

SUMMARY
Intracellular recordings in vivo remains the best technique to link single-neuron electrical properties to network function. Yet existing methods are limited in accuracy, throughput, and duration, primarily via washout, membrane damage, and movement-induced failure. Here, we introduce flexible quartz nanopipettes (inner diameters of 10–25 nm and spring constant of ~0.08 N/m) as nanoscale analogs of traditional glass microelectrodes. Nanopipettes enable stable intracellular recordings (seal resistances of 500 to ~800 MΩ, 5 to ~10 cells/nanopipette, and duration of ~1 hr) in anaesthetized and awake head-restrained mice, exhibit minimal diffusional flux, and facilitate precise recording and stimulation. When combined with quantum-dot labels and microprisms, nanopipettes enable two-photon targeted electrophysiology from both somata and dendrites, and even paired recordings from neighboring neurons, while permitting simultaneous population imaging across cortical layers. We demonstrate the versatility of this method by recording from parvalbumin-positive (Pv) interneurons while imaging seizure propagation, and we find that Pv depolarization block coincides with epileptic spread. Flexible nanopipettes present a simple method to procure stable intracellular recordings in vivo.
In Brief
Jayant et al. introduce flexible nanopipettes for minimally invasive intracellular electrical recordings in vivo and, in conjunction with two-photon imaging, show that these electrodes permit targeted recordings from both somata and thin dendrites. In addition, flexible nanopipettes facilitate targeted recordings across cortical depth and under different brain states.
Graphical Abstract
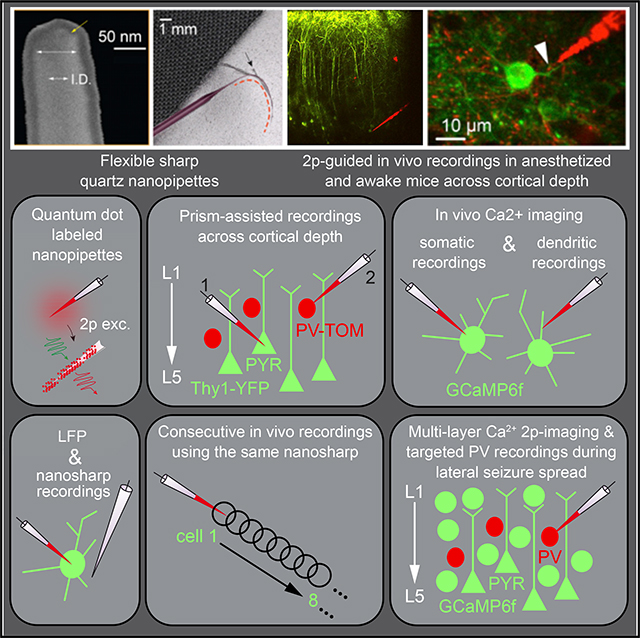
INTRODUCTION
Intracellular electrophysiological recordings in vivo are widely used to study synaptic integration (Petersen and Crochet, 2013; Svoboda et al., 1999), decipher local circuit interactions (Adesnik et al., 2012; Pluta et al., 2015; Polack et al., 2013), and elucidate the relationship between single-neuron dynamics and behavior (Houweling and Brecht, 2008). Yet limitations to this versatile technique have hindered more widespread use in vivo. The first pertains to electrode compliance, specifically the mechanical mismatch at the micropipette-neuron interface. Traditionally, single-cell electrophysiology in vivo is performed using sharp microelectrodes (Long et al., 2010; McIlwain and Creutzfeldt, 1967; Schneider et al., 2014; Svoboda et al., 1997, 1999; Yazaki-Sugiyama et al., 2009) or whole-cell patch pipettes (Kodandaramaiah et al., 2012; Lee et al., 2006, 2009; Margrie et al., 2002). Both techniques use glass microelectrodes, which have approximate tip diameters of ≥100 nm (sharp electrode) and 1–2 μm (patch electrode), as well as an abrupt conical taper with an outer diameter approaching ~1 mm within a few hundred microns of the tip. This geometry and design make the pipette stiff, which leads to low experimental throughput because of mechanical disruption, a critically important aspect when recording from soft tissues in awake moving animals (Lee et al., 2006). Furthermore, the large tip openings, coupled with the method of cell entry, can cause intracellular washout over time or, in the case of sharp microelectrodes, a permanent membrane shunt. Moreover, patch pipettes are not easily reusable in vivo, require continuous application of pressure while navigating through the brain, and exhibit variable access resistances with increased recording depths.
Another major limitation relates to the ability to perform targeted intracellular recordings from visually identified neurons deep in the living brain (Muñoz et al., 2014), a key requirement in modern circuit neuroscience (Petersen, 2017). Although targeted recordings can be performed using whole-cell patching under two-photon visual guidance, they are mainly relegated to superficial cortical regions because of scattering of light in deep tissue (Packer et al., 2015). However, conventional sharp electrodes are generally difficult to use under two-photon guidance due to poor visualization of the tip and inefficient pressure application, and they have only been used under two-photon visualization for targeted recordings in superficial brain regions and with larger pipette diameters (Svoboda et al., 1997). Furthermore, it is becoming increasingly important to be able to couple targeted recordings with simultaneous population calcium (Ca2+) imaging across layers, because it can help decode how single-neuron dynamics relates to network activity (Carrillo-Reid et al., 2016; Yuste and Katz, 1991). However, performing targeted in vivo electrophysiology applicable across any depth, along with multilayer population Ca2+ imaging (Yang et al., 2016), remains an unresolved technical challenge. Methods to overcome this limitation have treated these two components in isolation, solely focused on either improving the throughput of blind and targeted intracellular recordings in vivo (Kitamura et al., 2008; Kodandaramaiah et al., 2012; Long et al., 2015; Muñoz et al., 2014) or imaging neural activity in deeper regions of the cortex (Andermann et al., 2013; Horton et al., 2013; Theer et al., 2003; Tischbirek et al., 2015), but they have never sought to optimize them simultaneously, which is needed.
Ideally, to accomplish high-throughput intracellular electrophysiology in vivo, especially in awake animals, microelectrodes should be flexible, minimally invasive, and immune to movement, yet they also should be stiff enough to enter the brain, exhibit stable and high SNR (signal-to-noise ratio) recordings, be reusable, and be easily visualized under two-photon excitation to enable targeted recordings in vivo. Nanoelectrodes in the tens of nanometers range have gained widespread attention as minimally invasive intracellular voltage probes in vitro (Angle and Schaefer, 2012; Hai et al., 2010; Robinson et al., 2012; Tian et al., 2010). The nanoscale size, coupled with unique material properties, has been exploited to gain intracellular access with tight seals. However, measuring signals with stable resting membrane potentials (RMPs), negating the high impedance artifact (Brette et al., 2008; Jayant et al., 2017), and enabling precise stimulation is challenging, particularly in vivo. Flexible electrodes are mechanically more compatible with soft tissue and can be used to record for long periods in vivo, but they have only been realized for extracellular measurements to date because of their planar nature (Viventi et al., 2010). So could intracellular microelectrodes be modified to obtain a nanoscale flexible probe suitable in vivo? To address this challenge, we introduced electrolyte-filled quartz nanopipettes in vitro (Jayant et al., 2017), which have tips an order of magnitude smaller than conventional glass microelectrodes, exhibit stable and high SNR recordings, allow the measurement of direct current (DC) RMPs, and can be optimally compensated for electrode artifacts (electrode capacitance of ~8–10 pF). Here, we exploit the ultra-flexible property of these electrodes for in vivo use and demonstrate robust and stable measurements in the presence of animal movements. The ability to make long stable recordings, coupled with the benefit of being reusable, improves experimental throughput by allowing multiple cells to be probed by a single electrode without becoming clogged or causing washout. Furthermore, we demonstrate that flexible nanopipettes, in conjunction with quantum-dot labels and microprism-assisted two-photon imaging (Andermann et al., 2013), enable targeted recordings across all cortical layers, applicable in both the awake and the anaesthetized animal, with the advantage of being compatible with simultaneous population Ca2+ imaging.
RESULTS
Flexible Nanopipettes: Design and Characterization
From classical beam theory, the stiffness k of a hollow cylindrical tube of radius R and height h can be approximated by k = 3EI=L3 (cantilever approximation). Here, I ≈ πR3 tsignifies the geometric moment of inertia at the tip, L signifies the length of the taper, and E signifies Young’s modulus of the material. The product EI, which is the bending modulus, is a measure of how easily the structure will flex in response to a load. The stiffness k is directly proportional to the bending modulus and, through this, the cube of the radius, the thickness of pipette tip t, and inversely proportional to the cube of the length of the cylinder or taper. We used this relationship to optimize the form factor of fabricated quartz nanopipettes (each with an inner diameter [I.D.] of ~10–20 nm) (Figure 1A; STAR Methods) with long (7–10 mm), thin, and extremely flexible tapers (Figure 1B, left; Figure S1A) (radius curvature of ~2 mm and a bend of ~90 at the stem) that were nonetheless axially stiff to enter the brain without any change in tip resistance. Furthermore, the pipettes did not show signs of permanent deformation when bent (Figure 1B, middle) and were able to immediately regain their original shape thereafter (Figure 1B, right). We then scanned an atomic force microscope (AFM) tip across the long and narrow taper (Figure 1C) to measure the bending stiffness from the force distance curve (Figure S1B). We found bending stiffness values of ~0.08 N/m near the tip (black trace, Figure 1C) a value much lower than conventional flexible extracellular electrodes (Liu et al., 2015) and identical to the stiffness of cantilever probes used in AFM analysis of cell membranes (Qi et al., 2015). We compared this pipette design against quartz microelectrodes with shorter tapers but a nearly similar tip size ( ~50–100 nm outer diameter) (pink trace, Figure 1C) and found an order of magnitude difference in bending stiffness, which suggests that cone angle and taper length are key parameters in deciding flexibility. Because of material limitations, conventional borosilicate glass microelectrodes yield shorter tapers, wider cone angles, and larger tips, which render them less compliant than our long quartz nanopipette. Moreover, because of the nanoscale tip and the associated geometries achieved (Figure 1D), these electrodes are reusable, do not easily clog, and minimize intracellular washout. In addition, simulations based on the coupled Poisson-Nernst-Planck and Navier-Stokes equations (Lan et al., 2016) suggested minimal diffusional flux of ~0.1–1 fM/s for the geometries realized with resistances dependent on taper geometry and cytoplasmic diffusion constants (Figure 1E; Figure S1C; Methods S1). Electrolyte-filled (3 M KCl) nanopipettes were optimally compensated (wall capacitance of ~9–10 pF) in the extracellular bath before cell entry to reduce signal attenuation due to filtering (STAR Methods). Transmembrane entry was elicited either through complete tip insertion (type A entry, fully intracellular) (Figure 1F, left) or through an electroporation-induced nanopore (type B entry, quasi-intracellular) (Figure 1F, right) as previously described (Jayant et al., 2017). Clogging did not occur, and we hypothesize this resulted from the low flux and the presence of vortex flows at the tip of the nanopipette (Figure S1D), which preclude lipids from flowing into the orifice. Type B recordings can be characterized as one of two forms. In the first case, the recordings are similar to the quasi-intracellular or in-cell recording approach (Hai et al., 2010; Moore et al., 2017), in which the cell membrane engulfs the electrode tip, creating high seal resistance. Upon electroporation, the pore resistance (Rp) drops and thus dominates the capacitive contribution to the overall impedance of the membrane junction (i.e., Cp). The recordings in this case reflect attenuated intracellular signals, because the membrane voltage gets divided across the seal resistance (Rs) and Rp (Figure 1F, right). This attenuated signal can, if needed, be deconvolved or reestimated offline using measured equivalent circuit parameters (Jayant et al., 2017) (STAR Methods; Figure S2; Methods S1). However, in the absence of an electropore, and provided Rs is high, the waveform recorded by the nanopipette reflects a high-seal extracellular signal. This is because the C contribution to the impedance (Figure 1F, right) dominates over Rp (i.e., Cp dVm/dt dominates and Rp is very large). This form is similar to the cell-attached configuration in which the signal looks like the time derivative of the intracellular signal. The relative advantages of both forms of type B recording will be discussed in later sections, in which we describe recordings from dendrites and cell bodies with concomitant intracellular Ca2+ imaging. In this manuscript, we explicitly mention the particular cases in which type B recordings were employed and deconvolution or reestimation was used. All data presented in this manuscript are raw.
Figure 1. Flexible Nanopipette Design Characteristics.

(A) SEM of the nanopipette (left) and magnified view of the tip (right). The outer diameter-to-inner diameter (I.D.) ratio is 2:1. Note the outer 10 nm platinum coating layer required for SEM imaging.
(B) Images depicting the degree of nanopipette flexibility (left, middle, and right). The nanopipette (false colored; red dotted line) is immobilized on tape, and the taper is subsequently bent (left). The taper is bent farther (>60° ) (middle) and subsequently recovers to a taut axially stiff pipette (right) when removed from the tape without damage. The black arrows indicate imprints of the taper in the tape.
(C) AFM-based stiffness measurements of nanopipettes for varying taper lengths (black, blue, and red indicate a long nanopipette with small variations in taper length (~8 mm), and pink represents a short tapered conventional sharp microelectrode design). The inset depicts the AFM tip (red) scanning the tip of the nanopipette (yellow). The spring constant (stiffness) of the long tapered nanopipette close to the tip (~0.08 N/m) reveals a highly compliant structure.
(D) Geometry of the two types of nanopipettes commonly used: conical tip (upper left) and cylindrical tip with conical taper (upper right). The half cone angle (theta), length of the taper (Ltaper), diameter of the tip (dtip), and access resistance (Raccess) influence the net resistance, concentration gradient, and flux. Simulated heatmap of the concentration gradient at the tip of a purely conical nanopipette (bottom). Note the large gradient close to the tip.
(E) Numerical simulations of resistance versus theta (left y axis) and flux versus theta (right y axis) for the two geometries shown in (D) for a fixed diameter of 15 nm and 3 M KCl filling solution. Low flux values (<1 fM/s) can be achieved when the pipette tip is cylindrical (theta of ~0) near the end and the diffusion constant of ions at the tip (cytoplasmic fluid) is lower than bulk. Resistance versus theta was calculated analytically and corroborated numerically (see text for details).
(F) Equivalent circuit models showing the nanopipette cell membrane interface under full tip impalement (left, type A) and electroporation-induced or cell-attached entry (right, type B). Here, Re and Ce denote the distributed pipette resistance and capacitance, respectively. Rsh, Rs, and Rp represent the shunt, seal, and pore resistance, respectively. Rm represents the membrane resistance, Cm represents the membrane capacitance, Cp represents the pore capacitance, and Em represents the reversal potential of the leak channels. Depending on whether Rp or Cp dominates the junction contribution, and under a moderate to high Rs, the recorded signal Ve could reflect either a purely capacitive extracellular signal (Cp dominates, similar to cell-attached recording) or a tightly coupled and scaled intracellular signal (Rp dominates, similar to quasi-intracellular or in-cell recordings).
(G) CdSe/Cds/ZnS QDs label nanopipettes through adsorption, allowing clear visualization of the nanopipette relative to the patch pipette under two-photon visualization (bottom).
One important aspect that has traditionally precluded sharp microelectrodes from being used for targeted recordings in vivo is the poor visibility of the tip. Conventionally, whole-cell patch pipettes are filled with a dye, which allows the localization of the tip under two-photon illumination. Unfortunately, such dyes do not label the nanopipette shank and tip due to the miniature lumen, and they are prone to photobleaching, which makes it hard to visualize the tip (K.J., unpublished data). However, quantum dots (QDs) have a high two-photon absorption cross-section and are less prone to photobleaching, which renders them suitable labels for deep-layer in vivo studies (Andrásfalvy et al., 2014; Jayant et al., 2017). In this study, QD labeling of the nanopipette from the outside (Figure 1G; STAR Methods) enabled precise tip localization.
Targeted Recordings from Pyramidal Cells
To visualize the target cell and nanopipette in deep cortical layers and to enable precise registry of the QD-coated nanopipette to the target cell of interest, we used two-photon imaging through implanted microprisms (Figure 2A; Figure S3). We first targeted a layer 5 pyramidal cell in a lightly anesthetized thy1-YFP transgenic mouse (Feng et al., 2000) (STAR Methods) using flexible nanopipettes. Layer 5 somata with their main apical dendrites were visualized, and a target cell was identified by two-photon imaging through the prism (Figure 2B, left; Figures S3B and S3C). The QD-coated nanopipette was then guided to the target neuron under dual-color imaging (Figure 2B, right, inset; STAR Methods). Transmembrane access was elicited through type A, in which we clearly observed spontaneous action potentials (APs), sub-threshold activity, and cortical up states (Figure 2C, left, i) for durations routinely lasting more than 1 hr (statistics on recording durations are discussed in the section describing data from awake animals). Membrane shunting was negligible upon entry, which allowed unfiltered measurements of action potential (AP) amplitude with high temporal resolution. Moreover, with optimally compensated electrodes, we were able to induce AP firing activity via brief current injection (Figure 2C, right, ii). As a comparison, we imaged through the prism to guide whole-cell patch pipettes toward layer 5 pyramidal cells (850 μm below the surface of the pia) and performed stable (1.5 hr) targeted whole-cell patch-clamp recordings (Figure 2D). The SNR of whole-cell recordings (not corrected for junction potentials) was comparable in amplitude and timescale to nanopipette recordings (Figure S4), including similar baseline RMP values. Access resistances with whole-cell recordings in deep layers typically ranged between 13 and 80 MΩ Using simultaneous whole-cell patch and nanopipette recordings from somata of layer 5 pyramidal neurons in cortical slices, we measured the effective shunting-seal resistance of the nanopipette neuron interface. The input resistance of the cell was measured using the whole-cell patch electrode before and after nanopipette penetration (Figure 2E; STAR Methods). We found a mean seal resistance of ~600 MΩ (maximum of ~820 MΩ, n = 7 cells) (Figure 2E), a value much larger than the input resistance of most neurons. Such RMPs were obtained readily during entry in vivo (Figure 2C; Figure S4A); hence, measurement using type A entry did not need deconvolution or reestimation. We also found that AP properties—namely, RMP, AP rise time, decay time, and AP half-width recorded using nanopipettes—were nearly identical to those measured with whole-cell patch recordings in vivo (n = 5 cells) (Figure 2F). Specifically, the RMP and AP half-width measured using sharp nanopipettes (−67.99 ± 3.6 mV; 1.5 ± 0.3 ms; n = 5, mean ± SEM) were similar to those measured with whole-cell patch recordings (−68 ± 2 mV; 2.02 ± 0.46 ms; n = 5, mean ± SEM) indicating negligible shunting and the formation of a tight seal. Extending the application space and demonstrating the versatility of our flexible nanopipettes, we performed paired recordings from two pyramidal cells located one above the other in layer 5 (labeled 1 and 2) (Figure S4C).
Figure 2. Targeted Intracellular Recordings from Layer 5 Neurons in the Visual Cortex of Lightly Anaesthetized Mice.

(A) Illustration showing the targeted intracellular recordings technique applicable across all cortical layers. A glass microprism (height, 1 mm; width, 1 mm) with a reflective coating along the slanted edge is implanted into the brain of a head-fixed mouse. Two-photon imaging through the prism allows the visualization of labeled neurons and enables the precise guidance of the quantum-dot-coated sharp nanopipette to the target cell.
(B) Targeted nanopipette recordings (type A entry) from thy1-YFP-labeled layer 5 pyramidal neurons (top, left) using the technique outlined in (A). The prism enables the precise guidance of the nanopipette to the target cell (inset, right) and simultaneous visualization of the entire cortical column up to layer 1.
(C) (i) Intracellular measurements from the targeted pyramidal shown in (B) and typical AP characteristics (dotted green section). (ii) Membrane depolarization elicited through current injection across the sharp nanopipette in bridge mode. The electrode artifact is extremely low.
(D) Targeted whole-cell patch-clamp recordings from layer 5 pyramidal neurons. Note the high access resistances.
(E) Nanopipette shunt resistance (red) of 615.29 ± 59.69 MΩ (mean ± SEM, n = 7) measured using simultaneous whole-cell patch and nanopipette recordings (type A) from somas of neurons (inset) in vitro (see text for details). Gray dots reflect all measured values.
(F) Comparison of RMP, AP rise time, decay, and half-width measured using whole-cell patch pipettes (red) and nanopipettes (blue). Plots reflect mean ± SEM.
Targeted Recordings from Pv Interneurons
To expand the experimental repertoire of our combined flexible nanopipettes and microprism technique, we targeted superficial and deep-layer parvalbumin-positive (Pv) interneurons. We first recorded from layer 2/3 Pv interneurons (Pv1 and Pv2) (Figure 3A) and elicited entry via weak electroporation (type B). This mode of recording is easy to establish; occurs almost instantaneously upon contact with the cell membrane due to tight seals (either quasi-intracellular or extracellular); allows long recordings (the longest recording was ~2.5 hr), especially in the presence of movement or brain tissue pulsation; and can be easily reestablished in the event of recording loss. Specifically, the electroporated pore tends to reseal, evidenced by a return of the baseline to the prerecording condition, which can be electroporated again to achieve repeated intracellular access. However, type B entry, as described earlier, results in signal attenuation due to the voltage divider between the seal and the pore resistance (Figure 1F, right). This voltage divider will affect the absolute steady-state value but still permit detection of the AP transient (alternating current; AC). We clearly observed fast spiking activity of a labeled Pv interneuron (Pv1) (Figure 3B, gray box, top), based on the measured time constants and steady-state transients. These quasi-intracellular signals could be easily deconvolved or reestimated to reflect ~70 mV amplitudes (Pv1) (Figure 3B, gray box, bottom; Figure S2; Methods S1). We then used the same electrode and proceeded to record from another visually identified Pv interneuron along the same diagonal axis but located in a deeper layer. We again elicited transmembrane access via type B entry (Pv2) (Figure 3B, green box, right) and were able to observe fast spiking activity. The steady-state potentials (i.e., baseline) measured upon entry by the nanopipette were nearly identical in both cells (about −12 mV). This shows that the pipette did not clog, electroporation resulted in similar pore diameters, seal resistances were identical, and the tip potential (i.e., offsets due to charge at the tip, nominally about −2 to −4 mV) was unchanged across the traversed distance. The concept of a liquid junction potential does not affect these recordings, because the K+ and Cl− mobilities are nearly the same. A constant tip potential (due to the charge of the glass wall right at the tip and the nearly constant flux of ions) can be easily measured and subtracted from the overall RMP recording.
Figure 3. Targeted Intracellular Recordings from Pv Interneurons in the Visual Cortex of Lightly Anaesthetized Mice.

(A) Targeted recordings from tdTomato-labeled Pv interneurons (top, left). Inset 1 (top, right, Pv1) depicts targeted contact to a layer 2/3 Pv, while inset 2 (bottom, right, Pv2) depicts image-guided access to a layer 5 Pv using the same nanopipette. The nanopipette was advanced to layer 5 once the recordings in layer 2/3 were terminated. Here, nanopipette entry was elicited through electroporation (type B, see text for details) as opposed to full penetration (mechanical advancement with electrical stimulation). Electroporation-induced entry typically lasted several minutes until the pore resealed. Then, entry into the cell could again be evoked by application of an electroporation pulse.
(B) Raw type B recordings from the Pv interneurons shown in (A): layer 2/3 Pv (left box, marked 1) and layer 5 Pv (right box, marked 2). The steady-state potentials measured via electroporation from Pv1 and Pv2 are nearly identical, indicating little or no change in the tip potential of the nanopipette. Raw waveforms reflect scaled intracellular signals due to the high impedance junction (membrane voltage is divided across seal resistance and pore resis-tance). Deconvolved (reestimated) APs using the measured time constants and electroporation transients (see text for details) revealed full-scale AP spikes with a 0.5 ms half-width (bottom right of left box).
(C) Typical sharp nanopipette recordings (raw) from Pv interneurons in vivo elicited via type A entry.
(D) Spike-triggered average (STA) of the measured APs from (C).
(E) STA (left) of APs from a layer 4/5 Pv (raw, top) and simultaneously recorded local field potentials (LFPs; bottom; 150 mm depth) demonstrate that spikes coincided with brief extracellular potentials or putative network-wide up states. LFP recordings display a spike-locked increase in broadband power (middle) but specific phase locking in both theta-alpha (3–10 Hz) and gamma (35 Hz) bands (right and bottom, 100 trials; p values reflect Rayleigh statistics).
(F) Recordings from a layer 5 pyramidal neuron (raw) demonstrate similar spike-locked potentials and spectral power changes in the LFP, but phase locking was only present in the delta-theta band.
How do type B recordings compare with impalement-based (type-A) entry, and does electrolyte diffusion from the nanopipette affect Pv interneuron dynamics? To test this, we impaled Pv interneurons in vivo (Figure 3C). We observed that the AP amplitude, rise, decay, and RMPs were typical of Pv interneurons (Figure 3D) and were identical to the reestimated results in the cell-attached configuration (Pv1) (Figure 3B, gray box, bottom). This shows that type B recordings are physiological and can be used when full membrane impalement is not possible. Stable intracellular recording properties for ~1 hr and a calculated diffusional flux of ~0.1–1 fM/s proved that diffusion of KCl across the nanometer opening of the pipette does not affect cell physiology. Yet the lack of diffusion precludes diffusion of dye across the pipette tip, which makes it hard to backfill a neuron. To complete the overall characterization of the method, we studied the relation between intracellular Pv signals and network activity. To this effect, we combined intracellular recordings with local field potential (LFP) measurements (Figures 3E and 3F). We found that Pv intracellular APs clearly coincided with the LFP, demonstrating a spike-locked increase in broadband power (Figure 3E, top). Specific phase locking (Siapas et al., 2005; Womelsdorf et al., 2014) (Figure 3E, bottom) was clearly observed in both the theta-alpha band (3–12 Hz) and the gamma band (35 Hz), which is a hallmark of cortico-hippocampal Pv interneuron activity. In contrast, although intracellular recordings from targeted pyramidal cells showed similar spike-locked potentials and spectral power trends (Figure 3F, top), phase locking was only present in the delta-theta band (1–8 Hz) (Figure 3F, bottom), confirming that our targeted approach was specific to the cell visualized.
Robust and Stable Intracellular Recordings in Awake-Behaving Mice
We next performed intracellular recordings using flexible nanopipettes from awake head-fixed mice, allowed to move on a running wheel (Figure 4A). Although targeted recordings have been performed in awake head-fixed animals (Polack et al., 2013) and freely moving animals (Lee et al., 2006; Long and Lee, 2012), the mechanical mismatch between patch pipette and cell is a major contributor to recording disruption. Specifically, and as in the case of mice, body movements such as crouching, swaying from side to side, walking, and running are common (Figure 4A) and can easily lead to loss of recording. It is paramount that the electrode be able to both stimulate and record without loss of seal under these conditions. We first recorded from neurons ~800 μm below the pia (Figure 4B) using our flexible nanopipettes blindly. We elicited break-in (type A), with a stable baseline RMP (Figure 4B, black). Intracellular entry was established, and the movement of the mouse was monitored via a motion sensor on the wheel (Figure 4B, gray). A single transition in the motion sensor trace indicates subtle movement or single steps, while multiple transitions indicate running. We then injected brief current (Figure 4B, red) pulses into the cell to evoke an AP response and observed normal AP amplitude, rise time, and decay without loss of signal quality (Figure 4B, inset). Once the mouse stopped moving, spontaneous AP activity continued with the same stable RMP, demonstrating that the nanopipette was able to maintain a stable seal both upon entry and in the presence of locomotion. In a separate experiment we targeted Pv interneurons 600 μm below the surface of the pia, using flexible nanopipettes and microprisms (Figure 4C). We found a stable baseline, even during running (Figure 4C, gray), without loss of recording. We observed that the AP firing rate increased while running (Figure 4C, red), reduced when motion stopped (Figure 4C, blue), and increased again during subtle motion (Figure 4C, green). We clearly observed a slight depolarization in the RMP just as locomotion started (Figure 4C, green), which has been shown to influence brain state-dependent signal processing (Polack et al., 2013). The AP amplitude was nearly constant throughout the time the mouse was running, again indicative of stable and high seal resistance.
Figure 4. Targeted Intracellular Recordings from the Somatosensory Cortex in Awake Moving Mice.

(A) Schematic depicting blind and targeted intracellular recording using flexible nanopipettes in the awake head-fixed animal. The mouse is allowed to move on a circular wheel. The mouse exhibits various body movements: sway, crouch, walk, and run. The flexible nanopipette bends with this motion while maintaining intracellular access (inset).
(B) Typical blind recording performed ~800 μm deep in the visual cortex (V1). Intracellular break-in (type A) is elicited upon nanopipette stimulation (dark gray, shaded region) with a stable RMP of about −78 mV. Running and rest conditions (gray trace, black dots, digitized for clarity) are monitored by the motion sensor. Current injection (red trace) elicits clear AP activity (green dotted lines, shaded region) with a minimal increase in pipette resistance (residual unbridged resistance after cell entry). Note the stable intracellular baseline even in the presence of movement.
(C) Targeted intracellular recording (type A, black trace, top) from a layer 2/3 Pv interneuron (right, inset) in the motor cortex showing stable AP amplitude and steady RMP. Recordings last ~10 min to 1 hr in the presence of locomotion. Motion sensor (gray, raw data) indicates a rapid running movement (red region) followed by a gradual decrease, eventually coming to rest (blue region), and a subsequent movement again (green region, walk or run). Note the strong correlation between onset of movement and increase in AP firing rate (green trace).
(D) RMP for ten consecutive cells measured using a single nanopipette in each mouse. Entry in all cases was type A. Note the consistency in RMPs measured across multiple cells using the same electrode. The green dotted line indicates the −55 mV mark. This result clearly demonstrates that nanopipettes do not clog and can be repeatedly reused. The red dotted box shows the amplitude and stability of each recording from mouse 4 as an example.
(E) Duration of nanopipette recordings across 7 awake mice (n = 9 cells) compared to duration of recordings achieved using the whole-cell patch recordings in anaesthetized mice.
Data reflect mean ± SEM. Note the similarity in mean and maximal durations.
We then proceeded to demonstrate the stability (i.e., of RMP and AP amplitude), repeatability (i.e., number of cells per nanopipette), and robustness (i.e., duration of recordings) of the nanopipette under such conditions. We demonstrated repeatability concomitantly by recording blindly from multiple cells using a single nanopipette across ~900 μm of the cortex with both type A entry (n = 4 mice, 5–10 cells/pipette) (Figure 4D) and type B entry (Figure S5). We considered a recording successful if the RMP was more negative than −55mV and AP potential amplitude was at least 50–70mV. The stability of the RMPs indicated an unclogged pipette and healthy seal formation in every cell entered. This ability of the pipette to remain unclogged as it traverses across the brain was tested by recording both in the neocortex and in sub-cortical regions. We found that the pipette was essentially unchanged in resistance across depth and was able to record full-length APs, even 5 mm deep (Figure S6). The red dotted box (data from mouse 4 in Figure 4D) shows the waveforms recorded during this experiment in which every cell entered had stable baseline and AP characteristics. We held each of these cells for at least 3 min before proceeding to the next cell. This was done to demonstrate the reusable nature of the electrode. To demonstrate the stability of our recordings (i.e., the duration), we compared recording durations procured using the nanopipette across 4 awake mice with whole-cell recordings procured in anaesthetized animals. Whole-cell recordings in anaesthetized conditions present the best-case scenario in terms of recording durations, because they should be relatively immune to movement-induced failure. We were able to consistently maintain nanopipette intracellular access up to ~1 hr when exclusively testing for duration alone (n = 7 mice, 9 cells, and 6 nanopipettes) (Figure 4E). These durations performed in awake locomoting animals matched whole-cell patch recordings from anaesthetized mice (n = 5 mice and 5 cells), suggesting that the nanopipette recordings are similar in terms of stability. In contrast, type B recordings could last between 2.5 and 3.5 hr in an awake running animal and across multiple animals without loss of SNR (n = 9 cells from 4 mice) (Figure S5). Altogether, these results suggest that flexible nanopipettes can ensure minimally invasive recordings (if type B is used), facilitate high-throughput experimentation, and permit stable recordings.
Targeted Recordings from Dendrites and Simultaneous Ca2+ Imaging from Somata
In combination with Ca2+ imaging in vivo, intracellular recordings can potentially elucidate signal integration properties in neurons and sub-cellular dendritic processes (Palmer et al., 2014) and tie intracellular dynamics to population activity (Carrillo-Reid et al., 2016; Stosiek et al., 2003). To demonstrate the efficacy of the nanopipette, and the usefulness of the type B mode of recording, we performed simultaneous intracellular recordings and Ca2+ imaging from the soma and dendrites of labeled cells. As mentioned previously, depending on the nature of the pipette-membrane interface, type B recordings can result in either pure extracellular recordings (membrane capacitance dominates) or quasi-intracellular recordings (pore resistance dominates the capacitance contribution). First, we performed prism-assisted targeted electrophysiology from a pyramidal neuron 850 mm below the pial surface (Figure 5A) and simultaneous Ca2+ imaging from neighboring cell bodies in transgenic mice expressing GCaMP6f (Dana et al., 2014) (Figure 5B). Intracellular access was elicited using type B entry to avoid the formation of transmembrane shunt (Figure 5C, left). This approach resulted in attenuated spikes but could be easily reestimated offline (Figure 5C, right; Figure S2; Methods S1). Although these intracellular spikes were attenuated, the SNR was sufficiently high to observe a clear correspondence between individual APs and Ca2+ transients. Moreover, with electroporation-induced entry, reentry upon pore resealing could be reestablished without causing apparent damage to the cell. Then, in a separate experiment, we used our flexible nanopipettes to probe oblique dendrites close to the soma in both pyramidal cells and Pv interneurons (Figures 5D–5F). Targeted dendritic recordings were performed similarly to the targeted somatic recordings except that the pipette was moved more slowly when close to the membrane, readjusted by slow retraction if needed, and then renavigated to the dendrite. In GCaMP6f-expressing pyramidal neurons from layer 2/3, upon contact with the proximal basal dendrite (Figure 5D, left), which measured ~1 μm in diameter, we immediately noticed high SNR extracellular spikes (Figure 5D, right), which were accompanied by corresponding Ca2+ transients in the soma (Figures 5D and 5E). The SNR of these recordings increased as the recording ensued, suggesting that seal formation is a dynamic process. Such recordings typically lasted several minutes (n = 6 dendrites, maximum duration of ~9 min) before we lost contact due to motion, but contact could be reestablished by subsequent pipette readjustment. With recordings made from apical dendritic segments (Figure S7), we achieved better stability and were able to clearly procure type B quasi-intracellular signals along with Ca2+ transients. Thus, this approach can be considered a nanoscale analog of the classical cell-attached configuration using patch pipettes. With Pv interneurons exclusively labeled by tdTomato (Figure 5F, left), dendritic recordings (Figure 5F, right) also reflected a quasi-intracellular type B interface in which the recorded waveforms resembled attenuated intracellular potentials (Figure 5G). These signals were characterized by their faster rise times and half-widths typical of Pv dendritic recordings (Hu et al., 2010). Altogether, these results indicate that nanopipettes are minimally invasive and allow targeted recording from small, sub-cellular dendritic processes.
Figure 5. Simultaneous Two-Photon Ca2+ Imaging and Targeted Recordings from Somata and Dendrites in the Visual Cortex of Lightly Anaesthetized Mice.

(A) Simultaneous Ca2+ imaging and targeted intracellular recordings from GCaMP6f-labeled layer 5 pyramidal cells.
(B) Raw cellular Ca2+ transients for both the targeted cell and the two neighboring neurons.
(C) Simultaneous Ca2+ transients and intracellular potential dynamics from cell 1 (region outlined by a box in B). Intracellular entry was elicited through electroporation (type B, in-cell recordings), rather than impalement, to avoid shunting. Note the clear registry of Ca2+ transients to APs (inset, gray shaded region). Single APs could be reestimated offline (right, orange trace).
(D) Targeted electrical recordings from a basal dendrite of a GCaMP6f-labeled layer 2/3 pyramidal neuron (left). The raw waveforms reflect a type B recording (right) in which the nanopipette forms a tight seal with the dendrite and measures a capacitively coupled signal. Simultaneous Ca2+ transients recorded from the soma show a high degree of correlation with the AP spikes (bottom). The extracellular SNR improved over time, indicating better seal formation.
(E) Sections of the trace shown in (D), underlined in red or gray. The nanopipette is sensitive enough to measure single dendritic spikes that coincide with somatic Ca2+ transients.
(F) Targeted dendritic recordings from Pv interneurons (left and middle, white arrow, and inset). The two images show the nanopipette first approaching the target Pv (left, tip magnified in blue inset) and then in contact with the dendrite (middle). Raw AP waveforms reflect a type B entry (in-cell recordings) with a high seal. The steady-state voltage measured was close to the potential measured before contact with the dendrite, indicating no intracellular access. Here, the waveform resembles an attenuated intracellular signal, indicating a stronger resistive contribution at the junction between the pipette and the membrane.
(G) Typical dendritic AP waveform and STA of the waveforms (inset) recorded from the Pv dendrite.
Targeted Recordings from Pv Interneurons during Epileptic Seizures
Finally, to demonstrate the applicability of flexible nanopipettes to answer biological questions, we measured Pv interneuron dynamics in a pharmacological seizure model. Studies in brain slices and the intact brain have suggested that epileptiform activity is restrained by feedforward inhibition, the failure of which results in ictal progression (Trevelyan et al., 2006). Of the different inhibitory cell classes, Pv interneurons are considered an important source of this inhibition and have been reported to undergo firing impairment during propagation of epileptiform activity in vitro (Cammarota et al., 2013). However, how Pv interneurons affect local seizure progression and how they tie to ictal network activity in vivo remain poorly understood (Neumann et al., 2017; Wenzel et al., 2018). To address these questions, we performed prism-assisted targeted intracellular recordings from Pv interneurons, simultaneously measuring the LFP, and performed imaging Ca2+ activity in the local cortical network during seizure spread (Figure 6; STAR Methods). We induced epileptic seizures via brief surface application of 4-aminopyri-dine (4-AP) (1 mM), a well-established chemoconvulsive that leads to the acute formation of seizures, which are electrographically and behaviorally similar to focal onset seizures observed in humans (Wenzel et al., 2017). We found that the optical break-in of epileptiform activity into our field of view (Video S1) coincided with both the electrographic seizure and a Pv depolarization block (Figures 6C–6E). Moreover, before the cessation of firing, Pv neurons displayed sustained enhanced average firing rates (2 animals, 5 Pv interneurons, 15 preictal periods each, and 10.27 ± 1.956 spikes/s during the 5 s before seizure onset versus 2 animals, 5 Pv interneurons, 15 ictal periods, and 0.26 ± 0.13 spikes/s during the 5 s after seizure onset; p = 0.006) (Figure 6E). Confirming the inverse relation between Pv firing and ictal spread, toward the end of an electrographic seizure, the firing rate and RMP again stabilized (Figure 6D). Steady intracellular conditions were maintained even though the mouse at times experienced convulsions during seizures, an aspect we attribute to the flexible nature of the nanopipette. These results emphasize the critical role of Pv interneurons in restraining epileptic activity in the intact mammalian brain. Moreover, the recovery of Pv interneuron activity toward the end of seizures carries potential implications for the importance of Pv interneurons in seizure termination. For details on success rate and drawbacks, refer the Quantification and Statistical Analysis section.
Figure 6. Simultaneous Two-Photon Population Imaging and Targeted Intracellular Recordings during Epileptic Seizure Spread.

(A) Illustration showing simultaneous LFP recording, targeted intracellular recording, and dual-color two-photon imaging of both tdTomato-labeled Pv and GCaMP6f-expressing pyramidal cells from both the visual and the somatosensory cortex.
(B) Simultaneous intracellular (type A) recording from Pv interneurons (Pv1 and Pv2, red) approximately 400 μm below the surface of the pial layer, and population Ca2+ imaging (green) across the cortical column. The nanopipette was unchanged between recordings.
(C) Population Ca2+ transients from pyramidal cells (top, 5 individual cells in gray, population mean in blue) corresponded well with 4-AP-induced electrographic seizures (middle, LFP in red). Note the abrupt depolarization block in the Pv1 intracellular recording (bottom, black trace), which coincides with a sharp rise in population Ca2+ activity during seizure propagation into the field of view.
(D) Six consecutive seizures while recording from Pv1 and Pv2 (bottom, left and right, respectively, type A) depict the trend shown in (C). For each Pv, three consecutive optical break-ins of epileptic activity are superimposed (top, left and right, average population Ca2+ transient). Both Pv interneurons show consistent, immediate cessation of firing upon optical break-in of seizure activity, consistent with entering depolarization block. The intracellular membrane dynamics returned to the same baseline after each seizure period. No current was injected during voltage recordings. AP waveforms were not distorted, but firing rates changed after ictal break-in.
(E) Compared to baseline, Pv average firing rates increased during pending electrographic seizures (2 animals, 5 Pv interneurons, 15 preictal periods each, and 10.27 ± 1.956 spikes/s during the 5 s before seizure onset) but ceased upon optical break-in of ictal activity during electrographic seizure occurrence (2 animals, 5 Pv interneurons, 15 ictal periods, and 0.26 ± 0.13 spikes/s during the 5 s after seizure onset) (p = 0.006).
The concomitant RMP shift in (D) suggests a depolarization block.
DISCUSSION
Intracellular electrophysiology is a 6-decade-old foundational neuroscience technique (McIlwain and Creutzfeldt, 1967), and its basic methodology, because of its uncomplicated approach and elegance, has essentially remained unchanged. Traditionally performed using ~100 nm tip sharp glass microelectrodes (Denk and Detwiler, 1999; McIlwain and Creutzfeldt, 1967), a simple geometric modification of the pipette led to the whole-cell patch-clamp electrode widely used in vivo and in vitro (Margrie et al., 2003). Yet both types of electrodes are mechanically mismatched when applied to pliable and flexible cell membranes, which makes them susceptible to a loss of recordings in the presence of movement. Furthermore, intracellular dialysis, pipette clogging, and variable access resistances preclude high-throughput experimentation with a single pipette. Moreover, conventional sharp microelectrodes, with tip dimensions in the range of hundreds of nanometers, do not form high seals, cause a permanent shunt (Brette et al., 2008), and are prone to electrolyte leakage from the tip (Geisler et al., 1972). All these factors affect cell physiology and recording throughput and have prevented the widespread use of intracellular recordings in vivo from awake-behaving animals and more specifically with sharp electrodes. Here, we introduce and demonstrate the usability of flexible quartz nanopipettes, which alleviate these limitations and enable high SNR and high-throughput intracellular electrophysiology in the anaesthetized and the awake animal.
The nanopipette, a nanoscale analog of the glass sharp microelectrode, has small tips (~10–25 nm I.D.) with long narrow tapers that enable high seal resistances and tight cell-electrode coupling. As a consequence of the nanoscale form factor, these nanopipettes are ultra-flexible (~0.08 N/m), with stiffness values identical to the stiffness of soft AFM probes used to study cell mechanics (Qi et al., 2015). The narrow electrode profile also causes low electrolyte flux rates (0.1–1 fM/s) and renders the electrode reusable, thus improving recording throughput tremendously (5–10 cells/electrode) in awake moving animals. In addition to the nanopipette, and to enable targeted electrophysiology (Denk and Detwiler, 1999; Kitamura et al., 2008; Margrie et al., 2003) with single-cell resolution in vivo across any cortical depth, we combined it with two-photon microprism imaging. Our approach integrates two-photon guidance through an implanted microprism and intracellular recordings using QD-labeled nanopipettes. QDs coat the nanopipette from the outside, are not prone to photobleaching, and exhibit a high cross-section of absorption, thereby allowing easy visualization of a nanoscale tip that otherwise would be impossible to locate. Intracellular recordings are facilitated by QD-coated nanopipettes that are visually guided to the target cell via two-photon microprism imaging. Two-photon targeted patching (TPTP) has predominantly been performed in superficial cortical layers (Kitamura et al., 2008; Naka and Adesnik, 2016). This is partly because of the poor visualization of target cells and the recording patch pipette in deep scattering tissue. The microprism’s hypotenuse is coated with aluminum, which results in the laser beam being deflected by 90° and helps overcome two limitations that restrict in vivo imaging and cell targeting: visualizing deep cortical layers without scattering-imposed imaging limitations and concomitant imaging across cortical layers within a continuous, single field of view (Figure 6B; Figures S3B and S3C; Videos S1 and S2). Microprisms are inexpensive, easy to implant, and fully compatible with standard epifluorescence microscopy, two-photon imaging, and emerging methods, such as spatial light modulators (Yang et al., 2016), three-photon microscopy (Horton et al., 2013), and acousto-optic deflectors (Grewe et al., 2010). Moreover, prisms can be used with gradient index (GRIN) lenses, which are widely employed and well suited for deep-layer imaging (Ghosh et al., 2011).
Other methods have been developed for targeted whole-cell recordings in vivo. Shadow patching (Kitamura et al., 2008) was introduced to allow targeted electrophysiological recordings from unlabeled cells in vivo based on the negative contrast of the cell body. In principle, this method could be applicable to deeper regions of the cortex, but it becomes difficult when one is interested in recording from genetically defined cell populations, like interneurons subtypes, and still suffers from scattering in deep tissue. Moreover, because a large amount of dye is continuously perfused into the extracellular space to create clearly visible shadows—which has implications for tissue health with increasing experimental time—this approach often leads to large background fluorescence signals that can affect population imaging in deep cortical areas and limits the number of times a pipette can enter the brain. Options such as automated whole-cell patching have solely focused on improving the throughput of whole-cell recordings and have not yet demonstrated concomitant population Ca2+ imaging or suitability in awake moving animals. Moreover, automated TPTP still suffers from the known limitations related to light scattering in brain tissue. One viable approach, in contrast to TPTP, is optogenetically assisted whole-cell patching (Muñoz et al., 2014), which requires neurons to be transfected with channelrhodopsin-2 (ChR2) under a suitable promoter. Using the standard blind patching approach, glass electrodes (carrying an optical fiber inside for light stimulus delivery) are slowly advanced through the brain until a cell is encountered. ChR2-expressing neurons fire reliably upon light stimulation. Once a time-locked firing is recorded, whole-cell recordings are established using standard procedures. While this approach is technically not limited to any depth, the lack of optical monitoring makes it difficult to ascertain the precise location of the pipette relative to the cell. Moreover, a sparse target population will adversely affect the efficiency in finding the target cell of interest. In addition, because all preceding methods use conventional patch pipettes, intracellular recordings will still suffer from throughput limitations, clogging, and high access resistances with increased depth. Recently, tetrodes were used to record quasi-intracellular signals from dendrites in a moving animal (Moore et al., 2017). Although such an approach suffers from low success rates, is blind, and cannot be used at present for targeted intracellular recordings or with two-photon imaging, it suggests flexibility and a narrow profile as key design parameters to be considered when long-duration intracellular or quasi-intracellular recordings are paramount.
Using flexible nanopipettes and microprisms, we alleviate the preceding limitations. Specifically, we perform targeted recordings from mice under different brain states (locomotion and low anesthesia). We show that nanopipettes form a tight seal with neurons exhibiting either a purely intracellular or a quasi-intracellular mode (Jayant et al., 2017; McIlwain and Creutzfeldt, 1967; Moore et al., 2017). In addition, we demonstrate six advantages: (1) a flexible, reproducible, and stable intracellular nanoelectrode; (2) an ability to perform targeted recordings using nanopipettes across any depth in the brain; (3) an ability to perform targeted recordings from dendrites; (4) an ability to perform simultaneous recordings from pairs of neurons (Figure S4C); (5) an ability to perform simultaneous targeted recordings and cellular Ca2+ imaging across cortical layers; and (6) an ability to record in awake moving animals. In closing, the nanoelectrode and associated methods presented in this study improve the sharp microelectrode technique and can make intracellular electrophysiology routine in awake animals, where motion-invariant recordings are paramount, enabling its widespread use in circuit neuroscience in vivo.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Krishna Jayant (kj2346@columbia.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All experiments were conducted with care and in accordance with the Columbia University institutional animal care guidelines. Experiments were carried out on transgenic Thy1-YFP (Feng et al., 2000), PV-Cre::lsl-tdTomato, Thy1-GCaMP6f (Chen et al., 2013), and PV-Cre::lsl-tdTomato::Thy1-GCaMP6f animals (of both sexes) at postnatal age of 1–2 months. On the day of the experiment, mice were anesthetized with isoflurane (initial dose 2%–3% partial pressure in air, then reduction to 1.0%). A flap of skin above the skull was removed and a titanium head plate with a central foramen (7×7mm) was attached to the skull with dental cement. Then, a small craniotomy above the left hemisphere was carried out to gain access to left visual or somatosensory cortices. In brief, a t-shaped section of the skull was thinned using a dental drill until a small piece (circa 1×2×3 mm) of skull could be removed effortlessly with fine forceps (Figure 1A). In each experiment, a glass microprism (1×1×1 mm, Tower Optics) was implanted into the cortex similarly to previous reports (Andermann et al., 2013). To allow access for the sharp nanopipettes, LFP and 4-AP micropipettes during respective experiments, the 1×3 mm2 part of the craniotomy remained uncovered in front of the prism face (see Figure S6 for more details). The dura mater was further removed only at the pipette insertion sites to allow for ease of access for the nano-sharp, LFP and 4-AP micropipettes.
Slice preparation
For acute slice experiments, coronal sections of the neocortex of P7 to P20 old C57BL/6 mice of both sexes were prepared using a Leica VT1200S vibratome. The animal was decapitated (following deep anesthesia via inhalation of Isoflurane in case of animals older than P12), and the brain quickly removed. Slices of 300 mm thickness were prepared in ice-cold slicing solution containing (in mM): 93 N-Methyl-D-glucamine, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 Na-ascorbate, 3 Na-pyruvate, 10 MgSO4, 0.5 CaCl2, pH adjusted with HCl to 7.3, bubbled with 95% O2 and 5% CO2. After a short recovery period (4–8 min) in 35–37 C warm slicing solution, slices were kept at room temperature in ACSF until transferred into a recording chamber.
METHOD DETAILS
In vivo whole-cell electrophysiology
Electrophysiological recordings were performed in animals anaesthetized with isoflurane (2% v/v). Patch pipettes (5–7 MΩ) with a slightly long taper, were pulled from borosilicate glass capillaries (1.5mm O.D, 0.86mm I.D, Sutter instruments Co., USA) using a horizontal puller (DMZ-universal puller, Zeitz Instruments), and filled with internal solution containing (in mM): 120 K-gluconate, 3 KCl, 7 NaCl, 4 Mg-ATP, 0.3 Na-GTP, 20 HEPES, and 14 Tris-phosphocreatine, 0.025 Alexa-594, pH 7.3. During recording, and to avoid movement-related artifacts, 2% agarose gel in HEPES-based artificial cerebrospinal fluid (ACSF) containing (in mM): 150 NaCl, 2.5 KCl, 10 HEPES, 2 CaCl2, 1 MgCl2, pH 7.3 was used to fill the space between the objective and the brain. High positive pressure (200 mBar) was applied to the patch pipettes during entry into the cortex, and then reduced to 30 – 40 mBar when approaching a cell. Whole-cell access was then established using previously published techniques (Kitamura et al., 2008; Margrie et al., 2003). Access resistances varied between 13–115 MΩ dependent on cortical depth. The patch pipettes were visually guided by two-photon imaging through the prism. Signals were amplified (MultiClamp 700b Molecular Devices, USA), filtered (4 kHz Bessel-filter) and digitized at 10 kHz. Data was analyzed offline.
Nanopipette electrophysiology
Nanopipettes (inner tip diameters ~15–30nm) made from quartz capillaries (1mm O.D, 0.5mm I.D, Sutter instruments Co., USA), were fabricated using a P-2000 laser puller (Sutter Instrument Co., U.S.A). The general program parameter range used: Heat: 850–900; FIL: 5/4; VEL: 45–55; DEL: ~145; PUL: 175–200. It is important to note that program parameters were varied over time to obtain best results. These values vary from puller to puller and over time. The range of parameters used above result in a long flexible nanopipette. It should be noted that tip-diameters as small as 10 nm to 30 nm cannot be achieved using conventional borosilicate glass capillaries. Moreover, such fine tips cannot be pulled with a conventional Brown-Flaming style puller and would need a laser puller to melt quartz. The nanopipettes were first back-filled and then front-filled with 3M KCl (resistance range ~150–250MΩ), dipped in QD solution (3–5 times rapidly to ensure uniform coating) and carefully bridge and capacitance compensated prior to recording through the built in module in the multi-clamp 700B amplifiers (Molecular Devices, Union City, CA). Pipette bandwidth extraction was carried out both in the bath and after entry into the brain as previously described (Jayant et al., 2017). Briefly, a current pulse was injected. The resulting voltage response at the rising edge of the current pulse was fit to an exponential to extract the time constantτP. Dividing τP by the measured value of the pipette resistance Re, the pipette wall capacitance Ce was determined. The residual 3-dB bandwidth was calculated using the expression f3dB = 1/2πReCe. This calibration and measurement was performed as capacitance compensation was made optimal, and re-calibrated after withdrawal from a cell. Entry into cells was elicited through mechanical impalement or electroporation (“touch and buzz”) using the build in commands in the amplifier. Tip potential offsets (−2~−4mV) were zeroed prior to cell entry. Current clamp signals from both the nanopipette and patch pipette were filtered by a 4-pole Bessel filter (4 kHz) and acquired at 10kHz (NI-DAQ 6251, National Instruments, USA) using a PC equipped with custom software (PackIO) written in LabView (National Instruments, USA) (Packer, 2011). Tip diameters were measured by first coating the nanopipettes with ~10–15 nm of gold with a sputter coater (Cressington 108 Sputter Coater), then imaged with a scanning electron microscope (SEM, Hitachi S-4700).
For seal resistance measurements depicted in Figure 2E the procedure is as follows. First a whole-cell patch recording is established and the input resistance ascertained through a series of hyperpolarizing current pulses and the resultant voltage response (Rin_before). The nanopipette is then brought in close contact with the cell membrane and gently pushed while applying an electroporating pulse. This causes the membrane to rupture and the pipette tip to the enter the cell fully. This causes a shunt resistance to the external bath which is also the seal resistance of the nanopipette (Rshunt=seal). Once entry is established and the RMP settles, the input resistance from the patch pipette is once again ascertained (Rin_after). It is then relatively straightforward to show that Rin_after = Rin_before × Rshunt=seal /Rin_before + Rshunt/seal, from which (Rshunt/seal)can be extracted.
Targeted intracellular measurements using nanopipettes were carried out as follows. Target cells in vivo were identified through two-photon imaging across the prism. Nanopipettes were subsequently positioned vertically above the cells just above the brain surface, moved horizontally by approximately 2.5 times the vertical depth and then advanced into the brain at an angle of 30 degrees, which ensures the nanopipette is in prisms field of view (Figure S3B). Minor adjustments required the pipette to be completely retracted and then re-positioned since the pipette could not be moved laterally within deep tissue. The nanopipette resistance and residual uncompensated capacitance is periodically checked as the pipette is advanced and corrected if required. Brief electroporation (500ms) or short oscillatory pulses (50 μs) were applied to elicit entry using built in commands in the multiclamp700B. It is important to note that in comparison to conventional sharp electrode use, our method did not require silicone (English et al., 2014) or agarose for electrode stabilization, and did not require the application of negative current to stabilize the recording. This is an important difference as it helps improve the speed of experimentation. For local field potential (LFP) recordings, a glass micropipette (2–5 MΩ, 1.5mm O.D, 0.86mm I.D, Sutter instruments Co., USA) filled with saline and containing a silver chloride wire was carefully advanced into the cortex (30° angle) under visual control to a depth of around 100 μm beneath the pial surface. The pipette tip was positioned rostro-medially next to the imaged area. A reference electrode was positioned over the contralateral prefrontal cortex. LFP signals were amplified using a multiclamp 700B amplifier (Molecular Devices, Union City, CA), low-pass filtered (300Hz), digitized at 1 kHz (Bruker Instruments, USA) and recorded using Prairie View voltage recording software alongside with Ca2+ imaging. For induction of ictal events, another sharp glass micropipette containing 4-Aminopyridine (4-AP, 15mM, 500 nL [total amount delivered = 7.5μmol]) was slowly lowered (30° angle) into the cortex to a depth of 420–480 μm. The pipette tip was positioned at a distance of around 1.5 mm caudally to the imaged area. Correct positioning of the pipette tip was ensured by a diagonal dry-run in saline above the cortex preceding actual insertion. 4-AP was injected over the course of 10–15 min by use of a micro4 micro-syringe-pump controller (World Precision Instruments, USA). Electrographic seizure onset time points were determined mathematically by mean and standard deviation of LFP recordings. The first time point exceeding > 5 std from the interictal LFP rectified voltage was defined as seizure onset and confirmed by visual inspection. Blind recordings were performed in a similar manner except no target cell was chosen
Slice electrophysiology
Neuronal slices were visualized using an Olympus BX50WI microscope equipped with oblique illumination and a water immersion 40x/0.8 NA objective (Olympus). Labeled interneurons were first identified using epifluorescent illumination via a mercury lamp and appropriate emission filters prior to performing whole cell patch-clamp recordings. Whole cell recordings (pipette resistance 4–7MΩ) were obtained using pipettes pulled from borosilicate glass (1.5mm and 1mm O.D, 0.86mm and 0.5mm I.D, Sutter Instruments Co., USA) and established using a Multiclamp 700B amplifier (Molecular Devices, Union City, CA). The external bath comprised of artificial cerebral spinal fluid (ACSF) containing the following (in mM): 126 NaCl, 26 NaHCO3, 1.145 NaH2PO4, 10 glucose, 3 KCl, 2 MgSO4 and 2 CaCl2, Osmolarity ~300 mOsm. Patch pipettes were filled with internal solution containing (in mM): 130 K-gluconate, 5 NaCl, 2 MgSO4, 10 HEPES, 5 EGTA, 4 MgATP, 0.4 Na2GTP, 7 Na2-phospocreatine, 2 pyruvic acid, 0.002–0.01 alexa 488, pH adjusted to 7.2, ~280–290mOsm).
Quantum-dot synthesis and pipette coating
CdSe/CdS/ZnS nanocrystals were prepared and characterized as described previously (Jayant et al., 2017). QDs had a broad two-photon cross-section of absorption (800–1000 nm). Nanopipettes filled with electrolyte were briefly dipped into QD solution prior to entry into the brain. Upon entry into the brain, a brief voltage pulse is applied to ensure no clogging and resistance of the pipette is measured. QDs did not alter the resistance and capacitance properties of the nanopipette.
Local field potential (LFP) measurement
For LFP recordings, a glass micropipette (2–5 MΩ) filled with saline and containing a silver chloride wire was slowly advanced into the cortex (30° angle) under visual control and positioned close-by the imaged area at around 500 μm distance to the prism face. The reference electrode was positioned in the fluid solution between the objective and the brain surface. LFP signals were amplified using a Multiclamp 700B amplifier (Axon Instruments, Sunnyvale, CA), low-pass filtered (300Hz), digitized at 10 kHz (Bruker) and acquired simultaneously with intracellular nanopipette recordings and fluorescent imaging by Prairie View voltage recording software. Continuous data was digitally filtered with a 1Hz high-pass infinite impulse response (IIR; filter order 2) and 60Hz IIR notch filter (5Hz half-width). LFP data was aligned to spikes identified in the intracellular recordings and segmented in 1500 ms epochs.
Two-photon imaging in vivo
For all imaging experiments, a two-photon microscope (Bruker; Billerica, MA), a Ti:Sapphire laser (Chameleon Ultra II; Coherent), and a 25x objective (water immersion, N.A. 1,05, Olympus). were used. All imaging was carried out under light anesthesia (isoflurane at 0.5 – 1.0 partial pressure in air). The distance from the prism face to the focal plane ranged between 200 to 300 μm, maximizing tissue intactness while maintaining reasonable imaging parameters (laser power at the sample < 50mW/cm2) (Andermann et al., 2013). For structural Pv imaging, we used an excitation wavelength of 990 nm. In these experiments, the QD-coated nano-sharps were visualized using the same excitation wavelength. GCaMP6f was imaged at 940 nm. For multi-color imaging of YFP and QDs, an excitation wavelength of 860 nm was used. For multi-color imaging of GCaMP6f, tdTomato and QDs, we first used 990nm for targeting PV interneurons, and switched to 940nm for functional imaging of GCaMP6f once successful and stable electrical recording of individual Pv’s was achieved. For imaging of the optical break-in of epileptiform activity into the field of view during ictal propagation, images were acquired by resonant galvanometer scanning at an acquisition speed of 30.206 frames per second (512 3 512pixels). For all other experiments under physiological conditions, images were obtained through normal galvanometer scanning with the acquisition speeds depending on the chosen imaged field of view (~0.5–10 frames per second). All imaging parameters were controlled by Prairie View Imaging software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Image processing
Cells were visually identified in raw tiff movie files using ImageJ software. A list of cell centroid spatial coordinates was obtained and used to initialize the recently described constrained nonnegative matrix factorization algorithm (CNMF) to extract Ca2+ transients of all registered cells in MATLAB (Pnevmatikakis et al., 2016; Yang et al., 2016). The CNMF method identifies spatiotemporal components based on pixels of high covariance around defined cell centroids while accounting for background fluorescence and minimizing signal noise. Current GCaMP6 Ca2+ indicators exert a high dynamic range and baseline fluorescence in transgenic GCaMP6f animals was generally extremely low. Therefore, all Ca2+ transients in this paper are displayed as raw fluorescence (F) or ΔF.
Instantaneous amplitude/phase decomposition
LFP epochs were subjected to a moving sliding window fast-fourier transformation (FFT) using 500ms windows of LFP data (2Hz resolution) convolved with 5000 sample hanning-window (to minimize edge effects). Phase (angle) and power (absolute value) of the complex FFT output were computed for each time point 500 ms pre to 500 ms post spike.
Average power and phase locking characterization
Power was averaged across all trials. For each time-frequency (0–90hz; −500–500 ms) power was then divided by its baseline (−500 to −300ms), log10 transformed, and multiplied by 10 to yield decibels. Inter-spike phase locking (or inter-trial coherence, ITC) was calculated by dividing the complex output of the FFT for each time-frequency point by its absolute value on each trial, and then aver-aging across trials. The output is a circular measure of phase consistency, or the “Rayleigh” statistic (R), bound between 0 and 1 where 1 is perfect phase consistency across epochs/spikes. The significance of ITC values depends on the number of spikes. Significant ITC was determined at alpha-level 0.05 when p < 0.01 was observed for a time-frequency cluster larger than 8 contiguous time-frequency points. Average phase within a cluster for each spike was plotted as a circular histogram for descriptive purposes.
Atomic Force Microscopy (AFM) measurements
Nanopipette stiffness experiments were performed on a BioScope Catalyst (Bruker, USA) atomic force microscope (AFM) using SNL-D cantilevers (Bruker,USA), whose deflection sensitivity was first determined from force-distance curves acquired on glass. The nanopipette spring constant/stiffness (kP) was determined from force-distance curves (Figure S1B), in which the AFM tip approaches and retracts from the nanopipette while recording interaction forces over tip-sample distances. kP was calculated from the linear slope of the force-distance retract plot, after subtraction of the cantilever spring constant - determined via the thermal noise method. Distance from the nanopipette edge was determined from analysis of optical images or AFM stage movement, after identifying the pipette edge through topography images.
Multiphysics Simulations
Finite element simulations were carried out in COMSOL 5.2a software (COMSOL, USA). The pipette and bath was modeled as a 2D axisymmetric geometry with triangular mesh elements. The coupled set of differential equations were evaluated under varying ‘r-radius’, ‘z- height’, and ‘θ-cone angle’. The taper at the pipette end was modeled either as a pure cone or as a cone with a cylindrical tip, with varying radius and cone angle; the bulk fluid was modeled as a hemisphere of 1 mm radius, centered about the center of the nanopipette opening. The relevant geometries are depicted in Figure 1.
Success rate, drawbacks of flexible nanopipette blind and targeted recordings, and overall experimental ‘n’
Nanopipette recordings are relatively easy to perform, and the method can be easily adopted in any neuroscience laboratory. Once the pipette program is fixed in the puller, one can prepare a nanopipette in a few minutes, and resistances and tip diameters are consistent thereafter. Minor adjustments to puller parameters are required from time to time, but the characteristics of the pipette remain similar. Targeted and blind recordings were performed collectively from 14 mice. Target identification and positioning of the pipette to enter the brain usually took ~3–5 minutes depending on location of the target cell or process. Pipette entry into the brain occurred instantaneously upon contact provided the dura was snipped to allow easy access, and resistances remained unchanged. Nanopipettes were moved toward the target cell along the diagonal axis (Figure S3B) and manipulator increments were in micrometers. Time to reach target cell varied between a few minutes to 10’s of minutes, and depended critically on target depth, as the speed of traversing was slow. No buckling was observed. Weak electroporation or clearing pulses (50–100 μs oscillations) were then applied to elicit either type-A or type-B entry. Because of their small tip and long taper, diffusion (Figures 1D and 1E) from the pipette tip is very low (~0.1–1fM/s). While this is an advantage in that it precludes washout, it also precludes backfilling the neuron with dye, which is often useful in ascertaining target registry. Two-photon targeted recordings in this study were thus ascertained through either (i) small dimple formation in the target cell or process as the pipette was brought in contact with the membrane; (ii) matching the firing pattern and AP characteristic to the molecularly labeled cell, specifically with Pv’s; and (iii) phase-locking of the AP to the LFP for both pyramidals and Pv’s. Our targeted recording approach was further confirmed during the experiments with GCaMP experessing neurons, in which we measured concomitant Ca2+ activity and intracellular potentials in the targeted cell. In awake animals, flexible nanopipettes enabled multiple and consecutive blind intracellular recordings in all mice tested. Blind recordings occurred within a few minutes of entering the cortex and are were much faster to perform than targeted recordings. There are also limitations to our nanopipettes. Their small size and flexible nature allow easy navigation along the diagonal axis inside the brain, but does not render them amenable to lateral movements in deep tissue. In order to laterally move the nanopipette in the deep-cortex the pipette has to be completely retracted and the trajectory readjusted to ensure target entry. Moreover, since the QDs coat the nanopipette from the outside, they can desorb upon repeated navigation through tissue. This can complicate visualization of the tip over time. With improved grafting strategies, this issue can be resolved. Lastly, in the present study we did not examine nanopipette recording stability in the presence of particular motion such as licking. This aspect will be covered in future studies. We do however stress that the nanopipette due to its extremely small tip size and long flexible taper is able to sustain large body movements such as crouching, walking and running without the anchoring the pipette with silicone oil or agar, or special restraints for the animal.
Overall, we obtained recordings from 14 mice (7 anaesthetized and 7 awake) and performed several distinct types of experiments. A) Blind nanopipette recordings in awake running animals (51 cells with durations spanning a few minutes to hours). B) Targeted nanopipette recordings in anaesthetized mice under two-photon visualization (soma n = 12; dendrites n = 6); C) Targeted somatic recordings from pyramidals and PVs in awake animals (n = 3 targeted cells); D) Simultaneous Ca2+ imaging and intracellular recordings under epileptic conditions (n = 8 cells from 3 mice); E) simultaneous Ca2+ imaging and intracellular recordings from dendrites (n = 3 dendrites); F) long-duration recordings in awake animals 20 minutes-3.5 hours intracellular recording during locomotion (n = 9 cells; 7 mice, type-A entry, n = 9 cells, 4 mice type-B entry).
For details on COMSOL simulations used for ascertaining diffusion and flow profile of the nanopipettes and signal rescaling details, see Methods S1: Electrostatic simulations and transient entry modeling, related to STAR Methods
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| hexanes (98.5%) | Sigma Aldrich | 178918 |
| methanol (99.8%) | Sigma Aldrich | 179337 |
| toluene (99.5%) | Sigma Aldrich | 179418 |
| dioctylamine (98%) | Sigma Aldrich | (now available as: see below) |
| dioctylamine (97%) | Sigma Aldrich | D201146 |
| hexyl isothiocyanate (95%) | Sigma Aldrich | 475912 |
| 1-octadecene (90%) | Sigma Aldrich | O806 |
| octadecylamine (99%) | Sigma Aldrich | 74750 |
| oleic acid (99%) | Sigma Aldrich | O1008 |
| selenium pellets (>99.99%) | Sigma Aldrich | 209643 |
| zinc acetate dihydrate (>99%) | Sigma Aldrich | 96459 |
| cadmium oxide (99.99%) | Strem Chemicals | 93-4817 |
| tri-n-butylphosphine (99%) | Strem Chemicals | (now available as: see below) |
| tri-n-butylphosphine (min. 93%) | Strem Chemicals | 15-5801 |
| tri-n-octylphosphine (97%) | Strem Chemicals | 15-6655 |
| Alexa-594 | Sigma-Aldrich | N/A |
| 4-Aminopyridine | Sigma-Aldrich | Cat # 275875; CAS:504-24-5 |
| 3M KCl | Sigma-Aldrich | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 wildtype | Jackson Laboratory | RRID:IMSR_JAX:000664 |
| C57BL/6J-Tg(Thy1-GCaMP6f)GP5.11Dkim/J | Jackson Laboratory | RRID:IMSR_JAX:024276 |
| Thy-1 | Jackson Laboratory | RRID:IMSR_JAX:000406 |
| PV-Cre | Jackson Laboratory | RRID:IMSR_JAX:017320 |
| LSL-TOM | Jackson Laboratory | RRID:IMSR_JAX:007908 |
| Recombinant DNA | ||
| AAV1-Syn-GCaMP6s-WPRE-SV40 | U Penn Vector Core | Cat # AV-1-PV2824 |
| Software and Algorithms | ||
| ImageJ | https://imagej.nih.gov/ij/download.html | N/A |
| Moco | (Dubbs et al., 2016) | N/A |
| MATLAB(R2014b) | MathWorks | https://www.mathworks.com |
| COMSOL | COMSOL (USA) | N/A |
| Adobe Illustrator CS6 | Adobe | N/A |
| Custom lab view program for in vitro data acquisition | PackIO (Packer, 2011) | N/A |
| Priarie View Software for in vivo data acquisition | Bruker Corporation | N/A |
| Other | ||
| Custom built Ultima In Vivo two-photon microscope | Bruker Corporation | Model: U-IV-02 |
| Tunable Ti:Sapphire laser | Coherent | Model: Chameleon Ultra II |
| Pockels cell | Conoptics | Model # 350-160BK |
| Pockels cell driver | Conoptics | Model # 302 RM |
| MultiClamp 700B amplifier+commander | Molecular Devices | Model # 2500-0157 |
| Custom built Olympus In Vitro microscope | Olympus | N/A |
| Atomic Force Microscope | Bruker Corporation | Bruker Catalyst |
| P-2000 laser puller | Sutter Instruments, USA | P-2000 |
| Quartz capillaries 1mm * 0.5mm | Sutter Instruments, USA | N/A |
Highlights.
Flexible nanopipettes are minimally invasive intracellular nanoelectrodes
Quantum-dot coatings enable two-photon visualization of flexible nanopipettes
Two-photon targeted recordings from both somata and dendrites in vivo
PV+ interneurons go into depolarization block during seizure spread in vivo
ACKNOWLEDGMENTS
This work was supported by the NIMH (R01MH101218 and R01MH100561), NEI (DP1EY024503 and R01EY011787), and the DFG (WE 5517/1-1). This material is also based upon work supported by, or in part by, the U.S. Army Research Office under contract W911NF-12-1-0594 (MURI) and contract DARPA-BAA-16-17. K.J. was supported by the Kavli Institute of Brain Science at Columbia. We thank all members of the Yuste lab for help and useful comments.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures, two videos, and one methods files and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.12.019.
REFERENCES
- Adesnik H, Bruns W, Taniguchi H, Huang ZJ, and Scanziani M (2012). A neural circuit for spatial summation in visual cortex. Nature 490, 226–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andermann ML, Gilfoy NB, Goldey GJ, Sachdev RN, Wölfel M, McCormick DA, Reid RC, and Levene MJ (2013). Chronic cellular imaging of entire cortical columns in awake mice using microprisms. Neuron 80, 900–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrásfalvy BK, Galiñanes GL, Huber D, Barbic M, Macklin JJ, Susumu K, Delehanty JB, Huston AL, Makara JK, and Medintz IL (2014). Quantum dot-based multiphoton fluorescent pipettes for targeted neuronal electrophysiology. Nat. Methods 11, 1237–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angle MR, and Schaefer AT (2012). Neuronal recordings with solid-conductor intracellular nanoelectrodes (SCINEs). PLoS ONE 7, e43194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brette R, Piwkowska Z, Monier C, Rudolph-Lilith M, Fournier J, Levy M, Frégnac Y, Bal T, and Destexhe A (2008). High-resolution intracellular recordings using a real-time computational model of the electrode. Neuron 59, 379–391. [DOI] [PubMed] [Google Scholar]
- Cammarota M, Losi G, Chiavegato A, Zonta M, and Carmignoto G (2013). Fast spiking interneuron control of seizure propagation in a cortical slice model of focal epilepsy. J. Physiol. 591, 807–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo-Reid L, Yang W, Bando Y, Peterka DS, and Yuste R (2016). Imprinting and recalling cortical ensembles. Science 353, 691–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T-W, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, et al. (2013). Ultrasen-sitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana H, Chen T-W, Hu A, Shields BC, Guo C, Looger LL, Kim DS, and Svoboda K (2014). Thy1-GCaMP6 transgenic mice for neuronal population imaging in vivo. PLoS ONE 9, e108697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk W, and Detwiler PB (1999). Optical recording of light-evoked calcium signals in the functionally intact retina. Proc. Natl. Acad. Sci. USA 96, 7035–7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubbs A, Guevara J, and Yuste R (2016). moco: Fast motion correction for calcium imaging. Front. Neuroinform. 10, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English DF, Peyrache A, Stark E, Roux L, Vallentin D, Long MA, and Buzsáki G (2014). Excitation and inhibition compete to control spiking during hippocampal ripples: intracellular study in behaving mice. J. Neurosci. 34, 16509–16517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, and Sanes JR (2000). Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28, 41–51. [DOI] [PubMed] [Google Scholar]
- Geisler CD, Lightfoot EN, Schmidt FP, and Sy F (1972). Diffusion effects of liquid-filled micropipettes: a pseudobinary analysis of electrolyte leakage. IEEE Trans. Biomed. Eng. 19, 372–375. [DOI] [PubMed] [Google Scholar]
- Ghosh KK, Burns LD, Cocker ED, Nimmerjahn A, Ziv Y, Gamal AE, and Schnitzer MJ (2011). Miniaturized integration of a fluorescence microscope. Nat. Methods 8, 871–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewe BF, Langer D, Kasper H, Kampa BM, and Helmchen F (2010). High-speed in vivo calcium imaging reveals neuronal network activity with near-millisecond precision. Nat. Methods 7, 399–405. [DOI] [PubMed] [Google Scholar]
- Hai A, Shappir J, and Spira ME (2010). In-cell recordings by extracellular microelectrodes. Nat. Methods 7, 200–202. [DOI] [PubMed] [Google Scholar]
- Horton NG, Wang K, Kobat D, Clark CG, Wise FW, Schaffer CB, and Xu C (2013). In vivo three-photon microscopy of subcortical structures within an intact mouse brain. Nat. Photonics 7, 205–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houweling AR, and Brecht M (2008). Behavioural report of single neuron stimulation in somatosensory cortex. Nature 451, 65–68. [DOI] [PubMed] [Google Scholar]
- Hu H, Martina M, and Jonas P (2010). Dendritic mechanisms underlying rapid synaptic activation of fast-spiking hippocampal interneurons. Science 327, 52–58. [DOI] [PubMed] [Google Scholar]
- Jayant K, Hirtz JJ, Plante IJ, Tsai DM, De Boer WD, Semonche A, Peterka DS, Owen JS, Sahin O, Shepard KL, and Yuste R (2017). Targeted intracellular voltage recordings from dendritic spines using quantum-dot-coated nanopipettes. Nat. Nanotechnol. 12, 335–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura K, Judkewitz B, Kano M, Denk W, and Häusser M (2008). Targeted patch-clamp recordings and single-cell electroporation of unlabeled neurons in vivo. Nat. Methods 5, 61–67. [DOI] [PubMed] [Google Scholar]
- Kodandaramaiah SB, Franzesi GT, Chow BY, Boyden ES, and Forest CR (2012). Automated whole-cell patch-clamp electrophysiology of neurons in vivo. Nat. Methods 9, 585–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan W-J, Edwards MA, Luo L, Perera RT, Wu X, Martin CR, and White HS (2016). Voltage-rectified current and fluid flow in conical nanopores. Acc. Chem. Res. 49, 2605–2613. [DOI] [PubMed] [Google Scholar]
- Lee AK, Manns ID, Sakmann B, and Brecht M (2006). Whole-cell recordings in freely moving rats. Neuron 51, 399–407. [DOI] [PubMed] [Google Scholar]
- Lee AK, Epsztein J, and Brecht M (2009). Head-anchored whole-cell recordings in freely moving rats. Nat. Protoc. 4, 385–392. [DOI] [PubMed] [Google Scholar]
- Liu J, Fu T-M, Cheng Z, Hong G, Zhou T, Jin L, Duvvuri M, Jiang Z, Kruskal P, Xie C, et al. (2015). Syringe-injectable electronics. Nat. Nanotechnol. 10, 629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long MA, and Lee AK (2012). Intracellular recording in behaving animals. Curr. Opin. Neurobiol. 22, 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long MA, Jin DZ, and Fee MS (2010). Support for a synaptic chain model of neuronal sequence generation. Nature 468, 394–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long B, Li L, Knoblich U, Zeng H, and Peng H (2015). 3D image-guided automatic pipette positioning for single cell experiments in vivo. Sci. Rep. 5, 18426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margrie TW, Brecht M, and Sakmann B (2002). In vivo, low-resistance, whole-cell recordings from neurons in the anaesthetized and awake mammalian brain. Pflugers Arch. 444, 491–498. [DOI] [PubMed] [Google Scholar]
- Margrie TW, Meyer AH, Caputi A, Monyer H, Hasan MT, Schaefer AT, Denk W, and Brecht M (2003). Targeted whole-cell recordings in the mammalian brain in vivo. Neuron 39, 911–918. [DOI] [PubMed] [Google Scholar]
- McIlwain JT, and Creutzfeldt OD (1967). Microelectrode study of synaptic excitation and inhibition in the lateral geniculate nucleus of the cat. J. Neurophysiol. 30, 1–21. [Google Scholar]
- Moore JJ, Ravassard PM, Ho D, Acharya L, Kees AL, Vuong C, and Mehta MR (2017). Dynamics of cortical dendritic membrane potential and spikes in freely behaving rats. Science 355, eaaj1497. [DOI] [PubMed] [Google Scholar]
- Muñoz W, Tremblay R, and Rudy B (2014). Channelrhodopsin-assisted patching: in vivo recording of genetically and morphologically identified neurons throughout the brain. Cell Rep. 9, 2304–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naka A, and Adesnik H (2016). Inhibitory circuits in cortical layer 5. Front. Neural Circuits 10, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann AR, Raedt R, Steenland HW, Sprengers M, Bzymek K, Navratilova Z, Mesina L, Xie J, Lapointe V, Kloosterman F, et al. (2017). Involvement of fast-spiking cells in ictal sequences during spontaneous seizures in rats with chronic temporal lobe epilepsy. Brain 140, 2355–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer AM (2011). Understanding the nervous system as an information processing machine: dense, nonspecific, canonical microcircuit architecture of inhibition in neocortex and A neural circuit for angular velocity computation (Columbia University), PhD thesis. [Google Scholar]
- Packer AM, Russell LE, Dalgleish HWP, and Häusser M (2015). Simultaneous all-optical manipulation and recording of neural circuit activity with cellular resolution in vivo. Nat. Methods 12, 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LM, Shai AS, Reeve JE, Anderson HL, Paulsen O, and Larkum ME (2014). NMDA spikes enhance action potential generation during sensory input. Nat. Neurosci. 17, 383–390. [DOI] [PubMed] [Google Scholar]
- Petersen CCH (2017). Whole-cell recording of neuronal membrane potential during behavior. Neuron 95, 1266–1281. [DOI] [PubMed] [Google Scholar]
- Petersen CC, and Crochet S (2013). Synaptic computation and sensory processing in neocortical layer 2/3. Neuron 78, 28–48. [DOI] [PubMed] [Google Scholar]
- Pluta S, Naka A, Veit J, Telian G, Yao L, Hakim R, Taylor D, and Adesnik H (2015). A direct translaminar inhibitory circuit tunes cortical output. Nat. Neurosci. 18, 1631–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pnevmatikakis EA, Soudry D, Gao Y, Machado TA, Merel J, Pfau D, Reardon T, Mu Y, Lacefield C, Yang W, et al. (2016). Simultaneous denoising, deconvolution, and demixing of calcium imaging data. Neuron 89, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack P-O, Friedman J, and Golshani P (2013). Cellular mechanisms of brain state-dependent gain modulation in visual cortex. Nat. Neurosci. 16, 1331–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Andolfi L, Frattini F, Mayer F, Lazzarino M, and Hu J (2015). Membrane stiffening by STOML3 facilitates mechanosensation in sensory neurons. Nat. Commun. 6, 8512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Jorgolli M, Shalek AK, Yoon MH, Gertner RS, and Park H (2012). Vertical nanowire electrode arrays as a scalable platform for intracellular interfacing to neuronal circuits. Nat. Nanotechnol. 7, 180–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider DM, Nelson A, and Mooney R (2014). A synaptic and circuit basis for corollary discharge in the auditory cortex. Nature 513, 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siapas AG, Lubenov EV, and Wilson MA (2005). Prefrontal phase locking to hippocampal theta oscillations. Neuron 46, 141–151. [DOI] [PubMed] [Google Scholar]
- Stosiek C, Garaschuk O, Holthoff K, and Konnerth A (2003). In vivo two-photon calcium imaging of neuronal networks. Proc. Natl. Acad. Sci. USA 100, 7319–7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda K, Denk W, Kleinfeld D, and Tank DW (1997). In vivo dendritic calcium dynamics in neocortical pyramidal neurons. Nature 385, 161–165. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Helmchen F, Denk W, and Tank DW (1999). Spread of dendritic excitation in layer 2/3 pyramidal neurons in rat barrel cortex in vivo. Nat. Neurosci. 2, 65–73. [DOI] [PubMed] [Google Scholar]
- Theer P, Hasan MT, and Denk W (2003). Two-photon imaging to a depth of 1000 microm in living brains by use of a Ti:Al2O3 regenerative amplifier. Opt. Lett. 28, 1022–1024. [DOI] [PubMed] [Google Scholar]
- Tian B, Cohen-Karni T, Qing Q, Duan X, Xie P, and Lieber CM (2010). Three-dimensional, flexible nanoscale field-effect transistors as localized bio-probes. Science 329, 830–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischbirek C, Birkner A, Jia H, Sakmann B, and Konnerth A (2015). Deep two-photon brain imaging with a red-shifted fluorometric Ca2+ indicator. Proc. Natl. Acad. Sci. USA 112, 11377–11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevelyan AJ, Sussillo D, Watson BO, and Yuste R (2006). Modular propagation of epileptiform activity: evidence for an inhibitory veto in neocortex. J. Neurosci. 26, 12447–12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viventi J, Kim DH, Moss JD, Kim YS, Blanco JA, Annetta N, Hicks A, Xiao J, Huang Y, Callans DJ, et al. (2010). A conformal, bio-interfaced class of silicon electronics for mapping cardiac electrophysiology. Sci. Transl. Med. 2, 24ra22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel M, Hamm JP, Peterka DS, and Yuste R (2017). Reliable and elastic propagation of cortical seizures in vivo. Cell Rep. 19, 2681–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel M, Hamm JP, Peterka DS, and Yuste R (2018). Seizures start as silent microseizures by neuronal ensembles. bioRxiv. 10.1101/358903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womelsdorf T, Valiante TA, Sahin NT, Miller KJ, and Tiesinga P (2014). Dynamic circuit motifs underlying rhythmic gain control, gating and integration. Nat. Neurosci. 17, 1031–1039. [DOI] [PubMed] [Google Scholar]
- Yang W, Miller JE, Carrillo-Reid L, Pnevmatikakis E, Paninski L, Yuste R, and Peterka DS (2016). Simultaneous multi-plane imaging of neural circuits. Neuron 89, 269–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazaki-Sugiyama Y, Kang S, Câteau H, Fukai T, and Hensch TK (2009). Bidirectional plasticity in fast-spiking GABA circuits by visual experience. Nature 462, 218–221. [DOI] [PubMed] [Google Scholar]
- Yuste R, and Katz LC (1991). Control of postsynaptic Ca2+ influx in developing neocortex by excitatory and inhibitory neurotransmitters. Neuron 6, 333–344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.