

Abstract
Natural organisms use a four-letter genetic alphabet that makes available 64 triplet codons, of which 61 are sense codons used to encode proteins with the 20 canonical amino acids. We have shown that the unnatural nucleotides dNaM and dTPT3 pair to form an unnatural base pair (UBP) and allow for the creation of semi-synthetic organisms (SSOs) with additional sense codons. Here we report a systematic analysis of the unnatural codons. We identify nine unnatural codons that can produce unnatural protein with nearly complete incorporation of an encoded non-canonical amino acid (ncAA). We also show that at least three of the codons are orthogonal and can be simultaneously decoded in the SSO, affording the first 67-codon organism. The ability to site-specifically incorporate multiple, different ncAAs into a protein should now allow for the development of proteins with novel activities and possibly even SSOs with new forms and functions.
The natural genetic code consists of the 64 codons made possible by the four letters of the genetic alphabet. Three are used as stop codons, leaving 61 sense codons that are recognized by tRNAs charged by cognate tRNA synthetases (aaRSs) with one of the 20 proteogenic amino acids. While the canonical amino acids have enabled the remarkable diversity of living organisms, there are many chemical functionalities and associated reactivities that they do not provide. The ability to expand the genetic code to include non-canonical amino acids (ncAAs), perhaps with ncAAs selected to bestow the protein with a desired function or activity, would dramatically facilitate many known and emerging applications of proteins, such as for therapeutic development1. In 2001, it was demonstrated that the tyrosyl-tRNA from Methanocaldococcus (formerly Methanococcus) jannaschii could be recoded to suppress an amber stop codon, charged with an ncAA by an evolved variant of its cognate TyrRS synthetase, and used to expand the genetic code in Escherichia coli2. Since then, amber suppression has been extended to other orthogonal tRNA–aaRS pairs, most notably the pyrrolysyl pair from the Methanosarcina genus (e.g. M. mazei pyrrolysyl-tRNA (tRNAPyl))3, broadening the scope of ncAAs that may be incorporated (reviewed in Liu & Schultz 20104). The simultaneous use of a quadruplet codon and/or suppression of one or more stop codons allows for the production of proteins containing multiple different ncAAs5–7. However, protein yields are limited by competition with normal decoding, and while the deletion of release factor 1 (ΔprfA) eliminates competition for amber decoding8, it increases competition with near cognate tRNAs and thus reduces fidelity9.
An alternative approach to a potentially less restricted expansion of the genetic code is to create entirely new codons, unencumbered by native biological functions, via the expansion of the genetic alphabet from four letters and two base pairs to six letters and three base pairs10,11. Towards this goal, we have developed a fifth and sixth nucleotide, dNaM and dTPT3 (Fig. 1a), that selectively pair to form an unnatural base pair (UBP)12–14. By expression of the Phaeodactylum tricornutum nucleoside triphosphate transporter 2 (PtNTT2)15, E. coli cells can import the unnatural deoxynucleoside triphosphates (dNaMTP and dTPT3TP) and with their native replication machinery16,17, use them to replicate DNA containing the dNaM-dTPT3 UBP18. We have also shown that when provided with the requisite ribonucleoside triphosphates (NaMTP and TPT3TP), the resulting semi-synthetic organism (SSO) can transcribe DNA containing the unnatural nucleotides into both tRNAs and mRNAs, and along with cognate aaRSs, use the complementary unnatural codons and anticodons to produce protein containing an ncAA19. In principle, the UBP makes 152 unnatural codons available, but only two have been examined, and it is unclear whether others are functional, and importantly, whether any are orthogonal, which is required to simultaneously decode multiple, different ncAAs.
Fig. 1 |. Protein production in non-clonal SSOs using unnatural codons and anticodons.
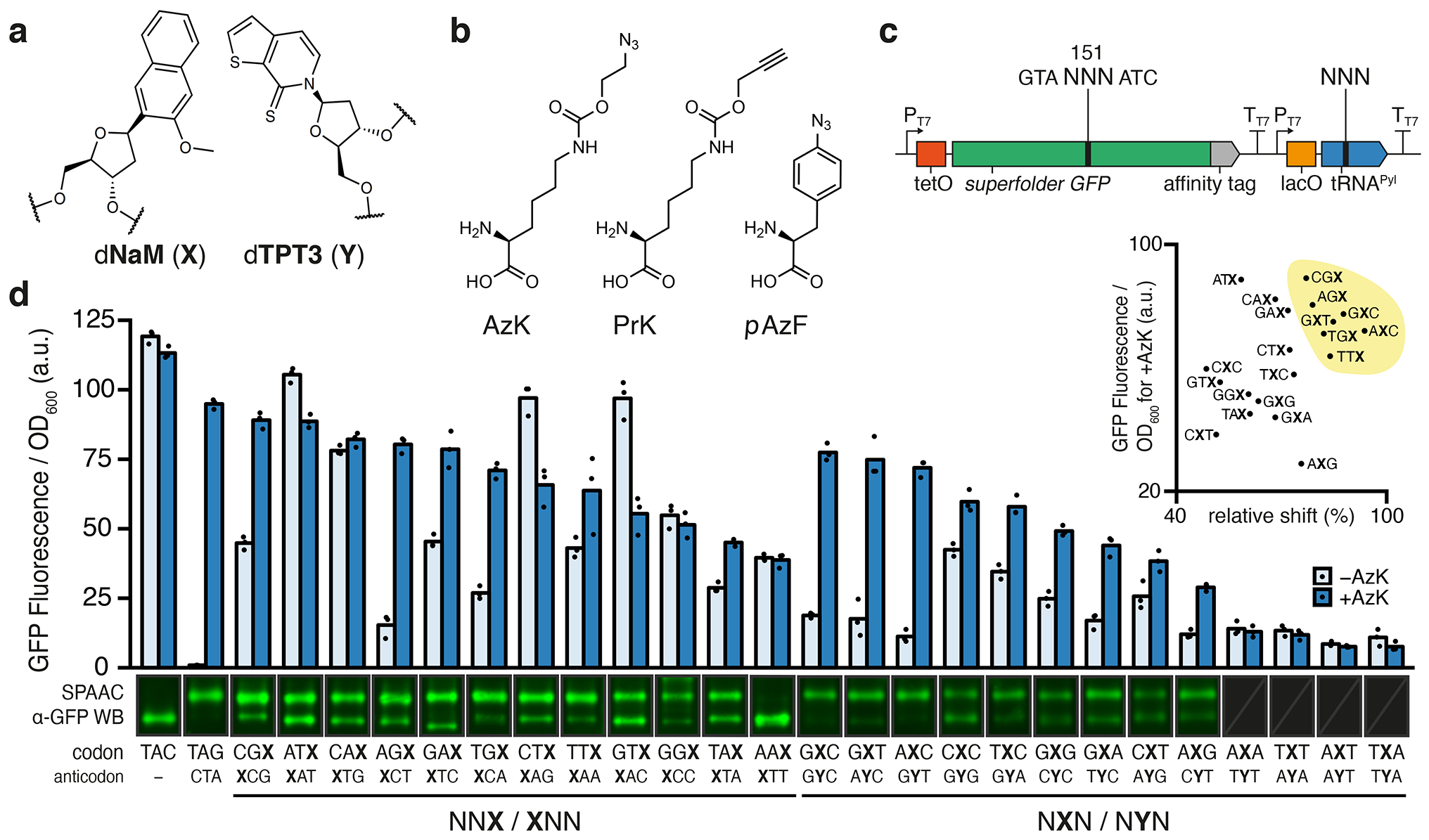
a, Chemical structure of the dNaM-dTPT3 UBP. b, Chemical structures of ncAAs, AzK, PrK, and pAzF. c, Schematic illustration of gene cassette used to express sfGFP151(NNN) and M. mazei tRNAPyl(NNN), where NNN refers to any specified codon or anticodon. d, Normalized fluorescence from non-clonal SSO cultures at the endpoint of protein expression (i.e. t = 180 min after addition of aTc) using specified codons and anticodons both with and without AzK in the media (n = 3, biological replicates; mean with individual data points shown). One representative cropped western blot of purified sfGFP, subjected to SPAAC with TAMRA-PEG4-DBCO, from SSO cultures shown above each codon and anticodon (only α-GFP channel). Triplicate western blots shown in Supplementary Figs. 2a and 9. d inset, Scatterplot of mean endpoint fluorescence in the presence of AzK (from d) versus mean of quantified relative protein shift induced by SPAAC (n = 3; biological replicates; Supplementary Fig. 2b). Seven top codons chosen for further analyses are highlighted (yellow).
Here we report the first systematic analysis of unnatural codons in an SSO. With the SSO replicating and transcribing the UBP in different sequence contexts, we explore the ability of codons with NaM or TPT3 at the first, second, or third position, and tRNAs with cognate unnatural anticodons, to mediate the production of proteins with ncAAs. While it appears that first position unnatural codons are not decoded efficiently, second position NaM codons are generally well decoded with cognate TPT3 anticodons, and finally that several third positions NaM codons are well decoded by self-pairing NaM anticodons. This analysis identifies nine unnatural codon/anticodon pairs that are stably encoded in DNA, are efficiently transcribed into mRNA and tRNA, and can efficiently mediate decoding at the ribosome, increasing the number of available codons from 64 to 73. Moreover, we examine three of the unnatural codon/anticodon pairs and find them to be orthogonal to each other, and we demonstrate the simultaneous decoding of three unnatural codons in the SSO, which to our knowledge is the first time 67 codons have been decoded in a living organism.
Results
Initial screening of codon function in non-clonal SSOs.
We and others have used green fluorescent protein and variants such as sfGFP20 as model systems for the study of ncAA incorporation, especially at position Y151, which tolerates a variety of natural and ncAA substitutions. We constructed plasmids containing two dNaM-dTPT3 UBPs, one positioned within codon 151 of sfGFP and the other positioned to encode the anticodon of M. mazei tRNAPyl (Fig. 1c), which is selectively charged by PylRS with the ncAA N6-(2-azidoethoxy)-carbonyl-L-lysine (AzK)21 (Fig. 1b). We constructed plasmids to examine the decoding of six codons, including two first position unnatural codons (XTC and XTG; X refers to dNaM), two second position unnatural codons (AXC and GXA), and two unnatural third position codons (AGX and CAX), as well as the opposite strand context codons (YTC, YTG, AYC, GYA, AGY, and CAY; Y refers to dTPT3).
While clonal populations of SSOs are able to produce larger quantities of pure unnatural protein, likely due to the elimination of plasmids that were misassembled during in vitro construction, to facilitate the initial codon screen we first explored protein expression with a non-clonal population of cells and assayed protein production immediately after transformation. Plasmids were used to transform E. coli ML2 (BL21(DE3) lacZYA::PtNTT2(66–575) ΔrecA polB++)17 that harbored an accessory plasmid encoding a chimeric pyrrolysyl-tRNA synthetase variant (chPylRSIPYE)22 and after growth to early stationary phase in selective media supplemented with dNaMTP and dTPT3TP, cells were transferred to fresh media. Following growth to mid-exponential phase, the culture was supplemented with NaMTP, TPT3TP, and AzK, and isopropyl-β-d-thiogalactoside (IPTG) was added to induce expression of T7 RNA polymerase (T7 RNAP), chPylRSIPYE, and tRNAPyl. After 1 h of additional growth, anhydrotetracycline (aTc) was added to induce expression of sfGFP, which was monitored by fluorescence.
First position codons showed no significant fluorescence in the absence or presence of AzK, regardless of whether decoding was attempted with the heteropairing or self-pairing anticodons (e.g. tRNAPyl(CAY) or tRNAPyl(CAX), respectively, for XTG) (Supplementary Fig. 1). Codons with dNaM at the second position showed little fluorescence in the absence of AzK, but in its presence showed significant fluorescence when decoded with tRNAPyl recoded with the heteropairing anticodons tRNAPyl(GYT) or tRNAPyl(TYC), but not with self-pairing anticodons tRNAPyl(GXT) or tRNAPyl(TXC). With dTPT3 at the second position, no fluorescence was observed with or without added AzK regardless of whether decoding was attempted with heteropairing or self-pairing tRNAs. The third position codons CAX and CAY showed high fluorescence in the absence of AzK, and surprisingly showed less with its addition, regardless of whether decoding was attempted with a heteropairing or self pairing tRNAPyl. This result suggests that the corresponding third position unnatural tRNAs nonproductively bind at the ribosome and block unnatural codon read-through by a natural tRNA. In the absence of AzK, AGX and AGY showed little fluorescence, and AGX with tRNAPyl(XCT) showed an increase in fluorescence with the addition of AzK.
As the first position codons did not appear promising, we next turned to a more comprehensive screen of second position codons. Because the initial analysis indicated potential decoding only with NaM in the codon and with TPT3 in the anticodon, we focused on the possible NXN codons with cognate tRNAPyl(NYN). Of the 16 possible codons, CXA, CXG, and TXG were excluded as we have found that the corresponding sequence context is poorly retained in the DNA of the SSO18. In agreement with previous results19, in the absence of AzK, the use of codons GXC and AXC resulted in little to no fluorescence, while in the presence of AzK, they resulted in significant fluorescence (Fig. 1d). Similarly, with the GXT, CXC, TXC, GXG, GXA, CXT, and AXG codons, the addition of AzK resulted in significant increases in fluorescence, relative to when AzK was withheld. The remaining four codons, AXA, AXT, TXA, and TXT, produced little fluorescence regardless of whether or not AzK was added, revealing a stringent requirement for at least one G-C pair.
To screen for unnatural protein production, sfGFP was purified via the C-terminal StrepII affinity tag and subjected to a strain-promoted azide-alkyne cycloaddition (SPAAC) reaction with dibenzocyclooctyne (DBCO) linked to a rhodamine dye (TAMRA) by four PEG units (TAMRA-PEG4-DBCO). As shown previously23, successful conjugation not only tags the proteins containing the ncAA with a detectable fluorophore, but also produces a detectable shift in electrophoretic mobility, allowing quantification of protein containing AzK relative to the total protein produced (i.e. fidelity of ncAA incorporation; Fig 1d, Supplementary Fig. 2). In agreement with previous results19, the use of codons GXC and AXC resulted in the production of significant amounts of sfGFP with the AzK residue. Remarkably, seven additional unnatural codons, GXT, CXC, TXC, GXG, GXA, CXT, and AXG, also yielded significant levels of unnatural protein (Fig. 1d, Supplementary Fig. 2).
Finally, we turned to a more comprehensive screen of third position codons. Because in the initial screen only AGX appeared to be decoded, and only then by the self-pairing tRNAPyl(XCT), we further screened codons with dNaM at the third position of the codon (NNX) with cognate self-pairing tRNAPyl(XNN) (Fig 1c). NCX codons were excluded as they result in sequence contexts of NCXA, which as noted above are not well retained in the DNA of the SSO18. In agreement with the initial analysis, in the absence of AzK these codons generally resulted in more fluorescence than we observed with the second position codons, but in the presence of AzK, variable increases in fluorescence were observed (Fig. 1d). Regardless, when protein was isolated and analyzed as described above, the use of CGX, ATX, CAX, AGX, GAX, TGX, CTX, TTX, GTX, or TAX all resulted in significant levels of unnatural protein production (Fig. 1d, Supplementary Fig. 2). Codon GGX produced multiple shifted species, suggesting that tRNAPyl(XCC) decodes one or more natural codons. No unnatural protein was detected when codon AAX was used.
Codon function and orthogonality in clonal SSOs.
To select the most promising codon/anticodon pairs identified in the above described codon screen, we compared the observed fluorescence in the presence of AzK and the induced mobility shift in isolated protein (Fig. 1d, inset). Based on this analysis, seven unnatural codon/anticodon pairs, GXC/GYC, GXT/AYC, AXC/GYT, AGX/XCT, CGX/XCG, TGX/XCA, TTX/XAA were selected for further characterization. These codon/anticodon pairs were examined in clonal SSOs, which eliminates cells that were transformed with misassembled plasmids or plasmids that had lost the UBP during in vitro construction. Clonal SSOs were obtained by streaking transformants onto solid growth media containing dNaMTP and dTPT3TP, selecting individual colonies, and confirming plasmid integrity and high UBP retention (see Methods). High retention clones were regrown and induced to produce protein as described above. Remarkably, the observed fluorescence indicates that each of the seven codon/anticodon pairs produces protein at a level that compares favorably with the amber suppression control, and moreover, the gel shift assay demonstrates that virtually all of the sfGFP contains the ncAA (Fig. 2a, left; Supplementary Fig. 3). Decoding using codon/anticodon pairs AGX/XCT, CGX/XCG, TGX/XCA, and TTX/XAA depended on NaMTP in the expression media and produced sfGFP with a similar AzK content both with and without TPT3TP added (Supplementary Fig. 4).
Fig. 2 |. Protein production and analyses of codon orthogonality in clonal SSOs.
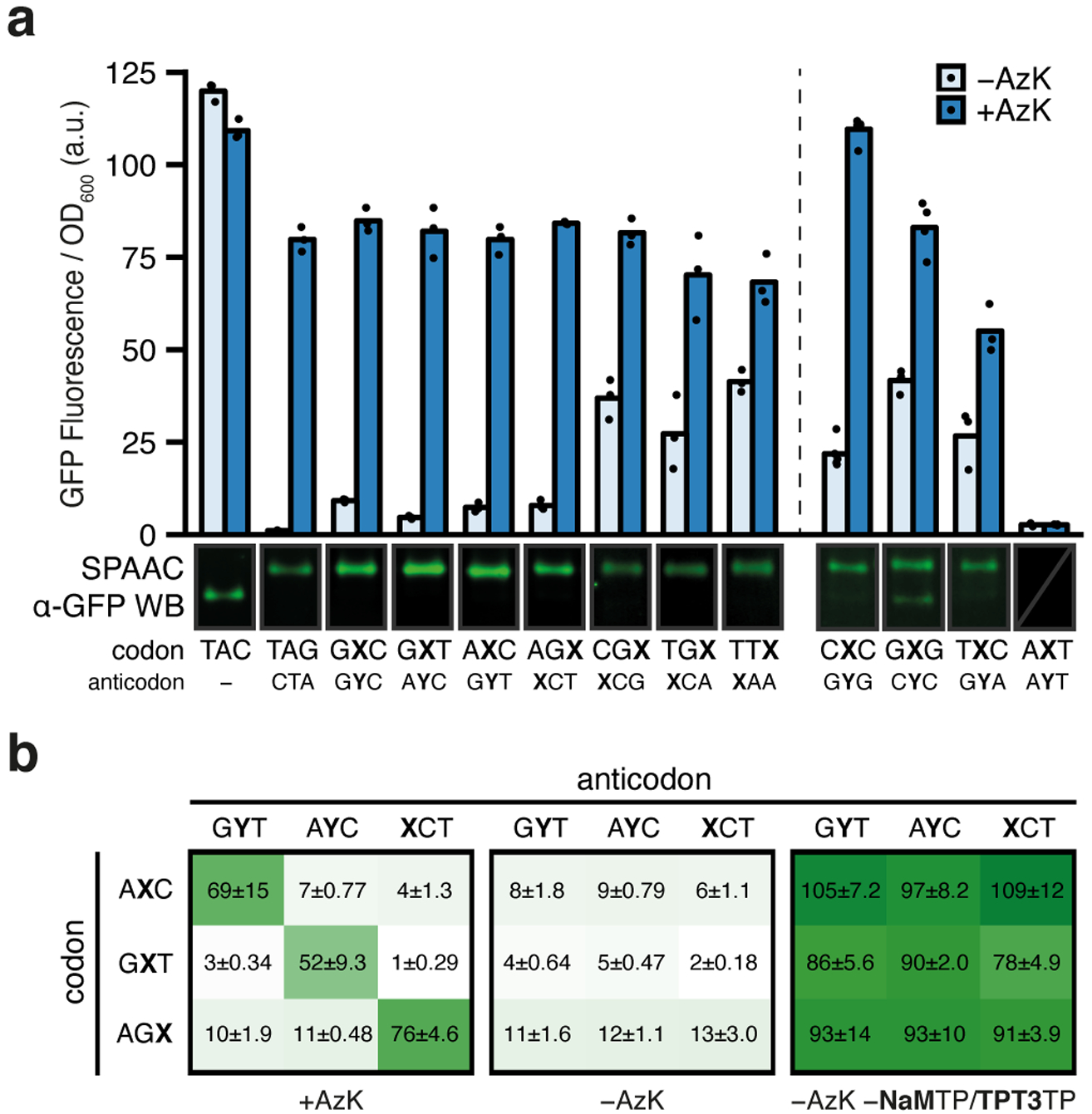
a, Normalized fluorescence from clonal SSOs at the endpoint of protein expression (i.e. t = 180 min after addition of aTc) for the seven top codons and anticodons (left) as well as the four other selected codons (right) both with and without AzK (left: n = 3, right: n = [5, 4, 3, 3]; biological replicates; mean with individual data points shown). The dashed line demarcates slight variations in how the clonal SSOs were prepared (see Methods). One representative cropped western blot of purified sfGFP, subjected to SPAAC with TAMRA-PEG4-DBCO from SSO cultures is shown (only α-GFP channel). All western blots shown in Supplementary Figs. 3 and 10. b, Normalized fluorescence from clonal SSO cultures at the endpoint of expression for all pairwise combinations of select codons and anticodons with and without AzK in media. Positive controls, without ribonucleoside triphosphates NaMTP and TPT3TP, which forces the incorporation of a natural ribonucleotide opposite the unnatural nucleotide in the template, were run to verify the integrity of the clone. Each culture was propagated from a single colony and mean ± standard deviation is indicated (black text; n = 3; biological replicates).
The seven unnatural codon/anticodon pairs analyzed above clearly mediate efficient decoding at the ribosome, however, it is possible that other codons from the preliminary non-clonal screen would also show efficient decoding if analyzed in clonal SSOs. Thus, we explored unnatural protein production in clonal SSOs with four codon/anticodon pairs that showed varying incorporation of AzK in the initial screen (CXC/GYG, GXG/CYC, TXC/GYA, and AXT/AYT; Fig. 1d inset). Despite high UBP retention (Supplementary Table 1) in the clonal SSO, AXT showed no fluorescence signal with or without AzK, further supporting the requirement for a G-C pair with the second position codons. Fluorescence with added AzK for CXC, GXG, and TXC was comparable to that of the seven initially characterized codons, although it was somewhat higher in the absence of AzK (Fig. 2a, right). SPAAC gel shift analysis revealed that CXC clearly results in significantly more shifted protein in the clonal SSO than observed in the preliminary screen with non-clonal SSOs, and GXG and TXC likely do as well, although the relatively larger error of the data from the preliminary screen precludes a quantitative comparison (Supplementary Figs. 2 and 3). The data suggest that for some codons, the suboptimal performance in the non-clonal screen resulted, at least in part, from sequence-dependent differences in in vitro plasmid construction. Regardless, the results identify two additional high-fidelity codons, CXC and TXC, and suggest that more viable codons may yet be identified.
To begin to evaluate the orthogonality of unnatural codon/anticodon pairs, we selected three, AXC/GYT, GXT/AYC, and AGX/XCT, and examined protein production in clonal SSOs with all pairwise combinations of unnatural codons and anticodons. With added AzK, significant fluorescence was observed when each unnatural codon was paired with a cognate unnatural anticodon, and virtually no increase over background was observed when paired with a non-cognate unnatural anticodon (Fig. 2b). Thus, AXC/GYT, GXT/AYC, and AGX/XCT are orthogonal and should be capable of simultaneous use in the SSO.
Simultaneous decoding of two unnatural codons.
To explore the simultaneous decoding of multiple codons, we first constructed a plasmid with the native sfGFP codons at position 190 and 200 replaced by GXT and AXC, respectively (sfGFP190,200(GXT,AXC)). In addition, the plasmid encoded both tRNAPyl(AYC) and M. jannaschii tRNApAzF, which is selectively charged by the M. jannaschii TyrRS variant pAzFRS (MjpAzFRS) with p-azido-L-phenylalanine (pAzF; Fig 1b)24, and whose anticodon was recoded to recognize AXC (tRNApAzF(GYT); Fig. 3a). E. coli ML2 harboring an accessory plasmid encoding both chPylRSIPYE and MjpAzFRS, was transformed with the UBP-containing plasmid and clonal SSOs were obtained, grown, and induced to produce sfGFP as described above. With both AzK and pAzF provided, increased cell fluorescence was observed within the same timescale as expression with single codon constructs (Fig. 3b, Supplementary Fig. 5) While the level of fluorescence with expression of sfGFP190,200(GXT,AXC) was somewhat less than half that observed with sfGFP190(GXT) or sfGFP200(AXC), it was significantly greater than that observed from an amber, ochre control (sfGFP190,200(TAA,TAG)) decoded with the corresponding suppressor tRNAs (Fig 3c, Supplementary Fig. 5). In both cases, when analyzed by SPAAC gel shift, no unshifted band was apparent and the mobility of the major band was further retarded compared with that observed for the incorporation of a single ncAA, suggesting that indeed two ncAAs had been incorporated (Fig. 3d). To confirm that both pAzF and AzK were incorporated, we analyzed purified protein using quantitative intact protein mass spectrometry (HRMS ESI-TOF). In agreement with the gel shift assay, this analysis revealed that 91 ± 1.1% of the isolated protein contained both pAzF and AzK, while 1.7 ± 0.4% contained a single pAzF and 7.5 ± 0.78% a single AzK (Supplementary Fig. 6). In both cases, the mass of the identified impurities correspond to the amino acid substitution consistent with a dX to dT mutation, which we have previously shown is the major mutation during replication18. Since the loss of UBP retention in the DNA (Supplementary Table 1) appears correlated with the impurities observed via HRMS, the majority of loss in ncAA incorporation fidelity likely results from loss of dNaM or dTPT3 during replication and is not due to errors during transcription or translation. However, we cannot rule out that some mis-incorporation results from transcription or translation errors. The SSO yielded 16 ± 3.2 μg·ml−1 of purified protein, whereas the amber, ochre suppression control yielded 6.8 ± 1.1 μg·ml−1 (i.e. total protein purified per volume of culture; unadjusted for fidelity). However, we note that the SSO culture grew to a lower density than the amber, ochre control cells (Supplementary Fig. 7), and when normalized for OD600, the SSO yielded 13 ± 1.6 μg·ml−1 of purified protein, whereas amber, ochre suppression yielded 2.8 ± 0.28 μg·ml−1, demonstrating that the SSO produces in excess of 4.5-fold more protein per OD600 (Supplementary Table 2). Thus, the SSO efficiently produces unnatural protein with two ncAAs.
Fig. 3 |. Simultaneous decoding of two unnatural codons.
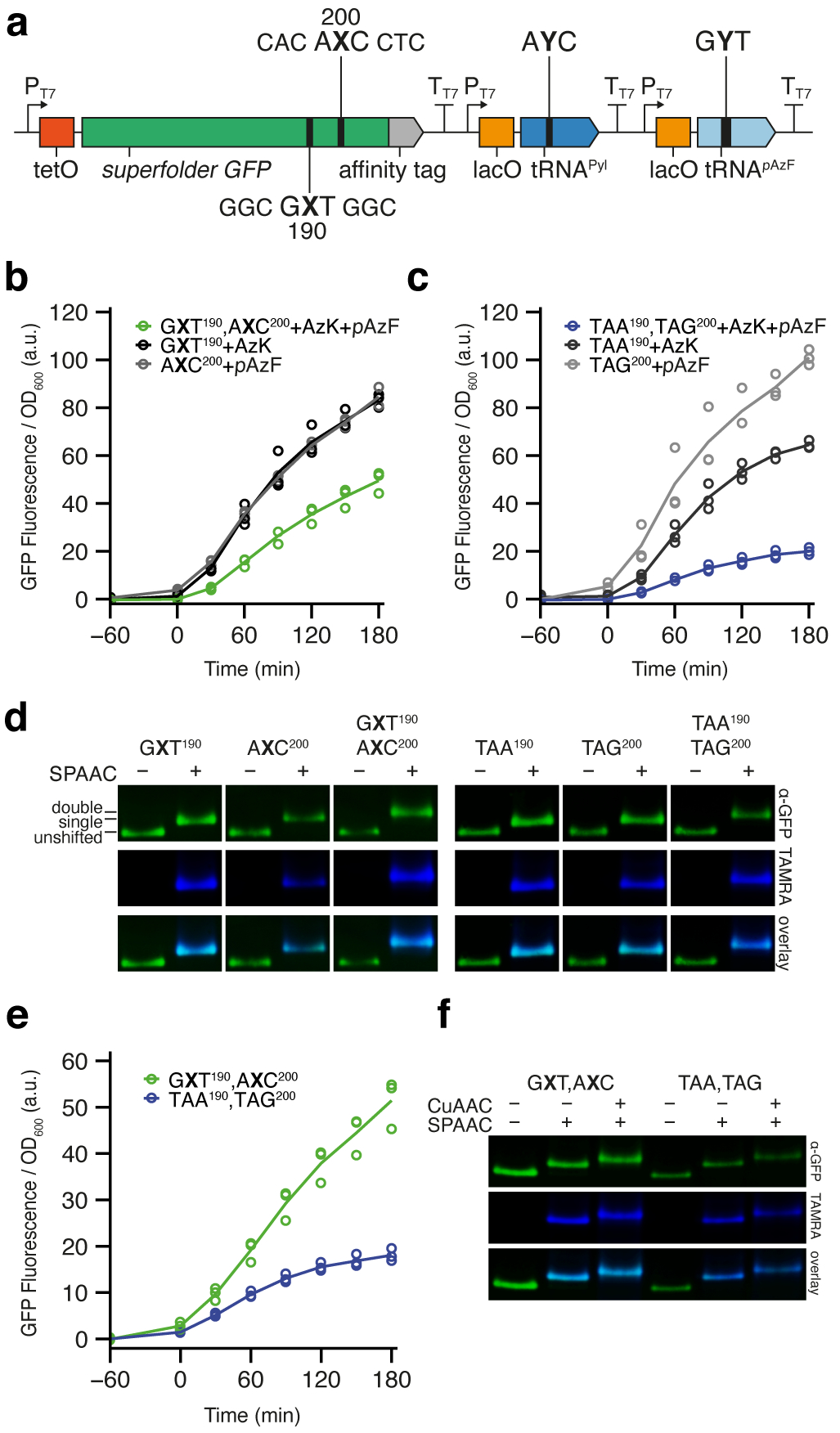
a, Schematic illustration of gene cassette containing sfGFP190,200(GXT,AXC), M. mazei tRNAPyl(AYC), and M. jannaschii tRNApAzF(GYT). b-c, Time-course plot of normalized fluorescence during sfGFP expression in the presence of denoted ncAAs (aTc added at t = 0; n = 3, biological replicates; mean and individual data points shown). b, Clonal SSO expression of the cassette in a as well as controls showing expression of cassettes containing only single codons with the appropriate tRNA. c, Clonal expression of a cassette containing sfGFP190,200(TAA,TAG), M. mazei tRNAPyl(TTA), and M. jannaschii tRNApAzF(CTA) also shown, as well as control cassettes containing the single stop-codons with the appropriate suppressor tRNA. d, Pseudocolored western blots (green) and TAMRA fluorescence scans (blue) of purified sfGFP from SSOs in b-c, with and without conjugation to TAMRA-PEG4-DBCO by SPAAC. Images are cropped from the same blots (UBP constructs and stop codon suppressors) but positioned to align the unshifted band in order compare electrophoretic migration. e, Time-course plot of normalized fluorescence during clonal expression of double codon/tRNA cassettes from b-c, with addition of PrK and pAzF. Mean and individual data points shown (n = 3, biological replicates). f, Pseudocolored western blots (green) and TAMRA fluorescence scans (blue) of purified sfGFP from SSOs in e, with and without conjugation to TAMRA-PEG4-DBCO by SPAAC and to TAMRA-PEG4-azide by CuAAC. Uncropped scans of western blots (d, f) shown in Supplementary figure 11.
To characterize expression of proteins with ncAAs with different functional groups, we expressed sfGFP190,200(GXT,AXC) in the SSO as described above but supplemented the growth medium with N6-(propargyloxy)-carbonyl-L-lysine (PrK, Fig. 1b)21, which is also recognized by chPylRSIPYE, instead of AzK. No substantial impact on expression was observed by fluorescence for either the SSO or the amber, ochre control (Fig. 3e). In each case, we verified the correct incorporation of both PrK and pAzF by SPAAC with TAMRA-PEG4-DBCO followed by copper-catalyzed alkyne-azide cycloaddition (CuAAC) using TAMRA-PEG4-azide, as both induce an observable shift in electrophoretic mobility. Protein produced by the SSO, as well as the amber, ochre control, showed the expected gel shifts and TAMRA signal (Fig. 3f).
Simultaneous decoding of three unnatural codons.
The two orthogonal tRNA/aaRS pairs employed above are among the most validated and are the only pairs where charging is also known to not involve significant anticodon recognition. Thus, to explore the simultaneous decoding of the three orthogonal unnatural codons, we employed the endogenous serine tRNASer, E. coli SerT, which is charged by endogenous SerRS without anticodon recognition25, and which we have previously recoded to decode an unnatural codon19. E. coli ML2 harboring an accessory plasmid encoding chPylRSIPYE and MjpAzFRS was transformed with a plasmid expressing sfGFP151,190,200(AXC,GXT,AGX) as well as tRNAPyl(XCT), tRNApAzF(GYT), and tRNASer(AYC) (Fig. 4a), and clonal SSOs were prepared, grown, and induced to produce protein as described above. With AzK and pAzF added to the media, significant fluorescence was observed, similar to results obtained above for simultaneous decoding of two codons (Fig. 4b, Supplementary Fig. 5). These cells yielded 12.1 ± 1.9 μg·ml−1 (7.8 ± 1.1 μg·ml−1·OD600−1) of isolated protein, which is only slightly less than the quantity isolated with the decoding of two unnatural codons (Supplementary Table 2). To confirm that pAzF, AzK, and Ser had all been incorporated, we analyzed purified protein via HRMS ESI-TOF, and found that 96 ± 0.63% of the isolated protein contained pAzF, AzK, and Ser, while the major impurity was sfGFP containing only AzK and Ser (3.5 ± 0.63%). Protein without Ser incorporation was almost undetectable (0.20 ± 0.087%), whereas a mass corresponding to protein containing only pAzF and Ser could not be detected (Fig. 4c, Supplementary Fig. 8). Additionally, we were unable to detect any impurities corresponding to the multiple insertion of either Ser, AzK, or pAzF.
Fig. 4 |. Simultaneous decoding of three unnatural codons.
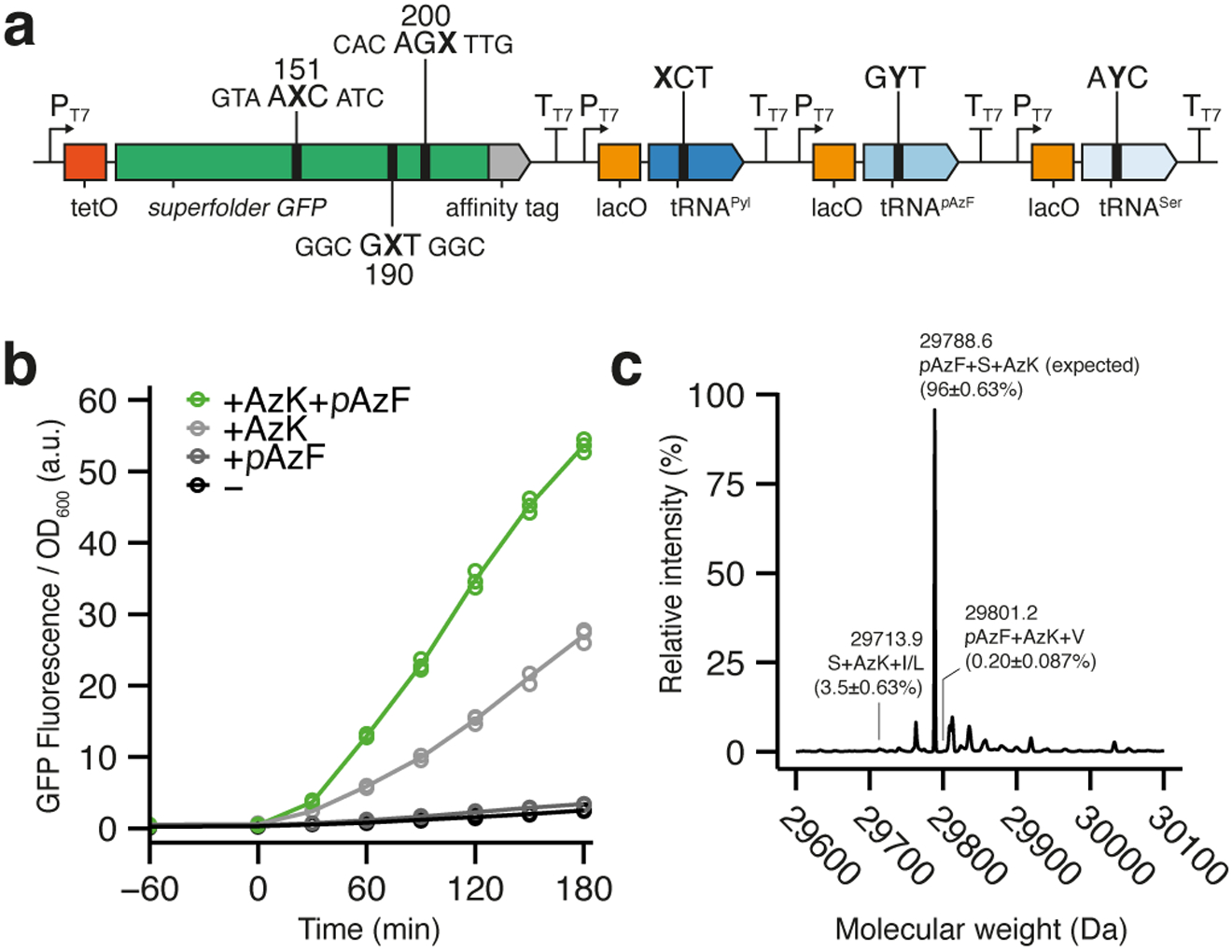
a, Schematic illustration of gene cassette containing sfGFP151,190,200(AXC,GXT,AGX), M. mazei tRNAPyl(XCT), M. jannaschii tRNApAzF(GYT), and E. coli tRNASer(AYC). b, Time-course plot of normalized fluorescence during sfGFP expression in the absence or presence of AzK and/or pAzF (aTc added at t = 0; n = 3, biological replicates; mean and individual data points shown). c, Representative deconvoluted mass spectrum from HRMS ESI-TOF analysis of intact sfGFP purified from SSOs in b. Peak labels denote molecular weight as well as quantification of each peak relative to other relevant species (Supplementary Fig. 8). Standard single-letter amino acid code used. Mean ± standard deviation shown (n = 3).
Discussion
Since at least the last common ancestor of all living things, cells have retrieved information via a genetic code made up of the 64 codons made available with the four natural letters of the genetic alphabet. Because each of the 61 sense codons is assigned to a canonical amino acid, this limits the diversity of proteins that life can produce. Codon reassignment can alleviate this limitation, but is challenged by competition with natural decoding, the ability to encode multiple different ncAAs, and/or the construction of strains with completely recoded genomes8,26,27. We have explored the use of SSOs with an expanded genetic alphabet to provide multiple new codons that can be dedicated to multiple ncAAs.
In general, we found that efficient decoding requires NaM at the second or third position of the codon and TPT3 or NaM, respectively, at the corresponding position of the anticodon. Poor decoding with an unnatural nucleotide at the first position may result from interrogation by the ribosome with a type I A-minor interaction, where a single adenine of the ribosome spans and hydrogen bonds with both nucleotides of the minihelix to select for correct Watson-Crick-like geometries28,29. While structural data demonstrates that the UBP forms a Watson-Crick-like structure during synthesis (i.e. in the polymerase active site), it adopts a cross-strand intercalated structure in free duplex DNA30,31, which would clearly be incapable of engaging the A-minor interaction. Thus, the data suggests that the UBP in the RNA minihelix, at least at the first position of the codon, either does not adopt an appropriate structure or does not present appropriate hydrogen bonding functionality to productively participate in the type I A-minor interaction. The decoding of codons containing hydrogen bonding unnatural base pairs, whose structures resemble a Watson-Crick geometry, at the first position has been reported in vitro32, 33, but it is unclear whether they could be decoded in vivo.
The structure of the second position base pair of the codon/anticodon minihelix is interrogated by a type II A-minor interaction, wherein each complementary nucleotide of the minihelix interacts with a different nucleotide of the ribosome (an adenine and a guanine). While the two interrogating nucleotides interact with each other via a hydrogen bond, at least some repositioning may be more feasible, allowing for greater flexibility in accommodating the structure or hydrogen-bonding potential of the unnatural nucleotides. Nonetheless, distortion from the optimal structure may result in decreased pairing stability, which may account for the apparent requirement for at least one G-C base pair when NaM is at this position. Consistent with this possibility, structural studies have shown that a G-C or C-G at the first position forms an additional hydrogen bond with the adenine mediating the A-minor interaction34. No A-minor interaction interrogates the base pair formed at the third position of the codon/anticodon minihelix, and this position, the wobble position, has long been known to accommodate a variety of non-canonical pairings. Recently, via transfection of HEK cells, it has been demonstrated that nucleobases that do not interact via hydrogen-bonding can participate in decoding when at the wobble position35. Nonetheless, it is currently unclear why decoding by the NaM self-pair is favored over decoding by the heteropair.
Regardless of the mechanistic underpinnings, 19 unnatural codons were decoded with at least moderate fidelity in non-clonal SSOs. We suspect the majority of unshifted protein originates from cells that received plasmids that have lost the UBP, as it is known that the fidelity of replication is higher in SSOs than during PCR36. However, we cannot rule out a contribution from erroneous decoding by endogenous tRNAs or by tRNAPyl that is charged by an endogenous aaRSs with a natural amino acid. Nonetheless, when expressed in clonal SSOs, nine codon-anticodon pairs, including the two previously reported, produced protein with little to no detectable contaminating natural protein. These nine codons and anticodons are well retained during replication, efficiently transcribed into mRNA and tRNA, and efficiently mediate protein synthesis at the ribosome.
In order for the expanded genetic alphabet to underlie an unrestricted expansion of the genetic code, the unnatural codon/anticodon pairs must be orthogonal to each other. Our initial efforts to explore this orthogonality focused on the AXC/GYT, GXT/AYC, and AGX/XCT codon/anticodon pairs and showed that each efficiently and orthogonally mediates protein synthesis. In fact, the simultaneous use of the GXT and AXC codons produced over 4-fold more protein per OD600 than an amber, ochre stop codon suppression control. While the SSO grew to lower density, the absolute amount of protein produced with two ncAAs was still more than 2-fold greater with the SSO than with the amber, ochre control. Protein production in the control can potentially yield higher titers in an RF1-deficient strain8, however, RF1 deletion does not consistently increased protein yields when only a single codon is suppressed37. Nevertheless, fluorescence per OD600 during protein production with two unnatural codons is ~40% that of its completely natural counterpart. Since incorporation of ncAAs is generally limited by tRNA charging19, 38, this suggests that decoding of both GXT and AXC is quite efficient.
Finally, we explored the simultaneous decoding of three unnatural codons. AXC was decoded with tRNApAzF(GYT), AGX was decoded with tRNAPyl(XCT), and because there are only two thoroughly validated orthogonal tRNA/aaRS pairs, GXT was decoded with tRNASer(AYC). Remarkably, virtually all of the protein produced (96%) contained all three of the encoded amino acids. We note that the protein yield is only marginally reduced relative to the amount produced via the decoding of two unnatural codons and that it compares favorably to yields reported for the suppression of multiple stop codons7. However, this comparison is complicated by the fact that the third unnatural codon was decoded as a natural amino acid, as natural aaRSs are generally more active than ones that recognize ncAAs38.
We have shown previously that the SSO is capable of storing the dNaM-dTPT3 UBP in a wide variety of sequence contexts17, 18, and we have now demonstrated that it is also capable of transcribing a wide variety of sequence contexts into mRNA and tRNA and using nine of the resulting codon/anticodon pairs to mediate efficient protein synthesis at the ribosome. The demonstration that (at least) three of these codons and cognate anticodons are orthogonal has to our knowledge enabled, for the first time, a cell to decode 67 codons. The continued exploration of unnatural codon/anticodon pairs made available by dNaM-dTPT3 and the continued optimization of the UBP itself will undoubtedly make available additional codon/anticodon pairs that may be simultaneously used in the SSO, further increasing its potential to store and retrieve unnatural information. With this success, efforts now must focus on the development of additional orthogonal tRNA/aaRS pairs with which to decode this newly available information. This work is in progress and promises to enable the creation of SSOs that produce proteins, and possibly even themselves acquire forms and functions, that are outside the scope of those previously available to living organisms.
ONLINE METHODS
Materials
A complete list of oligonucleotides used in this study can be found in Supplementary Table 3. Natural ssDNA oligonucleotides and gBlocks were purchased from IDT (San Diego, CA). Genewiz (San Diego, CA) performed sequencing. All purification of DNA was carried out using Zymo Research silica column kits. All cloning enzymes and polymerases were purchased from New England Biolabs (Ipswich, MA). All bioconjugation reagents were purchased from Click Chemistry Tools (Scottsdale, AZ). Unnatural nucleosides (dNaM, dTPT3, NaM, TPT3, dMMO2Bio, d5SICS) were commercially synthesized (WuXi AppTec) and triphosphorylated (TriLink BioTechnologies LLC and MyChem LLC; >98% purity by NMR and HPLC) for Synthorx, Inc. (La Jolla, CA) and generously gifted for this study. All nucleoside phosphoramidites and ssDNA dNaM templates used in this study were also gifted by Synthorx, Inc (La Jolla, CA). All ssDNA dNaM templates were also gifted by Synthorx, Inc. (La Jolla, CA), with the exception of sfGFP200(AGX), which was synthesized in-house as described previously39.
Growth conditions
All bacterial experiments were carried out in 300 μl 2×YT (Fischer Scientific) media supplemented with potassium phosphate (50 mM pH 7). Growth was done in flat-bottomed 48-well plates (CELLSTAR, Greiner Bio-One) with shaking at 200 rpm at 37 °C (Infors HT Minitron). Antibiotics were used at the following concentrations: chloramphenicol (5 μg/ml), carbenicillin (100 μg/ml), and zeocin (50 μg/ml). Unnatural nucleoside triphosphates were used at the following concentrations (unless otherwise noted): dNaMTP (150 μM), dTPT3TP (10 μM), NaMTP (250 μM), TPT3TP (30 μM). UBP media is defined as the buffered 2×YT media above containing dNaMTP and dTPT3TP.
Plasmid construction
Large insertions (>100 bp; e.g. insertion of MjpAzFRS or tRNAs) were done by Gibson assembly40 of PCR amplicons or gBlocks. Amplicons were treated with DpnI over night at room temperature before assembly for 1.5 h at 50 °C. Deletions or small insertions (<50 bp; e.g. anticodon/codon mutagenesis, removal of restriction sites, or introduction golden gate destination sites) were constructed by introducing the desired change into PCR primer overhangs, designed to amplify the entire plasmid. Primers were phosphorylated using T4 PNK before PCR, and the resulting PCR amplicon was treated with DpnI overnight at room temperature and recircularized using T4 DNA ligase. After initial assembly/ligation, plasmids were transformed into electrocompetent XL-10 Gold cells and grown on selective LB Lennox agar (BP Difco). Plasmids were isolated from individual colonies and were verified by Sanger sequencing before use. All plasmids used in this study are listed in Supplementary Table 4.
PCR of UBP oligos
Double-stranded DNA inserts with the UBP-containing sequence were obtained from PCR (1× OneTaq Standard Buffer, 0.025 units/μl OneTaq, 0.2 mM dNTPs, 0.1 mM dTPT3TP, 0.1 mM dNaMTP, 1.2 mM MgSO4, 1× SYBR Green I, 1.0 μM primers, ~20 pM template; cycling conditions: 96 °C 30 s followed by <24 cycles of [96 °C 30 s, 54 °C 30 s, 68 °C 4 min, fluorescence read]) using chemically synthesized dNaM containing ssDNA oligonucleotides as template (Supplementary Table 3). Inserts for position sfGFP190 and sfGFP200 were combined by overlap extension PCR using forward primer for the sfGFP190 template and the reverse primer for the sfGFP200 template. Conditions were as described above, except that both templates were added to 1 nM. Amplifications were monitored and reactions were put on ice as the SYBR green trace plateaued. Products were analyzed via native PAGE (6% acrylamide:bisacrylamide 29:1; SYBR Gold stain in 1× TBE) to verify single amplicons, purified on a spin-column (Zymo Research), and quantified using Qubit dsDNA BR (ThermoFisher).
Golden Gate assembly of SSO expression vectors
UBP-containing inserts were incorporated into the pSYN entry vector framework (Supplementary Table 4) via Golden Gate assembly (1× Cutsmart buffer, 1 mM ATP, 6.67 units/μl T4 DNA ligase, 0.67 units/μl BsaI-HFv2, 20 ng/μl entry vector DNA; cycling conditions: 37 °C 10 min followed by 39 cycles of [37 °C 5 min, 16 °C 5 min, 22 °C 2 min] then 37 °C 20 min, 55 °C 15 min, 80 °C 30 min) with 3:1 molar ratio of each insert to entry vector. BsaI-HF was used for experiments in Fig. 1. Residual linear DNA and undigested entry vector was digested with first KpnI-HF (0.33 units/μl, 1 h at 37 °C) followed by T5 exonuclease (0.17 units/μl, 30 min at 37 °C). Product was purified on a spin-column (Zymo Research) and quantified using Qubit dsDNA HS (ThermoFisher).
Preparation of competent starter cells
Strain ML2 (BL21(DE3) lacZYA::PtNTT2(66–575) ΔrecA polB++) was transformed with the accessory pGEX plasmid (Supplementary Table 4) and plated on LB Lennox agar with chloramphenicol and carbenicillin. Single colonies were picked and verified for PtNTT2 activity by uptake of radioactive [α−32P]dATP as previously described18. Competent cells for UBP replication and translation was prepared by growth in 2×YT media at 37 °C with shaking at 250 rpm. in a baffled culture flask until the OD600 reached 0.25–0.30. The cultures were transferred to pre-chilled 50 mL Falcon tubes and gently shaking in an ice-water bath for 2 min. Cells were pelleted by centrifugation and washed in cold sterile water, pelleted and washed again, before finally being pelleted and suspended in 50 μl cold 10% glycerol per 10 mL culture. The cells were either used immediately (e.g. for non-clonal SSO experiments) or frozen at −80 °C for later use.
Non-clonal SSO experiments
Freshly prepared competent cells were electroporated (2.5 kV) with ~0.4 ng Golden Gate assembly product and immediately suspended in 950 μl 2×YT supplemented with potassium phosphate (50 mM pH 7), whereof 10 μl was diluted into 40 μl of UBP media containing 1.25× dNaMTP and dTPT3TP without zeocin. After recovering the cells for 1 h at 37 °C, 15 μl cells were suspended in 285 μl UBP media with zeocin and grown at 37 °C with shaking in a 48-well plate. Cultures were transferred to ice before reaching stationary phase, at OD600 ~1, and stored overnight for before being assayed for protein expression.
Clonal SSO experiments
Competent cells were electroporated with Golden Gate assembly product (1–20 ng) and recovered as for non-clonal population experiments. Plating was carried out by spreading 10 μl recovery culture (and dilutions thereof) onto an agar droplets (250 μl 2×YT with 2% agar and 50 mM potassium phosphate) containing chloramphenicol, carbenicillin, zeocin, dNaMTP, and dTPT3TP. Colonies approximately 0.5 mm in diameter were picked and suspended into UBP media (300 μl) after growth on the plate (12–20 h; 37 °C). Each culture was transferred to pre-chilled tubes on ice before reaching stationary phase, at OD600 ~1, and stored overnight before being assayed for protein expression. Each culture was prescreened for 1) UBP retention using the streptavidin-biotin shift assay (as described below) and 2) qualitative sfGFP expression by mixing the culture 1:4 with media already containing the components for expression (ribonucleoside triphosphates, ncAAs, IPTG, and anhydrotetracycline). Colonies were discarded if they did not produce any fluorescent signal when the appropriate ncAA was added after 2 h of incubation at 37 °C or overnight at room temperature (λex 485 nm, λem 525 nm; TECAN Infinite M200 PRO). Additionally, colonies with <80% UBP retention in sfGFP were discarded. If more than three colonies satisfied these criteria, then only the three with highest UBP retention were chosen to limit material expenses. The data shown to the right of the dashed line in Fig. 2a was obtained through a related project (unpublished data, Vivian T. Dien & Floyd E. Romesberg) and therefore slightly different methods were used. Instead of prescreening colonies as described above, expression was carried out on numerous colonies but protein analyses was only performed for cultures that showed fluorescence during expression.
Precloned SSO expression vectors
In the experiments shown in Fig. 2b, 3, and 4, plasmids from prescreened colonies were isolated (Zymo Research) to serve as starting plasmid for precloned transformation in order to ease colony prescreening. Plasmids were prescreened (as described above) for qualitative fluorescence from sfGFP expression with the appropriate ncAA(s). Colonies for the data in fig. 2b were instead prescreened with and without rNaMTP and rTPT3TP in the presence of AzK to qualitative produce a dark and a fluorescent signal, respectively. All precloned plasmids were prescreened for UBP retention in sfGFP (>80%). Furthermore, all plasmids were sequenced to verify integrity of the natural sequence in the plasmid. This was done by Sanger sequencing of a PCR amplicon from standard OneTaq PCR (New England Biolabs) without unnatural nucleoside triphosphates, which force dX to dN mutations. Silent mutations were allowed in protein coding sequences.
SSO protein expression
Cultures were refreshed in UBP media to OD600 0.10–0.15 and incubated at 37 °C with shaking until OD600 0.5–0.8 at which time ribonucleoside triphosphates were added to 250 μM NaMTP and 30 μM TPT3TP, alongside ncAAs at 5 mM pAzF, 20 mM AzK, or 10 mM PrK. Only 10 mM AzK was used in experiments in Fig. 2a (right), 3 and 4 or controls thereof. After 20 min of further incubation, preinduction was initiated by adding IPTG (1 mM) and the cultures were incubated for an additional hour. Finally sfGFP expression was induced by derepression of tetO by adding anhydrotetracycline (100 ng/μl). OD600 and GFP fluorescence was monitored (every 30 min) using Perkin Elmer Envision 2103 Multilabel Reader (OD: 590/20 nm filter; sfGFP: λex 485/14 nm, λem 535/25 nm). After 3 h of expression, cultures were pelleted and stored at −80 °C for later analyses.
Streptavidin-biotin shift assay for UBP retention
UBP retention in plasmid DNA was determined by PCR amplification using unnatural nucleoside triphosphate d5SICSTP as well as the biotinylated dNaM analog dMMO2BioTP (as previously described17). Plasmids from SSOs were isolated via standard miniprep, resulting in a mixture of SSO expression plasmids (pSYN) and accessory plasmids (pGEX). A total of 2 ng of the plasmid mixture was used as a template in a 15 μl PCR reaction (1× OneTaq Standard Buffer, 0.018 units/μl OneTaq, 0.007 units/μl DeepVent, 0.4 mM dNTPs, 0.1 mM d5SICSTP, 0.1 mM dMMO2BioTP, 2.2 mM MgSO4, 1× SYBR Green I, 1.0 μM primers; cycling conditions: 96 °C 2:00 min followed by <24 cycles of [96 °C 30 s, 50 °C 10 s, 68 °C 4 min, fluorescence read, 68 °C 10 s]). Individual samples were removed during the last step of each cycle as the SYBR trace showed the amplification to plateau. The resulting biotinylated amplicons were supplemented with 10 μg streptavidin (Promega) per 1.5–2.0 μl crude PCR reaction. The streptavidin bound fraction was visualized as a shift by 6% native-PAGE and both shifted and unshifted bands were quantified by ImageStudioLite or Fiji to yield the relative raw percentage of shift. By normalizing the raw shift to a control shift, generated by templating the PCR reaction with the chemically synthesized oligonucleotide, we assessed the overall UBP retention. Normalization was not possible for tRNApAzF or tRNASer as faithful amplification was only possible with primers annealing outside the Golden Gate insert and thus did not anneal to the corresponding control oligonucleotide.
Protein purification
Cell pellets from protein expression experiments (200 μl) were lysed using BugBuster (100 μl; EMD Millipore; 15 min; room temperature; 220 rpm). Cell lysates were then diluted in Buffer W (50 mM HEPES pH 8, 150 mM NaCl, 1 mM EDTA) to a final volume equal to 500 μl minus the volume of affinity beads used. Magnetic Strep-Tactin XT beads (5% (v/v) suspension of MagStrep “type3” XT beads, IBA Lifesciences) were used at 20 μl for routine purification and 100 μl for estimation of total expression yield. Protein was bound to beads (30 min; 4 °C; gentle rotation) before beads were pulled down and washed with Buffer W (2×500 μl). In protein purification for HRMS ESI-TOF analysis or for the data in fig. 2a (right), Buffer W2 was used (50 mM HEPES pH 8, 1 mM EDTA) instead. Finally protein was eluted using 25 μl Buffer BXT (50 mM HEPES pH 8, 150 mM NaCl, 1 mM EDTA, 50 mM d-Biotin) for 10 min at room temperature with occasional vortexing. Protein was eluted with buffer BXT2 (50 mM HEPES pH 8, 1 mM EDTA, 50 mM d-Biotin) for HRMS ESI-TOF analysis. Qubit Protein Assay Kit (ThermoFisher) was used for quantification. Protein yields were determined by multiplying elution volume by the quantified concentration, which was then normalized to the volume of culture. When protein yield were normalized to OD600, the last read before harvesting cells was used. The numbers are not normalized to the fidelity observed in HRMS ESI-TOF.
Western blotting of TAMRA conjugated sfGFP
SPAAC was carried out by incubation of 33 ng/μl pure protein with 0.1 mM TAMRA-PEG4-DBCO (#A131, Click Chemistry Tools) over night at room temperature in darkness. The reactions were mixed 2:1 with SDS-PAGE loading dye (250 mM Tris-HCl pH 6, 30% glycerol, 5% β-mercaptoethanol, 0.02% bromophenol blue) and denatured for 5 min at 95 °C. For SDS-PAGE gels, 5% acrylamide stacking gels and 15% acrylamide resolution gel were used when analyzing the position sfGFP151 and 17% for when analyzing sfGFP190,200 (resolution gel: 15% or 17% acrylamide:bisacrylamide 29:1, 0.1% (w/v) APS, 0.04% TEMED, 0.375 M Tris-HCl pH 8.8, 0.1% (w/v) SDS; stacking: 5% acrylamide:bisacrylamide 29:1, 0.1% (w/v) APS, 0.1% TEMED, 0.125 M Tris-HCl pH 6.8, 0.1% (w/v) SDS). Electrophoresis was carried out for 15 min at 50 V before running at ~5 h at 120 V for 15% gels and ~6.5 h for 17% gels. Running buffer (25 mM Tris base, 200 mM glycine, 0.1% (w/v) SDS) was changed every 2 h. The resulting gel was blotted onto PVDF (EMD Millipore 0.45 μm PVDF-FL) using wet transfer in cold transfer buffer (20% (v/v) MeOH, 50 mM Tris base, 400 mM glycine, 0.0373% (w/v) SDS) for 1 h at 90 V. The membrane was blocked using 5% non-fat milk solution in PBS-T (PBS pH 7.4, 0.01% (v/v) Tween-20) overnight at 4 °C with gentle agitation. Primary antibodies (rabbit α-Nterm-GFP Sigma Aldrich #G1544) were applied in PBS-T (1:3,000) for 1 h (room temperature; gentle agitation). The blot was washed in PBS-T (5 min) before secondary antibodies (goat α-rabbit-Alexa Fluor 647-conjugated antibody, ThermoFisher #A32733) were applied in PBS-T (1:20,000) for 45 min (room temperature; gentle agitation). The blot was washed with PBS-T before (3×5 min) imaging using a Typhoon 9410 laser scanner at 50–100 μm resolution, scanning first for AlexaFluor 647 (λex 633 nm; λem 670/30 nm; PMT 500 V) and then TAMRA (λex 532 nm; λem 580/30 nm; PMT 400 V). Bands were quantified using ImageStudioLite. All scans of full PVDF membranes are supplied in Supplementary Figs. 9, 10, and 11.
Dual bioconjugation of PrK-pAzF labeled protein
Cell pellets from 1 mL of culture were lyzed using BugBuster (100 μl; EMD Millipore; 15 min at room temperature; 220 rpm). The lysate was diluted in Buffer W (600 μl) and Magnetic Strep-Tactin XT beads were added (200 μl) and allowed to bind (30 min; 4 °C; gentle rotation). The beads were pulled down using a magnet and washed with cold Buffer W (2×1000 μl) before being suspended in Buffer W (200 μl). SPAAC was carried out using half of this suspension with TAMRA-PEG4-DBCO (0.5 mM) for 12–16 h (room temperature; gentle rotation). The beads were washed with EDTA-free Buffer W (2× 500 μl; HEPES 50 mM pH 7.4, 150 mM NaCl) before being suspended in EDTA-free Buffer W (100 μl). CuAAC was carried out (1.5 h; room temperature; gentle rotation) using half of this suspension with TAMRA-PEG4-azide (0.2 mM; #AZ109, Click Chemistry Tools) as well as copper(II) sulphate (0.5 mM), Tris(benzyltriazolylmethyl)amine (2 mM; THPTA), and sodium ascorbate (15 mM). Beads were washed with Buffer W (2×500 μl) before elution with buffer BXT (10 min; room temperature; occasional vortexing).
Intact protein high-resolution mass spectrometry
Purified protein (5 μg) was desalted into HPLC grade water (4×500 μl) by four cycles of centrifugation through 10K Amicon Ultra Centrifugal filters (EMD Millipore) at 14,000g (3×10 min and then 1×18 min). After recovering the protein, 6 μl protein was injected into a Waters I-Class LC connected to a Waters G2-XS TOF. Flow conditions were 0.4 mL/min of 50:50 water:acetonitrile plus 0.1% formic acid. Ionization was done by ESI+ and data was collected for m/z 500–2000. A spectral combine was performed over the main portion of the mass peak and the combined spectrum was deconvoluted using Waters MaxEnt1. Analysis was carried out by automated peak integration as well as manual peak identification (Supplementary Figs. 6 and 8). Fidelity was calculated as the integral of expected mass relative to integrals of all masses identified to be either product or impurity without taking technical impurities into consideration (e.g. salt adducts, arginine oxidation). The method has previously been described and validated to be quantitative39.
Statistics and reproducibility
All data was collected from at least three biologically indenpendet cultures (i.e. biological replicates). In experiments with non-clonal SSOs, each replicate is derived from a different batch of competent SSO starter cells. In experiments with clonal SSOs, each replicate is derived from a different individual SSO colony grown on agar. The initial codon screens (Fig. 1d and Supplementary Fig. 1) were not replicated in full due to material constraints, owing to the amount of unnatural nucleoside triphosphates required for UBP media. Select codon/anticodon pairs from these experiments were replicated at least once and showed similar results. The data describing codon function in clonal SSOs (Fig. 2 and Supplementary Fig. 3) was replicated at least once but, in some cases, with slight variations in methods for preparation of SSOs or alternative expression schemes. Data for double or triple codons expression (Fig. 3b–f and Fig. 4b) were replicated in full three times and showed identical results. However, HRMS ESI-TOF analyses for these experiments were only replicated once. Technical replicates of western blots were performed at least three times for the initial codon screen (Supplementary Fig. 2a) and double codon experiments (Fig. 3d, f). All SSO cultures that only differ by additives to the media (e.g. ncAAs or unnatural nucleoside triphosphates) are derived from the same replicate. No statistical tests were perfomed.
Data availability
Annotated plasmid sequences from this study are available via Genbank (accession numbers MN882182–MN882190) as detailed in Supplementary Table 4. All data supporting the findings of this study is available within the paper and the supplementary information or from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (GM118178 to F.E.R, GM123735 to Y.Z., and GM128376 to R.J.K.). E.C.F. was supported by a Boehringer Ingelheim Fonds PhD Fellowship. K.H. was supported by a JSPS Overseas Research Fellowship. A.W.F. and M.P.L. were supported by a National Science Foundation Graduate Research Fellowship (NSF/DGE-1346837). R.K. was supported by NASA Exobiology (NNX14AP59G).
Footnotes
Competing interests
The authors declare the following competing financial interests: a patent application has been filed based on the use of UBPs in SSOs (PCT/US2018/041509).
Supplementary information is available for this paper.
References
- 1.Leader B, Baca QJ & Golan DE Protein therapeutics: a summary and pharmacological classification. Nat. Rev. Drug Discov 7, 21–39 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Wang L, Brock A, Herberich B & Schultz PG Expanding the genetic code of Escherichia coli. Science 292, 498–500 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Blight SK et al. Direct charging of tRNA(CUA) with pyrrolysine in vitro and in vivo. Nature 431, 333–335 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Liu CC & Schultz PG Adding new chemistries to the genetic code. Annu. Rev. Biochem 79, 413–444 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Neumann H, Wang K, Davis L, Garcia-Alai M & Chin JW Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature 464, 441–444 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Chatterjee A, Sun SB, Furman JL, Xiao H & Schultz PG A versatile platform for single- and multiple-unnatural amino acid mutagenesis in Escherichia coli. Biochemistry 52, 1828–1837 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Italia JS et al. Mutually orthogonal nonsense-suppression systems and conjugation chemistries for precise protein labeling at up to three distinct sites. J. Am. Chem. Soc 141, 6204–6212 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lajoie MJ et al. Genomically recoded organisms expand biological functions. Science 342, 357–360 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aerni HR, Shifman MA, Rogulina S, O’Donoghue P & Rinehart J Revealing the amino acid composition of proteins within an expanded genetic code. Nucleic Acids Res. 43, e8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamashima K, Kimoto M & Hirao I Creation of unnatural base pairs for genetic alphabet expansion toward synthetic xenobiology. Curr. Opin. Chem. Biol 46, 108–114 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Biondi E & Benner SA Artificially expanded genetic information systems for new aptamer technologies. Biomedicines 6, 53 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li L et al. Natural-like replication of an unnatural base pair for the expansion of the genetic alphabet and biotechnology applications. J. Am. Chem. Soc 136, 826–829 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morris SE, Feldman AW & Romesberg FE Synthetic biology parts for the storage of increased genetic information in cells. ACS Synth. Biol 6, 1834–1840 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seo YJ, Hwang GT, Ordoukhanian P & Romesberg FE Optimization of an unnatural base pair toward natural-like replication. J. Am. Chem. Soc 131, 3246–3252 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ast M et al. Diatom plastids depend on nucleotide import from the cytosol. Proceedings of the National Academy of Sciences of the United States of America 106, 3621–3626 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malyshev DA et al. A semi-synthetic organism with an expanded genetic alphabet. Nature 509, 385–388 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ledbetter MP, Karadeema RJ & Romesberg FE Reprograming the replisome of a semisynthetic organism for the expansion of the genetic alphabet. J. Am. Chem. Soc 140, 758–765 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y et al. A semisynthetic organism engineered for the stable expansion of the genetic alphabet. Proc. Natl. Acad. Sci. USA 114, 1317–1322 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y et al. A semi-synthetic organism that stores and retrieves increased genetic information. Nature 551, 644–647 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pedelacq JD, Cabantous S, Tran T, Terwilliger TC & Waldo GS Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol 24, 79–88 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Nguyen DP et al. Genetic encoding and labeling of aliphatic azides and alkynes in recombinant proteins via a pyrrolysyl-tRNA Synthetase/tRNA(CUA) pair and click chemistry. J. Am. Chem. Soc 131, 8720–8721 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Bryson DI et al. Continuous directed evolution of aminoacyl-tRNA synthetases. Nat. Chem. Biol 13, 1253–1260 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dien VT et al. Progress toward a semi-synthetic organism with an unrestricted expanded genetic alphabet. J. Am. Chem. Soc 140, 16115–16123 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chin JW et al. Addition of p-azido-L-phenylalanine to the genetic code of Escherichia coli. J. Am. Chem. Soc 124, 9026–9027 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Shimizu M, Asahara H, Tamura K, Hasegawa T & Himeno H The role of anticodon bases and the discriminator nucleotide in the recognition of some E. coli tRNAs by their aminoacyl-tRNA synthetases. J. Mol. Evol 35, 436–443 (1992). [DOI] [PubMed] [Google Scholar]
- 26.Fredens J et al. Total synthesis of Escherichia coli with a recoded genome. Nature 569, 514–518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ostrov N et al. Design, synthesis, and testing toward a 57-codon genome. Science 353, 819–822 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Nissen P, Ippolito JA, Ban N, Moore PB & Steitz TA RNA tertiary interactions in the large ribosomal subunit: the A-minor motif. Proc. Natl. Acad. Sci, USA 98, 4899–4903 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramakrishnan V Ribosome structure and the mechanism of translation. Cell 108, 557–572 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Betz K et al. Structural insights into DNA replication without hydrogen bonds. J. Am. Chem. Soc 135, 18637–18643 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Betz K et al. KlenTaq polymerase replicates unnatural base pairs by inducing a Watson-Crick geometry. Nat. Chem. Biol 8, 612–614 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirao I et al. An unnatural base pair for incorporating amino acid analogs into proteins. Nat. Biotechnol 20, 177–182 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Bain JD, Switzer C, Chamberlin AR & Benner SA Ribosome-mediated incorporation of a non-standard amino acid into a peptide through expansion of the genetic code. Nature 356, 537–539 (1992). [DOI] [PubMed] [Google Scholar]
- 34.Ogle JM et al. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science 292, 897–902 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Hoernes TP et al. Translation of non-standard codon nucleotides reveals minimal requirements for codon-anticodon interactions. Nat. Commun 9, 4865 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feldman AW & Romesberg FE In vivo structure-activity relationships and optimization of an unnatural base pair for replication in a semi-synthetic organism. J. Am. Chem. Soc 139, 11427–11433 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwark DG, Schmitt MA & Fisk JD Dissecting the contribution of release factor interactions to amber stop codon reassignment efficiencies of the Methanocaldococcus jannaschii orthogonal pair. Genes (Basel) 9, E546 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Donoghue P, Ling J, Wang YS & Soll D Upgrading protein synthesis for synthetic biology. Nat. Chem. Biol 9, 594–598 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feldman AW et al. Optimization of replication, transcription, and translation in a semi-synthetic organism. J. Am. Chem. Soc 141, 10644–10653 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gibson DG Enzymatic assembly of overlapping DNA fragments. Methods Enzymol. 498, 349–361 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Annotated plasmid sequences from this study are available via Genbank (accession numbers MN882182–MN882190) as detailed in Supplementary Table 4. All data supporting the findings of this study is available within the paper and the supplementary information or from the corresponding author upon reasonable request.