokines to growth. Am J Rhinol 2000;14:279–90. [DOI] [PubMed] [Google Scholar]
- 65.Takabayashi T, Kato A, Peters AT, Hulse KE, Suh LA, Carter R, et al. Excessive fibrin deposition in nasal polyps caused by fibrinolytic impairment through reduction of tissue plasminogen activator expression. Am J Respir Crit Care Med 2013;187:49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shi LL, Xiong P, Zhang L, Cao PP, Liao B, Lu X, et al. Features of airway remodeling in different types of Chinese chronic rhinosinusitis are associated with inflammation patterns. Allergy 2013;68:101–9. [DOI] [PubMed] [Google Scholar]
- 67.Van Bruaene N, Derycke L, Perez-Novo CA, Gevaert P, Holtappels G, De Ruyck N, et al. TGF-beta signaling and collagen deposition in chronic rhinosinusitis. J Allergy Clin Immunol 2009;124:253–9, 259.e1–2. [DOI] [PubMed] [Google Scholar]
- 68.Otto BA, Wenzel SE. The role of cytokines in chronic rhinosinusitis with nasal polyps. Curr Opin Otolaryngol Head Neck Surg 2008;16:270–4. [DOI] [PubMed] [Google Scholar]
- 69.Nonaka M, Pawankar R, Fukumoto A, Yagi T. Heterogeneous response of nasal and lung fibroblasts to transforming growth factor-beta 1. Clin Exp Allergy 2008; 38:812–21. [DOI] [PubMed] [Google Scholar]
- 70.Gevaert P, Van Bruaene N, Cattaert T, Van Steen K, Van Zele T, Acke F, et al. Mepolizumab, a humanized anti-IL-5 mAb, as a treatment option for severe nasal polyposis. J Allergy Clin Immunol 2011;128:989–95.e1–8. [DOI] [PubMed] [Google Scholar]
- 71.Bachert C, Han JK, Desrosiers M, Hellings PW, Amin N, Lee SE, et al. Efficacy and safety of dupilumab in patients with severe chronic rhinosinusitis with nasal polyps (LIBERTY NP SINUS-24 and LIBERTY NP SINUS-52): results from two multicentre, randomised, double-blind, placebo-controlled, parallel-group phase 3 trials. Lancet 2019;394:1638–50. [DOI] [PubMed] [Google Scholar]
- 72.Mitsui C, Kajiwara K, Hayashi H, Ito J, Mita H, Ono E, et al. Platelet activation markers overexpressed specifically in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol 2016;137:400–11. [DOI] [PubMed] [Google Scholar]
- 73.Laidlaw TM, Boyce JA. Platelets in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol 2015;135:1407–14; quiz 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Takabayashi T, Kato A, Peters AT, Hulse KE, Suh LA, Carter R, et al. Increased expression of factor XIII-A in patients with chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol 2013;132:584–92.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.de Boer JD, Majoor CJ, van’t Veer C, Bel EH, van der Poll T. Asthma and coagulation. Blood 2012;119:3236–44. [DOI] [PubMed] [Google Scholar]
- 76.Takabayashi T, Imoto Y, Sakashita M, Kato Y, Tokunaga T, Yoshida K, et al. Nattokinase, profibrinolytic enzyme, effectively shrinks the nasal polyp tissue and decreases viscosity of mucus. Allergol Int 2017;66:594–602. [DOI] [PubMed] [Google Scholar]
- 77.Jennewein C, Tran N, Paulus P, Ellinghaus P, Eble JA, Zacharowski K. Novel aspects of fibrin(ogen) fragments during inflammation. Mol Med 2011; 17:568–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Del Rosso M, Fibbi G, Pucci M, Margheri F, Serrati S. The plasminogen activation system in inflammation. Front Biosci 2008;13:4667–86. [DOI] [PubMed] [Google Scholar]
- 79.Hamblin SE, Furmanek DL. Intrapleural tissue plasminogen activator for the treatment of parapneumonic effusion. Pharmacotherapy 2010;30:855–62. [DOI] [PubMed] [Google Scholar]
- 80.Akassoglou K, Adams RA, Bauer J, Mercado P, Tseveleki V, Lassmann H, et al. Fibrin depletion decreases inflammation and delays the onset of demyelination in a tumor necrosis factor transgenic mouse model for multiple sclerosis. Proc Natl Acad Sci U S A 2004;101:6698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Takabayashi T, Tanaka Y, Susuki D, Yoshida K, Tomita K, Sakashita M, et al. Increased expression of L-plastin in nasal polyp of patients with nonsteroidal anti-inflammatory drug-exacerbated respiratory disease. Allergy 2019;74: 1307–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shimizu S, Ogawa T, Takezawa K, Tojima I, Kouzaki H, Shimizu T. Tissue factor and tissue factor pathway inhibitor in nasal mucosa and nasal secretions of chronic rhinosinusitis with nasal polyp. Am J Rhinol Allergy 2015;29:235–42. [DOI] [PubMed] [Google Scholar]
- 83.Owczarek D, Undas A, Foley JH, Nesheim ME, Jab1onski K, Mach T. Activated thrombin activatable fibrinolysis inhibitor (TAFIa) is associated with inflammatory markers in inflammatory bowel diseases: TAFIa level in patients with IBD. J Crohns Colitis 2012;6:13–20. [DOI] [PubMed] [Google Scholar]
- 84.Imoto Y, Kato A, Takabayashi T, Stevens W, Norton JE, Suh LA, et al. Increased thrombin-activatable fibrinolysis inhibitor levels in patients with chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol 2019;144: 1566–74.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bagoly Z, Katona E, Muszbek L. Factor XIII and inflammatory cells. Thromb Res 2012;129:S77–81. [DOI] [PubMed] [Google Scholar]
- 86.Krysko O, Holtappels G, Zhang N, Kubica M, Deswarte K, Derycke L, et al. Alternatively activated macrophages and impaired phagocytosis of S. aureus in chronic rhinosinusitis. Allergy 2011;66:396–403. [DOI] [PubMed] [Google Scholar]
- 87.Stevens WW, Ocampo CJ, Berdnikovs S, Sakashita M, Mahdavinia M, Suh L, et al. Cytokines in chronic rhinosinusitis: role in eosinophilia and aspirin-exacerbated respiratory disease. Am J Respir Crit Care Med 2015;192:682–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 2012;13:484–92. [DOI] [PubMed] [Google Scholar]
- 89.Nicodemus-Johnson J, Naughton KA, Sudi J, Hogarth K, Naurekas ET, Nicolae DL, et al. Genome-wide methylation study identifies an IL-13-induced epigenetic signature in asthmatic airways. Am J Respir Crit Care Med 2016; 193:376–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kidoguchi M, Noguchi E, Nakamura T, Ninomiya T, Morii W, Yoshida K, et al. DNA methylation of proximal PLAT promoter in chronic rhinosinusitis with nasal polyps. Am J Rhinol Allergy 2018;32:374–9. [DOI] [PubMed] [Google Scholar]
- 91.Kruithof EK, Dunoyer-Geindre S. Human tissue-type plasminogen activator. Thromb Haemost 2014;112:243–54. [DOI] [PubMed] [Google Scholar]
- 92.Sivaprakasam S, Prasad PD, Singh N. Benefits of short-chain fatty acids and their receptors in inflammation and carcinogenesis. Pharmacol Ther 2016;164: 144–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Imoto Y, Kato A, Takabayashi T, Sakashita M, Norton JE, Suh LA, et al. Short-chain fatty acids induce tissue plasminogen activator in airway epithelial cells via GPR41&43. Clin Exp Allergy 2018;48:544–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hu K, Wu C, Mars WM, Liu Y. Tissue-type plasminogen activator promotes murine myofibroblast activation through LDL receptor-related protein 1-mediated integrin signaling. J Clin Invest 2007;117:3821–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lemarchant S, Docagne F, Emery E, Vivien D, Ali C, Rubio M. tPA in the injured central nervous system: different scenarios starring the same actor? Neuropharmacology 2012;62:749–56. [DOI] [PubMed] [Google Scholar]
- 96.Yoshida K, Takabayashi T, Imoto Y, Sakashita M, Narita N, Fujieda S. Reduced nasal nitric oxide levels in patients with eosinophilic chronic rhinosinusitis. Allergol Int 2019;68:225–32. [DOI] [PubMed] [Google Scholar]
- 97.Maniscalco M, Sofia M, Pelaia G. Nitric oxide in upper airways inflammatory diseases. Inflamm Res 2007;56:58–69. [DOI] [PubMed] [Google Scholar]
- 98.Lindberg S, Cervin A, Runer T. Low levels of nasal nitric oxide (NO) correlate to impaired mucociliary function in the upper airways. Acta Otolaryngol 1997;117: 728–34. [DOI] [PubMed] [Google Scholar]
- 99.Wang X, Zhang N, Bo M, Holtappels G, Zheng M, Lou H, et al. Diversity of T H cytokine profiles in patients with chronic rhinosinusitis: a multicenter study in Europe, Asia, and Oceania. J Allergy Clin Immunol 2016;138:1344–53. [DOI] [PubMed] [Google Scholar]
- 100.Seshadri S, Lin DC, Rosati M, Carter RG, Norton JE, Suh L, et al. Reduced expression of antimicrobial PLUNC proteins in nasal polyp tissues of patients with chronic rhinosinusitis. Allergy 2012;67:920–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tieu DD, Peters AT, Carter RG, Carter RT, Suh L, Conley DB, et al. Evidence for diminished levels of epithelial psoriasin and calprotectin in chronic rhinosinusitis. J Allergy Clin Immunol 2010;125:667–75. [DOI] [PMC free article] [PubMed] [Google Scholar]