

In this study, Tsai et al. sought to understand the mechanisms controlling dysregulation of the DNA/RNA binding protein FUS, which causes certain subtypes of ALS/FTD. They demonstrate that in human cells overexpressing WT FUS or expressing mutant derivatives, the protein associates with multiple mRNAs, and these are enriched in mRNAs encoding mitochondrial respiratory chain components, showing that respiratory chain complex mRNA sequestration underlies the mitochondrial defects characteristic of ALS/FTD and contributes to the FUS toxic gain of function linked to this disease spectrum.
Keywords: ALS/FTD, FUS, respiratory chain complex mRNA
Abstract
Dysregulation of the DNA/RNA-binding protein FUS causes certain subtypes of ALS/FTD by largely unknown mechanisms. Recent evidence has shown that FUS toxic gain of function due either to mutations or to increased expression can disrupt critical cellular processes, including mitochondrial functions. Here, we demonstrate that in human cells overexpressing wild-type FUS or expressing mutant derivatives, the protein associates with multiple mRNAs, and these are enriched in mRNAs encoding mitochondrial respiratory chain components. Notably, this sequestration leads to reduced levels of the encoded proteins, which is sufficient to bring about disorganized mitochondrial networks, reduced aerobic respiration and increased reactive oxygen species. We further show that mutant FUS associates with mitochondria and with mRNAs encoded by the mitochondrial genome. Importantly, similar results were also observed in fibroblasts derived from ALS patients with FUS mutations. Finally, we demonstrate that FUS loss of function does not underlie the observed mitochondrial dysfunction, and also provides a mechanism for the preferential sequestration of the respiratory chain complex mRNAs by FUS that does not involve sequence-specific binding. Together, our data reveal that respiratory chain complex mRNA sequestration underlies the mitochondrial defects characteristic of ALS/FTD and contributes to the FUS toxic gain of function linked to this disease spectrum.
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are devastating neurodegenerative diseases often characterized by RNA-binding protein (RBP) aggregation within neural tissues. Such aggregation can contribute to ALS/FTD pathology in several ways, for example by producing misspliced transcripts due to sequestration of available splicing regulators (Lee et al. 2013; Conlon et al. 2016, 2018). Among ALS/FTD-linked RBPs, fused in sarcoma (FUS), a nuclear RBP participating in splicing and other cellular processes, forms cytoplasmic aggregates in a significant fraction of ALS/FTD patient brains (Kwiatkowski et al. 2009; Vance et al. 2009; Dormann and Haass 2011). Pathological mutations in FUS have also been identified in ∼5% of inherited ALS (Deng et al. 2014). Growing evidence has suggested that FUS cytoplasmic gain of function drives ALS/FTD progression. For example, mice expressing human FUS with a truncated nuclear localization signal show substantial motor neuron degeneration, while motor neuron-specific FUS knockout mice lack an ALS phenotype (Scekic-Zahirovic et al. 2016; Sharma et al. 2016). Mutant FUS has also been reported to retain SMN, a protein essential to motor neuron survival, in the cytoplasm and to impair axonal growth (Groen et al. 2013; Sun et al. 2015). Additionally, FUS cytoplasmic inclusions sequester motor protein kinesin-1 and disrupt RNA localization (Yasuda et al. 2017). Although FUS thus affects many processes when mislocalized to the cytoplasm, important questions regarding the origin of FUS toxicity remain to be answered. Especially significant is whether and how FUS damages important cellular organelles, and how this ultimately contributes to motor neuron degeneration.
Mitochondrial dysfunction is strongly associated with various neurodegenerative diseases including ALS/FTD (Lin and Beal 2006; Smith et al. 2017). Patient cells show elevated numbers of mitochondria with ruptured cristae, shortened length, and reduced ATP levels (Sasaki and Iwata 2007; Wang et al. 2016). Mutations in the mitochondrial gene CHCHD10 have been identified in ALS/FTD patients, which further suggests that mitochondrial defects may contribute to ALS/FTD pathogenicity (Bannwarth et al. 2014). FUS has also been reported to damage mitochondria when mislocalized to the cytoplasm. For example, wild-type and mutant FUS are recruited to mitochondria by HSP60 and can induce mitochondrial fragmentation (Deng et al. 2015). Also, overexpressed wild-type FUS or expression of mutant FUS has been shown to activate GSK3-β and disrupt ER–mitochondria contacts (Stoica et al. 2016). Intriguingly, not only does mislocalized mutant FUS induce mitochondrial dysfunction, but increased wild-type FUS expression can also cause similar effects. Indeed, FUS pathology has been identified in ∼10% of FTD patient brains, including increased FUS expression, cytoplasmic aggregates and mitochondrial damage, even in the absence of FUS mutations (Dormann and Haass 2011; Deng et al. 2015; Hofmann et al. 2018). One study identified mutations in the 3′UTR of FUS mRNA in a number of ALS patients, leading to twofold to fivefold increases in both FUS protein and transcript levels, and to FUS accumulation in the cytoplasm (Sabatelli et al. 2013). These studies together point to mitochondria as a vulnerable organelle upon increased FUS protein accumulation in the cytoplasm.
FUS has been well studied as an RNA/DNA-binding protein. The protein binds to long pre-mRNAs, noncoding RNAs and single-stranded DNA, and is predominantly nuclear-localized under normal conditions (Dormann et al. 2010; Lagier-Tourenne et al. 2012; Tan et al. 2012). Functions attributed to FUS in the nucleus are varied and include control of alternative splicing and alternative polyadenylation, a role in nuclear body formation, and functions in the DNA damage response and transcriptional regulation (Schwartz et al. 2012; Tan et al. 2012; Wang et al. 2013b; Zhou et al. 2013; Hennig et al. 2015; Masuda et al. 2015). Under stress conditions, such as hyperosmolar stress, FUS translocates to the cytoplasm and incorporates into stress granules with other RBPs to promote cell survival (Sama et al. 2013). Studies using cross-linking immunoprecipitation (CLIP) have shown that wild-type and mutant FUS are enriched on distinct regions of mRNAs, with wild-type primarily binding within introns and mutant binding to 3′UTRs or other exonic regions (Hoell et al. 2011; Nakaya and Maragkakis 2018). The distinct binding regions reflect the respective nuclear and cytoplasmic localization of wild-type and mutant FUS, respectively. Our previous study using fluorescence in situ hybridization (FISH) and biochemical fractionation showed that mutant FUS sequesters several mRNAs, including MeCP2 mRNA, in cytoplasmic granules and prevents MeCP2 translation (Coady and Manley 2015). Although MeCP2 has not been directly linked to ALS/FTD, these findings suggest a possible broad impact on expression of numerous FUS-sequestered cytoplasmic mRNAs.
Here we extended our analysis of FUS-sequestered mRNAs and investigated whether this sequestration might contribute to dysregulated gene expression and subcellular defects linked to ALS/FTD. To this end, we isolated and sequenced ALS mutant FUS-bound cytoplasmic mRNAs from FUS-R521C transfected cells. Unexpectedly, analysis of our RNA-seq data, together with CLIP-seq and RIP-seq data sets derived from independent studies, revealed that mRNAs encoding mitochondrial respiratory chain complex (RCC) proteins were preferentially associated with mutant FUS. In addition, we show that expression of the encoded proteins was sharply and specifically reduced, without significant changes in overall translation or mRNA expression levels. We also show that mitochondrial networks were disrupted and aerobic respiration inhibited in both cells overexpressing wild-type FUS or expressing ALS mutant derivatives, as well as in ALS patient-derived fibroblasts. Moreover, depletion of the same RCC proteins recapitulated FUS-induced mitochondrial dysfunction, suggesting that loss of respiratory chain function lies at the center of FUS-induced mitochondrial defects. In contrast, mitochondrial defects were not observed in FUS depleted cells, consistent with the mechanism being a toxic gain of function. Finally, we demonstrate that mutant FUS accumulates at mitochondria and interacts with mitochondrial—as well as nuclear—encoded respiratory chain mRNAs, suggesting a mechanism by which mutant/overexpressed FUS preferentially associates with these mRNAs. Together, our findings explain how overexpressed or mutant FUS brings about the mitochondrial dysfunction that occurs in ALS/FTD
Results
Mutant FUS associates with mRNAs encoding mitochondrial RCC proteins
Our previous experiments indicated that ALS mutant FUS can sequester certain mRNAs in cytoplasmic aggregates, leading to their translational silencing (Coady and Manley 2015). To extend these results, we first examined whether mutant FUS colocalizes with poly(A+) RNA in the cytoplasm by transfecting plasmids encoding GFP-tagged wild-type or R521C mutant FUS into HEK293T cells. Note that Western blot (WB) revealed down-regulation of endogenous FUS after overexpressing GFP-tagged wild-type FUS, but not R521C mutant FUS (Fig. 1A). This is consistent with previous studies showing that wild-type, but not mutant, FUS autoregulates endogenous FUS levels (Zhou et al. 2013), and provides evidence that the GFP tag does not compromise normal FUS functions. Indeed, R521C mutant FUS, visualized by GFP fluorescence, and poly(A+) RNA detected by oligo(dT) hybridization displayed striking colocalization (Fig. 1B,C), supporting the existence of FUS-mRNA cytoplasmic aggregates.
Figure 1.

Nuclear-encoded respiratory chain complex (RCC) mRNAs are preferentially sequestered by ALS mutant FUS. (A) GFP-tagged FUS was transiently expressed in HEK293T cells for 48 h followed by Western blot of endogenous and GFP-tagged FUS. (B) Immunofluorescent staining of GFP-FUS and poly(A+) RNAs. Scale bar, 15 µm. (C) Quantification of GFP-FUS-poly(A+) RNA granules per cell. Total cells analyzed: N = 153. Fifty or more cells were analyzed in each transfection condition. (***) P < 0.001, Mann-Whitney U-test. (D) Flag-tagged mutant FUS-R521C was transiently expressed in U87 cells followed by fractionation to generate soluble and insoluble fractions. The insoluble fraction was extracted with 100 mM urea and the supernatant was IPed with an antibody targeting Flag-tagged FUS-R521C. Precipitated RNAs were purified and used for 3′READS to sequence poly(A+) RNAs bound by FUS-R521C. (E) Scatter plot of all poly(A+) RNAs associated with insoluble FUS-R521C (IP) versus poly(A+) RNA expression levels in whole cell (WC). Standard curve was plotted using linear regression. R521C-enriched poly(A+) RNAs are shown in red. RNAs with enrichment below the standard curve are nonenriched transcripts (pale blue). The remainders are neither (dark gray). (F) GO overrepresentation analysis of R521C-enriched and nonenriched transcripts in FUS-R521C IP. GO enrichment scores are displayed in −log10 (P-value). (G) List of respiratory chain complex I genes identified in overrepresentation analysis of R521C-enriched poly(A+) RNAs. (H) Genome browser snapshot of example gene read clusters from two replicates. (Top) NDUFA5. (Bottom) INTS4. (pA) Poly(A) site. (I) Box plots showing IP enrichment of RCC mRNAs (rep1: 64 genes, rep2: 60 genes) and total poly(A+) RNAs associated with FUS-R521C. Statistics: Wilcoxcon rank sum test.
We next wished to identify globally the mRNAs present in the FUS aggregates. To this end, we used the procedure described in our previous study (see also Fig. 1D; Coady and Manley 2015). Briefly, we transfected a plasmid encoding FLAG-tagged R521C mutant FUS (FUS-R521C) into U87 glioma cells (used because they provide a brain-derived cell line amenable to these biochemical analyses), isolated an insoluble fraction containing FUS-R521C, and then gently resolubilzed the FUS complexes (with 0.1 M urea) and isolated them by immunoprecipitation (IP) with anti-Flag antibodies. RNA was extracted and subjected to 3′ region extraction and deep sequencing (3′READS) (Hoque et al. 2013) to identify FUS-bound poly(A+) RNAs. Poly(A+) RNA reads from the mutant FUS IP were then compared with input reads to identify mRNAs susceptible to possible FUS sequestration. The scatter plot shows that the abundance of poly(A+) RNAs associated with FUS-R521C was positively correlated with their expression levels (rep1: R = 0.928, rep2: R = 0.801, Pearson's correlation.) (Fig. 1E).
We next wished to investigate whether FUS-R521C associated preferentially with specific mRNAs, as was suggested by our previous work (Coady and Manley 2015). To this end, we normalized sequencing reads of RNA associated with FUS-R521C isolated from the insoluble fraction to the whole-cell reads and calculated the fold change of RNA read counts in the IP relative to the whole cell to obtain a “sequestration susceptibility” for each RNA (Fig. 1E). We designated RNAs with the top 5% greatest fold change above the standard curve as “R521C-enriched.” RNAs below the standard curve were designated as “nonenriched,” while the remainder were considered to be neither enriched nor nonenriched.
We next identified the R521C-enriched transcripts found in both of two replicates (236 RNAs) (Supplemental Table S1) and subjected them to gene ontology (GO) enrichment analysis. Interestingly, GO analysis revealed overrepresentation of mitochondrial inner membrane, mitochondrial respiratory chain and respiratory chain complex I (or Type I NADH dehydrogenase) proteins encoded by R521C-enriched mRNAs (Fig. 1F). These proteins are essential components of the RCC directly responsible for electron transport for ATP production (Fig. 1F,G), a process indeed suggested to be impaired in ALS/FTD patients (Wang et al. 2016; Pansarasa et al. 2018). We identified 11 transcripts encoding RCC subunits, which have been reported to be dysfunctional in, or associated with, other neurodegenerative diseases, such as mitochondrial complex I deficiency, Alzheimer's, Huntington's, and Parkinson's (Supplemental Fig. S1A). We confirmed that the R521C-enriched mRNAs were poly(A+) with reads clustered in the 3′UTR and possessed stronger signal peaks than in the whole-cell fraction (Fig. 1H; Supplemental Fig. S1B). In contrast, nonenriched transcripts did not contain stronger signal peaks (Fig. 1H; Supplemental Fig. S1C). To demonstrate that the GO terms overrepresented in the R521C-enriched RNAs were not identified by coincidence, we performed the same analysis both with all the nonenriched transcripts as well as with the 5% most depleted mRNAs (Fig. 1F; Supplemental Table S2). The results did not indicate any specific GO terms overrepresented in these mRNAs. In addition, we generated a negative control scatter plot using the two whole-cell input replicate samples and performed GO analysis on the top 5% outlier genes (Supplemental Fig. S1D). The results did not indicate any specific GO terms related to RCC components overrepresented in these transcripts. We also performed an unbiased enrichment analysis using total poly(A+) RNA associated with FUS-R521C, and as expected found mRNAs encoding RCC proteins to be significantly enriched compared with total poly(A+) RNA (Fig. 1I).
We next wished to obtain more evidence that mRNAs encoding RCC proteins are preferentially sequestered by ALS FUS mutants. We first reanalyzed wild-type and ALS mutant FUS-R495X CLIP-seq data sets from mouse ESCs-differentiated neurons (Nakaya and Maragkakis 2018) to identify mRNAs enriched in FUS CLIP (Supplemental Fig. S2A). Consistent with our analysis, RNA abundance in both wild-type and mutant FUS CLIP fractions positively correlated with their expression levels in mouse ESC-derived neurons (Supplemental Fig. S2A). We then selected the top 2% CLIP-enriched mRNAs, which yielded 187 transcripts. (Note that this number is comparable with the number of R521C-enriched transcripts [236] analyzed above.) Indeed, six RCC mRNAs were found in this cohort of FUS-R495X CLIP-enriched transcripts and mitochondrial RCC GO terms were significantly overrepresented (Supplemental Fig. S2B,C; Supplemental Table S3). In contrast, although three RCC mRNAs were bound by wild-type FUS within the top 2% CLIP-enriched mRNAs (Supplemental Fig. S2D), mitochondrial RCC GO terms were not overrepresented (Supplemental Fig. S2D; Supplemental Table S3). The same analysis performed with the 2% most depleted transcripts from the wild-type and FUS-R495X CLIP-seq analysis revealed no mitochondrial RCC GO terms overrepresented in these transcripts (Supplemental Table S3).
To extend this comparison to human cells, we overlapped the R521C-enriched mRNAs identified in our analysis with mRNAs bound by FUS-R521G/H but not by wild-type FUS in stably transformed HEK293 cells, determined by CLIP (Supplemental Fig. S2E; Hoell et al. 2011). Although the number of overlapped targets was modest, GO analysis of the overlapping mRNAs again revealed overrepresentation of mitochondrial RCC proteins (Supplemental Fig. S2E). This data provides additional evidence that mRNAs encoding mitochondrial RCC components are preferentially bound by mutant FUS. The combined analysis of RIP-seq, CLIP-seq and whole-cell RNA-seq data targeting wild-type or ALS mutant FUS thus demonstrates that mRNAs encoding RCC components are overrepresented among the enriched mutant FUS-bound transcripts.
Density of known wild-type FUS-binding sequences is not correlated with enrichment of mutant FUS-associated transcripts
We next wished to determine whether mutant FUS preferentially binds certain transcripts via specific sequence motifs. To this end, we first interrogated the density of three reported FUS-binding sites derived from multiple SELEX, RNAcompete and CLIP-seq studies (Lerga et al. 2001; Lagier-Tourenne et al. 2012; Ray et al. 2013; Wang et al. 2015b) in the R521C-enriched transcripts as well as in total FUS-R521C associated poly(A+) RNA. Possible co-occurrence of all three motifs described in the above studies was analyzed first. We found no significant correlation between enrichment by mutant FUS and the density of sequence motifs presented in total R521C-associated transcripts (Supplemental Fig. S3A). We also found no significant increase of FUS binding motifs in RCC mRNAs (Supplemental Fig. S3B) or in the R521C-enriched RNAs (Supplemental Fig. S3C). Evaluation of the three motifs separately again showed no significant differences in motif density between RCC mRNA and total FUS-R521C associated poly(A+) RNA (Supplemental Fig. S3D). These results together suggest that mutant FUS employs a mechanism to recognize target mRNAs distinct from binding the reported sequence motifs. This is consistent with previous studies of FUS binding specificity using gel mobility shift assays, which suggested that both wild-type and mutant FUS do not possess strong sequence specificity (Wang et al. 2015b).
Mutant and overexpressed wild-type FUS associate with RCC mRNAs in human cell lines and ALS patient cells
We next examined whether wild-type and mutant FUS bind mRNAs encoding RCC proteins with different apparent affinities. We transfected plasmids encoding GFP-tagged wild type or FUS-R521C into HEK293T cells for 36 h, fractionated the cells as described in Figure 1D and performed IP using anti-GFP antibodies (Fig. 2A). We first examined twelve RCC mRNAs from the R521C-enriched transcripts (Fig. 2B). The amount of each mRNA tested that copurified with the GFP-tagged FUS derivatives was then determined by RT-qPCR (Fig. 2C). In the soluble fraction, wild-type and mutant FUS associated with these mRNAs at comparable levels. Intriguingly, though, mutant FUS bound sixfold higher on average to the mRNAs isolated from the insoluble fraction than did wild-type FUS (Fig. 2C). To determine whether these transcripts were preferentially bound relative to nonenriched transcripts, we compared their enrichment relative to seven mRNAs (Fig. 2D) that were not among the R521C-enriched transcripts. In the soluble fraction, binding of the RCC mRNAs and nonenriched mRNAs was similar, and not significantly different in wild-type or mutant FUS expressing cells (Fig. 2E; Supplemental Fig. S4A). In contrast, in the insoluble fraction, while all mRNAs showed increased binding to mutant as opposed to wild-type FUS, RCC mRNAs were more enriched compared with control mRNAs, relative to their abundance in the input fraction (Fig. 2F; Supplemental Fig. S4B). These findings together suggest that mutant FUS as well as overexpressed wild-type FUS preferentially associate with mRNAs encoding a subset of mitochondrial proteins and drive them into FUS aggregates.
Figure 2.

RCC mRNAs are preferentially associated with ALS mutant FUS in transfected human cells and ALS patient fibroblasts. (A) HEK293T were transfected with GFP-tagged wild-type and R521C mutant FUS followed by fractionation. Western blot of GFP-FUS IP performed with soluble and insoluble fractions using GFP antibody. (B) Scatter plot depicts RCC mRNAs enriched in the insoluble mutant FUS IP and are linked to neurodegenerative disease pathways. NDUFS6 is outside the vertical axis range. (C) RT-qPCR of the listed 12 RCC genes in soluble and insoluble fractions. Each point represents mean RNA fold change in IP relative to input fraction, then normalized to GFP transfected control cells. Data are shown in mean ± SD from three independent experiments. (****) P < 0.0001, Mann-Whitney U-test. (D) Scatter plot depicts neither and nonenriched RNAs in the insoluble mutant FUS IP. mRNAs shown are INTS4, UPF1, UPF2, RPL12, SMG1, and PPP2CA. (E,F) RNAs associated with GFP-tagged wild-type and R521C mutant FUS were extracted from soluble (E) and insoluble (F) fractions followed by RT-qPCR. IP fold changes of 12 RCC transcripts are compared with seven control transcripts (INTS4, UPF1, UPF2, RPL12, SMG1, PPP2CA, and 18S) that are non-enriched or not expected to be preferentially bound by mutant FUS. Data are shown in mean ± SD from three independent experiments. (***) P < 0.001; (****) P < 0.0001, Mann-Whitney U-test. (G) Western blot of FUS IP from the soluble and insoluble fractions from wild-type and R495Efs527X mutant FUS patient fibroblasts using antibody against FUS. (H) RNAs associated with wild-type and R495Efs527X mutant FUS were extracted from soluble and insoluble fractions. RT-qPCR of the listed 12 RCC genes described in C. Each point represents mean RNA fold change in IP relative to input fraction. (*) P < 0.05; (****) P < 0.0001, Mann-Whitney U-test. (I) RNAs associated with wild-type and R495Efs527X mutant FUS were extracted from soluble fraction followed by RT-qPCR. IP fold changes of the 12 RCC transcripts are compared with seven control transcripts (IGFBP3, NSUN5, MST1, ZFAND4, AAMP, ABCA7, and NEFH) that are shown to be depleted in 3′READS analysis (Supplemental Table S2) or previously shown not to be selectively bound by mutant FUS (Coady and Manley 2015). Data are displayed in mean ± SD. (*) P < 0.05, Mann-Whitney U-test.
We next wished to determine whether the above findings extend to ALS patient cells containing FUS mutations. We performed the same cell fractionation procedure described above with control, normal fibroblasts and R495Efs527X mutant FUS ALS patient fibroblasts followed by IP using a FUS antibody, and analyzed the associated RNAs by RT-qPCR (Fig. 2G). Strikingly, in the soluble fraction, the amount of mRNAs encoding RCC proteins bound by R495Efs527X FUS was ∼10-fold greater than bound by wild-type FUS (Fig. 2H). In contrast, the insoluble fraction contained only very low levels of these transcripts, and little if any enrichment for them in the R495Efs527X mutant cells. This likely reflects the lack of detectable cytoplasmic aggregates, as the mutant protein showed only diffuse cytoplasmic localization (Supplemental Fig. S4C). We also compared amounts of seven nonenriched mRNAs and the 12 RCC mRNAs bound by soluble FUS (Fig. 2I). While we did not observe significant differences in control fibroblasts, the RCC mRNAs showed higher enrichment in the R495Efs527X mutant fibroblasts (Fig. 2I). Our data demonstrate that mutant FUS displays stronger binding to mRNAs encoding RCC proteins in both FUS transfected cells and patient fibroblasts.
RCC mRNAs are sequestered in mutant FUS aggregates
The experiments above demonstrate that RCC mRNAs associate with insoluble mutant FUS. We next investigated whether this association also occurs in intact cells. We expressed GFP-tagged wild-type or FUS-R521C in U87 and 293T cells and performed RNA-FISH targeting NDUFA5 mRNA (see the Materials and Methods for FISH procedure). As expected, NDUFA5 mRNA showed little overlap with the GFP signal in vector control and wild-type FUS transfected cells (Fig. 3A; Supplemental Fig. S5A). Notably, NDUFA5 mRNA formed large and irregularly shaped granules and showed marked colocalization with FUS-R521C cytoplasmic aggregates in both cell types (Fig. 3A,B; Supplemental Fig. S5A,B). This data provides direct evidence for RCC mRNA sequestration by mutant FUS. In contrast, IGFBP3 mRNA, which is not an R521C-enriched transcript and was previously shown not to be recruited to FUS-R521C aggregates (Coady and Manley 2015), displayed limited overlap with FUS-R521C aggregates (Fig. 3C; Supplemental Fig. S5C), suggesting that NDUFA5 mRNA was preferentially sequestered. Controls with cells incubated solely with fluorophore-conjugated probes gave no signals (Supplemental Fig. S5D). Altogether, this experiment demonstrates that an RCC mRNA can be preferentially sequestered in mutant FUS aggregates.
Figure 3.

NDUFA5 mRNA colocalizes with mutant FUS aggregates. (A) U87 cells were transfected with GFP-tagged wild-type or R521C mutant FUS for 48 h. Immunofluorescence-FISH targeting GFP-FUS protein and NDUFA5 mRNA. Scale bar, 10 µm. (B) Colocalization analysis of NDUFA5 mRNA and R521C mutant FUS aggregates. Arrows indicate the aggregates for intensity profile analysis. (Green) GFP-FUS; (red) NDUFA5 FISH. Scale bar in insets, 2 µm (C) Colocalization analysis of IGFBP3 mRNA and R521C mutant FUS aggregates. Arrows indicate the aggregates for intensity profile analysis. (Green) GFP-FUS; (red) IGFBP3 FISH. Scale bar in insets, 2 µm.
Exogenous expression of mutant or wild-type FUS leads to reduced RCC protein expression
We next tested the possibility that FUS-mediated sequestration of the RCC mRNAs resulted in reductions in expression of the corresponding proteins. For this, we determined levels of these proteins in GFP-tagged wild-type FUS and ALS mutant R521C, P525L, and R495X FUS-expressing 293T cells. Western blots revealed that levels of all four NADH dehydrogenase subunits were indeed significantly reduced, not only in mutant but also WT FUS transfected cells (Fig. 4A,B). This is consistent with the data described above (Fig. 2C) showing that overexpressed wild-type as well as mutant FUS bound to mRNAs encoding these proteins, in both the soluble and insoluble fractions. In contrast, several proteins encoded by mRNAs not among the R521C-enriched transcripts did not show changes in expression (Fig. 4C,D; Supplemental Fig. S1C). The observed reduction in mitochondrial protein levels in both wild-type and mutant FUS transfected cells is thus consistent with our hypothesis that binding of mRNAs, either by high levels of WT FUS or of ALS mutant derivatives, sequesters them and suppresses their translation.
Figure 4.

Expression of ALS mutant FUS or overexpression of wild-type FUS represses protein levels of target RCC transcripts. (A) GFP-FUS and indicated mutant derivatives were transiently expressed in HEK293T cells for 48 h. Western blot (WB) of FUS targeted NADH dehydrogenase subunits. (B) WB quantification of FUS targeted NADH dehydrogenase subunit levels in HEK293T expressing GFP-tagged wild-type FUS and mutant derivatives. Protein levels are normalized to actin. Data are shown in mean with SE from three independent experiments. (*) P < 0.05; (**) P < 0.01; (***)P < 0.001, unpaired t-test. (C) WB of proteins encoded by transcripts that are not enriched in the insoluble mutant FUS IP. (D) WB analysis of relative protein levels in C. Data are displayed in mean with SE from three independent experiments. Protein levels are normalized to GAPDH. Statistics: unpaired t-test. (E) Total RCC mRNA levels were determined by RT-qPCR. Relative mRNA levels are normalized to GAPDH. Data are displayed in mean with SD from three experiments. Statistics: unpaired t-test. (F) WB of NADH dehydrogenase subunit expression levels in HEK293T transfected with siFUS for 72 h. (G) WB quantification of NADH dehydrogenase subunits in F. Protein levels are normalized to actin. Data are shown in mean with SD from three independent experiments. Statistics: unpaired t-test.
We assume from the above data and our previous study (Coady and Manley 2015) that the translational repression we observed was specific to sequestered mRNAs. However, to rule out the possibility that wild-type or mutant FUS expression suppressed protein synthesis more globally (Kamelgarn et al. 2018), we performed puromycin incorporation assays with wild-type and ALS mutant FUS transfected 293T cells, using WB with antipuromycin antibodies to measure levels of newly synthesized proteins (Supplemental Fig. S6A). As expected, this assay revealed no reductions in global protein synthesis in the FUS expressing cells (Supplemental Fig. S6B). To extend these results, we reanalyzed ribosome sequencing GEO data sets obtained from mouse ESC-derived neurons transduced with wild-type or ALS mutant FUS-R495X (Nakaya and Maragkakis 2018). In agreement with our analyses, wild-type and FUS-R495X transduction resulted in reduced translation of many of the top 5% FUS CLIP fraction enriched mRNAs, with 74 out of 242 (31%) in wild-type and 207 out of 375 (56%) in the ALS mutant displaying reduced translation (Supplemental Fig. S6C). The increased number of translationally repressed genes in FUS-R495X transduced neurons supports the idea that cytoplasmic FUS suppresses expression of the associated transcripts.
We also wished to address the possibility that the lower complex I protein levels were due to reduced mRNA levels. Therefore, we extracted total RNA from wild-type and mutant FUS transfected cells and performed RT-qPCR, and found no significant reduction in transcript levels (Fig. 4E). These data suggest that the FUS-sequestered mRNAs were generally stable, and that the lower protein levels likely resulted from reduced translation reflecting their sequestration, as we described previously for MeCP2 (Coady and Manley 2015).
FUS possesses a strong ability to interact with other proteins including itself to form RNP granules (Schwartz et al. 2013; Patel et al. 2015). FUS with ALS mutations or elevated levels of wild-type FUS can promote irreversible RNP granule formation and thus impair the protein's normal functions (Murakami et al. 2015; Patel et al. 2015). Moreover, cytoplasmic mutant FUS sequesters wild-type FUS in RNP granules and prevents its nuclear entry (Vance et al. 2013). We indeed observed strong interactions between endogenous FUS and exogenously expressed GFP-tagged wild-type and mutant FUS (Supplemental Fig. S7A). Mutant FUS ALS patient fibroblasts showed a reduced nuclear to cytoplasmic FUS protein ratio relative to control fibroblasts (Fig. 5A), and FUS nuclear clearance was especially notable in early onset mutants R495Efs527X and P525L (Supplemental Fig. S7B). In light of the above, we considered the possibility that loss of nuclear FUS induced or contributed to the RCC protein expression we observed. To test directly whether reduced FUS levels affect RCC protein expression, we depleted FUS using siRNAs and examined complex I protein expression. Western blot analysis shows that complex I subunit levels were unaltered in FUS KD cells (Fig. 4F,G).
Figure 5.

Expression of ALS mutant FUS or overexpression of wild-type FUS induces mitochondrial dysfunction. (A) The indicated human fibroblasts were incubated with 100 nM MitoTracker Red CMXRos for 30 min followed by immunofluorescence to detect FUS proteins. Cytoplasmic to nuclear FUS protein ratio quantitation is shown in mean with error bar SD. N = 174. (*) P < 0.05; (***) P < 0.001, Mann-Whitney U-test. Scale bar, 15 µm. (B) Zoom-in images of mitochondrial network morphology of FUS wild-type and mutant fibroblasts. (C) Binary skeletonized images of mitochondrial network shown in B. (D) Mitochondrial network branch lengths and footprint/mass quantification in wild-type and mutant fibroblasts. N = 170. (*) P < 0.05; (**) P < 0.01; (****) P < 0.0001, one-way ANOVA test. (E) O2 consumption rate of WT and ALS mutant FUS transfected 293T cells were normalized to GFP vector control in percentage. Data are displayed in mean with SE from four independent experiments. Statistics: unpaired t-test. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. (AMA) Antimycin A.
Expression of mutant or overexpression of wild-type FUS disrupts mitochondrial networks and inhibits aerobic respiration
The effects of FUS on nuclear-DNA encoded mitochondrial gene expression prompted us to investigate mitochondrial network morphology and respiratory functions in cells expressing GFP-tagged mutant or overexpressing wild-type FUS. Mitochondrial networks are highly dynamic structures regulated by fusion/fission between individual mitochondrion (Youle and van der Bliek 2012). To obtain information on mitochondrial network integrity, we used the fluorescent dye mitotracker to stain functional mitochondria and quantify the number of mitochondrial networks in wild-type and mutant FUS transfected cells (Supplemental Fig. S8A,B). Mitochondria were evenly distributed in GFP vector transfected cells. In contrast, mitochondria became irregularly arranged in wild-type overexpressing and mutant FUS expressing cells (Supplemental Fig. S8A). The number of branched mitochondrial networks showed >50% decrease in both wild-type and mutant FUS transfected cells compared with the GFP vector control (Supplemental Fig. S8B).
Since we detected mitochondrial network disruption in FUS transfected cells, we next investigated whether this alteration has ALS pathological relevance. To address this, we compared mitochondrial network integrity in the control and FUS-ALS fibroblasts described above. Strikingly, mitochondrial network morphology in the ALS fibroblasts was irregularly arranged and shortened compared with non-ALS fibroblasts (Fig. 5B,C), with the effects in the R524W fibroblasts somewhat less than in the other two (quantification in Fig. 5D). Furthermore, we observed a decrease in mitochondrial mass in all three ALS patient fibroblasts (Fig. 5D). These data agree with our hypothesis that mutant FUS-induced mitochondrial network disruption is linked to ALS-associated mitochondrial dysfunction, and are consistent with mitochondrial fragmentation observed in mutant FUS expressing cells in previous reports (Deng et al. 2015, 2018; Naumann et al. 2018).
The observed alterations in mitochondrial morphology suggest functional changes in cellular respiration. Since aerobic respiration is the most efficient way to produce ATP, we examined aerobic respiratory capacity in FUS transfected cells by using an extracellular oxygen consumption rate assay (OCR assay). Importantly, we observed a significantly lower OCR in both wild-type and ALS mutant FUS transfected cells compared with GFP vector controls, consistent with reduced RCC protein expression and disrupted mitochondrial network morphology (Fig. 5E). In contrast to FUS overexpression, FUS KD did not suppress aerobic respiration (Supplemental Fig. S8C).
Elevated mitochondrial ROS has been reported to trigger a series of mitochondrial stress responses, including mitochondrial fragmentation (Wu et al. 2011) and mitophagy (Xiao et al. 2017), and is one of the characteristics of FUS ALS/FTD-linked mitochondrial defects (Deng et al. 2015). Indeed, FACS analysis revealed that mitochondrial ROS levels showed 20%–70% increases in both wild-type overexpressing and ALS mutant FUS-expressing cells compared with the GFP vector control cells (Supplemental Fig. S8D). The moderate mitochondrial ROS elevation in wild-type overexpressing cells appears to reflect their modestly repressed RCC protein levels (Fig. 4B).
Together, the above data suggest that FUS granule formation leads to a dominant gain-of-function phenotype in which sequestration of RCC mRNAs leads to multiple mitochondrial defects we and others have characterized.
Depletion of RCC proteins recapitulates FUS-induced mitochondrial dysfunction
We next wanted to provide direct evidence that reductions in the levels of one or more of the RCC proteins can cause the same mitochondrial defects we characterized above. We used siRNA-mediated knockdown (KD) of the NADH dehydrogenase complex I components analyzed above and quantified changes in mitochondrial network integrity, aerobic respiration, and mitochondrial ROS production. NDUFA5 and NDUFB9 KDs showed significant reduction in unbranched mitochondria and branched networks, similar to what was observed in the FUS transfected cells (Fig. 6A,B). We also knocked down a major mitochondrial fusion regulatory protein, MFN2, as a positive control for impaired mitochondrial network formation (Bach et al. 2003; Chen et al. 2003; Züchner et al. 2004), and recapitulated the reduced number of unbranched mitochondria and mitochondrial networks (Fig. 6A,B). Next, we investigated whether loss of NADH dehydrogenase subunits represses aerobic respiration. We found that NDUFV2 and NDUFB9 KD cells indeed displayed lower OCR, whereas OCR in NDUFA5 KD cells was unaltered (Fig. 6C). It has been reported that inherited NDUFV2 and NDUFB9 mutations are tightly linked to mitochondrial complex I deficiency, while NDUFA5 partial ablation in mice showed reduced mitochondrial complex I activity (Bénit et al. 2003; Haack et al. 2012; Peralta et al. 2014). We also determined mitochondrial ROS levels in NDUFV2, NDUFB9, and NDUFA5 KD cells using MitoSOX. All three NADH dehydrogenase subunit KD cells showed elevated mitochondrial ROS production (Fig. 6D). Together, these experiments confirm the link between FUS-induced mitochondrial defects and reduced RCC protein expression.
Figure 6.

Depletion of RCC proteins disrupts mitochondrial networks and inhibits aerobic respiration. (A) NADH dehydrogenase subunit siRNA knockdowns in HEK293T cells for 72 h, followed by Western blot (WB) and MitoTracker Red CMXRos (100 nM) incubation for 30 min. (B) Mitochondrial network images of siRNA transfected cells. Networks are shown in binary skeletonized images. Dot plots show the quantification of unbranched mitochondria (number of individuals) and branched mitochondrial networks (number of networks) of each cell. MitoTracker quantification: >300 cells are analyzed. Scale bar, 10 µm. (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001, Mann-Whitney U-test. (C) O2 consumption rate of NADH dehydrogenase subunit knockdown cells. (AMA) Antimycin A. Data are displayed in mean with SD from four independent experiments. (*) P < 0.05; (**) P < 0.01, unpaired t-test. (D) Mitochondrial ROS level in NADH dehydrogenase subunit knockdown cells. MitoSOX signal intensities were analyzed in 307 cells. (***) P < 0.001; (****) P < 0.0001, Mann-Whitney U-test.
Mutant FUS shows increased mitochondrial association and binds to mRNAs encoded by the mitochondrial genome
A number of studies have reported that both wild-type and mutant ALS-linked proteins such as SOD1, TDP-43, and FUS are imported into or associate with mitochondria via diverse mechanisms and induce mitochondrial damage (Igoudjil et al. 2011; Deng et al. 2015; Wang et al. 2016). Since all protein-coding mitochondrial RNAs (mt mRNAs) produce respiratory chain components (Anderson et al. 1981; Chomyn et al. 1985; Hanna and Nelson 1999) and defective mt mRNA metabolism results in neuromuscular disorders (Chatfield et al. 2015; Barchiesi and Vascotto 2019), we next asked whether mutant FUS, especially given its association with mitochondria (Deng et al. 2015), might also interact with mt mRNAs. To address this, we first transfected GFP-tagged wild-type or FUS-R521C into 293T cells and performed mitochondrial fractionation. Largely consistent with previous studies (Deng et al. 2015, 2018), we detected ∼0.6% of exogenously expressed wild-type FUS and 1.8% of FUS-R521C in the mitochondrial fraction (note that these are minimums as the calculation does not account for possible losses during fractionation) (Fig. 7A). We next asked whether wild-type and mutant FUS bound to mt mRNAs using the cell fractionation/IP protocol illustrated in Figure 1D. Strikingly, mutant FUS-R521C bound the four mt mRNAs tested, ND1, CYTB, COX1, and ATP6, greater than fivefold more than did wild-type FUS in the urea-extracted fraction (Fig. 7B). In contrast, wild type and FUS-R521C bound only weakly to mt mRNAs in the soluble fraction (Fig. 7B). Notably, we also detected mt mRNAs with mutant FUS in our 3′READS data (Supplemental Fig. S9). However, the 3′READS method captures stable mRNAs with long poly(A) tails, which is not always the case with mt mRNAs. To provide additional evidence that mutant FUS binds to mt mRNAs, we reanalyzed endogenous wild-type and P525L mutant FUS CLIP-seq data sets performed in iPSC-differentiated motor neurons (De Santis et al. 2019). Indeed, both wild-type and mutant FUS CLIP reads mapped to the mitochondrial genome (Fig. 7C). Mutant FUS though displayed ∼1.3 times more clusters compared with wild type, indicating increased mutant FUS localization to mitochondria and binding to mt mRNAs.
Figure 7.
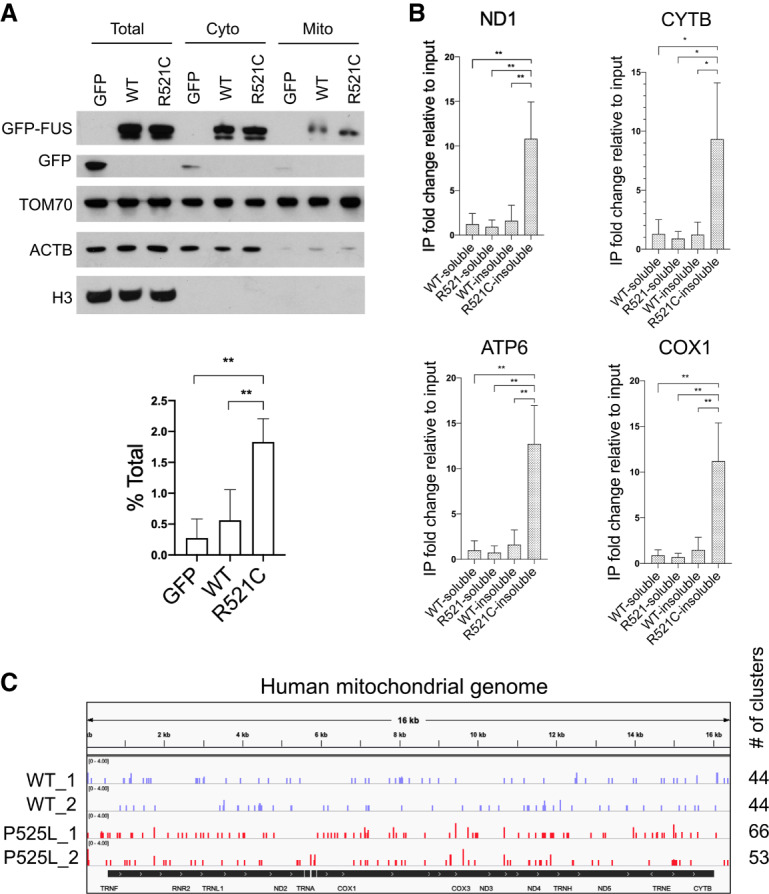
FUS associates with mitochondria and binds mitochondrial-encoded mRNAs. (A) HEK293T cells were transiently transfected with GFP-tagged wild-type and R521C mutant FUS for 48 h followed by mitochondrial fractionation. Western blot of mitochondrial fractionation. Purity of mitochondrial fractions was demonstrated by enrichment in mitochondrial protein TOM70 and absence of cytosolic protein beta actin and nuclear protein histone H3. Quantification of exogenously expressed GFP-tagged wild-type and FUS-R521C detected in mitochondrial fraction is displayed in percent of total protein. Data are shown in mean with SD from four independent experiments. (**) P < 0.01, unpaired t-test. (B) GFP-FUS IP followed by RT-qPCR analysis of RCC mRNAs encoded in mitochondrial genome from soluble fraction and urea-extracted insoluble fraction. Relative fold change is displayed in mean with SD from three independent experiments. (*) P < 0.05; (**) P < 0.01, unpaired t-test. (C) Realigned wild-type and P525L mutant FUS CLIP-seq data sets in iPSC-derived motor neurons (De Santis et al. 2019). Snapshot of wild-type and mutant FUS binding sites on mitochondrial-encoded transcripts. Number of read clusters aligned to mitochondrial genome was counted.
We next investigated whether the above findings extend to ALS patient fibroblasts. We performed immunofluorescence using FUS antibody and mitotracker to measure FUS mitochondrial localization in R495Efs527X mutant FUS patient fibroblasts. Since mutant FUS displayed diffuse cytoplasmic localization (Supplemental Fig. S4C), which likely overlaps with extensive mitochondrial networks in fibroblasts using conventional confocal microscopy, we performed airyscan confocal microscopy to visualize colocalization of mitochondrial and FUS signals with improved spatial resolution (Huff 2015). Indeed, mutant FUS and mitochondrial signals showed significant overlap (Supplemental Fig. S10B,C). As a negative control, we stained G3BP1, a stress granule marker that is diffuse in the cytoplasm under unstressed condition and does not colocalize with mutant FUS (Supplemental Fig. S10A). As expected, G3BP1 showed little overlap with mitochondria (Supplemental Fig. S10B,C).
FUS-associated RCC mRNAs also associate with mitochondria
The above results suggest a possible mechanism by which overexpressed wild-type and mutant FUS proteins associate with mitochondrial mRNAs. Specifically, nuclear-encoded mRNAs encoding respiratory chain components can localize to the mitochondrial surface to facilitate import of the translated proteins (Matsumoto et al. 2012; Lesnik et al. 2015). This localization would then bring these mRNAs into proximity with wild-type or mutant FUS that independently associates with mitochondria. We thus examined whether mRNAs encoding RCC proteins targeted by FUS-R521C were enriched in the mitochondrial fraction (Supplemental Fig. S11A), with GAPDH and ACTB cytoplasmic mRNAs serving as negative controls (Supplemental Fig. S11B), and ND1 mt mRNA serving as a positive control (Supplemental Fig. S11C). We extracted RNA from mitochondrial fractions of GFP-FUS transfected cells and performed RT-qPCR. Indeed, multiple FUS-R521C targeted RCC mRNAs, including NDUFV2, NDUFB9, NDUFA5, SOD1, and ATP5J, showed twofold to 20-fold elevation in the mitochondrial fraction compared with GAPDH and ACTB cytoplasmic mRNAs (Supplemental Fig. S11A,B), and this localization was independent of wild-type or mutant FUS overexpression. Therefore, as we discuss below, mitochondrial localization of nuclear transcribed RCC mRNAs may play an important role in facilitating their association with mutant FUS.
Discussion
FUS is a nucleocytoplasmic shuttling protein that functions in various physiological processes, largely through its RNA binding ability (Zinszner et al. 1997; Dormann and Haass 2011; Ederle et al. 2018). ALS/FTD-linked mutations in the FUS nuclear localization signal disturb its nuclear import and lead to cytoplasmic accumulation. A number of mechanisms have been proposed to address whether FUS nuclear loss of function or cytoplasmic gain of function underlies ALS/FTD pathology. Growing evidence in animal models, postmortem ALS/FTD patient tissues and experiments in human cell lines has argued in favor of gain of function as a primary source of toxicity while nuclear loss of function may exacerbate disease progression (Deng et al. 2015; Sun et al. 2015; Scekic-Zahirovic et al. 2016; Sharma et al. 2016; López-Erauskin et al. 2018; An et al. 2019). However, the mechanisms of FUS toxic gain of function remain unclear. Our previous study showed that interaction of mutant FUS proteins with specific mRNAs leads to aggregation and sequestration of the transcripts, preventing their translation (Coady and Manley 2015). Here we analyzed transcripts bound by mutant FUS globally, and found that mRNAs encoding mitochondrial RCC components are preferentially sequestered and translationally silenced, leading to mitochondrial dysfunction. Our findings were made in multiple systems, including FUS transfected human cells, ALS patient-derived mutant FUS fibroblasts, and online experimental data sets from several independent groups. These findings, which we discuss in detail below, greatly extend our previous study and demonstrate how abnormal RNA binding activity induces ALS/FTD-associated mitochondrial defects.
The majority of mitochondrial proteins are encoded in the nucleus (Eslamieh et al. 2017). These proteins are translated in the cytoplasm, imported into mitochondria, and sorted to designated compartments (Schmidt et al. 2010). The mitochondrial respiratory chain consists of five complexes located at the inner membrane, coupling with oxidative phosphorylation to produce ATP. Our data show that mRNAs encoding RCC components are overrepresented among the top-ranked mutant FUS-sequestered mRNAs. Many of these proteins are complex I subunits, which oxidizes NADH to NAD+ and transfers protons from the mitochondrial matrix to the intermembrane space to generate an electrochemical gradient (Hirst 2013). Complex I is the electron entry point of the respiratory chain and the rate-limiting site for ATP synthesis (Sharma et al. 2009). Deficiencies in complex I have been reported in various inherited neuromuscular disorders and Parkinson Disease due to mutations in 24 genes encoding mitochondrial complex I subunits (Fassone and Rahman 2012). More relevant to our work, several studies have linked complex I deficiency to ALS. For example, fibroblasts derived from ALS patients expressing mutant derivatives of the RBP TDP-43 show complex I disassembly and mitochondrial dysfunction, perhaps reflecting binding of TDP-43 to transcripts encoding two complex 1 subunits, ND3 and ND6 (Wang et al. 2016). Decreased complex I activity has also been observed in sporadic ALS patients and mutant SOD1 ALS rats (Wiedemann et al. 2002; Li et al. 2010; Ghiasi et al. 2012). Additionally, disruption of other respiratory chain complexes besides complex I have also been implicated in ALS and FTD. Overexpression of wild-type or mutant FUS in human cells has been shown to disrupt complex V assembly and inhibit ATP production, perhaps by direct binding to the complex V subunit ATP5B (Deng et al. 2018). Our results documenting reductions of complex I protein levels and aerobic respiration significantly extend these findings, most notably by providing a clear molecular mechanism.
Our data reveal that mRNAs encoding certain mitochondrial proteins are preferentially targeted by mutant FUS. This suggests that common features might exist in the top-ranked FUS-sequestered mRNAs. Although various techniques have been used to study FUS RNA-binding specificity, any consensus RNA features remain elusive. In fact, wild-type and mutant FUS were shown to bind RNA in a largely sequence-independent manner (Hoell et al. 2011; Wang et al. 2015b), while our analysis of the top-ranked mutant FUS-sequestered RNAs did not find increased density of any published FUS binding sequence motifs. Secondary structures were proposed to facilitate FUS RNA-binding specificity, although discrepant results regarding common secondary or sequence features require further investigation (Hoell et al. 2011; Loughlin et al. 2019). Additionally, our poly(A+) RNA-specific 3′READS data together with CLIP-seq exon analysis conducted by others (Nakaya and Maragkakis 2018) showed that RNA abundance in the FUS-bound fraction positively correlated with expression levels, suggesting that mutant FUS indeed binds most mature mRNAs in a largely sequence-independent manner.
One possible mechanism underlying FUS binding selectivity is that mRNA 3′UTRs contain signals for mRNA mitochondrial localization, which would then allow association of these transcripts with mitochondrial localized FUS (see below). In yeast, the pumilio protein Puf3 binds to an element located in the 3′UTRs of >100 mitochondrial mRNAs (Zhu et al. 2009) and promotes mRNA mitochondrial localization (Lesnik et al. 2015). Whether this occurs in humans is unknown, but PUF proteins are highly conserved across eukaryotes (Wang et al. 2018) and 3′UTRs are required for mRNA localization to mitochondria in both yeast and humans (Sylvestre et al. 2003). In any event, the colocalization of FUS and mitochondrial mRNAs at mitochondria might then enhance their interaction simply due to increased local concentration.
We demonstrated that mutant FUS shows increased mitochondrial association in cultured cells and in human ALS fibroblasts. This may facilitate its binding to mRNAs transcribed from the mitochondrial genome and to nuclear-encoded mitochondrial mRNAs associating with mitochondria, as suggested above. A mitochondrial targeting signal search using the published algorithm MitoFate (Fukasawa et al. 2015) did not reveal N-terminal cleavable or internal mitochondrial import signals in FUS. Nevertheless, previous studies using immunoEM and mitochondrial fractionation have shown that both wild-type and mutant FUS are recruited to mitochondria (Deng et al. 2015, 2018). Recruitment appears to be mediated by the chaperone HSP60, which assists proper folding and assembly of mitochondrial proteins destined to mitochondria (Cheng et al. 1989; Ostermann et al. 1989). It was suggested that FUS cytoplasmic localization due to pathogenic mutations or stress results in increased FUS–HSP60 interaction and FUS mitochondrial localization (Deng et al. 2015). Mutant FUS also shows increased interaction with the mitochondrial-associated chaperones HSP70 and HSP90 (Wang et al. 2015a), which deliver mitochondrial preproteins to the mitochondrial surface and facilitate their import (Faou and Hoogenraad 2012). In addition, wild-type and mutant TDP-43 have also been shown to localize to mitochondria in mouse models, transfected human cells and patient fibroblasts (Wang et al. 2013a, 2016; Davis et al. 2018). TDP-43 mitochondrial localization facilitates binding of the protein to RCC mRNAs, encoding ND3 and ND6 as noted above, thereby suppressing their expression. Notably, TDP-43 mitochondrial import is achieved by an import signal, and blocking it using synthetic peptides rescued mitochondrial defects (Wang et al. 2016). Therefore, although FUS and TDP-43 appear to localize to mitochondria via distinct mechanisms, the pathways converge at increased binding to mRNAs encoded in the mitochondrial genome.
Our data shows that overexpressed wild-type FUS, unlike mutant FUS, associates with mRNAs encoding mitochondrial proteins mostly in the soluble fraction. In contrast to mutant FUS-expressing cells, wild-type FUS-overexpressing cells had fewer large, visible aggregates. Nonetheless, wild-type FUS-overexpressing cells also showed moderate decreases in mitochondrial protein expression as well as mitochondrial defects. We suspect that the functional consequences of wild-type FUS-RNA interaction occurred without formation of large, visible aggregates. In addition, the ribosome profiling data sets of Nakaya and Maragkakis (2018) revealed reduced translation of 88 mitochondrial genes in wild-type FUS-overexpressing neurons. These results together indicate that translation of RCC subunit-encoding mRNAs is sensitive to cytoplasmic FUS levels. Thus, the effects of cytoplasmic FUS on translation may not require large aggregate formation. Consistent with this idea, our previous work analyzing ALS and FTD postmortem brains demonstrated that the wide-spread RNA splicing defects induced by RBP loss of function due to insolubility of multiple RBPs did not necessarily correlate with the near ubiquitous TDP-43 histopathology (Conlon et al. 2018). This agrees with our data showing that visible aggregates are rare in wild-type FUS overexpressing cells, but we nonetheless observed a significant portion of wild-type FUS in the urea-extracted insoluble fraction. It is thus possible that small cytosolic wild-type FUS granules interfere with the translation of RCC proteins. Also, wild-type or mutant FUS-induced mitochondrial stress activates mitochondrial quality control pathways (Deng et al. 2018), which could lead to translational repression and increased degradation of nuclear-encoded mitochondrial proteins (Fiorese and Haynes 2017). These findings together support the idea that increased insoluble RBP levels are sufficient to induce functional consequences, such as reduced translation or splicing defects, that are relevant to disease.
Oxidative stress and mitochondrial damage are risk factors associated with aging and are prevalent in various neurodegenerative diseases (Lin and Beal 2006). In response to oxidative stress, cells form phase-separated granules containing translationally repressed RNPs while up-regulating stress responsive genes (Spriggs et al. 2010; Molliex et al. 2015; Riback et al. 2017). Under normal conditions, the entire process is reversible when stress is relieved, and granules dissipate. In contrast, stress granule dynamics becomes aberrant and irreversible in ALS/FTD patient-derived cells (Ling et al. 2013; Deng et al. 2014). Comparison between normal and ALS iPS-derived motor neurons revealed a higher propensity for granule formation as well as altered protein composition and subcellular localization of stress granules in the diseased cells (Markmiller et al. 2018). Interestingly, it has been shown that mRNAs encoding mitochondrial proteins, especially the complex I respiratory chain components identified here, are depleted in oxidative stress-induced granules (Khong et al. 2017), suggesting that these transcripts are less likely to be sequestered in stress granules under stress conditions. Our data show that mutant FUS binds these transcripts with substantially higher enrichment than does wild-type FUS in the soluble fraction of ALS patient fibroblasts. Given ALS mutations in FUS result in static binding to RNA targets (Niaki et al. 2020), it is possible that under stress conditions, these mutant FUS-bound mitochondrial mRNAs would be sequestered by mutant FUS protein aggregation (Shelkovnikova et al. 2014; Patel et al. 2015).
Mitochondrial dysfunction and RNA metabolism defects are two factors suggested to play significant roles in ALS/FTD pathogenesis (Liu et al. 2017; Tank et al. 2018). Our previous studies have provided evidence that ALS pathological aggregates profoundly affect RNA metabolism including splicing, translation and degradation (Coady and Manley 2015; Conlon et al. 2016, 2018). Here, we revealed that mRNAs important for mitochondrial respiratory chain functions are sequestered with mutant FUS in insoluble aggregates, which impairs their translation, leading to reduced levels of the encoded proteins and to multiple defects in mitochondrial function. Our results highlight a new role for mutant FUS that connects RNA metabolism defects to mitochondrial dysfunction, and thus provides novel insights into the mechanisms underlying FUS toxic gain of function in ALS/FTD.
Materials and methods
Cell culture
Human skin fibroblasts, U87-MG, and HEK293T cells were cultured in DMEM (Gibco 11965092) supplemented with 10% FBS (Gemini 100-106) in a 37°C, 5% CO2 incubator.
Cloning of wild-type FUS and mutant derivatives
Flag-tagged FUS-R521C plasmid described previously (Coady and Manley 2015) was used in this study. The GFP-tagged wild-type and mutant FUS plasmids were constructed with pEGFP-C3 vector. ALS mutations were created using site-directed mutagenesis. All primers used are listed in Table 1.
Table 1.
Primer sequences

Plasmids and siRNAs transfection
Lipofectamine 2000 (Thermo 11668019) was used for plasmid transfections: Lipofectamine ratio of 1:3 in serum- and antibiotic-free OptiMEM medium (Gibco 31985070). The siRNAs were transfected with Dharmafect 1 (GE Dharmacon) at 20 nM for 48–72 h. Cells were harvested for subsequent assays. All siRNA sequences are listed in Table 2.
Table 2.
siRNA sequences

Cell fractionation and RNA immunoprecipitation
The cell fractionation and RNA-IP procedures were as described previously (Coady and Manley 2015). All lysis buffers were supplemented with 1x protease inhibitor cocktail (Roche 04693159001) and 100 U/mL RNase inhibitor (Promega N2518). Cells were lysed with ice-cold 1% NP-40 hypotonic buffer (10 mM Tris-HCl, 10 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40), added NaCl to final concentration of 150 mM, then centrifuged 16,000g for 10 min. The supernatant was designated as detergent soluble fraction. The pellet was then extracted with 100 mM urea buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 100 mM urea) for 1 h at 37°C in Autoblot rotator incubator (Thomas Scientific). The extracted urea supernatant was designated as the insoluble fraction. Antibody (2–4 µg) was incubated and rotated with soluble and insoluble fractions overnight at 4°C, and complexes were precipitated with Pierce Protein A/G magnetic beads (Thermo 88802). Proteins were eluted with 2% SDS sample buffer for subsequent Western blot analysis. RNAs were stored in TRIzol (Thermo 15596018) and extracted following the manufacturer's instructions. All antibodies used are listed in Table 3.
Table 3.
Antibodies

RT-qPCR
RNA samples were pretreated with DNase I to remove potential DNA contamination. One-hundred nanograms of RNA was used for reverse transcription with 0.3 µL Maxima RT enzyme (Thermo EP0742), 1 µM of oligo-dT, and 1 µM of random hexamers per reaction. Transcript levels were measured using SYBR Green qPCR (Applied Biosystems 4367659). Relative RNA levels in whole-cell lysates were determined by normalizing expression levels of the target genes to expression levels of GAPDH. Relative enrichment of RNAs in IP samples was determined by normalizing amount of target RNA in IP samples to amount in the input samples. Mitochondrial fractionation samples were also analyzed by normalizing target gene levels in the mitochondrial fraction to input fractions. All primer sequences used are listed in Table 1.
3′READS and gene ontology analysis
Total and FUS-R521C immunoprecipitated RNAs were purified as described above. RNA integrity was assessed using Agilent Bioanalyzer. The 3′READS procedure and reads alignment were done as described in (Hoque et al. 2013). In summary, the RNAs were fragmented by sonication and were captured using CU5T45 chimeric magnetic beads. The captured RNAs were then eluted from the beads and poly(A) tails removal by RNase H treatment. Eluted RNAs were then subjected to 5′ and 3′ adaptor ligation, RT-PCR, and sequencing (Illumina TruSeq LT library preparation kit). To obtain total poly(A+) RNA level expressed by a gene, all reads mapped to polyadenylation sites of a gene were counted and summed. Read counts derived from whole-cell (WC) and FUS-R521C immunoprecipitation (IP) were averaged as reads per million (rpm). All genes ≤1 rpm were discarded. Enrichment of each poly(A+) RNA was then determined by the fold change of read counts in the IP relative to read counts in the WC. Transcripts with top 5% greatest fold change above the standard curve are R521C-enriched. Transcripts with fold changes below the standard curve are nonenriched. The remainders are neither enriched nor nonenriched. The shared top 5% enriched targets from both replicates were subsequently used for gene ontology analysis. Gene ontology enrichment analysis was performed in Panther gene list analysis database (http://www.pantherdb.org) and DAVID bioinformatics resources (https://david.ncifcrf.gov).
CLIP-seq analysis
FUS CLIP-seq data sets of human iPSC-differentiated motor neurons (De Santis et al. 2019) were remapped to human mitochondrial genome NC012920 and hg38 reference genome using PARA suite PAR-CLIP analysis tool (Kloetgen et al. 2016).
Puromycin incorporation assay
The assay was performed as described (Ogami et al. 2017). Plasmid transfected HEK293T cells were treated with 1 µg/mL puromycin for 30 min to label nascent peptides. Control cells were incubated with 10 µg/mL cycloheximide for 10 min before puromycin treatment. The puromycin-labeled proteins were then resolved by SDS-PAGE and detected by Western blotting using antipuromycin antibody (Kerafast EQ 0001).
Immunofluorescence and FISH
Cells were fixed with 2% formaldehyde for 10 min at room temperature. After fixation, cells were permeabilized with 3% Triton X-100 for 20 min at 4°C. Blocking was done with 3% BSA dissolved in PBS for 30 min at room temperature before incubating primary antibodies overnight at 4°C. All primary antibodies were diluted 1:200 in 3% BSA. Cells were washed with 0.2% PBST at least three times to remove nonspecific background. For samples not used for FISH, secondary antibodies were directly applied for 1 h at room temperature, washed with 0.2% PBST and mounted onto frosted microscope slides (Fisher Scientific12-544-3; Fluoroshield mounting medium with DAPI [ab104139]). For poly(A) FISH, cells were preincubated with 10% formamide for 10 min at room temperature. Biotinylated oligo(dT)30 was diluted in 10% dextran sulfate FISH hybridization solution to final concentration of 0.1 µM oligo(dT)30. The cells were then incubated with oligo(dT)30 solution for 1 h at 37°C. Cells after FISH hybridization were washed twice with 10% formamide and incubated with fluorophore-conjugated streptavidin and secondary antibodies. Subsequent washing and mounting procedures were as above. Transcript-specific FISH procedure was adapted from smiFISH (Tsanov et al. 2016). We used unlabeled target-specific primary probes for first hybridization (100 ng/µL) for 3 h at 37°C. Each primary probe contains a 20-nt overhang at the 3’ end that does not hybridize with the targets. After two washes with 10% formamide containing buffer, we incubated the samples with fluorophore-conjugated secondary probes (10 ng/µL), which are the reverse complements of the 3’ overhangs at 37°C. After 2 h, samples were washed with 10% formamide containing buffers followed by 2xSSC and mounted for confocal imaging. FISH probe sequences are listed in Table 4.
Table 4.
FISH probes

Mitochondrial network and ROS staining
MitoTracker Red CMXRos (Thermo M7512) was diluted 1:10000 in prewarmed DMEM with 10% FBS cell culture medium and incubated with live cells in cell culture incubator for 30 min. Cells were then washed with PBS and fixed with 2% formaldehyde. The subsequent procedures are the same as immunofluorescence described above. To stain mitochondrial ROS, live cells were incubated with 5 µM MitoSOX Red (Thermo M36008) for 10 min at 37°C and washed with PBS. The stained samples were either subjected to flow cytometry for ROS quantification or fixed and mounted on microscope slides for confocal imaging.
Extracellular oxygen consumption rate assay
The OCR assay was performed by using extracellular oxygen consumption assay kit (Abcam ab197243), which contains fluorescent reagent that can be quenched by oxygen at the excited state. As the cells respire, intensity of the fluorescence would be inversely correlated with the oxygen concentration in the environment, thus positively correlated with the OCR of the cells. Approximately 7 × 104 cells were seeded into each well of a 96-well plate for 24 h. The extracellular oxygen consumption reagent was mixed with cell culture media and sealed with high-sensitivity mineral oil to prevent diffusion of ambient oxygen. The mineral oil-sealed 96-well plate was then subjected to fluorescence measurement using microplate reader (BioTek Synergy Neo2). The OCR was calculated based on linear portion of the changes in signal profile of a given time interval.
Mitochondrial fractionation
Cells were swelled on ice with hypotonic buffer (10 mM NaCl, 1.5 mM MgCl2, 10 mM Tris-HCl at pH 7.5) for 30 min and break the cells with Potter-Elvehjem homogenizer for 10–20 strokes. The lysates were supplemented with mannitol and sucrose (2.5× MS solution: 525 mM mannitol, 175 mM sucrose, 12.5 mM Tris-HCl at pH 7.5, 2.5 mM EDTA at pH 7.5) to reach final concentration of 210 mM and 70 mM respectively. Lysates were then centrifuged 1300g for 5 min to remove nuclei, cell debris, and unbroken cells. This step was performed three times to ensure that nuclei and other organelle contaminants were effectively removed. Final centrifugation was 17,000g for 15 min to precipitate mitochondria. The mitochondrial pellet was washed with mannitol-sucrose solution and dissolve in 2% SDS protein sample buffer or TRIzol for RNA analysis.
Confocal microscopy and imaging analysis
Immunofluorescence images were taken using Zeiss LSM800 confocal microscope with constant gain and laser excitation across experimental replicates. The images were analyzed with FIJI ImageJ software. Cytoplasmic aggregates were quantified based on number of local maximal pixel intensity of GFP channel per cell. Mitochondrial network parameters were obtained and analyzed using MiNA ImageJ macro tool (Valente et al. 2017). In brief, mitochondrial network signals are transformed into binary, skeletonized images. Unbranched mitochondria (individuals) are defined as rods, and branched networks (networks) are defined as rods joined by at least one junction node. The methods for quantifying individuals and networks are the number of objects in the image that fit the above definitions. Mitochondrial lengths were calculated as mean number of pixels consisted in each branch of a continuous network or an unbranched mitochondrion per cell. Mitochondrial footprint was defined as proportion of cell area occupied by mitochondrial signals.
Data and code availability
RNA sequencing data reported here were submitted to Gene Expression Omnibus (GEO) database under accession number GSE148550.
Supplementary Material
Acknowledgments
We thank Dr. Barbara Corneo of the Columbia Stem Cell Core Facility for providing ALS patient fibroblasts, and Dr. Robert H. Singer and Jeetayu Biswas of the Albert Einstein College of Medicine for invaluable advice on FISH experiments. This work is supported by National Institutes of Health (NIH) grant R35 GM118136 to J.L.M., Y.L.T. is supported by a Government Scholarship to Study Abroad, Ministry of Education, Taiwan R.O.C. B.T. and D.Z. are supported by NIH grants GM084089 and GM129069.
Author contributions: Y.L.T. designed the research, performed the experiments and data analysis with assistance from T.H.C., L.L., and I.A. Y.L.T. and J.L.M. wrote and edited the manuscript. D.Z. and B.T. provided 3′READS pipeline and edited the manuscript. N.A.S. provided neurology expertise and contributed to the manuscript preparation.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.335836.119.
References
- An H, Skelt L, Notaro A, Highley JR, Fox AH, La Bella V, Buchman VL, Shelkovnikova TA. 2019. ALS-linked FUS mutations confer loss and gain of function in the nucleus by promoting excessive formation of dysfunctional paraspeckles. Acta Neuropathol Commun 7: 7 10.1186/s40478-019-0658-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, et al. 1981. Sequence and organization of the human mitochondrial genome. Nature 290: 457–465. 10.1038/290457a0 [DOI] [PubMed] [Google Scholar]
- Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, Daugaard JR, Lloberas J, Camps M, Zierath JR, et al. 2003. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem 278: 17190–17197. 10.1074/jbc.M212754200 [DOI] [PubMed] [Google Scholar]
- Bannwarth S, Ait-El-Mkadem S, Chaussenot A, Genin EC, Lacas-Gervais S, Fragaki K, Berg-Alonso L, Kageyama Y, Serre V, Moore DG, et al. 2014. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 137: 2329–2345. 10.1093/brain/awu138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barchiesi A, Vascotto C. 2019. Transcription, processing, and decay of mitochondrial RNA in health and disease. Int J Mol Sci 20: 2221 10.3390/ijms20092221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bénit P, Beugnot R, Chretien D, Giurgea I, De Lonlay-Debeney P, Issartel JP, Corral-Debrinski M, Kerscher S, Rustin P, Rötig A, et al. 2003. Mutant NDUFV2 subunit of mitochondrial complex I causes early onset hypertrophic cardiomyopathy and encephalopathy. Hum Mutat 21: 582–586. 10.1002/humu.10225 [DOI] [PubMed] [Google Scholar]
- Chatfield KC, Coughlin CR, Friederich MW, Gallagher RC, Hesselberth JR, Lovell MA, Ofman R, Swanson MA, Thomas JA, Wanders RJ, et al. 2015. Mitochondrial energy failure in HSD10 disease is due to defective mtDNA transcript processing. Mitochondrion 21: 1–10. 10.1016/j.mito.2014.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HC, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. 2003. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160: 189–200. 10.1083/jcb.200211046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng MY, Hartl FU, Martin J, Pollock RA, Kalousek F, Neuper W, Hallberg EM, Hallberg RL, Horwich AL. 1989. Mitochondrial heat-shock protein hsp60 is essential for assembly of proteins imported into yeast mitochondria. Nature 337: 620–625. 10.1038/337620a0 [DOI] [PubMed] [Google Scholar]
- Chomyn A, Mariottini P, Cleeter MW, Ragan CI, Matsuno-Yagi A, Hatefi Y, Doolittle RF, Attardi G. 1985. Six unidentified reading frames of human mitochondrial DNA encode components of the respiratory-chain NADH dehydrogenase. Nature 314: 592–597. 10.1038/314592a0 [DOI] [PubMed] [Google Scholar]
- Coady TH, Manley JL. 2015. ALS mutations in TLS/FUS disrupt target gene expression. Genes Dev 29: 1696–1706. 10.1101/gad.267286.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon EG, Lu L, Sharma A, Yamazaki T, Tang T, Shneider NA, Manley JL. 2016. The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. Elife 5: e17820 10.7554/eLife.17820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon EG, Fagegaltier D, Agius P, Davis-Porada J, Gregory J, Hubbard I, Kang K, Kim D, The New York Genome Center ALS Consortium, Phatnani H, et al. 2018. Unexpected similarities between C9ORF72 and sporadic forms of ALS/FTD suggest a common disease mechanism. Elife 7: e37754 10.7554/eLife.37754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis SA, Itaman S, Khalid-Janney CM, Sherard JA, Dowell JA, Cairns NJ, Gitcho MA. 2018. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci Lett 678: 8–15. 10.1016/j.neulet.2018.04.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Gao K, Jankovic J. 2014. The role of FUS gene variants in neurodegenerative diseases. Nat Rev Neurol 10: 337–348. 10.1038/nrneurol.2014.78 [DOI] [PubMed] [Google Scholar]
- Deng J, Yang M, Chen Y, Chen X, Liu J, Sun S, Cheng H, Li Y, Bigio EH, Mesulam M, et al. 2015. FUS interacts with HSP60 to promote mitochondrial damage. PLoS Genet 11: e1005357 10.1371/journal.pgen.1005357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Wang P, Chen X, Cheng H, Liu J, Fushimi K, Zhu L, Wu JY. 2018. FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proc Natl Acad Sci 115: E9678–E9686. 10.1073/pnas.1806655115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santis R, Alfano V, de Turris V, Colantoni A, Santini L, Garone MG, Antonacci G, Peruzzi G, Sudria-Lopez E, Wyler E, et al. 2019. Mutant FUS and ELAVL4 (HuD) aberrant crosstalk in amyotrophic lateral sclerosis. Cell Rep 27: 3818–3831.e5. 10.1016/j.celrep.2019.05.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D, Haass C. 2011. TDP-43 and FUS: a nuclear affair. Trends Neurosci 34: 339–348. 10.1016/j.tins.2011.05.002 [DOI] [PubMed] [Google Scholar]
- Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, et al. 2010. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J 29: 2841–2857. 10.1038/emboj.2010.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ederle H, Funk C, Abou-Ajram C, Hutten S, Funk EBE, Kehlenbach RH, Bailer SM, Dormann D. 2018. Nuclear egress of TDP-43 and FUS occurs independently of Exportin-1/CRM1. Sci Rep 8: 7084 10.1038/s41598-018-25007-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eslamieh M, Williford A, Betrán E. 2017. Few nuclear-encoded mitochondrial gene duplicates contribute to male germline-specific functions in humans. Genome Biol Evol 9: 2782–2790. 10.1093/gbe/evx176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faou P, Hoogenraad NJ. 2012. Tom34: a cytosolic cochaperone of the Hsp90/Hsp70 protein complex involved in mitochondrial protein import. Biochim Biophys Acta 1823: 348–357. 10.1016/j.bbamcr.2011.12.001 [DOI] [PubMed] [Google Scholar]
- Fassone E, Rahman S. 2012. Complex I deficiency: clinical features, biochemistry and molecular genetics. J Med Genet 49: 578–590. 10.1136/jmedgenet-2012-101159 [DOI] [PubMed] [Google Scholar]
- Fiorese CJ, Haynes CM. 2017. Integrating the UPRmt into the mitochondrial maintenance network. Crit Rev Biochem Mol Biol 52: 304–313. 10.1080/10409238.2017.1291577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukasawa Y, Tsuji J, Fu SC, Tomii K, Horton P, Imai K. 2015. MitoFates: improved prediction of mitochondrial targeting sequences and their cleavage sites. Mol Cell Proteomics 14: 1113–1126. 10.1074/mcp.M114.043083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiasi P, Hosseinkhani S, Noori A, Nafissi S, Khajeh K. 2012. Mitochondrial complex I deficiency and ATP/ADP ratio in lymphocytes of amyotrophic lateral sclerosis patients. Neurol Res 34: 297–303. 10.1179/1743132812Y.0000000012 [DOI] [PubMed] [Google Scholar]
- Groen EJ, Fumoto K, Blokhuis AM, Engelen-Lee J, Zhou Y, van den Heuvel DM, Koppers M, van Diggelen F, van Heest J, Demmers JA, et al. 2013. ALS-associated mutations in FUS disrupt the axonal distribution and function of SMN. Hum Mol Genet 22: 3690–3704. 10.1093/hmg/ddt222 [DOI] [PubMed] [Google Scholar]
- Haack TB, Madignier F, Herzer M, Lamantea E, Danhauser K, Invernizzi F, Koch J, Freitag M, Drost R, Hillier I, et al. 2012. Mutation screening of 75 candidate genes in 152 complex I deficiency cases identifies pathogenic variants in 16 genes including NDUFB9. J Med Genet 49: 83–89. 10.1136/jmedgenet-2011-100577 [DOI] [PubMed] [Google Scholar]
- Hanna MG, Nelson IP. 1999. Genetics and molecular pathogenesis of mitochondrial respiratory chain diseases. Cell Mol Life Sci 55: 691–706. 10.1007/s000180050327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig S, Kong G, Mannen T, Sadowska A, Kobelke S, Blythe A, Knott GJ, Iyer KS, Ho D, Newcombe EA, et al. 2015. Prion-like domains in RNA binding proteins are essential for building subnuclear paraspeckles. J Cell Biol 210: 529–539. 10.1083/jcb.201504117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J. 2013. Mitochondrial complex I. Annu Rev Biochem 82: 551–575. 10.1146/annurev-biochem-070511-103700 [DOI] [PubMed] [Google Scholar]
- Hoell JI, Larsson E, Runge S, Nusbaum JD, Duggimpudi S, Farazi TA, Hafner M, Borkhardt A, Sander C, Tuschl T. 2011. RNA targets of wild-type and mutant FET family proteins. Nat Struct Mol Biol 18: 1428–1431. 10.1038/nsmb.2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann JW, Seeley WW, Huang EJ. 2018. RNA binding proteins and the pathogenesis of frontotemporal lobar degeneration. Annu Rev Pathol 14: 469–495. 10.1146/annurev-pathmechdis-012418-012955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque M, Ji Z, Zheng D, Luo W, Li W, You B, Park JY, Yehia G, Tian B. 2013. Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat Methods 10: 133–139. 10.1038/nmeth.2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff J. 2015. The Airyscan detector from ZEISS: confocal imaging with improved signal-to-noise ratio and super-resolution. Nat Methods 12: i–ii. 10.1038/nmeth.f.388 [DOI] [Google Scholar]
- Igoudjil A, Magrane J, Fischer LR, Kim HJ, Hervias I, Dumont M, Cortez C, Glass JD, Starkov AA, Manfredi G. 2011. In vivo pathogenic role of mutant SOD1 localized in the mitochondrial intermembrane space. J Neurosci 31: 15826–15837. 10.1523/JNEUROSCI.1965-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamelgarn M, Chen J, Kuang L, Jin H, Kasarskis EJ, Zhu H. 2018. ALS mutations of FUS suppress protein translation and disrupt the regulation of nonsense-mediated decay. Proc Natl Acad Sci 115: E11904–E11913. 10.1073/pnas.1810413115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khong A, Matheny T, Jain S, Mitchell SF, Wheeler JR, Parker R. 2017. The stress granule transcriptome reveals principles of mRNA accumulation in stress granules. Mol Cell 68: 808–820.e5. 10.1016/j.molcel.2017.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloetgen A, Borkhardt A, Hoell JI, McHardy AC. 2016. The PARA-suite: PAR-CLIP specific sequence read simulation and processing. PeerJ 4: e2619 10.7717/peerj.2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, et al. 2009. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323: 1205–1208. 10.1126/science.1166066 [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling SC, Liang TY, Mazur C, et al. 2012. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci 15: 1488–1497. 10.1038/nn.3230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughlin FE, Lukavsky PJ, Kazeeva T, Reber S, Hock EM, Colombo M, Von Schroetter C, Pauli P, Clery A, Muhlemann O, et al. 2019. The solution structure of FUS bound to RNA reveals a bipartite mode of RNA recognition with both sequence and shape specificity. Mol Cell 73: 490–504.e6. 10.1016/j.molcel.2018.11.012 [DOI] [PubMed] [Google Scholar]
- Lee YB, Chen HJ, Peres JN, Gomez-Deza J, Attig J, Štalekar M, Troakes C, Nishimura AL, Scotter EL, Vance C, et al. 2013. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep 5: 1178–1186. 10.1016/j.celrep.2013.10.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerga A, Hallier M, Delva L, Orvain C, Gallais I, Marie J, Moreau-Gachelin F. 2001. Identification of an RNA binding specificity for the potential splicing factor TLS. J Biol Chem 276: 6807–6816. 10.1074/jbc.M008304200 [DOI] [PubMed] [Google Scholar]
- Lesnik C, Golani-Armon A, Arava Y. 2015. Localized translation near the mitochondrial outer membrane: an update. RNA Biol 12: 801–809. 10.1080/15476286.2015.1058686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Vande Velde C, Israelson A, Xie J, Bailey AO, Dong MQ, Chun SJ, Roy T, Winer L, Yates JR, et al. 2010. ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc Natl Acad Sci 107: 21146–21151. 10.1073/pnas.1014862107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Beal MF. 2006. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443: 787–795. 10.1038/nature05292 [DOI] [PubMed] [Google Scholar]
- Ling SC, Polymenidou M, Cleveland DW. 2013. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79: 416–438. 10.1016/j.neuron.2013.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YJ, Tsai PY, Chern Y. 2017. Energy homeostasis and abnormal RNA metabolism in amyotrophic lateral sclerosis. Front Cell Neurosci 11: 126 10.3389/fncel.2017.00126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Erauskin J, Tadokoro T, Baughn MW, Myers B, McAlonis-Downes M, Chillon-Marinas C, Asiaban JN, Artates J, Bui AT, Vetto AP, et al. 2018. ALS/FTD-linked mutation in FUS suppresses intra-axonal protein synthesis and drives disease without nuclear loss-of-function of FUS. Neuron 100: 816–830.e7. 10.1016/j.neuron.2018.09.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markmiller S, Soltanieh S, Server KL, Mak R, Jin W, Fang MY, Luo EC, Krach F, Yang D, Sen A, et al. 2018. Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell 172: 590–604.e13. 10.1016/j.cell.2017.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda A, Takeda J, Okuno T, Okamoto T, Ohkawara B, Ito M, Ishigaki S, Sobue G, Ohno K. 2015. Position-specific binding of FUS to nascent RNA regulates mRNA length. Genes Dev 29: 1045–1057. 10.1101/gad.255737.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto S, Uchiumi T, Saito T, Yagi M, Takazaki S, Kanki T, Kang D. 2012. Localization of mRNAs encoding human mitochondrial oxidative phosphorylation proteins. Mitochondrion 12: 391–398. 10.1016/j.mito.2012.02.004 [DOI] [PubMed] [Google Scholar]
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP. 2015. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163: 123–133. 10.1016/j.cell.2015.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Qamar S, Lin JQ, Schierle GSK, Rees E, Miyashita A, Costa AR, Dodd RB, Chan FTS, Michel CH, et al. 2015. ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 88: 678–690. 10.1016/j.neuron.2015.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaya T, Maragkakis M. 2018. Amyotrophic Lateral Sclerosis associated FUS mutation shortens mitochondria and induces neurotoxicity. Sci Rep 8: 15575 10.1038/s41598-018-33964-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumann M, Pal A, Goswami A, Lojewski X, Japtok J, Vehlow A, Naujock M, Günther R, Jin M, Stanslowsky N, et al. 2018. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat Commun 9: 335 10.1038/s41467-017-02299-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niaki AG, Sarkar J, Cai X, Rhine K, Vidaurre V, Guy B, Hurst M, Lee JC, Koh HR, Guo L, et al. 2020. Loss of dynamic RNA interaction and aberrant phase separation induced by two distinct types of ALS/FTD-linked FUS mutations. Mol Cell 77: 82–94.e4. 10.1016/j.molcel.2019.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogami K, Richard P, Chen Y, Hoque M, Li W, Moresco JJ, Yates JR 3rd, Tian B, Manley JL. 2017. An Mtr4/ZFC3H1 complex facilitates turnover of unstable nuclear RNAs to prevent their cytoplasmic transport and global translational repression. Genes Dev 31: 1257–1271. 10.1101/gad.302604.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermann J, Horwich AL, Neupert W, Hartl FU. 1989. Protein folding in mitochondria requires complex formation with hsp60 and ATP hydrolysis. Nature 341: 125–130. 10.1038/341125a0 [DOI] [PubMed] [Google Scholar]
- Pansarasa O, Bordoni M, Drufuca L, Diamanti L, Sproviero D, Trotti R, Bernuzzi S, La Salvia S, Gagliardi S, Ceroni M, et al. 2018. Lymphoblastoid cell lines as a model to understand amyotrophic lateral sclerosis disease mechanisms. Dis Model Mech 11: dmm031625 10.1242/dmm.031625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, et al. 2015. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162: 1066–1077. 10.1016/j.cell.2015.07.047 [DOI] [PubMed] [Google Scholar]
- Peralta S, Torraco A, Wenz T, Garcia S, Diaz F, Moraes CT. 2014. Partial complex I deficiency due to the CNS conditional ablation of Ndufa5 results in a mild chronic encephalopathy but no increase in oxidative damage. Hum Mol Genet 23: 1399–1412. 10.1093/hmg/ddt526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A, et al. 2013. A compendium of RNA-binding motifs for decoding gene regulation. Nature 499: 172–177. 10.1038/nature12311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riback JA, Katanski CD, Kear-Scott JL, Pilipenko EV, Rojek AE, Sosnick TR, Drummond DA. 2017. Stress-triggered phase separation is an adaptive, evolutionarily tuned response. Cell 168: 1028–1040.e19. 10.1016/j.cell.2017.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatelli M, Moncada A, Conte A, Lattante S, Marangi G, Luigetti M, Lucchini M, Mirabella M, Romano A, Del Grande A, et al. 2013. Mutations in the 3′ untranslated region of FUS causing FUS overexpression are associated with amyotrophic lateral sclerosis. Hum Mol Genet 22: 4748–4755. 10.1093/hmg/ddt328 [DOI] [PubMed] [Google Scholar]
- Sama RR, Ward CL, Kaushansky LJ, Lemay N, Ishigaki S, Urano F, Bosco DA. 2013. FUS/TLS assembles into stress granules and is a prosurvival factor during hyperosmolar stress. J Cell Physiol 228: 2222–2231. 10.1002/jcp.24395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S, Iwata M. 2007. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 66: 10–16. 10.1097/nen.0b013e31802c396b [DOI] [PubMed] [Google Scholar]
- Scekic-Zahirovic J, Sendscheid O, El Oussini H, Jambeau M, Sun Y, Mersmann S, Wagner M, Dieterlé S, Sinniger J, Dirrig-Grosch S, et al. 2016. Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. EMBO J 35: 1077–1097. 10.15252/embj.201592559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt O, Pfanner N, Meisinger C. 2010. Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol 11: 655–667. 10.1038/nrm2959 [DOI] [PubMed] [Google Scholar]
- Schwartz JC, Ebmeier CC, Podell ER, Heimiller J, Taatjes DJ, Cech TR. 2012. FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2. Genes Dev 26: 2690–2695. 10.1101/gad.204602.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JC, Wang XY, Podell ER, Cech TR. 2013. RNA seeds higher-order assembly of FUS protein. Cell Rep 5: 918–925. 10.1016/j.celrep.2013.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma LK, Lu J, Bai Y. 2009. Mitochondrial respiratory complex I: structure, function and implication in human diseases. Curr Med Chem 16: 1266–1277. 10.2174/092986709787846578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Lyashchenko AK, Lu L, Nasrabady SE, Elmaleh M, Mendelsohn M, Nemes A, Tapia JC, Mentis GZ, Shneider NA. 2016. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat Commun 7: 10465 10.1038/ncomms10465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelkovnikova TA, Robinson HK, Southcombe JA, Ninkina N, Buchman VL. 2014. Multistep process of FUS aggregation in the cell cytoplasm involves RNA-dependent and RNA-independent mechanisms. Hum Mol Genet 23: 5211–5226. 10.1093/hmg/ddu243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EF, Shaw PJ, De Vos KJ. 2017. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci Lett 710: 132933 10.1016/j.neulet.2017.06.052 [DOI] [PubMed] [Google Scholar]
- Spriggs KA, Bushell M, Willis AE. 2010. Translational regulation of gene expression during conditions of cell stress. Mol Cell 40: 228–237. 10.1016/j.molcel.2010.09.028 [DOI] [PubMed] [Google Scholar]
- Stoica R, Paillusson S, Gomez-Suaga P, Mitchell JC, Lau DH, Gray EH, Sancho RM, Vizcay-Barrena G, De Vos KJ, Shaw CE, et al. 2016. ALS/FTD-associated FUS activates GSK-3β to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations. EMBO Rep 17: 1326–1342. 10.15252/embr.201541726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Ling SC, Qiu J, Albuquerque CP, Zhou Y, Tokunaga S, Li H, Qiu H, Bui A, Yeo GW, et al. 2015. ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat Commun 6: 6171 10.1038/ncomms7171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylvestre J, Margeot A, Jacq C, Dujardin G, Corral-Debrinski M. 2003. The role of the 3′ untranslated region in mRNA sorting to the vicinity of mitochondria is conserved from yeast to human cells. Mol Biol Cell 14: 3848–3856. 10.1091/mbc.e03-02-0074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan AY, Riley TR, Coady T, Bussemaker HJ, Manley JL. 2012. TLS/FUS (translocated in liposarcoma/fused in sarcoma) regulates target gene transcription via single-stranded DNA response elements. Proc Natl Acad Sci 109: 6030–6035. 10.1073/pnas.1203028109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tank EM, Figueroa-Romero C, Hinder LM, Bedi K, Archbold HC, Li X, Weskamp K, Safren N, Paez-Colasante X, Pacut C, et al. 2018. Abnormal RNA stability in amyotrophic lateral sclerosis. Nat Commun 9: 2845 10.1038/s41467-018-05049-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsanov N, Samacoits A, Chouaib R, Traboulsi AM, Gostan T, Weber C, Zimmer C, Zibara K, Walter T, Peter M, et al. 2016. smiFISH and FISH-quant—a flexible single RNA detection approach with super-resolution capability. Nucleic Acids Res 44: e165 10.1093/nar/gkw784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente AJ, Maddalena LA, Robb EL, Moradi F, Stuart JA. 2017. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem 119: 315–326. 10.1016/j.acthis.2017.03.001 [DOI] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, et al. 2009. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323: 1208–1211. 10.1126/science.1165942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Scotter EL, Nishimura AL, Troakes C, Mitchell JC, Kathe C, Urwin H, Manser C, Miller CC, Hortobágyi T, et al. 2013. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum Mol Genet 22: 2676–2688. 10.1093/hmg/ddt117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Li L, Lin WL, Dickson DW, Petrucelli L, Zhang T, Wang X. 2013a. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum Mol Genet 22: 4706–4719. 10.1093/hmg/ddt319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WY, Pan L, Su SC, Quinn EJ, Sasaki M, Jimenez JC, Mackenzie IR, Huang EJ, Tsai LH. 2013b. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat Neurosci 16: 1383–1391. 10.1038/nn.3514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Jiang X, Chen G, Xu J. 2015a. Interaction of amyotrophic lateral sclerosis/frontotemporal lobar degeneration-associated fused-in-sarcoma with proteins involved in metabolic and protein degradation pathways. Neurobiol Aging 36: 527–535. 10.1016/j.neurobiolaging.2014.07.044 [DOI] [PubMed] [Google Scholar]
- Wang X, Schwartz JC, Cech TR. 2015b. Nucleic acid-binding specificity of human FUS protein. Nucleic Acids Res 43: 7535–7543. 10.1093/nar/gkv679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wang L, Lu J, Siedlak SL, Fujioka H, Liang J, Jiang S, Ma X, Jiang Z, da Rocha EL, et al. 2016. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat Med 22: 869–878. 10.1038/nm.4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Ogé L, Perez-Garcia MD, Hamama L, Sakr S. 2018. The PUF protein family: overview on PUF RNA targets, biological functions, and post transcriptional regulation. Int J Mol Sci 19: 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann FR, Manfredi G, Mawrin C, Beal MF, Schon EA. 2002. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J Neurochem 80: 616–625. 10.1046/j.0022-3042.2001.00731.x [DOI] [PubMed] [Google Scholar]
- Wu S, Zhou F, Zhang Z, Xing D. 2011. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. FEBS J 278: 941–954. 10.1111/j.1742-4658.2011.08010.x [DOI] [PubMed] [Google Scholar]
- Xiao B, Goh JY, Xiao L, Xian H, Lim KL, Liou YC. 2017. Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin. J Biol Chem 292: 16697–16708. 10.1074/jbc.M117.787739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda K, Clatterbuck-Soper SF, Jackrel ME, Shorter J, Mili S. 2017. FUS inclusions disrupt RNA localization by sequestering kinesin-1 and inhibiting microtubule detyrosination. J Cell Biol 216: 1015–1034. 10.1083/jcb.201608022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. 2012. Mitochondrial fission, fusion, and stress. Science 337: 1062–1065. 10.1126/science.1219855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Liu S, Liu G, Öztürk A, Hicks GG. 2013. ALS-associated FUS mutations result in compromised FUS alternative splicing and autoregulation. PLoS Genet 9: e1003895 10.1371/journal.pgen.1003895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Stumpf CR, Krahn JM, Wickens M, Hall TM. 2009. A 5′ cytosine binding pocket in Puf3p specifies regulation of mitochondrial mRNAs. Proc Natl Acad Sci 106: 20192–20197. 10.1073/pnas.0812079106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H, Sok J, Immanuel D, Yin Y, Ron D. 1997. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci 110 (Pt 15): 1741–1750. [DOI] [PubMed] [Google Scholar]
- Züchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, et al. 2004. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet 36: 449–451. 10.1038/ng1341 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing data reported here were submitted to Gene Expression Omnibus (GEO) database under accession number GSE148550.