

In this study, Yamada et al. set out to define structures of resected double-strand breaks (DSBs) in mouse spermatocytes genome-wide. Using S1-seq, a novel method that maps meiotic DSB resection endpoints genome-wide at single-nucleotide resolution, the authors report previously undocumented characteristics of resection, including global patterns, locus-to-locus variation, and the genetic pathways that control resection. The authors also report novel structures that appear to be intermolecular recombination intermediates and unexpected properties of these intermediates in mice shed light on the mechanism of meiotic recombination and the structure of chromatin at sites undergoing recombination.
Keywords: meiosis, recombination, DNA double-strand breaks, resection, EXO1, ATM, PRDM9, chromatin
Abstract
Exonucleolytic resection, critical to repair double-strand breaks (DSBs) by recombination, is not well understood, particularly in mammalian meiosis. Here, we define structures of resected DSBs in mouse spermatocytes genome-wide at nucleotide resolution. Resection tracts averaged 1100 nt, but with substantial fine-scale heterogeneity at individual hot spots. Surprisingly, EXO1 is not the major 5′ → 3′ exonuclease, but the DSB-responsive kinase ATM proved a key regulator of both initiation and extension of resection. In wild type, apparent intermolecular recombination intermediates clustered near to but offset from DSB positions, consistent with joint molecules with incompletely invaded 3′ ends. Finally, we provide evidence for PRDM9-dependent chromatin remodeling leading to increased accessibility at recombination sites. Our findings give insight into the mechanisms of DSB processing and repair in meiotic chromatin.
Nucleolytic processing of double-strand break (DSB) ends, termed resection, generates the single-stranded DNA (ssDNA) that is used for homology search and strand invasion during recombination (Symington 2014). Despite a decades-long appreciation of resection's central role in DSB repair, however, we lack detailed understanding of resection mechanisms and the fine-scale structure of resected DNA ends in most species, including mammals. We address these issues here through genome-wide analysis of DSB resection during meiosis in mouse spermatocytes.
Meiotic recombination, which ensures homologous chromosome pairing and segregation and enhances genetic diversity, initiates with DSBs made by SPO11 via a covalent protein–DNA intermediate (Fig. 1A, panel i; Lam and Keeney 2014; Hunter 2015). Our current understanding, from the budding yeast Saccharomyces cerevisiae, is that endonucleolytic cleavage by Mre11–Rad50–Xrs2 (MRX) plus Sae2 nicks Spo11-bound strands, releasing Spo11 bound to a short oligonucleotide (Keeney et al. 1997; Neale et al. 2005) and providing entry points for modest 3′ → 5′ Mre11 exonuclease and robust 5′ → 3′ Exo1 exonuclease (Zakharyevich et al. 2010; Garcia et al. 2011; Keelagher et al. 2011; Cannavo and Cejka 2014; Mimitou et al. 2017). However, aside from yeast, meiotic resection mechanisms are unknown. In mice, for example, we do not even know whether EXO1 is required, and we have only a low-resolution population-average view of resection lengths that was deduced indirectly from sequencing of ssDNA bound by the strand exchange protein DMC1 (Lange et al. 2016).
Figure 1.

Nucleotide-resolution maps of meiotic DSB resection in mice. (A) Early recombination steps and S1-seq. (Panel i) SPO11 (magenta ellipses) cuts DNA via a covalent protein–DNA intermediate. SPO11-bound strands are nicked (arrowheads) by MRE11 and associated factors, providing an entry point(s) for exonucleolytic resection and release of SPO11-oligo complexes. Resected ends have 3′-ssDNA ends that serve as substrates for strand-exchange proteins DMC1 and RAD51, which search for a homologous duplex and carry out strand invasion. (Panel ii) In S1-seq, sequencing adaptors are linked to duplex ends generated by removal of ssDNA tails using S1 nuclease. (B) Strand-specific S1-seq (reads per million mapped reads [RPM]) at a representative DSB hot spot. Dmc1+/+ is a Dmc1-proficient control from the same breeding colony as Dmc1–/– null on a mixed background. ExoT-seq uses an exonuclease instead of S1 endonuclease to remove ssDNA tails. SPO11-oligo sequencing data here and throughout are from Lange et al. (2016). The baseline of the Y-axis for each plot is at 0. (C) S1-seq reads averaged around 13,960 SPO11-oligo hot spot centers in wild type and Spo11–/–. (D) Correlation (Pearson's r) of S1-seq read count with DSB intensity measured by SPO11-oligo sequencing. Each point is a SPO11-oligo hot spot, with S1-seq signal summed from −2000 to −250 bp (bottom strand) and +250 to +2000 bp (top strand) around hot spot centers. S1-seq signal was background-corrected by subtracting Spo11–/– signal from the wild-type signal. Hot spots with ≤0 corrected S1-seq tag counts (n = 4050) were excluded for Pearson's r calculation. Hot spots with ≤10−3 corrected S1-seq tag counts (n = 4071) were set as 10−3 for plotting purposes. (E) Stereotyped distribution of resection endpoints around DSB hot spots. (Top panels) Heat maps (data in 40-bp bins) show SPO11 oligos and strand-specific S1-seq or ExoT-seq reads around DSB hot spots. Each line is a hot spot, ranked from strongest at the top. Sequencing signals were clocally normalized by dividing each signal by the total signal in a 4001-bp window around each hot spot's center. Each hot spot thus has a total value of 1, so that spatial patterns can be compared between hot spots of different strengths. (Bottom panels) Locally normalized average profiles (data in 40-bp bins) were shown for all hot spots divided into four groups (q1–q4) based on hot spot heat. (F) Global average patterns of SPO11-specific S1-seq reads at hot spots. S1-seq signal was background-corrected by subtracting Spo11–/– signal from the wild-type signal. Top and bottom strand reads were then co-oriented and averaged. (G) Comparison of yeast (Mimitou et al. 2017) and mouse resection tract lengths. SPO11-specific S1-seq profiles were averaged across all SPO11-oligo hot spots (n = 13,960) and binned every 10 bp. An estimated background was removed by subtracting from all values the value of signal 2.5 kb away from the hot spot center. The signal close to and further away from the hot spot center was excluded by setting values of positions <100 bp and >2.5 kb to zero. Fractions of total signal were calculated every 100 bp and plotted. (H) Resection lengths measured by S1-seq are longer than those estimated using anti-DMC1 ChIP (SSDS) (Lange et al. 2016). Signal was smoothed with a 151-bp Hann filter in B, F, and H, and with a 401-bp Hann filter in C.
One advantage of using meiotic recombination as a paradigmatic context to study resection is that SPO11 generates numerous DSBs in a regulated fashion at a defined stage in prophase I after DNA replication (Padmore et al. 1991; Mahadevaiah et al. 2001; Murakami and Keeney 2014). Moreover, most DSBs form within narrow genomic segments called hot spots (typically <150 bp wide), which means that most DSB resection tracts emanate from relatively defined locations, facilitating their structural analysis (Lange et al. 2016; Mimitou et al. 2017).
A key determinant of hot spot locations in mice and humans is PRDM9, which contains a sequence-specific zinc finger DNA-binding domain and a PR/SET domain that methylates histone H3 on lysines 4 and 36 (Baudat et al. 2010; Myers et al. 2010; Brick et al. 2012; Wu et al. 2013). This histone methylation is required for nearby DNA cleavage (Diagouraga et al. 2018), although it remains unclear how SPO11 targeting occurs. Interestingly, PRDM9 action on the unbroken recombination partner appears to facilitate strand exchange and recombination (Davies et al. 2016; Hinch et al. 2019; Li et al. 2019), but how this is accomplished is also unknown.
In this study we mapped the endpoints of mouse meiotic resection tracts genome-wide and at single-nucleotide resolution. We uncover previously undocumented characteristics of resection, including global patterns, locus-to-locus variation, and the genetic pathways that control resection. Our genomic sequencing method (S1-seq) also detects structures that appear to be intermolecular recombination intermediates. Unexpected properties of these intermediates in mice shed light on the mechanism of meiotic recombination and the structure of chromatin at sites undergoing recombination.
Results
A nucleotide-resolution map of meiotic DSB resection endpoints
To directly survey the fine-scale molecular structure of resected DSBs, we adapted the S1-seq method (Mimitou et al. 2017; Mimitou and Keeney 2018) to mouse spermatocytes (Fig. 1A, panel ii; Supplemental Fig. S1A). Testicular cell suspensions were embedded in agarose to protect DNA from shearing, then DNA liberated by treatment with SDS and proteinase K was digested with ssDNA-specific S1 nuclease to remove ssDNA tails at DSB ends, making them blunt-ended at resection endpoints. The resulting DNA was ligated to biotinylated adaptors, fragmented by sonication, affinity purified using streptavidin, and ligated to separate adaptors at the opposite ends prior to amplification and deep sequencing. To minimize background and improve signal:noise ratio, we used testis samples from juvenile mice during the first semi-synchronous wave of spermatogenesis (12–16 d postpartum [dpp]); at these ages, postmeiotic cells have not yet formed and meiotic cells are at stages when DSBs are present (leptonema, zygonema, and early pachynema) (Bellve et al. 1977; Zelazowski et al. 2017).
True resection endpoints should yield sequencing reads of defined polarity: top strand reads for resection tracts moving away from the DSB toward the right; bottom strand reads for leftward resection (Fig. 1A, panel ii). As predicted, S1-seq reads of the correct polarity were enriched adjacent to DSB hot spots for wild-type C57BL/6J (B6) mice relative to congenic mice lacking SPO11 (Fig. 1B,C; Supplemental Fig. S1B). Read depth near hot spots showed good reproducibility (Pearson's r = 0.76 for biological replicates) (Supplemental Fig. S1C) and was correlated with local DSB activity as measured by sequencing of SPO11 oligos or DMC1-bound ssDNA (SSDS) (Fig. 1D; Supplemental Fig. S1D). No similar enrichment was observed around sites that can be targeted by versions of PRDM9 that are not present in these experimental mice (e.g., PRDM9 from Mus musculus castaneus (CAST/EiJ) or PRDM9 carrying a humanized Zn finger array (Supplemental Fig. S1E; Davies et al. 2016).
We conclude that S1-seq quantitatively captures bona fide resection endpoints. We also observed a prominent signal near hot spot centers with a polarity inconsistent with resection endpoints, but consistent with expectation for S1-sensitive, intermolecular strand-exchange intermediates (e.g., Fig. 1B). We revisit this DNA species in detail below. We showed previously that S1-seq faithfully captures DNA ends at single-nucleotide resolution in yeast (Mimitou et al. 2017; Mimitou and Keeney 2018), so we assume that the same resolution is true for mouse.
Resection is locally heterogeneous within globally stereotyped constraints
DSBs are highly clustered within hot spots in mice (Fig. 1E, left panel), confined mostly to the relatively nucleosome-depleted regions (NDR) enclosing the PRDM9 binding sites and, to a lesser degree, the linkers between the flanking methylated nucleosomes (Lange et al. 2016). Analogous to, but more dispersed than this clustering of DSB positions, resection endpoints showed a stereotyped distribution that was broadly similar across both sides of most hot spots, with most endpoints falling within zones 0.3–2 kb from hot spot centers (Fig. 1E). These spatial patterns were highly reproducible across biological replicate samples (Supplemental Fig. S2). We obtained similar resection endpoint distributions if we removed DSB 3′ ends with the ssDNA-specific 3′ → 5′ exonuclease ExoT (Canela et al. 2019) instead of S1 nuclease (Fig. 1B,E; Supplemental Fig. S1B,F). The greater signal-to-noise ratio of ExoT-seq showed that the stereotypical distribution of resection endpoints applied to even the weaker subset of hot spots (Fig. 1E).
Within these broader zones of resection endpoints, however, individual hot spots showed substantial heterogeneity; i.e., peaks and valleys in the S1-seq maps (Fig. 1B; Supplemental Fig. S1B). Although relative heights of the peaks varied substantially from sample to sample, presumably reflecting noise from sequence sampling error, the positions of peaks were reproducible between S1-seq data sets and between S1 and ExoT (Fig. 1B; Supplemental Fig. S1B,G–I). The reproducibility of peak positions suggests that the peaks are not solely caused by nuclease biases during library generation, but instead reflect heterogeneity in the resection endpoint locations themselves. Similar local heterogeneity in yeast reflects in part the underlying chromatin structure (Mimitou et al. 2017), but we observed no clear relation of resection endpoint peaks with preferred positions of nucleosomes (Supplemental Fig. S3A,B). This may indicate that chromatin structure has no effect on resection in mice. However, we note that a lack of correlation may be uninformative if nucleosome positions are variable between chromosomes in the population (see legend to Supplemental Fig. S3A,B).
Whatever the cause of the heterogeneity, we interpret that individual genomic locations have preferred regions for resection termination within broader zones that are defined by globally stereotyped minimal and maximal resection lengths. Previously, it was not possible to evaluate resection patterns at individual mouse hot spots because of limitations of available data (Lange et al. 2016). Our findings demonstrate the stochastic, probabilistic nature of resection termination and reveal that there is substantial variation from DSB to DSB within a cell, and even at the same hot spot between cells.
To precisely define global resection patterns, we generated a genome-wide average profile by subtracting the signal obtained in the Spo11–/– mutant and then averaging top and bottom strand reads after co-orienting them around hot spot centers (Fig. 1F). Alternative background-subtraction and normalization methods gave similar results (Supplemental Fig. S3C). Resection tracts averaged ∼1100 nt, but with a wide distribution (Fig. 1G). Similar results were obtained from ExoT-seq or if only stronger hot spots (with higher signal:noise ratios) were considered (Supplemental Fig. S3D–G). Of all DSB ends, 99% were resected at least 280 nt and only 1% were resected more than 1800 nt. This extent of resection predicts 440–660 kb of ssDNA per meiosis assuming 200–300 DSBs.
The distribution of mouse resection lengths appears less positively skewed than in yeast and mouse resection is more than one-third longer on average than in yeast (822 nt) (Fig. 1G). More importantly, resection is longer by ∼200 nt than our previous estimate based on coverage from sequencing of ssDNA bound by DMC1 (SSDS) (Fig. 1H; Lange et al. 2016). The difference between SSDS-based imputation and our direct measurement by S1-seq may reflect technical aspects of the SSDS method. For example, ssDNA sequence coverage is likely to be incomplete because of a foldback annealing step that requires intramolecular microhomology (Khil et al. 2012; Lange et al. 2016). However, it is also likely that DMC1 coats only a portion of the ssDNA that usually does not include the break-distal segment closest to the ssDNA–dsDNA junction. This interpretation is consistent with cytological data in yeast (Brown et al. 2015) and agrees with direct analysis of relative distributions of DMC1 and RAD51 by ChIP-seq and immunocytology in mice (AG Hinch, PW Becker, T Li, et al., in prep.).
Recombination intermediates close to DSB ends
We observed a prominent collection of S1-seq reads close to DSB hot spot centers in addition to the more distal reads from resection endpoints (Fig. 1B,E,F; Supplemental Fig. S1B). The hot spot-proximal reads were narrowly distributed across a region shifted to the side of DSB positions as defined by SPO11-oligo sequencing (modal shift of 35 bp) (Fig. 2A). Because of this shift, we can definitively conclude that these reads are not principally from unresected DSBs, which S1-seq readily detects in yeast (Supplemental Fig. S4A; Mimitou et al. 2017; Mimitou and Keeney 2018). Furthermore, the direction of the shift means that these reads have the wrong polarity to be resection tracts emanating from the DSB hot spots. These central reads made up 19.3% of the total signal around hot spots in B6 mice.
Figure 2.

Intermolecular recombination intermediates. (A) Spatial disposition of central (hot spot-proximal) S1-seq signal. The top panel shows the background-subtracted S1-seq signal (from Fig. 1F) and the bottom panel shows a zoom into the region immediately surrounding hot spot centers, showing the offset of the S1-seq signal from the SPO11-oligo distribution. (B) Lack of central S1-seq signal and hyperresection of DSB ends in the absence of DMC1. Wild-type and Dmc1–/– maps were prepared from animals from the same mixed-background breeding colony. (C) Absence of central S1-seq signal at hot spots (n = 571) on sex chromosomes. DSB sites in the pseudoautosomal region are not included because these are too broadly distributed to be able to define hot spot-proximal versus hot spot-distal locations. (D) Schematic illustrating how S1 cleavage of D-loops could explain the central signal. The vertical dashed line aligns the different elements of the cartoon by the 3′ end of the DSB ssDNA tail. S1 cleavage of the intact recombination partner at the DSB-proximal and DSB-distal boundaries of the joint molecules would be expected to yield sequencing reads mapping to the bottom and top strands, respectively. See also Supplemental Figure S4D and its legend for more details. (E) Comparison of predicted sensitivities of resected DSBs and D-loops to S1 versus ExoT nucleases. Average sequencing profiles are reproduced from Figure 1F and Supplemental Figure S1F. (F) S1-seq at asymmetric hot spots in B6xPWD F1 hybrid mice. The graphs show strand-specific signal that mapped to either the B6 or PWD chromosomes at highly asymmetric hot spots (called from B6 SPO11-oligo maps) where PRDM9B6 preferentially targets the PWD chromosomes (defined as those where the fraction of total SSDS reads that derived from the B6 chromosome was ≤0.1; n = 1220). S1-seq signal was background corrected with signal from F1 hybrid mice at 9 dpp, when few meiotic cells have yet formed. See also Supplemental Figure S4E. Signal was smoothed with a 151-bp Hann filter in A, E, and F (top panel) as well as B and C, and with a 51-bp Hann filter in A and F (bottom panel).
We reasoned that this central signal might be analogous to a “wrong polarity” signal in yeast that was proposed to come from recombination intermediates of as-yet undefined structure (Mimitou et al. 2017). To test this hypothesis, we examined physical properties and genetic dependencies for formation of this signal.
The central signal was not from other hot spots nearby (Supplemental Fig. S4B), was correlated with hot spot heat (Supplemental Fig. S4C), was specific for sites being actively targeted by PRDM9 (Supplemental Fig. S1E), and was SPO11-dependent (Fig. 1B,C,F; Supplemental Fig. S1B). We therefore conclude that the central signal requires meiotic DSB formation.
The central signal was largely or completely absent in Dmc1–/– mutants (Fig. 2B; Supplemental Fig. S2A), which lack a strand exchange protein essential for meiotic recombination (Bishop et al. 1992; Pittman et al. 1998). Remarkably, the signal also appeared specific for autosomes: Little or no central S1-seq signal was apparent at hot spots on the X and Y chromosomes (Fig. 2C). DSBs on nonhomologous portions of the sex chromosomes persist longer than on autosomes, presumably because of a temporary barrier to using the sister chromatid as the recombination partner (Moens et al. 1997; Mahadevaiah et al. 2001; Lange et al. 2016). The absence of the central S1-seq signal at sex chromosome hot spots did not appear to be solely a consequence of this delayed repair because S1-seq maps made from older juveniles also had little if any of the central signal at these hot spots (Fig. 2C). The dependence on DMC1 suggests that detection of the central S1-seq signal requires the ability to carry out recombination, and absence at sex chromosome hot spots suggests that presence of a homolog is required.
A straightforward hypothesis is that we are detecting intermolecular recombination intermediates such as D-loops (Fig. 2D; Supplemental Fig. S4D). In this model, sequencing reads are from the intact recombination partner that is rendered S1-sensitive when it is invaded by the end of the broken chromosome. If so, the central reads would be from the DSB-proximal ends of D-loops, with bottom strand reads arising from invasion by the right side of the DSB (i.e., the opposite polarity from resection endpoint reads). Sequencing reads that might come from the more distal ends of the D-loops would be expected to have the same polarity as resection endpoint reads, and might thus be masked by the larger number of resection reads (Fig. 2D; Supplemental Fig. S4D, panels i–iii).
One prediction from this hypothesis is that the central signal should require digestion with S1 or a similar endonuclease; i.e., it should be absent if sequencing libraries are prepared using an exonuclease (Fig. 2E). Indeed, as predicted, maps generated using ExoT (which requires a free DNA end) failed to display the central signal (Figs. 1B,E, 2E; Supplemental Fig. S1B,F). We note that the D-loop proposed in the canonical recombination model, in which the 3′ end of the ssDNA is fully invaded, would predict sequencing reads that line up with DSB positions (Supplemental Fig. S4D, panel ii). The shift that we observed instead can be simply explained if the 3′ end is not fully invaded (Fig. 2D; Supplemental Fig. S4D, panels i,iii); Brown and Bishop 2014; Chan et al. 2019). Further implications and additional features of this model are addressed in the Discussion.
We attempted to test whether the central signal reflects invasion of the homolog by generating S1-seq maps from B6xPWD F1 hybrid mice in which DSBs at certain hot spots occur only or predominantly on one of the parental chromosomes because of strain-of-origin differences in PRDM9 binding (Davies et al. 2016; Smagulova et al. 2016). However, at highly asymmetric hot spots where PRDM9B6 preferentially targets the PWD chromosomes, we observed a central signal derived only from the same chromosomes that were undergoing DSB formation (Fig. 2F). This is counter to expectation for interhomolog joint molecules. The same conclusion emerged from examination of hot spots where PRDM9PWD preferentially targets B6 chromosomes (Supplemental Fig. S4E).
The results suggest that at least some of the central signal reflects recombination intermediates involving invasion of the sister chromatid. Although we could not detect the predicted signature of interhomolog joint molecules, we cannot exclude the possibility that interhomolog recombination intermediates contribute to the central signal in B6 mice and at symmetric hot spots in the F1 hybrid (see the Discussion).
Mechanism of resection
To identify molecular pathways responsible for resection, we applied S1-seq to testis samples from mutant mice. To test the contribution of EXO1, we examined mice homozygous for a knock-in mutation changing an active site aspartate to alanine (D173A, hereafter Exo1DA), which eliminates nuclease activity (Lee et al. 2002; Zhao et al. 2018). In yeast, the equivalent exo1 mutation reduces resection tracts to less than one-third the normal length (Mimitou et al. 2017). Surprisingly, however, Exo1DA mice showed only a modest (∼10%) decrease in resection length (Fig. 3A). Thus, EXO1 is not a major participant in 5′ → 3′ resection in mice, or it is substantially redundant with one or more other resection activities.
Figure 3.

Genetic control of meiotic resection. (A) EXO1 exonuclease activity contributes only modestly to the full extent of resection length. (B) Hyperresection is greater in samples from older Dmc1–/– animals (16 dpp vs. 12 dpp), which are enriched for spermatocytes at later stages in prophase I. See also Supplemental Figure S2A,C. (C) Altered resection in Atm–/– spermatocytes. The bottom panel shows a zoom into the region around hot spot centers, showing coincidence of the central S1-seq signal with SPO11 oligos in Atm–/–, unlike in wild type. (D) Unlike in wild type, the central signal in ATM-deficient mutants (with or without Spo11 heterozygosity) is prominent at hot spots on sex chromosomes. (E) Spo11 heterozygosity modifies the resection defects caused by ATM deficiency. Signal was smoothed with a 151-bp Hann filter in A, B, and C–E (top panel), and with a 51-bp Hann filter in C–E (bottom panel).
In Dmc1–/–, DSBs were resected further than normal (Figs. 1B, 2B; Supplemental Fig. S1B). Moreover, more of the very long resection tracts were observed in testes from older mice (16 dpp versus 12 dpp), whereas resection tract lengths in wild type were similar across these ages (Fig. 3B). These results reveal that mice lacking DMC1 hyperresect their DSBs, and suggest that hyperresection continues progressively. Both properties are reminiscent of dmc1 mutant yeast (Bishop et al. 1992; Mimitou et al. 2017).
The DSB-responsive kinase ATM (ataxia telangiectasia mutated) is a key regulator of DSB formation, apparently controlling a negative feedback circuit whereby DSBs inhibit formation of other DSBs nearby on the same chromatid or its sister (Lange et al. 2011; Garcia et al. 2015; Lukaszewicz et al. 2018). The ATM ortholog in yeast (Tel1) also promotes meiotic resection (Joshi et al. 2015; Mimitou et al. 2017), so we asked whether this function is conserved.
S1-seq maps from Atm–/– mice revealed drastic changes (Figs. 1B, 3C; Supplemental Fig. S1B). Resection endpoints were spread over a much wider area, with many tracts shorter than in wild type and many tracts longer (Fig. 3C, top panel). This unexpected mix of hyporesection and hyperresection indicates that ATM controls, directly or indirectly, the extent of resection.
A central S1-seq signal was present in Atm–/– mice, but with an abnormal spatial disposition that was highly coincident with DSBs (Fig. 3C, lower panel). Unlike in wild type, the central signal in Atm–/– was readily apparent at hot spots on the sex chromosomes (Fig. 3D). These findings suggest that Atm–/– mutants accumulate unresected DSBs (as in tel1 yeast) (Joshi et al. 2015; Mimitou et al. 2017) and do not accumulate intermolecular recombination intermediates. The central signal was 14% of the total S1-seq signal around hot spots in Atm−/−.
We sought to determine which changes are more likely to reflect direct requirements for ATM by performing S1-seq in Spo11+/– Atm–/– mice. Atm–/– single mutants experience a strong increase in DSB formation, estimated at ≥10-fold based on quantification of SPO11-oligo complexes (Lange et al. 2011). Their spermatocytes also experience severe recombination defects and catastrophic meiotic failure resulting in pachytene arrest (Barlow et al. 1996; Barchi et al. 2005). However, reducing Spo11 gene dosage attenuates the increased DSB numbers (Lange et al. 2011) and suppresses many of the meiotic defects of Atm–/– mutants, allowing spermatocytes to proceed much further in meiotic prophase and apparently complete most recombination (Bellani et al. 2005; Barchi et al. 2008). This suppression suggests that at least some of the recombination problems in ATM-deficient spermatocytes are a secondary consequence of the massively increased DSB load rather than reflecting direct roles of ATM per se.
S1-seq patterns differed considerably in Spo11+/– Atm–/– mice compared with Atm–/– single mutants or wild type (Fig. 3E). Hyperresection was essentially eliminated, but a subset of DSBs remained underresected, giving an average resection length of 978 nt (88% of the average in wild type) (Fig. 3E, top panel). The distribution of the central signal also changed, giving an intermediate pattern between wild type and Atm–/–, which we interpret as a mixture of recombination intermediates and unresected DSBs (Fig. 3E, bottom panel). This interpretation is supported by the retention of a DSB-coincident central signal at sex chromosome hot spots (Fig. 3D).
Because reducing Spo11 gene dosage fully suppressed hyperresection and partially suppressed the defect in forming intermolecular joint molecules, we conclude that excessive exonucleolytic processing and apparently defective strand exchange in Atm–/– mutants are indirect consequences of a high DSB burden when ATM is absent. In contrast, the retention of a population of unresected DSBs and the decrease in resection tract length suggest that these defects are more directly tied to ATM function per se, especially given that meiotic prophase I progression and recombination are very substantially rescued by Spo11 heterozygosity. We therefore conclude that ATM is critical for both the initiation and extension of meiotic DSB resection in mice. Strikingly, these aspects of the Atm–/– mouse phenotype are the ones most reminiscent of tel1 mutant yeast (Mimitou et al. 2017), suggesting wide conservation of ATM/Tel1 functions in controlling resection, even if the resection machinery itself differs (e.g., role of EXO1).
PRDM9-dependent modulation of local chromatin accessibility
How recombination machinery accesses a chromatinized donor is not well understood (Neale and Keeney 2006; Kobayashi et al. 2016). Average profiles around hot spots of H3K4me3 and H3K36me3 chromatin immunoprecipitation (ChIP) sequencing data show peaks of methylated nucleosome coverage flanking PRDM9 binding sites and the main cluster of DSB positions (Fig. 4A; Baker et al. 2014; Lange et al. 2016; Powers et al. 2016; Yamada et al. 2017). An implication is that the strand exchange machinery frequently invades segments of the homologous donor DNA that contain these methylated nucleosomes (if PRDM9 acted on the donor as well as the broken chromosome) plus additional flanking nucleosomes further away (Fig. 4B). To more precisely define the chromatin structure at such recombination sites, we examined recently published data sets of bulk nucleosomes (ChIP input samples) liberated by micrococcal nuclease (MNase) from flow-sorted spermatocyte nuclei from B6 mice (Lam et al. 2019).
Figure 4.
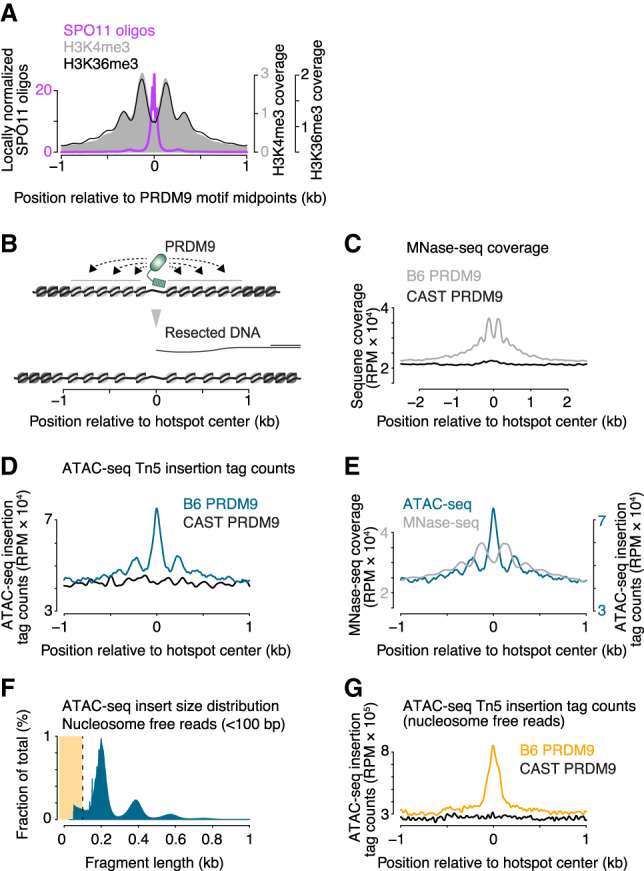
Increased chromatin accessibility at sites of PRDM9 activity. (A) Profiles of nucleosomes trimethylated at H3K4 or H3K36 around hot spot-enriched 12-bp PRDM9 motifs. Modified from Lange et al. (2016) with permission from Elsevier; methylated histone ChIP-seq data are from Baker et al. (2014) and Powers et al. (2016). (B) To-scale schematic illustrating how resected DNA of the broken chromosome compares with the zone over which PRDM9 influences chromatin structure. (C) MNase-seq coverage around hot spots from cells in leptonema through early pachynema of the first meiotic prophase. Shown are average profiles of sequence coverage from ChIP-seq input samples (before immunoprecipitation) prepared by MNase digestion of flow-sorted SYCP3 positive and histone H1t negative spermatocyte nuclei (sequencing data from Lam et al. 2019). Note that this represents total DNA liberated by MNase digestion, not just methylated nucleosomes. The profile around B6 hot spots was compared with the profile around hot spots not active in these strains (i.e., sites that can be targeted by the CAST version of PRDM9). (D) ATAC-seq cleavage profiles around active (PRDM9B6) and inactive (PRDM9CAST) hot spots. Note that Tn5 integration positions, not sequence coverage of the entire fragments, were plotted to show the local chromatin accessibility. (E) Comparison of ATAC-seq cleavage positions with MNase-seq coverage maps. (F) Fragment length distribution of ATAC-seq. Subnucleosomal reads were defined as those <100 bp in length (highlighted in yellow). (G) ATAC-seq cleavage profiles generated only with subnucleosomal reads around active (PRDM9B6) and inactive (PRDM9CAST) hot spots. Signal was smoothed with a 51-bp Hann filter in A, C–E, and G.
MNase-seq coverage from cells in leptonema through early pachynema of the first meiotic prophase displayed a series of peaks flanking PRDM9 sites and extending 1 kb (approximately six nucleosomes’ worth) on either side on average (Fig. 4C). At these stages, PRDM9-dependent methylation is present, DSBs are made, and recombination is occurring (Mahadevaiah et al. 2001; Moens et al. 2002; Guillon et al. 2005; Lam et al. 2019). This signature was also specific for sites of PRDM9 action (Fig. 4C).
Importantly, we noted that this signature represents extra sequence coverage above the local baseline, including within the valleys between peaks. At first glance, this pattern appears at odds with the model that SPO11 targets a PRDM9-dependent nucleosome-depleted region (NDR) (Baker et al. 2014; Lange et al. 2016). If nucleosomes were becoming well positioned where they were already present but randomly distributed in the population, we would instead expect the peaks and valleys to oscillate above and below the baseline; i.e., the area under the coverage curve would have been unchanged. The increased MNase-seq coverage we observed instead could mean that PRDM9-modified sites have higher nucleosome occupancy. Alternatively, it could be that greater chromatin accessibility allows MNase to more readily release nucleosomes, resulting in higher DNA fragment coverage in the sequencing libraries. In this interpretation, higher MNase-seq coverage paradoxically reflects more accessibility, not more nucleosome occupancy. However, available MNase-seq data cannot distinguish these possibilities because the degree of MNase treatment (i.e., complete or partial digestion) also affects MNase-seq coverage depth.
To test this idea, we assayed testis cell chromatin structure in B6 mice using ATAC-seq, which assesses how accessible DNA is to Tn5 transposase (Buenrostro et al. 2013). We observed total ATAC-seq read depth that was elevated above local baseline specifically around PRDM9 sites (Fig. 4D). Tn5 integration positions—enriched in hot spot centers and the presumed linkers between flanking nucleosomes—were anticorrelated with MNase-seq coverage, as expected (Fig. 4E). (Note: This read depth reports cleavage frequency rather than coverage depth for fragments in the sequencing library, unlike the MNase-seq maps.) Nevertheless, a substantial fraction of the total ATAC-seq signal was from nucleosome-sized fragments or larger (Fig. 4F), so considering the total ATAC-seq map is similar to examining the MNase-seq coverage map. Importantly, however, if we focus specifically on subnucleosome-sized ATAC-seq fragments, reads were highly enriched at centers of hot spots targeted by PRDM9B6 and not at control (PRDM9CAST) sites (Fig. 4G). Thus, these findings support the interpretation that there tends to be a relatively more open region at hot spot centers when PRDM9 has acted. We interpret that the relatively high MNase-seq coverage across hot spot centers (Fig. 4C) reflects substantial heterogeneity between cells in the population and between hot spots as to precisely where nucleosomes are positioned (e.g., Supplemental Fig. S3A).
We conclude that PRDM9 action causes changes in local chromatin structure that include a tendency toward positioning of nucleosomes but also greater overall accessibility. An earlier study using MNase-seq also argued in favor of this idea, but without supporting evidence (Baker et al. 2014). Because neither the MNase-seq nor ATAC-seq maps should have been able to pick up a nucleosomal sequencing signature on the ssDNA of resected DSB ends, we infer that the increased chromatin accessibility is occurring on intact chromosomes.
Discussion
Our results uncover both conserved and unique features of mouse meiotic resection. A surprisingly unique aspect was the minimal contribution of EXO1 to overall resection lengths, unlike in yeast. One possibility is that mouse meiocytes, similar to somatic cells, use additional resection activities that act redundantly with EXO1, such as DNA2 plus BLM or WRN helicase (Symington 2014).
Conserved features include a similar overall length scale for resection in yeast and mouse despite large differences in genome size, and an important role for DMC1 in limiting the extent of resection. It is likely that assembly of DMC1 and/or RAD51 nucleoprotein filaments limits access of DSB ends to the resection machinery (Shinohara et al. 1997; Henry et al. 2006). In this context, the correlation of hyperresection with absence of intermolecular recombination intermediates in Atm–/– (but not Spo11+/– Atm–/–) mutants points to defects in strand-exchange protein assembly and/or function affecting both resection and strand exchange in this mutant.
Even more striking is the conservation of roles of ATM in controlling resection. ATM promotes resection in some contexts in somatic cells as well, although the mechanism is not well understood (e.g., Lee et al. 2018). Our findings provide the insight that ATM can influence resection in at least two ways. First, accumulation of unresected DSBs in Atm–/– mutants suggests that ATM regulates MRE11 endonuclease activity, perhaps via phosphorylation of CtIP (Cartagena-Lirola et al. 2006). Second, the shorter resection tracts in Atm–/– mutants indicate that ATM controls the extension of resection once it has begun. One possibility is that ATM directly stimulates the resection machinery and/or down-regulates inhibitors of resection. A nonexclusive possibility is that ATM recruits chromatin remodeling activities to DSBs (Chakraborty et al. 2018).
An interesting feature of S1-seq maps is the hot spot-proximal signal that likely reflects intermolecular recombination intermediates. Our results analyzing hot spots that are asymmetrically bound by PRDM9 in F1 hybrid mice suggest that at least some of the putative recombination intermediate signal involves invasion of the sister chromatid. One possibility is that all of the central S1-seq signal at all hot spots (even symmetric ones) reflects only intersister joint molecules. However, if this interpretation is correct, it is puzzling that the central signal is absent at sex chromosome hot spots, for which a sister chromatid is present. One solution to this conundrum is to envision that intersister joint molecules are paradoxically more stable if (invisible) interhomolog invasion can also occur. Such a scenario might be consistent with a proposed “quiescent end” complex that inhibits recombination by one end of a DSB while the other end engages the homolog (Storlazzi et al. 2010).
An alternative, however, is that interhomolog joint molecules do contribute to the central signal at symmetric hot spots (e.g., in B6 mice) but are not detectable by S1-seq at asymmetric hot spots. This could happen if steady-state levels of interhomolog joints are low because of more dynamic turnover or a delay in formation relative to joint molecules at PRDM9-symmetric sites, and/or because D-loop boundaries become more variable and thus too blurred to detect in population average. Both effects (lower levels and altered distributions) can easily be envisioned to accompany a lack of PRDM9-dependent opening of chromatin structure on the intact homolog. In this context, it is important to emphasize that DSBs at asymmetric hot spots experience delayed recombination relative to symmetric hot spots (Davies et al. 2016; Hinch et al. 2019; Li et al. 2019). Thus, a caveat of our experiment is that the same polymorphisms that are required to discriminate intersister from interhomolog recombination may prevent interhomolog events from being detected. If so, this would mean that available methods are unable to define the chromosome of origin for the central signal.
If these are D-loops, the spatial disposition of the signal suggests that they consist of invasion of an internal segment of the ssDNA and usually do not extend all the way to the 3′ end (Supplemental Figs. S4D, S5; Brown and Bishop 2014; Chan et al. 2019). A simple way to account for this would be for SPO11-oligo complexes to cap the 3′ ends of resected DSBs, as we previously proposed (Supplemental Fig. S5; Neale et al. 2005). Potentially consistent with such a cap being biochemically stable, recombinant yeast Spo11 complexes bind very tightly (even without a covalent end-linkage) to DNA ends in vitro (Claeys Bouuaert et al. 2020). Moreover, the exonucleolytic END-seq method using ExoVII in addition to ExoT to process DSB ends with adducts (Canela et al. 2019) detects a sequencing signal in wild-type mouse testis cells that resembles unresected DSBs and that shares many features with the S1-seq central signal including autosome-specificity and requirement for DMC1 (Paiano et al. 2020). This result would be predicted for a SPO11-oligo-capped end on recombination intermediates.
If correct, an intriguing implication is that capping would provide a mechanism to maintain the strand-exchange intermediate in a poised state that retains potential to move either forward or backward. Displacing the SPO11-oligo complex (Neale et al. 2005; Murayama et al. 2011) or removing the uninvaded 3′ end by cleaving the flap (Peterson et al. 2020) would free a 3′-OH end to prime DNA synthesis and drive D-loop extension. Conversely, reversal of strand exchange would leave the broken end with a ssDNA gap that could be filled in by DNA synthesis primed from the 3′ end of the SPO11 oligo, potentially allowing nonhomologous end joining as a backup repair pathway.
Our studies uncover a previously undocumented remodeling of chromatin structure at sites of PRDM9 action. The more open chromatin structure observed with both MNase digestion and ATAC-seq may reflect wider linkers, displacement of linker histones, less tightly bound nucleosomal particles, and or dynamic nucleosome removal and redeposition such that a fraction of DNA molecules is free at steady state. Our findings agree with a recent study of PRDM9-dependent recruitment of the HELLS chromatin remodeler to hot spots (Spruce et al. 2020). Regardless of the source, this open chromatin structure seems likely to be important for SPO11 access. Moreover, a further key implication is that PRDM9 action on the uncut homolog generates a more accessible chromatin structure precisely where its broken partner needs to engage it for repair (Fig. 4B). Thus, this greater accessibility may provide a mechanistic explanation for observed repair differences between hot spots that are symmetrically versus asymmetrically bound by PRDM9 (Davies et al. 2016; Hinch et al. 2019; Li et al. 2019).
We previously documented a “wrong polarity” S1-seq signal in yeast that we ascribed to recombination intermediates (Mimitou et al. 2017). This yeast signal and the mouse central signal share a complete dependence on Dmc1, but the sequence reads in yeast are spread over a much wider area rather than being concentrated close to the DSB site. Given the different spatial arrangements, it is possible that the yeast and mouse S1-seq signals reflect completely different recombination intermediates. However, it is also possible that they are related molecular structures, in which case the distinct spatial patterns would suggest that the relevant strand exchange events in the two species engage different parts of the invading ssDNA. One simple possibility is that the localized PRDM9-dependent opening of chromatin structure on the donor in mice provides a window of accessibility that facilitates strand exchange but also tends to constrain it to occur near the DSB end. In contrast, because most yeast hot spots are in active gene promoters (Baudat and Nicolas 1997; Pan et al. 2011), perhaps yeast DSBs find a more broadly permissive chromatin structure on their recombination partner, such that strand exchange in this species is less spatially constrained than in mice. Such differences might contribute to the fact that tracts of heteroduplex DNA and of gene conversion are so much longer in yeast (∼2 kb) (Mancera et al. 2008; Martini et al. 2011; Marsolier-Kergoat et al. 2018) than in mice (∼500 bp for crossovers, ≤50 bp for noncrossovers) (Cole et al. 2014; Peterson et al. 2020).
Materials and methods
Mice
Experiments conformed to the US Office of Laboratory Animal Welfare regulatory standards and were approved by the Memorial Sloan Kettering Cancer Center Institutional Animal Care and Use Committee. Mice were maintained on regular rodent chow with continuous access to food and water until euthanasia by CO2 asphyxiation prior to tissue harvest. Previously described Spo11 (Baudat et al. 2000), Atm (Barlow et al. 1996), and Exo1DA (Zhao et al. 2018) mutations were maintained on a congenic B6 strain background. The Dmc1 mutation (Pittman et al. 1998) was maintained on a mixed (129/SV and B6) background. B6xPWD F1 hybrid mice (semifertile) were generated by crossing B6 (stock no. 000664) female mice and PWD/PhJ (stock no. 004660) male mice obtained from the Jackson Laboratory.
S1-seq
Testis dissociation
Unless otherwise stated, testis cells from12-dpp juvenile mice were obtained as described previously (Cole et al. 2014). Briefly, testes were decapsulated and incubated in Gey's balanced salt solution (GBSS) (Sigma) with 0.5 mg/mL collagenase type 1 (Worthington) for 15 min at 33°C. Seminiferous tubules were then rinsed three times and further treated with 0.5 mg/mL trypsin (Worthington) and 1 µg/mL DNase I (Sigma) for 15 min at 33°C. Trypsin was inactivated with 5% FCS and tubules were further dissociated by repeated pipetting. Cells were passed through a 70-µm cell strainer (BD Falcon) and washed three times in GBSS.
DNA extraction in plugs
Cells were embedded in plugs of 0.5% low-melting-point agarose (Lonza) in GBSS. One plug mold (Bio-Rad) was used per two testes (1 million to 2 million cells per plug). Plugs were incubated with 50 µg/mL proteinase K (Roche) in lysis buffer (0.5 M EDTA at pH 8.0, 1% N-lauroylsarcosine sodium salt) overnight. Plugs were washed five times with TE (10 mM Tris-HCl at pH 7.5, 1 mM EDTA at pH 8.0), and then incubated with 50 µg/mL RNase A (Thermo Fisher) for 3 h at 37°C. They were washed five times with TE and stored in TE at 4°C.
S1-seq library preparation
In-plug overhang removal with S1 and adaptor ligation were performed as described previously (Mimitou et al. 2017; Mimitou and Keeney 2018). For ExoT-seq, the S1 treatment step was replaced with the following: Plugs were washed in 1 mL of NEBuffer 4 three times for 15 min, treated with 75 U of exonuclease T in 100 µL of NEBuffer 4 for 90 min at 24°C and washed three times with 8 mL of TE for 15 min (Canela et al. 2019). After adaptor ligation, plugs were washed three times in TE and incubated in TE overnight at 4°C to diffuse excessive unligated adaptors out of plugs. Agarose was then digested by the Epicentre GELase Enzyme Digestion protocol. DNA was fragmented by vortex and further sheared to DNA fragment sizes ranging between 200–500 bp with a Bioruptor waterbath sonicator (Diagenode) for 40 cycles of 30 sec on/off at 4°C at the middle power setting. DNA was purified by ethanol precipitation and dissolved in 100 µL of TE. Fragments containing the biotinylated adaptor were purified with streptavidin, ligated to adaptors at sheared end and amplified by PCR as previously described (Mimitou et al. 2017; Mimitou and Keeney 2018). PCR products were purified with 0.9× AMPure XP beads (Beckman Coulter) to remove primer dimers. DNA was sequenced on the Illumina HiSeq platform in the Integrated Genomics Operation at Memorial Sloan Kettering Cancer Center. We obtained paired-end reads of 50 bp except for F1 hybrid experiments, for which we obtained 100-bp paired-end reads.
Mapping and preprocessing
Sequence reads were mapped onto the mouse reference genome (mm10) by bowtie2 version 2.2.1 (Langmead et al. 2009) with the argument –X 1000. Uniquely and properly mapped reads were counted, at which a nucleotide next to biotinylated adaptor DNA was mapped (this corresponds to a position of a resection endpoint). The mapping statistics are listed in Supplemental Table S1.
ATAC-seq
Testis cells were isolated from 12-dpp juvenile mice as described for S1-seq sample preparation. Subsequent ATAC-seq library preparation and sequencing were performed in the Integrated Genomics Operation at Memorial Sloan Kettering Cancer Center as described (Buenrostro et al. 2013) with slight modifications. Briefly, nuclei extracted from 50,000 testis cells were treated with Tn5 transposase for 45 min at 42°C. Libraries were amplified and sequenced on the Illumina HiSeq platform. Reads were mapped as described for S1-seq data processing. Uniquely and properly mapped reads were counted at which Tn5 insertion positions were mapped.
Other data sets and data availability
Raw and processed sequencing S1-seq and ATAC-seq data were deposited at the Gene Expression Omnibus (GEO) (accession number GSE141850). We used SPO11-oligo, SSDS, H3K4me3, MNase-seq, and yeast S1-seq data from GEO accession numbers GSE84689, GSE35498, GSE52628, GSE121760, and GSE85253, respectively (Brick et al. 2012; Baker et al. 2014; Lange et al. 2016; Mimitou et al. 2017; Lam et al. 2019). For SPO11-oligo maps, we used either “B6” or “Atm null 1” data set, which were, respectively, from wild-type mice from a pure B6 background, or Atm–/– mice from a mix of B6 and 129/Sv strain backgrounds, which carry the same Prdm9 allele. Unless otherwise stated, hot spots analyzed in this study were the 13,960 hot spots previously identified using uniquely mapped SPO11-oligo reads (Lange et al. 2016). The hot spot center was defined as the position of the smoothed peak in the SPO11-oligo density. MNase-seq sequence coverage was calculated by extending uniquely mapped reads to 150 nt to represent a mononucleosome.
Quantification and statistical analyses
Statistical analyses were performed using R versions 3.2.3 to 3.3.1 (http://www.r-project.org). Statistical parameters and tests are reported in the figures and legends.
Supplementary Material
Acknowledgments
We thank A. Viale, N. Mohibullah, and R. Patel (Memorial Sloan Kettering Cancer Center [MSKCC] Integrated Genomics Operation) for sequencing and ATAC-seq library preparation; E. Mimitou for advice during adaptation of the S1-seq method; S. Peterson and M. Jasin (MSKCC), P. Donnelly (University of Oxford), A. Nussenzweig, W. Wu, and J. Paiano (National Institutes of Health) for discussions and sharing unpublished data; P.C. Huang (MSKCC) for ExoT treatment optimization; P.C. Huang and H. Murakami (MSKCC) for help in analysis of recombination intermediates; and M. Neale (University of Sussex) and members of the Keeney and Jasin laboratories for discussions. MSKCC core facilities were supported by National Institutes of Health (NIH) Cancer Center Core Grant P30 CA008748. This work was supported by NIH grant R35 GM118092 (to S.K.).
Author contributions: S.Y. performed experiments. S.Y. analyzed S1-seq data with contributions from A.G.H. K.H. performed an initial ATAC-seq analysis. Y.Z. and W.E. generated Exo1DA mice. S.Y. and S.K. designed the study and wrote the paper with contributions from A.G.H.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Aritcle and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.336032.119.
References
- Baker CL, Walker M, Kajita S, Petkov PM, Paigen K. 2014. PRDM9 binding organizes hotspot nucleosomes and limits Holliday junction migration. Genome Res 24: 724–732. 10.1101/gr.170167.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barchi M, Mahadevaiah S, Di Giacomo M, Baudat F, de Rooij DG, Burgoyne PS, Jasin M, Keeney S. 2005. Surveillance of different recombination defects in mouse spermatocytes yields distinct responses despite elimination at an identical developmental stage. Mol Cell Biol 25: 7203–7215. 10.1128/MCB.25.16.7203-7215.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barchi M, Roig I, Di Giacomo M, de Rooij DG, Keeney S, Jasin M. 2008. ATM promotes the obligate XY crossover and both crossover control and chromosome axis integrity on autosomes. PLoS Genet 4: e1000076 10.1371/journal.pgen.1000076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, et al. 1996. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86: 159–171. 10.1016/S0092-8674(00)80086-0 [DOI] [PubMed] [Google Scholar]
- Baudat F, Nicolas A. 1997. Clustering of meiotic double-strand breaks on yeast chromosome III. Proc Natl Acad Sci 94: 5213–5218. 10.1073/pnas.94.10.5213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat F, Manova K, Yuen JP, Jasin M, Keeney S. 2000. Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol Cell 6: 989–998. 10.1016/S1097-2765(00)00098-8 [DOI] [PubMed] [Google Scholar]
- Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, Coop G, de Massy B. 2010. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327: 836–840. 10.1126/science.1183439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellani MA, Romanienko PJ, Cairatti DA, Camerini-Otero RD. 2005. SPO11 is required for sex-body formation, and Spo11 heterozygosity rescues the prophase arrest of Atm−/− spermatocytes. J Cell Sci 118: 3233–3245. 10.1242/jcs.02466 [DOI] [PubMed] [Google Scholar]
- Bellve AR, Cavicchia JC, Millette CF, O'Brien DA, Bhatnagar YM, Dym M. 1977. Spermatogenic cells of the prepuberal mouse. Isolation and morphological characterization. J Cell Biol 74: 68–85. 10.1083/jcb.74.1.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop DK, Park D, Xu L, Kleckner N. 1992. DMC1: a meiosis-specific yeast homolog of E. coli recA required for recombination, synaptonemal complex formation, and cell cycle progression. Cell 69: 439–456. 10.1016/0092-8674(92)90446-J [DOI] [PubMed] [Google Scholar]
- Brick K, Smagulova F, Khil P, Camerini-Otero RD, Petukhova GV. 2012. Genetic recombination is directed away from functional genomic elements in mice. Nature 485: 642–645. 10.1038/nature11089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Bishop DK. 2014. DNA strand exchange and RecA homologs in meiosis. Cold Spring Harb Perspect Biol 7: a016659 10.1101/cshperspect.a016659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Grubb J, Zhang A, Rust MJ, Bishop DK. 2015. Small Rad51 and Dmc1 complexes often co-occupy both ends of a meiotic DNA double strand break. PLoS Genet 11: e1005653 10.1371/journal.pgen.1005653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. 10.1038/nmeth.2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canela A, Maman Y, Huang SN, Wutz G, Tang W, Zagnoli-Vieira G, Callen E, Wong N, Day A, Peters JM, et al. 2019. Topoisomerase II-induced chromosome breakage and translocation is determined by chromosome architecture and transcriptional activity. Mol Cell 75: 252–266 e258. 10.1016/j.molcel.2019.04.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E, Cejka P. 2014. Sae2 promotes dsDNA endonuclease activity within Mre11–Rad50–Xrs2 to resect DNA breaks. Nature 514: 122–125. 10.1038/nature13771 [DOI] [PubMed] [Google Scholar]
- Cartagena-Lirola H, Guerini I, Viscardi V, Lucchini G, Longhese MP. 2006. Budding yeast Sae2 is an in vivo target of the Mec1 and Tel1 checkpoint kinases during meiosis. Cell Cycle 5: 1549–1559. 10.4161/cc.5.14.2916 [DOI] [PubMed] [Google Scholar]
- Chan YL, Zhang A, Weissman BP, Bishop DK. 2019. RPA resolves conflicting activities of accessory proteins during reconstitution of Dmc1-mediated meiotic recombination. Nucleic Acids Res 47: 747–761. 10.1093/nar/gky1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Pandita RK, Hambarde S, Mattoo AR, Charaka V, Ahmed KM, Iyer SP, Hunt CR, Pandita TK. 2018. SMARCAD1 phosphorylation and ubiquitination are required for resection during DNA double-strand break repair. iScience 2: 123–135. 10.1016/j.isci.2018.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claeys Bouuaert C, Tischfield SE, Pu S, Mimitou EP, Arias-Palomo E, Berger JM, Keeney S. 2020. Structural and functional characterization of the Spo11 core complex. bioRxiv 10.1101/2020.02.21.960211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole F, Baudat F, Grey C, Keeney S, de Massy B, Jasin M. 2014. Mouse tetrad analysis provides insights into recombination mechanisms and hotspot evolutionary dynamics. Nat Genet 46: 1072–1080. 10.1038/ng.3068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies B, Hatton E, Altemose N, Hussin JG, Pratto F, Zhang G, Hinch AG, Moralli D, Biggs D, Diaz R, et al. 2016. Re-engineering the zinc fingers of PRDM9 reverses hybrid sterility in mice. Nature 530: 171–176. 10.1038/nature16931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diagouraga B, Clement JAJ, Duret L, Kadlec J, de Massy B, Baudat F. 2018. PRDM9 methyltransferase activity is essential for meiotic DNA double-strand break formation at its binding sites. Mol Cell 69: 853–865 e856. 10.1016/j.molcel.2018.01.033 [DOI] [PubMed] [Google Scholar]
- Garcia V, Phelps SE, Gray S, Neale MJ. 2011. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 479: 241–244. 10.1038/nature10515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia V, Gray S, Allison RM, Cooper TJ, Neale MJ. 2015. Tel1(ATM)-mediated interference suppresses clustered meiotic double-strand-break formation. Nature 520: 114–118. 10.1038/nature13993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillon H, Baudat F, Grey C, Liskay RM, de Massy B. 2005. Crossover and noncrossover pathways in mouse meiosis. Mol Cell 20: 563–573. 10.1016/j.molcel.2005.09.021 [DOI] [PubMed] [Google Scholar]
- Henry JM, Camahort R, Rice DA, Florens L, Swanson SK, Washburn MP, Gerton JL. 2006. Mnd1/Hop2 facilitates Dmc1-dependent interhomolog crossover formation in meiosis of budding yeast. Mol Cell Biol 26: 2913–2923. 10.1128/MCB.26.8.2913-2923.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinch AG, Zhang G, Becker PW, Moralli D, Hinch R, Davies B, Bowden R, Donnelly P. 2019. Factors influencing meiotic recombination revealed by whole-genome sequencing of single sperm. Science 363: eaau8861 10.1126/science.aau8861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter N. 2015. Meiotic recombination: the essence of heredity. Cold Spring Harb Perspect Biol 7: 361–395. 10.1101/cshperspect.a016618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi N, Brown MS, Bishop DK, Borner GV. 2015. Gradual implementation of the meiotic recombination program via checkpoint pathways controlled by global DSB levels. Mol Cell 57: 797–811. 10.1016/j.molcel.2014.12.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keelagher RE, Cotton VE, Goldman AS, Borts RH. 2011. Separable roles for Exonuclease I in meiotic DNA double-strand break repair. DNA Repair (Amst) 10: 126–137. 10.1016/j.dnarep.2010.09.024 [DOI] [PubMed] [Google Scholar]
- Keeney S, Giroux CN, Kleckner N. 1997. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88: 375–384. 10.1016/S0092-8674(00)81876-0 [DOI] [PubMed] [Google Scholar]
- Khil PP, Smagulova F, Brick KM, Camerini-Otero RD, Petukhova GV. 2012. Sensitive mapping of recombination hotspots using sequencing-based detection of ssDNA. Genome Res 22: 957–964. 10.1101/gr.130583.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi W, Takaku M, Machida S, Tachiwana H, Maehara K, Ohkawa Y, Kurumizaka H. 2016. Chromatin architecture may dictate the target site for DMC1, but not for RAD51, during homologous pairing. Sci Rep 6: 24228 10.1038/srep24228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam I, Keeney S. 2014. Mechanism and regulation of meiotic recombination initiation. Cold Spring Harb Perspect Biol 7: a016634 10.1101/cshperspect.a016634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam KG, Brick K, Cheng G, Pratto F, Camerini-Otero RD. 2019. Cell-type-specific genomics reveals histone modification dynamics in mammalian meiosis. Nat Commun 10: 3821 10.1038/s41467-019-11820-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange J, Pan J, Cole F, Thelen MP, Jasin M, Keeney S. 2011. ATM controls meiotic double-strand-break formation. Nature 479: 237–240. 10.1038/nature10508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange J, Yamada S, Tischfield SE, Pan J, Kim S, Zhu X, Socci ND, Jasin M, Keeney S. 2016. The landscape of mouse meiotic double-strand break formation, processing, and repair. Cell 167: 695–708 e616. 10.1016/j.cell.2016.09.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BI, Nguyen LH, Barsky D, Fernandes M, Wilson DM III. 2002. Molecular interactions of human Exo1 with DNA. Nucleic Acids Res 30: 942–949. 10.1093/nar/30.4.942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Mand MR, Kao CH, Zhou Y, Ryu SW, Richards AL, Coon JJ, Paull TT. 2018. ATM directs DNA damage responses and proteostasis via genetically separable pathways. Sci Signal 11: eaan5598 10.1126/scisignal.aan5598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Bitoun E, Altemose N, Davies RW, Davies B, Myers SR. 2019. A high-resolution map of non-crossover events reveals impacts of genetic diversity on mammalian meiotic recombination. Nat Commun 10: 3900 10.1038/s41467-019-11675-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukaszewicz A, Lange J, Keeney S, Jasin M. 2018. Control of meiotic double-strand-break formation by ATM: local and global views. Cell Cycle 17: 1155–1172. 10.1080/15384101.2018.1464847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevaiah SK, Turner JM, Baudat F, Rogakou EP, de Boer P, Blanco-Rodriguez J, Jasin M, Keeney S, Bonner WM, Burgoyne PS. 2001. Recombinational DNA double-strand breaks in mice precede synapsis. Nat Genet 27: 271–276. 10.1038/85830 [DOI] [PubMed] [Google Scholar]
- Mancera E, Bourgon R, Brozzi A, Huber W, Steinmetz LM. 2008. High-resolution mapping of meiotic crossovers and non-crossovers in yeast. Nature 454: 479–485. 10.1038/nature07135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsolier-Kergoat MC, Khan MM, Schott J, Zhu X, Llorente B. 2018. Mechanistic view and genetic control of DNA recombination during meiosis. Mol Cell 70: 9–20 e26. 10.1016/j.molcel.2018.02.032 [DOI] [PubMed] [Google Scholar]
- Martini E, Borde V, Legendre M, Audic S, Regnault B, Soubigou G, Dujon B, Llorente B. 2011. Genome-wide analysis of heteroduplex DNA in mismatch repair-deficient yeast cells reveals novel properties of meiotic recombination pathways. PLoS Genet 7: e1002305 10.1371/journal.pgen.1002305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Keeney S. 2018. S1-seq assay for mapping processed DNA ends. Methods Enzymol 601: 309–330. 10.1016/bs.mie.2017.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Yamada S, Keeney S. 2017. A global view of meiotic double-strand break end resection. Science 355: 40–45. 10.1126/science.aak9704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moens PB, Chen DJ, Shen Z, Kolas N, Tarsounas M, Heng HHQ, Spyropoulos B. 1997. Rad51 immunocytology in rat and mouse spermatocytes and oocytes. Chromosoma 106: 207–215. 10.1007/s004120050241 [DOI] [PubMed] [Google Scholar]
- Moens PB, Kolas NK, Tarsounas M, Marcon E, Cohen PE, Spyropoulos B. 2002. The time course and chromosomal localization of recombination-related proteins at meiosis in the mouse are compatible with models that can resolve the early DNA–DNA interactions without reciprocal recombination. J Cell Sci 115: 1611–1622. [DOI] [PubMed] [Google Scholar]
- Murakami H, Keeney S. 2014. Temporospatial coordination of meiotic DNA replication and recombination via DDK recruitment to replisomes. Cell 158: 861–873. 10.1016/j.cell.2014.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama Y, Tsutsui Y, Iwasaki H. 2011. The fission yeast meiosis-specific Dmc1 recombinase mediates formation and branch migration of Holliday junctions by preferentially promoting strand exchange in a direction opposite to that of Rad51. Genes Dev 25: 516–527. 10.1101/gad.1997511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S, Bowden R, Tumian A, Bontrop RE, Freeman C, MacFie TS, McVean G, Donnelly P. 2010. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science 327: 876–879. 10.1126/science.1182363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale MJ, Keeney S. 2006. Clarifying the mechanics of DNA strand exchange in meiotic recombination. Nature 442: 153–158. 10.1038/nature04885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale MJ, Pan J, Keeney S. 2005. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 436: 1053–1057. 10.1038/nature03872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmore R, Cao L, Kleckner N. 1991. Temporal comparison of recombination and synaptonemal complex formation during meiosis in S. cerevisiae. Cell 66: 1239–1256. 10.1016/0092-8674(91)90046-2 [DOI] [PubMed] [Google Scholar]
- Paiano J, Wu W, Yamada S, Sciascia N, Callen E, Paola Cotrim A, Deshpande RA, Maman Y, Day A, Paull TT, et al. 2020. ATM and PRDM9 regulate SPO11-bound recombination intermediates during meiosis. Nat Commun 11: 857 10.1038/s41467-020-14654-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Sasaki M, Kniewel R, Murakami H, Blitzblau HG, Tischfield SE, Zhu X, Neale MJ, Jasin M, Socci ND, et al. 2011. A hierarchical combination of factors shapes the genome-wide topography of yeast meiotic recombination initiation. Cell 144: 719–731. 10.1016/j.cell.2011.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson SE, Keeney S, Jasin M. 2020. Mechanistic insight into crossing over during mouse meiosis. Mol Cell (in press). 10.1016/j.molcel.2020.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman DL, Cobb J, Schimenti KJ, Wilson LA, Cooper DM, Brignull E, Handel MA, Schimenti JC. 1998. Meiotic prophase arrest with failure of chromosome synapsis in mice deficient for Dmc1, a germline-specific RecA homolog. Mol Cell 1: 697–705. 10.1016/S1097-2765(00)80069-6 [DOI] [PubMed] [Google Scholar]
- Powers NR, Parvanov ED, Baker CL, Walker M, Petkov PM, Paigen K. 2016. The meiotic recombination activator PRDM9 trimethylates both H3K36 and H3K4 at recombination hotspots in vivo. PLoS Genet 12: e1006146 10.1371/journal.pgen.1006146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara A, Gasior S, Ogawa T, Kleckner N, Bishop DK. 1997. Saccharomyces cerevisiae recA homologues RAD51 and DMC1 have both distinct and overlapping roles in meiotic recombination. Genes Cells 2: 615–629. 10.1046/j.1365-2443.1997.1480347.x [DOI] [PubMed] [Google Scholar]
- Smagulova F, Brick K, Pu Y, Camerini-Otero RD, Petukhova GV. 2016. The evolutionary turnover of recombination hot spots contributes to speciation in mice. Genes Dev 30: 266–280. 10.1101/gad.270009.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruce C, Dlamini S, Ananda G, Bronkema N, Tian H, Paigen K, Carter GW, Baker CL. 2020. HELLS and PRDM9 form a pioneer complex to open chromatin at meiotic recombination hot spots. Genes Dev 34: 398–412. 10.1101/gad.333542.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storlazzi A, Gargano S, Ruprich-Robert G, Falque M, David M, Kleckner N, Zickler D. 2010. Recombination proteins mediate meiotic spatial chromosome organization and pairing. Cell 141: 94–106. 10.1016/j.cell.2010.02.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS. 2014. End resection at double-strand breaks: mechanism and regulation. Cold Spring Harb Perspect Biol 6: a016436 10.1101/cshperspect.a016436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Mathioudakis N, Diagouraga B, Dong A, Dombrovski L, Baudat F, Cusack S, de Massy B, Kadlec J. 2013. Molecular basis for the regulation of the H3K4 methyltransferase activity of PRDM9. Cell Rep 5: 13–20. 10.1016/j.celrep.2013.08.035 [DOI] [PubMed] [Google Scholar]
- Yamada S, Kim S, Tischfield SE, Jasin M, Lange J, Keeney S. 2017. Genomic and chromatin features shaping meiotic double-strand break formation and repair in mice. Cell Cycle 16: 1870–1884. 10.1080/15384101.2017.1361065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharyevich K, Ma Y, Tang S, Hwang PY, Boiteux S, Hunter N. 2010. Temporally and biochemically distinct activities of Exo1 during meiosis: double-strand break resection and resolution of double Holliday junctions. Mol Cell 40: 1001–1015. 10.1016/j.molcel.2010.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelazowski MJ, Sandoval M, Paniker L, Hamilton HM, Han J, Gribbell MA, Kang R, Cole F. 2017. Age-dependent alterations in meiotic recombination cause chromosome segregation errors in spermatocytes. Cell 171: 601–614 e613. 10.1016/j.cell.2017.08.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Zhang Y, Wilkins K, Edelmann W, Usdin K. 2018. MutLγ promotes repeat expansion in a Fragile X mouse model while EXO1 is protective. PLoS Genet 14: e1007719 10.1371/journal.pgen.1007719 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.