

Summary
Aspergillus fumigatus, a ubiquitous mold, is a common cause of invasive aspergillosis (IA) in immunocompromised patients. Host defense against IA relies on lung-infiltrating neutrophils and monocyte-derived dendritic cells (Mo-DCs). Here, we demonstrate that plasmacytoid dendritic cells (pDCs), which are prototypically antiviral cells, participate in innate immune crosstalk underlying mucosal antifungal immunity. Aspergillus-infected murine Mo-DCs and neutrophils recruited pDCs to the lung by releasing the CXCR3 ligands, CXCL9 and CXCL10, in a Dectin-1 and Card9- and type I and III interferon signaling-dependent manner, respectively. During aspergillosis, circulating pDCs entered the lung in response to CXCR3-dependent signals. Via targeted pDC ablation, we found that pDCs were essential for host defense in the presence of normal neutrophil and Mo-DC numbers. Although interactions between pDC and fungal cells were not detected, pDCs regulated neutrophil NADPH oxidase activity and conidial killing. Thus, pDCs act as positive feedback amplifiers of neutrophil effector activity against inhaled mold conidia.
Keywords: Aspergillus fumigatus, CXCR3, CXCL9, CXCL10, plasmacytoid DC, monocyte-derived dendritic cell, neutrophil, innate immunity, lung, fungus
Graphical Abstract
Highlights
-
•
Infected Mo-DCs and neutrophils secrete CXCL9 and -10 through Dectin-1 and IFN signaling
-
•
CXCL9 and -10 promote the entry of circulating CXCR3+ pDCs into the lung
-
•
Lung-infiltrating CXCR3+ pDCs are essential for host defense against A. fumigatus
-
•
pDCs activate neutrophil NADPH oxidase activity to promote sterilizing immunity
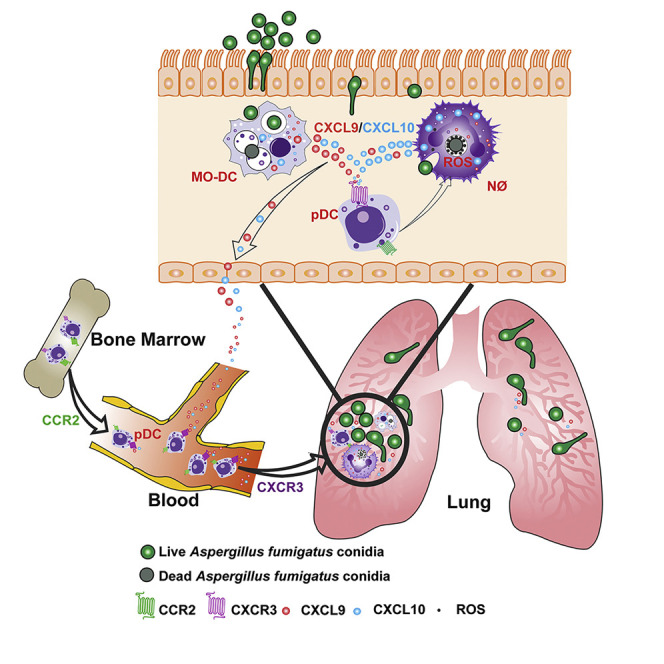
Guo et al. describe innate immune crosstalk in the lung during respiratory fungal infection. Infected neutrophils and Mo-DCs release CXCL9 and -10, recruiting CXCR3+ pDCs to the lung. In turn, pDCs enhance neutrophil antifungal activity and act as a positive feedback amplifier to promote the clearance of inhaled mold conidia.
Introduction
Aspergillus fumigatus forms airborne spores (conidia) that humans typically clear in a silent and asymptomatic manner. Due to the growing number of humans that live in immune compromised states, A. fumigatus is the most common and lethal agent of mold pneumonia worldwide (Brown et al., 2012, Latgé and Chamilos, 2019, Lionakis and Levitz, 2018, Tischler and Hohl, 2019). In humans and mice, sterilizing immunity against A. fumigatus conidia depends on the action of myeloid cells, primarily neutrophils, lung-infiltrating monocytes, and monocyte-derived dendritic cells (Mo-DCs), as well as alveolar macrophages; all lymphoid cells are redundant for host defense (Espinosa et al., 2014, Hohl et al., 2009, Latgé and Chamilos, 2019, Mircescu et al., 2009). Phagocyte NADPH oxidase is central to fungal clearance, as evidenced by a 40%–55% lifetime prevalence of IA in patients with chronic granulomatous disease (Marciano et al., 2015). Reactive oxygen species (ROS) induce a regulated cell death process in neutrophil-engulfed conidia and thereby protect the respiratory tract from tissue-invasive hyphae and fungal dissemination (Shlezinger et al., 2017).
Using targeted cell depletion strategies based on murine C-C chemokine receptor 2 (CCR2) promoter-dependent diphtheria toxin receptor (DTR) transgene expression (Espinosa et al., 2014, Hohl et al., 2009), recent studies implicated CCR2+ inflammatory monocytes and Mo-DCs in sterilizing antifungal immunity, both by direct fungal killing and by regulating the lung inflammatory milieu (Espinosa et al., 2017, Espinosa et al., 2014). Since depletion of CCR2+ cells diminished neutrophil antifungal activity, two models could explain these findings. First, CCR2+ monocytes and Mo-DCs could release mediators that act directly on neutrophils to enhance antifungal effector functions. Second, CCR2+ monocytes and Mo-DCs could mediate the recruitment or activation of a third cellular constituent that conditions the lung inflammatory milieu independent of contributions from CCR2+ monocytes and their derivatives. Here, we provide evidence for the second model and identify plasmacytoid DCs (pDCs) as a third leukocyte constituent that is essential for innate immune crosstalk and host defense against A. fumigatus in an otherwise immune competent host.
The mechanism by which pDCs contribute to antifungal immunity in the lung remains an open question. In vitro, human pDCs may spread over A. fumigatus hyphae to blunt fungal metabolic activity, and, in rare instances, undergo a cell death process that may result in extracellular traps (Loures et al., 2015, Ramirez-Ortiz et al., 2011). Mice treated with a mAb (i.e., anti-PDCA-1 or anti-CD317) that primarily targets pDCs in the steady state, but likely targets additional leukocyte subsets under inflammatory conditions (Blasius et al., 2006), are susceptible to A. fumigatus challenge. In this study, we integrate pDCs into a model of innate immune crosstalk that is critical for defense against A. fumigatus in the lung. We found that fungus-infected Mo-DCs and neutrophils utilize Dectin-1and Card9 signaling to release C-X-C chemokine-9 (CXCL9) and responded to type I and type III interferon (IFN) signaling to release C-X-C chemokine-10 (CXCL10). These C-X-C chemokine receptor 3 (CXCR3) ligands promoted pDC egress from the circulation into the infected lung. Lung CXCR3+ pDCs enhanced neutrophil NADPH oxidase activity and fungal killing, preventing the formation of tissue-invasive hyphae and promoting sterilizing immunity. These findings integrate antifungal pDCs into a model of mucosal immune defense against inhaled molds.
Results
Mo-DCs and Neutrophils Produce CXCL9 and CXCL10 during A. fumigatus Infection
To examine specific contributions of CCR2+ monocytes and Mo-DCs to the lung inflammatory milieu during A. fumigatus challenge and to eliminate potential contributions of CCR2+ natural killer (NK) cell and CD8+ T cell subsets, we crossed the CCR2-DTR transgene to Rag2 −/− interleukin-2 receptor γ chain−/− mice. Lung homogenates of diphtheria toxin (DT)-treated CCR2-DTR+/− Rag2 −/− interleukin-2 receptor γ chain−/− mice (CCR2-DTR+/− Rag2 −/− Il2rg −/−) contained less CXCL10 (IP-10), CCL5 (RANTES), CCL17 (TARC), and CCL20 (MIP-3α) compared with DT-treated non-transgenic Rag2 −/− Il2rg −/− littermates at 36 h post-infection (pi), as measured in a cytokine array (Figures S1A and S1B).
Because a CXCL10 polymorphism is implicated in IA susceptibility in hematopoietic cell transplant recipients (Fisher et al., 2017, Mezger et al., 2008), we measured lung levels of CXCL9 and CXCL10, both CXCR3 ligands, in naive and in fungus-infected C57BL6 mice. A. fumigatus challenge induced CXCL9 and CXCL10 expression in the lung, with a peak at 48 h pi (Figure 1 A). To validate the multiplex array data, we ablated CCR2+ monocytes and Mo-DCs in CCR2-DTR+/− Rag2 −/− Il2rg −/− mice and observed a 50%–70% reduction in lung CXCL9 and CXCL10 levels at 48 h pi (Figure S1C). Ablation of CCR2+ monocytes and Mo-DCs in conventional CCR2-DTR+/− (CCR2 depleter) mice that contain lymphoid lineage cells yielded nearly identical results (Figures 1B and 1C), consistent with the model that CCR2+ myeloid cells represent a major cellular source of CXCR3 ligands during acute A. fumigatus infection.
Figure 1.

CCR2+ Mo-DCs and Neutrophils Produce CXCL9 and CXCL10 during A. fumigatus Challenge
(A–C) Lung CXCL9 and CXCL10 levels in (A) C57BL6 mice (n = 5) at 0–72 h pi or (B and C) in DT-treated CCR2-DTR+/− and non-Tg (CCR2-DTR−/−) littermates (n = 5) at 48 h pi with 3 × 107 CEA10 conidia.
(D) Representative plots of RFP (CXCL9) and BFP (CXCL10) expression in indicated lung leukocytes isolated from Rex3 Tg → C57BL6.SJL BM chimeric mice at baseline (naive, top row) and 48 h pi with 3 × 107 CEA10 conidia (infected, bottom row). The blue and purple gates indicate the frequency of BFP+ (CXCL9+) and BFP+RFP+ (CXCL9+ CXCL10+) cells, respectively.
(E and F) (E) The graphs indicate the frequency of CXCL9− CXCL10+ and CXCL9+ CXCL10+ neutrophils, monocytes, Mo-DCs, and pDCs and (F) the cellular identity of all CXCL9− CXCL10+ (top) and CXCL9+ CXCL10+ lung leukocytes (bottom) at 48 h pi.
(G and H) Lung (G) CXCL9 and (H) CXCL10 levels in DT-treated ROSA26-iDTRMrp8-Cre → C57BL6.SJL (neutrophil depleter) or non-Cre ROSA26-iDTR littermates → C57BL 6.SJL (control) BM chimeric mice (n = 10) at 48 h pi with 3–4 × 107 heat-killed swollen CEA10 conidia. Data from two independent experiments were pooled.
(A–C, G, and H) Dots represent individual mice and data are presented as mean ± SEM. Statistical analysis: (A) Kruskal-Wallis test, time points compared with t = 0 h, (B, C, G, and H) Mann-Whitney test. See also Figure S1.
To visualize hematopoietic cellular sources of CXCL9 and CXCL10, we generated chimeric mice (REX3 Tg → C57BL6.SJL) in which radiosensitive hematopoietic cells encoded RFP and BFP transgenes that were driven by the Cxcl9 and Cxcl10 promoters, respectively (Groom et al., 2012). Following A. fumigatus challenge, Mo-DCs were the major cell types that expressed the RFP and BFP transgenes, consistent with the CCR2+ cell ablation data in the CCR2-DTR+/− Rag2 −/− Il2rg −/− and CCR2-DTR+/− backgrounds (Figures 1D–1F). The majority of Mo-DCs produced CXCL9 and CXCL10 simultaneously, whereas a minority were positive only for CXCL10 (Figures 1E and 1F). Monocytes expressed primarily the CXCL10 transgene, while CXCL9 transgene expression was undetectable at baseline and during fungal infection. Neutrophils expressed both reporter transgenes during respiratory fungal infection, albeit with a lower frequency compared with Mo-DCs. pDCs, CD4+ and CD8+ T cells, and NK cells expressed very low levels of the reporter transgenes, respectively (Figures 1D–1F, S1D, and S1E).
To ascertain that neutrophils contribute to CXCL9 and CXCL10 production, we generated bone marrow (BM) chimera mice that enabled DT-induced neutrophil depletion (ROSA26-iDTRMrp8-Cre → C57BL6.SJL mice, neutrophil depleter) or not (non-Cre ROSA26-iDTR littermates → C57BL6.SJL) and measured CXCL9 and CXCL10 levels following challenge with heat-killed swollen A. fumigatus conidia 48 h pi. This experimental setup was utilized to eliminate potential differences in fungal growth observed in neutrophil depletion studies (Bonnett et al., 2006, Espinosa et al., 2014, Mehrad et al., 1999, Mircescu et al., 2009). Neutrophil-depleted mice exhibited significantly lower CXCL10 lung levels compared with non-depleted littermates, though CXCL9 lung levels were similar (Figures 1G and 1H). Although CCR2+ Mo-DCs were the major cellular source of CXCL9 and CXCL10 during acute A. fumigatus challenge, neutrophils contributed to CXCL10 production as well.
Distinct Signaling Pathways Promote CXCL9 and CXCL10 Production by Fungus-Infected Myeloid Cells in the Lung
To examine whether fungal uptake by Mo-DCs and neutrophils drives CXCL9 and CXCL10 expression, we infected chimeric REX3 Tg → C57BL6 mice with Alexa 633 (AF633)-labeled conidia and compared RFP and BFP expression in fungus-engaged (AF633+) and bystander (AF633−) leukocytes. Fungus-engaged Mo-DCs and neutrophils had much higher levels of fluorescent transgene expression than corresponding bystander cells had, indicating that fungal uptake promotes Mo-DC CXCL9 and CXCL10 production, neutrophil CXCL10 production (Figures 2A–2E), and, to a lesser extent, monocyte CXCL9 production (Figures S2A and S2B).
Figure 2.

Fungal Uptake and Dectin-1 and Card9 and IFN Signaling Regulate Lung CXCL9 and CXCL10 Levels during A. fumigatus Infection
(A–E) (A) Representative flow cytometry plots and bar graphs (B–E) that indicate the frequency of RFP+ (CXCL9+; red bar in B and C), RFP− (CXCL9−; gray bar in B and C), BFP+ (CXCL10+; blue bar, in D and E), and BFP− (CXCL10−; gray bar in D and E) bystander and fungus-engaged leukocytes isolated from (A, left column) naive or (A, middle and right column, B, and E) infected Rex3 Tg → C57BL6.SJL BM chimeric mice (n = 7) with 3 × 107 AF633-labeled CEA10 conidia. The solid black gates in (A) indicate bystander neutrophils, whereas the solid blue gates indicate fungus-engaged leukocytes. The dashed line in the black and blue gate indicate CXCL9+ and CXCL9− (top quadrants) leukocytes and CXCL10+ and CXCL10− (bottom quadrants) leukocytes.
(F) Lung CXCL9 and (G) CXCL10 levels in naive wild-type (WT, n = 5) and in WT (n = 10), Clec7a−/− (n = 8), Card9−/− (n = 7), Ifnar1−/− (n = 9), and Ifnlr−/− (n = 9) mice 48 h pi with 3 × 107 CEA10 conidia. Data were pooled from two independent experiments.
(B–G) Dots represent individual mice and data are presented as mean ± SEM. Statistical analysis: Mann-Whitney test. See also Figure S2.
Dectin-1 is a major A. fumigatus recognition receptor by binding to β-glucan moieties that are exposed during conidial swelling, the first step in germination and hyphal formation (Brown et al., 2018, Hohl et al., 2005, Steele et al., 2005, Werner et al., 2009). Dectin-1 transduces signals via a cytoplasmic immunoreceptor tyrosine based activation motif (ITAM)-like motif that, upon tyrosine phosphorylation, activates spleen tyrosine kinase and Card9 (Gross et al., 2006, Hsu et al., 2007, Jhingran et al., 2015, Jia et al., 2014, Rogers et al., 2005). To determine whether Dectin-1 and Card9 signaling mediates CXCL9 and CXCL10 production, we infected gene-deficient mice and found that CXCL9, but not CXCL10 production, was significantly attenuated in both Clec7a −/− and Card9 −/− mice (Figures 2F and 2G), directly linking fungal recognition by C-type lectin receptor signaling to CXCL9 production in the lung.
Prior studies have demonstrated that CXCL10 expression can be induced in a variety of cells, including endothelial cells, keratinocytes, fibroblasts, mesangial cells, astrocytes, monocytes, and neutrophils by stimulation with IFN-α, IFN-β, IFN-γ, or lipopolysaccharide (LPS) and in T cells by antigen activation (Colvin et al., 2004, Luster and Ravetch, 1987, Ohmori and Hamilton, 1990, Qian et al., 2007). Since type I and type III IFN signaling are essential for host defense against A. fumigatus (Espinosa et al., 2017), we measured CXCL10 production in IFN-signaling-deficient mice. Unlike CXCL9, lung CXCL10 levels were significantly reduced in Ifnar1 −/− and in Ifnlr −/− mice (Figures 2F and 2G), indicating that distinct upstream signals couple fungal recognition to CXCL9 and CXCL10 production during acute A. fumigatus infection.
Since β-glucan recognition and IFN signaling may impact the number of Mo-DCs and neutrophils in A. fumigatus-infected lungs (Jhingran et al., 2015, Jhingran et al., 2012, Werner et al., 2009), we sorted Mo-DCs and neutrophils from gene-deficient and wild-type mice and quantified cxcl9 and cxcl10 mRNA expression. Preliminary studies that measured cxcl9 and cxcl10 mRNA expression in Mo-DCs and neutrophils isolated from infected wild-type, Clec7a −/−, Ifnar −/−, and Ifnlr −/− mice supported the concept that β-glucan recognition and IFN signaling directly regulate cxcl9 and cxcl10 transcription, respectively (data not shown).
CXCR3 Is Critical for Survival and Fungal Clearance
To assess the relevance of CXCR3 ligand production (Groom et al., 2012) for host outcomes, the survival of Cxcr3 −/− mice was compared with Cxcr3 +/+ controls following A. fumigatus challenge. Cxcr3 −/− mice were significantly more susceptible to challenge compared with controls (Figure 3 A). The heightened susceptibility correlated with an increase in lung fungal burden (Figures 3B, S3A, and S3B) and in lung tissue damage, as determined by comparisons of bronchoalveolar lavage fluid (BALF) lactate dehydrogenase (LDH) (Figure 3C) and albumin (Figure 3D) levels at 48 h pi or 72 h pi. These data linked A. fumigatus-triggered CXCL9 and CXCL10 production by Mo-DCs and neutrophils during acute A. fumigatus challenge to CXCR3-dependent protection and fungal clearance.
Figure 3.

CXCR3 Is Critical for Anti-Aspergillus Defense
(A) Kaplan-Meier survival of C57BL6 (n = 37) and Cxcr3−/− (n = 35) mice challenged with 4–8 × 107 CEA10 conidia. Data from three experiments were pooled.
(B–D) (B) Normalized lung fungal burden, (C) BALF LDH level, and (D) BALF albumin levels in C57BL6 and Cxcr3−/− mice 48 h pi with 3 × 107 CEA10 conidia. (A–D) Dots represent individual mice and data were pooled from 2–4 experiments and presented as mean ± SEM. Statistical analysis: Mann-Whitney test. See also Figure S3.
CXCR3- and CCR2-Dependent Signals Mediate pDC Lung Recruitment
To define the cellular target of CXCR3 signaling, we measured CXCR3 expression on a wide range of leukocytes. BM-resident pDCs from naive mice and lung pDCs from A. fumigatus-infected mice expressed CXCR3, unlike other myeloid cell subsets analyzed (Figure 4 A). As expected, CD4+ T cells, CD8+ T cells, and NK cells expressed CXCR3 (Figure S4A), but mice that lack the lymphoid lineage (Rag2 −/− Il2rg −/−) did not exhibit heightened susceptibility to A. fumigatus challenge (Figure S4B), as reported previously (Espinosa et al., 2014).
Figure 4.

CXCR3+ pDCs Traffic to A. fumigatus-Infected Lungs
(A) CXCR3 surface expression in BM (top row) and lung (bottom row) leukocytes isolated from WT (black lines) or Cxcr3−/− mice (purple lines).
(B) Lung (blue filled dots) and BM (open black dots) pDC numbers at baseline and indicated times pi with 3 × 107 CEA10 conidia (n = 5).
(C) Lung pDC numbers in WT (open black dots) and Cxcr3−/− mice (open purple dots) at 72 h pi (n = 10).
(D) CCR2 surface expression in BM and lung pDCs in WT (black lines) or Ccr2−/− mice (green lines).
(E) Lung pDC numbers in WT (open black dots) and Ccr2−/− mice (open green dots) at 72 h pi (n = 10).
(B, C, and E) Data from 2–3 experiments were pooled and expressed as mean ± SEM. Statistical analysis: Mann-Whitney test. See also Figure S4.
Based on these results and published work (Ramirez-Ortiz et al., 2011), we hypothesized that CXCR3-dependent pDC trafficking is critical for innate antifungal immunity. In prior studies, we did not identify pDCs in lung cell suspensions due to the inclusion of collagenase type IV. This preparation method degraded a pDC epitope utilized for their identification (Figure S4C). Using a collagenase-free method to prepare single-cell lung suspensions, we observed that pDCs started to infiltrate A. fumigatus-infected lungs at 48 h pi, with a peak at 72 h pi (Figure 4B). pDC lung infiltration followed the observed peak in lung CXCL9 and CXCL10 levels and the peak influx of CXCL9- and CXCL10-producing innate immune cells (Figures S4D–S4F). Cxcr3 −/− mice exhibited a ∼50% reduction in lung pDCs (Figure 4C), whereas the number of lung monocytes, Mo-DCs, and neutrophils were similar to control mice at 72 h pi (Figures S4G–S4I). CXCR3 surface expression was reduced on lung pDCs compared with circulating pDCs, consistent with the idea that CXCR3 surface expression is downregulated following engagement by CXCR3 ligands (Figure 4A). To examine whether CXCR3 acts in series or in parallel with other chemokine receptors to mediate pDC trafficking from the BM to the lung, we measured pDC surface expression of additional chemokine receptors in the resting and fungus-infected state. Consistent with prior studies (Fujimura et al., 2015, Sawai et al., 2013, Serbina and Pamer, 2006, Swiecki et al., 2017), we found that CCR2 is expressed by lung-infiltrating pDCs (Figure 4D). pDC lung recruitment was attenuated by 60%–70% in Ccr2 −/− mice at 72 h pi compared with controls (Figure 4E). As expected (Hohl et al., 2009), lung monocyte and Mo-DC numbers were significantly reduced in Ccr2 −/− mice (Figures S4J and S4K), while lung neutrophil accumulation was not affected (Figure S4L). In sum, these data indicate that CXCR3 and CCR2 both mediate pDC trafficking.
Sequential CCR2- and CXCR3-Dependent Signals Mediate pDC Lung Recruitment
To determine at which steps CXCR3 and CCR2 regulate pDC trafficking, we generated mixed BM chimeric mice to compare the trafficking of congenically marked gene-deficient (Cxcr3 −/− or Ccr2 −/−) and gene-sufficient (Cxcr3 +/+ or Ccr2 +/+) pDCs (Figure 5 A). In one model, CXCR3 and CCR2 may act in parallel at the same trafficking step. In this scenario, the ratio of gene-knockout to gene-sufficient pDCs would be increased in the same compartment for both chemokine receptors, prior to the dependent trafficking step. In an alternate model, CXCR3 and CCR2 may act in series at different trafficking steps. In this scenario, the ratio of gene-deficient to gene-sufficient pDCs would be elevated in distinct compartments.
Figure 5.

Sequential CCR2- and CXCR3-Dependent Signals Mediate Lung pDC Recruitment
(A) Experimental scheme to generate mixed BM chimeric mice and compare the trafficking of Cxcr3−/−, Ccr2−/−, and WT pDCs during A. fumigatus infection.
(B and C) Relative frequencies of (B) Cxcr3−/− (open purple bars) and Cxcr3+/+ (WT, open black bars) and (C) Ccr2−/− (open green bars) and Ccr2+/+ (WT, open black bars) pDCs in the BM, blood, and lung of mixed BM chimeric mice 72 h pi. Data from 2–3 experiments were pooled and expressed as mean ± SEM. Statistical analysis: Mann-Whitney test. See also Figure S5.
The ratio of Cxcr3 −/− to Cxcr3 +/+ pDCs was elevated in the blood compared with the BM and lung compartments (Figures 5B and S4A), both during infection and at baseline (Figures 5B and S5A). These data support a model in which CXCR3 primarily, but not exclusively, mediates pDCs trafficking from the circulation into the lung in A. fumigatus-infected mice. In contrast to pDCs, the ratio Cxcr3 −/− to Cxcr3 +/+ neutrophils and monocytes was similar in all compartments analyzed, consistent with CXCR3-independent trafficking (Figures S5B and S5C).
The ratio of Ccr2 −/− to Ccr2 +/+ pDCs was elevated in BM compared with the blood and lung under all conditions examined. These data are consistent with a CCR2-dependent defect in pDC exit from the BM during infection (Figure 5C) and in the steady state (Figure S5D). As anticipated, monocyte BM egress was highly dependent on CCR2 (Figure 5SE), whereas neutrophil BM egress was not (Figure S5F). In sum, these data show that CCR2- and CXCR3-dependent signals act in series to mediate pDC entry into the circulation and pDC egress from the circulation into the lung, respectively.
Lung pDCs Are Essential for Host Defense against A. fumigatus
To link CXCR3 signaling to pDC-dependent antifungal immunity, our model would predict that the chemokine receptor, CXCR3, and its cellular target, the pDC, are both essential for innate antifungal immunity. To test this conjecture, we infected BDCA2-DTR+/− (pDC depleter) mice in which pDCs are ablated at rest and under inflammatory conditions (Swiecki et al., 2010). Following DT treatment, pDCs were depleted fully in the steady state and at 72 h pi, whereas lung-infiltrating monocytes, Mo-DCs, neutrophils, and all other leukocyte subsets examined were not (Figures 6A–6E). In contrast to BDCA2-DTR+/− mice, CCR2-DTR+/− mice facilitated DT-induced ablation of pDCs in addition to other known CCR2+ cells, i.e., monocytes, Mo-DCs, and NK cells (Figures S6A–S6E) (Espinosa et al., 2014, Hohl et al., 2009).
Figure 6.

pDCs Are Critical for Anti-Aspergillus Defense
(A–E) (A) Representative flow cytometry plots of B220+Siglec-H+ pDCs and (B) lung pDC, (C) lung monocyte, (D) lung Mo-DC, and (E) lung neutrophil numbers in DT-treated pDC depleter mice (BDCA2-DTR; open red symbols) and non-Tg littermate controls (open black symbols) at 72 h pi with 3 × 107 CEA10 conidia.
(F) Kaplan-Meier survival of DT-treated pDC depleter (n = 20) and non-Tg littermate control mice (n = 19) infected with 3–6 × 107 CEA10 conidia.
(G and H) (G) BALF LDH levels and (H) fungal burden of DT-treated pDC depleter (open red symbols; n = 6–18) or non-Tg littermates (control, open black symbols; n = 6–17) at 72 h pi with 3 × 107 CEA10 conidia.
(I) Lung fungal burden at 72 h pi with 3 × 107 Af293 conidia.
(J) Lung histopathology (top row, hematoxylin and eosin stain, scale bar: 800 μm; middle row, Gomori methenamine silver stain, scale bar: 800 μm; bottom row, GMS, scale bar: 200 μm) of DT-treated pDC depleter (right column; n = 6) or non-Tg littermates (Control, left column; n = 6) at 72 h pi with 3 × 107 CEA10 conidia.
(K and L) (K) Lung CXCL9 and (L) lung CXCL10 levels in DT-treated pDC depleter mice (red symbols) and non-Tg littermate controls (black symbols) at 72 h pi with 3 × 107 CEA10 conidia.
(B–L) Data were pooled from 2–3 independent experiments and expressed as mean ± SEM. Statistical analysis: Mann-Whitney test. See also Figure S6.
To define the functional role of pDCs during A. fumigatus infection, we compared the mortality, lung LDH levels, lung fungal burden, and histopathology in pDC-depleted mice and in littermate controls. pDC-depleted mice were more susceptible to A. fumigatus challenge than control mice (Figure 6F), and mortality correlated with greater lung damage, as measured by BALF LDH levels (Figure 6G), an increased fungal burden following infection with two commonly used A. fumigatus isolates (Figures 6H and 6I), and a failure to control fungal germination in the lung at 72 h pi (Figure 6J). Importantly, pDCs did not contribute to the production of CXCL9 and CXCL10 in the lung during A. fumigatus challenge, as predicted by prior data (Figures 6K and 6L). In sum, CXCR3+ pDCs were essential for innate antifungal immunity in the lung after A. fumigatus challenge.
Lung pDCs Enhance Neutrophil Fungicidal Activity
To determine how pDCs shape innate antifungal immunity in the lung, we hypothesized that pDCs are critical for antifungal effector functions, since lung neutrophil and Mo-DC numbers were unaffected in pDC-depleted mice. To test this hypothesis, we utilized the fluorescent Aspergillus reporter (FLARE) conidia that encode a dsRed viability fluorophore and are labeled with an Alexa Fluor 633 tracer (AF633). FLARE conidia enable us to distinguish live (DsRed+AF633+) and dead (DsRed−AF633+) conidia during cellular interactions with leukocytes with single-encounter resolution (Figure 7 A). Using the FLARE strain, we quantified leukocyte conidial uptake and killing in pDC-depleted and littermate control mice. pDCs did not bind to or engulf A. fumigatus conidia, as measured by acquisition of AF633 fluorescence (Figures 7B and S7A). pDC ablation did not affect lung neutrophil fungal uptake at 72 h pi compared with lung neutrophils in pDC-sufficient controls (Figures 7C and 7D). However, the frequency of neutrophils that contained live conidia was substantially increased in pDC-depleted mice compared with control mice (Figures 7C and 7E). In other words, neutrophil-engulfed conidia were more likely to be killed in control mice than in pDC-depleted mice (Figures 7C and 7E). pDC ablation did not alter conidial uptake or significantly reduce conidial killing by monocytes and Mo-DCs at 72 h pi (Figures 7D and 7E). These findings indicate that pDCs enhanced neutrophil fungicidal activity.
Figure 7.

Lung pDCs Enhance Neutrophil Fungicidal Activity
(A) Schematic of FLARE strain and changes in fluorescence emission following fungal uptake and killing by host leukocytes.
(B) AF633 fluorescence intensity in indicated lung leukocytes 72 h pi with FLARE (blue line) or AF633-unlabeled conidia.
(C) Representative plots that display dsRed and AF633 fluorescence intensity of lung neutrophils in DT-treated pDC depleter (right panel) and non-Tg littermates (left panel) 72 h pi with 3 × 107 Af293 FLARE conidia. R1 denotes neutrophils that contain live conidia, R2 denotes neutrophils that contain killed conidia.
(D and E) The plots show neutrophil, monocyte, and Mo-DC (D) conidial uptake (R1 + R2) ± SEM and (E) conidial viability (R1/ (R1 + R2) ± SEM in indicated lung leukocytes isolated from DT-treated pDC depleter (red symbols) and non-Tg littermates (black symbols) 72 h pi with 3 × 107 FLARE conidia.
(F and G) (F) Representative plot and (G) mean ± SEM neutrophil ROS production in cells isolated from DT-treated pDC depleter (right panel) and non-Tg littermates (left panel) 72 h pi with 3 × 107 CEA10 conidia.
(C–E) Data from 2 experiments were pooled.
(F and G) Data are representative of two experiments.
(D, E, and G) Dots represent individual mice and data are expressed as mean ± SEM. Statistical analysis: Mann-Whitney test. See also Figure S7.
Neutrophils generate ROS via NADPH oxidase as a primary effector mechanism against A. fumigatus (Marciano et al., 2015). Neutrophil NADPH oxidase induces a regulated cell death process in A. fumigatus conidia (Shlezinger et al., 2017). To determine whether pDCs regulate the neutrophil oxidative burst in A. fumigatus-infected mice, we measured neutrophil ROS production in pDC-depleted mice. The ROS median fluorescence intensity in ROS+ lung neutrophils isolated from pDC-depleted mice was significantly reduced compared with ROS+ neutrophils isolated from littermate control mice at 72 h pi (Figures 7E and 7F). These data show that lung pDCs regulate neutrophil ROS generation during A. fumigatus challenge.
Discussion
Humans maintain lifelong sterilizing immunity to inhaled mold conidia. Breaches in the respiratory innate immune system lead to mold pneumonia, a life-threatening disease in patients with functional or numeric deficits in myeloid cell function, particularly with the loss of the antifungal properties of neutrophils. In this study, we demonstrate a three-cell circuit that involves pDCs, monocytes and derivative Mo-DCs, and neutrophils to regulate sterilizing immunity against Aspergillus conidia. This circuit acts as a feedforward amplification mechanism, by coupling fungal recognition and fungus-induced inflammation to CXCR3 signaling-dependent pDC recruitment in the lung. pDCs regulate neutrophil ROS induction and fungal killing to mediate sterilizing immunity at the respiratory barrier.
Our data demonstrate that conidial uptake by lung-infiltrating monocytes and Mo-DCs triggered Dectin-1- and CARD9-dependent production of CXCL9 and type I and type III IFN signaling-dependent production of CXCL10; the latter process was readily detectable in neutrophils as well. The relative contribution of CXCL9 and CXCL10 and potential molecular redundancy with regard to downstream CXCR3-dependent processes during fungal infection remains an open question for future studies. Prior studies identified a polymorphism in the Cxcl10 gene as a risk factor for IA in allogeneic hematopoietic cell transplant patients (Fisher et al., 2017, Mezger et al., 2008). In this cohort, patients with IA had higher CXCL10 serum levels than controls that did not develop IA, consistent with the idea that pulmonary fungal infection induces CXCL10, similar to findings in mice in this work. Furthermore, the high-risk haplotype was associated with reduced Cxcl10 mRNA expression by immature DCs (iDCs) co-cultured with Aspergillus germlings compared with wild-type iDCs (Mezger et al., 2008). Thus, human iDCs and murine lung Mo-DCs both responded to A. fumigatus challenge by transcribing Cxcl10 mRNA. Although this process is generally regulated by IFN signaling in both mammalian species (O'Connell et al., 2019, Ohmori et al., 1993), a recent study by Rivera and colleagues defined essential roles for type I and type III, but not type II, IFNs in murine host defense against A. fumigatus (Espinosa et al., 2017). One model to account for these findings is that an early wave of type I IFN signaling, detected as early as 6 h pi in the lung and produced primarily by CCR2+ lung-infiltrating monocytes and Mo-DCs (Espinosa et al., 2017), may drive CXCL10 release in the lung in a paracrine or autocrine manner, as observed in this study. In accordance with this model, CXCL10 lung levels that peak at 48 h pi are dependent on type I and type III IFN receptor signal transduction in the Aspergillus-infected murine lung.
Although IFN-dependent and -independent stimuli can induce CXCL9 during microbial infection (Forero et al., 2019, Groom and Luster, 2011), previous studies have not directly linked C-type lectin receptor signaling to fungus-induced CXCL9 production. In murine models of experimental cryptococcosis with a wild-type or a recombinant vaccine strain that expresses human IFN-γ fungal infection led to lung CXCL9 and CXCL10 production, though the identity of chemokine-producing cells was not investigated (Hole et al., 2016, Yamamoto et al., 2014). In this model, Card9 −/− mice had lower lung CXCL9 levels than control mice. This finding was attributed to reduced lung infiltration of IFN-γ-producing NK and CD4+ T cells because Cryptococcus-stimulated BM DCs failed to produce either cytokine in vitro (Yamamoto et al., 2014). In contrast, splenic and central nervous system (CNS)-resident Mo-DCs produced copious levels of CXCL9 and CXCL10 during murine cerebral malaria (Hirako et al., 2016). Because a significant fraction of CXCL9+CXCL10+ splenic Mo-DCs contained hemozoin, a malarial pigment, this study supported the idea that microbial uptake regulated CXCR3 ligand release either directly or indirectly in infected cells. The authors provided evidence for indirect CXCL9 and CXCL10 regulation since their secretion was highly dependent on intact IFN-γ receptor signaling. In the case of respiratory A. fumigatus infection, the dual regulation of CXCL9 by Dectin-1 and Card9 and of CXCL10 by type I and type III IFN signaling indicates both direct and indirect regulation of distinct CXCR3 ligands by a pathogen-associated molecular pattern receptor and soluble type I and type III IFNs.
CXCR3 ligands have been studied extensively in the context of CD4+ and CD8+ T cell trafficking in peripheral lymphoid and non-lymphoid tissues, in part due to high CXCR3 expression on effector and memory T cell subsets, in contrast to naive T cells (Qin et al., 2011). In draining lymph nodes (LNs), antigen-activated and transferred DCs express CXCL9 and CXCL10 and thereby regulate the frequency of IFN-γ+ CD4+ T cells, linking the CXCR3 signaling axis to the formation of effector Th1 cells (Groom et al., 2012). CXCR3 facilitated the entry of effector T cells into otherwise restricted sites, exemplified by CXCR3+ CD8+ T cells infiltration of the CNS or genital tract during murine malaria, West Nile virus, or herpes simplex virus 2 infection (Hirako et al., 2016, Nakanishi et al., 2009, Thapa and Carr, 2009, Thapa et al., 2008). During bacterial infection, a study reported that CXCR3 signaling regulated neutrophil influx to the infected cecum and thus controlled Salmonella extra-intestinal dissemination (Chami et al., 2017). In rodent models of viral (e.g., influenza virus or coronavirus) or chemical acute respiratory distress syndrome, CXCL10+ lung-infiltrating neutrophils and CXCL9+ macrophages mediated the recruitment of tissue-destructive CXCR3+ neutrophils; this pathologic process was ameliorated in Cxcr3 −/− or Cxcl10 −/− mice (Ichikawa et al., 2013). During respiratory fungal challenge, we did not observe CXCR3 expression on lung-infiltrating neutrophils and neutrophil recruitment was unaffected in Cxcr3 −/− mice compared with control mice. In more recent work in the Salmonella model, infected splenic macrophages reside in T cell-sparse granulomas but are surrounded by mononuclear phagocytes that release CXCL9 and CXCL10 to recruit antigen-specific CXCR3+ Th1 cells for bacterial containment (Goldberg et al., 2018). Thus, CXCR3 ligands target both innate and adaptive immune cells in a highly context-dependent manner; this process can result in microbial containment or microbe-elicited tissue damage.
pDCs originate in the BM, circulate in the blood, and traffic to lymphoid and non-lymphoid tissues under homeostatic and inflammatory conditions (Fujimura et al., 2015, Sozzani et al., 2010, Swiecki et al., 2017). However, the mechanism that couples microbial recognition to pDC accumulation at peripheral sites has remained poorly understood. During respiratory fungal infection, neither CCR2+ monocytes nor CCR2− neutrophils had the capacity to respond to CXCR3 ligands because these cells do not express CXCR3. Thus, the recruitment of lung-infiltrating monocytes and pDCs occurs via a pathway that bifurcated after the initial CCR2-dependent egress from the BM reservoir. Although the precise cues that regulate the CXCR3-independent entry of circulating monocytes into the fungus-infected lung remain unknown, integrin-mediated mechanisms enable monocytic influx into hepatic foci of Listeria monocytogenes during systemic infection (Shi et al., 2010).
During other types of lung infections, e.g., influenza and respiratory syncytial virus (RSV), pDCs traffic to the lungs and draining LNs of mice (Langlois and Legge, 2010, Smit et al., 2006, Wang et al., 2006) via pathways that are distinct compared with conventional DCs (Sawai et al., 2013, Sozzani et al., 2010). For example, pDC LN trafficking during murine influenza is regulated primarily by L-selectin (CD62L), with contributions from CXCR3 and CXCR4. Further pDC positioning within the LN is mediated by CCR7 and the release of pDC-derived CXCR3 ligands to enable the formation of clusters of pDCs that interact and activate cells of the adaptive immune system (Krug et al., 2002). In contrast, we did not observe pDC-dependent release of CXCL9 and CXCL10 in the Aspergillus-infected lung, consistent with the model that pDC recruitment cues depends on heterologous myeloid cells. Beyond CXCR3, L-selectin, and CXCR4, the E-selectin ligands, β1 and β2 integrins, CCR5, CCR7, and the chemerin receptor ChemR23 have been implicated in pDC trafficking during states of inflammation (Diacovo et al., 2005, Kohrgruber et al., 2004, Sozzani et al., 2010, Vanbervliet et al., 2003, Wendland et al., 2007), though it has remained unclear how individual cues cooperate to guide distinct pDC trafficking steps, and what the relative strength of individual cues is for defined tissue destinations and types of inflammatory stimuli. The sequential requirements for CCR2 and CXCR3 signaling in distinct pDC trafficking steps during respiratory A. fumigatus infection provides a combinatorial model, in which a more general cue for myeloid cell BM egress (i.e., CCR2 that acts on monocytes and pDCs) is combined with a more specific, pDC-targeted cue (i.e., CXCR3). The latter cue emanates primarily from CCR2+ Mo-DCs that were not equipped to control fungal tissue invasion or license the full spectrum of neutrophil antifungal activity per se.
pDCs were functionally characterized as mediators of antiviral defense (Assil et al., 2019, Swiecki and Colonna, 2010). Recent studies have expanded their role to antibacterial (e.g., Legionella pneumophila, Chlamydia pneumoniae, and Staphylococcus aureus) (Ang et al., 2010, Crother et al., 2012, Parcina et al., 2013) and to antifungal defense (Ang et al., 2010, Hole et al., 2016). pDC-mediated antiviral responses are stimulated upon physical contact with infected cells (Assil et al., 2019). pDCs adhere to infected cells via αLβ2 integrin and ICAM-1 interactions, during which viral RNA is transferred to pDCs, leading to IFN production via the nucleic acid sensor TLR7. This process activates type I IFN-dependent antiviral programs in infected tissues. During respiratory fungal infection, lung-infiltrating pDCs do not interact directly with Aspergillus conidia but instead regulate neutrophils ROS induction. Our study does not exclude the possibility that Aspergillus- or host cell-derived nucleic acids activate pDCs in the lung in situ. Prior work has demonstrated that Aspergillus-derived unmethylated CpG sequences can activate TLR9 signaling in vitro (Herbst et al., 2015, Ramirez-Ortiz et al., 2008). Alternatively, pDCs may recognize fungal Dectin-2 (Clec4n) or Dectin-3 (Clec4e) ligands (Loures et al., 2015, Maldonado and Fitzgerald-Bocarsly, 2017, Preite et al., 2018). In patients with systemic lupus erythematosus, neutrophil-derived extracellular traps (NETs) activate pDCs by releasing DNA-protein complexes, resulting in TLR9-dependent release of pathologic type I IFNs during autoimmunity (Garcia-Romo et al., 2011, Lande et al., 2011). While A. fumigatus conidia are poor inducers of NETosis, products of germination, specifically hyphae that are too large to be phagocytosed, are potent stimuli for NETosis (Bianchi et al., 2009, Bruns et al., 2010, Gibrat et al., 1989). Thus, pDC activation in the fungus-infected lung likely occurs by multiple, yet undefined mechanisms.
The molecular basis for pDC-neutrophil crosstalk represents an important area of future investigation. Two non-exclusive models are likely operative. First, pDCs may form direct contact with neutrophils after arriving at the site of infection. In this model, an individual pDC would interact with many neutrophils since neutrophils are ∼100 times more prevalent in the lung at 48 h pi compared with pDCs. The relative paucity of pDCs, compared with other infiltrating myeloid cells (i.e., neutrophils and Mo-DCs), makes it unlikely that direct pDC-mediated fungistasis, observed at high effector to target cell ratios in the test tube, is physiologically significant in the fungus-infected lung (Loures et al., 2015, Maldonado and Fitzgerald-Bocarsly, 2017). Second, either fungus-triggered or host cell-triggered pDC activation may result in the release of soluble mediators that boost neutrophil fungicidal activity. Previous studies have identified GM-CSF, type I IFN, and type III IFN as important regulators of neutrophil NADPH oxidase activity during respiratory A. fumigatus and B. dermatiditis challenge (Espinosa et al., 2017, Hernandez-Santos et al., 2019, Kasahara et al., 2016). The development of pDC-restricted Cre-lox candidate gene deletion strategies is essential to test these candidates formally.
In this study, we uncover essential functions for pDCs in innate antifungal defense that cannot be compensated by the presence of other myeloid cells, including neutrophils, monocytes, and Mo-DCs. In fact, fungus-engaged Mo-DCs and neutrophils harness pDCs as a feedforward amplification mechanism to enhance innate antifungal activity in the lung by coupling fungal recognition and fungus-induced inflammation to the CXCR3 signaling-dependent recruitment of pDCs into the fungus-infected lung. In the lung pDCs regulate neutrophil ROS induction, a process that induces a regulated cell death process in mold conidia (Shlezinger et al., 2017). Our findings indicate that pDC recovery following administration of chemotherapy and in BM transplant recipients may represent an important variable that affects infectious susceptibility not just to viral but also to fungal pathogens. Recent studies in BM transplant patients indicate that donor pDC reconstitution in the recipient is associated with favorable clinical outcomes, including non-relapse mortality from unrelated donors (Gonçalves et al., 2015, Waller et al., 2019, Waller et al., 2014). These data raise the possibility that pDC recovery in BM transplant patients may be important for controlling systemic fungal infections. Further studies to decipher the mechanistic role of pDCs in antifungal immunity is likely to inform strategies to harness these cells for prophylactic or therapeutic gain in vulnerable patient populations.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC Mouse anti-Mouse CD45.1 (clone A20) | BD Bioscience | Cat# 558701; RRID: AB_1645214 |
| PerCP-Cy™5.5 Mouse anti-Mouse CD45.2 (clone 104) | BD Bioscience | Cat# 552950; RRID: AB_394528 |
| APC-Cy™7 Rat anti-Mouse CD45 (clone 30-F11) | BD Bioscience | Cat# 557659; RRID: AB_396774 |
| PerCP-Cy™5.5 Rat anti-CD11b (clone M1/70) | BD Bioscience | Cat# 550993; RRID: AB_394002 |
| PE-Cy™7 Hamster anti-Mouse CD11c (clone HL3) | BD Bioscience | Cat# 558079; RRID: AB_647251 |
| BV 605™ anti-mouse CD11c Antibody (clone N418) | BioLegend | Cat# 117334; RRID: AB_117334 |
| PE-Cy™7 Rat Anti-Mouse Ly-6C (clone AL-21) | BD Bioscience | Cat# 560593; RRID: AB_1727557 |
| FITC Rat anti-Mouse Ly-6G (clone 1A8) | BD Bioscience | Cat# 551460; RRID: AB_394207 |
| FITC Rat anti-Mouse CD45R/B220 (clone RA3-6B2) | BD Bioscience | Cat# 553087; RRID: AB_394617 |
| PE anti-mouse CD183 Antibody (clone CXCR3-173) | BioLegend | Cat# 126505; RRID: AB_1027656 |
| BV421 Rat Anti-Mouse Siglec-F (clone E50-2440) | BD Bioscience | Cat# 562681; RRID: AB_2722581 |
| BV605 Rat Anti-Mouse Siglec-H (clone 440c) | BD Bioscience | Cat# 747673; RRID: AB_2744234 |
| Purified Rat Anti-Mouse CD16/CD32 (clone 2.4G2) | BD Bioscience | Cat# 553142; RRID: AB_394657 |
| eFluor 450 NK1.1 Monoclonal Antibody (clone PK136) | eBioscience | Cat# 48-5941-80; RRID: AB_2043878 |
| eFluor 450 CD19 Monoclonal Antibody (clone eBio1D3) | eBioscience | Cat# 48-0193-82; RRID: AB_2734905 |
| Alexa Fluor 700, I-A/I-E Monoclonal Antibody (clone M5/114.15.2) | eBioscience | Cat# 56-53-21; RRID: AB_494009 |
| FITC Mouse IgG1 kappa Isotype Control | eBioscience | Cat# 11-4714-42; RRID: AB_10596964 |
| PE Rat IgG2a kappa Isotype Control | eBioscience | Cat# 12-4321-42; RRID: AB_1518773 |
| PerCP-Cyanine5.5 IgG Isotype Control | eBioscience | Cat# 45-4888-80; RRID: AB_906260 |
| APC Rat IgG2b kappa Isotype Control | eBioscience | Cat# 17-4031-82; RRID: AB_470176 |
| APC-eFluor 780 Rat IgG2b kappa Isotype Control | eBioscience | Cat# 47-4031-80; RRID: AB_1272021 |
| Alexa Fluor 700 Rat IgG2b kappa Isotype Control | eBioscience | Cat# 56-4031-80; RRID: AB_837123 |
| PE-Cyanine7 Rat IgG2a kappa Isotype Control | eBioscience | Cat# 25-4321-82; RRID: AB_470200 |
| eFluor 450 Rat IgG1 kappa Isotype Control | eBioscience | Cat# 48-4301-82; RRID: AB_1271984 |
| Bacterial and Virus Strains | ||
| AF293 | Fungal Genetics Stock Center | Cat# A1100 |
| AF293 ds-Red | (Jhingran et al., 2012) | N/A |
| CEA10 (also known as CBS144.89) | (Girardin et al., 1993) | Received from Robert A. Cramer |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Alexa Fluor 633 succinimidyl ester | Invitrogen | Cat# S12375 |
| Collagenase IV | Thermo Fisher Scientific | Cat# LS004189 |
| RPMI-1640 | RPMI-1640 | 21870092 |
| Enrofloxacin | Bayer | Baytril 100 |
| Voriconazole | Pfizer | N/A |
| Tween-20 | Sigma | P9416 |
| Protease Inhibitor Cocktail | Roche | Cat# 11697498001 |
| DNase I, Grade II | Sigma | Cat# 10104159001 |
| BCG albumin assay kit | Sigma | MAK124 |
| Isoflurane | Henry Schein Animal Health | Cat# 29405 |
| Diphtheria toxin (DT) | List Biological Laboratories | Cat# 150 |
| Paraformaldehyde, 32% Solution | Thermo Fisher Scientific | Cat# 50-980-495 |
| CM-H2DCFDA | Thermo Fisher Scientific | Cat# C6827 |
| 10x HBSS | Thermo Fisher Scientific | 14065056 |
| TRIzol-LS | Invitrogen | 10296028 |
| Mouse CXCL9/MIG DuoSet ELISA | R&D Systems | DY492 |
| Mouse CXCL10/IP-10/CRG-2 DuoSet ELISA | R&D Systems | DY466 |
| CytoTox96 non-radioactive cytotoxicity assay kit | Promega | G1780 |
| High Capacity RNA-to-cDNA Kit | Applied Biosystems | 4387406 |
| Ribosomal RNA Control Reagents | Applied Biosystems | 4308329 |
| Cxcl9 ASSAY ID: Mm01345159_m1 | Thermo Fisher | 4448892 |
| Cxcl10 ASSAY ID: Mm00445235_m1 | Thermo Fisher | 4453320 |
| Experimental Models: Organisms/Strains | ||
| Cxcr3-/- | The Jackson Laboratory | JAX: 005796 |
| BDCA2-DTR | The Jackson Laboratory | JAX: 014176 |
| Clec7a-/- | (Saijo et al., 2007) | N/A |
| Card9-/- | (Hsu et al., 2007) | JAX: 028652 |
| Ccr2-/- | The Jackson Laboratory | JAX: 004999 |
| Ifnar1-/- | The Jackson Laboratory | JAX: 32045 |
| Ifnlr-/- | (Lin et al., 2016) | N/A |
| C57BL6 | The Jackson Laboratory | JAX: 000664 |
| C57BL6.SJL | Charles River Laboratories | Stock No. 564 |
| Rag2-/-Il2rg-/- | Taconic | Stock No. 4111 |
| MRP8-Cre-IRES/GFP | The Jackson Laboratory | JAX: 021614 |
| ROSA26 iDTR | The Jackson Laboratory | JAX: 007900 |
| CCR2-DTR | (Hohl et al., 2009) | N/A |
| REX3 Tg | (Groom et al., 2012) | N/A |
| Software and Algorithms | ||
| Prism 8 | Prism 8 | N/A |
| FlowJo 9.9.6 | FlowJo 9.9.6 | N/A |
Resource Availability
Lead Contact
Further information and requests for resources or reagents should be directed to the Lead Contact, Tobias M. Hohl (hohlt@mskcc.org).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate or analyze new datasets or codes.
Experimental Model and Subject Details
Mice
Cxcr3 -/-, C57BL6 (CD45.2+/+), BDCA2-DTR mice were purchased from Jackson Laboratories (JAX: 014176). Rag2 -/- Il2rg -/- mice (Stock No. 4111) were purchased from Taconic. Ccr2 -/- (JAX: 004999) (Serbina and Pamer, 2006), Ifnar1 -/- (JAX: 32045) (Jia et al., 2009), and Ifnlr -/-(Lin et al., 2016) mice were provided by Dr. Eric Pamer (University of Chicago) and Dr. Sergei Kotenko (Rutgers), respectively. Bones from REX3 Tg (CXCL9-Red Red fluorescent protein and CXCL10-Blue fluorescent protein reporter) mice (Groom et al., 2012) were provided by Dr. Andrew Luster (Massachusetts General Hospital). Clec7a -/ - mice (Saijo et al., 2007) were provided by Dr. Shinobu Saijo (University of Tokyo). Card9 -/- mice (Hsu et al., 2007) were provided by Dr. Xin Lin (Tsinghua University). MRP8-Cre-IRES/GFP and ROSA26 iDTR mice were crossed to generate ROSA26-iDTRMrp8-Cre mice. C57BL6 and C57BL6.SJL mice were crossed to generate CD45.1+CD45.2+ recipient mice for mixed BM chimeras. CCR2-DTR+/− (Hohl et al., 2009) and non-transgenic littermates on the Rag2 -/- Il2rg -/- background were maintained on amoxicillin- and Vitamin E-containing chow. All experiments with CCR2-DTR, CCR2-DTR Rag2 -/- Il2rg -/- and BDCA2-DTR mice used co-housed littermate controls. For experiments in which the breeding strategy did not yield littermate controls, gene-knockout mice were co-housed with C57BL6 mice for 14 days prior to infection, whenever possible. All mouse strains were bred and housed in the MSKCC or Rutgers Research Animal Resource Center under specific pathogen-free conditions. All animal experiments were conducted with sex- and age-matched mice and performed with MSKCC or Rutgers Institutional Animal Care and Use Committee approval. Animal studies complied with all applicable provisions established by the Animal Welfare Act and the Public Health Services Policy on the Humane Care and Use of Laboratory Animals.
Generation of Bone Marrow Chimeric Mice
For single BM chimeras, CD45.1+ C57BL6.SJL recipients were lethally irradiated (900cG), reconstituted with either 2-5 × 106 CD45.2+ Rex3-Tg, CD45.2+ ROSA26-iDTRMrp8-Cre, or CD45.2+ non-Cre iDTR littermates BM cells. For mixed BM chimeras, CD45.1+CD45.2+ recipients were irradiated and reconstituted with a 1:1 mixture of CD45.1+ C57BL6.SJL and CD45.2+ Cxcr3 -/- or CD45.2+ Ccr2 -/- BM cells. After BM transplantation, recipient mice received 400 μg/ml enrofloxacin in the drinking water for 21 days to prevent bacterial infections and rested for 6-8 weeks prior to experimental use.
Aspergillus fumigatus culture and Infection Model
A. fumigatus Af293, Af293-dsRed (Jhingran et al., 2012), and CEA10 (Girardin et al., 1993) strains were cultured on glucose minimal medium slants at 37°C for 4-7 days prior to harvesting conidia for experimental use. To generate FLARE conidia, briefly, 7 × 108 Af293-dsRed conidia were rotated in 10 μg/ml Biotin XX, SSE in 1 ml of 50 mM carbonate buffer (pH 8.3) for 2 hr at 4°C, incubated with 20 μg/ml Alexa Fluor 633 succinimidyl ester at 37°C for 1 h, resuspended in PBS and 0.025% Tween 20 for use within 24 hr (Heung et al., 2015, Jhingran et al., 2016, Jhingran et al., 2012). To generate morphologically uniform heat-killed swollen conidia, 5 × 106/ml conidia were incubated at 37°C for 14 hours in RPMI-1640 and 0.5 μg/ml voriconazole and heat killed at 100°C for 30 minutes (Hohl et al., 2005). To infect mice with 30-60 million live or heat-killed A. fumigatus cells, conidia were resuspended in PBS, 0.025% Tween-20 at a concentration of 0.6-1.2 × 109 cells and 50 μl of cell suspension was administered via the intranasal route to mice anesthetized by isoflurane inhalation.
Experimental Details
Analysis of In Vivo and In Vitro Conidial Uptake and Killing
To analyze of conidia uptake and killing, FLARE conidia were used to infect mice (in vivo) or co-culture with BM leukocyte (in vitro). In data analyses for a given leukocyte subset, conidial uptake refers to the frequency of fungus-engaged leukocytes, i.e. the sum of dsRed+AF633+ and dsRed-AF633+ leukocytes. Conidial viability within a specific leukocyte subset refers to the frequency of leukocytes that contain live conidia (dsRed+AF633+) divided by the frequency of all fungus-engaged leukocytes (dsRed+AF633+ and dsRed-AF633+).
In Vivo Cell Depletion
To ablate specific cells, CCR2-DTR, CCR2-DTR, Rag2 -/- Il2rg -/-, ROSA26-iDTRMrp8-Cre, BDCA2-DTR, and non-transgenic littermate controls were injected i.p. with 10 ng/g body weight DT on Day -1, Day 0, and Day +2 pi (Espinosa et al., 2017, Swiecki et al., 2010), unless noted otherwise.
Analysis of Infected Mice
Single cell lung suspensions were prepared for flow cytometry as described in (Hohl et al., 2009), with minor modifications. Briefly, perfused murine lungs were placed in a gentle MACS™ C tube and mechanically homogenized in 5 ml RPMI-1640, 10% FBS, and 0.1 mg/ml DNAse using a gentle MACS™ Octo Dissociator (Miltenyi Biotecc) in the absence of collagenase. Lung cell suspensions were lysed of RBCs, enumerated, and stained with fluorophore-conjugated antibodies prior to flow cytometric analysis on a BD LSR II or flow cytometric sorting on a BD Aria, flow plot analysis was performed using FlowJo v.9.6.6 software.
Neutrophils were identified as CD45+ CD11b+ Ly6Clo Ly6G+ cells, inflammatory monocytes as CD45+ CD11b+ CD11c- Ly6G- Ly6Chi cells, Mo-DCs as CD45+ CD11b+ CD11c+ Ly6G- Ly6Chi MHC class II+ cells, and pDCs as CD45+ CD11cint SiglecF- CD19- NK1.1- CD11b- B220+ SiglecH+ cells.
To analyze the lung fungal burden, perfused murine lungs were homogenized using a PowerGen 125 homogenizer (Fisher) in 2 mL PBS, 0.025% Tween-20, and plated on Sabouraud dextrose agar. To analyze cytokine levels by ELISA, whole lungs were weighed and mechanically homogenized in 2 mL PBS containing protease inhibitor. To analyze cytokine levels by qRT-PCR, total RNA from cells was extracted with TRIzol (Invitrogen). One to two micrograms of total RNA were reverse-transcribed using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). TaqMan Fast Universal Master Mix (2×), TaqMan probes (Applied Biosystems) for each gene were used and normalized to glyceraldehyde-3-phosphate dehydrogenase. Gene expression was calculated using the ΔΔCt method relative to the naïve sample. For histology, perfused lungs were fixed in 4% paraformaldehyde, embedded in paraffin, sectioned in 4 μm slices, stained with hematoxylin and eosin (H&E) or modified Gomori methenamine silver (GMS), and digitally scanned using a Zeiss Mirax Desk Scanner. Images were captured from whole slide images, acquired with an Aperio ScanScope (Aperio Technologies) using 5× and 20× objectives at the Molecular Cytology Core Facility (MSKCC). BAL LDH and albumin levels were measured with a CytoTox96 non-radioactive cytotoxicity assay kit and a BCG albumin assay kit, respectively.
Reactive Oxygen Production
Intracellular ROS levels were measured in cells using CM-H2DCFDA [5-(and 6-) chloromethyl-2,7-dichlorodihydrofluorescein diacetate, acetyl ester] as described in (Espinosa et al., 2017, Hackstein et al., 2012). Briefly, single cell lung suspensions were incubated with 1μM CM-H2DCFDA in Hanks’ balanced salt solution at 37° C for 45 min according to manufacturer’s instruction and analyzed by flow cytometry.
Quantification and Statistical Analysis
Data are representative of at least 2 independent experiments, as indicated. All results are expressed as mean (± SEM), unless stated otherwise. The Mann-Whitney test was used for comparisons of two groups, unless noted otherwise. The Kruskal-Wallis test was used for multi-group comparisons, unless noted otherwise. Survival data was analyzed by long-rank test. All statistical analyses were performed with GraphPad Prism software, v8.2.0.
Acknowledgments
We thank Eric Pamer (University of Chicago) for sharing Ccr2 −/− and Ifnar1 −/− mice, Xin Lin (Tsinghua University) for Card9 −/− mice, and Robert A. Cramer (Dartmouth College) for sharing the A.fumigatus CEA10 strain and for numerous conversations and insights into this study. We thank Franck Barrat, Iliyan Iliev (both Weill Cornell Medical College), and members of the Hohl laboratory for insightful discussions. The studies were supported by Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Diseases Awards (T.M.H. and A.R.) and by NIH grants P30 CA 008748 (to MSKCC), R01 AI 093808 (T.M.H.), R01 AI 139632 (T.M.H.), R01 CA 204028 (A.D.L.), R01 AI 114747 (A.R.), and R01 AI 141368 (A.R.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of manuscript.
Author Contributions
Conceptualization, T.M.H., Y.G., and S.K.; Methodology, T.M.H., Y.G., and S.K.; Investigation, Y.G., S.K., A.J., B.Z., M.A.A., K.A.M.M, V.E., and A.R.; Writing – Original Draft, Y.G. and T.M.H.; Writing – Review & Editing, Y.G. and T.M.H.; Funding Acquisition, T.M.H.; Resources, A.D.L., V.E., and A.R.
Declaration of Interests
The authors declare no competing interests.
Published: June 1, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.chom.2020.05.002.
Supplemental Information
References
- Ang D.K.Y., Oates C.V.L., Schuelein R., Kelly M.K., Sansom F.M., Bourges D., Boon L., Hertzog P.J., Hartland E.L., van Driel I.R. Cutting edge: pulmonary Legionella pneumophila is controlled by plasmacytoid dendritic cells but not type I IFN. J. Immunol. 2010;184:5429–5433. doi: 10.4049/jimmunol.1000128. [DOI] [PubMed] [Google Scholar]
- Assil S., Coléon S., Dong C., Décembre E., Sherry L., Allatif O., Webster B., Dreux M. Plasmacytoid dendritic cells and infected cells form an interferogenic synapse required for antiviral responses. Cell Host Microbe. 2019;25:730–745.e6. doi: 10.1016/j.chom.2019.03.005. [DOI] [PubMed] [Google Scholar]
- Bianchi M., Hakkim A., Brinkmann V., Siler U., Seger R.A., Zychlinsky A., Reichenbach J. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood. 2009;114:2619–2622. doi: 10.1182/blood-2009-05-221606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasius A.L., Giurisato E., Cella M., Schreiber R.D., Shaw A.S., Colonna M. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J. Immunol. 2006;177:3260–3265. doi: 10.4049/jimmunol.177.5.3260. [DOI] [PubMed] [Google Scholar]
- Bonnett C.R., Cornish E.J., Harmsen A.G., Burritt J.B. Early neutrophil recruitment and aggregation in the murine lung inhibit germination of Aspergillus fumigatus conidia. Infect. Immun. 2006;74:6528–6539. doi: 10.1128/IAI.00909-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown G.D., Denning D.W., Gow N.A., Levitz S.M., Netea M.G., White T.C. Hidden killers: human fungal infections. Sci. Transl. Med. 2012;4:165rv13. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- Brown G.D., Willment J.A., Whitehead L. C-type lectins in immunity and homeostasis. Nat. Rev. Immunol. 2018;18:374–389. doi: 10.1038/s41577-018-0004-8. [DOI] [PubMed] [Google Scholar]
- Bruns S., Kniemeyer O., Hasenberg M., Aimanianda V., Nietzsche S., Thywissen A., Jeron A., Latgé J.P., Brakhage A.A., Gunzer M. Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathog. 2010;6:e1000873. doi: 10.1371/journal.ppat.1000873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chami B., Yeung A., Buckland M., Liu H., M Fong G., Tao K., Bao S. CXCR3 plays a critical role for host protection against salmonellosis. Sci. Rep. 2017;7:10181. doi: 10.1038/s41598-017-09150-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin R.A., Campanella G.S.V., Sun J.T., Luster A.D. Intracellular domains of CXCR3 that mediate CXCL9, CXCL10, and CXCL11 function. J. Biol. Chem. 2004;279:30219–30227. doi: 10.1074/jbc.M403595200. [DOI] [PubMed] [Google Scholar]
- Crother T.R., Ma J., Jupelli M., Chiba N., Chen S., Slepenkin A., Alsabeh R., Peterson E., Shimada K., Arditi M. Plasmacytoid dendritic cells play a role for effective innate immune responses during Chlamydia pneumoniae infection in mice. PLoS One. 2012;7:e48655. doi: 10.1371/journal.pone.0048655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diacovo T.G., Blasius A.L., Mak T.W., Cella M., Colonna M. Adhesive mechanisms governing interferon-producing cell recruitment into lymph nodes. J. Exp. Med. 2005;202:687–696. doi: 10.1084/jem.20051035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa V., Dutta O., McElrath C., Du P., Chang Y.J., Cicciarelli B., Pitler A., Whitehead I., Obar J.J., Durbin J.E. Type III interferon is a critical regulator of innate antifungal immunity. Sci. Immunol. 2017;2:eaan5357. doi: 10.1126/sciimmunol.aan5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa V., Jhingran A., Dutta O., Kasahara S., Donnelly R., Du P., Rosenfeld J., Leiner I., Chen C.C., Ron Y. Inflammatory monocytes orchestrate innate antifungal immunity in the lung. PLoS Pathog. 2014;10:e1003940. doi: 10.1371/journal.ppat.1003940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher C.E., Hohl T.M., Fan W., Storer B.E., Levine D.M., Zhao L.P., Martin P.J., Warren E.H., Boeckh M., Hansen J.A. Validation of single nucleotide polymorphisms in invasive aspergillosis following hematopoietic cell transplantation. Blood. 2017;129:2693–2701. doi: 10.1182/blood-2016-10-743294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forero A., Ozarkar S., Li H., Lee C.H., Hemann E.A., Nadjsombati M.S., Hendricks M.R., So L., Green R., Roy C.N. Differential activation of the transcription factor IRF1 underlies the distinct immune responses elicited by type I and Type III interferons. Immunity. 2019;51:451–464.e6. doi: 10.1016/j.immuni.2019.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura N., Xu B., Dalman J., Deng H., Aoyama K., Dalman R.L. CCR2 inhibition sequesters multiple subsets of leukocytes in the bone marrow. Sci. Rep. 2015;5:11664. doi: 10.1038/srep11664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Romo G.S., Caielli S., Vega B., Connolly J., Allantaz F., Xu Z., Punaro M., Baisch J., Guiducci C., Coffman R.L. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 2011;3:73ra20. doi: 10.1126/scitranslmed.3001201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibrat R., Grouzis J.P., Rigaud J., Galtier N., Grignon C. Electrostatic analysis of effects of ions on the inhibition of corn root plasma membrane Mg2+-ATPase by the bivalent orthovanadate. Biochim. Biophys. Acta. 1989;979:46–52. doi: 10.1016/0005-2736(89)90521-x. [DOI] [PubMed] [Google Scholar]
- Girardin H., Latgé J.P., Srikantha T., Morrow B., Soll D.R. Development of DNA probes for fingerprinting Aspergillus fumigatus. J. Clin. Microbiol. 1993;31:1547–1554. doi: 10.1128/jcm.31.6.1547-1554.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg M.F., Roeske E.K., Ward L.N., Pengo T., Dileepan T., Kotov D.I., Jenkins M.K. Salmonella persist in activated macrophages in T cell-sparse granulomas but are contained by surrounding CXCR3 ligand-positioned Th1 cells. Immunity. 2018;49:1090–1102.e7. doi: 10.1016/j.immuni.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves M.V., Yamamoto M., Kimura E.Y., Colturato V.A., de Souza M.P., Mauad M., Ikoma M.V., Novis Y., Rocha V., Ginani V.C. Low counts of plasmacytoid dendritic cells after engraftment are associated with high early mortality after allogeneic stem cell transplantation. Biol. Blood Marrow Transplant. 2015;21:1223–1229. doi: 10.1016/j.bbmt.2015.03.010. [DOI] [PubMed] [Google Scholar]
- Groom J.R., Luster A.D. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol. Cell Biol. 2011;89:207–215. doi: 10.1038/icb.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom J.R., Richmond J., Murooka T.T., Sorensen E.W., Sung J.H., Bankert K., von Andrian U.H., Moon J.J., Mempel T.R., Luster A.D. CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity. 2012;37:1091–1103. doi: 10.1016/j.immuni.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross O., Gewies A., Finger K., Schäfer M., Sparwasser T., Peschel C., Förster I., Ruland J. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature. 2006;442:651–656. doi: 10.1038/nature04926. [DOI] [PubMed] [Google Scholar]
- Hackstein H., Wachtendorf A., Kranz S., Lohmeyer J., Bein G., Baal N. Heterogeneity of respiratory dendritic cell subsets and lymphocyte populations in inbred mouse strains. Respir. Res. 2012;13:94. doi: 10.1186/1465-9921-13-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst S., Shah A., Mazon Moya M., Marzola V., Jensen B., Reed A., Birrell M.A., Saijo S., Mostowy S., Shaunak S., Armstrong-James D. Phagocytosis-dependent activation of a TLR9-BTK-calcineurin-NFAT pathway co-ordinates innate immunity to Aspergillus fumigatus. EMBO Mol. Med. 2015;7:240–258. doi: 10.15252/emmm.201404556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Santos N., Wiesner D.L., Fites J.S., McDermott A.J., Warner T., Wuthrich M., Klein B.S. Lung epithelial cells coordinate innate lymphocytes and immunity against pulmonary fungal infection. Cell Host Microbe. 2019;25:630. doi: 10.1016/j.chom.2019.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heung L.J., Jhingran A., Hohl T.M. Deploying FLAREs to visualize functional outcomes of host-pathogen encounters. PLoS Pathog. 2015;11:e1004912. doi: 10.1371/journal.ppat.1004912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirako I.C., Ataide M.A., Faustino L., Assis P.A., Sorensen E.W., Ueta H., Araújo N.M., Menezes G.B., Luster A.D., Gazzinelli R.T. Splenic differentiation and emergence of CCR5+CXCL9+CXCL10+ monocyte-derived dendritic cells in the brain during cerebral malaria. Nat. Commun. 2016;7:13277. doi: 10.1038/ncomms13277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohl T.M., Rivera A., Lipuma L., Gallegos A., Shi C., Mack M., Pamer E.G. Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe. 2009;6:470–481. doi: 10.1016/j.chom.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohl T.M., Van Epps H.L., Rivera A., Morgan L.A., Chen P.L., Feldmesser M., Pamer E.G. Aspergillus fumigatus triggers inflammatory responses by stage-specific beta-glucan display. PLoS Pathog. 2005;1:e30. doi: 10.1371/journal.ppat.0010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hole C.R., Leopold Wager C.M., Mendiola A.S., Wozniak K.L., Campuzano A., Lin X., Wormley F.L., Jr. Antifungal activity of plasmacytoid dendritic cells against Cryptococcus neoformans in vitro requires expression of Dectin-3 (CLEC4D) and reactive oxygen species. Infect. Immun. 2016;84:2493–2504. doi: 10.1128/IAI.00103-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu Y.M., Zhang Y., You Y., Wang D., Li H., Duramad O., Qin X.F., Dong C., Lin X. The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat. Immunol. 2007;8:198–205. doi: 10.1038/ni1426. [DOI] [PubMed] [Google Scholar]
- Ichikawa A., Kuba K., Morita M., Chida S., Tezuka H., Hara H., Sasaki T., Ohteki T., Ranieri V.M., dos Santos C.C. CXCL10-CXCR3 enhances the development of neutrophil-mediated fulminant lung injury of viral and nonviral origin. Am. J. Respir. Crit. Care Med. 2013;187:65–77. doi: 10.1164/rccm.201203-0508OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhingran A., Kasahara S., Hohl T.M. Flow cytometry of lung and bronchoalveolar lavage fluid cells from mice challenged with fluorescent Aspergillus Reporter (FLARE) conidia. Bio Protoc. 2016;6:e1927. doi: 10.21769/BioProtoc.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhingran A., Kasahara S., Shepardson K.M., Junecko B.A., Heung L.J., Kumasaka D.K., Knoblaugh S.E., Lin X., Kazmierczak B.I., Reinhart T.A. Compartment-specific and sequential role of MyD88 and CARD9 in chemokine induction and innate defense during respiratory fungal infection. PLoS Pathog. 2015;11:e1004589. doi: 10.1371/journal.ppat.1004589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhingran A., Mar K.B., Kumasaka D.K., Knoblaugh S.E., Ngo L.Y., Segal B.H., Iwakura Y., Lowell C.A., Hamerman J.A., Lin X. Tracing conidial fate and measuring host cell antifungal activity using a reporter of microbial viability in the lung. Cell Rep. 2012;2:1762–1773. doi: 10.1016/j.celrep.2012.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia T., Leiner I., Dorothee G., Brandl K., Pamer E.G. MyD88 and Type I interferon receptor-mediated chemokine induction and monocyte recruitment during Listeria monocytogenes infection. J. Immunol. 2009;183:1271–1278. doi: 10.4049/jimmunol.0900460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia X.M., Tang B., Zhu L.L., Liu Y.H., Zhao X.Q., Gorjestani S., Hsu Y.M., Yang L., Guan J.H., Xu G.T., Lin X. CARD9 mediates Dectin-1-induced ERK activation by linking Ras-GRF1 to H-Ras for antifungal immunity. J. Exp. Med. 2014;211:2307–2321. doi: 10.1084/jem.20132349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara S., Jhingran A., Dhingra S., Salem A., Cramer R.A., Hohl T.M. Role of granulocyte-macrophage colony-stimulating factor signaling in regulating neutrophil antifungal activity and the oxidative burst During respiratory fungal challenge. J. Infect. Dis. 2016;213:1289–1298. doi: 10.1093/infdis/jiw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohrgruber N., Gröger M., Meraner P., Kriehuber E., Petzelbauer P., Brandt S., Stingl G., Rot A., Maurer D. Plasmacytoid dendritic cell recruitment by immobilized CXCR3 ligands. J. Immunol. 2004;173:6592–6602. doi: 10.4049/jimmunol.173.11.6592. [DOI] [PubMed] [Google Scholar]
- Krug A., Uppaluri R., Facchetti F., Dorner B.G., Sheehan K.C., Schreiber R.D., Cella M., Colonna M. IFN-producing cells respond to CXCR3 ligands in the presence of CXCL12 and secrete inflammatory chemokines upon activation. J. Immunol. 2002;169:6079–6083. doi: 10.4049/jimmunol.169.11.6079. [DOI] [PubMed] [Google Scholar]
- Lande R., Ganguly D., Facchinetti V., Frasca L., Conrad C., Gregorio J., Meller S., Chamilos G., Sebasigari R., Riccieri V. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 2011;3:73ra19. doi: 10.1126/scitranslmed.3001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois R.A., Legge K.L. Plasmacytoid dendritic cells enhance mortality during lethal influenza infections by eliminating virus-specific CD8 T cells. J. Immunol. 2010;184:4440–4446. doi: 10.4049/jimmunol.0902984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latgé J.P., Chamilos G. Aspergillus fumigatus and Aspergillosis in 2019. Clin. Microbiol. Rev. 2019;33 doi: 10.1128/CMR.00140-18. e00140-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.D., Feng N., Sen A., Balan M., Tseng H.C., McElrath C., Smirnov S.V., Peng J., Yasukawa L.L., Durbin R.K. Distinct roles of type I and Type III interferons in intestinal immunity to homologous and heterologous Rotavirus infections. PLoS Pathog. 2016;12:e1005600. doi: 10.1371/journal.ppat.1005600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionakis M.S., Levitz S.M. Host Control of Fungal Infections: Lessons from basic studies and human cohorts. Annu. Rev. Immunol. 2018;36:157–191. doi: 10.1146/annurev-immunol-042617-053318. [DOI] [PubMed] [Google Scholar]
- Loures F.V., Röhm M., Lee C.K., Santos E., Wang J.P., Specht C.A., Calich V.L., Urban C.F., Levitz S.M. Recognition of Aspergillus fumigatus hyphae by human plasmacytoid dendritic cells is mediated by dectin-2 and results in formation of extracellular traps. PLoS Pathog. 2015;11:e1004643. doi: 10.1371/journal.ppat.1004643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luster A.D., Ravetch J.V. Biochemical characterization of a gamma interferon-inducible cytokine (IP-10) J. Exp. Med. 1987;166:1084–1097. doi: 10.1084/jem.166.4.1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado S., Fitzgerald-Bocarsly P. Antifungal activity of plasmacytoid dendritic cells and the impact of chronic HIV infection. Front. Immunol. 2017;8:1705. doi: 10.3389/fimmu.2017.01705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciano B.E., Spalding C., Fitzgerald A., Mann D., Brown T., Osgood S., Yockey L., Darnell D.N., Barnhart L., Daub J. Common severe infections in chronic granulomatous disease. Clin. Infect. Dis. 2015;60:1176–1183. doi: 10.1093/cid/ciu1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrad B., Strieter R.M., Moore T.A., Tsai W.C., Lira S.A., Standiford T.J. CXC chemokine receptor-2 ligands are necessary components of neutrophil-mediated host defense in invasive pulmonary aspergillosis. J. Immunol. 1999;163:6086–6094. [PubMed] [Google Scholar]
- Mezger M., Steffens M., Beyer M., Manger C., Eberle J., Toliat M.R., Wienker T.F., Ljungman P., Hebart H., Dornbusch H.J. Polymorphisms in the chemokine (C-X-C motif) ligand 10 are associated with invasive aspergillosis after allogeneic stem-cell transplantation and influence CXCL10 expression in monocyte-derived dendritic cells. Blood. 2008;111:534–536. doi: 10.1182/blood-2007-05-090928. [DOI] [PubMed] [Google Scholar]
- Mircescu M.M., Lipuma L., van Rooijen N., Pamer E.G., Hohl T.M. Essential role for neutrophils but not alveolar macrophages at early time points following Aspergillus fumigatus infection. J. Infect. Dis. 2009;200:647–656. doi: 10.1086/600380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi Y., Lu B., Gerard C., Iwasaki A. CD8(+) T lymphocyte mobilization to virus-infected tissue requires CD4(+) T-cell help. Nature. 2009;462:510–513. doi: 10.1038/nature08511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell P., Pepelyayeva Y., Blake M.K., Hyslop S., Crawford R.B., Rizzo M.D., Pereira-Hicks C., Godbehere S., Dale L., Gulick P. SLAMF7 is a critical negative regulator of IFN-alpha-mediated CXCL10 production in chronic HIV infection. J. Immunol. 2019;202:228–238. doi: 10.4049/jimmunol.1800847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmori Y., Hamilton T.A. A macrophage LPS-inducible early gene encodes the murine homologue of IP-10. Biochem. Biophys. Res. Commun. 1990;168:1261–1267. doi: 10.1016/0006-291x(90)91164-n. [DOI] [PubMed] [Google Scholar]
- Ohmori Y., Wyner L., Narumi S., Armstrong D., Stoler M., Hamilton T.A. Tumor necrosis factor-alpha induces cell type and tissue-specific expression of chemoattractant cytokines in vivo. Am. J. Pathol. 1993;142:861–870. [PMC free article] [PubMed] [Google Scholar]
- Parcina M., Miranda-Garcia M.A., Durlanik S., Ziegler S., Over B., Georg P., Foermer S., Ammann S., Hilmi D., Weber K.J. Pathogen-triggered activation of plasmacytoid dendritic cells induces IL-10-producing B cells in response to Staphylococcus aureus. J. Immunol. 2013;190:1591–1602. doi: 10.4049/jimmunol.1201222. [DOI] [PubMed] [Google Scholar]
- Preite N.W., Feriotti C., Souza de Lima D., da Silva B.B., Condino-Neto A., Pontillo A., Calich V.L.G., Loures F.V. The Syk-coupled C-type lectin receptors Dectin-2 and Dectin-3 are involved in Paracoccidioides brasiliensis recognition by human plasmacytoid dendritic cells. Front. Immunol. 2018;9:464. doi: 10.3389/fimmu.2018.00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian C., An H., Yu Y., Liu S., Cao X. TLR agonists induce regulatory dendritic cells to recruit Th1 cells via preferential IP-10 secretion and inhibit Th1 proliferation. Blood. 2007;109:3308–3315. doi: 10.1182/blood-2006-08-040337. [DOI] [PubMed] [Google Scholar]
- Qin X., He Z., Zhao D., Li L., Yuan L. The RANTES gene promoter polymorphisms are associated with the risk of atherothrombotic cerebral infarction in Northern Han Chinese. Clin. Chim. Acta. 2011;412:1112–1115. doi: 10.1016/j.cca.2011.02.033. [DOI] [PubMed] [Google Scholar]
- Ramirez-Ortiz Z.G., Lee C.K., Wang J.P., Boon L., Specht C.A., Levitz S.M. A nonredundant role for plasmacytoid dendritic cells in host defense against the human fungal pathogen Aspergillus fumigatus. Cell Host Microbe. 2011;9:415–424. doi: 10.1016/j.chom.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Ortiz Z.G., Specht C.A., Wang J.P., Lee C.K., Bartholomeu D.C., Gazzinelli R.T., Levitz S.M. Toll-like receptor 9-dependent immune activation by unmethylated CpG motifs in Aspergillus fumigatus DNA. Infect. Immun. 2008;76:2123–2129. doi: 10.1128/IAI.00047-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers N.C., Slack E.C., Edwards A.D., Nolte M.A., Schulz O., Schweighoffer E., Williams D.L., Gordon S., Tybulewicz V.L., Brown G.D. Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity. 2005;22:507–517. doi: 10.1016/j.immuni.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Saijo S., Fujikado N., Furuta T., Chung S.H., Kotaki H., Seki K., Sudo K., Akira S., Adachi Y., Ohno N. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat. Immunol. 2007;8:39–46. doi: 10.1038/ni1425. [DOI] [PubMed] [Google Scholar]
- Sawai C.M., Sisirak V., Ghosh H.S., Hou E.Z., Ceribelli M., Staudt L.M., Reizis B. Transcription factor Runx2 controls the development and migration of plasmacytoid dendritic cells. J. Exp. Med. 2013;210:2151–2159. doi: 10.1084/jem.20130443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serbina N.V., Pamer E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- Shi C., Velázquez P., Hohl T.M., Leiner I., Dustin M.L., Pamer E.G. Monocyte trafficking to hepatic sites of bacterial infection is chemokine independent and directed by focal intercellular adhesion Molecule-1 expression. J. Immunol. 2010;184:6266–6274. doi: 10.4049/jimmunol.0904160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlezinger N., Irmer H., Dhingra S., Beattie S.R., Cramer R.A., Braus G.H., Sharon A., Hohl T.M. Sterilizing immunity in the lung relies on targeting fungal apoptosis-like programmed cell death. Science. 2017;357:1037–1041. doi: 10.1126/science.aan0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit J.J., Rudd B.D., Lukacs N.W. Plasmacytoid dendritic cells inhibit pulmonary immunopathology and promote clearance of respiratory syncytial virus. J. Exp. Med. 2006;203:1153–1159. doi: 10.1084/jem.20052359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sozzani S., Vermi W., Del Prete A., Facchetti F. Trafficking properties of plasmacytoid dendritic cells in health and disease. Trends Immunol. 2010;31:270–277. doi: 10.1016/j.it.2010.05.004. [DOI] [PubMed] [Google Scholar]
- Steele C., Rapaka R.R., Metz A., Pop S.M., Williams D.L., Gordon S., Kolls J.K., Brown G.D. The beta-glucan receptor dectin-1 recognizes specific morphologies of Aspergillus fumigatus. PLoS Pathog. 2005;1:e42. doi: 10.1371/journal.ppat.0010042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiecki M., Colonna M. Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance. Immunol. Rev. 2010;234:142–162. doi: 10.1111/j.0105-2896.2009.00881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiecki M., Gilfillan S., Vermi W., Wang Y.M., Colonna M. Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity. 2010;33:955–966. doi: 10.1016/j.immuni.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiecki M., Miller H.L., Sesti-Costa R., Cella M., Gilfillan S., Colonna M. Microbiota induces tonic CCL2 systemic levels that control pDC trafficking in steady state. Mucosal Immunol. 2017;10:936–945. doi: 10.1038/mi.2016.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapa M., Carr D.J. CXCR3 deficiency increases susceptibility to genital herpes simplex virus type 2 infection: uncoupling of CD8+ T-cell effector function but not migration. J. Virol. 2009;83:9486–9501. doi: 10.1128/JVI.00854-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapa M., Welner R.S., Pelayo R., Carr D.J. CXCL9 and CXCL10 expression are critical for control of genital herpes simplex virus type 2 infection through mobilization of HSV-specific CTL and NK cells to the nervous system. J. Immunol. 2008;180:1098–1106. doi: 10.4049/jimmunol.180.2.1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischler B.Y., Hohl T.M. Menacing mold: recent advances in Aspergillus pathogenesis and host defense. J. Mol. Biol. 2019;431:4229–4246. doi: 10.1016/j.jmb.2019.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanbervliet B., Bendriss-Vermare N., Massacrier C., Homey B., de Bouteiller O., Brière F., Trinchieri G., Caux C. The inducible CXCR3 ligands control plasmacytoid dendritic cell responsiveness to the constitutive chemokine stromal cell-derived factor 1 (SDF-1)/CXCL12. J. Exp. Med. 2003;198:823–830. doi: 10.1084/jem.20020437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller E.K., Logan B.R., Fei M., Lee S.J., Confer D., Howard A., Chandrakasan S., Anasetti C., Fernando S.M., Giver C.R. Kinetics of immune cell reconstitution predict survival in allogeneic bone marrow and G-CSF-mobilized stem cell transplantation. Blood Adv. 2019;3:2250–2263. doi: 10.1182/bloodadvances.2018029892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller E.K., Logan B.R., Harris W.A., Devine S.M., Porter D.L., Mineishi S., McCarty J.M., Gonzalez C.E., Spitzer T.R., Krijanovski O.I. Improved survival after transplantation of more donor plasmacytoid dendritic or naive T cells from unrelated-donor marrow grafts: results from BMTCTN 0201. J. Clin. Oncol. 2014;32:2365–2372. doi: 10.1200/JCO.2013.54.4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Peters N., Schwarze J. Plasmacytoid dendritic cells limit viral replication, pulmonary inflammation, and airway hyperresponsiveness in respiratory syncytial virus infection. J. Immunol. 2006;177:6263–6270. doi: 10.4049/jimmunol.177.9.6263. [DOI] [PubMed] [Google Scholar]
- Wendland M., Czeloth N., Mach N., Malissen B., Kremmer E., Pabst O., Förster R. CCR9 is a homing receptor for plasmacytoid dendritic cells to the small intestine. Proc. Natl. Acad. Sci. USA. 2007;104:6347–6352. doi: 10.1073/pnas.0609180104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner J.L., Metz A.E., Horn D., Schoeb T.R., Hewitt M.M., Schwiebert L.M., Faro-Trindade I., Brown G.D., Steele C. Requisite role for the dectin-1 beta-glucan receptor in pulmonary defense against Aspergillus fumigatus. J. Immunol. 2009;182:4938–4946. doi: 10.4049/jimmunol.0804250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H., Nakamura Y., Sato K., Takahashi Y., Nomura T., Miyasaka T., Ishii K., Hara H., Yamamoto N., Kanno E. Defect of CARD9 leads to impaired accumulation of gamma interferon-producing memory phenotype T cells in lungs and increased susceptibility to pulmonary infection with Cryptococcus neoformans. Infect. Immun. 2014;82:1606–1615. doi: 10.1128/IAI.01089-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate or analyze new datasets or codes.