

Abstract
By uniformly analyzing 723 RNA-seq data from 91 tissues and cell types, we built a comprehensive gene atlas and studied tissue specificity of genes in cattle. We demonstrated that tissue-specific genes significantly reflected the tissue-relevant biology, showing distinct promoter methylation and evolution patterns (e.g., brain-specific genes evolve slowest, whereas testis-specific genes evolve fastest). Through integrative analyses of those tissue-specific genes with large-scale genome-wide association studies, we detected relevant tissues/cell types and candidate genes for 45 economically important traits in cattle, including blood/immune system (e.g., CCDC88C) for male fertility, brain (e.g., TRIM46 and RAB6A) for milk production, and multiple growth-related tissues (e.g., FGF6 and CCND2) for body conformation. We validated these findings by using epigenomic data across major somatic tissues and sperm. Collectively, our findings provided novel insights into the genetic and biological mechanisms underlying complex traits in cattle, and our transcriptome atlas can serve as a primary source for biological interpretation, functional validation, studies of adaptive evolution, and genomic improvement in livestock.
Over the last decade, genome-wide association studies (GWAS) have been successful at discovering trait-/disease-associated genomic variants (Visscher et al. 2012, 2017). However, such studies provided limited information about novel molecular mechanisms underlying complex traits and diseases, partly due to the lack of knowledge of in what tissues or cell types those genomic variants would act. Recently, researchers have been actively pursuing a comprehensive map of functional elements, aiming to identify which genes and regulatory factors (e.g., promoters and enhancers) are functional or active in a large range of tissues and cell types—for example, Roadmap Epigenomics (Roadmap Epigenomics Consortium et al. 2015), GETx (The GTEx Consortium 2017), and Cell Atlas (Regev et al. 2017) projects in human, as well as the Functional Annotation of Animal Genomes (FAANG) project in livestock (Andersson et al. 2015). Integrative analyses of functional genome information with large-scale GWAS data provide unprecedented potential to discover trait-/disease-relevant tissues or cell types, which is crucial for understanding the molecular underpinnings of complex traits and diseases (Finucane et al. 2018; Hormozdiari et al. 2018). For instance, the Roadmap Epigenomics Consortium (2015) showed that GWAS hits of many traits and diseases are significantly enriched in epigenomic marks (e.g., H3K4me1) of trait-/disease-relevant tissues and cell types in humans. Finucane et al. (2018) recently explored disease-relevant tissues and cell types for various human diseases by examining their heritability enrichments among diverse tissues and cell types, such as inhibitory neurons for bipolar disorder (Finucane et al. 2018).
In livestock, due to the limited amount of functional genome data available (Fang et al. 2019), to our knowledge no previous publication has systematically reported the causal tissues or cell types for complex traits and diseases of economic importance. A comprehensive map linking complex traits with their specifically relevant tissues will offer valuable information for fine-mapping causal genes/variants, for functionally validating of GWAS hits (i.e., selecting the “right” tissues and cell types), and for understanding of adaptive evolution (Quiver and Lachance 2018), as well as for the design of genome editing experiments (Ruan et al. 2017). Additionally, a better understanding of the genetic architecture underlying complex traits may make a contribution to the genetic improvement programs among livestock species (Goddard and Hayes 2009; Georges et al. 2019). For instance, Fang et al. (2017a,b) reported improved genomic prediction accuracy for mastitis and milk production traits in cattle by incorporating biological priors and gene expression information relevant to bacterial infection into genomic prediction models (Fang et al. 2017a,b).
Here, we uniformly assembled and analyzed 723 (156 newly generated and 567 existing) RNA-seq data sets to build a new gene atlas in cattle (Supplemental Code), which included 91 tissues and cell types from 447 individuals (http://cattlegeneatlas.roslin.ed.ac.uk). We summarized the global design of this study in Supplemental Figure S1. We first detected genes that were highly and specifically expressed in each tissue or cell type and then explored their biological characteristics in terms of biological function, DNA methylation, and evolution. We detected relevant tissues/cell types and candidate genes for 45 complex traits of economic importance in cattle, including 18 body conformation, six milk production, 12 reproduction, eight health, and one feed efficiency traits, by integrating those tissue-specific genes with large-scale (n = 27,214 bulls) GWAS data. We validated our findings by analyzing whole-genome DNA methylation data across major somatic tissues and sperm in cattle. In addition, we tested whether the tissue-specificity information of genes can improve genomic prediction. Our results, for the first time, systemically establish connections at the RNA level between tissue/cell types and complex traits in livestock and provide an important starting point for post-GWAS functional experiments to explore genotype-phenotype relationships in livestock.
Results
Summary of cattle gene atlas
Using a uniform pipeline of bioinformatics analysis, we obtained 18,468,126,120 clean reads from 723 RNA-seq data sets with an averaged uniquely mapping rate of 94.18%. We summarized details of sample information in Supplemental Table S1. We determined the normalized expression levels (i.e., fragments per kilobase per million mapped reads, FPKM) for all 24,616 Ensembl genes among 723 samples. In general, we found an average of 15,864 genes (median = 16,086, ranging from 7807 to 18,258) expressed (FPKM > 0) across 91 tissues and cell types, of which the majority (n = 14,682 on average) were protein-coding genes (Supplemental Fig. S2). Despite differences in experimental conditions and sample characteristics, samples from similar tissues and cell types clustered together based on their gene expression profiles (Fig. 1A), validating the potential of our data for studying the specificity of tissue expression. For instance, we found that samples from 14 adult brain regions (central neural system, CNS) clustered together with those from fetus brain and four other brain endocrine tissues (stalk median eminence [SME], anterior pituitary, posterior pituitary, and pineal gland). All samples from seven blood/immune tissues and cell types clustered together, including CD4 cells, CD8 cells, white-blood cells, lymphocyte, spleen, thymus, and lymph nodes (Fig. 1A).
Figure 1.

General characteristics of the cattle gene atlas. (A) Clustering analysis of all 723 RNA-seq samples using t-SNE (t-Distributed Stochastic Neighbor Embedding) procedure. (CNS) Central neural system. (B) Examples of tissue-specific genes in brain (GRM5), liver (SLC22A9), white blood cells (FCRL3), uterus (TDGF1), and testes (TRIM9). The y-axis is the raw gene expression, that is, fragments per kilobase per million mapped reads (FPKM). (C) Gene Ontology enrichment analysis of tissue-specific genes (the top 5% of genes based on t-statistics). The value in each bar is the fold of enrichment. (D) The enrichment analysis of cattle tissue-specific genes with human tissue-specific genes. The P-value is obtained using a hypergeometric test.
Detection and functional characterization of tissue-/ cell type–specific genes
We calculated a t-statistic to measure the specific expression of a gene in a given tissue/cell type (Methods). We found that tissues and cell types within the same system highly positively correlated based on these t-statistics (Supplemental Fig. S3), indicating the high similarity of their tissue-specific expression. Of special interest, we found that mammary gland highly negatively correlated with corpus luteum and endometrium (Pearson's r = −0.88 and −0.85, respectively) (Supplemental Fig. S3). This may reflect the well-known, antagonistic relationship between milk yield and fertility in dairy animals (Veerkamp et al. 2001; Berry et al. 2003). Additionally, liver and rumen epithelial cells negatively correlated with several immune tissues and cell types, including CD4 cells, CD8 cells, white blood cells, and thymus (the averaged Pearson's r = −0.62) (Supplemental Fig. S3). This may support the observed connections between feed efficiency and immune responses in cattle (Hou et al. 2012). However, the underlying molecular mechanisms of these negative correlations are largely unknown and require further investigations.
We detected tissue-specific genes for each tested tissue based on the rank of t-statistics (i.e., top 5%). We showed the top tissue-specific genes in brain (GRM5), liver (SLC22A9), white blood cell (FCRL3), uterus (TDGF1), and testis (TRIM69) as examples in Figure 1B. The functional annotation of tissue-specific genes validated the known tissue-relevant biology (Fig. 1C; Supplemental Table S2). For instance, brain-specific genes significantly enriched for nervous system development (FDR = 1.67 × 10−48, enrichment fold = 3.24), liver for organic acid metabolism process (FDR = 1.33 × 10−51, enrichment fold = 4.92), white blood cell for regulation of immune system (FDR = 7.81 × 10−48, enrichment fold = 3.85), uterus for embryonic morphogenesis (FDR = 1.97 × 10−20, enrichment fold = 3.63), and testis for male gamete generation (FDR = 2.94 × 10−28, enrichment fold = 5.08) (Fig. 1C). In addition, we confirmed that promoters of tissue-specific genes in liver and muscle had specifically low DNA methylation in the corresponding tissues (Supplemental Fig. S4), consistent with promoter methylation being negatively correlated with gene expression (Smith and Meissner 2013). For instance, the promoter methylation of SLC22A9 (liver-specific expression), which is an important hepatic transport protein (Riedmaier et al. 2016), was significantly lower in liver when compared to other tissues (Supplemental Fig. S5). Moreover, our motif enrichment analysis of tissue-specific genes revealed potential master regulators (transcriptional factors) (Supplemental Fig. S6; Supplemental Table S3), which could contribute to regulation of gene activity and differentiations of cell types and tissues (Spitz and Furlong 2012). As shown in Supplemental Figure S6, we found that STAT1 was significantly (FDR < 0.05) enriched in CD4 cells and lymph nodes, which have crucial roles in multiple immune responses (Shuai and Liu 2003; Hu and Ivashkiv 2009), whereas ZFX, which participates in neuronal differentiation, was significantly enriched in hippocampus and cerebral cortex (Harel et al. 2012; Burney et al. 2013).
To explore the evolutionary conservation of tissue-specific genes among mammals, we first compared the cattle tissue-specific genes with human tissue-specific genes among 10 major tissues. We found that tissue-specific genes significantly overlapped in the matched tissues between cattle and human (Fig. 1D). We further explored dN/dS ratios of orthologous genes between cattle and five other mammals (i.e., human, mouse, dog, pig, and sheep). We consistently observed that genes specific for brain regions had the significantly lowest dN/dS ratios, whereas genes specific for male reproductive tissues (e.g., testes and sperm) and blood/immune system (e.g., lymph nodes) had the significantly highest dN/dS ratios (Fig. 2; Supplemental Fig. S7). We then correlated the expression of all orthologous genes among major tissues in both cattle versus human and cattle versus sheep comparisons and confirmed that testes had the lowest correlation, whereas brain showed a relatively higher one (Supplemental Fig. S8). Our findings demonstrated that, in constrained tissues (e.g., brain), tissue-specific genes tended to evolve slowly, whereas in the relaxed tissues (e.g., testes), tissue-specific genes evolved more rapidly, revealing the importance of tissue-driven evolution.
Figure 2.

Comparison of dN/dS ratios of tissue-specific genes across all the 91 tissues and cell types between human and cattle. The red line represents the averaged dN/dS ratio of all orthologous genes between human and cattle. For each tissue, we compared tissue-specific genes of this tissue against the remaining genes using a two-tailed t-test; (*) P < 0.01.
Detection of tissues and cell types relevant with 45 agronomic traits
By integrating tissue-specific genes with large-scale GWAS, we revealed a comprehensive genetic relationship between 91 tissues/cell types and 45 complex traits of economic importance in cattle, providing novel insights into the molecular underpinnings of such economically important traits (Fig. 3). To validate our findings, we repeated GWAS signal enrichment analyses using tissue-specific DNA methylation regions instead of tissue-specific genes. We found GWAS enrichments from DNA methylation highly correlated with those from gene expression across all 45 traits among multiple tissues, for example, sperm (Pearson's r = 0.67; P = 4.41 × 10−7) and lung (Pearson's r = 0.65; P = 1.60 × 10−6) (Fig. 4). We summarized details of all 288 significant (FDR < 0.1) associations between traits and tissues in Supplemental Table S4. In addition, we summarized the top three expressed tissues of all 525 fine-mapped genes across all complex traits in Supplemental Table S5. The details of fine-mapped genes were described previously (Jiang et al. 2019).
Figure 3.

The relationships between 45 complex traits and 91 tissues and cell types. The color corresponds to enrichment degrees (i.e., −log10P) that are computed using a sum-based GWAS signal enrichment analysis based on the top 5% tissue-specific genes and a 50-kb extension. (*) Corrected-P (FDR) < 0.1.
Figure 4.
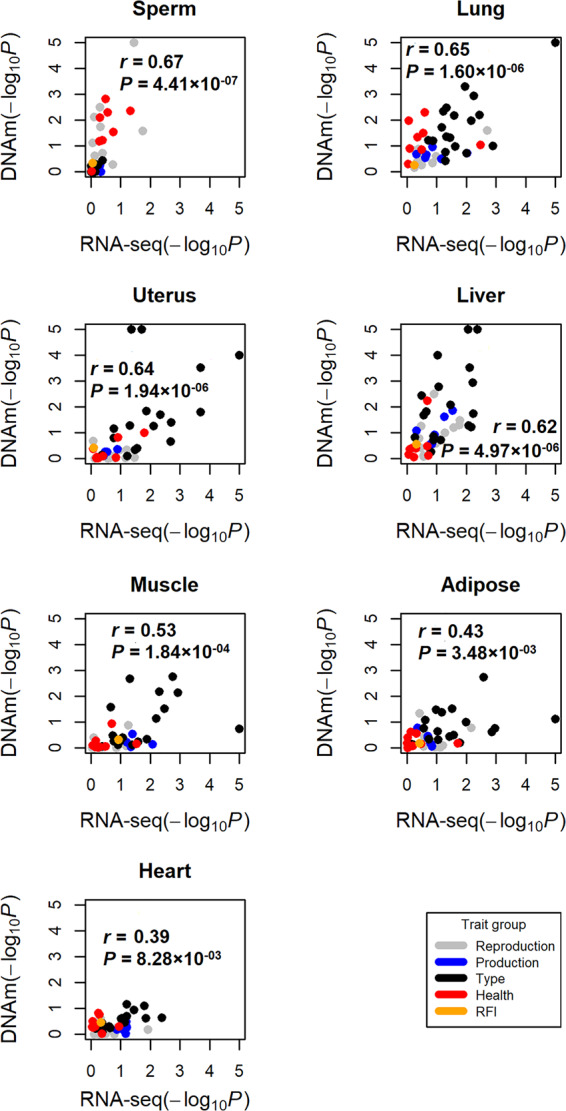
Validation of trait-tissue associations using DNA methylation data across seven tissues. Each dot represents a trait. The y-axis is for GWAS signal enrichments (−log10P) obtained using tissue-specific DNA methylated regions, whereas the x-axis is for GWAS signal enrichments obtained using tissue-specific expressed genes. The r is for Pearson's correlation.
Milk production traits
Generally, we observed that milk production traits were significantly associated with a few tissues and cell types (Fig. 3), indirectly supporting their highly polygenic architecture (Cole et al. 2009; Kemper and Goddard 2012). Of note, we found that mammary gland was the most significant (P = 2 × 10−4) tissue for protein yield (Supplemental Fig. S9A) and validated this by demonstrating that two of its fine-mapped genes (i.e., CSN1S1 with the posterior probability of causality (PPC) = 1, and PAEP with PPC = 0.84) were highly specifically expressed in mammary gland and milk cells (Supplemental Fig. S9B). Mammary gland was also the top significant tissue (P = 1.5 × 10−3) for lifetime net merit, which is an economic index including multiple traits that is used to rank animals for selection, suggesting the importance of a “good” mammary gland in the dairy industry. Moreover, we found two fine-mapped genes, MRTFA (previously known as MKL1) (PPC = 1) and NCF4 (PPC = 0.55), for milk/protein yields and protein percentage, respectively, which specifically expressed in blood/immune system (Supplemental Fig. S10). This provides evidence of the underlying genetic correlations between milk production and immune disorders (e.g., mastitis) in cattle. Although brain tissues showed no significant enrichments for milk production (Fig. 3), we noticed that they indeed exhibited a significantly higher enrichment for milk production as compared to other types of traits, except for feed efficiency (i.e., residual feed intake [RFI]) (Fig. 5A). We found two fine-mapped genes, TRIM46 (PPC = 0.59) and RAB6A (PPC = 0.79), for protein percentage and milk yield, respectively, which highly specifically expressed in brain regions (Fig. 5B). By examining quantitative trait loci (QTL) of 19 milk-relevant traits in cattle QTLdb, we confirmed that brain-specific genes were significantly enriched for genes (i.e., closest genes to the lead SNPs) associated with milk production traits (e.g., milk yield and fat/protein percentage) but not for certain milk content traits (e.g., such as milk iron and zinc contents) (Supplemental Fig. S11). To further explore which brain regions were relevant to milk production traits, we pinpointed tissue-specific genes within 11 brain regions and another four brain endocrine tissues. We observed that anterior pituitary, cerebellum, and temporal cortex were significantly (FDR < 0.05) associated with protein yield (Fig. 5C). To our knowledge, no previous publication has reported such relationships between the brain and milk production by an integrative analysis of genomic and transcriptomic data.
Figure 5.

Relationships between milk production traits and brain regions. (A) Milk production traits have a significantly higher GWAS signal enrichments (−log10P) than other types of traits in 14 brain regions (CNS), except for feed efficiency (i.e., residual feed intake [RFI]). We calculate P-values between groups using Student's t-test. (B) Two fine-mapped genes, TRIM46 (top; posterior probability of causality [PPC] = 0.59) and RAB6A (bottom; PPC = 0.79), for protein percentage and milk yield, respectively, are specifically highly expressed in CNS compared with all other tissues and cell types. (C) The associations of milk production traits with brain regions and four brain endocrine tissues (i.e., stalk median eminence [SME], anterior pituitary, posterior pituitary, and pineal gland) based on the GWAS signal enrichments of tissue-specific genes detected within these brain-relevant tissues. (*) Corrected-P (FDR) < 0.1.
Body conformation traits
Body conformation (type) traits were significantly associated with many tissues and cell types, except for brain regions (Fig. 3), similar to findings in human height (Finucane et al. 2018), reflecting their highly polygenetic architectures. We used cattle stature as an example and found that three of its fine-mapped genes with PPC = 1, FGF6, CCND2, and TCP11, were highly specifically expressed in fetal muscle, rumen epithelial cell, and testes, respectively (Supplemental Fig. S9C,D). By examining heritability enrichments of human height among 33 tissues (Finucane et al. 2018), we observed that these tissues significantly positively correlated (Pearson's r = 0.64; P = 6.82 × 10−5) between human height and cattle stature in terms of GWAS signal enrichment (i.e., −log10P) (Supplemental Fig. S12). Uterus and aorta were the top significantly enriched tissues for both human height and cattle stature, and they significantly associated with many other body type traits in cattle as well, such as rump width and rump angle (Fig. 3). All these findings support the view that mammals shared similar molecular mechanisms underlying body size.
Reproduction and health traits
We noticed that immune/blood system was significantly associated with multiple reproduction traits (Fig. 3). Overall, reproduction traits showed a significantly higher enrichment in immune/blood system when compared to other types of traits (Fig. 6A). The lymph node was the most significant tissue for both sire conception rate (P < 10−5) and sire still birth (P < 10−5) (Fig. 6B; Supplemental Table S4). We found a fine-mapped gene (i.e., CCDC88C with PPC = 1) of DFB (days to first breeding, a measurement of fertility ability), which highly specifically expressed in both blood/immune system and infundibulum (Fig. 6C). CCDC88C plays important roles in the regulation of T cells maturation during bacterial inflammation (Kennedy et al. 2014). Additionally, we found that immune/blood system was significantly associated with several health traits (Fig. 3). For instance, thymus was the top relevant (P = 2.70 × 10−3) tissue for ketosis (KETO) (Fig. 6B). We also found C6, a fine-mapped gene (PPC = 1) for somatic cell score (SCS) in milk, highly specifically expressed in liver and duodenum (Fig. 6C). SCS is an important indicator of mastitis in dairy cattle (Heringstad et al. 2006). Because the small intestine system exhibited immune functions (Santaolalla and Abreu 2012) and showed significant associations with multiple reproduction traits and health traits (Fig. 3), we further pinpointed tissue-specific genes within blood/immune system and four intestine parts, including ileum, duodenum, jejunum, and caecum. We found that thymus showed the highest and most significant enrichments for multiple health and reproduction traits, including daughter still birth, daughter calving ease, SCS, KETO, DFB, and displaced abomasum (DSAB). CD8 cells were significantly associated with daughter calving ease and daughter still birth, whereas CD4 cells were significantly associated with cow conception rate and SCS (Fig. 6D).
Figure 6.

Associations of male reproduction and health traits with blood and immune tissues and cell types. (A) Reproduction traits have a significantly higher GWAS signal enrichment (−log10P) than other types of traits in blood/immune tissues. We calculate P-values between groups using Student's t-test. (B) Enrichments of tissues and cell types with sire conception rate (SCR) and ketosis (KETO) disease. (C) Expression patterns of two fine-mapped genes across all 91 tissues and cell types, C6 (top; PPC = 1 for somatic cell score [SCS]) and CCDC88C (bottom; PPC = 1 for day of first birth [DFB]). (D) Associations of male reproduction and health traits with blood/immune tissues and cell types and four intestinal parts based on the GWAS signal enrichments of tissue-specific genes detected within these immune-relevant tissues and cell types. (*) Corrected-P (FDR) < 0.05.
Feed efficiency
We observed that the alimentary canal was the top relevant system for feed efficiency (i.e., RFI), among which rumen was the most significant (P = 5.70 × 10−3) tissue (Supplemental Fig. S13). We also found that brain pons was associated (P = 2.19 × 10−2) with RFI, which suggests the important role of the gut-brain axis in feed intake (Konturek et al. 2004). Nasal mucosa was another tissue associated (P = 1.06 × 10−2) with RFI, in line with the fact that olfactory receptors are known to be associated with RFI in cattle (Seabury et al. 2017).
A further application of this gene atlas is to explore whether the tissue specificity of genes could enhance genomic improvement in dairy cattle. We focused on three milk production traits (milk, fat, and protein yields). To reduce the redundancy and computational burdens, we clustered 91 tissues and cell types into 20 categories (Supplemental Fig. S14A). For each category, we then fitted SNPs within tissue-specific genes of this category and those in the remaining genome into a two-component Bayesian prediction model. By comparing with a two-component model (i.e., all genes vs. the remaining genome), whose prediction accuracy was similar to that of a single-component model (including all SNPs), we found that there is no improvement in genomic prediction accuracy on average (Supplemental Fig. S14B). We showed that the number of SNPs within tissue-specific genes did not bias prediction accuracy of models (Supplemental Fig. S15). However, we found the category consisting of immune/blood system and liver resulted in an increase of 0.041, 0.032, and 0.015 in prediction accuracy for fat yield, milk yield, and protein yield, respectively (Supplemental Fig. S14B). Another category consisting of salivary gland, larynx cartilage, tongue, chorid plexus, and muscle also increased the prediction accuracy across the three milk traits, that is, 0.044, 0.028, and 0.003 for fat, milk, and protein yields, respectively. We also observed that brain regions led to an increase of 0.033 and 0.016 in prediction accuracy for fat yield and milk yield, respectively, but not for protein yield (Supplemental Fig. S14B). Of note was that DGAT1, a well-known milk and fat gene of large effect (Grisart et al. 2002), was not in those categories, implying that multiple loci of small effects are enriched in these tissue-specific genes.
Discussion
In this study, we built a cattle gene atlas by analyzing 723 RNA-seq data uniformly across 91 tissues and cell types, which also allows for phenome-wide association analysis of genes of interest in humans. Compared to the previous gene expression atlas based on the reference Hereford cow in 2010 (Harhay et al. 2010), we produced ∼70 times more data using RNA-seq (100-bp average length) instead of 3′ tag sequencing (20 bp), which enabled us to examine more genes (n = 22,243) than before (n = 16,517). To increase the statistical power for detecting tissue-specific genes, we generated another 51 new RNA-seq data from 14 major somatic tissues and sperm in Holstein, as well as uniformly analyzed other 567 public RNA-seq data of high quality. Using this newly built gene atlas, we identified relevant tissues/cell types and candidate genes for 45 complex traits of economic importance and further applied it in genomic prediction. Of interest, we observed that brain was associated with milk production traits, and two brain-specific genes, TRIM46 and RAB6A, were fine-mapped genes for protein percentage and milk yield, respectively. TRIM46 plays key roles in neuronal polarity and axon specification (van Beuningen et al. 2015), whereas RAB6A is a key regulator of membrane traffic from the Golgi apparatus toward the endoplasmic reticulum (Matsuto et al. 2015). PheWAS based on both Gene atlas (http://geneatlas.roslin.ed.ac.uk/region-phewas/) (Canela-Xandri et al. 2018) and GWAS atlas (https://atlas.ctglab.nl/PheWAS) (Watanabe et al. 2019) showed that TRIM46 was significantly associated with many metabolic traits (e.g., blood urea nitrogen and impedance of leg), whereas RAB6A was significantly associated with both neurological and metabolic traits (e.g., cingulum axial diusivities and whole body fat-free mass). Our cattle gene atlas will serve as a valuable source for the livestock science community to interpret GWAS findings, to design follow-up validation experiments through choosing the “right” tissues and cell types, as well as to enhance genomic improvement in livestock. With more molecular phenotypes becoming available across diverse tissues in livestock in the near future, for instance, from the on-going FAANG project (Andersson et al. 2015), our current research strategy will help gain more novel insights into the genetic and biological mechanisms underpinning agronomic traits and thus enhance genomic improvement programs.
We noticed some limitations in our current study. Our basic assumption here was that genomic variants ultimately regulated complex traits by altering gene expression in the relevant tissues and cell types. Previous studies showed that the majority of expression quantitative trait loci (eQTL) were cis-variants (The GTEx Consortium 2017). We therefore focused on cis-regulators of tissue-specific genes by extending certain distances (i.e., 10 kb, 20 kb, and 50 kb) around such genes. In order to study trans-eQTLs, we need a large amount of samples for each tissue and cell type due to their relatively small effects (Grundberg et al. 2012). The cell type composition of tissues could confound our interpretation of results. As we showed in Figure 6D, CD4 cells and CD8 cells had distinct enrichments across 19 reproduction and health traits. Therefore, pure bulk cells and/or single-cell expression data may help further detect which cell types are causal in a trait-relevant tissue. Additionally, tissues sharing similar expression patterns with causal tissues could hinder us from detecting the “drivers” among multiple “passengers,” which is similar to the situation with GWAS results, wherein we can only interpret the significant tissues and cell types as the “best proxy” for the causative one. We are also limited by the availability of transcriptomic data, thus potentially ignoring trait-relevant tissues and cell types, which are only biological important for the given traits in certain physiological stages or environmental conditions.
Due to the large amount of linkage disequilibrium (LD) among genomic markers within a single cattle breed (e.g., Holstein), traditional single-component prediction models (e.g., GBLUP and BayesA), which assume that all markers are drawn from the same prior distribution, work quite well within breed (Meuwissen et al. 2001). Such high LD within breed and the highly polygenic architecture of economic traits also make it hard to partition genomic variance into distinct components accurately in a linear mixed model framework, due to the potential high correlations among components. When incorporating tissue-specific genes into the extended prediction models, we thus observed a limited increase in prediction accuracy within Holstein compared to the traditional model, consistent with our previous findings (Fang et al. 2017a,b). However, this functional information may contribute much more to genomic prediction in other scenarios where reduced relatedness is observed between reference and target populations, such as multiple breeds and over generations (Liu et al. 2015; MacLeod et al. 2016; Fang et al. 2017a,b). In addition, when a large range of biological priori information is available in the future, we may use GWAS enrichment analysis as a guide to choose the most “relevant” biological priori information for genomic prediction, as genomic prediction is often computationally intensive (Fang et al. 2017a).
Methods
Bioinformatics analysis of second-generation sequencing data
In this study, we collected all 156 samples under the approval of the U.S. Department of Agriculture Agricultural Research Services Institutional Animal Care and Use Committee under the Protocol 16-016. We provided references where RNA-seq data were retrieved (i.e., SRP042639, PRJNA177791, PRJNA379574, PRJNA416150, PRJNA305942, PRJNA392196, PRJNA428884, PRJNA298914, PRJEB27455, PRJNA268096, and PRJNA446068) and summarized details of all 723 analyzed RNA-seq samples in Supplemental Table S1. Among the newly generated data, we collected 51 from six Holstein cows (GSE137943, GSE148707), 94 from the sequenced Hereford cow (L1 Dominette 01449) and its relatives (GSE128075) using a similar list as described before (Harhay et al. 2010), five from sperm of a Holstein bull (GSE131851), and six from rumen epithelial cells of Holstein calves (GSE129423) (Fang et al. 2019). Briefly, we extracted the total RNA from snap-frozen tissues using TRIzol (Thermo Fisher Scientific) according to the manufacturer's instructions. We measured the quantity and purity of RNA using a NanoDrop 8000 Spectrophotometer (NanoDrop Technologies) and Agilent 2100 Bioanalyzer System (Agilent). We sequenced these RNA samples using the Illumina HiSeq 2000 platform (Illumina) with paired-end (100- to 150-bp) reads for most of them and single-end reads for the rest (Supplemental Table S1).
We analyzed all 723 RNA-seq data uniformly using the following bioinformatics pipeline. First, we removed contaminating adapter molecules, reads containing ploy(N), and low-quality reads using Trimmomatic (version 0.38) (Bolger et al. 2014), obtaining a total of 18,468,126,120 clean reads. We then mapped clean reads to the cattle reference genome UMD3.1.1 using HISAT2 (version 2.1.0) (Kim et al. 2015), resulting in an averaged uniquely mapping rate of 94.18% (Supplemental Table S1). We used Ensembl genes (release 94) as the gene annotation file, including 24,616 genes. We determined gene expression levels (i.e., FPKM) using StringTie (version 1.3.4) (Pertea et al. 2015), while accounting for differences in sequence depth and gene length across samples.
Based on known biology (Harhay et al. 2010), we classified 91 tissues and cell types into 17 biological categories. In order to detect tissue-/cell-specific genes, we computed a t-statistic for each gene in a given tissue using the following approach (Finucane et al. 2018), by excluding tissues and cell types in the same biological category while accounting for known covariates (i.e., age, sex, and study) (Supplemental Table S1). We scaled the log2-transformed expression (i.e., log2FPKM) of genes to have a mean of zero and variance of one within each tissue and cell type.
where y is the scaled log2FPKM, μ is the intercept, X is the dummy variable for tissue, where samples of the tested tissue (e.g., CD4 cells) were denoted as ‘1’, whereas samples outside the same category (e.g., non-blood/immune tissues and cell types) were denoted as ‘−1’, b is the corresponding tissue effect, and Z is the matrix for covariables, including age, sex, and study effects. is the corresponding covariable effects, and e is the residual effect. We fitted this model for each gene in each tissue using the ordinary least-squares approach, as implemented in R (R Core Team 2018), and then obtained the t-statistic (i.e., the coefficient, b, divided by its standard error) for each gene to measure its expression specificity in the corresponding tissue. We employed the same approach to pinpoint tissue-specific genes within a biological category of interest (e.g., brain-regions and blood/immune system). We ranked genes in each tissue according to their t-statistic and chose the top 3%, 5%, and 10% of genes as tissue-specific genes, respectively. We conducted all subsequent analyses using these three cut-offs and obtained similar results. Therefore, we only presented results from the top 5% in Results.
We conducted the functional enrichment analyses for tissue-specific genes using a hypergeometric test with GO database, as implemented in PANTHER 14.0 (Mi et al. 2012). We obtained the tissue-specific genes of 10 major tissues in humans (https://www.proteinatlas.org/humanproteome/tissue/tissue+specific) and then tested their enrichments with cattle tissue-specific genes among the matched tissues using a hypergeometric test. We conducted the motif enrichment analyses for the promoter regions (i.e., 1500 bp upstream of and 500 bp downstream from the transcriptional start sites [TSSs]) of tissue-specific genes using MEME software (Bailey et al. 2009). For enrichment analyses with cattle QTLdb (Release 36, Aug. 22, 2018) (Hu et al. 2012), we chose QTLs for 19 milk-relevant traits and then arbitrarily considered the gene closest to the lead SNP in each corresponding QTL as the “causal” gene. We thus obtained a list of “causal” genes for each of the 19 milk-relevant traits. We conducted the QTL enrichment analysis for tissue-specific genes using the same hypergeometric test like the GO enrichment analysis. For DNA methylation data (Zhou et al. 2018), we also mapped them to the cattle reference genome UMD3.1.1 using Bismark v0.19.0 (Krueger and Andrews 2011). We only kept CpG sites with at least fivefold coverage for subsequent analyses. We employed an entropy-based framework to determine tissue-specific DNA methylation regions, as implemented in the SMART2 software (Liu et al. 2015). We only considered tissue-specific hypomethylated regions to validate our results of trait-relevant tissues obtained by using tissue-specific genes, because hypomethylation is generally related to gene activation (Jones 2012). We determined the promoter methylation level for each gene as the average methylation of CpG sites within its promoter region as defined above. We then obtained the adjusted promoter methylation in a tissue by adjusting for the averaged methylation over the entire genome in this particular tissue. For comparing gene expression among cattle, sheep, and human, we retrieved multitissue gene expression for human from GTEx v6 https://gtexportal.org/home/datasets, and for sheep from https://doi.org/10.1371/journal.pgen.1006997.s004. We obtained the ortholog genes among mammals from Ensembl v94 (https://www.ensembl.org/info/website/archives/index.html).
Single-marker GWAS and fine-mapping results
We previously reported details of the single-marker GWAS and fine-mapping analyses for body type, reproduction, production, and health traits from 27,214 U.S. Holstein bulls (Jiang et al. 2019; Freebern et al. 2020 ) and for feed efficiency (i.e., RFI) from 3947 Holstein cows (Li et al. 2019). Briefly, we used de-regressed breeding values (predicted transmitting abilities [PTA]) of Holstein bulls as phenotypes. We have adjusted such phenotypes for all known systematic effects, including herd, year, season, and parity (Norman et al. 2009). For feed efficiency, we corrected for the dry matter intake for milk yield, metabolic body weight, body weight change, and several environmental effects to obtain RFI (Lu et al. 2015). We used the high-density genotypes (777K) and imputed sequence markers (n = 2,619,418) with an imputation accuracy of 96.7% (Vanraden et al. 2017), minor allele frequency (MAF) > 0.01, and Hardy-Weinberg Equilibrium (HWE) test (P > 10−6) to conduct GWAS analyses for RFI and the remaining traits, respectively. We employed the following linear mixed model, implemented in MMAP software (https://mmap.github.io/), to test for association of genomic variants with all complex traits except for RFI:
where y is the de-regressed PTA, μ is the overall mean, X is the genotype of a genomic marker (coded as 0, 1, or 2), b is the marker effect, is the polygenic effect accounting for familial relationship and population structure, and is the residual. G is the genomic relationship matrix (Vanraden 2008), built using HD markers with MAF > 0.01. R is a diagonal matrix with , where is the reliability of phenotype for the ith individual. For RFI, we used a single-step method to conduct GWAS analysis, which was implemented in the BLUPF90 (version 2018) (Wang et al. 2012; Li et al. 2019).
GWAS signal enrichment analysis
Because complex traits being studied here are highly polygenic (Cole et al. 2009; Kemper and Goddard 2012; Boyle et al. 2017), we applied the following sum-based marker-set test approach, as implemented in the QGG package (Rohde et al. 2019), to determine whether GWAS signals were enriched in tissue-specific genes. We added 10-kb, 20-kb, and 50-kb windows around gene regions to include the potential cis-regulatory variants. Previous studies showed that this approach had at least equal power when compared to other commonly used GWAS signal enrichment methods in humans (Rohde et al. 2016), Drosophila melanogaster (Sørensen et al. 2017), and livestock (Sarup et al. 2016; Fang et al. 2017a,c), especially for the highly polygenic traits.
where mf is the number of genomic markers within a list of tissue-specific genes, and b is the marker effect from single-marker GWAS. We controlled marker-set sizes and LD patterns among markers through applying the following genotype cyclical permutation strategy (Rohde et al. 2016; Sørensen et al. 2017). Briefly, we first ordered marker effects (i.e., b2) using their chromosome positions (i.e., , , ⋯ , ). We then randomly selected one marker (i.e., ) from this vector as the first place and shifted the remaining ones to new positions, while retaining their original orders (i.e., , , ⋯ , , ⋯ ) to maintain correlation patterns among markers. We calculated a new summary statistic for given tissue-specific genes using their original chromosome locations. To obtain an empirical P-value for a list of tissue-specific genes, we repeated this permutation procedure 10,000 times and employed a one-tailed test of the proportion of random summary statistics greater than that observed.
For comparison, we also employed the following count-based approach that focused on the top variants passing a certain genome-wide significance level:
where mf is the number of markers in the tested tissue-specific gene list, pi is the P value for the ith marker from single-marker GWAS, p0 is an arbitrarily selected significant threshold, and I is an indicator function that takes value one when pi < p0, and value zero otherwise. Here, we chose p0 = 0.01 as the significant cut-off. Under the null hypothesis, we assumed that Tcount follows a hypergeometric distribution (Sørensen et al. 2017). We observed that results from these two GWAS enrichment approaches showed a positive correlation of 0.68 (Supplemental Fig. S16A). We here focused on results of a sum-based method. As results of 10-kb and 20-kb extensions were similar to those of 50-kb (Supplemental Fig. S16B), we only showed results of the 50-kb extension.
SNP-set-based genomic prediction analysis
We divided the entire Holstein cattle population into the reference population (n = 19,575) and the validation population (n = 3983) according to the year of birth (Vanraden et al. 2017). We applied the SNP-set-based genomic prediction (SSGP) software to incorporate tissue-specific genes into genomic prediction (https://sites.google.com/view/ssgp), which allows us to split genomic markers into different groups with group-specific effect variance.
where y is the phenotype vector (i.e., PTA), μ is the population mean, and p denotes the number of genetic components in the model. Here, we choose p = 2, corresponding to random effects for markers within the category-specific genes and the remaining genome, respectively. The random effects within the hth component are assumed to follow a multivariate normal distribution: , where Wh is a predefined diagonal matrix to weight each of the random effects, and is assumed to follow an inverse-gamma distribution with component-specific parameters , are shape and scale parameters, respectively. Kh is the corresponding design matrix. e is the residual effect following normal distribution , where R is the diagonal matrix with a predefined weight for error variance. To make models comparable (i.e., the same number of parameters to be estimated), we fitted another model with two genomic components as a null model, where all 24,616 genes as the first component and the rest of the genome as the second component. We also fitted a single-component model that was equivalent to the GBLUP model. We determined the prediction accuracy as the correlation between predicted PTAs and true PTAs in the validation population.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE128075, GSE137943, GSE147087, GSE147184, and GSE148707. The GWAS summary statistics for all complex traits have been submitted to Figshare, that is, body type, production, and reproduction traits under https://figshare.com/s/ea726fa95a5bac158ac1, and the remaining ones under https://figshare.com/s/94540148512dddf7ed32. All scripts and source codes can be found as Supplemental Code, as well as at the Cattle Gene Atlas (http://cattlegeneatlas.roslin.ed.ac.uk) and GitHub (https://github.com/LingzhaoFang1/Cattle-GeneAtlas).
Competing interest statement
The authors declare no competing interests.
Supplementary Material
Acknowledgments
We thank Reuben Anderson, Ransom L. Baldwin, Erin E. Connor, Daniel Jordan de Abreu Santos, Alexandre Dimtchev, Ellen Freebern, and Yang Zhou for technical assistance, sample collection, and early data access. We thank the 1000 Bull Genomes Project for global sequence data, the Council on Dairy Cattle Breeding for genotype, phenotype, and pedigree data, Interbull for global trait evaluations, and the anonymous reviewers for many helpful comments. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. The USDA is an equal opportunity provider and employer. This work was supported in part by Agriculture and Food Research Initiative (AFRI) grant numbers 2013-67015-20951, 2016-67015-24886, and 2019-67015-29321 from the USDA National Institute of Food and Agriculture (NIFA) Animal Genome and Reproduction Programs and BARD grant number US-4997-17 from the US-Israel Binational Agricultural Research and Development (BARD) Fund. B.L. was supported in part by an appointment to the Agriculture Research Service (ARS) Research Participation Program, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and ARS. G.E.L., B.D.R., S.G.S., and C.P.V.T. were partially supported by appropriated project 8042-31000-001-00-D, “Enhancing Genetic Merit of Ruminants Through Improved Genome Assembly, Annotation, and Selection”; J.B.C., P.M.V., and C.P.V.T. were partially supported by appropriated project 8042-31000-002-00-D, “Improving Dairy Animals by Increasing Accuracy of Genomic Prediction, Evaluating New Traits, and Redefining Selection Goals”; and C.-j.L. was partially supported by appropriated project 8042-31310-078-00-D, “Improving Feed Efficiency and Environmental Sustainability of Dairy Cattle through Genomics and Novel Technologies” of the Agricultural Research Service of the U.S. Department of Agriculture. A.T. acknowledges funding from the Biotechnology and Biological Sciences Research Council (BBSRC) through program grants BBS/E/D/10002070 and BBS/E/D/30002275, Medical Research Council (MRC) research grant MR/P015514/1, and Health Data Research UK (HDR-UK) award HDR-9004. L.F. was partially funded through HDR-UK award HDR-9004 and the Marie Skłodowska-Curie grant agreement No. [801215]. O.C.-X. was supported by MR/R025851/1. L.J.A. is a retired USDA ARS employee.
Author contributions: L.F., L.M., and G.E.L. conceived and designed the project. L.F., W.C., S.L., O.C.-X., Y.G., J.J., and B.L. performed data analyses. O.C.-X, K.R., S.G.S., B.D.R., C.-j.L., T.S.S., L.J.A., C.P.V.T., P.M.V., J.B.C., Y.Y., S.Z., and A.T. contributed to the sample collection and resource generation. L.F., L.M., and G.E.L. wrote the paper. All authors read and approved the final manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.250704.119.
References
- Andersson L, Archibald AL, Bottema CD, Brauning R, Burgess SC, Burt DW, Casas E, Cheng HH, Clarke L, Couldrey C, et al. 2015. Coordinated international action to accelerate genome-to-phenome with FAANG, the functional annotation of animal genomes project. Genome Biol 16: 57 10.1186/s13059-015-0622-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208. 10.1093/nar/gkp335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. 2010. The NIH Roadmap Epigenomics Mapping Consortium. Nature Biotechnol 28: 1045–1048. 10.1038/nbt1010-1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry D, Buckley F, Dillon P, Evans R, Rath M, Veerkamp R. 2003. Genetic relationships among body condition score, body weight, milk yield, and fertility in dairy cows. J Dairy Sci 86: 2193–2204. 10.3168/jds.S0022-0302(03)73809-0 [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle EA, Li YI, Pritchard JK. 2017. An expanded view of complex traits: from polygenic to omnigenic. Cell 169: 1177–1186. 10.1016/j.cell.2017.05.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik-Sullivan BK, Loh P-R, Finucane HK, Ripke S, Yang J, Schizophrenia Working Group of the Psychiatric Genomics Consortium, Patterson N, Daly MJ, Price AL, Neale BM. 2015. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 47: 291–295. 10.1038/ng.3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burney MJ, Johnston C, Wong KY, Teng SW, Beglopoulos V, Stanton LW, Williams BP, Bithell A, Buckley NJ. 2013. An epigenetic signature of developmental potential in neural stem cells and early neurons. Stem Cells 31: 1868–1880. 10.1002/stem.1431 [DOI] [PubMed] [Google Scholar]
- Canela-Xandri O, Rawlik K, Tenesa A. 2018. An atlas of genetic associations in UK Biobank. Nat Genet 50: 1593–1599. 10.1038/s41588-018-0248-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole J, Vanraden P, O'Connell J, Van Tassell C, Sonstegard T, Schnabel R, Taylor J, Wiggans G. 2009. Distribution and location of genetic effects for dairy traits. J Dairy Sci 92: 2931–2946. 10.3168/jds.2008-1762 [DOI] [PubMed] [Google Scholar]
- Fang L, Sahana G, Ma P, Su G, Yu Y, Zhang S, Lund MS, Sørensen P. 2017a. Exploring the genetic architecture and improving genomic prediction accuracy for mastitis and milk production traits in dairy cattle by mapping variants to hepatic transcriptomic regions responsive to intra-mammary infection. Genet Sel Evol 49: 44 10.1186/s12711-017-0319-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Sahana G, Ma P, Su G, Yu Y, Zhang S, Lund MS, Sørensen P. 2017b. Use of biological priors enhances understanding of genetic architecture and genomic prediction of complex traits within and between dairy cattle breeds. BMC Genomics 18: 604 10.1186/s12864-017-4004-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Sahana G, Su G, Yu Y, Zhang S, Lund MS, Sørensen P. 2017c. Integrating sequence-based GWAS and RNA-Seq provides novel insights into the genetic basis of mastitis and milk production in dairy cattle. Sci Rep 7: 45560 10.1038/srep45560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Liu S, Liu M, Kang X, Lin S, Li B, Connor EE, Baldwin RL, Tenesa A, Ma L, et al. 2019. Functional annotation of the cattle genome through systematic discovery and characterization of chromatin states and butyrate-induced variations. BMC Biol 17: 68 10.1186/s12915-019-0687-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane HK, Reshef YA, Anttila V, Slowikowski K, Gusev A, Byrnes A, Gazal S, Loh P-R, Lareau C, Shoresh N, et al. 2018. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat Genet 50: 621–629. 10.1038/s41588-018-0081-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freebern E, Santos D, Fang L, Jiang J, Parker KG, Liu G, Vanraden P, Maltecca C, Cole J, Ma L. 2020. GWAS and fine-mapping of livability and six disease traits in Holstein cattle. BMC Genomics 21: 41 10.1186/s12864-020-6461-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georges M, Charlier C, Hayes B. 2019. Harnessing genomic information for livestock improvement. Nat Rev Genet 20: 135–156. 10.1038/s41576-018-0082-2 [DOI] [PubMed] [Google Scholar]
- Goddard ME, Hayes BJ. 2009. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat Rev Genet 10: 381–391. 10.1038/nrg2575 [DOI] [PubMed] [Google Scholar]
- Grisart B, Coppieters W, Farnir F, Karim L, Ford C, Berzi P, Cambisano N, Mni M, Reid S, Simon P, et al. 2002. Positional candidate cloning of a QTL in dairy cattle: identification of a missense mutation in the bovine DGAT1 gene with major effect on milk yield and composition. Genome Res 12: 222–231. 10.1101/gr.224202 [DOI] [PubMed] [Google Scholar]
- Grundberg E, Small KS, Hedman ÅK, Nica AC, Buil A, Keildson S, Bell JT, Yang TP, Meduri E, Barrett A, et al. 2012. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat Genet 44: 1084–1089. 10.1038/ng.2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The GTEx Consortium. 2017. Genetic effects on gene expression across human tissues. Nature 550: 204–213. 10.1038/nature24277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harel S, Tu EY, Weisberg S, Esquilin M, Chambers SM, Liu B, Carson CT, Studer L, Reizis B, Tomishima MJ. 2012. ZFX controls the self-renewal of human embryonic stem cells. PLoS One 7: e42302 10.1371/journal.pone.0042302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harhay GP, Smith TP, Alexander LJ, Haudenschild CD, Keele JW, Matukumalli LK, Schroeder SG, Van Tassell CP, Gresham CR, Bridges SM, et al. 2010. An atlas of bovine gene expression reveals novel distinctive tissue characteristics and evidence for improving genome annotation. Genome Biol 11: R102 10.1186/gb-2010-11-10-r102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heringstad B, Gianola D, Chang Y, Ødegård J, Klemetsdal G. 2006. Genetic associations between clinical mastitis and somatic cell score in early first-lactation cows. J Dairy Sci 89: 2236–2244. 10.3168/jds.S0022-0302(06)72295-0 [DOI] [PubMed] [Google Scholar]
- Hormozdiari F, Gazal S, van de Geijn B, Finucane HK, Ju CJT, Loh PR, Schoech A, Reshef Y, Liu X, O'Connor L, et al. 2018. Leveraging molecular quantitative trait loci to understand the genetic architecture of diseases and complex traits. Nat Genet 50: 1041–1047. 10.1038/s41588-018-0148-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Bickhart DM, Chung H, Hutchison JL, Norman HD, Connor EE, Liu GE. 2012. Analysis of copy number variations in Holstein cows identify potential mechanisms contributing to differences in residual feed intake. Funct Integr Genomics 12: 717–723. 10.1007/s10142-012-0295-y [DOI] [PubMed] [Google Scholar]
- Hu X, Ivashkiv LB. 2009. Cross-regulation of signaling pathways by interferon-γ: implications for immune responses and autoimmune diseases. Immunity 31: 539–550. 10.1016/j.immuni.2009.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu ZL, Park CA, Wu XL, Reecy JM. 2012. Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res 41: D871–D879. 10.1093/nar/gks1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Cole JB, Freebern E, Da Y, Vanraden PM, Ma L. 2019. Functional annotation and Bayesian fine-mapping reveals candidate genes for important agronomic traits in Holstein bulls. Commun Biol 2: 212 10.1038/s42003-019-0454-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA. 2012. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13: 484–492. 10.1038/nrg3230 [DOI] [PubMed] [Google Scholar]
- Kemper KE, Goddard ME. 2012. Understanding and predicting complex traits: knowledge from cattle. Hum Mol Genet 21: R45–R51. 10.1093/hmg/dds332 [DOI] [PubMed] [Google Scholar]
- Kennedy JM, Fodil N, Torre S, Bongfen SE, Olivier J-F, Leung V, Langlais D, Meunier C, Berghout J, Langat P, et al. 2014. CCDC88B is a novel regulator of maturation and effector functions of T cells during pathological inflammation. J Exp Med 211: 2519–2535. 10.1084/jem.20140455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360. 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konturek S, Konturek P, Pawlik T, Brzozowski T. 2004. Brain-gut axis and its role in the control of food intake. J Physiol Pharmacol 55: 137–154. [PubMed] [Google Scholar]
- Krueger F, Andrews SR. 2011. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27: 1571–1572. 10.1093/bioinformatics/btr167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Fang L, Null DJ, Hutchison JL, Connor EE, Vanraden PM, VandeHaar MJ, Tempelman RJ, Weigel KA, Cole JB. 2019. High-density genome-wide association study for residual feed intake in Holstein dairy cattle. J Dairy Sci 102: 11067–11080. 10.3168/jds.2019-16645 [DOI] [PubMed] [Google Scholar]
- Liu H, Zhou H, Wu Y, Li X, Zhao J, Zuo T, Zhang X, Zhang Y, Liu S, Shen Y, et al. 2015. The impact of genetic relationship and linkage disequilibrium on genomic selection. PLoS One 10: e0132379 10.1371/journal.pone.0132379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Liu X, Zhang S, Lv J, Li S, Shang S, Jia S, Wei Y, Wang F, Su J, et al. 2016. Systematic identification and annotation of human methylation marks based on bisulfite sequencing methylomes reveals distinct roles of cell type-specific hypomethylation in the regulation of cell identity genes. Nucleic Acids Res 44: 75–94. 10.1093/nar/gkv1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Vandehaar M, Spurlock D, Weigel K, Armentano L, Staples C, Connor E, Wang Z, Bello N, Tempelman R. 2015. An alternative approach to modeling genetic merit of feed efficiency in dairy cattle. J Dairy Sci 98: 6535–6551. 10.3168/jds.2015-9414 [DOI] [PubMed] [Google Scholar]
- MacLeod I, Bowman P, Vander Jagt C, Haile-Mariam M, Kemper K, Chamberlain A, Schrooten C, Hayes B, Goddard M. 2016. Exploiting biological priors and sequence variants enhances QTL discovery and genomic prediction of complex traits. BMC Genomics 17: 144 10.1186/s12864-016-2443-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuto M, Kano F, Murata M. 2015. Reconstitution of the targeting of Rab6A to the Golgi apparatus in semi-intact HeLa cells: a role of BICD2 in stabilizing Rab6A on Golgi membranes and a concerted role of Rab6A/BICD2 interactions in Golgi-to-ER retrograde transport. Biochim Biophys Acta 1853: 2592–2609. 10.1016/j.bbamcr.2015.05.005 [DOI] [PubMed] [Google Scholar]
- Meuwissen T, Hayes B, Goddard M. 2001. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157: 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Thomas PD. 2012. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res 41: D377–D386. 10.1093/nar/gks1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman H, Wright J, Kuhn M, Hubbard S, Cole J, Vanraden P. 2009. Genetic and environmental factors that affect gestation length in dairy cattle. J Dairy Sci 92: 2259–2269. 10.3168/jds.2007-0982 [DOI] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. 2015. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33: 290–295. 10.1038/nbt.3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiver MH, Lachance J. 2018. Adaptive eQTLs reveal the evolutionary impacts of pleiotropy and tissue-specificity, while contributing to health and disease in human populations. bioRxiv 10.1101/444737 [DOI] [PMC free article] [PubMed]
- R Core Team. 2018. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna: https://www.R-project.org/. [Google Scholar]
- Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, Bodenmiller B, Campbell P, Carninci P, Clatworthy M, et al. 2017. Science forum: the human cell atlas. eLife 6: e27041 10.7554/eLife.27041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedmaier AE, Burk O, van Eijck B, Schaeffeler E, Klein K, Fehr S, Biskup S, Müller S, Winter S, Zanger U, et al. 2016. Variability in hepatic expression of organic anion transporter 7/SLC22A9, a novel pravastatin uptake transporter: impact of genetic and regulatory factors. Pharmacogenomics J 16: 341–351. 10.1038/tpj.2015.55 [DOI] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium. 2015. Integrative analysis of 111 reference human epigenomes. Nature 518: 317–330. 10.1038/nature14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde PD, Demontis D, Cuyabano BCD, Børglum AD, Sørensen P, Group G. 2016. Covariance association test (CVAT) identifies genetic markers associated with schizophrenia in functionally associated biological processes. Genetics 203: 1901–1913. 10.1534/genetics.116.189498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde PD, Sørensen IF, Sørensen P. 2019. qgg: an R package for large-scale quantitative genetic analyses. Bioinformatics 36: 2614–2615. 10.1093/bioinformatics/btz955 [DOI] [PubMed] [Google Scholar]
- Ruan J, Xu J, Chen-Tsai RY, Li K. 2017. Genome editing in livestock: Are we ready for a revolution in animal breeding industry? Transgenic Res 26: 715–726. 10.1007/s11248-017-0049-7 [DOI] [PubMed] [Google Scholar]
- Santaolalla R, Abreu MT. 2012. Innate immunity in the small intestine. Curr Opin Gastroenterol 28: 124–129. 10.1097/MOG.0b013e3283506559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarup P, Jensen J, Ostersen T, Henryon M, Sørensen P. 2016. Increased prediction accuracy using a genomic feature model including prior information on quantitative trait locus regions in purebred Danish Duroc pigs. BMC Genet 17: 11 10.1186/s12863-015-0322-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seabury CM, Oldeschulte DL, Saatchi M, Beever JE, Decker JE, Halley YA, Bhattarai EK, Molaei M, Freetly HC, Hansen SL, et al. 2017. Genome-wide association study for feed efficiency and growth traits in U.S. beef cattle. BMC Genomics 18: 386 10.1186/s12864-017-3754-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuai K, Liu B. 2003. Regulation of JAK–STAT signalling in the immune system. Nat Rev Immunol 3: 900–911. 10.1038/nri1226 [DOI] [PubMed] [Google Scholar]
- Smith ZD, Meissner A. 2013. DNA methylation: roles in mammalian development. Nat Rev Genet 14: 204–220. 10.1038/nrg3354 [DOI] [PubMed] [Google Scholar]
- Sørensen IF, Edwards SM, Rohde PD, Sørensen P. 2017. Multiple trait covariance association test identifies gene ontology categories associated with chill coma recovery time in Drosophila melanogaster. Sci Rep 7: 2413 10.1038/s41598-017-02281-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F, Furlong EE. 2012. Transcription factors: from enhancer binding to developmental control. Nature Rev Genet 13: 613–626. 10.1038/nrg3207 [DOI] [PubMed] [Google Scholar]
- van Beuningen SF, Will L, Harterink M, Chazeau A, van Battum EY, Frias CP, Franker MA, Katrukha EA, Stucchi R, Vocking K, et al. 2015. TRIM46 controls neuronal polarity and axon specification by driving the formation of parallel microtubule arrays. Neuron 88: 1208–1226. 10.1016/j.neuron.2015.11.012 [DOI] [PubMed] [Google Scholar]
- Vanraden PM. 2008. Efficient methods to compute genomic predictions. J Dairy Sci 91: 4414–4423. 10.3168/jds.2007-0980 [DOI] [PubMed] [Google Scholar]
- Vanraden PM, Tooker ME, O'Connell JR, Cole JB, Bickhart DM. 2017. Selecting sequence variants to improve genomic predictions for dairy cattle. Genet Sel Evol 49: 32 10.1186/s12711-017-0307-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veerkamp R, Koenen E, De Jong G. 2001. Genetic correlations among body condition score, yield, and fertility in first-parity cows estimated by random regression models. J Dairy Sci 84: 2327–2335. 10.3168/jds.S0022-0302(01)74681-4 [DOI] [PubMed] [Google Scholar]
- Visscher PM, Brown MA, McCarthy MI, Yang J. 2012. Five years of GWAS discovery. Am J Hum Genet 90: 7–24. 10.1016/j.ajhg.2011.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, Yang J. 2017. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet 101: 5–22. 10.1016/j.ajhg.2017.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Misztal I, Aguilar I, Legarra A, Muir W. 2012. Genome-wide association mapping including phenotypes from relatives without genotypes. Genet Res 94: 73–83. 10.1017/S0016672312000274 [DOI] [PubMed] [Google Scholar]
- Watanabe K, Stringer S, Frei O, Mirkov MU, de Leeuw C, Polderman TJC, van der Sluis S, Andreassen OA, Neale BM, Posthuma D. 2019. A global overview of pleiotropy and genetic architecture in complex traits. Nat Genet 51: 1339–1348. 10.1038/s41588-019-0481-0 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Connor EE, Bickhart DM, Li C, Baldwin RL, Schroeder SG, Rosen BD, Yang L, Van Tassell CP, Liu GE. 2018. Comparative whole genome DNA methylation profiling of cattle sperm and somatic tissues reveals striking hypomethylated patterns in sperm. Gigascience 7: giy039 10.1093/gigascience/giy039 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.