

Abstract
Fluorescence resonance energy transfer (FRET) is a distance-dependent process by which energy is transferred from an excited donor fluorophore to an acceptor molecule when the donor and acceptor are in close proximity to each other. Depending on the assay design, FRET can provide a real-time measurement of structural integrity and dynamics of biomacromolecules in solution and is particularly suitable for studying G-quadruplex (G4) nucleic acids and their ligand interactions. FRET-based assays are ideally suited for high throughput screening (HTS) methodology because they are simple, sensitive, and easily automated. G4s are stable nucleic acid structures involved in important regulatory roles in gene replication, transcription, and genomic instability. Four-stranded G4s are promising drug targets as these non-canonical structures are enriched in oncogene promoters, 5′ UTRs, and telomeres, and have been linked to regulation of gene expression in cancer and other diseases. Although molecules that bind to G4s, with subsequent influence on gene expression, have been well documented, the identification of new chemical scaffolds that potently and selectively bind to G4s and control specific gene expression are still much less common. Here, we describe a detailed protocol of a FRET-based HTS methodology to identify novel G4 ligands.
Keywords: FRET, G-quadruplex, Drug target, Promoter, Telomere, Ligand
1. Introduction
Four-stranded G-quadruplexes (G4s) are a family of nucleic acid secondary structures consisting of stacked G-tetrad planes stabilized by Hoogsteen hydrogen bonds and monovalent cations such as Na+ and K+ [1, 2]. There is growing evidence indicating that G4-forming sequences are concentrated at biologically relevant regions and play important regulatory roles in gene replication, transcription, and genomic instability [3, 4]. More recently, G4s have been visualized both in DNA and RNA of human cells, and further marked in human regulatory chromatin [5–7]. Such structures have been implicated in the regulation of genes, some of which are necessary for disease pathogenesis, leading to increased attention as potential novel targets for anticancer and anti-HIV agents [8–11]. To date, molecules that bind to G4s and stabilize the G4 secondary structure have been shown to significantly influence transcription and translation of the G4 associated genes [8–14]. The results illustrate the utility of such molecules for disease therapy and emphasize the importance of continued discovery of new chemical scaffolds that potently and selectively for specific G4s [2, 4, 12–15].
Fluorescence resonance energy transfer (FRET) is a dipole-dipole coupling process by which the excited-state energy of a fluorescent donor molecule is non-radiatively transferred to an acceptor molecule [16, 17]. As this effect is distance dependent, generally limited to 10–80 Å, optimization is necessary for placement of donor and acceptor fluorophores to maximize the FRET signal. However, once this step has been performed the assay provides a powerful tool in biomedical research and drug discovery to monitor integrity and dynamics of the target system in real-time [17, 18]. FRET is especially useful for tracking G4 folding and unfolding processes, which can be used to discover G4-interactive small-molecule ligands. For these experiments, a G4-forming nucleic acid is labeled at the 5′ and 3′ ends by a donor and an acceptor fluorophore, with the requirement that the fluorescence emission spectrum of the donor probe overlaps the excitation spectrum of the acceptor probe. Common FRET pairs include a 6-carboxyfluorescein (FAM) donor and the Black Hole Quencher 1 (BHQ1) or 6-carboxy tetramethylrhodamine (TAMRA) as acceptors. Alternative FRET pairs that display desired spectroscopic properties also include FAM-Cy3, Cy3-Cy5, and FAM-rhodamine dyes. The fluorescence intensity of the probe depends on the distance from the fluorophore to the quencher (or from the FRET acceptor) and this distance is a function of the G-quadruplex folding/unfolding process. In solution, without G4-favorable cations K+ or Na+, the G4-forming nucleic acid mainly exists as a single-strand, but the population of the folded G4 can dramatically increase upon addition of G4-interactive small molecules (Fig. 1) or K+ cations. Therefore, in the context of the FAM-BHQ1 pair, the ability to induce G4 formation of small molecules will be reflected by the decrease in fluorescence intensity of FAM relative to the negative control group (probe-only group). This FRET intensity-based assay is a rapid and convenient method suitable for high-throughput screening. Herein, we describe a detailed protocol for high-throughput screening of G4 ligands that are capable of inducing G4 formation from chemical libraries by FRET assay.
Fig. 1.
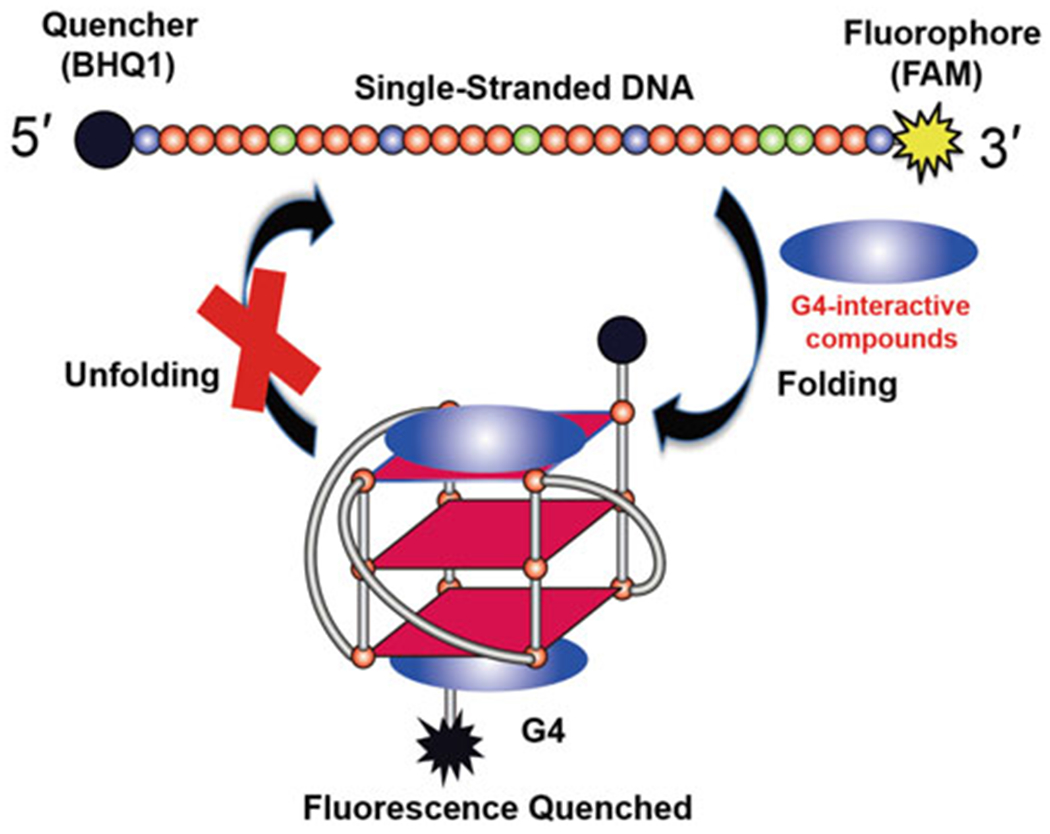
Principle of G4-interactive compounds screening by FRET assay
2. Materials
Oligonucleotide: 5′-BHQ1-TGGGGAGGGTGGGGAGGGTGGGGAAGGTT-FAM-3′ (named Bpu28F) (see Note 1).
384-well optical bottom black plates.
50 mL conical sterile polypropylene centrifuge tubes.
Precise compound transfer equipment (Echo/Access Workstation, see Note 2).
Liquid dispenser and strip washer (MultiFlo, see Note 3).
Plate reader (Synergy Neo2, see Note 4).
Centrifuges.
pH meter.
Diversity-oriented small molecules library (see Note 5). Known drugs, bioactive and natural products library (see Note 6).
100 mM Tris-acetate buffer, pH 7.0.
2 M potassium chloride (KCl) solution.
MilliQ water was used for the preparation of all solutions.
3. Methods
3.1. Sample Preparation and Method Validation
Set the parameters of fluorescence intensity measurement (Synergy Neo2 plate reader; Excitation: 490 nm, with a 10 nm bandwidth; Emission: 520 nm, with a 10 nm bandwidth; Gain: 80; Light source: xenon flash; Data point: 10; Temperature 25 °C; see Note 7). These settings were used for all consecutive scans.
Before the large-scale screening, a small-scale test of DNA probe quality and the instrument conditions was carried out each day to maintain robustness of the assay. Steps 3-7 describe the small-scale test.
The dual labeled Bpu28F probe (0.04 μmol) was dissolved in water to a stock concentration of 100 μM (0.1 mL), which was then diluted in 100 mM Tris-acetate buffer, pH 7.0, to a working concentration of 1 μM in a 10 mL final volume. The 1 μM probe solution was allowed to stand for 1 h at room temperature to reach equilibrium.
The potassium cation that stabilizes G4 structure served as a positive control. A stock of 2 M KCl was diluted in water to a working concentration of 200 mM.
In a black 384-well plate, 10 μL of 1 μM probe solution was added to columns 1 and 2 by MultiFlo. The column 1 (negative control group) was diluted with 10 μL MilliQ water (final well probe concentration/volume: 0.5 μM in 20 μL of 50 mM Tris-acetate buffer, pH 7). The column 2 (positive control group) was diluted with 10 μL 200 mM KCl (final buffer components: 50 mM Tris-acetate, pH 7, KCl 100 mM in 20 μL volume). The plate was centrifuged and incubated for 30 min at room temperature to reach equilibrium.
The fluorescence intensity was measured according to parameters listed in step 1 using the Synergy Neo2 plate reader.
Data analysis was done in Excel with calculation of the mean fluorescence and standard deviation (S.D.) for positive and negative controls, respectively. To illustrate, in this case the negative control group had a mean fluorescence value of 873.8 ± 15.2, and the positive control (100 mM KCl) group had values of 553.1 ± 9.0, respectively. The fluorescence intensity value of the negative control group was normalized to 100%, and a relative fluorescence intensity value of 63.3% was obtained for the positive control group. From our experience, the values of the positive control group ranging from 61% to 65% are acceptable given the day-to-day variation of reagents (Z′ = 0.7). If the small-scale experimental results are acceptable, then a large-scale screen of chemical libraries may be performed using the same reagents and instrument settings. We selected 50,000 screening decks from different libraries.
3.2. High-Throughput Screening of 50,000 Compounds from Different Chemical Libraries
We selected a total of 50,000 compounds from two major chemical libraries (the diversity-oriented small molecule library and the known drugs, bioactive and natural products library). The stock concentration of compounds in the chemical libraries was 1 mM in DMSO.
Add 9.8 μL MilliQ water to columns 3–22 of the black 384-well plates by MultiFlo. Add 10 μL MilliQ water and 200 mM KCl solution into columns 1–2 and 23–24 for negative and positive control groups, respectively (Fig. 2, see Notes 8 and 9).
Using the Access Workstation integrated with an Echo system (Labcyte), transfer 0.2 μL of each compound from libraries to columns 3-22 of the pre-prepared black 384-well plates for a total solution volume of 10 μL. Columns 1–2 and 23–24 are compound-free wells that serve as internal references for each plate.
Centrifuge the plates and incubate for 10 min at room temperature, then measure the fluorescence intensity on the Synergy Neo2 plate reader. The collected data serves as a compound-only control (see Note 10).
Add 10 μL 1 μM Bpu28F probe in 100mM Tris-acetate buffer, pH 7, to the entire black 384-well plates using MultiFlo, then incubate 30 min at room temperature for equilibrium. Final well concentrations/volumes: (1) Columns 1–2 (negative control group), 0.5 μM Bpu28F in 50 mM Tris-acetate, pH 7; (2) Columns 3–22 (sample group), 10 μM small molecule sample, 0.5 μM Bpu28F in 50 mM Tris-acetate, pH 7, 1% v/v DMSO (see Note 11); (3) Columns 23–24 (positive control group), 0.5 μM Bpu28F in 50 mM Tris-acetate, pH 7, 100 mM KCl.
Measure the fluorescence intensity using Synergy Neo2 plate reader.
Data analysis was done in Excel. Average the fluorescence intensity values of negative control groups (columns 1–2) and the positive control groups (columns 23–24) and calculate the standard deviation. Normalize the negative control to 100% fluorescence signal. Compare fluorescence intensity of all sample wells (columns 3–22) to normalized negative control value to obtain percentage of relative fluorescence intensity values for each molecule tested. Hits were defined molecules that provided relative fluorescence intensity values less than 70% compared to negative control. Using this criterion, we identified 329 hits (see Notes 12 and 13). A representative result from one plate using this high-throughput screening method is shown in Fig. 3.
Repeat the FRET experiment for these 329 hit compounds, in triplicate, to exclude the technical errors.
Fig. 2.
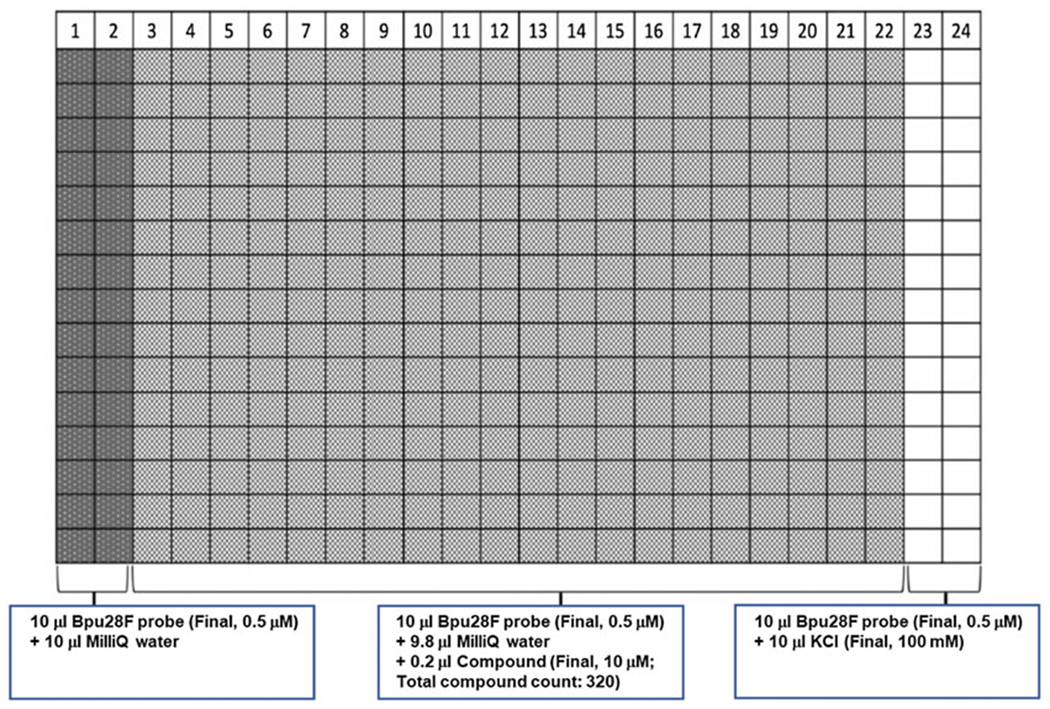
Plate design for the FRET experiment to screen G4-interactive compounds. The first two columns, 1–2, did not contain any compound or KCl, and served as negative controls. Columns 3–22 had different compound in each well. The last two columns, 23–24, had 100 mM KCl and served as positive controls. All wells contained 0.5 μM Bpu28F probe
Fig. 3.
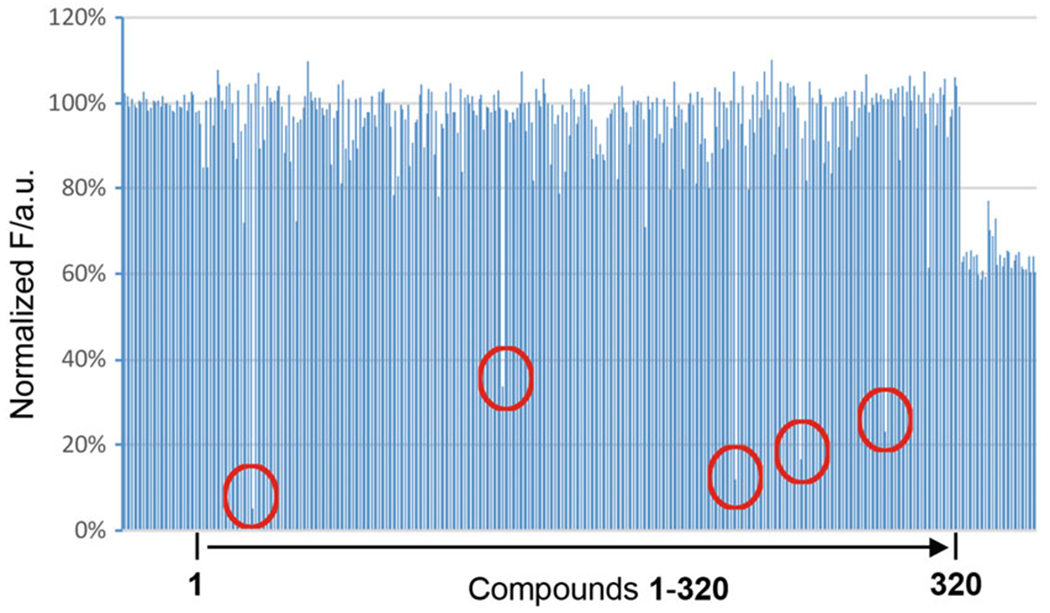
Representative result from one 384-well plate using the high-throughput screening method. Each plate contains 320 compounds. Wells in the first and last two columns (left and right ends in the figure) are negative and positive controls, respectively. There are 5 hits in this plate that are marked by red circles
4. Notes
We synthesized the dual-labeled Bpu28F DNA oligonucleotide, but the labeled DNAs are available from commercial sources. FAM is a common FRET donor. Black Hole Quencher-1 (BHQ-1) is classified as a dark quencher (a non-fluorescent chromophore), which is frequently used as the quencher moiety in a variety of FRET DNA detection probes. The advantages of using BHQ1 in a FRET probe are: (1) low background fluorescence (and thus better signal-to-noise ratio); (2) ease of synthesis for FRET probes with a dark quencher (due to dark quenchers being resistant to degradation during the oligo deprotection step) [19].
Echo uses acoustic energy to precisely transfer droplets (2.5 nL per droplet) of liquid from any well of a source plate to any well of a destination plate in a contact-free manner. The Access Workstation integrates Echo with a series of other devices, such as a plate hotel, a microplate centrifuge, a plate peeler, sealer and a washer/dispenser, so that a complete cycle of preparing an assay plate can be done on one workstation with one controlling program. From our screen, we use the Access Workstation to transfer library compounds to our assay plates.
MultiFlo (Biotek) is a housekeeping liquid dispenser and strip washer which can dispense up to three different reagents without changing dispensing cassettes so that it can avoid switching reagents and thus saving reagents for priming.
Synergy Neo2 HTS multimode microplate reader (Biotek).
The Chemical Genomics Facility at Purdue Institute for Drug Discovery Facility holds a total collection of about 430,000 diversity-oriented small molecules which are comprised by ChemDiv and ChemBridge libraries. The compounds in these libraries are structurally diverse that characterized by “drug-like” and good ADME profiles.
This collection includes LOPAC 1280 (known drugs and bioactives from Sigma), Spectrum 2400 (known drugs, and natural products from MicroSource), 1000 pure natural products extracted from plants and microorganisms, and 5000 semi-synthetic compounds that were synthesized based on the scaffold of natural products but with trackable chemistry.
If the parameters for FRET pairs are not well-established, one needs do excitation wavelength scans and emission wavelength scans to obtain the optimized excitation and emission wavelengths for the FRET pairs.
We recommend using two separate dispensing cassettes to dispense water and KCl. KCl is relatively high in concentration and hard to clean completely from the transfer consumables, and contamination by KCl will dramatically affect the G4 folding process and can cause failure of the experiments.
One can handle as many plates as desired in one day; however, we recommend 30 plates per day.
The compound-only fluorescence intensity control is needed because some compounds may have intrinsic fluorescence that overlaps with the spectrum of the probes. In this case, the detected fluorescence at 520 nM can be polluted by the fluorescence contribution from the test molecule and lead to a false-negative.
We determined that the 1% v/v DMSO did not affect our FRET system, so we did not include DMSO control groups in our experiment for convenience.
One can select the desired hits by defining the hit criteria based on their goals. In our case, we used 70% fluorescence intensity compared to negative controls for hit criteria. This criteria was roughly 5–10% higher than the 100 mM K+ positive control group.
Beside the technical artifacts, there are some cases that are not suitable for the FRET assay described here: (1) as mentioned in Note 10, the intrinsic absorption or fluorescence of test ligands may dramatically affect the FRET screening results. (2) The ligand molecule may interact with FAM rather than the G4 structure. In this case, a decrease of the fluorescence intensity of FAM reflects an interaction with the fluorescent dye, not the G4 structure, generating false-positive ligands. One needs to confirm the G4-interaction with a non-labeled oligonucleotide, using fluorescence independent techniques such as CD, gel electrophoresis, ITC, SPR, and NMR. (3) The small molecule compound may interact with BHQ1, which can hinder the identification of the G4-interactive compounds due to loss the function of BHQ1 acceptor. (4) One may encounter intermediate cases, in which the binding to the labeled oligonucleotide is different than that to the unlabeled oligonucleotide: the presence of fluorescent tags may affect the accessibility of the ligand. Regardless, all hit compounds should be subjected to rigorous secondary assays that are not related to the primary assay for hit validation. It is also suggested to screen all hits with cheminformatics filters to triage any known problematic scaffolds such as pan-assay interference compounds (PAINS) or known aggregators.
Acknowledgments
This research was supported by the National Institutes of Health (R01CA177585 (DY), and P30CA023168 (Purdue Center for Cancer Research)). We thank Dr. Clement Lin, Dr. Buket Onel, and Dr. Jonathan Dickerhoff for helpful discussion and proofreading the manuscript.
References
- 1.Chen Y, Yang D (2012) Sequence, stability, and structure of G-quadruplexes and their interactions with drugs. Curr Protoc Nucleic Acid Chem. Chapter 17: Unit 17.5. 10.1002/0471142700.nc1705s50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neidle S (2017) Quadruplex nucleic acids as targets for anticancer therapeutics. Nat Rev Chem 1(5):0041 [Google Scholar]
- 3.Bochman ML, Paeschke K, Zakian VA (2012) DNA secondary structures: stability and function of G-quadruplex structures. Nat Rev Genet 13(11):770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hänsel-Hertsch R, Di Antonio M, Balasubramanian S (2017) DNA G-quadruplexes in the human genome: detection, functions and therapeutic potential. Nat Rev Mol Cell Biol 18 (5):279. [DOI] [PubMed] [Google Scholar]
- 5.Hänsel-Hertsch R, Beraldi D, Lensing SV, Marsico G, Zyner K, Parry A, Di Antonio M, Pike J, Kimura H, Narita M (2016) G-quadruplex structures mark human regulatory chromatin. Nat Genet 48(10):1267. [DOI] [PubMed] [Google Scholar]
- 6.Biffi G, Tannahill D, McCafferty J, Balasubramanian S (2013) Quantitative visualization of DNA G-quadruplex structures in human cells. Nat Chem 5(3):182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biffi G, Di Antonio M, Tannahill D, Balasubramanian S (2014) Visualization and selective chemical targeting of RNA G-quadruplex structures in the cytoplasm of human cells. Nat Chem 6(1):75–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neidle S (2016) Quadruplex nucleic acids as novel therapeutic targets. J Med Chem 59 (13):5987–6011 [DOI] [PubMed] [Google Scholar]
- 9.Balasubramanian S, Hurley LH, Neidle S (2011) Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat Rev Drug Discov 10(4):261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang K-B, Li D-H, Hu P, Wang W-J, Lin C, Wang J, Lin B, Bai J, Pei Y-H, Jing Y-K (2016) A series of β-carboline alkaloids from the seeds of Peganum harmala show G-quadruplex interactions. Org Lett 18(14):3398–3401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amrane S, Kerkour A, Bedrat A, Vialet B, Andreola M-L, Mergny J-L (2014) Topology of a DNA G-quadruplex structure formed in the HIV-1 promoter: a potential target for anti-HIV drug development. J Am Chem Soc 136(14):5249–5252 [DOI] [PubMed] [Google Scholar]
- 12.Felsenstein KM, Saunders LB, Simmons JK, Leon E, Calabrese DR, Zhang S, Michalowski A, Gareiss P, Mock BA, Schneekloth JS Jr (2015) Small molecule microarrays enable the identification of a selective, quadruplex-binding inhibitor of MYC expression. ACS Chem Biol 11(1):139–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang H-J, Cui Y, Yin H, Scheid A, Hendricks WP, Schmidt J, Sekulic A, Kong D, Trent JM, Gokhale V (2016) A pharmacological chaperone molecule induces cancer cell death by restoring tertiary DNA structures in mutant hTERT promoters. J Am Chem Soc 138 (41):13673–13692 [DOI] [PubMed] [Google Scholar]
- 14.Qin H, Zhao C, Sun Y, Ren J, Qu X (2017) Metallo-supramolecular complexes enantioselectively eradicate cancer stem cells in vivo. J Am Chem Soc 139(45):16201–16209 [DOI] [PubMed] [Google Scholar]
- 15.Xu H, Di Antonio M, McKinney S, Mathew V, Ho B, O’Neil NJ, Dos Santos N, Silvester J, Wei V, Garcia J (2017) CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat Commun 8:14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Főrster T (1959) 10th Spiers Memorial Lecture. Transfer mechanisms of electronic excitation. Discuss Faraday Soc 27:7–17 [Google Scholar]
- 17.Masuko M, Ohuchi S, Sode K, Ohtani H, Shimadzu A (2000) Fluorescence resonance energy transfer from pyrene to perylene labels for nucleic acid hybridization assays under homogeneous solution conditions. Nucleic Acids Res 28(8):e34–e00 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Juskowiak B, Takenaka S (2006) Fluorescence resonance energy transfer in the studies of guanine quadruplexes In: Fluorescent energy transfer nucleic acid probes. Springer, pp 311–341 [DOI] [PubMed] [Google Scholar]
- 19.Yeung AT, Holloway BP, Adams PS, Shipley GL (2004) Evaluation of dual-labeled fluorescent DNA probe purity versus performance in real-time PCR. BioTechniques 36(2):266–275 [DOI] [PubMed] [Google Scholar]