

Abstract
From protein interactions to signal transduction, from metabolism to the nervous system: Virtually all processes in health and disease rely on the careful orchestration of a large number of diverse individual components ranging from molecules to cells and entire organs. Networks provide a powerful framework for describing and understanding these complex systems in a wholistic fashion. They offer a unique combination of a highly intuitive, qualitative description, and a plethora of analytical, quantitative tools. Here we provide a brief introduction to the emerging field of network medicine. After an overview of the core concepts for connecting network characteristics to biological functions, we review commonly used networks, ranging from the molecular interaction networks that form the basis of all biological processes in the cell to the global transportation networks that govern the spread of global epidemics. Lastly, we highlight current conceptual and practical challenges.
Keywords: Complex network, Disease module, Functional networks, Physical networks, Protein-protein interaction (PPI)
Glossary
- *ome
Suffix that is commonly used to emphasize that a collection of individual elements is considered as a whole. The entirety of all genes, for example, forms the genome. Compare with interactome and exposome.
- Complex network
Complex networks provide holistic models of systems that consist of many interacting elements. Each individual element is represented as a node (also called ‘vertex’) and each interaction between two elements as a link (or ‘edge’). The connection patterns within complex networks are typically neither completely regular, nor completely random.
- Disease module
Genes associated with a particular disease are not scattered randomly within molecular networks, but aggregate in certain neighborhoods or ‘disease modules.’
- Exposome
Organisms are constantly exposed to a multitude of biological and chemical factors through their environment, collectively referred to as ‘exposome.’ The dynamic combination of the internal biological state (homeostatis) and external influx (exposome) results in health or disease.
- Functional networks
Networks in which links represent indirect relationships, for example correlated activity patterns in functional brain region networks or shared biological processes in gene similarity networks. Compare with physical networks.
- Herd effect
Phenomenon observed in epidemiology that the immunization of one part of a population also decreases the risk of infection for the other part that is not immune. The herd effect is important to protect sub-populations that cannot be treated (e.g., immunocompromised people) and allows for the overall success of immunization campaigns even under partial compliance.
- Interactome
In analogy to the genome, the interactome represents the complex network of all molecular interactions within a biological system. More specifically, the term often refers to physical protein-protein interactions within the cell. Over the last two decades the interactomes of several organisms have been mapped out systematically, ranging from model organisms to human.
- P4 medicine
Predictive, preventive, personalized and participative medicine. This approach aims to improve healthcare by progressively reducing the need for one-size-fits-all palliative treatments by leveraging modern therapeutic solutions and personal patient data such as genome or microbiome sequencing.
- Physical networks
Networks in which links represent physical interactions, such as physical binding between proteins in interactome networks, or synapses in neural networks. Compare with functional networks.
- Protein-Protein Interaction (PPI)
Cellular processes rely on the coordinated interaction between different proteins. Several experimental methods are available to detect such interactions, for example yeast two-hybrid approaches or affinity purification combined with mass spectrometry.
Introduction
In the 19th century, brilliant pioneers the like of Mendel, Darwin and Semmelweis required little more than meticulous observation of their surroundings for developing their theories on inheritance, evolution and infectiology that would become the foundations of modern biology and medicine. Since then, a range of sophisticated technologies have been developed that now allow us to observe biological systems at molecular resolution. However, our rapidly growing knowledge at the molecular level also revealed the fundamental limitations of traditional reductionist approaches that aim to understand complex biological systems by dissecting their individual elements (Greene and Loscalzo, 2017; Stéphanou et al., 2018). It is becoming increasingly clear that many system-wide phenomena cannot be understood in this fashion, and that often the ‘whole is more than the sum of its parts.’ It is thus essential to systematically study not only the isolated elements of these systems, but also their interactions. These interactions are key to understand the emergence of novel properties and behaviors, in particular when moving across different scales, i.e., from molecules to cells, tissues, organs and organisms or entire populations (Fig. 1 ). Complex networks provide a natural framework for systematically investigating the various relationships between the constituents of biological systems within and between scales.
Fig. 1.
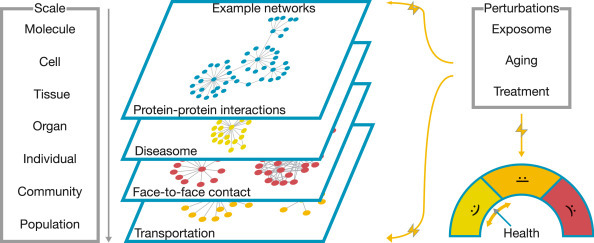
The processes involved in human health and disease range from the molecular to the population scale. Networks provide a unifying framework for describing and investigating the complex interactions that occur both within and across scales: They can be used to describe the molecular interactions forming the basis of all biological processes within the cell, as well as the social interactions that form the basis for the spread of infectious diseases. Diseases can be conceptualized as perturbations of these intricate systems. These perturbations may be internal, for example genetic mutations, or external, for example environmental exposures.
In the following we aim to provide an overview of how tools and concepts from network theory may help address important fundamental and practical challenges in biology and medicine. We start by reviewing key relationships between structural network properties and functional characteristics of the represented biological system. We then introduce frequently used networks, from the molecular level of protein-protein interactions within the cell, all the way to the level of transportation networks that span the globe. We conclude by highlighting a few future challenges in this highly active and dynamic research field.
From Network Structure to Biological Function
Network theory provides a versatile and general toolbox for investigating complex systems composed of interacting elements. In the most generic case, each element of the system is represented by a node and each interaction between a pair of nodes by a link (Fig. 2 ). In mathematical terms, the collection of all nodes and links is also called a ‘graph.’ This simple definition can be extended, for example by adding weighted or directed links, by including time-dependence or different layers of connectivity between the nodes, resulting in so-called weighted, directed, temporal or multi-layer networks, respectively.
Fig. 2.

(A) Illustration of basic network characteristics for a protein-chemical interaction network (data from the BioGRID database (Oughtred et al., 2019), obtained and laid out using the NDEx platform (Pratt et al., 2015)). Green and blue nodes represent chemicals and proteins, respectively, that interact with each other. Several network characteristics are highlighted: network motifs are small recurrent connection patterns; hubs are nodes that have a large number of neighbors; densely interconnected groups of nodes are called ‘communities’ (also ‘modules’ or ‘clusters’); high centrality indicates that the respective nodes/edges are of structural importance to the network. (B) Overview of different biological networks and their basic characteristics (data from the BIOSNAP repository (Leskovec and Sosič, 2016; Zitnik et al., 2018)). The average clustering coefficient is a measure for the overall local density of a network; the diameter of a network is given by the greatest distance between any pair of nodes.
Networks can be characterized at different levels, ranging from the level of individual nodes (e.g., their number of connections, or their centrality within the network) to the level of groups of nodes (e.g., their connection density), to the global level of the entire network (e.g., the distribution of the number of connections per node across all nodes in the network). The finding that these properties can be associated with important biological characteristics makes network theory a valuable tool in biology and medicine. For example, proteins that are located at a highly central position within molecular interaction networks have been shown to perform important roles in the cell, whereas more peripheral proteins are often less essential (Piñero et al., 2016; Costanzo et al., 2019). Densely interconnected groups of nodes correspond to functionally closely related groups of proteins (Barabási et al., 2011). Similarly, genes that are associated to the same disease tend to aggregate in specific disease modules within molecular networks (Menche et al., 2015; Ghiassian et al., 2015).
Random Networks as Reference Models
In order to assess the magnitude and statistical significance of an observed network characteristic, suitable random controls are needed. Network theory provides a wide range of well-studied random graph models that can be used as reference (Albert and Barabási, 2002; Piñero et al., 2016). The most basic model is the classic random graph, in which a given number of links is distributed randomly among a given number of nodes (Erdős and Rényi, 1960). In contrast to many real world networks, including most molecular networks, random graphs do not contain highly connected nodes, so-called hubs, underlying their importance as they could not have emerged by chance alone. More advanced reference models can be introduced by keeping additional properties of the original network constant during the randomization procedure. Rewiring algorithms, for example, keep the number of links per node fixed and have been used to uncover basic design principles of gene regulatory networks, such as the tendency of highly connected hubs to avoid each other (Maslov and Sneppen, 2002) or the discovery of network motifs, i.e., recurrent connectivity patterns among small groups of nodes (Milo et al., 2002).
Also dynamic aspects of networks, such as their growth over time, can be assessed through random models. An important class of networks that are characterized by the presence of hubs are so-called scale-free networks (Albert and Barabási, 2002). These networks emerge by iteratively adding new nodes to the network, such that they have a tendency to form links with already highly connected nodes. The basic model can be extended by adding a latent fitness to each node (Bianconi and Barabási, 2001) to investigate the role of evolutionary processes and positive selection of genes with high fitness in shaping the structure of molecular networks.
Molecular Interaction Networks
The first layer of information transfer and transformation from genotype to phenotype is mediated by molecular networks within the cell. In analogy to the genome representing the blueprint for all molecular components, the collection of all their interactions is referred to as the interactome. The interactome thus represents the blueprint for the collective functions that emerge from interactions between individual components. Most commonly, the term interactome is used specifically for protein-protein interaction (PPI) networks. Over the last two decades, genome-scale PPI networks have become available for a variety of species (Alanis-Lobato et al., 2017; Oughtred et al., 2019). PPIs can be mapped out systematically using yeast two-hybrid approaches (Rolland et al., 2014) or mass spectrometry based methods (Huttlin et al., 2017). The most comprehensive PPI networks also incorporate results compiled from numerous small-scale experiments from the literature (Oughtred et al., 2019) or computational predictions (Kovács et al., 2019).
PPIs are not the only molecular interactions within the cell that are biologically relevant and can be experimentally assessed. Other important molecular networks include metabolic networks and signaling cascades (Choudhary and Mann, 2010; Fabregat et al., 2016). Moreover, links may also represent indirect relations, for example in gene regulatory networks, where one gene can act on another via transcribed RNA or a translated protein, resulting from the binding of transcription factors and regulatory elements to the genetic material, which can be assessed experimentally through chromosome conformation capture techniques (Babaei et al., 2015).
Over the last two decades, numerous relationships have been uncovered between the structural characteristics of molecular networks and the function of the systems that they represent. In PPI networks, for example, connection patterns such as the number of interaction partners, network distance between proteins or densely interconnected network neighborhoods are directly related to biological functions in both healthy and disease states (Barabási et al., 2011; Caldera et al., 2017; Meyer et al., 2018). In a network context, disease states can often be identified with localized perturbations of the underlying molecular network. Such perturbations may be internal, for example genetic mutations associated with severe hereditary diseases (Köhler et al., 2008), or external, such as chemical or other environmental exposures (Kalia et al., 2019). Collectively, the set of all such external factors is called the exposome. Given the broad nature of this term, it is unclear whether a comprehensive mapping of the exposome and its impact on the interactome is at all achievable. First attempts in this direction focus on specific exposures, for example the impact of toxicants on metabolite networks (Kalia et al., 2019; Veneman et al., 2017). These approaches enable on the one hand the inference of which chemicals a system has encountered, and offer on the other hand an opportunity to elucidate the response mechanisms following a particular exposure.
Beyond Physical Interactions
The links in the molecular networks discussed above represent physical interactions that can be directly measured. In addition to these physical networks, we can also construct functional networks, where links represent more indirect relationships or similarities. The most commonly used functional networks are co-expression networks, where two genes are linked if their expression levels were found to be correlated across different experimental conditions (Saha et al., 2017). Other important examples are genetic interaction networks, where a link between two genes indicates that the phenotype of their combined knock-out deviates from the expectation based on the individual knockouts (Costanzo et al., 2016; Rauscher et al., 2018), drug-drug interaction networks, in which links connect non-additive drugs (Caldera et al., 2019) or chemical networks, in which compounds are linked based on structural similarity (Lo and Torres, 2016).
Functional networks may also contain various types of nodes, connecting for example genes and drugs: A genome-wide screen in Saccharomyces cerevisiae has recently been used to map out the interactions between 1377 chemical compounds and 177 genes (Piotrowski et al., 2017). Systematically exploring pairwise combinations of cellular perturbations has great potential for functionally annotating individual components, such as genes, drugs or environmental factors, as well as for identifying the involved molecular pathways and, more generally, for elucidating the fundamental rules that underlie the cellular response to combinations of perturbations.
Similarity networks were further used to study individual exposomes, by connecting co-occurring species or chemicals, revealing temporal and environmental patterns such as compounds that were released simultaneously during rainy days (Jiang et al., 2018). This exemplifies the potential to investigate different aspects of a biological concept (here the exposome) through complementary network approaches.
From Molecules to Organisms
While biological processes span a wide range from molecules to cells, tissues, organs and whole organisms, the networks at the molecular level are the most studied and best understood at this point. This reflects their importance as the primary interface between genotype and phenotype, but also the fact that they are more easily accessible experimentally compared to other relevant networks.
At the level of cellular organization, the neural networks that constitute the nervous system have probably received most attention (Bullmore and Sporns, 2009). Considerable efforts are made to systematically map out neural networks, in humans, as well as in model organisms. Similar to the different types of molecular networks introduced above, neural networks may also either represent direct cellular networks, where nerve cells are connected through synapses, or functional networks, in which regions of the brain are linked if they show correlated patterns of activity. The first complete direct neural network was resolved as early as 1986 for the worm Caenorhabditis elegans (White et al., 1986). For higher organisms, only partial maps are available to date, for example in mice (Briggman et al., 2011; Bock et al., 2011), but also in human (Glasser et al., 2016), if only at a very coarse grained level. Mapping out the complete human ‘connectome’ of all our brain cells will likely remain out of reach for many years due to its staggering size (Sporns, 2013).
A similarly complex and important biological system is the immune system, whose primary objective is to maintain the normal function of an organism under constant threat by internal and external challenges, ranging from tumor cells to viral infections. Given the diversity of participating organs, cell types and molecules, it has been proposed to conceptualize the immune system as a multi-layered network (Bergthaler and Menche, 2017; Rieckmann et al., 2017; Kveler et al., 2018). The nodes in this network represent cells, links represent communication through signaling molecules, such as cell-surface receptors or secreted molecules. Different layers may represent different contexts, such as organs, tissues or activation status.
Global Networks in Epidemiology
An effective response to a viral or bacterial infection is not only critical for individual organism, but may also be seen in the much larger, potentially world-wide, context of epidemics. To accurately model the spread of a contagious disease, we must understand both the social networks of personal interactions, as well as the local and global transportation networks along which people travel (Pastor-Satorras et al., 2015). Interestingly, it was shown that not only infectious diseases are transmitted across networks of social interactions, but also other sociological and health-related conditions, such as smoking behavior (Christakis and Fowler, 2008), weight gain (Christakis and Fowler, 2007) or happiness (Fowler and Christakis, 2009).
Mathematical models have a long history in epidemiology and date back to the early 20th century (Kermack and McKendrick, 1927). Classical models divide a population into three compartments, in which people are either susceptible to an infection (S), currently infected (I), or recovered (i.e., immunized) or otherwise removed (R) from the susceptible pool. The temporal dynamics of these SIR models can be described by differential equations. Early models typically assumed ‘uniform mixing,’ i.e., an equal probability for any infected individual to contaminate any susceptible individual. More recently, these models have been significantly improved by considering the relevant social and transportation networks that underly the disease spreading process (Wang et al., 2017). For instance, the structure of the face-to-face contact network has a profound impact on how fast and how far a contagious disease may spread among a population (Pastor-Satorras et al., 2015). Likewise, the efficiency of different immunization strategies can only be fully understood when taking these networks into account.
Smallpox is currently the only human infectious disease that was successfully eradicated through immunization. There are two key aspects for the success of an immunization campaign: First, the effective access to immunization, which includes the availability of a vaccine, but also the individual willingness to get vaccinated. The latter may decline along with the disease prevalence, even in countries where immunization is compulsory. It has been shown that a better understanding of the herd effect improves the adherence to such programs (Brockmann, 2017; Betsch et al., 2017). Second, the impact that an immunization of a certain subpopulation has on the spreading of the disease, i.e., how it reduces the contagion rate in total, as well as within smaller subcommunities. This aspect can be studied from a network theory point of view. The structure of the social face-to-face contact network determines whether bottlenecks (such as high centrality nodes or links) could prevent a disease outbreak and whether some communities are more at risk than others. This enables the design of more efficient quarantine strategies with maximal impact on the social network connectivity. In the past, epidemiological modeling was essential for example in handling the avian influenza outbreak in 2005 (Longini et al., 2005). More recently, great efforts have been made to profile and constrain the spread of the 2019-nCov virus, including charting the phylogeny of viral samples (Hadfield et al., 2018) and mapping the spreading risk based on global transportation networks, which allow for predicting disease spread much more accurately than maps based on geographic distance (Brockmann and Helbing, 2013).
Summary and Outlook
Networks provide a powerful framework for investigating biological systems ranging from the molecular to the global scale. A key factor for the success of network theory in biomedical applications is that many structural network characteristics can be related to functional properties of the respective biological system. In molecular networks, for example, densely connected node communities often correspond to proteins involved in a particular cellular process. Likewise, disease associated processes can be identified with specific connectivity patterns between groups of perturbed nodes.
An important open question in this context is how exactly different network perturbations influence each other. For example, it has been found that a network overlap between a drug-induced perturbation and a disease associated perturbation may either indicate an effective treatment of the respective disease, but also the opposite, namely that the disease may be a side effect of the treatment (Cheng et al., 2019; Guney et al., 2016). This highlights an important methodological and conceptual limitation of current network approaches: We are still lacking a systematic understanding of the combined effect of independent perturbations, in particular when considering complex phenotypes. To date, most large-scale experimental efforts for elucidating the combined effect of perturbations relied on relatively simple, one-dimensional readouts, such as growth assays, for example in the characterization of genetic interactions (Kuzmin et al., 2018) and drug-gene interactions in yeast (Piotrowski et al., 2017). More recently, more informative readouts have been employed as well, such as high-content imaging or next-generation sequencing, which allow for a much more detailed assessment of the interactions between different perturbations. Using these high-dimensional readouts it is possible to identify different types of interactions between perturbations (positive, negative), as well as their direction. Furthermore, high-dimensional readouts allow for the identification of interactions that lead to the emergence of entirely new phenotypes. The first studies aiming to map out such high-resolution ‘perturbome’ networks were based on morphological changes induced by combinations of genetic perturbations in a model organism (Fischer et al., 2015) and combinations of drug perturbations in cell lines (Caldera et al., 2019), respectively.
Another major focus of recent network-based biomedical research is the integration of the diverse data describing different levels of biological organization. While combinations of different ‘omics’ data, e.g., genomics, transcriptomics, proteomics, metabolomics and microbiome data, are becoming more and more common in basic research, their translation into clinical applications is still scare (Karczewski and Snyder, 2018), despite their potential for applications in P4 medicine being widely recognized (Apweiler et al., 2018). Network approaches can offer valuable contributions to solving current technical and conceptual challenges in integrating multi-omics and multi-scale data (McGillivray et al., 2018). Indeed, concrete translational impact is the ultimate ambition of network biology and network medicine.
See also
Metabolic Systems Structure and Function in Complex Biological Networks
Biographies

Loan Vulliard studied bioinformatics in Lyon (France), Daejon (South Korea), Copenhagen (Denmark), and Montpellier (France) and graduated in 2017 with a Master׳s degree in bioscience from INSA Lyon, France. Interested in systems biology and computational methods for analyzing large biological datasets, he then joined the group of Dr. Jörg Menche at the CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences in Vienna (Austria) as a doctoral candidate. His work involves planning and interpreting large-scale high-content screening experiments using network science. His main focus are drug-gene interactions and how intrinsic and extrinsic factors can combine and affect cellular morphology.

Dr. Jörg Menche is a theoretical and computational physicist by training. During his PhD at the Max-Planck-Institute for Colloids and Interfaces in Potsdam (Germany), he specialized in network science and afterward went to work with one of the world׳s leading experts in this field, Albert-László Barabási at Northeastern University in Boston (USA). Collaborating closely with Joseph Loscalzo from Harvard Medical School and Marc Vidal from Dana Farber Cancer Institute, he laid out the basic theoretical framework for how the interactome can be understood as a map to study human disease. Since 2015, he is a principal investigator at the CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences in Vienna (Austria). Major research areas of his group are network-based approaches to rare diseases, understanding the basic principles of how perturbations of biological systems influence each other and developing novel virtual reality (VR)-based technologies for analyzing large genomic data.
References
- Alanis-Lobato G., Andrade-Navarro M.A., Schaefer M.H. HIPPIE v2.0: enhancing meaningfulness and reliability of protein-protein interaction networks. Nucleic Acids Research. 2017;45(D1):D408–D414. doi: 10.1093/nar/gkw985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert R., Barabási A.-L. Statistical mechanics of complex networks. Reviews of Modern Physics. 2002;74(1):47–97. [Google Scholar]
- Apweiler R., Beissbarth T., Berthold M.R., Blüthgen N., Burmeister Y., Dammann O., Deutsch A., Feuerhake F., Franke A., Hasenauer J., Hoffmann S., Höfer T., Jansen P.L., Kaderali L., Klingmüller U., Koch I., Kohlbacher O., Kuepfer L., Lammert F., Maier D., Pfeifer N., Radde N., Rehm M., Roeder I., Saez-Rodriguez J., Sax U., Schmeck B., Schuppert A., Seilheimer B., Theis F.J., Vera J., Wolkenhauer O. Whither systems medicine? Experimental & Molecular Medicine. 2018;50(3) doi: 10.1038/emm.2017.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babaei S., Mahfouz A., Hulsman M., Lelieveldt B.P.F., de Ridder J., Reinders M. Hi-C chromatin interaction networks predict co-expression in the mouse cortex. PLoS Computational Biology. 2015;11(5) doi: 10.1371/journal.pcbi.1004221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabási A.-L., Gulbahce N., Loscalzo J. Network medicine: A network-based approach to human disease. Nature Reviews Genetics. 2011;12(1):56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergthaler A., Menche J. The immune system as a social network. Nature Immunology. 2017;18(5):481–482. doi: 10.1038/ni.3727. [DOI] [PubMed] [Google Scholar]
- Betsch C., Böhm R., Korn L., Holtmann C. On the benefits of explaining herd immunity in vaccine advocacy. Nature Human Behaviour. 2017;1(3):1–6. [Google Scholar]
- Bianconi G., Barabási A.-L. Competition and multiscaling in evolving networks. Europhysics Letters (EPL) 2001;54(4):436–442. [Google Scholar]
- Bock D.D., Lee W.-C.A., Kerlin A.M., Andermann M.L., Hood G., Wetzel A.W., Yurgenson S., Soucy E.R., Kim H.S., Reid R.C. Network anatomy and in vivo physiology of visual cortical neurons. Nature. 2011;471(7337):177. doi: 10.1038/nature09802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggman K.L., Helmstaedter M., Denk W. Wiring specificity in the direction-selectivity circuit of the retina. Nature. 2011;471(7337):183. doi: 10.1038/nature09818. [DOI] [PubMed] [Google Scholar]
- Brockmann D. Public health: This message must be herd. Nature Human Behaviour. 2017;1(3):0065. [Google Scholar]
- Brockmann D., Helbing D. The hidden geometry of complex, network-driven contagion phenomena. Science. 2013;342(6164):1337–1342. doi: 10.1126/science.1245200. [DOI] [PubMed] [Google Scholar]
- Bullmore E., Sporns O. Complex brain networks: Graph theoretical analysis of structural and functional systems. Nature Reviews Neuroscience. 2009;10(3):186–198. doi: 10.1038/nrn2575. [DOI] [PubMed] [Google Scholar]
- Caldera M., Buphamalai P., Müller F., Menche J. Interactome-based approaches to human disease. Current Opinion in Systems Biology. 2017;3:88–94. [Google Scholar]
- Caldera M., Müller F., Kaltenbrunner I., Licciardello M.P., Lardeau C.-H., Kubicek S., Menche J. Mapping the perturbome network of cellular perturbations. Nature Communications. 2019;10(1):5140. doi: 10.1038/s41467-019-13058-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F., Kovács I.A., Barabási A.-L. Network-based prediction of drug combinations. Nature Communications. 2019;10(1):1197. doi: 10.1038/s41467-019-09186-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C., Mann M. Decoding signalling networks by mass spectrometry-based proteomics. Nature Reviews Molecular Cell Biology. 2010;11(6):427–439. doi: 10.1038/nrm2900. [DOI] [PubMed] [Google Scholar]
- Christakis N.A., Fowler J.H. The spread of obesity in a large social network over 32 years. New England Journal of Medicine. 2007;357(4):370–379. doi: 10.1056/NEJMsa066082. [DOI] [PubMed] [Google Scholar]
- Christakis N.A., Fowler J.H. The collective dynamics of smoking in a large social network. New England Journal of Medicine. 2008;358(21):2249–2258. doi: 10.1056/NEJMsa0706154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M., VanderSluis B., Koch E.N., Baryshnikova A., Pons C., Tan G., Wang W., Usaj M., Hanchard J., Lee S.D., Pelechano V., Styles E.B., Billmann M., Van Leeuwen J., Van Dyk N., Lin Z.Y., Kuzmin E., Nelson J., Piotrowski J.S., Srikumar T., Bahr S., Chen Y., Deshpande R., Kurat C.F., Li S.C., Li Z., Usaj M.M., Okada H., Pascoe N., Luis B.J.S., Sharifpoor S., Shuteriqi E., Simpkins S.W., Snider J., Suresh H.G., Tan Y., Zhu H., Malod-Dognin N., Janjic V., Przulj N., Troyanskaya O.G., Stagljar I., Xia T., Ohya Y., Gingras A.C., Raught B., Boutros M., Steinmetz L.M., Moore C.L., Rosebrock A.P., Caudy A.A., Myers C.L., Andrews B., Boone C. A global genetic interaction network maps a wiring diagram of cellular function. Science. 2016;353(6306):aaf1420. doi: 10.1126/science.aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M., Kuzmin E., van Leeuwen J., Mair B., Moffat J., Boone C., Andrews B. Global genetic networks and the genotype-to-phenotype relationship. Cell. 2019;177(1):85–100. doi: 10.1016/j.cell.2019.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdős P., Rényi A. On the evolution of random graphs. Publication of the Mathematical Institute of the Hungarian Academy of Sciences. 1960;5(1):17–60. [Google Scholar]
- Fabregat A., Sidiropoulos K., Garapati P., Gillespie M., Hausmann K., Haw R., Jassal B., Jupe S., Korninger F., McKay S., Matthews L., May B., Milacic M., Rothfels K., Shamovsky V., Webber M., Weiser J., Williams M., Wu G., Stein L., Hermjakob H., D’Eustachio P. The reactome pathway knowledgebase. Nucleic Acids Research. 2016;44(D1):D481–D487. doi: 10.1093/nar/gkv1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer B., Sandmann T., Horn T., Billmann M., Chaudhary V., Huber W., Boutros M. A map of directional genetic interactions in a metazoan cell. eLife. 2015;4 doi: 10.7554/eLife.05464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler J.H., Christakis N.A. Dynamic spread of happiness in a large social network: Longitudinal analysis over 20 years in the Framingham Heart Study. BMJ (Online) 2009;338(7685):23–26. doi: 10.1136/bmj.a2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiassian S.D., Menche J., Barabási A.-L. A DIseAse MOdule Detection (DIAMOnD) Algorithm Derived from a systematic analysis of connectivity patterns of disease proteins in the human interactome. PLoS Computational Biology. 2015;11(4) doi: 10.1371/journal.pcbi.1004120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasser M.F., Coalson T.S., Robinson E.C., Hacker C.D., Harwell J., Yacoub E., Ugurbil K., Andersson J., Beckmann C.F., Jenkinson M., et al. A multi-modal parcellation of human cerebral cortex. Nature. 2016;536(7615):171–178. doi: 10.1038/nature18933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene J.A., Loscalzo J. Putting the patient back together—Social medicine, network medicine, and the limits of reductionism. New England Journal of Medicine. 2017;377(25):2493–2499. doi: 10.1056/NEJMms1706744. [DOI] [PubMed] [Google Scholar]
- Guney E., Menche J., Vidal M., Barábasi A.-L. Network-based in silico drug efficacy screening. Nature Communications. 2016;7(1):10331. doi: 10.1038/ncomms10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield J., Megill C., Bell S.M., Huddleston J., Potter B., Callender C., Sagulenko P., Bedford T., Neher R.A. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 2018;34(23):4121–4123. doi: 10.1093/bioinformatics/bty407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin E.L., Bruckner R.J., Paulo J.A., Cannon J.R., Ting L., Baltier K., Colby G., Gebreab F., Gygi M.P., Parzen H., Szpyt J., Tam S., Zarraga G., Pontano-Vaites L., Swarup S., White A.E., Schweppe D.K., Rad R., Erickson B.K., Obar R.A., Guruharsha K.G., Li K., Artavanis-Tsakonas S., Gygi S.P., Harper J.W. Architecture of the human interactome defines protein communities and disease networks. Nature. 2017;545(7655):505–509. doi: 10.1038/nature22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C., Wang X., Li X., Inlora J., Wang T., Liu Q., Snyder M. Dynamic human environmental exposome revealed by longitudinal personal monitoring. Cell. 2018;175(1):277–291. doi: 10.1016/j.cell.2018.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia V., Jones D.P., Miller G.W. Networks at the nexus of systems biology and the exposome. Current Opinion in Toxicology. 2019;16:25–31. [Google Scholar]
- Karczewski K.J., Snyder M.P. Integrative omics for health and disease. Nature Reviews Genetics. 2018;19(5):299–310. doi: 10.1038/nrg.2018.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kermack W.O., McKendrick A.G. A contribution to the mathematical theory of epidemics. Proceedings of the Royal Society A: Mathematical, Physical and Engineering Sciences. 1927;115(772):700–721. [Google Scholar]
- Köhler S., Bauer S., Horn D., Robinson P.N. Walking the interactome for prioritization of candidate disease genes. The American Journal of Human Genetics. 2008;82(4):949–958. doi: 10.1016/j.ajhg.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovács I.A., Luck K., Spirohn K., Wang Y., Pollis C., Schlabach S., Bian W., Kim D.-K., Kishore N., Hao T., Calderwood M.A., Vidal M., Barabási A.-L. Network-based prediction of protein interactions. Nature Communications. 2019;10(1):1–8. doi: 10.1038/s41467-019-09177-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmin E., VanderSluis B., Wang W., Tan G., Deshpande R., Chen Y., Usaj M., Balint A., Usaj M.M., Leeuwen J.v., Koch E.N., Pons C., Dagilis A.J., Pryszlak M., Wang J.Z.Y., Hanchard J., Riggi M., Xu K., Heydari H., Luis B.-J.S., Shuteriqi E., Zhu H., Dyk N.V., Sharifpoor S., Costanzo M., Loewith R., Caudy A., Bolnick D., Brown G.W., Andrews B.J., Boone C., Myers C.L. Systematic analysis of complex genetic interactions. Science. 2018;360(6386) doi: 10.1126/science.aao1729. eaao1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kveler K., Starosvetsky E., Ziv-Kenet A., Kalugny Y., Gorelik Y., Shalev-Malul G., Aizenbud-Reshef N., Dubovik T., Briller M., Campbell J., Rieckmann J.C., Asbeh N., Rimar D., Meissner F., Wiser J., Shen-Orr S.S. Immune-centric network of cytokines and cells in disease context identified by computational mining of PubMed. Nature Biotechnology. 2018;36(7):651–659. doi: 10.1038/nbt.4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leskovec J., Sosič R. SNAP: A general-purpose network analysis and graph-mining library. ACM Transactions on Intelligent Systems and Technology (TIST) 2016;8(1):1. doi: 10.1145/2898361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Y.-C., Torres J.Z. Special Topics in Drug Discovery. InTech; 2016. Chemical similarity networks for drug discovery. [Google Scholar]
- Longini I.M., Nizam A., Xu S., Ungchusak K., Hanshaoworakul W., Cummings D.A.T., Halloran M.E. Containing pandemic influenza at the source. Science (New York, N.Y.) 2005;309(5737):1083–1087. doi: 10.1126/science.1115717. [DOI] [PubMed] [Google Scholar]
- Maslov S., Sneppen K. Specificity and stability in topology of protein networks. Science. 2002;296(5569):910–913. doi: 10.1126/science.1065103. [DOI] [PubMed] [Google Scholar]
- McGillivray P., Clarke D., Meyerson W., Zhang J., Lee D., Gu M., Kumar S., Zhou H., Gerstein M.B. Network analysis as a grand unifier in biomedical data science. Annual Review of Biomedical Data Science. 2018;1(1):153–180. [Google Scholar]
- Menche J., Sharma A., Kitsak M., Ghiassian S.D., Vidal M., Loscalzo J., Barabási A.L. Uncovering disease-disease relationships through the incomplete interactome. Science. 2015;347(6224):841. doi: 10.1126/science.1257601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M.J., Beltrán J.F., Liang S., Fragoza R., Rumack A., Liang J., Wei X., Yu H. Interactome INSIDER: A structural interactome browser for genomic studies. Nature Methods. 2018;15(2):107–114. doi: 10.1038/nmeth.4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milo R., Shen-Orr S., Itzkovitz S., Kashtan N., Chklovskii D., Alon U. Network motifs: simple building blocks of complex networks. Science. 2002;298(5594):824–827. doi: 10.1126/science.298.5594.824. [DOI] [PubMed] [Google Scholar]
- Oughtred R., Stark C., Breitkreutz B.-J., Rust J., Boucher L., Chang C., Kolas N., O’Donnell L., Leung G., McAdam R., Zhang F., Dolma S., Willems A., Coulombe-Huntington J., Chatr-Aryamontri A., Dolinski K., Tyers M. The BioGRID interaction database: 2019 update. Nucleic Acids Research. 2019;47(D1):D529–D541. doi: 10.1093/nar/gky1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor-Satorras R., Castellano C., Van Mieghem P., Vespignani A. Epidemic processes in complex networks. Reviews of Modern Physics. 2015;87(3):925–979. [Google Scholar]
- Piñero J., Berenstein A., Gonzalez-Perez A., Chernomoretz A., Furlong L.I. Uncovering disease mechanisms through network biology in the era of Next Generation Sequencing. Scientific Reports. 2016;6(1):24570. doi: 10.1038/srep24570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotrowski J.S., Li S.C., Deshpande R., Simpkins S.W., Nelson J., Yashiroda Y., Barber J.M., Safizadeh H., Wilson E., Okada H., Gebre A.A., Kubo K., Torres N.P., LeBlanc M.A., Andrusiak K., Okamoto R., Yoshimura M., DeRango-Adem E., van Leeuwen J., Shirahige K., Baryshnikova A., Brown G.W., Hirano H., Costanzo M., Andrews B., Ohya Y., Osada H., Yoshida M., Myers C.L., Boone C. Functional annotation of chemical libraries across diverse biological processes. Nature Chemical Biology. 2017;13(9):982–993. doi: 10.1038/nchembio.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt D., Chen J., Welker D., Rivas R., Pillich R., Rynkov V., Ono K., Miello C., Hicks L., Szalma S., et al. Ndex, the network data exchange. Cell Systems. 2015;1(4):302–305. doi: 10.1016/j.cels.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauscher B., Heigwer F., Henkel L., Hielscher T., Voloshanenko O., Boutros M. Toward an integrated map of genetic interactions in cancer cells. Molecular Systems Biology. 2018;14(2) doi: 10.15252/msb.20177656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieckmann J.C., Geiger R., Hornburg D., Wolf T., Kveler K., Jarrossay D., Sallusto F., Shen-Orr S.S., Lanzavecchia A., Mann M., Meissner F. Social network architecture of human immune cells unveiled by quantitative proteomics. Nature Immunology. 2017;18(5):583–593. doi: 10.1038/ni.3693. [DOI] [PubMed] [Google Scholar]
- Rolland T., Taşan M., Charloteaux B., Pevzner S.J., Zhong Q., Sahni N., Yi S., Lemmens I., Fontanillo C., Mosca R., Kamburov A., Ghiassian S.D., Yang X., Ghamsari L., Balcha D., Begg B.E., Braun P., Brehme M., Broly M.P., Carvunis A.R., Convery-Zupan D., Corominas R., Coulombe-Huntington J., Dann E., Dreze M., Dricot A., Fan C., Franzosa E., Gebreab F., Gutierrez B.J., Hardy M.F., Jin M., Kang S., Kiros R., Lin G.N., Luck K., Macwilliams A., Menche J., Murray R.R., Palagi A., Poulin M.M., Rambout X., Rasla J., Reichert P., Romero V., Ruyssinck E., Sahalie J.M., Scholz A., Shah A.A., Sharma A., Shen Y., Spirohn K., Tam S., Tejeda A.O., Trigg S.A., Twizere J.C., Vega K., Walsh J., Cusick M.E., Xia Y., Barabási A.L., Iakoucheva L.M., Aloy P., De Las Rivas J., Tavernier J., Calderwood M.A., Hill D.E., Hao T., Roth F.P., Vidal M. A proteome-scale map of the human interactome network. Cell. 2014;159(5):1212–1226. doi: 10.1016/j.cell.2014.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A., Kim Y., Gewirtz A.D., Jo B., Gao C., McDowell I.C., Engelhardt B.E., Battle A., Battle A. Co-expression networks reveal the tissue-specific regulation of transcription and splicing. Genome Research. 2017;27(11):1843–1858. doi: 10.1101/gr.216721.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporns O. The human connectome: origins and challenges. NeuroImage. 2013;80:53–61. doi: 10.1016/j.neuroimage.2013.03.023. [DOI] [PubMed] [Google Scholar]
- Stéphanou A., Fanchon E., Innominato P.F., Ballesta A. Systems biology, systems medicine, systems pharmacology: The what and the why. Acta Biotheoretica. 2018:1–21. doi: 10.1007/s10441-018-9330-2. [DOI] [PubMed] [Google Scholar]
- Veneman W.J., Spaink H.P., Brun N.R., Bosker T., Vijver M.G. Pathway analysis of systemic transcriptome responses to injected polystyrene particles in zebrafish larvae. Aquatic Toxicology. 2017;190:112–120. doi: 10.1016/j.aquatox.2017.06.014. [DOI] [PubMed] [Google Scholar]
- Wang Z., Moreno Y., Boccaletti S., Perc M. Vaccination and epidemics in networked populations–An introduction. Chaos, Solitons and Fractals. 2017;103:177–183. [Google Scholar]
- White J.G., Southgate E., Thomson J.N., Brenner S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philosophical Transactions of the Royal Society B: Biological Sciences. 1986;314(1165):1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- Zitnik M., Sosič R., Maheshwari S., Leskovec J. BioSNAP Datasets: Stanford Biomedical Network Dataset Collection. 2018. http://snap.stanford.edu/biodata